

SUMMARY
The ability to edit plant genomes through gene targeting (GT) requires efficient methods to deliver both sequence-specific nucleases (SSNs) and repair templates to plant cells. This is typically achieved using Agrobacterium T-DNA, biolistics or by stably integrating nuclease-encoding cassettes and repair templates into the plant genome. In dicotyledonous plants, such as Nicotinana tabacum (tobacco) and Solanum lycopersicum (tomato), greater than 10-fold enhancements in GT frequencies have been achieved using DNA virus-based replicons. These replicons transiently amplify to high copy numbers in plant cells to deliver abundant SSNs and repair templates to achieve targeted gene modification. In the present work, we developed a replicon-based system for genome engineering of cereal crops using a deconstructed version of the wheat dwarf virus (WDV). In wheat cells, the replicons achieve a 110-fold increase in expression of a reporter gene relative to non-replicating controls. Furthermore, replicons carrying CRISPR/Cas9 nucleases and repair templates achieved GT at an endogenous ubiquitin locus at frequencies 12-fold greater than non-viral delivery methods. The use of a strong promoter to express Cas9 was critical to attain these high GT frequencies. We also demonstrate gene-targeted integration by homologous recombination (HR) in all three of the homoeoalleles (A, B and D) of the hexaploid wheat genome, and we show that with the WDV replicons, multiplexed GT within the same wheat cell can be achieved at frequencies of ~1%. In conclusion, high frequencies of GT using WDV-based DNA replicons will make it possible to edit complex cereal genomes without the need to integrate GT reagents into the genome.
Keywords: CRISPR/Cas9, multiplexed gene targeting, genome editing, homologous recombination, DNA replicons, Wheat, technical advance
INTRODUCTION
Methods to precisely edit plant genomes have advanced remarkably in the last few years with the development of highly efficient and programmable sequence-specific nucleases (SSNs), namely meganucleases (Puchta et al., 1993; Salomon and Puchta, 1998), zinc-finger nucleases (ZFNs; Kim et al., 1996; Townsend et al., 2009), transcription activator-like effector nucleases (TALENs; Bogdanove and Voytas, 2011; Christian et al., 2010) and the clustered regularly interspaced short palindromic repeat (CRISPR) Cas9 system (Hwang et al., 2013; Shan et al., 2013). SSNs are used to introduce a double-strand break (DSB) in the target locus to be modified, which is repaired by one of the two primary pathways: non-homologous end joining (NHEJ) or homologous recombination (HR). In NHEJ, the ends of the broken chromosome are rejoined, sometimes imprecisely, which introduces small insertions or deletions (indels) at the break site (Gorbunova and Levy, 1997). When indels occur in coding sequences, they often create frame-shift mutations that disrupt gene function. In HR, or gene targeting (GT), the DSB is repaired using a template with homology to the break site. The repair template can be the sister chromatid, a homologous or homeologous (in the case of polyploid species) chromosome, or exogenous templates containing specific sequence modifications to be incorporated into the break site. SSNs have been used to modify plant genomes by NHEJ and HR, including important cereal crops such as Zea mays (corn; Liang et al., 2014; Shukla et al., 2009), Oryza sativa (rice; Li et al., 2012; Shan et al., 2013), Triticum sp. (wheat; Shan et al., 2013) and Hordeum vulgare (barley; Wendt et al., 2013).
The efficient delivery of genome engineering reagents to plant cells is necessary to achieve targeted genome modification, particularly GT, which requires the delivery of both an SSN expression cassette and a repair template. Reagent delivery is often accomplished using Agrobacterium-mediated transformation or particle bombardment, both of which transform only a subset of cells in treated tissues. In some cases, mutagenesis and GT can be achieved by stably integrating GT reagents into the plant genome (in planta gene modification; Fauser et al., 2012; Schiml et al., 2014). As all cells in the transgenic plant carry the SSN expression cassette and repair template, the hope is that targeted modification will occur in cells that give rise to the germline, allowing the recovery of seeds with the targeted DNA sequence modification. In planta approaches, however, are time consuming and necessarily require a transgenic intermediate, which may trigger governmental regulation and thereby add extra costs to deregulate crop varieties prior to commercialization. Whereas high-efficiency transformation can be achieved in protoplasts, most plant species, especially monocots, cannot be regenerated from individual cells. More efficient methods to deliver genome engineering reagents are clearly needed if GT is to become routine, especially in difficult to transform crop species such as cereals.
Autonomously replicating virus-based vectors provide an alternative means to deliver genome-engineering reagents to plant cells. Among these are the RNA viruses, which for monocots include wheat streak mosaic virus (WSMV; Choi et al., 2000; Tatineni et al., 2011) and barley stripe mosaic virus (BSMV; Lee et al., 2012). These viruses can be modified to carry heterologous coding sequences, and protein expression has been achieved using WSMV in wheat, barley, corn, Avena sativa (oat), and Secale cereale (rye; Choi et al., 2000); BSMV has successfully expressed heterologous proteins in barley and oat (Haupt et al., 2001; Lawrence and Jackson, 2001). One of the main advantages of RNA virus-based vectors is that RNA does not integrate in the plant genome, and plants modified with RNA viruses expressing SSNs are non-transgenic. RNA viruses have limitations in the amount of heterologous DNA that they can carry, however: excess cargo results in the loss of systemic movement or recombinational loss of the cargo DNA (Shivprasad et al., 1999; Choi et al., 2000). This limits their use for delivering TALEN or Cas9 expression cassettes, which typically exceed the virus’ cargo capacity; however, for relatively small molecules such as ZFNs, meganucleases and sgRNAs, the positive-strand RNA virus tobacco rattle virus (TRV) has proven an effective delivery agent, achieving mutagenesis in somatic and germ cells of tobacco and petunia (Marton et al., 2010; Ali et al., 2015a,b).
DNA viruses, such as the geminiviruses, have also been engineered as vectors for the expression of heterologous proteins in plants. Whereas the cargo capacity of these viruses is quite restricted, they can be converted into non-infectious replicons by replacing genes important for infection and cell-to-cell movement with heterologous sequences, such as SSN expression cassettes and repair templates (Lazarowitz et al., 1989; Ugaki et al., 1991). Geminiviruses replicate by rolling circle replication (RCR; Figure 1), and consequently, viral replicons achieve high copy number, increasing the transient expression of the SSN and abundance of the repair template. Such a deconstructed version of bean yellow dwarf virus (BeYDV) was used to efficiently deliver ZFNs and repair templates to tobacco cells to achieve gene targeting at an integrated reporter gene (Baltes et al., 2014). The frequency of GT at the reporter was 10- to 100-times higher than when using conventional T-DNA delivery (Baltes et al., 2014). Furthermore, the BeYDV replicons have considerable cargo capacity and could deliver mutagenic TALENs and CRISPR/Cas9 reagents (Baltes et al., 2014). Recently, BeYDV replicons were used to achieve the targeted knock-in of a strong promoter upstream of a tomato gene that regulates anthocyanin synthesis; GT frequencies were ~12-fold higher than what could be achieved using standard Agrobacterium T-DNA delivery (Čermák et al., 2015). Three characteristics of the BeYDV replicon-based system (also applicable to other geminiviruses) appear to be key factors for their success as GT vectors: (i) the viral DNA genome can be used as repair template; (ii) the replicon (including the repair template) replicates to high copy number; and (iii) the expression of Rep and RepA viral proteins enhances HR because of the interaction with proteins in the plant cell that promote progression into S phase (Baltes et al., 2014). Geminivirus-based replicons have been modified to express heterologous proteins in monocots, including wheat dwarf virus (WDV) and maize streak virus (MSV) (Lazarowitz et al., 1989; Shen and Hohn, 1994, 1995), suggesting that they might be effective for genome engineering in cereals.
Figure 1.
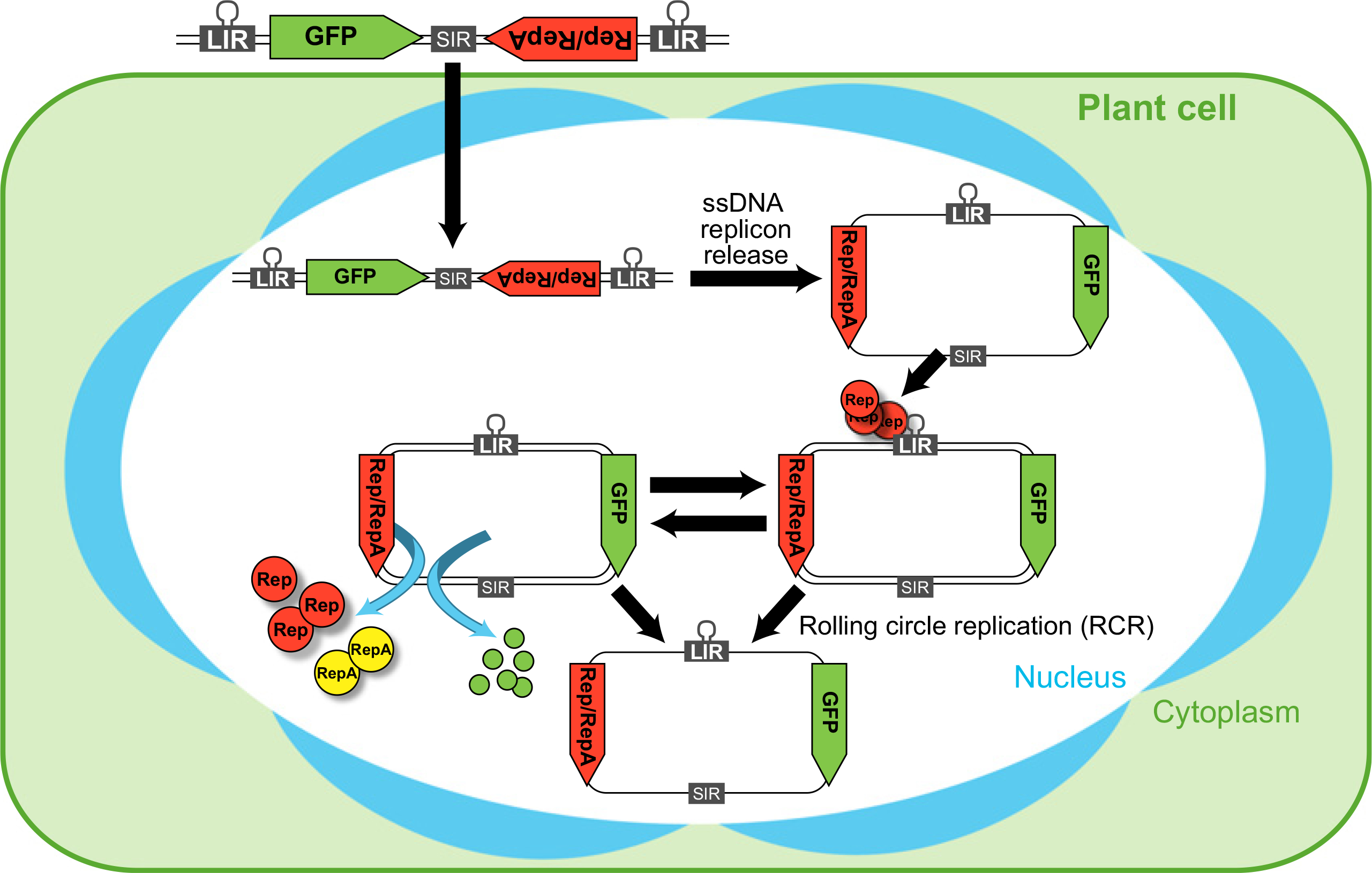
Replication cycle of wheat dwarf virus (WDV)-derived replicons in transformed plant cells. For genome engineering purposes, the WDV-derived replicon is delivered into plant cell nuclei by either particle bombardment (double-stranded DNA, dsDNA) or Agrobacterium-mediated transformation (single-stranded DNA, ssDNA). Then, an ssDNA replicon is released and converted into dsDNA by host polymerases. The replication initiation protein (Rep) recognizes a domain in the large intergenic region (LIR) and nicks the DNA at a 9-nt conserved site found on the hairpin structure of the LIR to promote rolling circle replication (RCR). As a result, newly synthesized ssDNA replicons are formed and converted again into dsDNA replicons. The new dsDNA replicons can be used to either express the encoded proteins (Rep, RepA and heterologous proteins such as GFP) or start a new RCR cycle.
In the present work, we developed replicons based on the WDV for cereal genome engineering. We show that our WDV-derived replicons amplify and express heterologous proteins in wheat, corn and rice. We characterize the replication and protein expression of the WDV system in wheat cells, and compare different replicon architectures to optimize WDV as a vector for delivering CRISPR/Cas reagents and donor templates. We also show that WDV replicons increases GT efficiency greater than 10-fold in wheat cells, and they are also able to promote multiplexed GT, achieving within the same cell targeted integration of different reporter genes in different loci of the polyploid wheat genome.
RESULTS AND DISCUSSION
Design of DNA replicons for genome editing in monocots
Two different geminiviruses – WDV and tomato leaf curl virus (ToLCV) – were deconstructed to create autonomous replicons that function in plant cells. WDV is an ssDNA virus (Mastrevirus) that infects a variety of grasses, including most cereals. WDV-derived replicons have previously been used to express foreign proteins in wheat and maize cells (Matzeit et al., 1991; Ugaki et al., 1991; Suarez-Lopez and Gutierrez, 1997). ToLCV is also an ssDNA virus (Begomovirus), and although its natural hosts are normally Solanaceous species, ToLCV-derived replicons efficiently replicate and express GFP in rice (Pandey et al., 2009). The movement protein and coat protein (CP) coding sequences were removed from both WDV and ToLCV, thereby eliminating the possibility of cell-to-cell movement as well as plant-to-plant insect-mediated transmission. Notably, the lack of the CP increases the copy number of dsDNA replicon intermediates (Padidam et al., 1999), probably because CP is not available to sequester and package ssDNA into virions, and the loss of CP/Rep interactions represses viral replication (Malik et al., 2005). A GFP coding sequence was inserted in both vectors such that expression would be driven from the endogenous viral promoters (giving rise to pWDV2-GFP and ToLCV-GFP; Figure S1).
Both pWDV2-GFP and ToLCV-GFP were used to transform wheat calli by biolistics. As a control, calli were also transformed with a BeYDV replicon carrying a GFP cassette (pBeYDV-GFP; Baltes et al., 2014). Only cells transformed with pWDV2-GFP showed evidence of GFP expression (Figure S1). pWDV2-GFP was also able to replicate and express GFP in rice (Figure S2) and corn (Figure S3). Consequently, we decided to use WDV-derived replicons as the platform for the targeted modification of monocot genomes, focusing on wheat. Two other WDV-derived replicons were made (Figure S4): pWDV1 has the maize ubiquitin1 promoter (ZmUbi) downstream of the left long intergenic region (LIR) to drive expression of heterologous protein-coding sequences; pWDV3 is a replicase-deficient version of pWDV2 that has a premature stop codon in the Rep/ RepA gene (C → G substitution at position 11 of the nucleotide coding sequence).
Time course of replicon amplification and transcript level
Wheat calli were transformed with either the pWDV2-GFP or the Ubi-GFP control to study transcript level and replication over a 2-week time course (Figure 2a). Relative transcript levels of GFP and Rep/RepA were monitored by quantitative real-time PCR (qRT-PCR). We observed a peak in the Rep/RepA transcript abundance at around 3 days post-bombardment (dpb). GFP transcripts expressed by the pWDV2-GFP peaked at 3–7 dpb, and then decreased rapidly. This decrease in the GFP transcripts may be a consequence of viral DNA methylation (Seemanpillai et al., 2003; Bian et al., 2006; Yadav and Chattopadhyay, 2011), and/or post-transcriptional gene silencing mediated by the RNAi defense mechanism (Vanitharani et al., 2005; Rodriguez-Negrete et al., 2009; Yadav and Chattopadhyay, 2011). In addition, we found that at 5 dpb, the copy number of the pWDV2-GFP replicon was ~80 times higher than the Ubi-GFP control plasmid (Figure 2b); the GFP transcript level was ~30 times higher (Figure 2a). Consequently, the optimal time frame for collecting samples in genome engineering experiments was estimated to be 5–7 days. These results are consistent with our previous experiments using the BeYDV in infiltrated tobacco plants (Baltes et al., 2014). In that case, the maximum copy number occurred around 5 days after transformation, with around 6000 copies of the replicon per single-copy gene, which resulted in high expression of the heterologous sequence on the replicon.
Figure 2.
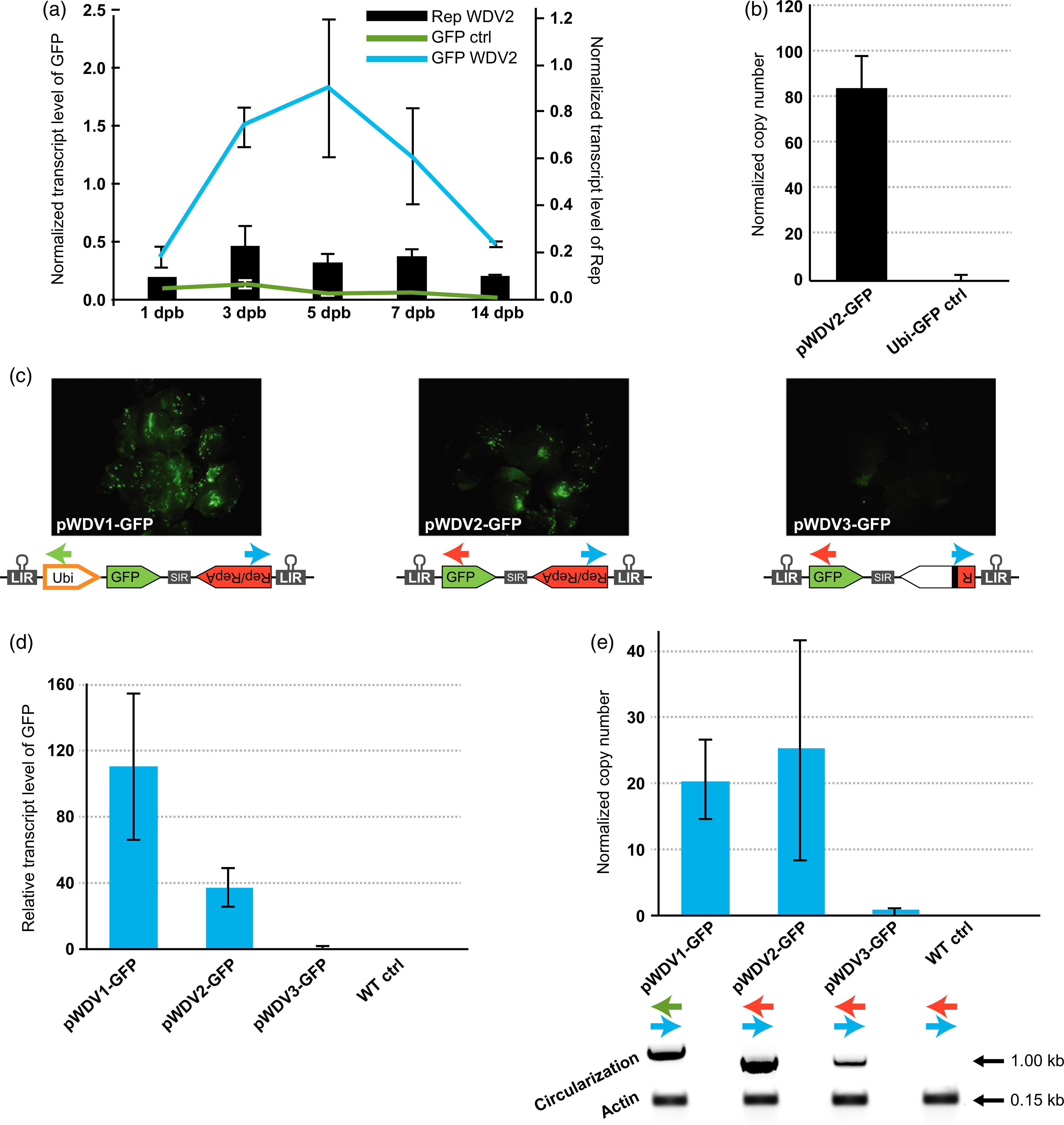
Wheat dwarf virus (WDV) replicon replication and performance of different WDV architectures designed for genome engineering of cereal species. (a) Time course of WDV replication and relative transcript level of GFP and Rep/RepA proteins in wheat calli at different days post bombardment (dpb). (b) Normalized copy number of the GFP-containing replicon relative to the Ubi-GFP control at 5 dpb. (c) GFP expression in wheat calli using the different WDV architectures: WDV1-GFP (left panel), WDV2-GFP (central panel) and WDV3-GFP (right panel). (d) qRT-PCR quantification of transcript level normalized to the non-replicating pWDV3-GFP. (e) Replicon copy number normalized to the non-replicating pWDV3-GFP and detection of circularization of the replicon and the actin PCR control. The colored arrows in (c) indicate the primers used to detect circularization in each replicon variant. Error bars represent the standard errors (SEs) of three independent biological replicates (n = 6 wheat calli per replicate) transformed by particle bombardment. Gold particles with no DNA were used to transform the wild-type (WT) control.
Different architectures of WDV show differences in replication and the expression of the heterologous proteins
To find the best promoter for heterologous protein expression, GFP transcript level and copy number of the replicon were monitored by qRT-PCR in wheat calli transformed with the different replicon architectures (Figure 2c–e). The level of GFP expression from the LIR bidirectional promoter in pWDV2-GFP was compared with the expression from the ZmUbi promoter in the pWDV1-GFP plasmid in transformed calli at 5 dpb. The pWDV3-GFP vector was used in the experiment as a non-replicating control. GFP expression and replicon circularization were observed in calli transformed with the three different constructs, although in the non-replicating control (pWDV3-GFP), expression was observed in only a few cells (Figure 2c). The quantification of RNA abundance by qRT-PCR showed a 110-fold increase in the expression of GFP with pWDV1-GFP and a 37-fold increase with pWDV2-GFP, when normalized to the non-replicating control (pWDV3-GFP; Figure 2d). We then analyzed the copy number of each of the plasmids (Figure 2e), and found a very similar increase in both pWDV1-GFP (20-fold) and pWDV2-GFP (26-fold) compared with the non-replicating control (WDV3-GFP). Together, these results show that the ZmUbi promoter (~2 kb) positively influenced the expression of the heterologous protein in the replicon without compromising replicon replication. Timmermans et al. (1992) reported lower copy numbers of the WDV vectors in maize cells when they increased the size of the replicon to ~3 kb; however, in our system the extra ~2 kb of the ZmUbi promoter did not significantly affect replication. Consequently, we selected the pWDV1 architecture as the most amenable for the gene-targeting experiments, as for this purpose we require both high expression of the nuclease and a high copy number of the donor template.
WDV-induced targeted mutagenesis
To test whether the WDV-derived replicons enable targeted mutagenesis, we designed a 20-nt chimeric single-guide RNA (sgRNA) that recognizes the third exon of a ubiquitin gene (sgUbi1). Wheat protoplasts were transformed with the different WDV constructs expressing the CRISPR/Cas reagents, namely sgUbi1 and a wheat codon-optimized Cas9 (TaCas9; vectors pWDV1-CR, pWDV2-CR, and pWDV3-CR; Figure 3a). Total DNA was isolated 2 days after transfection, and a 533-bp region encompassing the cleavage site was PCR-amplified and digested with HaeIII (present in the seed sequence of the sgUbi1). Mutation frequencies ranged from 12.9 to 20.7% (Figure 3b). Undigested bands were then purified and sequenced and found to contain mostly small (2–6 bp) deletions at the predicted cleavage site (Figure 3c). Interestingly, the pWDV2-CR appeared to be most effective for targeted mutagenesis of the ubiquitin locus. These results demonstrate that the WDV replicon system is compatible with the CRISPR/Cas9 nucleases and can be used to enable targeted mutagenesis in wheat.
Figure 3.
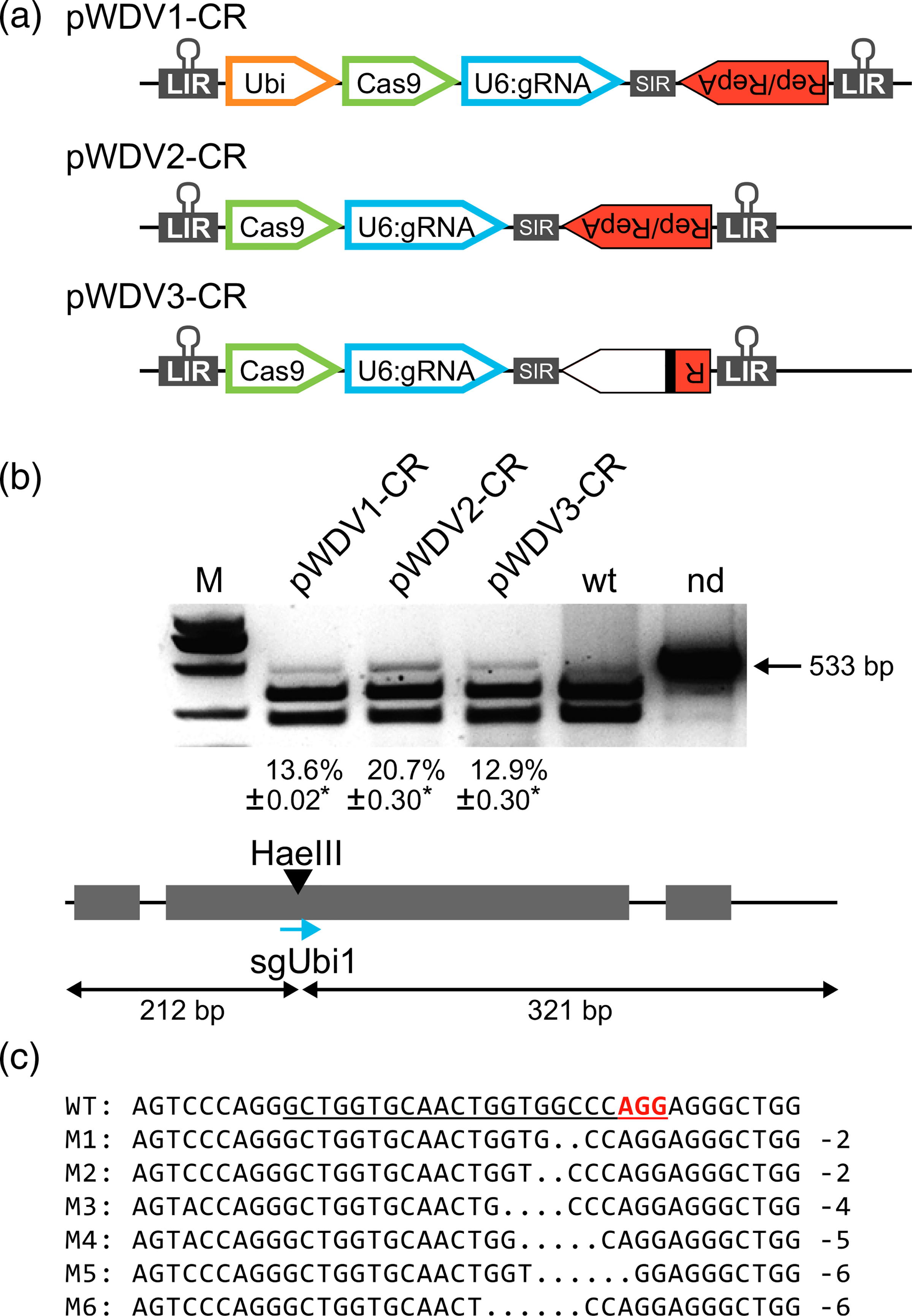
Wheat dwarf virus (WDV) replicon-mediated expression of the CRISPR/Cas9 system for targeted mutagenesis in wheat cells. (a) CRISPR/ Cas reagents expressed from the different architectures of the WDV replicons. (b) PCR/restriction enzyme assay to detect mutations in the ubiquitin gene induced with the sgUbi1 expressed in the different vectors shown in (A); *percentages of NHEJ (% ±SEs) represent the average of two different transfections and have been normalized to the transfection efficiency (40 and 60%, respectively). (c) Sequences of resistant bands obtained by NHEJ of the ubiquitin gene mediated by the pWDV1-CR vector.
High-efficiency gene targeting in wheat cells mediated by WDV replicons
Having identified a functional nuclease targeting the ubiquitin locus, we next tested whether the WDV replicons can induce GT in wheat cells. First, we used wheat protoplasts to compare the different WDV architectures for their ability to knock-in a promoter-less T2A:gfp sequence (Figures 4 and S5) into the third exon of the ubiquitin gene. CRISPR/ Cas9 reagents (TaCas9 and sgUbi1) and the T2A:gfp donor template were delivered by: (i) pWDV1.CR.GFP, pWDV1 with the ZmUbi promoter expressing TaCas9; (ii) pWDV2.CR.GFP, the pWDV2 version with the LIR expressing TaCas9; or (iii) pWDV3.CR.GFP, the replicase-deficient pWDV3 version with the LIR expressing TaCas9. We observed GFP expression only in protoplasts transfected with pWDV1.CR.GFP and pWDV2.CR.GFP; pWDV1.CR.GFP showed the highest GT efficiency (3.8%; Figure 4a). Targeted integration of the T2A:gfp fragment was detected only in samples transfected with pWDV1.CR.GFP (Figure 4b). Both 5' and 3' junctions of the targeted T2A:gfp fragment were sequenced to confirm that GT had occurred at the expected site (Figure 4c). We found integration of the T2A:gfp sequence in the three homoeoalleles (A, B and D) of the ubiquitin gene, indicating that the sgUbi1 was active in the three sites, and that the identity between the homology arms of the donor template (cloned from the D genome) and the A and B homoeoalleles (94.9 and 95.1%, respectively; Table S1) was sufficient to promote GT. No mutations were observed in the sequenced products, suggesting the repair was perfect. The difference in GT efficiencies between pWDV1 and pWDV2 is most likely the result of a higher expression level of TaCas9, as both replicons have similar copy numbers. These results confirm that the WDV system can be used to specifically target a heterologous sequence by HR in the wheat genome.
Figure 4.
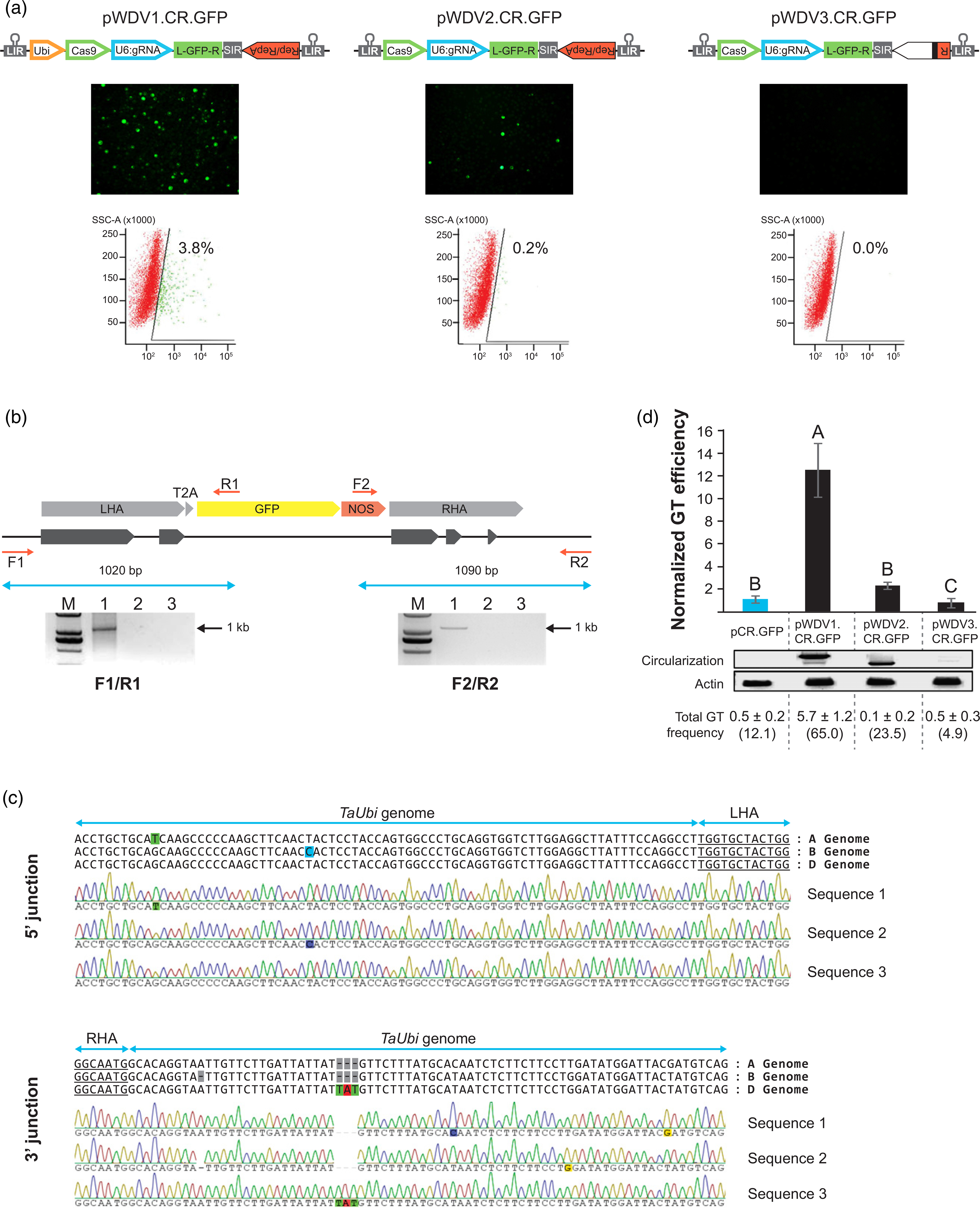
High-efficiency gene targeting (GT) mediated by wheat dwarf virus (WDV)-derived replicons expressing CRISPR/Cas9 reagents in wheat cells. (a) GT-mediated expression of GFP in wheat protoplasts 2 days after transfection with the WDV vectors carrying the CRISPR/Cas9 reagents and the T2A:gfp donor template. Quantification of the number of GFP-positive cells was carried out by flow cytometry. (b) Schematic of the expected integration of the T2A:gfp sequence in the genomic ubiquitin locus and molecular characterization by PCR using specific primers. Lane numbers in the gel denote the following constructs: 1, pWDV1.CR.GFP; 2, pWDV2.CR.GFP; 3, pWDV3.CR.GFP; (c), DNA sequences for the 5' and 3' junctions of the integrated T2A:gfp fragment obtained with the pWDV1.CR.GFP vector. (d) Enhancement of GT efficiency in wheat scutella mediated by the WDV-derived replicons. Black bars denote the normalized GT efficiency compared with the non-viral control (pCR.GFP, blue bar). Error bars represent the standard error (SE) of five different transformation experiments (n = 132 scutella for each treatment). Analysis of variance (ANOVA) was performed. Different letters in the bar graph indicate significant differences (P < 0.05), as determined by the ‘least significant difference’ (LSD) post-hoc all-pairwise comparison test. Replicon circularization and endogenous actin gene control were detected by PCR. Total GT frequency (% ±SE) represents the percentage of cells with GT events relative to the total number of transformed cells (as calculated with the pWDV1-GFP control in each individual experiment). The average percentage of scutella showing at least one GT event is shown in parenthesis.
We next quantified the efficiency of GT in wheat scutella with the different replicon architectures, and compared them with a non-viral control (pCR.GFP), in which TaCas9 is driven by the ZmUbi promoter. We transformed wheat scutella by particle bombardment, because this is one of the most frequently used approaches for creating transgenic wheat plants. GT events were calculated by counting the number of cells expressing GFP at 7 dpb (Figures 4d and S6). We observed a significant increase (~12-fold) in the number of GFP-positive cells with the pWDV1.CR.GFP compared with the pCR.GFP non-replicating control and the two other architectures (pWDV2.CR.GFP and pWDV3.CR.GFP). Of the total number of scutella transformed with pWDV1.CR.GFP, 65% showed GT events, whereas only 12.1% showed GT when using the pCR.GFP control. In addition, when we normalized the total number of GT events with the transformation efficiency (total number of GFP-expressing cells in the pWDV1-GFP control), the overall GT frequency of the pWDV1.CR.GFP was 5.74%; the pCR.GFP control was 0.50% (Figure 4d).
We also synthesized a vector, designated pWDV1.CR.BFP, that contains a sgRNA complementary to the Mildew Locus O (MLO) gene (sgMLO1), as well as a donor template, designated P2A-bfp, designed to knock-in a promoter-less bfp coding sequence by HR (Figure S7). We transfected wheat protoplasts with pWDV1.CR.BFP and obtained a 6.4% GT frequency (Figure 5a). Left (5') and right (30) junctions were amplified by PCR (Figure 5b), cloned, and sequenced (Figure 5c). We only detected integration of the P2A-bfp into the MLO homoeoallele on chromosome D. The identity between the homology arms of the P2A-bfp and the A and B homoeoalleles (83 and 80.4%, respectively) might explain why GT of the P2A:bfp occurred preferentially at the D homoeoallele (100% identity). We believe that GT may also take place at the A and B homoeoalleles, but at a lower frequency. These results demonstrate that the WDV system significantly enhances GT frequencies in wheat scutella, particularly when Cas9 is expressed by the strong and constitutive ZmUbi promoter.
Figure 5.
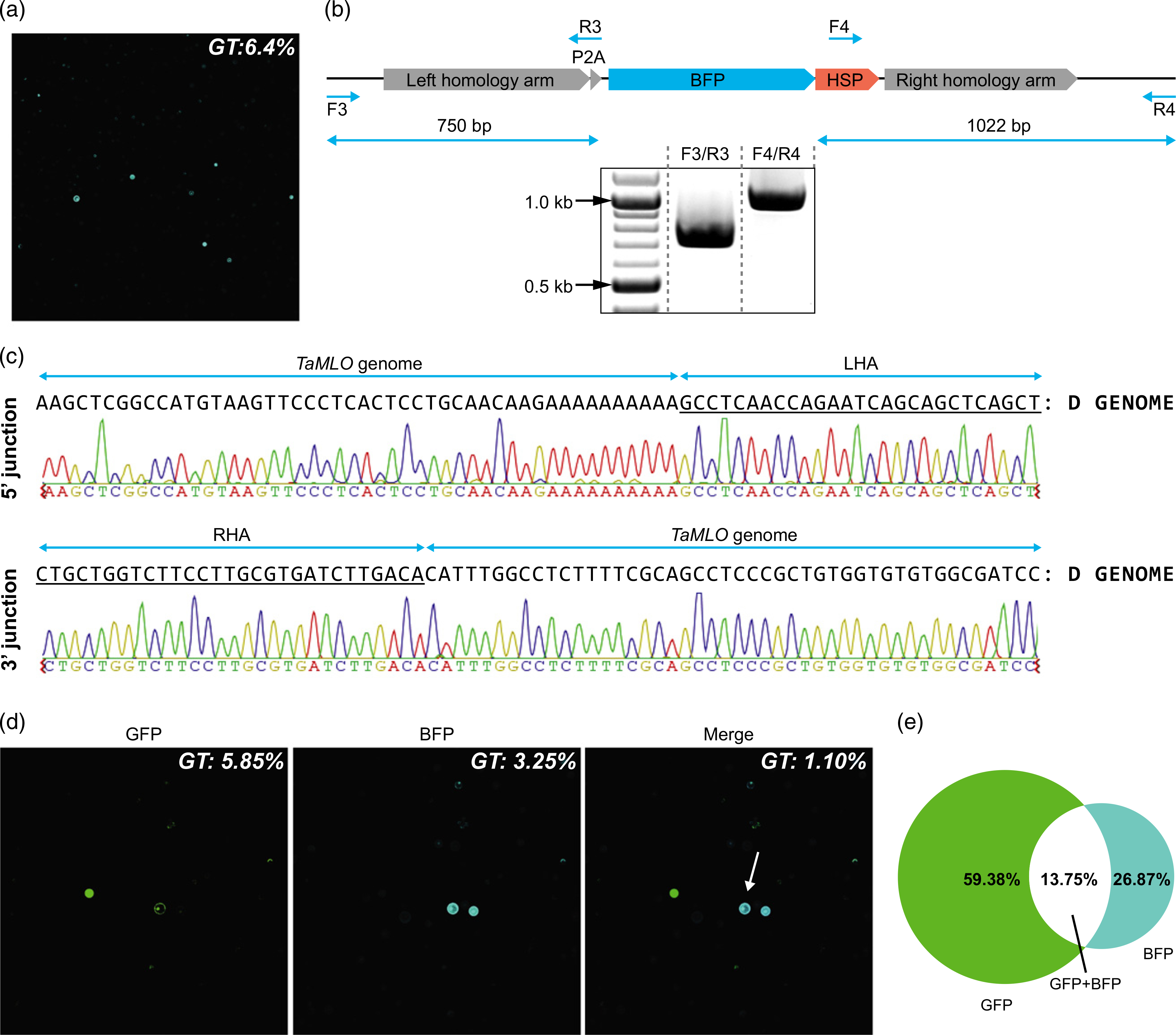
Single-cell multiplexed gene targeting (GT) mediated by the wheat dwarf virus (WDV)-CRISPR/Cas9 system in wheat cells. (a) GT-mediated expression of BFP in wheat protoplasts 2 days after transfection with the WDV vectors carrying the CRISPR/Cas9 reagents and the P2A:bfp donor template. Quantification of the BFP-positive cells was carried out by image quantification. (b) Schematic of the expected integration of the P2A:bfp sequence in the genomic MLO locus and PCR amplification of the 5' and 3' junctions using specific primers. (c) DNA sequences are shown for the 5' and 3' junctions, indicating the knock-in of the P2A:bfp into the MLO homoeoallele in the D genome. (d) Multiplexed GT of the promoter-less T2A:gfp and P2A:bfp sequences in wheat protoplasts. GFP and BFP channels are shown in the left-hand and center pictures, respectively. The right-hand picture is a merged image of the two single-channel pictures. The arrow in the right picture denotes a single cell expressing both GFP and BFP. (e) Diagram of GFP, BFP and GFP+BFP knock-in frequencies relative to the total number of cells that have undergone GT.
Multiplexed gene targeting in wheat cells
Next, we examined the ability of the WDV replicons to achieve multiplexed GT within the same cell. Our experiment was designed to simultaneously target the integration of T2A-gfp and P2A-bfp into the ubiquitin and MLO loci, respectively. We transfected wheat protoplasts with both pWDV1.CR.GFP and pWDV1.CR.BFP, and observed GT frequencies of 5.85 and 3.25% for the GFP and BFP reporters, respectively. The GT frequency was 1.1% for simultaneous integration of both reporters (Figure 5d). This means that 13.75% of the cells that have undergone gene targeting contain both events (Figure 5e).
Multiplexed GT was also accomplished with a single vector (pWDV1.CR.GFP+dsRed) designed to simultaneously modify the ubiquitin and the 5-enolypyruvylshikimate-3-phosphate synthase (EPSPS) loci. pWDV1. CR.GFP+dsRed has the sgUbi1 sgRNA and T2A:gfp donor template described above for GT into the ubiquitin gene. It also carries the sgEPS1 sgRNA and a donor template designed for in-frame integration of dsRed into the EPSPS coding sequence (P2A:dsRed; Figure S8). We transformed wheat scutella by biolistics and observed multiplexed integration of both reporters in 0.4% of the cells showing GT (Figure S9a,b). The low frequency of GT with the P2A: dsRed (only 4.7% of the GT cells) might be explained by the low activity of the sgRNA used, the short length of the left homology arm (210 bp) or a combination of both factors. We detected integration of the P2A-dsRed sequence in two of the EPSPS homoeoalleles (A and D genomes; Figure S9c). Collectively, these results indicate that multiplexed GT in wheat cells can be achieved at high frequency using WDV replicons that deliver active sgRNAs and donor templates with homology arms of proper length. This may let us knock-in a selectable marker to allow cells that have undergone GT to be identified. Such resistant cells could then be screened for GT at a second locus of interest, which may not provide a selectable or screenable phenotype.
In conclusion, GT is still challenging in plants, because both the SSNs and the repair template must be simultaneously delivered to cells. Consequently, there are very few examples of GT at genomic loci using reagents such as CRISPR/Cas9 (Li et al., 2013; Schiml et al., 2014; Shan et al., 2014). We employed the use of geminivirus-based DNA replicons to create a facile and transient system for cereal genome engineering. We demonstrated that WDV-derived replicons increase GT frequencies 12-fold over standard methods of DNA delivery. We further show that the Cas9 promoter was critical for achieving high-efficiency gene targeting: the ZmUbi promoter significantly enhanced GT frequencies compared with the viral LIR promoter. In addition, we demonstrate multiplexed, targeted integration by HR in all three of the homoeoalleles (A, B and D) in hexaploid wheat cells using CRISPR/Cas9. Our reagents offer considerable potential for genome editing of staple cereal crop genomes, including complex polyploid genomes such as wheat.
EXPERIMENTAL PROCEDURES
Vector construction
The replication elements of WDV (LIR, SIR and Rep/RepA) were PCR-amplified from pWI11 (Ugaki et al., 1991) and cloned by Gibson assembly (New England Biolabs) into the multi-cloning site of the pCLEAN-G185 binary vector in a LIR-SIR-Rep-LIR configuration (Figure S4). The final vector included the Gateway attR1 and attR2 sites (Invitrogen, now ThermoFisher Scientific, https://www.thermofisher.com), either between the ZmUbi promoter and the SIR region (vector pWDV1) or between the LIR and SIR regions (vectors pWDV2 and pWDV3). The Gateway sites make it possible to clone heterologous sequences on the virion sense strand. The bidirectional LIR promoter was included in all pWDV vectors to allow the release of the circular replicon. The replicase proteins (Rep/RepA) were expressed in all cases by the LIR promoter. The LIR promoter was used to express heterologous sequences in pWDV2 and pWDV3.
GFP was cloned into the Gateway site of the different WDV replicons, resulting in pWDV1-GFP, pWDV2-GFP and pWDV3-GFP. For the expression experiments, the vector Ubi-GFP (Figures S1 and S2) was used as non-geminivirus control, and has the ZmUbi promoter driving expression of GFP. For the directed mutagenesis and GT experiments, CRISPR/Cas9 reagents were cloned into the Gateway site. A wheat codon-optimized Cas9 sequence (TaCas9) was synthesized using gBLOCKs (Integrated DNA Technlogies, http://www.idtdna.com; Figure S10). The TaCas9 coding sequence contains an N-terminal 3x Flag and N- and C-terminal nuclear localization signals (NLSs) from the simian vacuolating virus 40 (SV40) and nucleoplasmin, respectively. For the expression of the sgRNAs, we used the Triticum aestivum U6 RNA polymerase III promoter (TaU6; Figure S11). sgRNAs were designed to recognize 20 nt (G-N19) in the target sites, and a Protospacer Adjacent Motif (PAM) sequence (NGG) was adjacent to the 3' end. sgUbi1 (5'-GCTGGTGCAACTGGTGGCCC-3') is completely complementary to the third exon of the ubiquitin homoeoalleles on chromosomes 1AL and 1DL; it has one mismatch at position 6 (from the PAM sequence) with the 1BL target site. The sgMLO1 (5'-GAACTGGTATTCCAAGGAGG-3') is complementary to the 5AL and 4DL homoeoalleles of the MLO gene, and has one mismatch at position 6 from the PAM sequence to the homeoallele on chromosome 4BL; the sgEPS1 (5'-GTAATGCTGGAACTGCAATG-3') has complete complementarity with exon 1 of the EPSPS alleles in both the 4AL and 7DS chromosomes.
Donor templates for GT experiments were generated to knock-in the different fluorescence reporters into the ubiquitin, MLO and EPSPS loci. GT into the ubiquitin gene was performed using a promoter-less ‘T2A-gfp-nos terminator’ cassette (referred as T2A:gfp; Figure S5). The cassette was flanked by left and right homology arms (PCR-amplified from the 1DL homoeoallele) of 747 and 773 bp, respectively. For GT into the MLO gene, we synthesized the ‘P2A-bfp-HSP terminator’ sequence (referred to as P2A:bfp; Figure S7), which was flanked by 674 -and 647-bp homology arms. The homology arms were PCR-amplified from the 4DL homoeoallele. For the GT experiments into the EPSPS gene, we synthesized a promoter-less ‘P2A-dsRed-nos terminator’ (referred to as P2A:dsRed; Figure S8), which was flanked by 210- and 646-bp homology arms cloned from the 4AL homoeoallele. Silent mutations in the PAM sequence of each designed sgRNA were introduced, when necessary, into the donor templates to avoid the cleavage by the CRISPR/Cas reagents.
Plant material
Wheat (T. aestivum cv. Bobwhite) plants were grown at 20°C day and 14°C night temperatures, with a relative air humidity of 60% under a 16-h photoperiod. Plants of the maize (Z. mays) Hi II hybrid genotype were grown in the glasshouse at 28°C with light supplementation for a 12-h photoperiod. Rice (O. sativa cv. Nipponbare) grains were dehulled and sterilized with 75% ethanol and 2.5% sodium hypochlorite, and then plated on half-strength MS solid medium in a round glass cup, and covered with sterilized plastic film. Then, plants were grown at 28°C with a photoperiod of 16 h light and 8 h dark for about 14–20 days in a growth chamber. Wheat and rice protoplast isolation was carried out from wheat and rice leaves as described by Shan et al. (2013).
Biolistic transformation
Immature wheat scutella (0.5–1.5 mm in diameter) were isolated from primary tillers harvested 16 days after anthesis, and were used for biolistic transformation ~1 h after isolation or were cultured for 2–3 weeks to induce callus. Scutella isolation and culture conditions were as described by Gil-Humanes et al. (2011), with an osmotic treatment applied between 1 h before and 2 h after bombardment. F2 immature zygotic embryos (1.5–2.0 mm) of corn were aseptically dissected from ears harvested 10–13 days after pollination. Immature corn embryos were isolated as described by Ishida et al. (2007) and placed with the embryo axis facing down in culture medium to induce cell division and callus formation for 2–3 weeks. Biolistic bombardment of immature embryos or calli of the different species was carried out using a PDS-1000 gene gun. Equimolar quantities (1 pmol DNA mg−1 of gold) of each plasmid were used for each experiment, with 60 μg of gold particles (0.6 μm in diameter) per shot. GFP images of transformed tissue were taken using a camera mounted on a Nikon microscope (http://www.nikon.com).
Genomic DNA and total RNA isolation from wheat callus and protoplasts
Total genomic DNA was isolated from ~200 mg of wheat callus with a 20 mM Tris-HCl (pH 7.5), 250 mM NaCl, 25 mM EDTA, 0.5% SDS extraction solution that included RNase. A 2% cetyl trimethylammonium bromide (CTAB) solution was used for total genomic DNA isolation from leaves (~50 mg of tissue) and protoplasts (~200 000 cells). RNA from wheat callus (~200 mg) was isolated using TRIzol reagent (Invitrogen) according to the manufacturer’s instructions and treated with TURBO DNase (Ambion, now ThermoFisher Scientific, http://www.thermofisher.com) to eliminate DNA contamination. RNA (500 ng) was converted to cDNA using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, now ThermoFisher Scientific, http://www.thermofisher.com).
Detecting WDV replicon circularization
Circularization of the replicon was detected by PCR using the Expand Long Template system (Roche, http://www.roche.com). Specific primers were designed to detect circularization of the different pWDV constructs expressing GFP or TaCas9 (Table S2). PCR conditions in all the cases were: 50–75 ng of DNA template, 0.15 μM of each primer, 1× Expand Long Template Buffer 1 and 1.87 U of the enzyme mix in a 25-μl reaction. Cycling conditions consisted on an initial denaturation step of 94°C for 5 min followed by 30 cycles of 94°C for 30 sec, 55°C for 30 sec and 68°C for 1 min, and then a final extension step of 68°C for 5 min.
Quantitative real-time PCR
qRT-PCR was used to assess both DNA copy number and transcript level from the replicons. qRT-PCR was performed using the FastStart Universal SYBR Green Mix kit (Roche) on the LightCycler® 480 Instrument (Roche). To carry out the copy number and relative transcript level experiments, we designed primers for the GFP coding sequence within the WDV constructs (GFP1_F and GFP1_R), for the Ubi-GFP control plasmid (GFP2_F and GFP2_R), as well as for the Rep and RepA coding sequence (Rep_F and Rep_R) (Table S2). The actin gene (actin_F and actin_R) and the RLI (similar to A. thaliana RNase L inhibitor protein) gene (RLI_F and RLI_R) were used as references to normalize replicon copy number and transcript levels. qRT-PCR conditions were: 0.3 μM of each primer and 1× FastStart Universal SYBR Green Mix in a final volume of 10 μl, with either cDNA obtained from 20 ng of total RNA or 50 ng of total genomic DNA for quantification of gene expression and copy number, respectively. Primer efficiencies and Cq values were determined using LINGREGPCR 2013.0 software (Ruijter et al., 2009). Normalized copy number and gene expression were calculated using the equation described in Hellemans et al. (2007) for multiple reference genes, and the results were standardized using an adapted version of the Microsoft Excel Qgene template (Muller et al., 2002). Three technical replicates were performed for each sample. Error bars represent standard errors of three different biological replicates (DNA or RNA from calli or protoplasts transformed independently).
Molecular characterization of targeted mutagenesis
A PCR/restriction enzyme assay was performed to detect mutations induced by NHEJ in the ubiquitin gene in transfected protoplasts. Insertions or deletions at the DSB induced by sgUbi1 would produce the loss of a HaeIII restriction site located just upstream of the PAM sequence. A 533-bp fragment containing the sgUbi1 target site was PCR-amplified simultaneously from the 1AL, 1BL and 1DL alleles using the pair of primers Ubi_F and Ubi_R (Table S2). PCR conditions were 50 ng of DNA template, 0.5 μM of each primer, 1× Q5 Reaction Buffer (NEB) and 0.5 U of the Q5 polymerase (NEB) in a 25-μl reaction. Cycling conditions consisted of an initial denaturation step of 98°C for 30 sec, followed by 40 cycles of 98°C for 10 sec, 64°C for 20 sec and 72°C for 20 sec, and a final extension step of 72°C for 5 min. The PCR product of each reaction was digested with HaeIII for 3 h and resolved on a 2% agarose gel. The frequency of mutations was estimated by quantifying the intensity of the undigested and digested bands with IMAGEJ (Schneider et al., 2012), as described in Shan et al. (2014). Cleavage-resistant amplicons were gel purified, cloned into pJET1.2 (ThermoFisher Scientific) and sequenced.
Molecular characterization of GT
Gene targeting of each of the GFP, BFP and dsRed reporters was detected by PCR. One primer in the genomic flanking region and one primer in the donor template were combined in each case to detect the 5' and 3' junctions of the insertion (Table S2). PCR conditions were: 150 ng of DNA template, 0.5 μM of each primer, 1× Q5 Reaction Buffer (NEB) and 0.5 U of the Q5 polymerase (NEB) in a 25-μl reaction. Cycling conditions consisted of an initial denaturation step of 98°C for 30 sec, followed by 40 cycles of 98°C for 10 sec, a variable annealing temperature for 20 sec and 72°C for 45 sec, with a final extension step of 72°C for 5 min. Amplicons were separated on a 1% agarose gel, purified and cloned into pJET1.2 (ThermoFisher Scientific) for sequencing. Around 10 colonies of each transformation event were sequenced by Sanger sequencing and analyzed.
Quantifying multiplexed GT in wheat protoplasts
Multiplexed GT was calculated by dividing the number of protoplasts expressing GFP and BFP by the total number of cells, and normalizing to the transformation efficiency of each experiment. We used a Nikon A1 Spectral Confocal Microscope to collect random photos with the different filters. IMAGEJ was used to count the number of cells (GFP- and/or BFP-expressing cells and total number of cells) in 10 random pictures for each treatment and experiment. Transformation efficiency was estimated with a replicon-based control plasmid expressing GFP (pWDV2-GFP).
Supplementary Material
Table S1. Nucleotide sequence identity of the homology arms in the DNA donor templates relative to the A, B and D homoeoalleles of the ubiquitin and MLO genes.
Table S2. List of primers.
Figure S1. GFP expression mediated by different geminiviruses in wheat calli.
Figure S2. GFP expression and circularization of the WDV vectors in transfected rice protoplasts.
Figure S3. GFP expression and circularization of the WDV vectors in transformed corn calli.
Figure S4. Schematic of the different architectures of the WDV.
Figure S5. Complete sequence of the T2A:gfp donor template.
Figure S6. Gene targeting of the T2A:gfp sequence into wheat scutella.
Figure S7. Complete sequence of the P2A:bfp donor template.
Figure S8. Complete sequence of the P2A:dsRed donor template.
Figure S9. Single-cell multiplexed GT of GFP and dsRed coding sequences mediated by the WDV-CRISPR/Cas9 system in wheat scutella.
Figure S10. Complete sequence of the wheat codon-optimized Cas9 (TaCas9).
Figure S11. Complete sequence of the sgRNA expression cassette.
ACKNOWLEDGEMENTS
Authors thank Dr Joachim Messing (Rutgers, The State University of New Jersey) for the pWI-11 plasmid containing the WDV genome and Dr Toni Wendt (Aarhus University, Denmark) for the Ubi-GFP plasmid. We acknowledge Dr Kan Wang (Iowa State University) for providing the corn ears for the isolation of immature embryos and Jade Mathre for technical assistance. We also thank Kit Leffler for editing and preparing the figures. This work was supported in part by grants to D.F.V. from the National Science Foundation (IOS-1444511and IOS-1339209), and to C.G. from the National Natural Science Foundation of China (31420103912). Javier Gil-Humanes acknowledges the Fundación Alfonso Martin Escudero for his post-doctoral fellowship.
Footnotes
CONFLICT OF INTEREST
D.F.V. and N.J.B. are named inventors on a patent application filed by the University of Minnesota on the use of geminivirus replicons for plant genome engineering and licensed to Cellectis S.A. D.F.V. serves as Chief Science Officer for Calyxt, a wholly-owned subsidiary of Cellectis S.A. J.G.H. and N.J.B. serve currently as Research Scientists for Calyxt.
SUPPORTING INFORMATION
Additional Supporting Information may be found in the online version of this article.
REFERENCES
- Ali Z, Abul-faraj A, Li L et al. (2015a) Efficient virus-mediated genome editing in plants using the CRISPR/Cas9 system. Mol. Plant, 8, 1288–1291. In press. [DOI] [PubMed] [Google Scholar]
- Ali Z, Abul-faraj A, Piatek M and Mahfouz MM (2015b) Activity and specificity of TRV-mediated gene editing in plants. Plant Signal. Behav. 10, e1044191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baltes NJ, Gil-Humanes J, Cermak T, Atkins PA and Voytas DF (2014) DNA replicons for plant genome engineering. Plant Cell, 26, 151–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bian XY, Rasheed MS, Seemanpillai MJ and Ali Rezaian M (2006) Analysis of silencing escape of tomato leaf curl virus: an evaluation of the role of DNA methylation. Mol. Plant Microbe Interact. 19, 614–624. [DOI] [PubMed] [Google Scholar]
- Bogdanove AJ and Voytas DF (2011) TAL effectors: customizable proteins for DNA targeting. Science, 333, 1843–1846. [DOI] [PubMed] [Google Scholar]
- Čermáak T, Baltes NJ, Čegan R, Zhang Y and Voytas DF (2015) High-frequency, precise modification of the tomato genome. Genome Biol. 16, 232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi IR, Stenger DC, Morris TJ and French R (2000) A plant virus vector for systemic expression of foreign genes in cereals. Plant J. 23, 547–555. [DOI] [PubMed] [Google Scholar]
- Christian M, Cermak T, Doyle EL, Schmidt C, Zhang F, Hummel A, Bogdanove AJ and Voytas DF (2010) Targeting DNA double-strand breaks with TAL effector nucleases. Genetics, 186, 757–U476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fauser F, Roth N, Pacher M, Ilg G, Sanchez-Fernandez R, Biesgen C and Puchta H (2012) In planta gene targeting. Proc. Natl Acad. Sci. USA, 109, 7535–7540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil-Humanes J, Ozuna CV, Marín S, León E, Barro F and Pistón F (2011) Genetic transformation of wheat: advances in the transformation method and applications for obtaining lines with improved bread-making quality and low toxicity in relation to celiac disease. In Genetic Transformation (Alvarez M ed). InTech, pp. 135–150. [Google Scholar]
- Gorbunova V and Levy AA (1997) Non-homologous DNA end joining in plant cells is associated with deletions and filler DNA insertions. Nucleic Acids Res. 25, 4650–4657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haupt S, Duncan GH, Holzberg S and Oparka KJ (2001) Evidence for symplastic phloem unloading in sink leaves of barley. Plant Physiol. 125, 209–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellemans J, Mortier G, De Paepe A, Speleman F and Vandesompele J (2007) qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol. 8, R19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang WY, Fu Y, Reyon D, Maeder ML, Tsai SQ, Sander JD, Peterson RT, Yeh JRJ and Joung JK (2013) Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat. Biotechnol. 31, 227–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida Y, Hiei Y and Komari T (2007) Agrobacterium-mediated transformation of maize. Nat. Protoc. 2, 1614–1621. [DOI] [PubMed] [Google Scholar]
- Kim YG, Cha J and Chandrasegaran S (1996) Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proc. Natl Acad. Sci. USA, 93, 1156–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence DM and Jackson AO (2001) Requirements for cell-to-cell movement of Barley stripe mosaic virus in monocot and dicot hosts. Mol. Plant Pathol. 2, 65–75. [DOI] [PubMed] [Google Scholar]
- Lazarowitz SG, Pinder AJ, Damsteegt VD and Rogers SG (1989) Maize streak virus genes essential for systemic spread and symptom development. EMBO J. 8, 1023–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WS, Hammond-Kosack KE and Kanyuka K (2012) Barley stripe mosaic virus-mediated tools for investigating gene function in cereal plants and their pathogens: virus-induced gene silencing, host-mediated gene silencing, and virus-mediated overexpression of heterologous protein. Plant Physiol. 160, 582–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Liu B, Spalding MH, Weeks DP and Yang B (2012) High-efficiency TALEN-based gene editing produces disease-resistant rice. Nat. Biotechnol. 30, 390–392. [DOI] [PubMed] [Google Scholar]
- Li JF, Norville JE, Aach J, McCormack M, Zhang D, Bush J, Church GM and Sheen J (2013) Multiplex and homologous recombination-mediated genome editing in Arabidopsis and Nicotiana benthamiana using guide RNA and Cas9. Nat. Biotechnol. 31, 688–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Z, Zhang K, Chen K and Gao C (2014) Targeted mutagenesis in Zea mays using TALENs and the CRISPR/Cas system. J. Genet. Genomics, 41, 63–68. [DOI] [PubMed] [Google Scholar]
- Malik PS, Kumar V, Bagewadi B and Mukherjee SK (2005) Interaction between coat protein and replication initiation protein of Mung bean yellow mosaic India virus might lead to control of viral DNA replication. Virology, 337, 273–283. [DOI] [PubMed] [Google Scholar]
- Marton I, Zuker A, Shklarman E, Zeevi V, Tovkach A, Roffe S, Ovadis M, Tzfira T and Vainstein A (2010) Nontransgenic genome modification in plant cells. Plant Physiol. 154, 1079–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matzeit V, Schaefer S, Kammann M, Schalk HJ, Schell J and Gronenborn B (1991) Wheat dwarf virus vectors replicate and express foreign genes in cells of monocotyledonous plants. Plant Cell, 3, 247–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller PY, Janovjak H, Miserez AR and Dobbie Z (2002) Processing of gene expression data generated by quantitative real-time RT-PCR. Biotechniques, 32, 1372–1379. [PubMed] [Google Scholar]
- Padidam M, Beachy RN and Fauquet CM (1999) A phage single-stranded DNA (ssDNA) binding protein complements ssDNA accumulation of a geminivirus and interferes with viral movement. J. Virol. 73, 1609–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey P, Choudhury NR and Mukherjee SK (2009) A geminiviral amplicon (VA) derived from Tomato leaf curl virus (ToLCV) can replicate in a wide variety of plant species and also acts as a VIGS vector. Virol. J. 6, 152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puchta H, Dujon B and Hohn B (1993) Homologous recombination in plant cells is enhanced by in vivo induction of double strand breaks into DNA by a site-specific endonuclease. Nucleic Acids Res. 21, 5034–5040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Negrete EA, Carrillo-Tripp J and Rivera-Bustamante RF (2009) RNA silencing against geminivirus: complementary action of post-transcripitional gene silencing and transcriptional gene silencing in host recovery. J. Virol. 83, 1332–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruijter JM, Ramakers C, Hoogaars WM, Karlen Y, Bakker O, van den Hoff MJ and Moorman AF (2009) Amplification efficiency: linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Res. 37, e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomon S and Puchta H (1998) Capture of genomic and T-DNA sequences during double-strand break repair in somatic plant cells. EMBO J. 17, 6086–6095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiml S, Fauser F and Puchta H (2014) The CRISPR/Cas system can be used as nuclease for in planta gene targeting and as paired nickases for directed mutagenesis in Arabidopsis resulting in heritable progeny. Plant J. 80, 1139–1150. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS and Eliceiri KW (2012) NIH Image to ImageJ: 25 years of image analysis. Nat. Methods, 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seemanpillai M, Dry I, Randles J and Rezaian A (2003) Transcriptional silencing of geminiviral promoter-driven transgenes following homologous virus infection. Mol. Plant Microbe Interact. 16, 429–438. [DOI] [PubMed] [Google Scholar]
- Shan Q, Wang Y, Li J et al. (2013) Targeted genome modification of crop plants using a CRISPR-Cas system. Nat. Biotechnol. 31, 686–688. [DOI] [PubMed] [Google Scholar]
- Shan Q, Wang Y, Li J and Gao C (2014) Genome editing in rice and wheat using the CRISPR/Cas system. Nat. Protoc. 9, 2395–2410. [DOI] [PubMed] [Google Scholar]
- Shen W-H and Hohn B (1994) Amplification and expression of the b-glucuronidase gene in maize plants by vectors based on maize streak virus. Plant J. 5, 227–236. [Google Scholar]
- Shen WH and Hohn B (1995) Vectors based on maize streak virus can replicate to high copy numbers in maize plants. J. Gen. Virol. 76(Pt 4), 965–969. [DOI] [PubMed] [Google Scholar]
- Shivprasad S, Pogue GP, Lewandowski DJ, Hidalgo J, Donson J, Grill LK and Dawson WO (1999) Heterologous sequences greatly affect foreign gene expression in tobacco mosaic virus-based vectors. Virology, 255, 312–323. [DOI] [PubMed] [Google Scholar]
- Shukla VK, Doyon Y, Miller JC et al. (2009) Precise genome modification in the crop species Zea mays using zinc-finger nucleases. Nature, 459, 437–441. [DOI] [PubMed] [Google Scholar]
- Suarez-Lopez P and Gutierrez C (1997) DNA replication of wheat dwarf geminivirus vectors: effects of origin structure and size. Virology, 227, 389–399. [DOI] [PubMed] [Google Scholar]
- Tatineni S, McMechan AJ, Hein GL and French R (2011) Efficient and stable expression of GFP through Wheat streak mosaic virus-based vectors in cereal hosts using a range of cleavage sites: formation of dense fluorescent aggregates for sensitive virus tracking. Virology, 410, 268–281. [DOI] [PubMed] [Google Scholar]
- Timmermans MC, Das OP and Messing J (1992) Trans replication and high copy numbers of wheat dwarf virus vectors in maize cells. Nucleic Acids Res. 20, 4047–4054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend JA, Wright DA, Winfrey RJ, Fu F, Maeder ML, Joung JK and Voytas DF (2009) High-frequency modification of plant genes using engineered zinc-finger nucleases. Nature, 459, 442–U161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugaki M, Ueda T, Timmermans MC, Vieira J, Elliston KO and Messing J (1991) Replication of a geminivirus derived shuttle vector in maize endosperm cells. Nucleic Acids Res. 19, 371–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanitharani R, Chellappan P and Fauquet CM (2005) Geminiviruses and RNA silencing. Trends Plant Sci. 10, 144–151. [DOI] [PubMed] [Google Scholar]
- Wendt T, Holm PB, Starker CG, Christian M, Voytas DF, Brinch-Pedersen H and Holme IB (2013) TAL effector nucleases induce mutations at a pre-selected location in the genome of primary barley transformants. Plant Mol. Biol. 83, 279–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav RK and Chattopadhyay D (2011) Enhanced viral intergenic region-specific short interfering RNA accumulation and DNA methylation correlates with resistance against a geminivirus. Mol. Plant Microbe Interact. 24, 1189–1197. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Nucleotide sequence identity of the homology arms in the DNA donor templates relative to the A, B and D homoeoalleles of the ubiquitin and MLO genes.
Table S2. List of primers.
Figure S1. GFP expression mediated by different geminiviruses in wheat calli.
Figure S2. GFP expression and circularization of the WDV vectors in transfected rice protoplasts.
Figure S3. GFP expression and circularization of the WDV vectors in transformed corn calli.
Figure S4. Schematic of the different architectures of the WDV.
Figure S5. Complete sequence of the T2A:gfp donor template.
Figure S6. Gene targeting of the T2A:gfp sequence into wheat scutella.
Figure S7. Complete sequence of the P2A:bfp donor template.
Figure S8. Complete sequence of the P2A:dsRed donor template.
Figure S9. Single-cell multiplexed GT of GFP and dsRed coding sequences mediated by the WDV-CRISPR/Cas9 system in wheat scutella.
Figure S10. Complete sequence of the wheat codon-optimized Cas9 (TaCas9).
Figure S11. Complete sequence of the sgRNA expression cassette.