

Abstract
DNA damage can be cytotoxic and mutagenic, and it is directly linked to aging, cancer, and other diseases. To counteract the deleterious effects of DNA damage, cells have evolved highly conserved DNA repair pathways. Many commonly used DNA repair assays are relatively low throughput and are limited to analysis of one protein or one pathway. Here, we have explored the capacity of the CometChip platform for parallel analysis of multiple DNA repair activities. Taking advantage of the versatility of the traditional comet assay and leveraging micropatterning techniques, the CometChip platform offers increased throughput and sensitivity compared to the traditional comet assay. By exposing cells to DNA damaging agents that create substrates of Base Excision Repair, Nucleotide Excision Repair, and Non-Homologous End Joining, we show that the CometChip is an effective method for assessing repair deficiencies in all three pathways. With these applications of the CometChip platform, we expand the utility of the comet assay for precise, high-throughput, parallel analysis of multiple DNA repair activities.
Keywords: DNA damage, DNA repair, comet assay, CometChip
INTRODUCTION
DNA damage promotes cancer, aging, neurological disorders and other diseases. Exposure to genotoxins is unavoidable, as DNA damaging agents are ubiquitous both in our environment and within our cells. To prevent adverse effects associated with damage to the genome, all species have evolved defenses for removing and repairing damaged DNA (1, 2). Over 200 human proteins have been categorized as bona fide DNA repair proteins (3-5) (12 January 2021, date last accessed). Most fall into the five classic DNA Repair pathways of Base Excision Repair (BER), Mismatch Repair (MMR), Nucleotide Excision Repair (NER), Homologous Recombination (HR) or Non-Homologous End-Joining (NHEJ). It is now well established that deficiencies in some DNA repair proteins promote genomic instability and can lead to carcinogenesis (1, 2, 6-12). Importantly, DNA damaging agents are also frequently used as chemotherapeutics to target rapidly dividing tumor cells (8, 10, 13). Therefore, understanding DNA repair capacity and the coordination of DNA repair pathways is important not only in revealing disease etiology, but also in predicting chemotherapeutic efficacy (11).
A wide variety of methods are used to measure DNA damage and repair, but we have limited strategies to assess multiple DNA repair pathways in parallel (14). For example, whereas DNA sequencing can identify DNA repair gene variants and rare mutations, the consequences are not always clear (15). Transcriptional profiling is often used to evaluate expression of DNA repair genes, but transcript levels do not always reflect DNA repair capacity (16). Direct activity assays using cell lysates are highly effective and have been used in population studies (17-21), but they are not always conducive to analysis of multiple pathways in parallel (22-27). To enable analysis of multiple DNA repair pathways, a fluorescence-based multiplex flow-cytometric host cell reactivation assay (FM-HCR) was recently developed (28). Here, we describe an alternative approach, which is to measure the activity of multiple DNA repair pathways using the single cell electrophoresis assay, known as the comet assay.
The comet assay can be used to measure the formation and repair of strand breaks and alkali labile sites, which arise from exposure to DNA damaging agents or from endogenous cellular processes. This assay was first developed by Ostling, Johansen, and Singh more than 30 years ago (29, 30), and it has been improved by many laboratories (31-34). The comet assay is based on the principle that, in an agarose gel under an electrophoretic field, damaged DNA migrates farther than undamaged DNA. In this assay, cells are exposed to test conditions, embedded in agarose, subjected to electrophoresis, and stained with a nucleic acid dye. DNA damage can be directly quantified by analyzing the extent of DNA migration. Multiple versions of the comet assay have been developed to measure repair of different kinds of damage in the genomes of live cells (35, 36). While the traditional comet assay has gained increasing popularity for studies of DNA damage and repair (37-46), it has not been widely adopted for multi-pathway analysis, in part due to low throughput, high inter-experimental and inter-laboratory variability, and the requirement for laborious analysis (47-54).
We have previously exploited microfabrication technologies to develop the CometChip, a mammalian cell array format that increases comet assay throughput and reproducibility (Figure 1) (55, 56). The technology uses photolithography to fabricate PDMS molds that have arrayed microposts to cast microwells in agarose (Figure 1A). Each microwell can be tuned to the size of a single cell. Cells are then loaded in the microwells by gravity and encapsulated with low melting point agarose (Figure 1B). By patterning cells into arrays, the technology minimizes the area required per sample, prevents overlapping comets, and places comets on a shared focal plane, such that only 1 or 2 images are required, rather than hundreds of images as is currently the standard for the traditional comet assay. Comets are then analyzed using freely available in-house software (55-57). Whereas in the traditional comet assay each sample requires its own slide, the CometChip fits each sample into a single well of a standard 96-well format (Figure 1). Previous studies show that, when compared to the traditional comet assay, the CometChip platform increases throughput by more than two orders of magnitude, and reduces noise (55, 57). The CometChip, therefore, allows for sensitive measurement of DNA damage and repair in live cells in a high throughput fashion.
Figure 1.

Patterning cells in the 96-well CometChip. (A) Procedures for making the 96-well CometChip using microfabricated PDMS mold. (B) Zoomed in view of cell patterning procedures in one well of the 96-well CometChip.
DNA damage and repair responses to genotoxic stress involve a complex network of multiple proteins and pathways. Two main pathways for excision of damaged DNA nucleotides are BER (for single base lesions) and NER (for bulky lesions and intrastrand DNA crosslinks). Both pathways have strong relevance to human health. People with BER gene polymorphisms and deficiencies have been shown to have increased cancer susceptibility (2, 25, 58-74) and show a link to degenerative diseases (75-78). Similarly, reduced NER capacity is also associated with increased risk of cancer (33, 79-91). DNA double strand breaks (DSBs) are repaired primarily by NHEJ and HR. In the case of NHEJ, polymorphisms in essential genes predispose individuals to many types of cancer including that of the breast, lung, brain, and liver (1, 92-96). Taken together, DNA repair is increasingly recognized as being critical for suppressing cancer, making a multi-pathway analysis platform relevant for learning more about DNA repair, performing epidemiological studies, and personalized medicine.
In order to leverage the CometChip for broad assessment of DNA repair capacity, we set out to analyze its utility for assessing repair of lesions from DNA damaging agents that create substrates for three major DNA repair pathways: BER, NER, and NHEJ. Using genetically defined cell lines depleted for key DNA repair enzymes, we evaluated the potential efficacy of the comet assay for assessment of these repair deficiencies using model DNA damaging agents. In order to study the kinetics of multiple DNA repair pathways, we assessed multiple time points using cell lines with varied DNA repair capacity in triplicate for each experiment and performed each experiment three times. Altogether, over 1,500 samples were analyzed (equivalent to 150,000 comets), which is far greater than what is feasible using the traditional comet assay, both because of the requisite labor, and also because of noise from sample to sample and from experiment to experiment. Our results demonstrate that the CometChip enables detection of a variety of DNA lesions and support its use for high throughput applications that require assessment of multiple cell types, chemical conditions, and repair time points.
MATERIAL AND METHODS
Cells and cell culture
TK6 human lymphoblastoid cells were a gift from Dr. William Thilly. TK6 and MCL-5 cells (97) were cultured in suspension in 1x RPMI 1640 medium with L-glutamine (Invitrogen) supplemented with 10% horse serum (Invitrogen) and 100 units/ml penicillin-streptomycin (Invitrogen) (98, 99). Blood from one anonymous healthy donor collected in a sodium heparin Vacutainer collection tube was purchased from Research Blood Components, Brighton, MA. Primary lymphocytes (peripheral blood mononuclear cells, PBMCs) were isolated from the blood using the standard Ficoll gradient density centrifugation. The PBMC pellet was suspended in freezing medium (40% RPMI-1640 + 50% HI-FBS + 10% DMSO). Vials were stored at −80°C. Cryopreserved lymphocytes were rapidly thawed at 37°C and resuspended in stimulation medium (RMPI-1640 + 20 % HI-FBS + 100 U/mL Pen-Strep + 5 μg/mL PHA-L) and T-lymphocytes were stimulated for 3 days at 37°C and 5% CO2. Wildtype, AagTg, Aag−/−, and Polβ−/− mouse embryonic fibroblast (MEF) cell lines were generated as described in (100, 101).
XPCS1RO and XPCS1RO + XPG human fibroblasts were a gift from Dr. Orlando D. Schärer (Chemical & Cancer Biology Branch, Institute of Basic Science – Center for Genomic Integrity, Ulsan, South Korea (102)). MEFs and human fibroblasts were cultured in DMEM (Invitrogen) supplemented with 10% FBS (Atlanta Biologicals, Atlanta, GA) and 100 units/ml penicillin-streptomycin (Invitrogen). Human glioblastoma cell lines M059J and M059K (from American Type Culture Collection) were cultured in DMEM/F12 medium (Invitrogen) supplemented with 10% FBS (Atlanta Biologicals, Atlanta, GA), MEM Non-Essential Amino Acids (Invitrogen) and 100 units/ml penicillin-streptomycin (Invitrogen). LN428 glioblastoma cell lines were described previously (103).
CometChip Fabrication
Figure 1A illustrates the 96-well CometChip fabrication using a PDMS mold. Briefly, 15 mL of molten normal melting point agarose was applied to GelBond® (Lonza, Switzerland) in a rectangular one-well plate (VWR). The agarose was allowed to gelate with the PDMS mold on top. Phosphate buffered saline (PBS) was added to aid in removal of the mold, which revealed patterned microwells across the surface of the gel. The gel was sandwiched between a glass plate and a bottomless 96-well plate and the setup was secured with 2” binder clips to create the 96-well CometChip. At least 10,000 cells were added to each macrowell and allowed to settle by gravity in complete growth media at 37°C, 5% CO2. Excess cells were washed off after 15-30 min and the bottomless plate was removed in order to enclose the arrayed cells in a layer of 1% low melting point agarose (Figure 1B). Cells were then treated with DNA damaging agents before proceeding to the remaining steps of the comet assay. See treatment details below comet assay description.
Alkaline Comet Assay
The alkaline comet assay was performed as described previously (104). Briefly, for dose-response experiments cells embedded in agarose gels were lysed in lysis buffer (10 mM Tris-HCl, 100 mM Na2EDTA, 2.5 M NaCl, pH 10 with 1% Triton X-100 added 20 min before use) overnight at 4°C. After lysis, the gels were transferred to an electrophoresis chamber filled with alkaline electrophoresis buffer (0.3 M NaOH and 1 mM Na2EDTA, pH>13) for 40 min at 4°C. Electrophoresis was conducted with the same buffer at 4°C for 30 min at 1 V/cm and 300 mA. Percent tail DNA was used to quantify DNA damage for alkaline comet assay.
Neutral Comet Assay
The neutral comet assay was performed using a modified version of the neutral comet protocol as previously described (104). Briefly, following treatments with DNA damaging agents, cells embedded in agarose gels were lysed for 4 hours at 43°C (2.5 M NaCl, 100 mM Na2EDTA, 10 mM Tris, 1% N-Laurylsarcosine, pH 9.5 with 0.5% Triton X-100 and 10% DMSO added 20 min before use). For evaluation of repair kinetics after treatments with DNA damaging agents, the cells were lysed by adding lysis buffer to the appropriate wells of the multiwell plate at 37°C while the repair kinetics time course was carried out and then transferred to 43°C overnight to complete the lysis step. After lysis, the gels were washed three times for 30 min with the electrophoresis buffer (90 mM Tris, 90 mM Boric Acid, 2 mM Na2EDTA, pH 8.5). Electrophoresis was conducted at 4°C for 1 hour at 0.6 V/cm and 6 mA. Tail length was used to quantify DNA damage for neutral comet assay.
Fluorescence Imaging and Comet Analysis
After electrophoresis, agarose gels were neutralized in 0.4 M Tris, pH 7.5 neutralizing buffer twice for 15 min and then stained with 1x SYBR Gold diluted in PBS (Invitrogen). Images were captured at 40x magnification using an epifluorescence microscope (Nikon Eclipse 80i, Nikon Instruments, Inc., Melville, NY, USA) with a 480 nm excitation filter. Comet images were automatically analyzed using custom software written in MATLAB (The Mathworks, Inc., Natick, MA, USA) (56). Outputs from Guicometanalyzer were processed by Comet2Excel, an in-house software developed in Python (Python Software Foundation, Python version 2.7.10). All software is freely available upon request. At least 100 comets were analyzed per condition.
Chemical Treatments
Hydrogen peroxide (H2O2) (Sigma) was diluted in cold PBS to the desired dose immediately before use. Encapsulated cells were immersed in ice cold H2O2 and incubated for 20 min prior to lysis. Methyl methanesulfonate (MMS) (Sigma) was diluted in PBS and cells were treated for 30 min at 37°C. Negative control cells were treated with complete media with the corresponding DMSO concentrations. After treatment, cells were either replenished with fresh media to allow measurement of repair kinetics or trypsinized and resuspended in media to load onto the CometChip for dose response experiments. After loading and encapsulation, gels were immediately lysed and alkaline comet assay was performed. For dead cell comet analysis, cells embedded in the CometChip were lysed for 4 hours at 4°C, washed with 1x PBS, and then exposed to H2O2.
UV Irradiation and Repair
TK6 cells were harvested and resuspended in culture medium supplemented with 10 mM L-glutathione (L-GSH) (Sigma). Following loading in the CometChip and encapsulation with 1% low melting point agarose, gels were cut into pieces containing 3 x 3 macrowells and individually exposed to UV light using a 254 nm UVC lamp (UVG-11, UVP, Upland, CA) calibrated with a UVX radiometer (UVP, Upland, CA) to 0.5 ± 0.1 J/m2/s. Exposure was performed in the dark at 4°C and cells were transferred to culture media with 10 mM L-GSH to allow for repair at 37°C. Cell lysis and alkaline comet assay were performed subsequently.
Enzyme Treatments
Following chemical treatment with H2O2 or MMS or UV treatment, encapsulated cells were lysed overnight. H2O2-treated cells were lysed overnight, washed in Fpg reaction buffer (40 mM HEPES, 0.1 M KCl, 0.5 mM EDTA, 0.2 mg/mL BSA, pH 8.0) three times for 15 min, incubated with 0.8 units/mL Fpg enzyme (New England Biolabs) in Fpg reaction buffer for 40 min at 37°C, and washed with PBS before incubation in lysis buffer. MMS-treated cells were lysed overnight, washed in hAAG enzyme reaction buffer (20 mM Tris-HCl, 10 mM (NH4)2SO4, 10 mM KCl, 2 mM MgSO4, 0.1% Triton®X-10, pH 8.8 at 25°C) three times for 15 min, incubated with 25 units/mL hAAG enzyme (New England Biolabs) in hAAG enzyme reaction buffer at 37°C for 15 min, and washed with PBS before incubation in lysis buffer. For controls, lysed cells were incubated with reaction buffer or with PBS under the same conditions. UV-exposed cells were incubated in working medium supplemented with 10 mM GSH for one hour, four hours, and 24 hours. Cells embedded in CometChip were lysed overnight in the alkaline lysis buffer. The CometChip was then washed three times with the enzyme reaction buffer (1 mM EDTA, 100 mM NaCl, 25 mM Na2HPO4, 100 μg/mL BSA, pH 7.2) by submerging for 15 min each time. The enzyme reaction was performed by incubating the CometChip with 50 U/mL bacterial T4 Endonuclease V (M0308S, New England BioLabs, Ipswich, MA) in the enzyme reaction buffer for 15 min at 37°C. Afterward, the CometChip was placed in the alkaline lysis buffer and processed following the remaining steps of the alkaline comet assay.
Ionizing Radiation Exposure
Cells were first loaded into the CometChip and overlaid with low melting point agarose. The gels were then cut and exposed in smaller pieces. Irradiation was performed using Gammacell 220E (MDS Nordion, Canada), which delivers ionizing radiation (IR) from a Co60 source at an approximate rate of 100 Gy/min (depending on source age). Cells were irradiated at room temperature. To evaluate repair kinetics, media was added to wells to allow cells to repair damage for varying time intervals, after which each well was filled with cell lysis buffer to terminate repair.
RESULTS
Detecting BER activity
The basis for the comet assay is that during electrophoresis, damaged DNA migrates through a matrix more readily than undamaged DNA. DNA is normally highly supercoiled, and thus resistant to DNA migration. When strand breaks are introduced by exposure to a DNA damaging agent, there is a loss of superhelical tension, enabling migration of long loops of DNA and fragmented DNA to create what looks like a comet (Figure 2A) (29). The comet assay detects both single strand breaks (SSBs) and double strand breaks (DSBs). Note that SSBs predominate in the cell under most conditions and as a result, the detection of DSBs is usually negligible under the alkaline conditions used for SSB detection. The assay also detects abasic sites as well as other alkali sensitive moieties that are converted to SSBs under alkaline conditions (29-32, 36). Importantly, base modifications caused by exposure to reactive oxygen species or alkylating agents are often not directly detectable by the assay due to their stability under alkaline conditions. However, in live cells, BER enzymes quickly recognize and excise oxidized and alkylated bases, creating BER intermediates that can be detected by the comet assay (Figure 2B and C). It is interesting that SSBs were not observed in H2O2 treated non-viable cells (e.g., cells that were exposed to detergent under alkaline conditions using standard lysis buffer before H2O2 treatment, see materials and methods). One possibility is that H2O2-induced SSBs are predominantly created by BER enzymes, which requires live cells. It is also noteworthy that SSBs can be formed directly under oxidative conditions (105-109), and so it was unexpected that DNA damage was not detected in non-viable cells. It is possible that the doses of H2O2 used were too low to detect directly-induced strand breaks and/or that formation of hydroxyl radical requires Fenton chemistry, which may be absent without the presence of iron in non-viable cells.
Figure 2.

Measuring DNA BER of oxidative lesions. (A) Representative arrayed comet images. (B) Schematic of the mammalian BER pathway. (C) H2O2 dose response curves when exposed to multiple doses of H2O2 for 20 min at 4°C for live and lysed (dead or non-viable) TK6 lymphoblasts. (D) H2O2 dose response curves for MCL-5 cells without Fpg, with Fpg buffer, and with Fpg enzyme + Fpg buffer. (E) MMS dose response curves for MCL-5 cells without hAAG, with hAAG buffer, and with hAAG enzyme + hAAG buffer. *p < 0.05, Student’s t-test (one-tailed, paired), comparing to WT at each time point (black stars show hAAG compared to WT, grey stars show buffer only compared to WT).
Although damaged DNA bases do not affect DNA migration and thus cannot be detected using the comet assay, using conditions that promote DNA cleavage at sites of unrepaired DNA base lesions is an effective means of unveiling their presence. For example, oxidized bases can be revealed when DNA is incubated with a bifunctional DNA glycosylase, which not only recognizes and removes the damaged base, but also cleaves the backbone to introduce an SSB. For 8-oxoguanine (8oxoG), cells can be treated with H2O2, lysed, rinsed and then incubated with purified formamidopyrimidine DNA glycosylase (Fpg) from E. coli, which removes oxidized purines, including 8oxoG, and for which the lyase activity introduces a nick. In essence, Fpg converts undetectable oxidized base lesions into detectable strand breaks. This is a commonly used approach (36, 51, 110-117) and we have demonstrated here that this is also an effective approach for MCL-5 lymphoblastoid cells using the CometChip (Figure 2D).
We demonstrate that this approach can be applied to reveal the presence of alkylated DNA bases as well. Cells were exposed to the alkylating agent MMS, which produces DNA alkylation lesions, such as 3-methyladenine (3MeA) and 7-methylguanine (7MeG). After exposure, cells were lysed and incubated with the purified human BER glycosylase hAAG, which excises alkylated DNA bases. Although hAAG is a monofunctional glycosylase that does not cleave the backbone, we were nevertheless able to detect AAG-induced abasic sites, most likely because ring-opened abasic sites are readily converted into strand breaks under alkaline conditions (Figure 2E) (118-120). These results are consistent with prior results by the Azqueta group (121).
To more closely examine the role of BER proteins in the repair of oxidative and alkylation damage using the CometChip, we used knockout mouse embryonic fibroblasts (MEFs). Specifically, we used the CometChip to assess the H2O2-induced DNA damage in MEFs with deficiencies in BER proteins. For oxidative damage, we compared the levels of BER intermediates in wild-type, Ogg1 null, and Polβ null cells. OGG1 is the main glycosylase responsible for removing oxidized purines (e.g., 8oxoG) and Polβ is a key polymerase involved in gap filling for short-patch BER (Figure 2B). For Polβ−/− cells, we observed more DNA damage after exposure to H2O2 (Figure 3A). These results indicate that BER intermediates are more persistent when Polβ is absent, which is consistent with prior studies showing that BER substrates accumulate in cells with a slow Polβ mutant (122). We also observed suppression of detectable DNA damage in Ogg1−/− cells exposed to H2O2 (Figure 3B). This result is indicative of reduced initiation of the BER pathway. As such, there is reduced conversion of undetectable base lesions into detectable BER intermediates.
Figure 3.
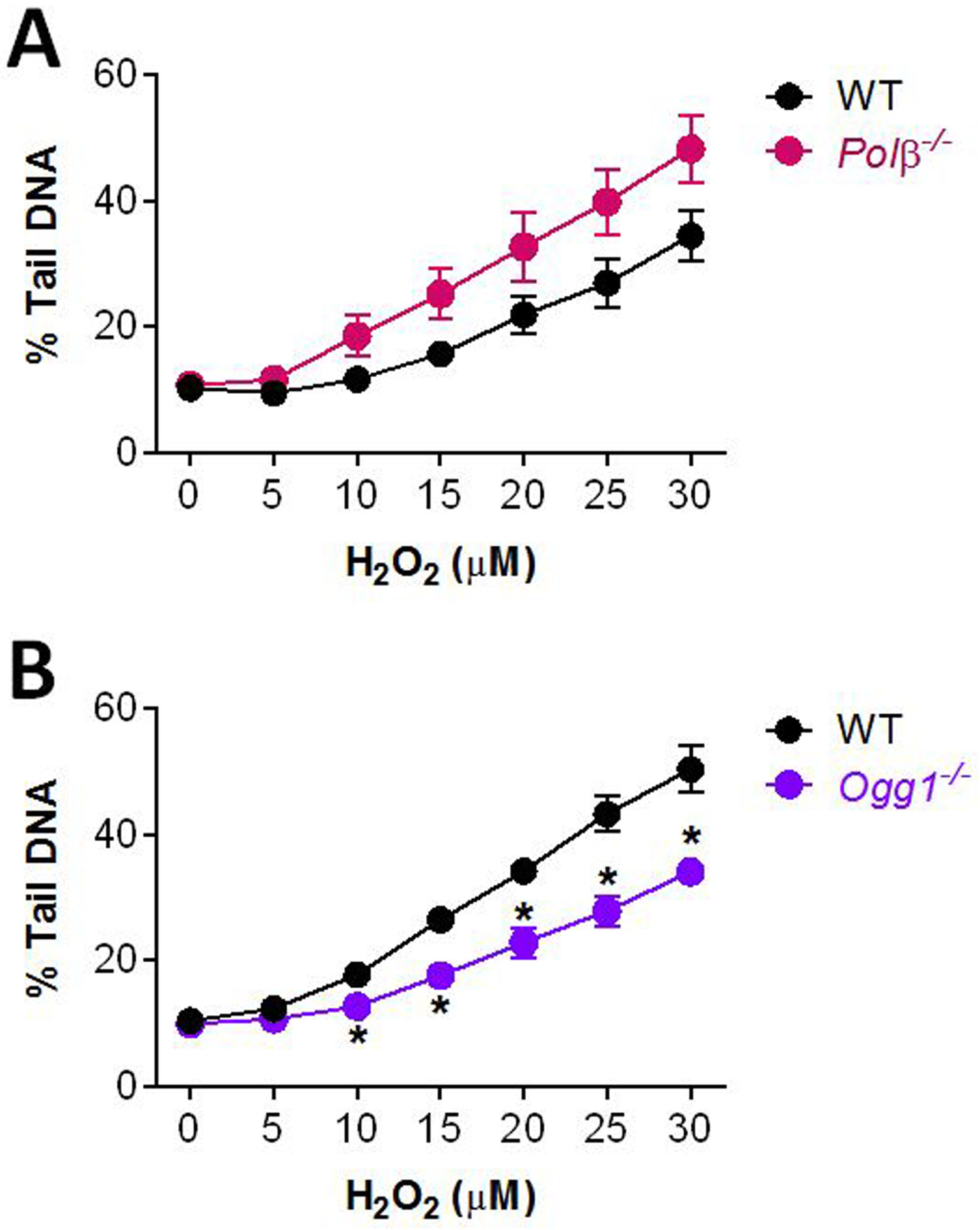
Measuring DNA damage in the absence of BER proteins. DNA damage in Polβ−/− (A) and Ogg1−/− (B) MEFs after treatment with H2O2 at 4°C for 20 min. All NT cells were exposed to 1x PBS as control. Data points and error bars represent averages and standard errors of the mean (SEM), respectively, of three independent experiments (n ≥ 3) with at least 100 comets scored for each condition in each experiment. *p < 0.05, Student’s t-test (one-tailed, paired), comparing to WT at each time point.
Detection of DNA repair using the CometChip
To learn more about the efficacy of the comet assay for studies of BER enzymes involved in repair of alkylation damage, we analyzed the impact of AAG and Polβ on repair kinetics for MMS-induced base lesions. After incubation with MMS for 30 min at 37°, we see significant induction of DNA damage in WT cells (Figure 4A). However, the kinetics of repair of MMS-induced DNA damage are relatively slow, with significant damage remaining at 6 h post exposure (Figure 4B). From prior studies, we know that the kinetics of 3MeA removal by BER are very rapid (<30 minutes), and repair of 7meG is substantially slower (t1/2 = 18 h), at least in embryonic stem cells (Smith and Engelward, 2000). Since the vast majority (~80%) of MMS-induced base lesions are 7MeG (Beranek, 1990), results shown in Figure 4B likely reflect the kinetics of 7MeG repair.
Figure 4.

Measuring DNA BER of alkylation damage. (A) Damage levels in MEFs for non-treated (NT) cells and immediately after 30 min of treatment with 1 mM MMS at 37°C. (B) Repair kinetics of MEFs after treatment with 1 mM MMS at 37°C for 30 min. NT and 0 min data replotted from (A). Repair was performed in media at 37°C. Data points and error bars represent averages and standard errors of the mean (SEM), respectively, of three independent experiments (n ≥ 3) with at least 100 comets scored for each condition in each experiment. *p < 0.05, Student’s t-test (one-tailed, paired), comparing to WT at each time point.
AAG is the major DNA glycosylase for repair of MMS-induced base lesions; specifically, AAG is responsible for repairing 7MeG and 3MeA, which together comprise the majority of the MMS-induced base lesions. We compared cells with WT levels of AAG, cells lacking AAG (Aag−/−), and cells that overexpress the AAG through a transgene (AagTg) (100). For the AagTg cells, we observed higher levels of damage immediately after exposure to MMS (Figure 4A and B), indicative of increased initiation of BER and conversion of undetectable methylated bases into detectable BER intermediates. However, repair was not significantly different between WT and AagTg, possibly because Polβ is rate limiting (101, 123, 124). We expected to see the opposite effect for AAG null cells, since there should be reduced initiation of BER, and thus reduced BER intermediates. Although there appears to be a slight suppression of BER intermediates in Aag−/− cells just after MMS exposure, the effect was not statistically significant for any of the time points (Figure 4A and B). This may be due to inefficiency of AAG to remove the dominant lesion, 7MeG (Smith and Engelward, 2000). We next studied Polβ, which is the primary polymerase for repair synthesis following initiation of BER by AAG (125). We found that the Polβ null cells have increased initial damage (Figure 4A), and that this is the case for all time points post exposure to MMS, which is consistent with inhibition of completion of the BER pathway (Figure 4B). Taken together, for both oxidative damage and alkylation damage, these results show that the comet assay is an effective approach for studying the BER pathway, but that the assay is less sensitive to differences in the levels of DNA glycosylases.
Analysis of Nucleotide Excision Repair Capacity
NER is the major repair pathway for bulky DNA lesions, such as UV-induced pyrimidine dimers (126-129). To remove such damage, XPF/ERCC1 and XPG cut upstream and downstream of the lesion respectively, releasing an oligonucleotide that contains the damaged base(s) (Figure 5A). NER is a highly coordinated pathway, wherein about a dozen proteins are required to assemble into a complex prior to cleavage of the DNA. As such, single-stranded intermediates are short-lived compared to BER intermediates (130), making it difficult to measure NER activity. As has been shown previously (33, 112, 131, 132), one approach for studying NER is to exploit T4 Endonuclease V (T4 EndoV) (132, 133), which cleaves the DNA 5’ to the pyrimidine dimer, thus converting undetectable damage into DNA strand breaks that can be detected using the comet assay (Figure 5A).
Figure 5.

Measuring DNA NER Kinetics in Live Cells. (A) Schematic of the mammalian NER pathway with the blue triangle representing a bulky lesion. (B) UV-irradiated TK6 cells were lysed overnight, and the level of CPDs was measured by incubating the exposed nuclear DNA with T4 EndoV. Buffer control is T4 EndoV reaction buffer with no enzyme added. Data points represent averages of three independent experiments. (C) TK6 and PHA-stimulated T-lymphocytes (PBMCs) after exposure to UV light. Repair was performed in media at 37°C. (D) Repair of human skin fibroblast cell lines, XPG−/− and WT. Data points represent averages of triplicate wells in one experiment. Error bars represent SEM. *p < 0.05, Student’s t-test (one-tailed, paired), comparing to WT at each time point.
We measured percent tail DNA for TK6 cells exposed to various doses of UV, but did not observe a dose-response at time 0 (Figure 5B, left). This is consistent with the fact that initial unrepaired UV-induced base lesions (predominantly cyclobutane pyrimidine dimers) are not detectable using the comet assay. Additionally, this is consistent with the possibility that NER has not yet commenced at time 0. To reveal unrepaired base lesions, DNA from lysed cells was incubated with T4EndoV. We observed a high level of damage just after exposure to UV (0 hr), indicative of the presence of UV dimers (Figure 5B, right). Twenty-four hours post exposure, there was a significant reduction in the levels of damage, which is consistent with clearance of UV dimers by NER.
NER activity can be highly variable in different individuals. For example, some people with Xeroderma Pigmentosum (XP) have severe defects in NER, which makes them extremely sensitive to sunlight-induced skin cancer (134, 135). NER also plays a role in removing DNA lesions induced by chemotherapeutic agents, such as cisplatin (136-139). Therefore, knowledge about DNA repair capacity in people has relevance both in terms of susceptibility to cancer as well as susceptibility to the toxic effects of chemotherapy. We explored the utility of the CometChip for studies of NER capacity in human cells. Specifically, we monitored repair of UV-induced damage in both TK6 cells and PBMCs. Using T4 EndoV, we observed a steep increase in DNA damage immediately after exposing cells to UV, followed by repair over the course of the subsequent 24 hours (Figure 5C). These results show that it is feasible to study repair kinetics for UV-induced DNA damage in primary cells and thus opens doors to more extensive molecular epidemiology studies.
To further explore the specificity of the CometChip-T4 EndoV analysis method, we studied cells with normal DNA repair and cells from an XPG-deficient patient. Since XPG is required for pre-assembly of the NER complex (140, 141), cells lacking XPG do not initiate NER and strand cleavage does not occur. As a result, UV dimers are anticipated to be persistent. We tested this model by treating WT and XPG-null cells with UV, and then monitoring the disappearance of T4 EndoV sensitive sites over time (Figure 5D). Both cell types accumulated the same amount of DNA damage and in WT cells, substrates of T4 EndoV are cleared over time. In contrast, DNA damage persists in the XPG-null cells. These results show that the T4 EndoV approach specifically detects substrates of NER, providing an effective approach for testing NER status in mammalian cells, as has been shown previously (33, 112, 131, 132).
Measuring Non-Homologous End-Joining
Unlike the alkaline comet assay (which detects SSBs and DSBs, abasic sites and alkali sensitive sites), the neutral comet assay can specifically detect DSBs (142). DSBs are repaired primarily by NHEJ and HR, and NHEJ is the primary DSB repair pathway for cells in G1 (143-147). NHEJ involves recognition of the double stranded DNA ends by the Ku70/80 heterodimer, followed by recruitment of DNA protein kinase catalytic subunit (DNA-PKcs), which brings the DNA ends together in the DNA-PK complex. Ends are then processed to create blunt ends or small overhangs that are substrates for the XRCC4/LigIV/XLF complex, which ligates the DSB ends together. To demonstrate the utility of the CometChip for studies of NHEJ, we assayed DSB repair kinetics both for cells lacking the essential NHEJ protein DNA-PKcs and for cells treated with a chemical inhibitor of LigIV (namely, Scr7) (148, 149). We exploited the human glioma cell line, M059J, which was developed from human tumors and lacks DNA-PKcs. Its sister cell line, M059K, was derived from the same human tumor and expresses wild type DNA-PKcs. Another glioma cell line expressing WT DNA-PKcs, LN428, was used for Scr7 treatment. After irradiation and allowing time to repair, both DNA-PKcs deficient cells and cells incubated with Scr7 demonstrated significant impairment of DSB repair (Figures 6A and B, respectively). These results are consistent with the importance of DNA-PKcs and LigIV in NHEJ repair, and agree with data indicating high potency Scr7 inhibition of LigIV (148). It is noteworthy that the tail length immediately post treatment was similar for both 50 Gy IR (Figure 6A) or 100 Gy IR (Figure 6B) treatment, suggesting that these were saturated doses for studying repair kinetics. Nevertheless, these data show that it is feasible to use the CometChip to detect reduced DSB repair kinetics.
Figure 6.

Analysis of DSB repair. (A) Repair kinetics of Glioblastoma M059K (WT) and M059J (DNA-PKcs deficient) cells after treatment with 50 Gy IR. Repair was performed in media at 37°C. (B) Repair kinetics of LN428 cells after exposure to 100 Gy IR with and without Ligase IV inhibition. LN428 cells were pre-incubated for 24 hours in media with 200 μM Scr7, a Ligase IV inhibitor. Following exposure to IR, cells were allowed to repair in media at 37°C with and without Scr7. Data and error bars represent averages and SEM of three independent experiments, respectively. At least 100 comets were scored for each condition in each experiment. *p < 0.05, Student’s t-test (one-tailed, paired), comparing to WT at each time point.
DISCUSSION
Here we show that the CometChip platform enables assessment of multiple cell lines, DNA damaging agents, doses, and time points in parallel in a highly sensitive and rapid fashion. Being able to collect dense data sets with multiple time points makes it possible to assess the kinetics of DNA repair. Further, by varying the type of DNA damaging agent and/or genetic factors, it is possible to study the repair kinetics for three major repair pathways: BER, NER, and NHEJ. Unlike the traditional comet assay, with the enhanced throughput of the CometChip, it is now feasible to query the impact of multiple repair deficiencies in a broad range of cell types, including primary, knock-out, knock-down, mutant, and chemically-inhibited cells. The CometChip thus has the potential to augment a broad range of applications in both the laboratory and the clinic.
Many excellent DNA repair assays exist and can provide information about a specific repair enzyme or pathway. However, historically there have been few assays usable for parallel analyses of both damage induction and clearance to provide detailed information on repair kinetics in living cells. Few, if any, assays have the ability to assess an agent’s potential to damage DNA and a cell’s response to the damage without prior knowledge of the agent’s mechanism of action. For example, oligo cleavage assays are good for measuring glycosylase activity but not the activity of the whole repair pathway. Other than the comet assay, most currently available assays developed for BER are often limited to in vitro analysis of the activity of particular proteins (22, 150), and thus do not reflect the repair capacity of the pathway on the whole in living cells. Available in vivo assays, such as unscheduled DNA synthesis (UDS) and sister chromatid exchange (SCE) are useful for NER and HR (151, 152), respectively, but they are not readily integrated into a multi-pathway platform, and they can be technically challenging. The development of assays for repair foci (e.g., γ-H2AX) can give real-time information on repair activity, but they are again restricted to analyses of specific repair-associated proteins and do not reflect physical DNA damage, but rather signaling events downstream of DNA damage that can be affected by multiple non-repair factors (153, 154). Further, γ-H2AX measures a response to DNA damage but does not directly measure DNA repair kinetics. For example, we previously assayed for DSBs in parallel with γ-H2AX and found that physical DSBs were quickly cleared, while a γ-H2AX signal persisted for more than 6 hours afterwards (55).
Recently, the development of the FM-HCR multiplexed platform enables assessment of multiple repair pathway capacities in parallel by introducing exogenously damaged DNA into living cells (28). It is noteworthy that DNA damaging agents generally induce a range of DNA lesions that are repaired by multiple DNA repair pathways, and thus DNA repair kinetics may vary by chemical agent. By analyzing physical strand breaks, the comet assay offers a complementary approach to FM-HCR in its ability to measure agent-specific DNA repair kinetics in a natural chromatin context. Importantly, the CometChip platform incorporates well-established and validated comet protocols (32, 36, 155, 156). In combination with the micropatterning technology that is the basis for the CometChip, the studies presented here show that this is a fast, simple, and reliable method to study the global repair response. Both FM-HCR and the CometChip have the strengths of versatility and adaptability to measure multiple repair pathways in living cells. In conclusion, we have demonstrated that the CometChip is an effective platform for assessing repair kinetics across several classes of DNA damage addressed by three major DNA repair pathways in living cells, namely BER, NER and NHEJ. As such, this platform opens doors to a deeper mechanistic understanding of DNA repair and discovery of multi-functionality of key DNA repair proteins. Clinically, the CometChip platform can also be used to understand how candidate drugs work mechanistically, to study on- and off-target effects, and to optimize treatment regimens. Finally, for public health, the CometChip can be a potential biomonitoring device for studying effects of environmental genotoxins and for evaluating variation in repair responses among people and among populations. Taken together, the versatility, simplicity, high throughput capacity, and low cost of the CometChip together make this platform valuable for basic, clinical and public health research.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr. Zachary Nagel and Dr. Norah Owiti for their critical comments. We thank Dr. Orlando Scharer for fibroblast cell lines.
FUNDING
This work was supported by grants from the National Institute of Health (NIH) National Cancer Institute R01 ES022872 [L.D.S.] and the National Institute of Environmental Health Sciences Superfund Basic Research Program, P42 ES027707. Additional support was provided by NIH grants R01 CA148629, GM087798 [R.W.S] and ES02116 [R.W.S. and B.P.E.]. This work was also supported by the MIT Center for Environmental Health Sciences P30-ES002109. S.R.F. was supported by Burroughs Wellcome Fund Career Award for Medical Scientists 1010922.
Footnotes
CONFLICT OF INTEREST
B.P.E. and D.M.W. are co-inventors on a patent for the CometChip.
REFERENCES
- 1.Errol CF, Graham CW, Wolfram S, Richard DW, Roger AS, Tom E. DNA Repair and Mutagenesis, Second Edition: American Society of Microbiology; 2006. [Google Scholar]
- 2.Chatterjee N, Walker GC. Mechanisms of DNA damage, repair, and mutagenesis. Environ Mol Mutagen. 2017;58(5):235–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wood RD, Mitchell M, Lindahl T. Human DNA repair genes, 2005. Mutat Res. 2005;577(1-2):275–83. [DOI] [PubMed] [Google Scholar]
- 4.Wood RD, Mitchell M, Lindahl T. Human DNA Repair Genes 2020. [ [DOI] [PubMed] [Google Scholar]
- 5.Wood RD, Mitchell M, Sgouros J, Lindahl T. Human DNA repair genes. Science. 2001;291(5507):1284–9. [DOI] [PubMed] [Google Scholar]
- 6.Alberts B Redefining cancer research. Science. 2009;325(5946):1319. [DOI] [PubMed] [Google Scholar]
- 7.Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461(7267):1071–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lord CJ, Ashworth A. The DNA damage response and cancer therapy. Nature. 2012;481(7381):287–94. [DOI] [PubMed] [Google Scholar]
- 9.Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40(2):179–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Torgovnick A, Schumacher B. DNA repair mechanisms in cancer development and therapy. Front Genet. 2015;6:157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Andre T, Shiu KK, Kim TW, Jensen BV, Jensen LH, Punt C, et al. Pembrolizumab in Microsatellite-Instability-High Advanced Colorectal Cancer. N Engl J Med. 2020;383(23):2207–18. [DOI] [PubMed] [Google Scholar]
- 12.Latham A, Srinivasan P, Kemel Y, Shia J, Bandlamudi C, Mandelker D, et al. Microsatellite Instability Is Associated With the Presence of Lynch Syndrome Pan-Cancer. J Clin Oncol. 2019;37(4):286–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Butler LR, Gilad O, Brown EJ. The DNA Damage Response: Roles in Cancer Etiology and Treatment. In: Pollard J, Curtin N, editors. Targeting the DNA Damage Response for Anti-Cancer Therapy. Cham: Springer International Publishing; 2018. p. 11–33. [Google Scholar]
- 14.Valdiglesias V, Pasaro E, Mendez J, Laffon B. Assays to determine DNA repair ability. J Toxicol Environ Health A. 2011;74(15-16):1094–109. [DOI] [PubMed] [Google Scholar]
- 15.Lo HS, Wang Z, Hu Y, Yang HH, Gere S, Buetow KH, et al. Allelic variation in gene expression is common in the human genome. Genome Res. 2003;13(8):1855–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nagel ZD, Chaim IA, Samson LD. Inter-individual variation in DNA repair capacity: a need for multi-pathway functional assays to promote translational DNA repair research. DNA Repair (Amst). 2014;19:199–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Paz-Elizur T, Krupsky M, Blumenstein S, Elinger D, Schechtman E, Livneh Z. DNA repair activity for oxidative damage and risk of lung cancer. J Natl Cancer Inst. 2003;95(17):1312–9. [DOI] [PubMed] [Google Scholar]
- 18.Paz-Elizur T, Ben-Yosef R, Elinger D, Vexler A, Krupsky M, Berrebi A, et al. Reduced repair of the oxidative 8-oxoguanine DNA damage and risk of head and neck cancer. Cancer Res. 2006;66(24):11683–9. [DOI] [PubMed] [Google Scholar]
- 19.Paz-Elizur T, Leitner-Dagan Y, Meyer KB, Markus B, Giorgi FM, O'Reilly M, et al. DNA Repair Biomarker for Lung Cancer Risk and its Correlation With Airway Cells Gene Expression. JNCI Cancer Spectr. 2020;4(1):pkz067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Somuncu B, Keskin S, Antmen FM, Saglican Y, Ekmekcioglu A, Ertuzun T, et al. Non-muscle invasive bladder cancer tissues have increased base excision repair capacity. Sci Rep. 2020;10(1):16371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Edwards SK, Ono T, Wang S, Jiang W, Franzini RM, Jung JW, et al. In Vitro Fluorogenic Real-Time Assay of the Repair of Oxidative DNA Damage. Chembiochem. 2015;16(11):1637–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Svilar D, Vens C, Sobol RW. Quantitative, real-time analysis of base excision repair activity in cell lysates utilizing lesion-specific molecular beacons. J Vis Exp. 2012(66):e4168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Geng H, Du C, Chen S, Salerno V, Manfredi C, Hsieh P. In vitro studies of DNA mismatch repair proteins. Anal Biochem. 2011;413(2):179–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Georgiadis P, Polychronaki N, Kyrtopoulos SA. Progress in high-throughput assays of MGMT and APE1 activities in cell extracts. Mutat Res. 2012;736(1-2):25–32. [DOI] [PubMed] [Google Scholar]
- 25.Leitner-Dagan Y, Sevilya Z, Pinchev M, Kramer R, Elinger D, Roisman LC, et al. N-methylpurine DNA glycosylase and OGG1 DNA repair activities: opposite associations with lung cancer risk. J Natl Cancer Inst. 2012;104(22):1765–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Redaelli A, Magrassi R, Bonassi S, Abbondandolo A, Frosina G. AP endonuclease activity in humans: development of a simple assay and analysis of ten normal individuals. Teratog Carcinog Mutagen. 1998;18(1):17–26. [PubMed] [Google Scholar]
- 27.Zhong Q, Boyer TG, Chen PL, Lee WH. Deficient nonhomologous end-joining activity in cell-free extracts from Brca1-null fibroblasts. Cancer Res. 2002;62(14):3966–70. [PubMed] [Google Scholar]
- 28.Nagel ZD, Margulies CM, Chaim IA, McRee SK, Mazzucato P, Ahmad A, et al. Multiplexed DNA repair assays for multiple lesions and multiple doses via transcription inhibition and transcriptional mutagenesis. Proc Natl Acad Sci U S A. 2014;111(18):E1823–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ostling O, Johanson KJ. Microelectrophoretic study of radiation-induced DNA damages in individual mammalian cells. Biochem Biophys Res Commun. 1984;123(1):291–8. [DOI] [PubMed] [Google Scholar]
- 30.Singh NP, McCoy MT, Tice RR, Schneider EL. A simple technique for quantitation of low levels of DNA damage in individual cells. Exp Cell Res. 1988;175(1):184–91. [DOI] [PubMed] [Google Scholar]
- 31.Hartmann A, Agurell E, Beevers C, Brendler-Schwaab S, Burlinson B, Clay P, et al. Recommendations for conducting the in vivo alkaline Comet assay. 4th International Comet Assay Workshop. Mutagenesis. 2003;18(1):45–51. [DOI] [PubMed] [Google Scholar]
- 32.Olive PL, Banath JP. The comet assay: a method to measure DNA damage in individual cells. Nat Protoc. 2006;1(1):23–9. [DOI] [PubMed] [Google Scholar]
- 33.Azqueta A, Slyskova J, Langie SA, O'Neill Gaivao I, Collins A. Comet assay to measure DNA repair: approach and applications. Front Genet. 2014;5:288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Langie SA, Knaapen AM, Brauers KJ, van Berlo D, van Schooten FJ, Godschalk RW. Development and validation of a modified comet assay to phenotypically assess nucleotide excision repair. Mutagenesis. 2006;21(2):153–8. [DOI] [PubMed] [Google Scholar]
- 35.Collins A, Dusinska M, Franklin M, Somorovska M, Petrovska H, Duthie S, et al. Comet assay in human biomonitoring studies: reliability, validation, and applications. Environ Mol Mutagen. 1997;30(2):139–46. [DOI] [PubMed] [Google Scholar]
- 36.Collins AR. The comet assay for DNA damage and repair: principles, applications, and limitations. Mol Biotechnol. 2004;26(3):249–61. [DOI] [PubMed] [Google Scholar]
- 37.Witte I, Plappert U, de Wall H, Hartmann A. Genetic toxicity assessment: employing the best science for human safety evaluation part III: the comet assay as an alternative to in vitro clastogenicity tests for early drug candidate selection. Toxicol Sci. 2007;97(1):21–6. [DOI] [PubMed] [Google Scholar]
- 38.Gajski G, Geric M, Zivkovic Semren T, Tariba Lovakovic B, Orescanin V, Pizent A. Application of the comet assay for the evaluation of DNA damage from frozen human whole blood samples: Implications for human biomonitoring. Toxicol Lett. 2020;319:58–65. [DOI] [PubMed] [Google Scholar]
- 39.Gajski G, Langie S, Zhanataev A. Recent applications of the Comet Assay: A report from the International Comet Assay Workshop 2019. Toxicol Lett. 2020;333:1–3. [DOI] [PubMed] [Google Scholar]
- 40.Hobbs CA, Recio L, Winters J, Witt KL. Use of Frozen Tissue in the Comet Assay for the Evaluation of DNA Damage. J Vis Exp. 2020(157). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bankoglu EE, Kodandaraman G, Stopper H. A systematic review of the use of the alkaline comet assay for genotoxicity studies in human colon-derived cells. Mutat Res. 2019;845:402976. [DOI] [PubMed] [Google Scholar]
- 42.Kruszewski M, Sikorska K, Meczynska-Wielgosz S, Grzelak A, Sramkova M, Gabelova A, et al. Comet assay in neural cells as a tool to monitor DNA damage induced by chemical or physical factors relevant to environmental and occupational exposure. Mutat Res. 2019;845:402990. [DOI] [PubMed] [Google Scholar]
- 43.Misik M, Nersesyan A, Ropek N, Huber WW, Haslinger E, Knasmueller S. Use of human derived liver cells for the detection of genotoxins in comet assays. Mutat Res. 2019;845:402995. [DOI] [PubMed] [Google Scholar]
- 44.Anderson D, Dhawan A, Laubenthal J. The Comet Assay in Human Biomonitoring. Methods Mol Biol. 2019;2031:259–74. [DOI] [PubMed] [Google Scholar]
- 45.Vodicka P, Vodenkova S, Opattova A, Vodickova L. DNA damage and repair measured by comet assay in cancer patients. Mutat Res. 2019;843:95–110. [DOI] [PubMed] [Google Scholar]
- 46.Collins A, Milic M, Bonassi S, Dusinska M. The comet assay in human biomonitoring: Technical and epidemiological perspectives. Mutat Res. 2019;843:1–2. [DOI] [PubMed] [Google Scholar]
- 47.Forchhammer L, Brauner EV, Folkmann JK, Danielsen PH, Nielsen C, Jensen A, et al. Variation in assessment of oxidatively damaged DNA in mononuclear blood cells by the comet assay with visual scoring. Mutagenesis. 2008;23(3):223–31. [DOI] [PubMed] [Google Scholar]
- 48.Moller P, Friis G, Christensen PH, Risom L, Plesner G, Kjaersgaard J, et al. Intra-laboratory comet assay sample scoring exercise for determination of formamidopyrimidine DNA glycosylase sites in human mononuclear blood cell DNA. Free Radic Res. 2004;38(11):1207–14. [DOI] [PubMed] [Google Scholar]
- 49.Forchhammer L, Ersson C, Loft S, Moller L, Godschalk RW, van Schooten FJ, et al. Inter-laboratory variation in DNA damage using a standard comet assay protocol. Mutagenesis. 2012;27(6):665–72. [DOI] [PubMed] [Google Scholar]
- 50.Azqueta A, Meier S, Priestley C, Gutzkow KB, Brunborg G, Sallette J, et al. The influence of scoring method on variability in results obtained with the comet assay. Mutagenesis. 2011;26(3):393–9. [DOI] [PubMed] [Google Scholar]
- 51.Collins AR, El Yamani N, Lorenzo Y, Shaposhnikov S, Brunborg G, Azqueta A. Controlling variation in the comet assay. Front Genet. 2014;5:359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ersson C, Moller P, Forchhammer L, Loft S, Azqueta A, Godschalk RW, et al. An ECVAG inter-laboratory validation study of the comet assay: inter-laboratory and intra-laboratory variations of DNA strand breaks and FPG-sensitive sites in human mononuclear cells. Mutagenesis. 2013;28(3):279–86. [DOI] [PubMed] [Google Scholar]
- 53.Forchhammer L, Johansson C, Loft S, Moller L, Godschalk RW, Langie SA, et al. Variation in the measurement of DNA damage by comet assay measured by the ECVAG inter-laboratory validation trial. Mutagenesis. 2010;25(2):113–23. [DOI] [PubMed] [Google Scholar]
- 54.Azqueta A, Ladeira C, Giovannelli L, Boutet-Robinet E, Bonassi S, Neri M, et al. Application of the comet assay in human biomonitoring: An hCOMET perspective. Mutat Res. 2020;783:108288. [DOI] [PubMed] [Google Scholar]
- 55.Weingeist DM, Ge J, Wood DK, Mutamba JT, Huang Q, Rowland EA, et al. Single-cell microarray enables high-throughput evaluation of DNA double-strand breaks and DNA repair inhibitors. Cell Cycle. 2013;12(6):907–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wood DK, Weingeist DM, Bhatia SN, Engelward BP. Single cell trapping and DNA damage analysis using microwell arrays. Proc Natl Acad Sci U S A. 2010;107(22):10008–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ge J, Chow DN, Fessler JL, Weingeist DM, Wood DK, Engelward BP. Micropatterned comet assay enables high throughput and sensitive DNA damage quantification. Mutagenesis. 2015;30(1):11–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Goode EL, Ulrich CM, Potter JD. Polymorphisms in DNA repair genes and associations with cancer risk. Cancer Epidemiol Biomarkers Prev. 2002;11(12):1513–30. [PubMed] [Google Scholar]
- 59.Hakem R DNA-damage repair; the good, the bad, and the ugly. EMBO J. 2008;27(4):589–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sliwinski T, Ziemba P, Morawiec Z, Kowalski M, Zadrozny M, Blasiak J. Polymorphisms of the DNA polymerase beta gene in breast cancer. Breast Cancer Res Treat. 2007;103(2):161–6. [DOI] [PubMed] [Google Scholar]
- 61.Sweasy JB, Lauper JM, Eckert KA. DNA polymerases and human diseases. Radiat Res. 2006;166(5):693–714. [DOI] [PubMed] [Google Scholar]
- 62.Nagel IACaZD. In: DMW, editor. The Base Excision Repair Pathway 2017. p. 557–607. [Google Scholar]
- 63.Cheadle JP, Sampson JR. MUTYH-associated polyposis--from defect in base excision repair to clinical genetic testing. DNA Repair (Amst). 2007;6(3):274–9. [DOI] [PubMed] [Google Scholar]
- 64.AlMutairi F, Pathan AA, Alanazi M, Shalaby M, Alabdulkarim HA, Alamri A, et al. Association of DNA Repair Gene APE1 Asp148Glu Polymorphism with Breast Cancer Risk. Dis Markers. 2015;2015:869512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li M, Zang W, Wang Y, Ma Y, Xuan X, Zhao J, et al. DNA polymerase beta mutations and survival of patients with esophageal squamous cell carcinoma in Linzhou City, China. Tumour Biol. 2014;35(1):553–9. [DOI] [PubMed] [Google Scholar]
- 66.Chaim IA, Nagel ZD. Assessing BER Capacity in the Human Population. The Base Excision Repair Pathway 2017. p. 557–607. [Google Scholar]
- 67.Sevilya Z, Leitner-Dagan Y, Pinchev M, Kremer R, Elinger D, Lejbkowicz F, et al. Development of APE1 enzymatic DNA repair assays: low APE1 activity is associated with increase lung cancer risk. Carcinogenesis. 2015;36(9):982–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sevilya Z, Leitner-Dagan Y, Pinchev M, Kremer R, Elinger D, Rennert HS, et al. Low integrated DNA repair score and lung cancer risk. Cancer Prev Res (Phila). 2014;7(4):398–406. [DOI] [PubMed] [Google Scholar]
- 69.Leitner-Dagan Y, Sevilya Z, Pinchev M, Kremer R, Elinger D, Rennert HS, et al. Enzymatic MPG DNA repair assays for two different oxidative DNA lesions reveal associations with increased lung cancer risk. Carcinogenesis. 2014;35(12):2763–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Vodenkova S, Jiraskova K, Urbanova M, Kroupa M, Slyskova J, Schneiderova M, et al. Base excision repair capacity as a determinant of prognosis and therapy response in colon cancer patients. DNA Repair (Amst). 2018;72:77–85. [DOI] [PubMed] [Google Scholar]
- 71.Li Q, Ma R, Zhang M. XRCC1 rs1799782 (C194T) polymorphism correlated with tumor metastasis and molecular subtypes in breast cancer. Onco Targets Ther. 2018;11:8435–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Azevedo AP, Silva SN, Reichert A, Lima F, Junior E, Rueff J. Effects of polymorphic DNA genes involved in BER and caspase pathways on the clinical outcome of myeloproliferative neoplasms under treatment with hydroxyurea. Mol Med Rep. 2018;18(6):5243–55. [DOI] [PubMed] [Google Scholar]
- 73.Wallace SS, Murphy DL, Sweasy JB. Base excision repair and cancer. Cancer Lett. 2012;327(1-2):73–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Marsden CG, Dragon JA, Wallace SS, Sweasy JB. Base Excision Repair Variants in Cancer. Methods Enzymol. 2017;591:119–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Allocca M, Corrigan JJ, Fake KR, Calvo JA, Samson LD. PARP inhibitors protect against sex- and AAG-dependent alkylation-induced neural degeneration. Oncotarget. 2017;8(40):68707–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ebrahimkhani MR, Daneshmand A, Mazumder A, Allocca M, Calvo JA, Abolhassani N, et al. Aag-initiated base excision repair promotes ischemia reperfusion injury in liver, brain, and kidney. Proc Natl Acad Sci U S A. 2014;111(45):E4878–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kisby GE, Fry RC, Lasarev MR, Bammler TK, Beyer RP, Churchwell M, et al. The cycad genotoxin MAM modulates brain cellular pathways involved in neurodegenerative disease and cancer in a DNA damage-linked manner. PLoS One. 2011;6(6):e20911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Meira LB, Moroski-Erkul CA, Green SL, Calvo JA, Bronson RT, Shah D, et al. Aag-initiated base excision repair drives alkylation-induced retinal degeneration in mice. Proc Natl Acad Sci U S A. 2009;106(3):888–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Slyskova J, Naccarati A, Polakova V, Pardini B, Vodickova L, Stetina R, et al. DNA damage and nucleotide excision repair capacity in healthy individuals. Environ Mol Mutagen. 2011;52(7):511–7. [DOI] [PubMed] [Google Scholar]
- 80.Thoms KM, Kuschal C, Emmert S. Lessons learned from DNA repair defective syndromes. Exp Dermatol. 2007;16(6):532–44. [DOI] [PubMed] [Google Scholar]
- 81.Berndt SI, Platz EA, Fallin MD, Thuita LW, Hoffman SC, Helzlsouer KJ. Genetic variation in the nucleotide excision repair pathway and colorectal cancer risk. Cancer Epidemiol Biomarkers Prev. 2006;15(11):2263–9. [DOI] [PubMed] [Google Scholar]
- 82.Huang WY, Berndt SI, Kang D, Chatterjee N, Chanock SJ, Yeager M, et al. Nucleotide excision repair gene polymorphisms and risk of advanced colorectal adenoma: XPC polymorphisms modify smoking-related risk. Cancer Epidemiol Biomarkers Prev. 2006;15(2):306–11. [DOI] [PubMed] [Google Scholar]
- 83.He BS, Xu T, Pan YQ, Wang HJ, Cho WC, Lin K, et al. Nucleotide excision repair pathway gene polymorphisms are linked to breast cancer risk in a Chinese population. Oncotarget. 2016;7(51):84872–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chang JS, Wrensch MR, Hansen HM, Sison JD, Aldrich MC, Quesenberry CP Jr., et al. Nucleotide excision repair genes and risk of lung cancer among San Francisco Bay Area Latinos and African Americans. Int J Cancer. 2008;123(9):2095–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhu J, Fu W, Jia W, Xia H, Liu GC, He J. Association between NER Pathway Gene Polymorphisms and Wilms Tumor Risk. Mol Ther Nucleic Acids. 2018;12:854–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yan Y, Xu J, Xu B, Wen Q, Zhou J, Zhang L, et al. Effects of Xeroderma pigmentosum group C polymorphism on the likelihood of prostate cancer. J Clin Lab Anal. 2020;34(9):e23403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tian Y, Lin X, Yang F, Zhao J, Yao K, Bian C. Contribution of xeroderma pigmentosum complementation group D gene polymorphisms in breast and ovarian cancer susceptibility: A protocol for systematic review and meta analysis. Medicine (Baltimore). 2020;99(21):e20299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Dai Y, Song Z, Zhang J, Gao W. Comprehensive assessment of the association between XPC rs2228000 and cancer susceptibility based on 26835 cancer cases and 37069 controls. Biosci Rep. 2019;39(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.He XF, Liu LR, Wei W, Liu Y, Su J, Wang SL, et al. Association between the XPG Asp1104His and XPF Arg415Gln polymorphisms and risk of cancer: a meta-analysis. PLoS One. 2014;9(5):e88490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Su J, Zhu Y, Dai B, Yuan W, Song J. XPG Asp1104His polymorphism increases colorectal cancer risk especially in Asians. Am J Transl Res. 2019;11(2):1020–9. [PMC free article] [PubMed] [Google Scholar]
- 91.Li YK, Xu Q, Sun LP, Gong YH, Jing JJ, Xing CZ, et al. Nucleotide excision repair pathway gene polymorphisms are associated with risk and prognosis of colorectal cancer. World J Gastroenterol. 2020;26(3):307–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sishc BJ, Davis AJ. The Role of the Core Non-Homologous End Joining Factors in Carcinogenesis and Cancer. Cancers (Basel). 2017;9(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sharma R, Lewis S, Wlodarski MW. DNA Repair Syndromes and Cancer: Insights Into Genetics and Phenotype Patterns. Front Pediatr. 2020;8:570084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bacon CM, Wilkinson SJ, Spickett GP, Barge D, Lucraft HH, Jackson G, et al. Epstein-Barr virus-independent diffuse large B-cell lymphoma in DNA ligase 4 deficiency. J Allergy Clin Immunol. 2013;131(4):1237–9, 9 e1. [DOI] [PubMed] [Google Scholar]
- 95.Staines Boone AT, Chinn IK, Alaez-Versón C, Yamazaki-Nakashimada MA, Carrillo-Sánchez K, García-Cruz MdlLH, et al. Failing to Make Ends Meet: The Broad Clinical Spectrum of DNA Ligase IV Deficiency. Case Series and Review of the Literature. Frontiers in Pediatrics. 2019;6(426). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kager L, Jimenez Heredia R, Hirschmugl T, Dmytrus J, Krolo A, Muller H, et al. Targeted mutation screening of 292 candidate genes in 38 children with inborn haematological cytopenias efficiently identifies novel disease-causing mutations. Br J Haematol. 2018;182(2):251–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Crespi CL, Gonzalez FJ, Steimel DT, Turner TR, Gelboin HV, Penman BW, et al. A metabolically competent human cell line expressing five cDNAs encoding procarcinogen-activating enzymes: application to mutagenicity testing. Chem Res Toxicol. 1991;4(5):566–72. [DOI] [PubMed] [Google Scholar]
- 98.Liber HL, Thilly WG. Mutation assay at the thymidine kinase locus in diploid human lymphoblasts. Mutat Res. 1982;94(2):467–85. [DOI] [PubMed] [Google Scholar]
- 99.Skopek TR, Liber HL, Penman BW, Thilly WG. Isolation of a human lymphoblastoid line heterozygous at the thymidine kinase locus: possibility for a rapid human cell mutation assay. Biochem Biophys Res Commun. 1978;84(2):411–6. [DOI] [PubMed] [Google Scholar]
- 100.Sobol RW, Kartalou M, Almeida KH, Joyce DF, Engelward BP, Horton JK, et al. Base excision repair intermediates induce p53-independent cytotoxic and genotoxic responses. J Biol Chem. 2003;278(41):39951–9. [DOI] [PubMed] [Google Scholar]
- 101.Sobol RW, Watson DE, Nakamura J, Yakes FM, Hou E, Horton JK, et al. Mutations associated with base excision repair deficiency and methylation-induced genotoxic stress. Proc Natl Acad Sci U S A. 2002;99(10):6860–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ellison AR, Nouspikel T, Jaspers NG, Clarkson SG, Gruenert DC. Complementation of transformed fibroblasts from patients with combined xeroderma pigmentosum-Cockayne syndrome. Exp Cell Res. 1998;243(1):22–8. [DOI] [PubMed] [Google Scholar]
- 103.Fang Q, Inanc B, Schamus S, Wang XH, Wei L, Brown AR, et al. HSP90 regulates DNA repair via the interaction between XRCC1 and DNA polymerase beta. Nat Commun. 2014;5:5513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ge J, Prasongtanakij S, Wood DK, Weingeist DM, Fessler J, Navasummrit P, et al. CometChip: a high-throughput 96-well platform for measuring DNA damage in microarrayed human cells. J Vis Exp. 2014(92):e50607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Schuchmann H The chemical basis of radiation biology : by C. von Sonntag; published by Taylor and Francis, London, 1987; 515 pp.; price U.S. $98; ISBN 0 85066 375 X. Journal of Photochemistry and Photobiology B-biology. 1989;3:465. [Google Scholar]
- 106.Mello Filho AC, Hoffmann ME, Meneghini R. Cell killing and DNA damage by hydrogen peroxide are mediated by intracellular iron. Biochem J. 1984;218(1):273–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Mello Filho AC, Meneghini R. In vivo formation of single-strand breaks in DNA by hydrogen peroxide is mediated by the Haber-Weiss reaction. Biochim Biophys Acta. 1984;781(1-2):56–63. [DOI] [PubMed] [Google Scholar]
- 108.Ward JF, Blakely WF, Joner EI. Mammalian cells are not killed by DNA single-strand breaks caused by hydroxyl radicals from hydrogen peroxide. Radiat Res. 1985;103(3):383–92. [PubMed] [Google Scholar]
- 109.Pryor WA. Oxy-radicals and related species: their formation, lifetimes, and reactions. Annu Rev Physiol. 1986;48:657–67. [DOI] [PubMed] [Google Scholar]
- 110.Gedik CM, Collins A, Escodd. Establishing the background level of base oxidation in human lymphocyte DNA: results of an interlaboratory validation study. FASEB J. 2005;19(1):82–4. [DOI] [PubMed] [Google Scholar]
- 111.Azqueta A, Arbillaga L, Lopez de Cerain A, Collins A. Enhancing the sensitivity of the comet assay as a genotoxicity test, by combining it with bacterial repair enzyme FPG. Mutagenesis. 2013;28(3):271–7. [DOI] [PubMed] [Google Scholar]
- 112.Azqueta A, Collins AR. The essential comet assay: a comprehensive guide to measuring DNA damage and repair. Arch Toxicol. 2013;87(6):949–68. [DOI] [PubMed] [Google Scholar]
- 113.Sisto R, Cavallo D, Ursini CL, Fresegna AM, Ciervo A, Maiello R, et al. Direct and Oxidative DNA Damage in a Group of Painters Exposed to VOCs: Dose - Response Relationship. Front Public Health. 2020;8:445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ursini CL, Fresegna AM, Ciervo A, Maiello R, Del Frate V, Folesani G, et al. Occupational exposure to graphene and silica nanoparticles. Part II: pilot study to identify a panel of sensitive biomarkers of genotoxic, oxidative and inflammatory effects on suitable biological matrices. Nanotoxicology. 2020:1–15. [DOI] [PubMed] [Google Scholar]
- 115.Selbach MT, Scotti AS, Feistel CC, Nicolau CC, Dalberto D, Dos Santos NG, et al. Evaluation of the cytotoxic and genotoxic effects of Sida planicaulis Cav extract using human neuroblastoma cell line SH-SY5Y. J Toxicol Environ Health A. 2021:1–11. [DOI] [PubMed] [Google Scholar]
- 116.Bankoglu EE, Gerber J, Kodandaraman G, Seyfried F, Stopper H. Influence of bariatric surgery induced weight loss on oxidative DNA damage. Mutat Res. 2020;853:503194. [DOI] [PubMed] [Google Scholar]
- 117.Ge J, Wood DK, Weingeist DM, Prasongtanakij S, Navasumrit P, Ruchirawat M, et al. Standard fluorescent imaging of live cells is highly genotoxic. Cytometry A. 2013;83(6):552–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hartwig A, Dally H, Schlepegrell R. Sensitive analysis of oxidative DNA damage in mammalian cells: use of the bacterial Fpg protein in combination with alkaline unwinding. Toxicol Lett. 1996;88(1-3):85–90. [DOI] [PubMed] [Google Scholar]
- 119.Prakash AS, Gibson NW. Sequence-selective depurination, DNA interstrand cross-linking and DNA strand break formation associated with alkylated DNA. Carcinogenesis. 1992;13(3):425–31. [DOI] [PubMed] [Google Scholar]
- 120.Mattes WB, Hartley JA, Kohn KW. Mechanism of DNA strand breakage by piperidine at sites of N7-alkylguanines. Biochim Biophys Acta. 1986;868(1):71–6. [DOI] [PubMed] [Google Scholar]
- 121.Muruzabal D, Sanz-Serrano J, Sauvaigo S, Gutzkow KB, Lopez de Cerain A, Vettorazzi A, et al. Novel approach for the detection of alkylated bases using the enzyme-modified comet assay. Toxicol Lett. 2020;330:108–17. [DOI] [PubMed] [Google Scholar]
- 122.Nemec AA, Murphy DL, Donigan KA, Sweasy JB. The S229L colon tumor-associated variant of DNA polymerase beta induces cellular transformation as a result of decreased polymerization efficiency. J Biol Chem. 2014;289(20):13708–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Horton JK, Watson M, Stefanick DF, Shaughnessy DT, Taylor JA, Wilson SH. XRCC1 and DNA polymerase beta in cellular protection against cytotoxic DNA single-strand breaks. Cell Res. 2008;18(1):48–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Srivastava DK, Berg BJ, Prasad R, Molina JT, Beard WA, Tomkinson AE, et al. Mammalian abasic site base excision repair. Identification of the reaction sequence and rate-determining steps. J Biol Chem. 1998;273(33):21203–9. [DOI] [PubMed] [Google Scholar]
- 125.Sobol RW, Horton JK, Kuhn R, Gu H, Singhal RK, Prasad R, et al. Requirement of mammalian DNA polymerase-beta in base-excision repair. Nature. 1996;379(6561):183–6. [DOI] [PubMed] [Google Scholar]
- 126.Gillet LC, Scharer OD. Molecular mechanisms of mammalian global genome nucleotide excision repair. Chem Rev. 2006;106(2):253–76. [DOI] [PubMed] [Google Scholar]
- 127.Scharer OD. Nucleotide excision repair in eukaryotes. Cold Spring Harb Perspect Biol. 2013;5(10):a012609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hanawalt PC. Subpathways of nucleotide excision repair and their regulation. Oncogene. 2002;21(58):8949–56. [DOI] [PubMed] [Google Scholar]
- 129.Marteijn JA, Lans H, Vermeulen W, Hoeijmakers JH. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat Rev Mol Cell Biol. 2014;15(7):465–81. [DOI] [PubMed] [Google Scholar]
- 130.Staresincic L, Fagbemi AF, Enzlin JH, Gourdin AM, Wijgers N, Dunand-Sauthier I, et al. Coordination of dual incision and repair synthesis in human nucleotide excision repair. EMBO J. 2009;28(8):1111–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Collins AR, Dobson VL, Dusinska M, Kennedy G, Stetina R. The comet assay: what can it really tell us? Mutat Res. 1997;375(2):183–93. [DOI] [PubMed] [Google Scholar]
- 132.Gedik CM, Ewen SW, Collins AR. Single-cell gel electrophoresis applied to the analysis of UV-C damage and its repair in human cells. Int J Radiat Biol. 1992;62(3):313–20. [DOI] [PubMed] [Google Scholar]
- 133.Collins AR, Mitchell DL, Zunino A, de Wit J, Busch D. UV-sensitive rodent mutant cell lines of complementation groups 6 and 8 differ phenotypically from their human counterparts. Environ Mol Mutagen. 1997;29(2):152–60. [PubMed] [Google Scholar]
- 134.DiGiovanna JJ, Kraemer KH. Shining a light on xeroderma pigmentosum. J Invest Dermatol. 2012;132(3 Pt 2):785–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kraemer KH, Lee MM, Andrews AD, Lambert WC. The role of sunlight and DNA repair in melanoma and nonmelanoma skin cancer. The xeroderma pigmentosum paradigm. Arch Dermatol. 1994;130(8):1018–21. [PubMed] [Google Scholar]
- 136.Huang JC, Zamble DB, Reardon JT, Lippard SJ, Sancar A. HMG-domain proteins specifically inhibit the repair of the major DNA adduct of the anticancer drug cisplatin by human excision nuclease. Proc Natl Acad Sci U S A. 1994;91(22):10394–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Reardon JT, Vaisman A, Chaney SG, Sancar A. Efficient nucleotide excision repair of cisplatin, oxaliplatin, and Bis-aceto-ammine-dichloro-cyclohexylamine-platinum(IV) (JM216) platinum intrastrand DNA diadducts. Cancer Res. 1999;59(16):3968–71. [PubMed] [Google Scholar]
- 138.Zamble DB, Mu D, Reardon JT, Sancar A, Lippard SJ. Repair of cisplatin--DNA adducts by the mammalian excision nuclease. Biochemistry. 1996;35(31):10004–13. [DOI] [PubMed] [Google Scholar]
- 139.Wang D, Hara R, Singh G, Sancar A, Lippard SJ. Nucleotide excision repair from site-specifically platinum-modified nucleosomes. Biochemistry. 2003;42(22):6747–53. [DOI] [PubMed] [Google Scholar]
- 140.Evans E, Moggs JG, Hwang JR, Egly JM, Wood RD. Mechanism of open complex and dual incision formation by human nucleotide excision repair factors. EMBO J. 1997;16(21):6559–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Mu D, Wakasugi M, Hsu DS, Sancar A. Characterization of reaction intermediates of human excision repair nuclease. J Biol Chem. 1997;272(46):28971–9. [DOI] [PubMed] [Google Scholar]
- 142.Olive PL, Wlodek D, Banath JP. DNA double-strand breaks measured in individual cells subjected to gel electrophoresis. Cancer Res. 1991;51(17):4671–6. [PubMed] [Google Scholar]
- 143.Takata M, Sasaki MS, Sonoda E, Morrison C, Hashimoto M, Utsumi H, et al. Homologous recombination and non-homologous end-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. EMBO J. 1998;17(18):5497–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Rothkamm K, Kruger I, Thompson LH, Lobrich M. Pathways of DNA double-strand break repair during the mammalian cell cycle. Mol Cell Biol. 2003;23(16):5706–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Mao Z, Bozzella M, Seluanov A, Gorbunova V. DNA repair by nonhomologous end joining and homologous recombination during cell cycle in human cells. Cell Cycle. 2008;7(18):2902–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Hinz JM, Yamada NA, Salazar EP, Tebbs RS, Thompson LH. Influence of double-strand-break repair pathways on radiosensitivity throughout the cell cycle in CHO cells. DNA Repair (Amst). 2005;4(7):782–92. [DOI] [PubMed] [Google Scholar]
- 147.Karanam K, Kafri R, Loewer A, Lahav G. Quantitative live cell imaging reveals a gradual shift between DNA repair mechanisms and a maximal use of HR in mid S phase. Mol Cell. 2012;47(2):320–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Srivastava M, Nambiar M, Sharma S, Karki SS, Goldsmith G, Hegde M, et al. An inhibitor of nonhomologous end-joining abrogates double-strand break repair and impedes cancer progression. Cell. 2012;151(7):1474–87. [DOI] [PubMed] [Google Scholar]
- 149.Vartak SV, Swarup HA, Gopalakrishnan V, Gopinatha VK, Ropars V, Nambiar M, et al. Autocyclized and oxidized forms of SCR7 induce cancer cell death by inhibiting nonhomologous DNA end joining in a Ligase IV dependent manner. FEBS J. 2018;285(21):3959–76. [DOI] [PubMed] [Google Scholar]
- 150.Li J, Svilar D, McClellan S, Kim JH, Ahn EE, Vens C, et al. DNA Repair Molecular Beacon assay: a platform for real-time functional analysis of cellular DNA repair capacity. Oncotarget. 2018;9(60):31719–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Latimer JJ, Kelly CM. Unscheduled DNA synthesis: the clinical and functional assay for global genomic DNA nucleotide excision repair. Methods Mol Biol. 2014;1105:511–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Wojcik A, Bruckmann E, Obe G. Insights into the mechanisms of sister chromatid exchange formation. Cytogenet Genome Res. 2004;104(1-4):304–9. [DOI] [PubMed] [Google Scholar]
- 153.Kinner A, Wu W, Staudt C, Iliakis G. Gamma-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic Acids Res. 2008;36(17):5678–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Nakamura A, Sedelnikova OA, Redon C, Pilch DR, Sinogeeva NI, Shroff R, et al. Techniques for gamma-H2AX detection. Methods Enzymol. 2006;409:236–50. [DOI] [PubMed] [Google Scholar]
- 155.Tice RR, Strauss GH. The single cell gel electrophoresis/comet assay: a potential tool for detecting radiation-induced DNA damage in humans. Stem Cells. 1995;13Suppl 1:207–14. [PubMed] [Google Scholar]
- 156.Sykora P, Witt KL, Revanna P, Smith-Roe SL, Dismukes J, Lloyd DG, et al. Next generation high throughput DNA damage detection platform for genotoxic compound screening. Sci Rep. 2018;8(1):2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.