

Abstract
Mitochondrial structures were probably observed microscopically in the 1840s, but the idea of oxidative phosphorylation (OXPHOS) within mitochondria did not appear until the 1930s. The foundation for research into energetics arose from Meyerhof’s experiments on oxidation of lactate in isolated muscles recovering from electrical contractions in an O2 atmosphere. Today, we know that mitochondria are actually reticula and that the energy released from electron pairs being passed along the electron transport chain from NADH to O2 generates a membrane potential and pH gradient of protons that can enter the molecular machine of ATP Synthase to resynthesize ATP. Lactate stands at the crossroads of glycolytic and oxidative energy metabolism. Based on reported research and our own modeling in silico, we contend that lactate is not directly oxidized in the mitochondrial matrix. Instead, the interim glycolytic products (pyruvate and NADH) are held in cytosolic equilibrium with the products of the lactate dehydrogenase (LDH) reaction and the intermediates of the malate-aspartate and glycerol 3-phosphate shuttles. This equilibrium supplies the glycolytic products to the mitochondrial matrix for OXPHOS. LDH in the mitochondrial matrix is not compatible with the cytoplasmic/matrix redox gradient; its presence would drain matrix reducing power and substantially dissipate the proton motive force. OXPHOS requires O2 as the final electron acceptor, but O2 supply is sufficient in most situations, including exercise and often acute illness. Recent studies suggest that atmospheric normoxia may constitute a cellular hyperoxia in mitochondrial disease. As research proceeds appropriate oxygenation levels should be carefully considered.
Keywords: Dysoxia, glycolysis, hypoxia, lactic acid, mitochondria, modeling in silico, NADH shuttles, oxidative phosphorylation, oxygen
Graphical Abstract
Credit for the discovery of what would become known as mitochondria is given to Rudolf Albrecht von Kölliker in 1857; these structures were subsequently described in greater detail by Richard Altmann. In 1898, Benda used a derivation of the Greek words for “thread” and “granule” to name these structures “mitochondria”. In 1907, Fletcher and Hopkins reported the disappearance of lactate in the presence of O2 in previously stimulated muscles. Approximately two decades later, Meyerhof’s work on O2 consumption and lactate (La−) resynthesis into glycogen during the recovery of isolated skeletal muscles from prior contractions was an early hint at the intersection of glycolysis and aerobic phosphorylation. Warburg related these phenomena to the metabolic physiology of cancer. Research by both Meyerhof and Emden led to discovery of the glycolytic pathway. In the 1930s, the work of Lundsgaard, Krebs, Kalckar, the Coris, Belitzer and Szent-Gyorgi, and subsequently Lipmann, Ochoa, Bensley & Hoerr and Claude in the 1940s led to establishing the bioenergetics of glycolysis and the TCA cycle and compounds of high phosphoryl transfer potential. The 1950s heralded the age of research using isolated, functioning mitochondria to explore bioenergetics, and featured prominently the work of Lehninger, Estabrook & Saktor, and Chance & Williams. In the 1960s, Peter Mitchell first proposed the chemiosmotic theory of oxidative phosphorylation, for which he was awarded the Nobel Prize. During this same decade, work by Borst clarified the malate-aspartate shuttle, wherein the exchange of anionic aspartate for undissociated glutamate (one negative charge exported from the matrix per exchange) is driven by the membrane potential (ΔΨ). Work by Skulachev in this decade and beyond further clarified mitochondrial bioenergetics and mitochondrial morphology. Boyer elucidated the nature of the ATP synthase, ultimately winning the Nobel Prize for his work. In the 1980s, David Nicholls further clarified mitochondrial bioenergetics, and the work of George Brooks initiated the era of the Cell-to-Cell Lactate Shuttle. Starting in the 1990s, research emerged suggesting that mitochondria are capable of transporting La− across the inner membrane and oxidizing it without the support of the cytosolic-mitochondrial electron shuttles (i.e., the malate-aspartate and glycerol-3-phosphate shuttles). The ultimate combustion of La− obviously takes place in the mitochondria; there is no question about that simple conclusion. However, our view is that La− is not directly oxidized by LDH in the mitochondrial matrix, but rather La− must first be converted to pyruvate (Pyr−) in the cytosol or intermembrane space. Rationale for this view includes the high activity of the near-equilibrium enzyme LDH, which exceeds glycolytic capacity, the highly oxidized NAD+/NADH ratio relative to the mitochondrial matrix, and the thermodynamic necessity for an energy-driven accumulation of shuttle species (e.g., ΔΨ–dependent aspartate-glutamate exchanger). Modeling in silico demonstrates that an active LDH in the matrix would render mitochondria nearly incapable of oxidizing Pyr−, a result which is inconsistent with decades of studies from hundreds of laboratories using both isolated mitochondria and permeabilized cells in which the mitochondrial reticulum remains intact. Healthy mitochondria function well, even at low O2 levels such that dysoxia is rare and low O2 is likely a minor factor in the increasing concentrations of La− typical with exercise or even many acute critical care situations.
Brief history of glycolytic and oxidative metabolism
For a detailed understanding of mitochondrial lactate metabolism one needs to appreciate the interlinkage of research into glycolysis and oxidative phosphorylation (OXPHOS) and the landmark historical events that led to the discovery of these pathways. Various aspects of the historical events of glycolysis and lactate metabolism have been presented previously (Brooks & Gladden, 2003; Ferguson et al., 2018). Prebble (Prebble, 2010) offers insights and interpretations into these discoveries and Needham (Needham, 1971) provides details of the studies that revealed the entirety of these pathways. A succinct overview of these advances follows.
Glycolysis.
As noted elsewhere (Ferguson et al., 2018), early research that ultimately led to our understanding of glycolysis arose from experiments that were mainly performed using yeast and skeletal muscle. Lehninger (Lehninger, 1970) provided a brief summary, focusing on a handful of important findings out of a multitude that culminated in the elucidation of the glycolytic pathway by 1938. First, Buchner (Buchner, 1897) reported that a cell-free extract of yeast could ferment glucose to ethanol. Subsequently, Meyerhof demonstrated that cell-free extracts of skeletal muscle could metabolize glucose to lactate (Meyerhof, 1927). Another early milestone was the report of Harden and colleagues in 1906 (Harden et al., 1906) describing that alcoholic fermentation in yeast extracts required phosphate and formed a compound that was later identified as fructose 1,6-bisphosphate. Under other experimental conditions this compound was itself utilized, illustrating that it was likely an intermediate in overall fermentation. Harden and coworkers also separated components of the fermentation process that were later identified as: 1) required enzymes, 2) nicotinamide adenine dinucleotide (NAD+), and 3) a mixture of adenine nucleotides, namely AMP, ADP, and ATP. Later experiments by others showed that inhibition of glycolysis by fluoride caused an accumulation of 3-phosphoglycerate and 2-phosphoglycerate, while inhibition of glycolysis by iodoacetate caused an accumulation of fructose 1,6-bisphosphate and triose phosphates. Otto Warburg determined some of the mechanisms of steps in glycolysis as well as the structure of NAD+ (Warburg & Christian, 1936) while Carl and Gerty Cori traced the pathway from glycogen to glucose 6-phosphate (Cori & Cori, 1936; Cori et al., 1938). In the midst of this plethora of findings by numerous researchers, the work of Embden and Meyerhof in separate laboratories has received the greatest recognition. Embden hypothesized the mechanism of fructose 1,6-bisphosphate cleavage and the steps involving NAD+/NADH (Embden et al., 1933), while Meyerhof isolated some of the glycolytic enzymes and determined the reaction sequence from 3-phosphoglycerate to lactate (Meyerhof, 1942). As a result, the glycolytic pathway is also known as the Embden-Meyerhof pathway (Kresge et al., 2005).
From glycolysis to oxidative phosphorylation.
The derivation of oxidative metabolism research from studies of lactate/glycolysis are clearly discernible from Meyerhof’s studies in the 1920s. This is evidenced by his proposition that respiratory oxidation in some unknown process provided the energy for glycogen resynthesis from lactate in isolated muscles following contractions (Meyerhof, 1927; Prebble, 2010). This of course followed from the classic paper of Fletcher and Hopkins in 1907 (Fletcher & Hopkins, 1907) which reported the disappearance of lactate in the presence of O2 in previously stimulated muscles. This lactate disappearance in an O2 atmosphere was in sharp contrast to a further increase in muscle lactate concentration when the muscles were incubated in an anaerobic atmosphere. Concurrently with Meyerhof’s work, several groups discovered the phosphagens which were ultimately identified as phosphocreatine and ATP (Fiske & Subbarow, 1927, 1929; Lohmann, 1929, 1934). By 1930, Lundsgaard (Lundsgaard, 1930), translation quoted in (Prebble, 2010), asserted that the energy for muscle contraction came directly from the splitting of phosphagen and that lactate formation was an anaerobic process that led to resynthesis of phosphagen. This forced Meyerhof to completely reinterpret his concepts of the role of lactate in metabolism. Significantly, Lundsgaard also noted that phosphagen resynthesis could take place aerobically in the absence of lactate formation (Lundsgaard, 1932). As a result, some have said that Lundsgaard was the “first to herald oxidative phosphorylations” (Prebble, 2010). In summary, at the dawn of the 1930s, scientists researching metabolism were aware that carbohydrate synthesis from lactate was powered by aerobic processes and further that the same was true for the phosphorylation of creatine (Prebble, 2010).
In subsequent years, Engelhardt studied metabolism in both mammalian and avian red blood cells (e.g., (Engelhardt & Ljubimova, 1930)). The mammalian cells were entirely glycolytic while the avian cells were capable of aerobic respiration because they apparently contain mitochondria (Stier et al., 2013). Engelhardt concluded that ATP synthesis occurred during respiration and that it was stimulated by the breakdown products of ATP (Engelhardt, 1932). However, it is not clear that Engelhardt distinguished between glycolytic and respiratory oxidation (Prebble, 2010). Later work by Kalckar, the Coris, and Belitzer provided evidence that OXPHOS was separate from glycolytic phosphorylation; this work included the measurement of the P:O ratio by Kalckar (Kalckar, 1937). By the mid-1940s, via the work of Lipmann and Ochoa (Lipmann, 1941; Prebble, 2010), OXPHOS became recognized as a key part of cell metabolism for the provision of energy by way of ATP synthesis (Prebble, 2010).
In summary, the interest and research that led to studies of OXPHOS arose from Meyerhof’s work on O2 consumption and lactate resynthesis into glycogen during the recovery of isolated skeletal muscles from prior contractions. Beyond this, most of the researchers who ultimately established aerobic phosphorylation were in some manner, either directly or indirectly, influenced by Meyerhof’s laboratory (Prebble, 2010). Therefore, as Prebble (Prebble, 2010) concludes, the concept of aerobic phosphorylation arose among a group of biochemists whose primary interest was in skeletal muscle glycolysis.
The next giant step in understanding OXPHOS had its beginnings in the isolation of subcellular particles by Bensley and Hoerr in 1934 (Bensley & Hoerr, 1934), followed by Claude and others in the 1940s (Claude & Fullam, 1945; Prebble, 2010). However, it was Lehninger (Kennedy & Lehninger, 1949; Lehninger, 1964; Prebble, 2010) who refined the isolation of mitochondria via cell fractionation and began the study of OXPHOS at the organelle level. A second major insight was provided by cell biologists using electron micrograph techniques; mitochondria were surrounded by two membranes with the real metabolic action occurring in the inner membrane! A group of biochemists became enamored with these organelles and were called “mitochondriacs” by their colleagues (Prebble, 2010).
As early as the 1910s it was recognized that biological oxidations were closely related to insoluble cellular structures (Ernster & Schatz, 1981). Using extracts of guinea pig liver, Warburg (Warburg, 1913) reported that respiration was linked to particles whose role was to increase the activity of an iron-containing “respiratory enzyme”. Slightly more than 10 years later, Keilin (Keilin, 1925) discovered the cytochromes which presaged recognition of a respiratory chain. In 1946, with technical advancements, Hogeboom and collaborators (Hogeboom et al., 1946) confirmed that succinoxidase and cytochrome oxidase were confined to mitochondria. In the early 1960s, researchers in the Green laboratory isolated four complexes that were later confirmed to reside in the inner mitochondrial membrane (Ernster & Schatz, 1981). These protein assemblies were called Complex I (Hatefi et al., 1961), II (Ziegler & Doeg, 1962), III (Hatefi et al., 1962b), and IV (Fowler et al., 1962). In the same time frame, Hatefi and colleagues (Hatefi et al., 1962a) reconstituted the four complexes in the presence of cytochrome c. This led to today’s current understanding of four complexes that pass electrons from NADH and FADH2 into the electron transport chain (ETC) and ultimately to O2, accompanied by the expulsion of protons from the mitochondrial matrix into the intermembrane space. Recent research into the structure of these complexes suggest that they may interact with each other to form supercomplexes (Guo et al., 2016; Letts & Sazanov, 2017).
Krebs cycle.
A key part of mitochondrial metabolism is the Krebs cycle, or tricarboxylic acid (TCA) cycle (Krebs & Johnson, 1937), and the history of its discovery is succinctly outlined by Lehninger (Lehninger, 1970). First, it was known from the 1910s that anaerobic suspensions of minced animal tissues contained dehydrogenases that transferred hydrogen atoms. In the 1930s, various researchers noted that minced tissue suspensions consumed O2 as they oxidized succinate, fumarate, malate, and citrate to CO2 and H2O. Then, in 1935, Szent-Gyorgyi and co-workers (Annau et al., 1935; Lehninger, 1970) made the vital observation that when small amounts of fumarate, malate, or succinate were added to minced muscle suspensions, O2 consumption was enhanced far in excess of the amount needed to oxidize the added acids to CO2 and H2O. From these results, they concluded that these intermediates were able to stimulate the oxidation of some endogenous substrate within the tissue. From here, Lehninger (Lehninger, 1970) outlines seven key experimental results on minced pigeon flight muscles that led Krebs to postulate the TCA cycle as the major pathway for carbohydrate oxidation in skeletal muscle (Krebs & Johnson, 1937):
The skeletal muscle suspensions oxidized only specific dicarboxylic (succinic, fumaric, malic, oxaloacetic, and α-ketoglutaric) and tricarboxylic (citric, isocitric, and cis-aconitic) acids at very high rates.
Oxidation of endogenous carbohydrate or added pyruvate by the skeletal muscle suspensions was specifically catalyzed by small amounts of succinate, fumarate, malate, oxaloacetate, citrate, cis-aconitate, isocitrate, and 2-oxoglutarate.
When succinate dehydrogenase was inhibited by malonate, stimulation of the oxidation of pyruvate by any of the specific acids denoted in #2 above was also completely blocked.
Small amounts of citrate were formed when the skeletal muscle suspensions were incubated with oxaloacetate and pyruvate under anaerobic conditions; this led Krebs to postulate that the combination of pyruvate and oxaloacetate to form citrate was a missing link in the completion of a cycle of reactions involving the dicarboxylic and tricarboxylic acids.
When succinate dehydrogenase was blocked by malonate followed by addition of citrate, isocitrate, cis-aconitate, or 2-oxoglutarate; succinate accumulated.
Incubation of malonate-poisoned skeletal muscle suspensions also accumulated succinate in the presence of fumarate, malate, or oxaloacetate, meaning that there must be an oxidative pathway for fumarate to be converted to succinate when succinate dehydrogenase is blocked. This suggested a cycle of reactions.
Finally, in malonate-poisoned suspensions, Krebs also found that one molecule of oxaloacetate disappeared for each pyruvate molecule consumed whereas in the uninhibited condition, one molecule of oxaloacetate could stimulate the oxidation of many pyruvate molecules because oxaloacetate is regenerated with each set of reactions; the entire set of reactions constitutes a cycle!
In 1949, Kennedy and Lehninger (Kennedy & Lehninger, 1949) made the critical confirmation that isolated mitochondria could carry out all the above reactions with an O2 consumption rate that accounted for the entire respiration rate of cells.
Control of OXPHOS.
As noted earlier, Engelhardt studied metabolism in both mammalian (entirely glycolytic) and avian (capable of aerobic respiration) red blood cells (e.g., (Engelhardt & Ljubimova, 1930)). Even more importantly in the context of energetics control, he also demonstrated that aerobic ATP synthesis was stimulated by the breakdown products of ATP (Engelhardt, 1932). The importance of this finding was not immediately appreciated (Slater, 1981). By the early 1950s, it was possible to isolate structurally well-preserved, functional mitochondria (Ernster & Schatz, 1981). Subsequently in the early years of that decade, “respiratory control”, defined as control of isolated mitochondrial rate via the availability of inorganic phosphate and ADP, was demonstrated by several laboratories (Ernster & Schatz, 1981). Then, in a series of papers in 1955, Chance and Williams (Chance & Williams, 1955a, b, c, d; Chance et al., 1955) established states 1-5 of the energetic condition of isolated mitochondria. The various states were defined on the basis of the medium to which the mitochondria were exposed and in particular to the rate-limiting factor for O2 consumption and ATP production (e.g., ADP, substrate, O2, or the respiratory chain itself). The respiratory control ratio is specifically defined as the ratio of the respiration rate in state 3 divided by the respiration rate in state 4 and has historically been considered a preferred index of the quality of isolated mitochondrial function (Brand & Nicholls, 2011). State 3 is the respiration rate of mitochondria when substrate is present in the medium, followed by addition of a limited amount of ADP; this elicits an elevated mitochondrial respiratory O2 flux (JO2) which slows as the ADP is phosphorylated to ATP. State 4 is the JO2 once all the added ADP of state 3 has been phosphorylated. Among muscle and exercise physiologists, state 3 is often equated to exercise (muscular contractions; elevated [ADP]; higher JO2) while state 4 is equated to resting conditions (low [ADP]; low JO2).
Chemiosmotic theory.
Given the link between O2 consumption and ATP synthesis, the major remaining question was, “How?”; i.e., the mechanism by which the ATP was formed in OXPHOS. With the understanding of substrate phosphorylation in glycolysis, much investigation was focused on finding a chemical intermediate that stored the energy of oxidation and coupled it to phosphorylation (Prebble, 2001). However, no such intermediate was ever found. Instead, “without a shred of experimental evidence”, Peter Mitchell proposed the chemiosmotic theory (Slater, 1994). In fact, none of the three key tenets of his original proposal had been tested (Prebble, 2001). To be fair, Mitchell’s training (e.g., with Danielli, a proposer of the phospholipid bilayer of biological membranes) and his own research on active transport across membranes, helped him develop underlying ideas of an analogy between translocation and enzyme-catalyzed, group-transfer reactions as well as the notion of a vectorial/directional component to reactions (Slater, 1994). After its proposal in 1961 (Mitchell, 1961), Mitchell’s theory slowly gained acceptance and he was awarded the Nobel Prize in 1978. It is now accepted knowledge (Nicholls & Ferguson, 2013) that the energy of electrons passed down the electron transport chain from NADH and FADH2 in the inner membrane of mitochondria to O2 powers the transfer of protons outwards across the membrane, establishing an electrochemical gradient (pH and electrical membrane potential). Subsequently, the energy of this gradient (a proton motive force) is used to resynthesize ATP.
ATP synthase.
To complete the process, a link was needed between the electrochemical gradient across the inner mitochondrial membrane and the actual resynthesis of ATP. Beginning in the 1950s, Paul Boyer became interested in an enzyme in the mitochondrial membrane, ATP synthase (Allchin, 2002). Along the way, he claimed to have found the long-sought missing intermediate, but he turned out to be wrong (Allchin, 2002), later stating, “I became overly enthusiastic…” and “I should have been more cautious”. In the 1970s, Boyer maintained a conformational hypothesis for oxidative ATP synthesis while discounting Mitchell’s chemiosmotic theory, later calling himself a “holdout”. Ultimately, however, Boyer found that chemiosmotic and conformational concepts were not mutually exclusive but were indeed complementary. Accordingly, in 1997 he shared the Nobel prize with John Walker for discovering the ATP synthase, “A Splendid Molecular Machine” (Boyer, 1997, 2002), so it is now accepted that the ATP synthase is a molecular, rotary engine that mints out ATP as H+ ions move through its turbine across the inner mitochondrial membrane into the mitochondrial matrix.
NADH shuttles.
Most of fuel metabolism results in NADH formation within the mitochondrial matrix where its electrons can be passed readily to Complex I of the electron transport chain. However, this is not the case for NADH that is formed in the cytosol; i.e., from glycolysis. The inner mitochondrial membrane is impermeable to NADH as shown by Lehninger (Lehninger, 1951; Lehninger, 1970). As described by Dawson (Dawson, 1979), given this impermeability, there must be a way to re-oxidize cytosolic NADH within the same compartment by transferring its reducing equivalents to another acceptor. Moreover, the cytosol is orders of magnitude more oxidized than the mitochondrial matrix (Williamson et al., 1967). This means that reducing equivalents on cytosolic NADH must “go uphill” energetically when they are transferred from the cytosol into the matrix. In other words, cytosol-to-matrix electron shuttling requires the input of a substantial thermodynamic driving force. These energetic realities led to the concept of energy driven shuttle systems. While several such shuttles have been proposed (Dawson, 1979), the most well-established of these are the glycerol-3-phosphate shuttle (Estabrook & Sacktor, 1958) and the malate-aspartate shuttle (proposed by Borst in 1963 (Borst, 1963b, a, 2006)). In the malate-aspartate shuttle, the exchange of anionic aspartate for undissociated glutamate (one negative charge exported from the matrix per exchange) is driven by the membrane potential (ΔΨ) (Bremer & Davis, 1975), which at rest is roughly 180 mV, matrix-negative (Nicholls & Ferguson, 2013). In the glycerol-3-phosphate shuttle, the energy driving cytosolic electrons on NADH into the mitochondrion arises from the fact that a cytosolic NAD+-linked oxidation/reduction with a midpoint potential of −320 mV transfers electrons to the ubiquinone pool of the ETC, with a much less negative midpoint potential close to zero (Nicholls & Ferguson, 2013). The glycerol-3-phosphate shuttle therefore transfers electrons into the ETC beyond the 4 protons pumped by Complex I; electron advancement toward O2 is thus not subjected to the immense “backpressure” energy equivalent of 4 protons. The lower P:O yield of the glycerol-3-phosphate shuttle energetically pays for this steep redox gradient favoring electron transfer from cytosol to mitochondrion.
History of mitochondrial structure.
In mitochondria, as in all of physiology and biochemistry, structure informs function. In the 1840s, not long after the discovery of the cell nucleus, there are records of intracellular structures that were probably mitochondria (Ernster & Schatz, 1981). However, Rudolf Albrecht von Kölliker is given credit for discovering mitochondria in 1857 (Schatz, 2013) and they were subsequently described in greater detail by Richard Altmann in 1890 (Altmann, 1890; Ernster & Schatz, 1981; Schatz, 2013). Specifically, Altmann noted their widespread appearance in different cells and referred to them as “bioblasts.” That same year, Retzius coined the term “sarcosomes” to refer specifically to these structures in cardiac muscle (Cleland & Slater, 1953). In 1898, Benda called these structures “mitochondria,” based on a Greek derivation from “mitos” meaning “thread” and “chondros” meaning “granule” (Ernster & Schatz, 1981). In Benda’s view, this was how the mitochondria appeared during spermatogenesis (Benda, 1898). Subsequently, based on electron micrographs, mitochondria were considered to be the elliptical (Kirkwood et al., 1986) “bean-shaped” organelles that permeate illustrations of cell structure and remain today in most biochemistry textbooks. However, between the late 1960’s and 1980s, several researchers had reported that mitochondria appeared as a reticulum, or network, in numerous tissues, including the liver, nephron, and myocardium (summarized by (Kirkwood et al., 1986)), and it was proposed, presciently, that connected mitochondrial networks could facilitate energy distribution throughout the cell in the form of mitochondrial membrane potential (Skulachev, 1969; Skulachev, 1990). Indeed, Skulachev’s group reported in 1978 (Bakeeva et al., 1978) that mitochondria could form physically connected networks across the entire width of the rat diaphragm muscle fiber, and several years later Brooks’s lab (Kirkwood et al., 1986) also observed that mitochondria appeared as a reticulum in rat limb skeletal muscle. However, while Kayar and coworkers (Kayar et al., 1988) found evidence of connected mitochondrial networks in some horse hindlimb muscles, they concluded that mitochondria did not exist as a continuously connected reticulum and suggested that more quantitative assessments of mitochondrial connectivity were needed to adequately address distribution of cellular energy.
Origin, controversy, and debate surrounding the intracellular lactate shuttle
Classic dogma of mitochondrial lactate metabolism.
The currently taught view (Nelson & Cox, 2017) of mitochondrial lactate metabolism follows. First, pyruvate is incorrectly viewed as the end product of glycolysis instead of lactate (Rogatzki et al., 2015; Ferguson et al., 2018). This misconception is derived from a failure to appreciate the extremely high activity of lactate dehydrogenase (LDH) and the fact that the LDH reaction equilibrium is significantly in the direction of lactate (Quistorff & Grunnet, 2011; Rogatzki et al., 2015; Bak & Schousboe, 2017; Ferguson et al., 2018). Second, lactate formation is incorrectly considered to be the primary fate of pyruvate only when O2 levels are low; i.e., hypoxia; this permits the regeneration of NAD+ and the continuation of glycolysis in the presence of O2-limited mitochondrial function. Remarkably, some modern biochemistry textbooks do not even consider the idea of lactate oxidation by mitochondria and instead view lactate disposal to be handled almost entirely via the Cori cycle (Nelson & Cox, 2017). Also, according to current dogma, when the O2 supply is sufficient, pyruvate is transported across the inner mitochondrial membrane by the pyruvate transporter (Bricker et al., 2012; Divakaruni & Murphy, 2012; Herzig et al., 2012) while reducing equivalents (electrons) are transferred across the inner membrane via the malate-aspartate and glycerol-3-phosphate shuttles, with the contribution of each varying according to the tissue type. Pyruvate is subsequently converted to acetyl-CoA for entry into the Krebs cycle while the shuttled cytosolic reducing equivalents enter the mitochondrial respiratory chain. There is no mention of an intracellular lactate shuttle or of the possibility of LDH in the mitochondrial matrix according to this classic dogma.
The basics of an LDH near equilibrium condition.
LDH catalyzes the reversible oxidation/reduction of lactate/pyruvate. Written in the direction of pyruvate (Pyr−) reduction to lactate (La−) the reaction is as follows:
| (1) |
Because myocyte cytosolic pH remains very close to pH 7.0 from resting through moderate exercise intensity conditions, the remainder of this basic introduction will be restricted to pH 7.0, in which case the proton can be omitted:
| (2) |
Written in this direction, the mass action ratio (MAR) of the reaction is:
| (3) |
If the reaction is at equilibrium, then the MAR is equal to the equilibrium constant, Keq:
| (4) |
Krebs and coworkers (Williamson et al., 1967) determined the value of the LDH Keq under physiological conditions and pH 7.0 to be about 9000. The actual free energy change (ΔG) of the reaction can be written as:
| (5) |
where R is the gas constant and T is the absolute temperature. Equation 5 clearly shows that an MAR equal to the Keq gives a ΔG of zero. However, if the MAR < Keq, then ΔG is negative and the reaction can spontaneously advance in the direction of Equation 2 if a pathway exists: pyruvate → lactate. Conversely, if MAR > Keq then a positive ΔG value results and the reaction as we have written it will proceed spontaneously in the opposite direction: lactate → pyruvate. In other words, the LDH reaction can be easily pushed to either lactate → pyruvate or pyruvate → lactate without the requirement for any special process or mechanism.
LDH is a very high activity enzyme.
LDH is a very high activity cytosolic enzyme in many tissues, skeletal muscle being a prime example. The maximal velocity of reaction (Vmax) of LDH in mammalian skeletal muscle is routinely measured at around 500 mmol·min−1·kg muscle−1 (Rasmussen et al., 2002). This immense catalytic potential ensures the maintenance of near equilibrium (MAR ≈ Keq) at rest and during moderate exercise. We will see below that aerobic glycolytic flux in resting human muscle after an overnight fast may be in the vicinity of 5 μmol glucosyl units·min−1·kg−1 (thus, pyruvate and cytosolic NADH each produced at 10 μmol·min−1·kg−1). The LDH Vmax exceeds this flux by 500/0.01 = 50,000-fold! During moderate knee extension exercise aerobic glycolytic flux rises dramatically, by 150-fold, but LDH catalytic potential is still about 340 times higher. Not surprisingly, abundant evidence supports the contention that LDH maintains a near equilibrium state under most conditions (Donovan & Brooks, 1983; Connett, 1987; Katz & Sahlin, 1988; Wolfe et al., 1988). An obvious feature of very high activity, near-equilibrium steps like LDH is that net flux in either direction will promptly and strongly advance in response to small changes in substrate or product concentrations; i.e., small changes in the MAR (Kushmerick, 1998).
Cytosolic lactate/pyruvate and NAD+/NADH ratios.
Routine assays of [La−] and [Pyr−] in resting skeletal muscle yields a ratio (lactate/pyruvate) of about 10-15 (Katz & Sahlin, 1988). Assuming LDH equilibrium and lactate/pyruvate = 10, we can therefore rearrange Eq (4) to estimate the cytosolic redox state:
| (6) |
In marked contrast, the NAD+/NADH in the mitochondrial matrix is closer to 1.0 (Katz & Sahlin, 1988); i.e., the matrix is much more reduced than the cytosol (corresponding redox potentials are: cytosol = −229 mV and matrix = −320 mV). The transfer of electron pairs from the cytosol into the matrix, an obligatory step of aerobic glycolysis, therefore must overcome a substantial thermodynamic “hill” of −91 mV: ΔEh = −320 – (−229) = −91 mV, which, using the Faraday constant converts to a substantial and unfavorable free energy change of +4.2 kcal per mol e− pair. What driving force “pushes” electron pairs up this steep redox gradient? In the malate-aspartate electron shuttle, anionic aspartate is electrophoretically driven from the matrix by the membrane potential, ΔΨ, in exchange for uncharged glutamic acid. In this example, a ΔΨ value of 182 mV (matrix-negative) would provide the minimum required −4.2 kcal of driving force. The exported aspartate transaminates with 2-oxoglutarate, generating cytosolic oxaloacetate, which oxidizes cytosolic NADH to NAD+ at cytosolic malate dehydrogenase and forms malate (Figure 1). The electroneutral exchange of this malate for 2-oxoglutarate export completes the malate-aspartate shuttle. Thus, ΔΨ-driven aspartate thermodynamically accounts for the existence of the redox gradient across the inner membrane and provides the driving force for the uphill uptake of electron pairs produced by the glycolytic pathway (Nicholls & Ferguson, 2013).
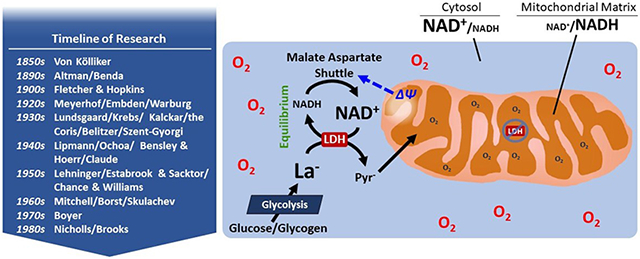
Figure 1.

Schematic representation of the interaction among mitochondrial electron shuttles and mitochondrial lactate (La−) oxidation (Kane, 2014; Ferguson et al., 2018). Mono- and dicarboxylate anions can move between the cytosol and the mitochondrial intermembrane space by crossing the outer mitochondrial membrane via the voltage-dependent anion channels (VDAC). Due to the action of the glycerol phosphate and malate-aspartate shuttles, the cytosolic NAD+/NADH ratio can be orders of magnitude greater than the mitochondrial matrix, but decreases during exercise along with decreasing mitochondrial membrane potential (ΔΨ) spanning the inner membrane (inset; (Sahlin et al., 1987)). The electrogenic transport of glutamate (Glu2−) across the inner mitochondrial membrane via the aspartate-glutamate exchanger (AGE) is a key regulator of mitochondrial lactate oxidation vis-à-vis aerobic glycolysis and the malate-aspartate shuttle. The putative mitochondrial lactate oxidation complex comprised of mLDH, CD147, cytochrome c oxidase, and monocarboxylate transporter is depicted (Hashimoto et al., 2006), as is a matrix mLDH (Brooks et al., 1999). In the text, we argue against the likelihood of LDH in the mitochondrial matrix and suggest that the necessity and/or role of the lactate oxidation complex requires further study. Abbreviations: 2-OG2− 2-oxoglutarate, I Complex I/NADH oxidoreductase of the mitochondrial electron system, II Complex II/succinate dehydrogenase of the mitochondrial electron system, III Complex III of the mitochondrial electron transport system, IV/COX complex IV/cytochrome c oxidase, AAT aspartate aminotransferase, AGE Aspartate-glutamate exchanger, Asp2− aspartate, C cytochrome c, cG3P DH cytosolic glycerol 3-phosphate dehydrogenase, CoA coenzyme A, DHAP2− dihydroxyacetone phosphate, G3P2− glycerol 3-phosphate, Glu2− glutamate, LDH L-lactate dehydrogenase, Mal2− malate, MCT monocarboxylate transporter, MDH malate dehydrogenase, mG3P DH mitochondrial glycerol 3-phosphate dehydrogenase, mLDH mitochondrial lactate dehydrogenase, MOE malate-2-oxoglutarate exchanger, MPC mitochondrial pyruvate carrier, OAA2− oxaloacetate, PDH pyruvate dehydrogenase complex, Pyr− pyruvate, Q quinone, TCA cycle tricarboxylic acid cycle
The transition from rest to heavy exercise demands a number of coordinated adjustments. Mitochondria must consume O2 and produce ATP at rates proportional to the energy demand. Unlike heart, in skeletal muscle this coupled adjustment requires a fall in cellular energy state (ATP free energy, ΔGATP) (Connett & Honig, 1989). While energetic driving forces are falling, however, glycolytic flux dramatically rises, requiring commensurately increased rates of cytosol-mitochondria electron shuttling. It is therefore not surprising that the lactate/pyruvate ratio begins to rise as activation of the glycolytic pathway dramatically accelerates. Rising lactate/pyruvate, and hence falling cytosolic NAD+/NADH, means a more negative cytosolic redox potential, thus diminishing the magnitude of the energetic hill that electron transfer must overcome (Katz & Sahlin, 1988; Connett et al., 1990).
Aerobic glycolysis.
We define aerobic glycolysis to mean that the products of the glycolytic pathway are oxidized to CO2 and H2O by mitochondria. These products are: 1) the electron pairs collected on NAD+ to form cytosolic NADH at glyceraldehyde-3-phosphate dehydrogenase and 2) lactate formed by LDH. However, there is a continuous reversal from lactate back to pyruvate; aerobic glycolysis therefore simply describes a match between the cytosolic net formation rates of these glycolytic products and the mitochondrial rates of their uptake and oxidation. Any mechanism(s) proposed to account for this metabolic coordination must be consistent with the kinetics and thermodynamics of the participating reactions.
Dynamic range of aerobic glycolysis.
After an overnight fast, human skeletal muscle rests at an O2 consumption rate of roughly 0.1 mmol O2·min−1·kg−1 with a respiratory quotient less than 0.80 (Andres et al., 1956), which mathematically converts to an aerobic glycolytic flux of slightly less than 5 μmol glucosyl units·min−1·kg−1. This carbon flow would be attended by a cytosolic-to-mitochondrial electron shuttling rate of roughly 10 μmol NADH electron pairs·min−1·kg−1. Contractile activity imposes severe challenges to the maintenance of this cytosolic-mitochondrial NADH transport. Skeletal muscle possesses an expansive aerobic scope (>100-fold) and myocytes switch fuel selection toward greater reliance on carbohydrate as the ATP turnover rate rises. These two factors multiply to yield an extremely large dynamic range for aerobic glycolysis. For example, the quadriceps of untrained healthy males performing exercise on a knee extension ergometer at 65% of peak aerobic power (~11 W·kg−1 external power output or ~ 167 ml O2·min−1·kg active muscle−1) have glycolytic fluxes around 735 μmol glucosyl units·min−1·kg−1, with net lactate efflux of essentially zero (Helge et al., 2007). With these data we can estimate that mitochondria in untrained skeletal muscle can adjust their rates of pyruvate and cytosolic electron pair uptake and oxidation across at least a 150-fold dynamic range from rest to heavy exercise.
The intracellular lactate shuttle hypothesis.
Abundant data support the contention that the initial products of glycolysis (i.e., pyruvate and cytosolic NADH) equilibrate with lactate and NAD+ more or less completely at the cytosolic LDH reaction; in fact, we contend that lactate rather than pyruvate is the final end product of glycolysis (Rogatzki et al., 2015). Thus, the ultimate combustion of lactate obviously takes place in the mitochondria; there is no question about that simple conclusion. What has raised this issue to the level of controversy in modern times was the published hypothesis and supporting data (Brooks et al., 1999) that mitochondria are capable of transporting lactate across the inner membrane and oxidizing it without the support of the cytosolic-mitochondrial electron shuttles (i.e., the malate-aspartate and glycerol-3-phosphate shuttles). Thus, as articulated by Brooks et al. (Brooks et al., 1999) for striated muscle, this intracellular lactate shuttle posits that lactate can be oxidized by LDH in the mitochondrial matrix. In the intracellular lactate shuttle, neither the malate-aspartate shuttle nor the glycerol-3-phosphate shuttle (Figure 1) would be necessary. Lactate transport into the mitochondrial matrix would simultaneously deliver both pyruvate and cytosolic reducing equivalents from the cytosol into the mitochondrial matrix. In that report (Brooks et al., 1999), mitochondria isolated from rodent skeletal muscle and heart given only saturating ADP, lactate, and malate to prime the TCA cycle, consumed O2 at rates greater than that of pyruvate + malate. Indeed, it was reported that the lactate + malate O2 consumption rate exceeded that of pyruvate + malate by roughly the increment expected on the basis of the additional NADH electron pair from the (matrix) LDH reaction. To be clear, no NAD+ was added to the respiration medium and, thus the only NAD+ pool present in these experiments would be expected to be limited to the mitochondrial matrix. Moreover, in both skeletal and cardiac muscle, oxamate, a well-known LDH inhibitor, both blocked lactate + malate O2 consumption and increased O2 consumption of pyruvate + malate (as would be predicted if matrix LDH were in competition with pyruvate dehydrogenase in a highly reduced compartment, such as the mitochondrial matrix). These data, which provided a new context and implied significance for the term “mitochondrial lactate oxidation,” were not simply surprising and unprecedented; they were fundamentally at odds with contemporary understanding of how glycolysis and mitochondria interact.
Experimental approaches to study the intracellular lactate shuttle in isolated mitochondria.
As early as 1971, the possibility of such a lactate shuttle was suggested by Baba and Sharma (Baba & Sharma, 1971) following the observations of a histochemical association of LDH with mitochondria in heart and skeletal muscle. However, they tempered their conclusions by writing “We observed … no [LDH] activity in the mitochondrial matrix,” and “Permeability of the mitochondria to lactate has not been well-demonstrated, and the lactate shuttle remains a pure speculation.” Subsequently, despite some evidence-based conclusions about the existence of mitochondrial matrix LDH in heart and skeletal muscle (reviewed in (Ferguson et al., 2018)), most groups have failed to identify support for mitochondrial matrix LDH congruent with an intracellular lactate shuttle (Baba & Sharma, 1971; Brandt et al., 1987; Rasmussen et al., 2002; Sahlin et al., 2002; Ponsot et al., 2005; Yoshida et al., 2007; Elustondo et al., 2013; Jacobs et al., 2013; Fulghum et al., 2019; Altinok et al., 2020).
One key issue that is often not clear in these studies is the definition of “mitochondrial LDH”, particularly with respect to whether it means specifically that LDH resides in the mitochondrial matrix. Unfortunately, some studies which have concluded the existence or absence of mitochondrial LDH, and implied the physiological relevance thereof, base such claims on experiments involving the continuous monitoring of added NADH autofluorescence in isolated mitochondria (Valenti et al., 2002; De Bari et al., 2004; Atlante et al., 2007; De Bari et al., 2010; Pizzuto et al.,2012; Passarella et al., 2014; Paventi et al., 2017). As discussed previously (Ferguson et al., 2018). aerobic re-oxidation of externally added NADH has been consistently observed in isolated mitochondria of diverse origin without, or before the addition of any other substrates, including lactate (Rasmussen, 1969; Bernardi & Azzone, 1981; Szczesna-Kaczmarek et al., 1984; Jorgensen et al., 1985; Rasmussen & Rasmussen, 1985; Nohl, 1987; Atlante et al., 1999; Rasmussen et al., 2001; Rasmussen et al., 2003a. b; Abbrescia et al., 2012)). While some of the earlier investigators suggested that this represents structural alterations of the mitochondrial (outer) membrane with isolation (e.g., (Lehninger, 1951; Chance & Williams, 1955c; Maley, 1957)), others have even suggested a physiological role for the pathway of “external NADH oxidation” itself (e.g., (Deshpande et al., 1961; Sottocasa et al., 1967; Rasmussen, 1969; Lofrumento et al., 1991). Ironically, this pathway of exogenous NADH oxidation has been observed to interact with the process of lactate metabolism involving LDH in isolated mitochondria (Deshpande et al., 1961; Szczesna-Kaczmarek et al., 1984). specifically, by reoxidizing NADH for the LDH reaction. Because the mitochondrial inner membrane is impermeable to NAD(+)(H), the observed increase in lactate oxidation with exogenous NADH implies that this LDH is operating outside of the matrix. With respect to the relevance in vivo however, it should be noted that a consistently observed characteristic among lactate-consuming cells is their high malate-aspartate shuttle activity (reviewed in (Kane, 2014)). Indeed, thermodynamic rationale and evidence from modeling in silico are presented below which echo experimental observations linking the malate-aspartate shuttle to aerobic glycolysis.
Subsequent to the proposal of the intracellular lactate shuttle, the Brooks laboratory (Hashimoto et al., 2006), using the techniques of confocal laser scanning microscopy and immunoblotting after immunoprecipitation in L6 skeletal muscle cells, postulated the presence of a lactate oxidation complex in mitochondria. Specifically, they reported evidence suggesting that LDH, monocarboxylate transporter 1, the single-span transmembrane glycoprotein CD147, and cytochrome oxidase are colocalized in the inner mitochondrial membrane. Importantly, they indicated that mitochondrial LDH resides on the outer surface of the inner membrane, a location that does not lead to the criticisms raised for a mitochondrial matrix location for LDH (Figure 1). Unfortunately, the major impact of this nuance has often been overlooked (Chen et al., 2016; Paventi et al., 2017; Young et al., 2020).
Direct uptake and oxidation of lactate by mitochondria at rates equal to or even greater than pyruvate as reported by Brooks et al. (Brooks et al., 1999) was contradictory to prior knowledge. Notably, these results obviated a role for the electron shuttles, in the process eliminating the thermodynamic basis for the large redox gradient known to exist across the inner membrane (Veech et al., 1970). Specifically, the presence of high activities of LDH on both sides of the inner membrane, along with transmembrane transporters for pyruvate and lactate provides no driving force to account for the observed steep redox gradient across the inner membrane. In fact, this intracellular lactate shuttle would create a short-circuit pathway to dissipate the cytosolic-matrix redox gradient generated by the malate-aspartate and glycerol-3-phosphate shuttles, as proposed by Sahlin and colleagues (Sahlin et al., 2002). The reality is that the transmembrane redox gradient is, in fact, observed, and its existence was an important motivating factor to explore the malate-aspartate and glycerol-3-phosphate electron shuttles and their energy dependence. Direct mitochondrial lactate oxidation is thermodynamically inconsistent with well-known observations.
Insights from modeling in silico.
To further explore the thermodynamic consequences of matrix LDH activity, we have adapted the computational model of Wu and coworkers (Wu et al., 2007). We simulated the oxidation of two substrate combinations, pyruvate + malate (10 mM + 2.5 mM) and lactate + malate (10 mM + 2.5 mM). The mitochondrial matrix LDH Vmax parameter was varied from zero to 100% of the pyruvate dehydrogenase (PDH) Vmax (LDH Vmax/PDH Vmax values of 0, 0.01, 0.025, 0.05, 0.10, 0.25, 0.50, 0.75, and 1.0). A simple two substrate - two product rate expression was used to simulate matrix LDH activity with the following KM values for pyruvate, NADH, lactate, and NAD+, respectively: 20 μM, 15 μM, 3.2 mM, and 20 μM. A lactate-specific monocarboxylate transporter with kinetics identical to the pyruvate carrier was also installed in the inner membrane. We emphasize that this approach in silico simulated the behavior of isolated skeletal muscle mitochondria during conventional polarographic assessment of respiration. The respiration buffer contained mitochondria, substrates (malate plus either pyruvate or lactate), and then ADP was added to induce maximum respiration. No NAD+ and no enzymes were present in the respiration buffer medium
All simulations used the same protocol: Mitochondrial protein, 0.1 mg, was pre-incubated with substrates for 180 sec in 2.0 ml of respiration medium. A 1.0 μmol bolus of ADP (0.5 mM final concentration) was then added and model output was followed for an additional 490 sec. In the first simulation, shown in Figure 2A, matrix LDH activity was set to zero and pyruvate + malate substrates were added. As can be seen in Figure 2A, the model accurately simulates what is routinely observed in laboratories around the world: Highly functional mitochondria with state 3 respiration in the vicinity of 400 nmol O2·min−1·mg−1, P:O of roughly 2.5, and respiratory control ratio close to 10. In the second simulation, shown in Figure 2B, lactate + malate is the substrate and matrix LDH Vmax is set equal to PDH Vmax. The model predicts that hypothetical mitochondria with matrix LDH activity equal to pyruvate dehydrogenase would indeed have state 3 rates that exceed pyruvate + malate, as was reported by Brooks et al. in 1999 (Brooks et al., 1999). Further, P:O and respiratory control ratio values would also equal or exceed pyruvate + malate.
Figure 2.

Results of modeling isolated mitochondrial energetics in silico. A) Typical experiment with Pyruvate + Malate, 10 mM + 2.5 mM and zero LDH in the mitochondrial matrix; B) Experiment with Lactate + Malate, 10 mM + 2.5 mM and matrix LDH activity set to equal PDH Vmax activity; C) Experiment with Pyruvate + Malate, 10 mM + 2.5 mM and matrix LDH activity set to equal PDH Vmax activity; D) Simulated state 3 rates with either Pyruvate + Malate or Lactate + Malate as substrates as matrix LDH activity is titrated from 0% to 100% of PDH Vmax. Complete details of the results are provided in the text. Abbreviations: LDH lactate dehydrogenase, PDH pyruvate dehydrogenase, P:O ratio of ATP synthesized to atomic oxygen consumed, RCR respiratory control ratio, JO2 mitochondrial respiratory O2 flux, Vmax maximal reaction velocity
However, the third simulation (Figure 2C) shows that matrix LDH activity dramatically impairs the oxidation of pyruvate + malate! In this simulation, matrix LDH Vmax equals PDH Vmax and pyruvate + malate are the added substrates. In this case, state 3 is only 85 nmol O2 ·min−1·mg−1 or about 23% of what is routinely observed in actual mitochondria experiments (i.e., when LDH activity equals zero, as simulated in Figure 2A). Due to this extremely low rate, P:O and respiratory control ratio cannot be determined. Figure 2D shows the results of a complete titration of matrix LDH/PDH catalytic potential on simulated state 3 rates with either pyruvate + malate or lactate + malate addition. Simply stated, Figure 2D shows that pyruvate and lactate oxidation are mutually exclusive; progressively higher matrix LDH activity would increase direct mitochondrial lactate oxidation, but it would also progressively impair the capacity to oxidize pyruvate to rates far below those routinely measured in laboratories. The simulation results therefore strongly reject the idea of matrix LDH activity in skeletal muscle mitochondria.
Figure 3 is a schematic diagram illustrating the effect of hypothetical matrix LDH activity on the metabolism of lactate (Figure 3A) and pyruvate (Figure 3B). In both scenarios it is important to remember the thermodynamics of the LDH reaction. Because the LDH equilibrium constant (Keq) favors lactate and NAD+ formation, net lactate → pyruvate flux can only proceed when matrix [Pyr−] and [NADH] are sufficiently low. This requirement is not a problem during state 3 respiration when the extramitochondrial energy state is unphysiologically low (saturating ADP) and there is therefore much less “backpressure” opposing oxidative phosphorylation. As a result, as shown in Figure 3A, the LDH reaction advances toward net pyruvate and NADH formation and these products, in turn, fuel flux through pyruvate dehydrogenase, the TCA cycle, the ETC, and the synthesis/export of ATP. Moreover, the low matrix [Pyr−] minimizes the loss of pyruvate carbon to the buffer via the mitochondrial pyruvate carrier. In dramatic contrast, Figure 3B illustrates the effect that matrix LDH would have when pyruvate is the fuel added to the buffer. In this case, the LDH Keq dictates the accumulation of extremely high matrix [La−] and [NAD+]. As a result, a substantial reduction of pyruvate to lactate proceeds at the expense of NADH generated by the TCA cycle. When pyruvate is the added fuel, the overall impact of matrix LDH is therefore the oxidation of the matrix (decreased “redox pressure” down the ETC) and the export of pyruvate carbon as lactate via the monocarboxylate transporter. These de-energizing effects of matrix LDH are especially evident when the buffer ATP/ADP ratio is maintained in a physiologically relevant range; i.e., the region of control between states 3 and 4 (not shown). Overall, these results in silico demonstrate that an active LDH in the matrix would render mitochondria nearly incapable of oxidizing pyruvate, a result which is inconsistent with decades of studies from hundreds of laboratories using both isolated mitochondria and permeabilized cells in which the mitochondrial reticulum remains intact.
Figure 3.

A) Schematic diagram showing the effect of hypothetical matrix LDH on the oxidation of Lactate. Green lines show the path of energy conservation and ATP production. Red line shows small loss of matrix pyruvate to buffer via the mitochondrial pyruvate carrier. B) Schematic diagram showing the effect of hypothetical matrix LDH on the oxidation of pyruvate. Green lines show the path of energy conservation and ATP production. Red lines show loss of matrix redox pressure (LDH catalyzes the oxidation of NADH produced by the TCA cycle) and export of pyruvate carbon as lactate due to LDH activity and a monocarboxylate transporter. Abbreviations:, C V Complex V, ETC electron transport chain, LDH lactate dehydrogenase, MCT monocarboxylate transporter, MPC mitochondrial pyruvate carrier, PDH pyruvate dehydrogenase, TCA tricarboxylic acid cycle
Modern view of mitochondrial structure and its implications.
Recently, networks of many adjacent, physically connected mitochondria were reported in mouse heart, and oxidative and glycolytic skeletal muscles and researchers have meticulously shown that these networks of mitochondria allow rapid cellular energy distribution through conduction of the mitochondrial membrane potential (Glancy et al., 2015; Glancy et al., 2017; Bleck et al., 2018). However, in these studies, connectivity of the mitochondrial power grid varied by cell type. For example, oxidative skeletal muscle fibers have fewer, larger reticula and are more connected than glycolytic fibers (Bleck et al., 2018). These differences likely reflect diverse approaches to balancing the energetic support system relative to the overall cell functions. The specific configuration of mitochondrial networks is critical to the cellular energy conversion process as the ability of a mitochondrion to provide the energy needed to support cellular function depends, in part, on its capacity to receive necessary substrates (e.g., fuel and O2) and to deliver its products (e.g., ATP) where needed. Thus, the amount, size, shape, and location of mitochondria all play a key role in determining mitochondrial functional capacity by regulating the spatial interactions among energetic sources and sinks within the cell. Mitochondrial content (the total amount of mitochondria in the cell, measured as volume density or number in imaging-based methods) results from a balance between mitochondrial biogenesis (Wu et al., 1999), mitophagy (Lemasters, 2005), and the import and degradation of individual proteins and lipids, the latter of which is the dominant mechanism in striated muscles (Karunadharma et al., 2015). The size of individual mitochondria is regulated in part by the balance between the well-known mitochondrial fission and fusion mechanisms (Nunnari et al., 1997) which split or merge mitochondria, respectively, and the frequency of these dynamic events can vary greatly by cell type. The infrequent mitochondrial fusion in adult muscle fibers, for instance, occurs ten-fold less often than in developing myotubes (Eisner et al., 2014). In the context of this review, larger, healthy mitochondria would, in theory, increase the capacity for mitochondrial lactate oxidation per mitochondrion.
How mitochondrial shape is regulated is not yet clear; simple fission and fusion cannot account for the large variation in mitochondrial shapes found within cells. Mitochondria can be observed as large or small spheres or as thick or thin tubules that are either straight or branched, sometimes all within the same cell (Bleck et al., 2018). Irregular mitochondrial structures such as nanotubes (Vincent et al., 2017) or donuts (Bleck et al., 2018) have been reported, though the functional implications are currently not well understood. Elongated mitochondrial shapes have relatively greater surface area-to-volume ratios than more compact shapes, and thus, are better equipped to interact with the surrounding environment which may be important for interactions between mitochondria and glycolytic outputs such as lactate. Mitochondria isolated from cells are spherical in nature (Hackenbrock, 1966) suggesting that cellular components such as the cytoskeleton may be involved in regulating the more complex mitochondrial shapes found within cells. Indeed, mitochondria have been shown to interact with microtubules and motor proteins for trafficking around the cell (Wang & Schwarz, 2009) and to undergo a calcium-dependent mitochondrial shape transition (Nemani et al., 2018) independent of fission or fusion.
Interactions among mitochondria and with other organelles or cellular structures are also likely to regulate mitochondrial function within cells (Murley & Nunnari, 2016). When two mitochondria come in close proximity with one another they can form specialized intermitochondrial junctions (Bakeeva et al., 1978; Glancy et al., 2015) which may involve cristae alignment of both mitochondria (Picard et al., 2015) and have been suggested to facilitate conduction of the mitochondrial membrane potential (Bakeeva et al., 1978; Glancy et al., 2015; Bleck et al., 2018). Mitochondrial interactions with endoplasmic reticulum and other organelles are known to occur at dedicated contact sites between the membranes of each organelle and have been shown to permit direct exchange and communication of signals and metabolites (Murley & Nunnari, 2016). Glycolytic enzymes such as hexokinase, enolase, and glyceraldehyde 3-phosphate dehydrogenase can also anchor to mitochondria though this appears to occur as a means to regulate apoptosis rather than to directly channel glycolytic flux into mitochondria (Majewski et al., 2004; Tristan et al., 2011; Gao et al., 2014). Enzymes of the cytosolic portion of the malate-aspartate and the glycerol-3-phosphate shuttles are located outside of the mitochondria (Figure 1) though cytosolic malate dehydrogenase is listed as part of MitoCarta 2.0 just as are LDH and four other glycolytic enzymes (hexokinase, glyceraldehyde 3-phosphate dehydrogenase, triose phosphate isomerase, and pyruvate kinase (Calvo et al., 2016).
The internal structure of a mitochondrion also plays a large part in determining capacity for mitochondrial energy conversion and is likely the structural aspect most relevant to mitochondrial lactate oxidation. The mitochondrial outer membrane contains voltage-dependent anion channels and other transporters that permit the flux of pyruvate, lactate, and NAD(+)(H) into and out of the intermembrane space, and its size and shape is regulated largely as described above. Within the mitochondrion is an inner membrane containing both tortuous cristae which can extend along the width of the mitochondrion and an inner boundary membrane which is closely associated to the outer mitochondrial membrane (Frey & Mannella, 2000). The inner membrane is the site of OXPHOS and glycerol 3-phosphate dehydrogenase, and also contains transporters such as the mitochondrial pyruvate carrier and putative monocarboxylate transporter (Ferguson et al., 2018). However, an intact inner membrane is not permeable to NAD(+)(H). Regulation of mitochondrial inner membrane structure is performed by the mitochondrial contact site and cristae organizing system complex of proteins and functions in tandem with the mitochondrial intermembrane space bridging complex (Kozjak-Pavlovic, 2017). Inside the inner membrane is the mitochondrial matrix which contains the mitochondrial components of the malate-aspartate shuttle and pyruvate dehydrogenase and the enzymes of the TCA cycle and beta oxidation which generate NADH and FADH2. It is currently unknown how, or if, changes in mitochondrial ultrastructure influence mitochondrial lactate oxidation specifically.
Summary of mitochondrial lactate bioenergetics.
In our view, lactate is the end product of glycolysis, and the final metabolic fate of lactate in vivo is often oxidation by mitochondria; i.e., aerobic glycolysis. Glycolytic enzymes produce pyruvate and NADH. Three cytosolic enzymes (LDH, malate dehydrogenase, and aspartate amino transferase, all possessing very high maximal reaction velocities (Vmax) and catalyzing fully reversible reactions, act to bring pyruvate, lactate, NAD+, NADH, glutamate, aspartate, 2-oxoglutarate, malate, and oxaloacetate into near equilibrium in the cytosol (Figure 1). All of the participating substrates can then generally diffuse freely through the voltage-dependent anion channels. These substrates then have access to inner membrane proteins: one transporter (the mitochondrial pyruvate carrier) and two exchangers (the glutamate/aspartate exchanger and the malate/2-oxoglutarate exchanger). One of these, the glutamate/aspartate exchanger, comes into near equilibrium with ΔΨ and accounts for the steep NAD+/NADH gradient that exists between the cytosol and the matrix. Negatively charged aspartate is exported from the matrix by nearly 200 mV of electrical potential. Outside the inner mitochondrial membrane, aspartate transaminates with 2-oxoglutarate to become oxaloacetic acid; the Keq of aminotransferases is close to unity. Oxaloacetic acid is the oxidized redox partner of malate in the malate dehydrogenase reaction. As a result, at rest, the oxaloacetate/malate and therefore the NAD+/NADH ratio in the cytosol, is orders of magnitude higher than the corresponding ratio in the matrix (Jong & Davis, 1983). The malate-aspartate shuttle operates near equilibrium, but it is kinetically challenged by two factors described elsewhere in this review: 1) the extremely high metabolic scope of aerobic glycolysis, and 2) the fact that a higher mitochondrial OXPHOS rate requires a fall in ΔΨ. Rising cytosolic lactate/pyruvate is therefore a predicted consequence of rising ATP turnover, hence, aerobic glycolytic flux.
Cytosolic NADH electron pairs can also enter the ETC via the glycerol-3-phosphate shuttle. In this case, electrons enter the ETC at the level of the ubiquinone pool. Electrons from glycerol-3-phosphate therefore entirely bypass Complex I and are not subjected to the "backpressure" of the 4 protons that Complex I pumps. The glycerol-3 phosphate shuttle therefore always operates far from equilibrium and would oxidize the cytosol even more than the malate-aspartate shuttle, were it not for kinetic control (mainly Ca2+-mediated) on mitochondrial glycerol-3-phosphate dehydrogenase (Mracek et al., 2013).
To be fair, essentially all experimental models (isolated mitochondria, permeabilized muscle fibers) tend to display at least a minor amount of mitochondrial lactate oxidation and the Mitocarta (2.0) continues to list LDH as a mitochondrial protein in a variety of tissues (Calvo et al., 2016). Perhaps LDH (and malate dehydrogenase) localize to the cytosol-facing surface of the outer mitochondrial membrane (Hung et al., 2017). As previously noted (Gladden, 2008), Skilleter and Kun (Skilleter & Kun, 1972) employed submitochondrial fractionation and concluded that LDH in intact mitochondria “is probably on the outer side of the inner membrane” in liver. Deimann et al. (Deimann et al., 1981) used scanning transmission electron microscopy and found the reaction product for LDH “clearly identified in the intermembranous space of mitochondria” in rabbit glycolytic skeletal muscle. Using proteolytic disruption of isolated liver mitochondria, Kline et al. (Kline et al., 1986) concluded that LDH is “mainly in the outer membrane and [intermembrane] space.” Brandt et al. (Brandt et al., 1987) used fractionated mitochondria isolated from rat heart, kidney, liver, and lymphocytes with digitonin; and reported that “the mitochondrial LDH is located primarily in the [intermembrane] space.” Using confocal microscopy to view immunohistochemically stained LDH and inner mitochondrial membrane proteins in skeletal muscle, Elustondo and collaborators (Elustondo et al., 2013) also found evidence of proximity between LDH and the mitochondrial inner membrane. Depending on the integrity of the outer mitochondrial membrane, LDH in an intermembrane location (including on the outer face of the inner mitochondrial membrane) might be more or less protected. LDH, with a molecular weight of 134,000 would unlikely pass through an intact outer mitochondrial membrane that is impermeable to molecules larger than a molecular weight of 5000 whereas NAD+/NADH at a molecular weight of approximately 664 moves through readily. LDH within the intermembrane space would also be protected from destruction by proteases used in mitochondrial isolation (trypsin, molecular weight ≈23,000; nagarse, molecular weight ≈27,000) (Gladden, 2008). Taken together, these facts lead us to question the bioenergetic relevance of an intermembrane LDH in skeletal muscle in vivo.
While our review and modeling refer specifically to skeletal muscle, any model which includes LDH on both sides of an inner mitochondrial membrane equipped with a lactate transporter must identify the driving force that is thermodynamically competent to account for the steep redox gradient known to exist across the inner mitochondrial membrane. In fact, matrix LDH has the opposite effect. Simulations with our computational model clearly indicate that matrix LDH dissipates the redox gradient established by the malate-aspartate shuttle (not shown). The higher the matrix LDH Vmax, the more rapid the dissipation.
Whether or not a putative lactate oxidation complex located on the outer surface of the inner mitochondrial membrane (Hashimoto et al., 2006) has a significant role in this process remains to be determined. Further experimentation is required which may or may not lead to a debate akin to that related to the role of creatine kinase isoforms in the phosphocreatine shuttle (Meyer et al., 1984).
Mitochondrial lactate metabolism in health (exercise) and disease
Exercise.
Our view is that lactate is not directly oxidized by mitochondria, but rather lactate must first be converted to pyruvate in the cytosol or intermembrane space. Nevertheless, mitochondrial density, adequate mitochondrial function, sufficient reducing equivalents, and O2 are relevant to lactate metabolism. In healthy individuals, exercise is typically the primary condition in which [La−] increases. Much attention has been devoted to the idea that increasing exercise intensity creates a hypoxic (anaerobic) environment that limits mitochondrial oxidation of fuels, creating an exaggerated dependence on glycolysis with subsequent lactate accumulation; i.e., an anaerobic threshold is reached (Wasserman & McIlroy, 1964; Wasserman et al., 1973). However, it is well-established that mitochondria are able to work at maximal OXPHOS rates down to very low O2 levels (i.e., PO2 = ≈2 mmHg), and as reviewed extensively by several authors (e.g., (Ferguson et al., 2018)), exercising muscles are unlikely to reach those limiting PO2 values at work rates eliciting significant increases in muscle and blood [La−]. Therefore, the term “anaerobic threshold” is inappropriate and should be replaced by “lactate threshold”. Similarly, indirect assessments of the lacta te threshold via gas exchange should be labeled as such (e.g., “ventilatory threshold”, or “gas exchange threshold”).
Lactate metabolism during exercise has been reviewed extensively (e.g., (Clanton et al., 2013; Ferguson et al., 2018)) and will not be discussed in detail here. However, it should be noted that if the concept of a mitochondrial reticular power grid (Glancy et al., 2015) is valid, this would further diminish the possibility of dysoxia (an O2 tension that is sufficiently low enough to limit cytochrome turnover in the electron transport chain (Connett et al., 1990) as a cause of increased muscle and blood [La−] during most exercise intensities. Clanton (Clanton, 2019) has also proposed the idea of a myoglobin/nitric oxide “shield” working in combination with the mitochondrial power grid to reduce the potential for low PO2 to limit mitochondrial OXPHOS activity. While decreasing PO2 in exercising muscles can cause an increase in lactate production ((Lundin & Strom, 1947; Hogan et al., 1983; Wasserman & Koike, 1992) and see Figure 27 in (Clanton et al., 2013)), this O2 dependency is not due to frank dysoxia, and O2 limitation of mitochondrial function is usually a minor player among the causes of lactate production; e.g., increasing stimulation of glycogenolysis by catecholamines (see relevance to disease below).
Overall, we see skeletal muscle mitochondrial density as the major factor in the relationship between exercise intensity and increases in muscle and blood [La−]. When lactate production by the glycolytic pathway accelerates, [La−] will increase unless there are sufficient mitochondria to siphon off pyruvate and NADH via the mitochondrial pyruvate carrier and NADH shuttles, respectively. Lactate concentration will always be a balance between the glycolytic rate and the subsequent mitochondrial metabolism of pyruvate and NADH. A greater volume density of healthy mitochondria will permit lower [La−] at higher glycolytic rates (engendered by higher exercise intensities). Mammalian myocytes can respond to endurance training with nearly 2-fold increases in mitochondrial abundance (e.g., (Holloszy & Coyle, 1984; Granata et al., 2018)) leading to significant decreases in lactate production, increased removal, and decreased net accumulation. This concept can be extended to include the whole body in lactate removal via mitochondrial oxidation of pyruvate and NADH. Intracellularly, the LDH equilibrium is maintaining [La−] higher than pyruvate concentration such that pyruvate transfer from cellular locations where glycolysis is occurring, to the mitochondrial reticula for subsequent metabolism is via the cytosol-to-mitochondria shuttle (see Figure 10 in (Ferguson et al., 2018)).
Disease – Acute Care.
Clinicians treating illness and injury are forced to make decisions based on interpretation of the best available data. Unfortunately, this has led to multiple misunderstandings of O2 uptake, lactate kinetics, and/or the role of “anaerobic metabolism” in the clinical setting. While a measurable increase in [La−] has remained a reliable predictor of poor outcomes in the clinical setting (Claridge et al., 2000), the mechanisms behind this have not been fully elucidated. Although an O2 limitation or intrinsic mitochondrial derangement would lead to increases in [La−], these remain the exception rather than the rule in clinical situations (Goodwin et al., 2019). By understanding the metabolic causes that are likely responsible for the elevation of [La−] observed in various common disease states, clinicians may not only improve current treatments but also devise broad strategies for more widespread implementation in the trauma bay, intensive care unit, or perioperative setting (Brooks, 2018).
Although a full history of lactate in the clinical setting is well beyond the scope of this review, key early developments must be noted. After Berzelius noted elevated [La−] in the muscles of hunted stags (Ferguson et al., 2018), over 100 years of research commenced, much of it with the overarching theme that lactate was a waste product formed due to the hypoxia of exhaustive contractions, cardiac or respiratory insufficiency, or other illness, or some combination thereof (Ferguson et al., 2018). Hypoxia, or more accurately dysoxia, is often an unlikely clinical scenario, yet many clinicians often treat an elevated [La−] as if by definition there is an O2 limitation. It is under this misunderstanding that the term “occult hypoperfusion” entered the literature (Mizock, 1989). This term was introduced in the trauma literature to explain conditions in which trauma patients were resuscitated as indicated by all measurable means (e.g., hemodynamics, urine output, etc.),yet an elevated blood [La−] persisted, often for hours. Occult hypoperfusion is the best explanation for clinicians who understand an elevation of blood [La−] solely as the end result of poor perfusion/oxygenation. Although some clinicians believe that an unmeasured, visceral hypoperfusion drives this elevation in blood [La−], strong supporting data are lacking. For example, in experiments utilizing a series of stepwise clamps on the superior mesenteric artery to induce visceral ischemia in pigs, venous [La−] increased locally, but arterial [La−] remained unchanged, even as mesenteric artery flow was completely occluded (Tenhunen et al., 2001). This directly contrasts with the clinical setting often encountered, where a trauma or intensive care unit patient may have an elevated [La−] for hours to days without signs of hemodynamic insufficiency or gut ischemia. Arguments have been put forth that some limitation exists at the level of the microcirculation. However, in studies designed to specifically test this proposition, correction of microperfusion deficits did not alter lactate responses (Trzeciak et al., 2008; Puskarich et al., 2016).
While the dangers of hypoxemia/dysoxia are obvious, there are also potential downsides to hyperoxemia. High levels of inspired O2 can cause increased formation of superoxides and free radicals resulting in lung injury (Ferguson, 2016; Damiani et al., 2018). As reviewed by Damiani and colleagues (Damiani et al., 2018), resorption atelectasis can also result from breathing hyperoxic gas mixtures. Further, animal models suggest that the increased oxidative stress of hyperoxemia may extend to systemic effects including an increase in inflammatory cytokines that may lead to more widespread infection and an increased incidence of multiple organ dysfunction (Damiani et al., 2018). There are also potential negative consequences for systemic blood flow control, coronary blood flow, and myocardial O2 consumption (Damiani et al., 2018). It is not surprising, then, that clinical reports of potential adverse effects of aggressive O2 therapy are beginning to appear. Specifically, Girardis et al. (Girardis et al., 2016) investigated the outcomes of critically ill patients with an intensive care unit length of stay of 72 hours or longer, and reported that intensive care unit mortality was lower for patients treated with a conservative protocol (maintenance of PaO2 between 70 and 100 mmHg or arterial oxyhemoglobin saturation (SaO2) between 94% and 98% vs. conventional therapy (PaO2 up to 150 mmHg or SaO2 between 97% and 100%)). Similarly, in a systematic review and meta-analysis, Chu and colleagues (Chu et al., 2018) reported problems with liberal O2 therapy in the acute care setting. Across trials that included 16,037 patients, liberal O2 therapy was defined with a median FIO2 of 0.52 for a median duration of 8 h in comparison to conservative therapy with a median FIO2 of 0.21. The patient groups included those with sepsis, critical illness, stroke, trauma, myocardial infarction, cardiac arrest, and emergency surgery. In these acutely ill adult patients, mortality was actually increased in the liberal O2 condition without evidence of improving other important patient outcomes. These concerns about hyperoxygenation have led to some rapid recommendations from an international panel of experienced clinicians (Siemieniuk et al., 2018). Specifically, the following recommendations were made:
| Strong recommendation: | If supplemental O2 is administered, the maximum peripheral capillary O2 saturation should be limited to 96%. |
| Strong recommendation: | For patients with myocardial infarction or stroke, do not initiate supplemental O2 if the initial peripheral capillary O2 saturation is greater than 92%. |
| Weak recommendation: | For patients with myocardial infarction or stroke, do not initiate supplemental O2 if the initial peripheral capillary O2 saturation is in the range of 90-92%. |
From a mitochondrial perspective, it is important to remember that intracellular PO2 in contracting muscles during exercise has been estimated to be in the range of 2-4 mmHg (Richardson et al., 1998; Richardson et al., 2001) with a metabolic rate that is many times greater than at rest. Yes, there is lactate efflux but it is not caused by the low intracellular PO2 (Richardson et al., 1998). The relationship to clinical conditions is that it may be necessary to have severely low cellular PO2 before there is a tissue limitation or a hypoxic/dysoxic stimulus for increased lactate production or decreased lactate removal, unless there is a mitochondrial abnormality. An important caveat is that none of this discussion obviates the clinical concern that often accompanies a declining arterial O2 saturation. Also, whether other tissues in addition to skeletal muscle are similarly functional with low cellular PO2 deserves further study.
Disease - Catecholamines and lactate.
If dysoxia is not always the cause of relevant elevations in blood [La−] in the clinic/hospital, what are other alternatives? Perhaps the most obvious contributor is circulating catetcholamines (Goodwin et al., 2019). Earlier investigated by Mazzeo with regard to the lactate threshold during exercise (Mazzeo & Marshall, 1989), the lactate response has been shown to closely mirror the catecholamine response. In the trauma and critical care setting, this link explains how an elevation in blood [La−] can persist in the face of restored hemodynamics/O2 measures. Circulating catecholamines provide a mechanism by which skeletal muscle glycogen can be broken down and enter the blood stream as lactate, to be circulated and used as a fuel where needed. This critical component of the cell-to-cell lactate shuttle (Brooks, 2018) allows remarkable whole-body coordination in times of fight or flight. From the viewpoint of the individual skeletal muscle, stored muscle glycogen is convenient in that it is available only to that muscle cell. However, from an organismal view, this can be overridden when the life of the organism demands, as circulating catecholamines bind β-receptors on skeletal muscle cells and lead to muscle glycogen breakdown to lactate, which then equilibrates with the plasma and red blood cells via monocarboxylate transporters and then ultimately with distant sites of usage. Goodwin et al. (Goodwin et al., 2019) summarize key evidence of the potential role of catecholamines in eliciting elevated [La−]s in certain clinical situations.
Despite the mechanistic misunderstanding by many, an elevated [La−] remains a harbinger of poor clinical outcomes. In the trauma setting, an elevated blood [La−] on presentation to the trauma bay or a persistent elevation in blood [La−] that is not trending downward within the first day portends a poor prognosis. For example, patients who presented to the trauma bay with arterial [La−] > 4.0mM had a mortality rate approaching 20% (Odom et al., 2013). Within that group, those who showed improvement within the first 6 hours ([La−] decreased by at least 60%) had a mortality rate of 7.5%, while those with less than a 30% [La−] reduction had a mortality rate approaching 30%! Other studies have examined the same phenomenon but with absolute cut offs. As an example, trauma patients who did not have an absolute [La−] of 2.5 mM or less at 24 hours post-admission experienced a poorer survival rate (Blow et al., 1999). This relationship between an elevated [La−] in critical conditions and poor prognosis has remained when examined across trauma, critical care, and perioperative conditions (Crowl et al., 2000; Grey et al., 2013; Venkatesan et al., 2015; Richards et al., 2016). As emphasized by Brooks (Brooks, 2018), perhaps a better appreciation that lactate is not simply a deadend waste product of anaerobic glycolysis but is instead an important intermediate that shuttles among tissues even in the presence of adequate O2, will lead to better treatments in acute care situations.
Disease – Mitochondriopathy.
While there are several causes of abnormal mitochondrially linked disease, a primary cause is mutations of mitochondrial DNA (mtDNA). Such mutations of mtDNA usually cause disruptions in respiratory chain function (Schapira, 2012; Gorman et al., 2016). In other cases, mitochondrial dysfunction may be secondary to biochemical abnormalities induced by other disorders (Schapira, 2012). Given that disease-causing mutations have been reported in more than 230 different genes, mitochondriopathy describes a heterogeneous group of diseases with a variety of clinical phenotypes (Koopman et al., 2012; Schapira, 2012; Rotig, 2014; Gorman et al., 2016). Most of these mutations affect the mitochondrial respiratory chain (Gorman et al., 2015; Mootha & Chinnery, 2018). The overall severity of the disease is likely to depend on the degree of heteroplasmy; that is, the mutated mtDNA versus wild-type DNA in the individual cells of a person (Gorman et al., 2016). In severe cases such as Leigh syndrome, children can exhibit developmental delay and die of respiratory failure in their first few years of life (Ferrari et al., 2017).
Despite the heterogeneity of the disease, there are some physiological/biochemical characteristics that are generally associated with mitochondrial malfunction. At rest, blood [La−] is elevated (e.g., 1.4-5.0 mM vs. ≤ 1.0 mM in controls) but the magnitude of the elevation varies considerably and is not a sensitive indicator of the extent of disease (Taivassalo et al., 2003; Robinson, 2006; Grassi et al., 2007; Gorman et al., 2016; Delaney et al., 2017). During exercise, blood [La−] of mitochondrial disease patients increases in a pattern that is similar to that of controls, but at much lower work rates (Taivassalo et al., 2003; Grassi et al., 2007; Delaney et al., 2017). In other words, for any given work rate, the patients have a higher blood [La−]. While some patients overlap with healthy controls, in general, individuals with mitochondrial disease have a lower peak O2 uptake (); for example, 16 vs. 32 ml O2·kg−1·min−1 (Taivassalo et al., 2003). However, despite a lower peak work rate and lower , the peak cardiac output of mitochondrial disease patients is similar to that of controls (Taivassalo et al., 2003; Delaney et al., 2017). Additionally, these patients have a decreased exercise efficiency, slower pulmonary on-kinetics, and a greater slow component (Porcelli et al., 2016; Grassi et al., 2019).
Given the normal cardiac output () at peak exercise in combination with a reduced , the Fick equation () illustrates that O2 extraction (i.e., arteriovenous O2 concentration difference = a-vO2d) is impaired in mitochondrial disease (Taivassalo et al., 2003; Delaney et al., 2017). In fact, Taivassalo et al. (Taivassalo et al., 2003) reported a linear correlation between and peak systemic a-vO2d, concluding that exercise intolerance in mitochondrial disease patients is directly correlated with the severity of impaired muscle OXPHOS as reflected by the peak O2 extraction. Grassi and colleagues (Grassi et al., 2007; Grassi et al., 2019) used near infrared spectroscopy to interrogate the relative deoxygenation of skeletal muscle hemoglobin and myoglobin during exercise, arriving at similar conclusions. In general, deoxygenation was less at any given work rate, suggesting an “exaggerated” (wording of (Grassi et al., 2019)) cardiovascular response relative to the metabolic demand in the mitochondrial disease patients. The notion of higher tissue oxymyoglobin and oxyhemoglobin saturations along with higher [La−] is consistent with first principles. Dysfunctional mitochondria with limited respiratory capacity would engender elevated stimuli for increased cardiac output and local active muscle vasodilation. At the same time, stimuli for OXPHOS (e.g., [ADP]•[Pi]/[ATP]) would likely be elevated, providing “extra” stimulation of the glycolytic pathway and increased lactate production. It should be noted that the acidosis accompanying elevated blood [La−] is not entirely negative. Acidosis shifts the oxyhemoglobin dissociation curve rightward, which in the periphery would assist in maintaining a higher mean capillary PO2 and thereby a greater driving gradient for O2 into tissues, thus improving diffusion. Without this acidosis effect, cardiac output might be even more hyperperfusive.
The evidence above favors a supposition of an overabundance of O2 in the tissues of those with mitochondrial disease. This concept segues to tantalizing new information relative to the role of O2 in this disease. Mootha and Chinnery (Mootha & Chinnery, 2018) note that high-flow O2 (i.e., rapid flow rate of supplemental O2 via a mask) is the typical response when a compromised mitochondrial disease patient shows up at the emergency room or intensive care unit. However, a screen for clustered regularly interspaced short palindromic repeats (CRISPR) by Jain et al. (Jain et al., 2016) directed attention to elements of the hypoxia response pathway. Subsequently, they (Jain et al., 2016; Mootha & Chinnery, 2018) altered the environmental O2 level (FIO2) in a genetic mouse model of Leigh syndrome, the most common pediatric form of mitochondrial disease. The results were striking; when these mice inspired 11% O2, as opposed to the typical ambient level of 21% O2, they regained body weight, achieved normothermia, and lived longer. To the contrary and unlike wild-type, normal mice, when the Leigh syndrome mice breathed 55% O2, they died within days (Jain et al., 2016; Mootha & Chinnery, 2018). Further research revealed that hypoxia-treated knockout mice died of neurodegeneration at about 270 days in comparison to about 60 days for normoxiatreated mice (Ferrari et al., 2017). Further, less hypoxic regimens, such as 17% O2, did not prevent neuropathology, whereas 11% hypoxia appeared to reverse the neurological disease even in the late stages (Ferrari et al., 2017). What mechanism is at work? Mootha and Chinnery (Mootha & Chinnery, 2018) speculate that the relative abundance of O2 in the diseased mice might be limiting the activation of glycolysis and/or be directly toxic due to formation of reactive O2 species and enzyme inactivation. Currently, whether high supplemental O2 is detrimental to human patients with mitochondrial disease or whether hypoxia might be a treatment for such patients is unknown (Mootha & Chinnery, 2018; Jain et al., 2019).
Using the results described above as a springboard, Ast and Mootha (Ast & Mootha, 2019) note the almost universal discrepancy between tissue O2 levels in vivo and the O2 levels in routine mammalian cell culture. While cell culture incubators typically maintain a PO2 of about 140 mmHg, O2 tension in human organs tends to be much lower; for example, large intestine (3-11 mmHg), uterus (15-19 mmHg), liver (30-55 mmHg) (Ast & Mootha, 2019), resting skeletal muscle (34 mmHg; (Richardson et al., 2006)), and exercising muscle (2-4 mmHg; (Richardson et al., 1998; Richardson et al., 2001)). The PO2 in the core of untreated tumors might be on the order of 2 mmHg (McKeown, 2014). We have raised this concern about cellular O2 levels in vitro previously (Ferguson et al., 2018) and echo the sentiments of Ast and Mootha (Ast & Mootha, 2019) which are now more supported by actual data.
Cancer, lactate, and mitochondrial dysfunction.
The role of lactate metabolism in cancer remains an area of intense debate. Cancers have long been associated with deranged glucose and lactate metabolism, with many of these aberrations implicating lactate as “tumorigenic” (Goodwin et al., 2014; Brooks, 2018; Gladden, 2019). Dating back to Warburg and the Cori’s in the early 1920’s, this aberrant behavior of increased glucose utilization and lactate production despite adequate O2 has been termed “the Warburg effect” (Warburg et al., 1927; Otto, 2016). Early work centered on the possibility of severe mitochondrial dysfunction driving the onset of cancer (Otto, 2016), although it is now well known that most cancers do have normally or near-normally functioning mitochondria (Martin et al., 1998; Moreno-Sanchez et al., 2007; Jose et al., 2011; Vander Heiden & DeBerardinis, 2017). Cancers are now recognized to have altered metabolism upstream from the mitochondria (e.g., glycolytic enzymes are elevated severalfold in some tumors (Moreno-Sanchez et al., 2007; San-Millan & Brooks, 2017)), often altering their behavior based on substrate availability, the local microenvironment, metabolic demands, and vascularity, although some degree of mitochondrial dysfunction may exist in particular cancers (Avagliano et al., 2019).
As one example of how mitochondrial function is directly affected in cancers, evidence supporting the role of “mitochondrial reprogramming” in various cancer types continues to mount. Wang et al. (Wang et al., 2019) used various breast cancer cell lines to demonstrate a significant uptake of glutamine during hypoxia, leading to inhibition of the electron transport chain and accumulation of reducing equivalents (NADH), in turn impacting mitochondrial respiration. Currently, mitochondria are increasingly being investigated for their role in tumorigenesis, although data elucidating specific mechanisms are not conclusive.
Finally, it should be noted that these investigations into metabolism of various cancers are further confounded by the difficulty in replicating the complex tumor microenvironment in vivo under laboratory conditions in vitro (Muir et al., 2018). We have already noted issues about O2 levels above. Additional confounding occurs due to the wide variety of metabolic behaviors that have been observed in cancer types. While lactate is now seen as “tumorigenic” in most cancers (San-Millan & Brooks, 2017) some tumors seem to produce lactate in a traditional Warburg manner, while others seem to metabolize lactate, taking advantage of its characteristics both as a potent fuel and signaling molecule (Gladden et al., 2011; Goodwin et al., 2014), driving vascular endothelial growth factor and vascularity. For full reviews of the role of lactate in cancer, the reader is encouraged to see the following (San-Millan & Brooks, 2017; Vander Heiden & DeBerardinis, 2017; Brooks, 2018; Ferguson et al., 2018; Gillies et al., 2019; Goodwin et al., 2019; Payen et al., 2019; Pennington et al., 2019).
Conclusion
In summary, we return to the early 20th century reports of lactate removal via O2 consumption by Meyerhof (Meyerhof, 1927) in recovering, previously stimulated amphibian skeletal muscle. As detailed early in the current review, these studies presaged the discovery of OXPHOS in the mitochondrial reticulum. Numerous studies over the intervening period of almost a century have shown that lactate is not merely a dead end waste product of anaerobic metabolism, but is instead a continuously produced and removed metabolite that circulates among essentially all cells of the body; i.e., the cell-to-cell lactate shuttle as so brilliantly deduced by Brooks (Brooks, 2018). Lactate is indeed oxidized by the mitochondrial reticulum but the weight of evidence indicates that it is first converted to pyruvate via the LDH reaction, which then moves across the inner membrane into the mitochondrial matrix via the mitochondrial pyruvate carrier. The electron pairs resulting from glycolysis are transferred across the inner membrane via the long-known malate-aspartate and glycerol-3-phosphate shuttles, both of which have nonequilibrium steps to maintain the large redox gradient between the mitochondrial matrix and the cytoplasm. Transmission of pyruvate and NADH from active intracellular glycolytic sites to mitochondrial consumption sites is likely facilitated by an LDH equilibrium throughout the cell which essentially uses lactate as the transmitted species; i.e., the cytosol-to-mitochondria shuttle (Rogatzki et al., 2015; Ferguson et al., 2018). This means that lactate is the end product of glycolysis in the cytosol, but it combines with NAD+ to yield pyruvate and NADH in locations near the mitochondrial reticulum, but outside the inner membrane. Lactate metabolism is intimately tied to mitochondrial function and volume density because of the competition of mitochondrial components (mitochondrial pyruvate carrier and the NAD+/NADH shuttles) with glycolytic rate and the LDH reaction.
Blood and muscle [La−]s increase as exercise intensity increases and intramuscular PO2 declines to the range of 2-4 mmHg. However, healthy mitochondria function well even at these low O2 levels such that dysoxia is rare and low O2 is likely a minor factor in the increasing [La−]. The exercise lactate response is very much influenced by skeletal muscle mitochondrial volume density. A greater presence of mitochondria allows them to compete favorably with the LDH reaction for the interim products of glycolysis (pyruvate and NADH), thus minimizing lactate accumulation.
While lactic acidosis is indeed a harbinger of dire consequences in acute and critical care situations, numerous studies show that high [La−] often remains even after adequate O2 supply has been assured., Therefore, clinicians should look to mounting evidence that the elevated [La−] may be due to a stress response that is expressed by elevated catecholamines that stimulate glycolysis. Intriguingly, recent data suggest that tissue O2 supply may be in surplus in at least some mitochondrial diseases. For example, initial experiments in a mouse model of Leigh syndrome show that low O2 breathing (11% O2) improves the animals’ health and increases their life span. Cancer cells are voracious consumers of glucose and avid producers of lactate even in the presence of sufficient O2. Nevertheless, PO2 can be quite low at the core of tumors (e.g., 2 mmHg). These results from various lines of research spotlight the role (or not) of O2 in metabolism. A specific take-home message is that researchers studying isolated cells in vitro should carefully consider not only the degree to which experimental substrate concentrations and spatial constraints replicate the environments encountered in vivo, but also whether the level of O2 exposure is appropriate for the question being studied. In most cases, atmospheric air is hyperoxic for cells.
Funding.
No funding was received for this work.
List of Abbreviations
- a-vO2d
arteriovenous O2 difference
- CRISPR
clustered regularly interspaced short palindromic repeats
- ΔG
free energy change
- ΔΨ
mitochondrial inner membrane electrical potential
- ETC
electron transport chain
- FIO2
inspired fraction of O2
- JO2
mitochondrial respiratory O2 flux
- Keq
equilibrium constant
- KM
Michaelis-Menten constant
- [La−]
lactate concentration
- LDH
lactate dehydrogenase
- MAR
mass action ratio
- OXPHOS
oxidative phosphorylation
- PDH
pyruvate dehydrogenase
- Pi -
inorganic phosphate
- P:O
ratio of ATP synthesized to atomic oxygen consumed
- PaO2
arterial partial pressure of O2
- PO2
partial pressure of O2
- [Pyr−]
pyruvate concentration
cardiac output
- SaO2
arterial oxyhemoglobin saturation
- TCA
tricarboxylic acid cycle
- Vmax
maximal reaction velocity
O2 uptake/consumption
peak O2 uptake/consumption
Biography
![]()
Brian Glancy (far left) is an Earl Stadtman Investigator at the National Heart, Lung, and Blood Institute and National Institute of Arthritis and Musculoskeletal and Skin Diseases. Dan Kane (second from left) is an Associate Professor of Exercise Physiology at St Francis Xavier University. Andreas Kavazis (third from left) is a Professor in the School of Kinesiology at Auburn University. Matthew Goodwin (third from right) is a Spine Surgeon and Assistant Professor of Orthopedic and Neurological Surgery at Washington University in Saint Louis. Wayne Willis (second from right) is an Associate Professor of Medicine at the University of Arizona. Bruce Gladden (far right) is a Professor in the School of Kinesiology at Auburn University. Together they are interested in mitochondrial energetics, ranging from how mitochondria meet rapid changes in energy demand upon onset of muscle contraction to novel metabolic targets of spinal tumors, to thermodynamic control of oxidative phosphorylation.
Footnotes
Competing Interests. None of the authors has any conflict of interest regarding the submission of this manuscript.
Publisher's Disclaimer: This is an Accepted Article that has been peer-reviewed and approved for publication in the The Journal of Physiology, but has yet to undergo copy-editing and proof correction.
REFERENCES
- DI Abbrescia, La Piana G & Lofrumento NE. (2012). Malate-aspartate shuttle and exogenous NADH/cytochrome c electron transport pathway as two independent cytosolic reducing equivalent transfer systems. Arch Biochem Biophys 518, 157–163. [DOI] [PubMed] [Google Scholar]
- Allchin D. (2002). To err and win a nobel prize: Paul Boyer, ATP synthase and the emergence of bioenergetics. J Hist Biol 35, 149–172. [DOI] [PubMed] [Google Scholar]
- Altinok O, Poggio JL, Stein DE, Bowne WB, Shieh AC, Snyder NW & Orynbayeva Z. (2020). Malate-aspartate shuttle promotes l-lactate oxidation in mitochondria. J Cell Physiol 235, 2569–2581. [DOI] [PubMed] [Google Scholar]
- Altmann R. (1890). Die Elementarorganismen und ihre Beziehungen zu den Zellen. Veit, Leipzig. [Google Scholar]
- Andres R, Cader G & Zierler KL. (1956). The quantitatively minor role of carbohydrate in oxidative metabolism by skeletal muscle in intact man in the basal state; measurements of oxygen and glucose uptake and carbon dioxide and lactate production in the forearm. J Clin Invest 35, 671–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annau E, Banga I, Gözsy B, Huszák S, Laki K, Straub B & Szent-Györgryi A. (1935). Über die bedeutung der fumarsaure fur die tierische gewabsatmung. Z Physiol Chem 235, 1–68. [Google Scholar]
- Ast T & Mootha VK. (2019). Oxygen and mammalian cell culture: are we repeating the experiment of Dr. Ox? Nature Metabolism 1, 858–860. [DOI] [PubMed] [Google Scholar]
- Atlante A, de Bari L, Bobba A, Marra E & Passarella S. (2007). Transport and metabolism of L-lactate occur in mitochondria from cerebellar granule cells and are modified in cells undergoing low potassium dependent apoptosis. Biochim Biophys Acta 1767, 1285–1299. [DOI] [PubMed] [Google Scholar]
- Atlante A, Gagliardi S, Marra E, Calissano P & Passarella S. (1999). Glutamate neurotoxicity in rat cerebellar granule cells involves cytochrome c release from mitochondria and mitochondrial shuttle impairment. J Neurochem 73, 237–246. [DOI] [PubMed] [Google Scholar]
- Avagliano A, Ruocco MR, Aliotta F, Belviso I, Accurso A, Masone S, Montagnani S & Arcucci A. (2019). Mitochondrial Flexibility of Breast Cancers: A Growth Advantage and a Therapeutic Opportunity. Cells 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba N & Sharma HM. (1971). Histochemistry of lactic dehydrogenase in heart and pectoralis muscles of rat. J Cell Biol 51, 621–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bak LK & Schousboe A. (2017). Misconceptions regarding basic thermodynamics and enzyme kinetics have led to erroneous conclusions regarding the metabolic importance of lactate dehydrogenase isoenzyme expression. J Neurosci Res 95, 2098–2102. [DOI] [PubMed] [Google Scholar]
- Bakeeva LE, Chentsov Yu S & Skulachev VP. (1978). Mitochondrial framework (reticulum mitochondriale) in rat diaphragm muscle. Biochim Biophys Acta 501, 349–369. [DOI] [PubMed] [Google Scholar]
- Benda C. (1898). Ueber die Spermatogenese der Vertebraten und höherer Evertebraten, II Theil: Die Histiogenese der Spermien. Arch Anat Physiol 73, 393–398. [Google Scholar]
- Bensley RR & Hoerr NL. (1934). Studies on cell structure by the freezing-drying method VI. The preparation and properties of mitochondria. Anatomical Record 60, 449–455. [Google Scholar]
- Bernardi P & Azzone GF. (1981). Cytochrome c as an electron shuttle between the outer and inner mitochondrial membranes. J Biol Chem 256, 7187–7192. [PubMed] [Google Scholar]
- Bleck CKE, Kim Y, Willingham TB & Glancy B. (2018). Subcellular connectomic analyses of energy networks in striated muscle. Nature communications 9, 5111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blow O, Magliore L, Claridge JA, Butler K & Young JS. (1999). The golden hour and the silver day: detection and correction of occult hypoperfusion within 24 hours improves outcome from major trauma. J Trauma 47, 964–969. [DOI] [PubMed] [Google Scholar]
- Borst P. (1963a). Hydrogen transport and transport metabolites. In Funktionelle und morphologische Organisation der Zelle, ed. Karlson P. Springer Verlag, Heidelberg. [Google Scholar]
- Borst P. (1963b). Interrelations between cytoplasmic and mitochondrial diphosphopyridine nucleotide in Ehrlich ascites tumor cells. Proc 5th Intern Congr Biochem II, 233–247. [Google Scholar]
- Borst P. (2006). How I became a biochemist. Iubmb Life 58, 177–182. [DOI] [PubMed] [Google Scholar]
- Boyer PD. (1997). The ATP synthase--a splendid molecular machine. Annu Rev Biochem 66, 717–749. [DOI] [PubMed] [Google Scholar]
- Boyer PD. (2002). A research journey with ATP synthase. J Biol Chem 277, 39045–39061. [DOI] [PubMed] [Google Scholar]
- Brand MD & Nicholls DG. (2011). Assessing mitochondrial dysfunction in cells. Biochem J 435, 297–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt RB, Laux JE, Spainhour SE & Kline ES. (1987). Lactate dehydrogenase in rat mitochondria. Arch Biochem Biophys 259, 412–422. [DOI] [PubMed] [Google Scholar]
- Bremer J & Davis EJ. (1975). Studies on the active transfer of reducing equivalents into mitochondria via the malate-aspartate shuttle. Biochim Biophys Acta 376, 387–397. [DOI] [PubMed] [Google Scholar]
- Bricker DK, Taylor EB, Schell JC, Orsak T, Boutron A, Chen YC, Cox JE, Cardon CM, Van Vranken JG, Dephoure N, Redin C, Boudina S, Gygi SP, Brivet M, Thummel CS & Rutter J. (2012). A mitochondrial pyruvate carrier required for pyruvate uptake in yeast, Drosophila, and humans. Science 337, 96–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks GA. (2018). The Science and Translation of Lactate Shuttle Theory. Cell Metab 27, 757–785. [DOI] [PubMed] [Google Scholar]
- Brooks GA, Dubouchaud H, Brown M, Sicurello JP & Butz CE. (1999). Role of mitochondrial lactate dehydrogenase and lactate oxidation in the intracellular lactate shuttle. Proc Natl Acad Sci U S A 96, 1129–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks GA & Gladden LB. (2003). The metabolic systems: anaerobic metabolism (glycolytic and phosphagen). In Exercise physiology people and ideas, ed. CET, pp. 322–360. Oxford University Press, NewYork. [Google Scholar]
- Buchner E. (1897). Alkoholische Gährung ohne Hefezellen. Ber deut chem Ges 30, 117–124. [Google Scholar]
- Calvo SE, Clauser KR & Mootha VK. (2016). MitoCarta2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res 44, D1251–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chance B & Williams GR. (1955a). Respiratory enzymes in oxidative phosphorylation. I. Kinetics of oxygen utilization. J Biol Chem 217, 383–393. [PubMed] [Google Scholar]
- Chance B & Williams GR. (1955b). Respiratory enzymes in oxidative phosphorylation. II. Difference spectra. J Biol Chem 217, 395–407. [PubMed] [Google Scholar]
- Chance B & Williams GR. (1955c). Respiratory enzymes in oxidative phosphorylation. III. The steady state. J Biol Chem 217, 409–427. [PubMed] [Google Scholar]
- Chance B & Williams GR. (1955d). Respiratory enzymes in oxidative phosphorylation. IV. The respiratory chain. J Biol Chem 217, 429–438. [PubMed] [Google Scholar]
- Chance B, Williams GR, Holmes WF & Higgins J. (1955). Respiratory enzymes in oxidative phosphorylation. V. A mechanism for oxidative phosphorylation. J Biol Chem 217, 439–451. [PubMed] [Google Scholar]
- Chen YJ, Mahieu NG, Huang X, Singh M, Crawford PA, Johnson SL, Gross RW, Schaefer J & Patti GJ. (2016). Lactate metabolism is associated with mammalian mitochondria. Nat Chem Biol 12, 937–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu DK, Kim LH, Young PJ, Zamiri N, Almenawer SA, Jaeschke R, Szczeklik W, Schunemann HJ, Neary JD & Alhazzani W. (2018). Mortality and morbidity in acutely ill adults treated with liberal versus conservative oxygen therapy (IOTA): a systematic review and meta-analysis. Lancet 391, 1693–1705. [DOI] [PubMed] [Google Scholar]
- Clanton TL. (2019). Managing the power grid: how myoglobin can regulate PO2 and energy distribution in skeletal muscle. J Appl Physiol (1985) 126, 787–790. [DOI] [PubMed] [Google Scholar]
- Clanton TL, Hogan MC & Gladden LB. (2013). Regulation of cellular gas exchange, oxygen sensing, and metabolic control. Compr Physiol 3, 1135–1190. [DOI] [PubMed] [Google Scholar]
- Claridge JA, Crabtree TD, Pelletier SJ, Butler K, Sawyer RG & Young JS. (2000). Persistent occult hypoperfusion is associated with a significant increase in infection rate and mortal ity in major trauma patients. J Trauma 48, 8–14; discussion 14-15. [DOI] [PubMed] [Google Scholar]
- Claude A & Fullam EF. (1945). An Electron Microscope Study of Isolated Mitochondria : Method and Preliminary Results. J Exp Med 81, 51–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleland KW & Slater EC. (1953). Respiratory granules of heart muscle. Biochem J 53, 547–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connett RJ. (1987). Cytosolic pH during a rest-to-work transition in red muscle: application of enzyme equilibria. J Appl Physiol (1985) 63, 2360–2365. [DOI] [PubMed] [Google Scholar]
- Connett RJ & Honig CR. (1989). Regulation of VO2 in red muscle: do current biochemical hypotheses fit in vivo data? Am J Physiol 256, R898–906. [DOI] [PubMed] [Google Scholar]
- Connett RJ, Honig CR, Gayeski TE & Brooks GA. (1990). Defining hypoxia: a systems view of VO2, glycolysis, energetics, and intracellular PO2. J Appl Physiol (1985) 68, 833–842. [DOI] [PubMed] [Google Scholar]
- Cori C & Cori G. (1936). Mechanism of formation of hexosemonophosphate in muscle and isolation of a new phosphate ester. Proc Soc Exp Biol Med 34, 702–705. [Google Scholar]
- Cori G, Colowick S & Cori C. (1938). The enzymatic conversion of glucose-1-phosphoric ester to 6-ester in tissue extracts. J Biol Chem 124, 543–555. [Google Scholar]
- Crowl AC, Young JS, Kahler DM, Claridge JA, Chrzanowski DS & Pomphrey M. (2000). Occult hypoperfusion is associated with increased morbidity in patients undergoing early femur fracture fixation. J Trauma 48, 260–267. [DOI] [PubMed] [Google Scholar]
- Damiani E, Donati A & Girardis M. (2018). Oxygen in the critically ill: friend or foe? Curr Opin Anaesthesiol 31, 129–135. [DOI] [PubMed] [Google Scholar]
- Dawson AG. (1979). Oxidation of Cytosolic Nadh Formed during Aerobic Metabolism in Mammalian - Cells. Trends Biochem Sci 4, 171–176. [Google Scholar]
- De Bari L, Atlante A, Valenti D & Passarella S. (2004). Partial reconstruction of in vitro gluconeogenesis arising from mitochondrial l-lactate uptake/metabolism and oxaloacetate export via novel L-lactate translocators. Biochem J 380, 231–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bari L, Chieppa G, Marra E & Passarella S. (2010). L-lactate metabolism can occur in normal and cancer prostate cells via the novel mitochondrial L-lactate dehydrogenase. Int J Oncol 37, 1607–1620. [DOI] [PubMed] [Google Scholar]
- Deimann W, Freeman R & Fahimi HD. (1981). Improved contrast in cytochemistry of dehydrogenases by scanning transmission electron microscopy. J Histochem Cytochem 29, 678–681. [DOI] [PubMed] [Google Scholar]
- Delaney NF, Sharma R, Tadvalkar L, Clish CB, Haller RG & Mootha VK. (2017). Metabolic profiles of exercise in patients with McArdle disease or mitochondrial myopathy. Proc Natl Acad Sci U S A 114, 8402–8407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande PD, Hickman DD & Von Korff RW. (1961). Morphology of isolated rabbit heart muscle mitochondria and the oxidation of extramitochondrial reduced diphosphopyridine nucleotide. J Biophys Biochem Cytol 11, 77–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Divakaruni AS & Murphy AN. (2012). Cell biology. A mitochondrial mystery, solved. Science 337, 41–43. [DOI] [PubMed] [Google Scholar]
- Donovan CM & Brooks GA. (1983). Endurance training affects lactate clearance, not lactate production. Am J Physiol 244, E83–92. [DOI] [PubMed] [Google Scholar]
- Eisner V, Lenaers G & Hajnoczky G. (2014). Mitochondrial fusion is frequent in skeletal muscle and supports excitation-contraction coupling. J Cell Biol 205, 179–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elustondo PA, White AE, Hughes ME, Brebner K, Pavlov E & Kane DA. (2013). Physical and functional association of lactate dehydrogenase (LDH) with skeletal muscle mitochondria. J Biol Chem 288, 25309–25317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Embden G, Deuticke H & Kraft G. (1933). Über die intermediären Vorgänge bei der Glykolyse in der Muskulatur. Klin Wochensch 12, 213–215. [Google Scholar]
- Engelhardt V. (1932). Die beziehungen zwischen atmung und pyrophosphatumatz in vogelerythrocyten. Biochem Z 251, 343–368. [Google Scholar]
- Engelhardt V & Ljubimova MN. (1930). Glycolyse und Phosphorsäureumsatz in den Blutzellen verschiedener Tiere. Biochem Z 227, 6–15. [Google Scholar]
- Ernster L & Schatz G. (1981). Mitochondria: a historical review. J Cell Biol 91, 227s–255s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estabrook RW & Sacktor B. (1958). alpha-Glycerophosphate oxidase of flight muscle mitochondria. J Biol Chem 233, 1014–1019. [PubMed] [Google Scholar]
- Ferguson BS, Rogatzki MJ, Goodwin ML, Kane DA, Rightmire Z & Gladden LB. (2018). Lactate metabolism: historical context, prior misinterpretations, and current understanding. Eur J Appl Physiol 118, 691–728. [DOI] [PubMed] [Google Scholar]
- Ferguson ND. (2016). Oxygen in the ICU: Too Much of a Good Thing? JAMA 316, 1553–1554. [DOI] [PubMed] [Google Scholar]
- Ferrari M, Jain IH, Goldberger O, Rezoagli E, Thoonen R, Cheng KH, Sosnovik DE, Scherrer-Crosbie M, Mootha VK & Zapol WM. (2017). Hypoxia treatment reverses neurodegenerative disease in a mouse model of Leigh syndrome. Proc Natl Acad Sci U S A 114, E4241–E4250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiske CH & Subbarow Y. (1927). The Nature of the "Inorganic Phosphate" in Voluntary Muscle. Science 65, 401–403. [DOI] [PubMed] [Google Scholar]
- Fiske CH & Subbarow Y. (1929). Phosphorus Compounds of Muscle and Liver. Science 70, 381–382. [DOI] [PubMed] [Google Scholar]
- Fletcher WM & Hopkins FG. (1907). Lactic acid in amphibian muscle. J Physiol 35, 247–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler LR, Richardson SH & Hatefi Y. (1962). A rapid method for the preparation of highly purified cytochrome oxidase. Biochim Biophys Acta 64, 170–173. [DOI] [PubMed] [Google Scholar]
- Frey TG & Mannella CA. (2000). The internal structure of mitochondria. Trends Biochem Sci 25, 319–324. [DOI] [PubMed] [Google Scholar]
- Fulghum KL, Rood BR, Shang VO, McNally LA, Riggs DW, Zheng YT & Hill BG. (2019). Mitochondria-associated lactate dehydrogenase is not a biologically significant contributor to bioenergetic function in murine striated muscle. Redox Biol 24, 101177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao S, Li H, Cai Y, Ye JT, Liu ZP, Lu J, Huang XY, Feng XJ, Gao H, Chen SR, Li M & Liu PQ. (2014). Mitochondrial binding of alpha-enolase stabilizes mitochondrial membrane: its role in doxorubicin-induced cardiomyocyte apoptosis. Arch Biochem Biophys 542, 46–55. [DOI] [PubMed] [Google Scholar]
- Gillies RJ, Pilot C, Marunaka Y & Fais S. (2019). Targeting acidity in cancer and diabetes. Biochim Biophys Acta Rev Cancer 1871, 273–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girardis M, Busani S, Damiani E, Donati A, Rinaldi L, Marudi A, Morelli A, Antonelli M & Singer M. (2016). Effect of Conservative vs Conventional Oxygen Therapy on Mortality Among Patients in an Intensive Care Unit: The Oxygen-ICU Randomized Clinical Trial. JAMA 316, 1583–1589. [DOI] [PubMed] [Google Scholar]
- Gladden LB. (2008). 200th anniversary of lactate research in muscle. Exerc Sport Sci Rev 36, 109–115. [DOI] [PubMed] [Google Scholar]
- Gladden LB. (2019). Lactate as a key metabolic intermediate in cancer. Ann Transl Med 7, 210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gladden LB, Goodwin ML, McDonald JR & Nijsten MW. (2011). Fuel for cancer cells? Cell Cycle 10, 2421–2422. [DOI] [PubMed] [Google Scholar]
- Glancy B, Hartnell LM, Combs CA, Femnou A, Sun J, Murphy E, Subramaniam S & Balaban RS. (2017). Power Grid Protection of the Muscle Mitochondrial Reticulum. Cell Rep 19, 487–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glancy B, Hartnell LM, Malide D, Yu ZX, Combs CA, Connelly PS, Subramaniam S & Balaban RS. (2015). Mitochondrial reticulum for cellular energy distribution in muscle. Nature 523, 617–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin ML, Jin H, Straessler K, Smith-Fry K, Zhu JF, Monument MJ, Grossmann A, Randall RL, Capecchi MR & Jones KB. (2014). Modeling alveolar soft part sarcomagenesis in the mouse: a role for lactate in the tumor microenvironment. Cancer Cell 26, 851–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin ML, Pennington Z, Westbroek EM, Cottrill E, Ahmed AK & Sciubba DM. (2019). Lactate and cancer: a "lactatic" perspective on spinal tumor metabolism (part 1). Ann Transl Med 7, 220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorman GS, Chinnery PF, DiMauro S, Hirano M, Koga Y, McFarland R, Suomalainen A, Thorburn DR, Zeviani M & Turnbull DM. (2016). Mitochondrial diseases. Nat Rev Dis Primers 2, 16080. [DOI] [PubMed] [Google Scholar]
- Gorman GS, Schaefer AM, Ng Y, Gomez N, Blakely EL, Alston CL, Feeney C, Horvath R, Yu-Wai-Man P, Chinnery PF, Taylor RW, Turnbull DM & McFarland R. (2015). Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann Neurol 77, 753–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granata C, Jamnick NA & Bishop DJ. (2018). Training-Induced Changes in Mitochondrial Content and Respiratory Function in Human Skeletal Muscle. Sports Med 48, 1809–1828. [DOI] [PubMed] [Google Scholar]
- Grassi B, Marzorati M, Lanfranconi F, Ferri A, Longaretti M, Stucchi A, Vago P, Marconi C & Morandi L. (2007). Impaired oxygen extraction in metabolic myopathies: detection and quantification by near-infrared spectroscopy. Muscle Nerve 35, 510–520. [DOI] [PubMed] [Google Scholar]
- Grassi B, Porcelli S & Marzorati M. (2019). Translational Medicine: Exercise Physiology Applied to Metabolic Myopathies. Med Sci Sports Exerc 51, 2183–2192. [DOI] [PubMed] [Google Scholar]
- Grey B, Rodseth RN & Muckart DJ. (2013). Early fracture stabilisation in the presence of subclinical hypoperfusion. Injury 44, 217–220. [DOI] [PubMed] [Google Scholar]
- Guo R, Gu J, Wu M & Yang M. (2016). Amazing structure of respirasome: unveiling the secrets of cell respiration. Protein Cell 7, 854–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackenbrock CR. (1966). Ultrastructural bases for metabolically linked mechanical activity in mitochondria. I. Reversible ultrastructural changes with change in metabolic steady state in isolated liver mitochondria. J Cell Biol 30, 269–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harden A, Young W & Martin C. (1906). The alcoholic ferment of yeast-juice. Proc R Soc Lond B 77, 405–420. [Google Scholar]
- Hashimoto T, Hussien R & Brooks GA. (2006). Colocalization of MCT1, CD147, and LDH in mitochondrial inner membrane of L6 muscle cells: evidence of a mitochondrial lactate oxidation complex. Am J Physiol Endocrinol Metab 290, E1237–1244. [DOI] [PubMed] [Google Scholar]
- Hatefi Y, Haavik AG, Fowler LR & Griffiths DE. (1962a). Studies on the electron transfer system. XLII. Reconstitution of the electron transfer system. J Biol Chem 237, 2661–2669. [PubMed] [Google Scholar]
- Hatefi Y, Haavik AG & Griffiths DE. (1962b). Studies on the electron transfer system. XLI. Reduced coenzyme Q (QH2)-cytochrome c reductase. J Biol Chem 237, 1681–1685. [PubMed] [Google Scholar]
- Hatefi Y, Haavik AG & Jurtshuk p. (1961). Studies on the electron transport system. XXX. DPNH-cytochrome c reductase I. Biochim Biophys Acta 52, 106–118. [DOI] [PubMed] [Google Scholar]
- Helge JW, Stallknecht B, Richter EA, Galbo H & Kiens B. (2007). Muscle metabolism during graded quadriceps exercise in man. J Physiol 581, 1247–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzig S, Raemy E, Montessuit S, Veuthey JL, Zamboni N, Westermann B, Kunji ER & Martinou JC. (2012). Identification and functional expression of the mitochondrial pyruvate carrier. Science 337, 93–96. [DOI] [PubMed] [Google Scholar]
- Hogan MC, Cox RH & Welch HG. (1983). Lactate accumulation during incremental exercise with varied inspired oxygen fractions. J Appl Physiol Respir Environ Exerc Physiol 55, 1134–1140. [DOI] [PubMed] [Google Scholar]
- Hogeboom GH, Claude A & Hotch-Kiss RD. (1946). The distribution of cytochrome oxidase and succinoxidase in the cytoplasm of the mammalian liver cell. J Biol Chem 165, 615–629. [PubMed] [Google Scholar]
- Holloszy JO & Coyle EF. (1984). Adaptations of skeletal muscle to endurance exercise and their metabolic consequences. J Appl Physiol Respir Environ Exerc Physiol 56, 831–838. [DOI] [PubMed] [Google Scholar]
- Hung V, Lam SS, Udeshi ND, Svinkina T, Guzman G, Mootha VK, Carr SA & Ting AY. (2017). Proteomic mapping of cytosol-facing outer mitochondrial and ER membranes in living human cells by proximity biotinylation. Elife 6, e24463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs RA, Meinild AK, Nordsborg NB & Lundby C. (2013). Lactate oxidation in human skeletal muscle mitochondria. Am J Physiol Endocrinol Metab 304, E686–694. [DOI] [PubMed] [Google Scholar]
- Jain IH, Zazzeron L, Goldberger O, Marutani E, Wojtkiewicz GR, Ast T, Wang H, Schleifer G, Stepanova A, Brepoels K, Schoonjans L, Carmeliet P, Galkin A, Ichinose F, Zapol WM & Mootha VK. (2019). Leigh Syndrome Mouse Model Can Be Rescued by Interventions that Normalize Brain Hyperoxia, but Not HIF Activation. Cell Metab 30, 824–832 e823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain IH, Zazzeron L, Goli R, Alexa K, Schatzman-Bone S, Dhillon H, Goldberger O, Peng J, Shalem O, Sanjana NE, Zhang F, Goessling W, Zapol WM & Mootha VK. (2016). Hypoxia as a therapy for mitochondrial disease. Science 352, 54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jong YS & Davis EJ. (1983). Reconstruction of steady state in cell-free systems. Interactions between glycolysis and mitochondrial metabolism: regulation of the redox and phosphorylation states. Arch Biochem Biophys 222, 179–191. [DOI] [PubMed] [Google Scholar]
- Jorgensen BM, Rasmussen HN & Rasmussen UF. (1985). Ubiquinone reduction pattern in pigeon heart mitochondria. Identification of three distinct ubiquinone pools. Biochem J 229, 621–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jose C, Bellance N & Rossignol R. (2011). Choosing between glycolysis and oxidative phosphorylation: a tumor's dilemma? Biochim Biophys Acta 1807, 552–561. [DOI] [PubMed] [Google Scholar]
- Kalckar H. (1937). Phosphorylation in kidney tissue. Enzymologia 2, 47–52. [Google Scholar]
- Kane DA. (2014). Lactate oxidation at the mitochondria: a lactate-malate-aspartate shuttle at work. Front Neurosci 8, 366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karunadharma PP, Basisty N, Chiao YA, Dai DF, Drake R, Levy N, Koh WJ, Emond MJ, Kruse S, Marcinek D, Maccoss MJ & Rabinovitch PS. (2015). Respiratory chain protein turnover rates in mice are highly heterogeneous but strikingly conserved across tissues, ages, and treatments. FASEB J 29, 3582–3592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz A & Sahlin K. (1988). Regulation of lactic acid production during exercise. J Appl Physiol (1985) 65, 509–518. [DOI] [PubMed] [Google Scholar]
- Kayar SR, Hoppeler H, Mermod L & Weibel ER. (1988). Mitochondrial size and shape in equine skeletal muscle: a three-dimensional reconstruction study. Anat Rec 222, 333–339. [DOI] [PubMed] [Google Scholar]
- Keilin D. (1925). On cytochrome, a respiratory pigment common to animals, yeast and higher plants. Proc R Soc B 98, 312–339. [Google Scholar]
- Kennedy EP & Lehninger AL. (1949). Oxidation of fatty acids and tricarboxylic acid cycle intermediates by isolated rat liver mitochondria. J Biol Chem 179, 957–972. [PubMed] [Google Scholar]
- Kirkwood SP, Munn EA & Brooks GA. (1986). Mitochondrial reticulum in limb skeletal muscle. Am J Physiol 251, C395–402. [DOI] [PubMed] [Google Scholar]
- Kline ES, Brandt RB, Laux JE, Spainhour SE, Higgins ES, Rogers KS, Tinsley SB & Waters MG. (1986). Localization of L-lactate dehydrogenase in mitochondria. Arch Biochem Biophys 246, 673–680. [DOI] [PubMed] [Google Scholar]
- Koopman WJ, Willems PH & Smeitink JA. (2012). Monogenic mitochondrial disorders. N Engl J Med 366, 1132–1141. [DOI] [PubMed] [Google Scholar]
- Kozjak-Pavlovic V. (2017). The MICOS complex of human mitochondria. Cell Tissue Res 367, 83–93. [DOI] [PubMed] [Google Scholar]
- Krebs HA & Johnson WA. (1937). The role of citric acid in intermediate metabolism in animal tissues. Enzymologia 4, 148–156. [DOI] [PubMed] [Google Scholar]
- Kresge N, Simoni RD & Hill RL. (2005). Otto Fritz Meyerhof and the elucidation of the glycolytic pathway. J Biol Chem 280, e3. [PubMed] [Google Scholar]
- Kushmerick MJ. (1998). Energy balance in muscle activity: simulations of ATPase coupled to oxidative phosphorylation and to creatine kinase. Comp Biochem Physiol B Biochem Mol Biol 120, 109–123. [DOI] [PubMed] [Google Scholar]
- Lehninger AL. (1951). Phosphorylation coupled to oxidation of dihydrodiphosphopyridine nucleotide. J Biol Chem 190, 345–359. [PubMed] [Google Scholar]
- Lehninger AL. (1964). The mitochondrion. Benjamin, New York. [Google Scholar]
- Lehninger AL. (1970). Biochemistry. Worth Publishers, Inc., New York. [Google Scholar]
- Lemasters JJ. (2005). Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res 8, 3–5. [DOI] [PubMed] [Google Scholar]
- Letts JA & Sazanov LA. (2017). Clarifying the supercomplex: the higher-order organization of the mitochondrial electron transport chain. Nat Struct Mol Biol 24, 800–808. [DOI] [PubMed] [Google Scholar]
- Lipmann F. (1941). Metabolic generation and utilization of phosphate bond energy. Adv Enzymol Rel S Bi 1, 99–162. [Google Scholar]
- Lofrumento NE, Marzulli D, Cafagno L, La Piana G & Cipriani T. (1991). Oxidation and reduction of exogenous cytochrome c by the activity of the respiratory chain. Arch Biochem Biophys 288, 293–301. [DOI] [PubMed] [Google Scholar]
- Lohmann K. (1929). Uber die pyrophosphatfraktion im muske! Naturwissenschaften 17, 624–625. [Google Scholar]
- Lohmann K. (1934). Uber die enzymatische augspaltung der kreatinphosphorsaure: zugleich ein beitrag zum chemismus der muskelkontraktion. Biochem Z 271, 264–277. [Google Scholar]
- Lundin G & Strom G. (1947). The concentration of blood lactic acid in man during muscular work in relation to the partial pressure of oxygen of the inspired air. Acta Physiol Scand 13, 253–266. [DOI] [PubMed] [Google Scholar]
- Lundsgaard E. (1930). Weitere untersuchungen über muskelkontraktionen ohne milchsäurebildung. Biochem Z 227, 51–83. [Google Scholar]
- Lundsgaard E. (1932). The significance of the phenomenon ‘alactacid muscle contraction’ for an interpretation of the chemistry of muscle contraction. Danske Hospitalstidende 75, 84–95. [Google Scholar]
- Majewski N, Nogueira V, Bhaskar P, Coy PE, Skeen JE, Gottlob K, Chandel NS, Thompson CB, Robey RB & Hay N. (2004). Hexokinase-mitochondria interaction mediated by Akt is required to inhibit apoptosis in the presence or absence of Bax and Bak. Mol Cell 16, 819–830. [DOI] [PubMed] [Google Scholar]
- Maley GF. (1957). Phosphorylations associated with the oxidation of external reduced diphosphopyridine nucleotide by rat liver mitochondria. J Biol Chem 224, 1029–1038. [PubMed] [Google Scholar]
- Martin M, Beauvoit B, Voisin PJ, Canioni P, Guerin B & Rigoulet M. (1998). Energetic and morphological plasticity of C6 glioma cells grown on 3-D support; effect of transient glutamine deprivation. J Bioenerg Biomembr 30, 565–578. [DOI] [PubMed] [Google Scholar]
- Mazzeo RS & Marshall P. (1989). Influence of plasma catecholamines on the lactate threshold during graded exercise. J Appl Physiol (1985) 67, 1319–1322. [DOI] [PubMed] [Google Scholar]
- McKeown SR. (2014). Defining normoxia, physoxia and hypoxia in tumours-implications for treatment response. Br J Radiol 87, 20130676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer RA, Sweeney HL & Kushmerick MJ. (1984). A simple analysis of the "phosphocreatine shuttle". Am J Physiol 246, C365–377. [DOI] [PubMed] [Google Scholar]
- Meyerhof O. (1927). Recent Investigations on the Aerobic and an-Aerobic Metabolism of Carbohydrates. J Gen Physiol 8, 531–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyerhof O. (1942). Intermediary carbohydrate metabolism. In A symposium on respiratory enzymes, pp. 3–15. University of Wisconsin Press, Madison. [Google Scholar]
- Mitchell P. (1961). Coupling of phosphorylation to electron and hydrogen transfer by a chemi - osmotic type of mechanism. Nature 191, 144–148. [DOI] [PubMed] [Google Scholar]
- Mizock BA. (1989). Lactic acidosis. Dis Mon 35, 233–300. [DOI] [PubMed] [Google Scholar]
- Mootha VK & Chinnery PF. (2018). Oxygen in mitochondrial disease: can there be too much of a good thing? J Inherit Metab Dis 41, 761–763. [DOI] [PubMed] [Google Scholar]
- Moreno-Sanchez R, Rodriguez-Enriquez S, Marin-Hernandez A & Saavedra E. (2007). Energy metabolism in tumor cells. FEBS J 274, 1393–1418. [DOI] [PubMed] [Google Scholar]
- Mracek T, Drahota Z & Houstek J. (2013). The function and the role of the mitochondrial glycerol -3- phosphate dehydrogenase in mammalian tissues. Biochim Biophys Acta 1827, 401–410. [DOI] [PubMed] [Google Scholar]
- Muir A, Danai LV & Vander Heiden MG. (2018). Microenvironmental regulation of cancer cell metabolism: implications for experimental design and translational studies. Dis Model Mech 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murley A & Nunnari J. (2016). The Emerging Network of Mitochondria-Organelle Contacts. Mol Cell 61, 648–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Needham DM. (1971). Machina carnis: the biochemistry of muscular contraction in its historical development. Cambridge University Press, Cambridge. [Google Scholar]
- Nelson DL & Cox MM. (2017). Lehninger Principles of Biochemistry. Macmillan Learning, New York. [Google Scholar]
- Nemani N, Carvalho E, Tomar D, Dong Z, Ketschek A, Breves SL, Jana F, Worth AM, Heffler J, Palaniappan P, Tripathi A, Subbiah R, Riitano MF, Seelam A, Manfred T, Itoh K, Meng S, Sesaki H, Craigen WJ, Rajan S, Shanmughapriya S, Caplan J, Prosser BL, Gill DL, Stathopulos PB, Gallo G, Chan DC, Mishra P & Madesh M. (2018). MIRO-1 Determines Mitochondrial Shape Transition upon GPCR Activation and Ca(2+) Stress. Cell Rep 23, 1005–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls DG & Ferguson SJ. (2013). Bioenergetics 4. Elsevier, New York. [Google Scholar]
- Nohl H. (1987). Demonstration of the existence of an organo-specific NADH dehydrogenase in heart mitochondria. Eur J Biochem 169, 585–591. [DOI] [PubMed] [Google Scholar]
- Nunnari J, Marshall WF, Straight A, Murray A, Sedat JW & Walter P. (1997). Mitochondrial transmission during mating in Saccharomyces cerevisiae is determined by mitochondrial fusion and fission and the intramitochondrial segregation of mitochondrial DNA. Mol Biol Cell 8, 1233–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odom SR, Howell MD, Silva GS, Nielsen VM, Gupta A, Shapiro NI & Talmor D. (2013). Lactate clearance as a predictor of mortality in trauma patients. J Trauma Acute Care Surg 74, 999–1004. [DOI] [PubMed] [Google Scholar]
- Otto AM. (2016). Warburg effect(s)-a biographical sketch of Otto Warburg and his impacts on tumor metabolism. Cancer Metab 4, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passarella S, Paventi G & Pizzuto R. (2014). The mitochondrial L-lactate dehydrogenase affair. Front Neurosci 8, 407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paventi G, Pizzuto R & Passarella S. (2017). The occurrence of l-lactate dehydrogenase in the inner mitochondrial compartment of pig liver. Biochem Biophys Res Commun 489, 255–261. [DOI] [PubMed] [Google Scholar]
- Payen VL, Mina E, Van Hee VF, Porporato PE & Sonveaux P. (2019). Monocarboxylate transporters in cancer. Mol Metab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennington Z, Goodwin ML, Westbroek EM, Cottrill E, Ahmed AK & Sciubba DM. (2019). Lactate and cancer: spinal metastases and potential therapeutic targets (part 2). Ann Transl Med 7, 221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard M, McManus MJ, Csordas G, Varnai P, Dorn GW 2nd, Williams D, Hajnoczky G & Wallace DC. (2015). Trans-mitochondrial coordination of cristae at regulated membrane junctions. Nat Commun 6, 6259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizzuto R, Paventi G, Porcile C, Sarnataro D, Daniele A & Passarella S. (2012). l-Lactate metabolism in HEP G2 cell mitochondria due to the l-lactate dehydrogenase determines the occurrence of the lactate/pyruvate shuttle and the appearance of oxaloacetate, malate and citrate outside mitochondria. Biochim Biophys Acta 1817, 1679–1690. [DOI] [PubMed] [Google Scholar]
- Ponsot E, Zoll J, N'Guessan B, Ribera F, Lampert E, Richard R, Veksler V, Ventura-Clapier R & Mettauer B. (2005). Mitochondrial tissue specificity of substrates utilization in rat cardiac and skeletal muscles. J Cell Physiol 203, 479–486. [DOI] [PubMed] [Google Scholar]
- Porcelli S, Marzorati M, Morandi L & Grassi B. (2016). Home-based aerobic exercise training improves skeletal muscle oxidative metabolism in patients with metabolic myopathies. J Appl Physiol (1985) 121, 699–708. [DOI] [PubMed] [Google Scholar]
- Prebble JN. (2001). The philosophical origins of Mitchell's chemiosmotic concepts: the personal factor in scientific theory formulation. J Hist Biol 34, 433–460. [DOI] [PubMed] [Google Scholar]
- Prebble JN. (2010). The discovery of oxidative phosphorylation: a conceptual off-shoot from the study of glycolysis. Stud Hist Philos Biol Biomed Sci 41, 253–262. [DOI] [PubMed] [Google Scholar]
- Puskarich MA, Shapiro NI, Massey MJ, Kline JA & Jones AE. (2016). Lactate Clearance in Septic Shock Is Not a Surrogate for Improved Microcirculatory Flow. Acad Emerg Med 23, 690–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quistorff B & Grunnet N. (2011). The isoenzyme pattern of LDH does not play a physiological role; except perhaps during fast transitions in energy metabolism. Aging (Albany NY) 3, 457–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen HN, van Hall G & Rasmussen UF. (2002). Lactate dehydrogenase is not a mitochondrial enzyme in human and mouse vastus lateralis muscle. J Physiol 541, 575–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen UF. (1969). The oxidation of added NADH by intact heart mitochondria. FEBS Lett 2, 157–162. [DOI] [PubMed] [Google Scholar]
- Rasmussen UF, Krustrup P, Bangsbo J & Rasmussen HN. (2001). The effect of high-intensity exhaustive exercise studied in isolated mitochondria from human skeletal muscle. Pflugers Arch 443, 180–187. [DOI] [PubMed] [Google Scholar]
- Rasmussen UF, Krustrup P, Kjaer M & Rasmussen HN. (2003a). Experimental evidence against the mitochondrial theory of aging. A study of isolated human skeletal muscle mitochondria. Exp Gerontol 38, 877–886. [DOI] [PubMed] [Google Scholar]
- Rasmussen UF, Krustrup P, Kjaer M & Rasmussen HN. (2003b). Human skeletal muscle mitochondrial metabolism in youth and senescence: no signs of functional changes in ATP formation and mitochondrial oxidative capacity. Pflugers Arch 446, 270–278. [DOI] [PubMed] [Google Scholar]
- Rasmussen UF & Rasmussen HN. (1985). The NADH oxidase system (external) of muscle mitochondria and its role in the oxidation of cytoplasmic NADH. Biochem J 229, 631–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards JE, Matuszewski PE, Griffin SM, Koehler DM, Guillamondegui OD, O'Toole RV, Bosse MJ, Obremskey WT & Evans JM. (2016). The Role of Elevated Lactate as a Risk Factor for Pulmonary Morbidity After Early Fixation of Femoral Shaft Fractures. J Orthop Trauma 30, 312–318. [DOI] [PubMed] [Google Scholar]
- Richardson RS, Duteil S, Wary C, Wray DW, Hoff J & Carlier PG. (2006). Human skeletal muscle intracellular oxygenation: the impact of ambient oxygen availability. J Physiol 571, 415–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson RS, Newcomer SC & Noyszewski EA. (2001). Skeletal muscle intracellular PO(2) assessed by myoglobin desaturation: response to graded exercise. J Appl Physiol (1985) 91, 2679–2685. [DOI] [PubMed] [Google Scholar]
- Richardson RS, Noyszewski EA, Leigh JS & Wagner PD. (1998). Lactate efflux from exercising human skeletal muscle: role of intracellular PO2. J Appl Physiol (1985) 85, 627–634. [DOI] [PubMed] [Google Scholar]
- Robinson BH. (2006). Lactic acidemia and mitochondrial disease. Mol Genet Metab 89, 3–13. [DOI] [PubMed] [Google Scholar]
- Rogatzki MJ, Ferguson BS, Goodwin ML & Gladden LB. (2015). Lactate is always the end product of glycolysis. Front Neurosci 9, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotig A. (2014). Genetics of mitochondrial respiratory chain deficiencies. Rev Neurol (Paris) 170, 309–322. [DOI] [PubMed] [Google Scholar]
- Sahlin K, Fernstrom M, Svensson M & Tonkonogi M. (2002). No evidence of an intracellular lactate shuttle in rat skeletal muscle. J Physiol 541, 569–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahlin K, Katz A & Henriksson J. (1987). Redox state and lactate accumulation in human skeletal muscle during dynamic exercise. Biochem J 245, 551–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- San-Millan I & Brooks GA. (2017). Reexamining cancer metabolism: lactate production for carcinogenesis could be the purpose and explanation of the Warburg Effect. Carcinogenesis 38, 119–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schapira AH. (2012). Mitochondrial diseases. Lancet 379, 1825–1834. [DOI] [PubMed] [Google Scholar]
- Schatz G. (2013). Getting mitochondria to center stage. Biochem Biophys Res Commun 434, 407–410. [DOI] [PubMed] [Google Scholar]
- Siemieniuk RAC, Chu DK, Kim LH, Guell-Rous MR, Alhazzani W, Soccal PM, Karanicolas PJ, Farhoumand PD, Siemieniuk JLK, Satia I, Irusen EM, Refaat MM, Mikita JS, Smith M, Cohen DN, Vandvik PO, Agoritsas T, Lytvyn L & Guyatt GH. (2018). Oxygen therapy for acutely ill medical patients: a clinical practice guideline. BM J 363, k4169. [DOI] [PubMed] [Google Scholar]
- Skilleter DN & Kun E. (1972). The oxidation of L-lactate by liver mitochondria. Arch Biochem Biophys 152, 92–104. [DOI] [PubMed] [Google Scholar]
- Skulachev VP. (1969). Energy Accumulation in the Cell. Nauka Press, Moscow [Google Scholar]
- Skulachev VP. (1990). Power transmission along biological membranes. J Membr Biol 114, 97–112. [DOI] [PubMed] [Google Scholar]
- Slater E. (1981). The discovery of oxidative phosphorylation. Trends Biochem Sci 6, 226–227. [Google Scholar]
- Slater EC. (1994). Peter Dennis Mitchell. Biographical memoirs of fellows of the Royal Society, London 40, 281–305. [Google Scholar]
- Sottocasa GL, Kuylenstierna B, Ernster L & Bergstrand A. (1967). An electron-transport system associated with the outer membrane of liver mitochondria. A biochemical and morphological study. J Cell Biol 32, 415–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stier A, Bize P, Schull Q, Zoll J, Singh F, Geny B, Gros F, Royer C, Massemin S & Criscuolo F. (2013). Avian erythrocytes have functional mitochondria, opening novel perspectives for birds as animal models in the study of ageing. Front Zool 10, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczesna-Kaczmarek A, Litwinska D & Popinigis J. (1984). Oxidation of NADH via an "external" pathway in skeletal-muscle mitochondria and its possible role in the repayment of lactacid oxygen debt. Int J Biochem 16, 1231–1235. [DOI] [PubMed] [Google Scholar]
- Taivassalo T, Jensen TD, Kennaway N, DiMauro S, Vissing J & Haller RG. (2003). The spectrum of exercise tolerance in mitochondrial myopathies: a study of 40 patients. Brain 126, 413–423. [DOI] [PubMed] [Google Scholar]
- Tenhunen JJ, Jakob SM & Takala JA. (2001). Gut luminal lactate release during gradual intestinal ischemia. Intensive Care Med 27, 1916–1922. [DOI] [PubMed] [Google Scholar]
- Tristan C, Shahani N, Sedlak TW & Sawa A. (2011). The diverse functions of GAPDH: views from different subcellular compartments. Cell Signal 23, 317–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trzeciak S, McCoy JV, Phillip Dellinger R, Arnold RC, Rizzuto M, Abate NL, Shapiro NI, Parrillo JE, Hollenberg SM, Microcirculatory Alterations in R & Shock i. (2008). Early increases in microcirculatory perfusion during protocol-directed resuscitation are associated with reduced multi-organ failure at 24 h in patients with sepsis. Intensive Care Med 34, 2210–2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenti D, de Bari L, Atlante A & Passarella S. (2002). L-Lactate transport into rat heart mitochondria and reconstruction of the L-lactate/pyruvate shuttle. Biochem J 364, 101–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG & DeBerardinis RJ. (2017). Understanding the Intersections between Metabolism and Cancer Biology. Cell 168, 657–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veech RL, Raijman L & Krebs HA. (1970). Equilibrium relations between the cytoplasmic adenine nucleotide system and nicotinamide-adenine nucleotide system in rat liver. Biochem J 117, 499–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesan M, Smith RP, Balasubramanian S, Khan A, Uzoigwe CE, Coats TJ & Godsiff S. (2015). Serum lactate as a marker of mortality in patients with hip fracture: A prospective study. Injury 46, 2201–2205. [DOI] [PubMed] [Google Scholar]
- Vincent AE, Turnbull DM, Eisner V, Hajnoczky G & Picard M. (2017). Mitochondrial Nanotunnels. Trends Cell Biol 27, 787–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X & Schwarz TL. (2009). The mechanism of Ca2+ -dependent regulation of kinesin-mediated mitochondrial motility. Cell 136, 163–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Bai C, Ruan Y, Liu M, Chu Q, Qiu L, Yang C & Li B. (2019). Coordinative metabolism of glutamine carbon and nitrogen in proliferating cancer cells under hypoxia. Nat Commun 10, 201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O & Christian W. (1936). Pyridin, der wasserstoffü bertragende Bestandteil von Gärungsfermenten. BiochemZ 287, 291–328. [Google Scholar]
- Warburg O, Wind F & Negelein E. (1927). The Metabolism of Tumors in the Body. J Gen Physiol 8, 519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg OH. (1913). Über sauerstoffatmende kömchen aus leberzellen und über sauerstoffatmong in berfefeld-filtraten wässriger leberextrakte. Pflüger’s Archiv für Gesammte Physiologie des Menschen und der Thiere 154, 599–617. [Google Scholar]
- Wasserman K & Koike A. (1992). Is the anaerobic threshold truly anaerobic? Chest 101, 211S–218S. [DOI] [PubMed] [Google Scholar]
- Wasserman K & McIlroy MB. (1964). Detecting the Threshold of Anaerobic Metabolism in Cardiac Patients during Exercise. Am J Cardiol 14, 844–852. [DOI] [PubMed] [Google Scholar]
- Wasserman K, Whipp BJ, Koyl SN & Beaver WL. (1973). Anaerobic threshold and respiratory gas exchange during exercise. J Appl Physiol 35, 236–243. [DOI] [PubMed] [Google Scholar]
- Williamson DH, Lund P & Krebs HA. (1967). The redox state of free nicotinamide-adenine dinucleotide in the cytoplasm and mitochondria of rat liver. Biochem J 103, 514–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe RR, Jahoor F & Miyoshi H. (1988). Evaluation of the isotopic equilibration between lactate and pyruvate. Am J Physiol 254, E532–535. [DOI] [PubMed] [Google Scholar]
- Wu F, Yang F, Vinnakota KC & Beard DA. (2007). Computer modeling of mitochondrial tricarboxylic acid cycle, oxidative phosphorylation, metabolite transport, and electrophysiology. J Biol Chem 282, 24525–24537. [DOI] [PubMed] [Google Scholar]
- Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC & Spiegelman BM. (1999). Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 98, 115–124. [DOI] [PubMed] [Google Scholar]
- Yoshida Y, Holloway GP, Ljubicic V, Hatta H, Spriet LL, Hood DA & Bonen A. (2007). Negligible direct lactate oxidation in subsarcolemmal and intermyofibrillar mitochondria obtained from red and white rat skeletal muscle. J Physiol 582, 1317–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young A, Oldford C & Mailloux RJ. (2020). Lactate dehydrogenase supports lactate oxidation in mitochondria isolated from different mouse tissues. Redox Biol 28, 101339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler DM & Doeg KA. (1962). Studies on the electron transport system XLIII. The isolation of a succinic-coenzyme Q reductase from beef heart mitochondria. Arch Biochem Biophys 97, 41–50. [Google Scholar]