

Abstract
Medial arterial calcification (MAC) is a chronic systemic vascular disorder distinct from atherosclerosis, frequently, but not always associated with diabetes mellitus, chronic kidney disease, and aging. MAC is also a part of more complex phenotypes in a number of less common diseases. The hallmarks of MAC include disseminated and progressive precipitation of calcium phosphate (CaP) within the medial layer, prolonged clinically silent course, and compromise of hemodynamics associated with chronic limb threatening ischemia. MAC increases the risk of complications during vascular interventions and mitigates their outcomes. With the exception of rare monogenetic defects affecting ATP metabolism, MAC pathogenesis remains unknown and causal therapy is not available. Implementation of genetics and omics-based approaches in research recognizing the critical importance of CaP thermodynamics holds promise to unravel MAC molecular pathogenesis and to provide guidance to therapy. The current state of knowledge concerning MAC is reviewed and future perspectives are outlined.
Keywords: Medial arterial calcification, vascular calcifications, atherosclerosis, arterial revascularization, genetics, omics
Condensed Abstract:
Medial arterial calcification (MAC) is a chronic systemic vascular disorder distinct from atherosclerosis frequently associated with diabetes mellitus, chronic kidney disease, aging and a large number of less common diseases. The hallmarks of MAC include disseminated and progressive precipitation of calcium phosphate (CaP) within the medial layer and prolonged clinically silent course often leading to chronic limb threatening ischemia. MAC increases the risk of vascular interventions and mitigates their outcomes. With the exception of rare monogenetic defects affecting ATP metabolism MAC pathogenesis remains unknown and causal therapy is not available. The current state of knowledge concerning MAC is provided and future perspectives are outlined.
Introduction
Medial arterial calcification (MAC) is a systemic vascular disorder distinct from atherosclerosis associated with increase in arterial stiffness (1), diastolic heart failure (2), impaired perfusion of a high blood flow organs such as brain, kidney and liver (3) and chronic limb threatening ischemia (CLTI) (4–7). MAC is frequently, but not always (8) associated with diabetes mellitus (DM) (9, 10), chronic kidney disease (CKD) (11, 12), aging (13). Furthermore, MAC is a part of more complex phenotypes in a large number of less common disorders (s. Supplement A). MAC interferes with revascularization procedures and mitigates their outcomes (14–16).
MAC is characterized by accumulation of calcium phosphate (CaP) with formation of hydroxyapatite (HAP) crystals (17) resulting in progressive petrification of the medial layer of the arterial wall. In some cases ectopic vascular osteogenesis may also develop (18).
The discovery of bone morphogenetic protein in samples of calcified human atherosclerotic lesions launched the biological hypothesis of arterial calcifications (AC) (19). However, to date with the exception of the monogenic disorders (20–23) the molecular pathogenesis of AC is poorly understood (24) and no causal treatment is available (25). The slow progress in unraveling the pathogenesis stems from confounding distinct AC entities, lack of experimental models replicating MAC (26) and a deficient appreciation of the thermodynamic principles governing CaP precipitations (27).
Here, we review the clinical and research signatures of MAC, address some of the perceived current shortcomings and suggest directions for future research.
Epidemiology
The true prevalence of MAC is not known. MAC frequently represents incidental findings or it is misdiagnosed as atherosclerosis and may therefore escape clinical attention. The prevalence of MAC, based on ankle-brachial index (ABI) >1.3, has been estimated to be ~0.5% of adults, with a male-to-female ration of 3:2 (28), however this is not an accurate metric for diagnosis (29). MAC is found in 17 – 42% of type 2 DM (T2DM) patients (9, 10), in 27–40% of patients with advanced CKD (11, 12) and in up to 72% in patients with critical limb threatening ischemia (CLTI) (30, 31).
The prevalence of MAC in coronary arteries is unknown. However, intravascular ultrasound (IVUS) imaging revealed isolated deep and deep combined with superficial calcifications in 28% and 24%, respectively; potentially corresponding to MAC and atherosclerotic intimal calcifications (IAC) (32).
The prevalence of MAC in the thoracic aorta is currently unknown even though the distinction between MAC and atherosclerotic IAC is clinically important in patients with porcelain aorta (33). In patients with acute and chronic Stanford A or B dissection of the thoracic aorta MAC was found in 22% and 52%, respectively (34).
Clinical implications
In the past MAC was considered an innocent bystander however, studies now demonstrate that MAC can be considered the silent killer of the cardiovascular system.
In a 10-year follow-up study of 133 patients with T2DM MAC was found to be a powerful and independent risk factor for cardiovascular mortality and substantially stronger than the impact of IAC (9). MAC was found to be a strong independent predictor of total cardiovascular mortality, and predicts the risk of future coronary events in diabetic patients (10). Similarly, MAC is closely associated with the duration of hemodialysis and CaP disorders (11). In patients with T2DM the finding of MAC in foot arteries has been associated with below-the-knee (BTK) artery disease and foot ulcers (35, 36). A recent meta-analysis of BTK MAC demonstrated a strong association between infrapopliteal MAC and lower limb amputation risk (37).
These clinical data provide definite proof that MAC is an independent cause of peripheral artery disease (PAD) shadowing the role of atherosclerotic PAD in predisposed patients and clearly justifies systematic studies targeting detection and clinical relevance of MAC in the arteries large and small.
Pathophysiology
The current hypotheses of AC, including both, MAC and IAC, span a wide range of molecular biology pathways found in inflammation, apoptosis, disruptions of CaP homeostasis, matrix vesicle (MV) extrusions, osteogenic transformations, extracellular matrix (ECM) degeneration and genetic aberrations. While these processes overlap, the initial drivers specific to MAC are unclear. Figure 1 provides an overview of some of the proposed pathogenetic principles to explain AC.
Figure 1. Mechanisms Contributing to MAC Pathogenesis.
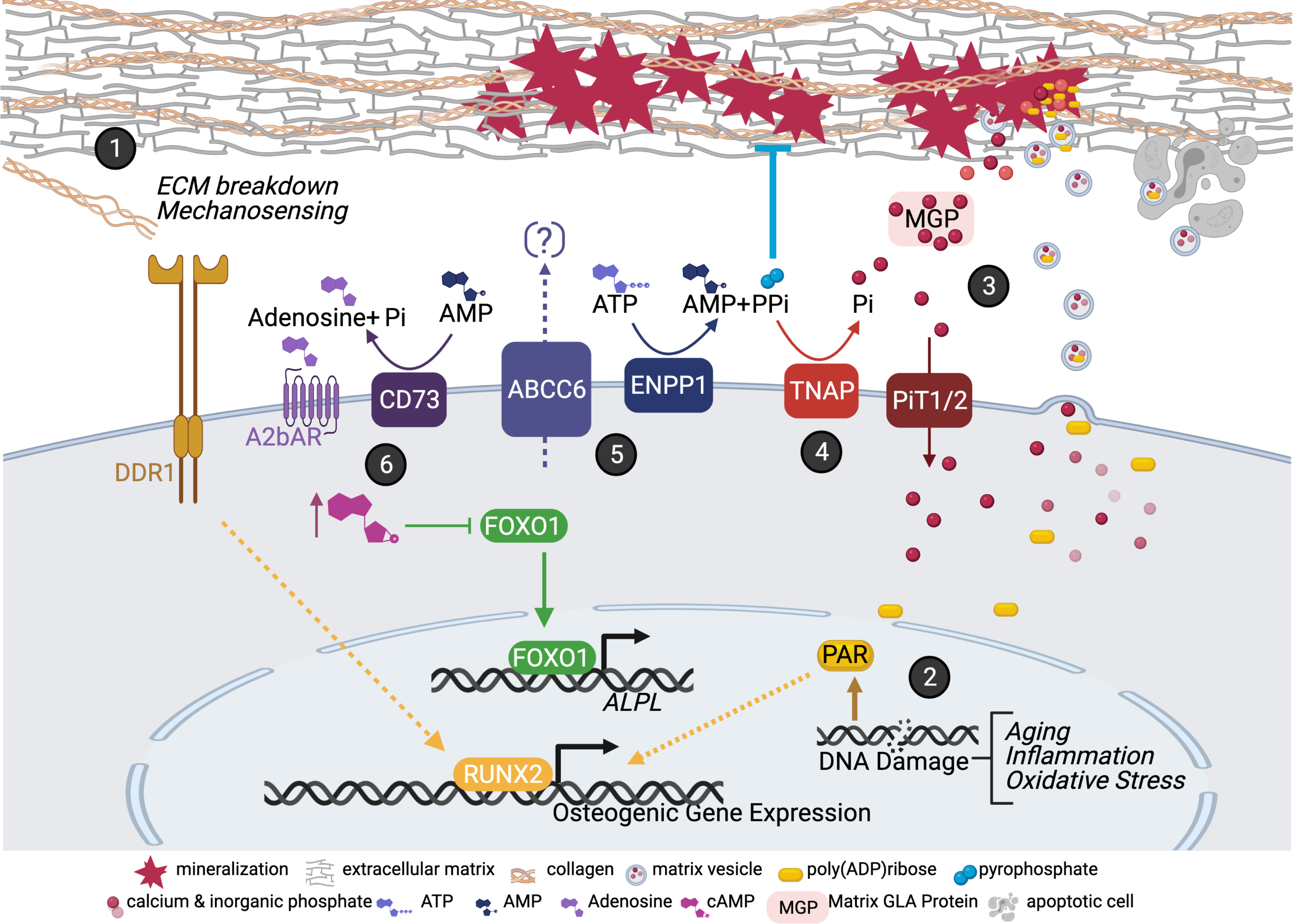
(1) Extracellular matrix (ECM) cues are detected via DDR1 receptor promoting the activation of osteogenic genes. (2) DNA damage response pathway leads to the accumulation of poly (AMP) ribose (PAR) molecules. PAR are secreted in matrix vesicles (MVs) and act as niduses for calcification. (3) Calcium/phosphate minerals can nucleate in the ECM, but this is inhibited via MGLA. PiT1/2 transporter removes inorganic phosphate (Pi) from the extracellular environment. (4) TNAP converts the endogenous inhibitor of mineral nucleation, pyrophosphate (PPi), into the mineral building block, Pi. (5) PPi and AMP are produced from the activity of ENPP1 in the extracellular ATP metabolic pathway. The substrate for ABCC6 is thought to also be ATP but currently not known. (6) CD73-mediated A2b adenosine receptor signaling functions to suppress MAC via cAMP-mediated repression of FOXO1 gene. With a lack of this signaling FOXO1 upregulates ALPL/TNAP. Created
Cellular and molecular considerations
It is assumed that, with the proper building blocks and milieu conditions, CaP ions are prone to nucleate. Thus, it has been proposed that to prevent ectopic nucleation within the ECM, certain proteins such as matrix gamma-carboxyglutamic acid (MGLA) protein may neutralize the crystallization process in a vitamin K-dependent manner (38). While studies have shown that patients with T2DM have a higher serum level of the inactive form of MGLA that correlated with BTK MAC (39) a recent systematic review found that “no single gamma-carboxyglutamic protein species has demonstrated a significant association with VC” (40).
In humans, inorganic phosphate serum levels are tightly regulated within the range of 2.5 to 4.5 mg/dl (0.81–1.45mmol/L). In patients with CKD profound impairment of phosphate homeostasis promotes the development of MAC (41, 42). Extracellular levels of phosphate are sensed and taken up by the type III sodium-dependent phosphate cotransporters PiT1 and PiT2, as well as the calcium sensing receptor. Mutations in PiT2 are found in idiopathic basal ganglia calcification, where calcification has been hypothesized to be due to excess accumulation of extracellular phosphate (43). More recent studies have shown that these proteins not only transport phosphate, but also appear to trigger intracellular signaling events that suppress osteogenic programs (44, 45) as deficiencies in PiT-mediated signaling induce the upregulation of osteogenic genes and exacerbates MAC in a murine CKD model (46, 47). Nevertheless, the applicability of the experimental data to MAC evolution in humans has been questioned (48–50) and the controversy concerning the recipe molding the ingredients into a plausible molecular pathways’ hypothesis persists.
The calcium-sensing receptor (CaSR) regulates CaP homeostasis and is also expressed within arteries with altered CaSR function impairing the transdifferentiation of vascular smooth muscle cells (VSMCs) to mineralizing cells (51). The CaSR is also expressed in monocytes, which have been shown to prevent arterial calcification in-vitro (52). Monocyte CaSR expression is decreased in CKD and this is associated with impaired ability of monocytes to inhibit AC (52).
Other minerals besides CaP ions may influence the development of MAC. In animal and laboratory experiments, magnesium has been shown to modulate the development of phosphate-induced calcification in a dose-dependent manner through the downregulation of promoters and upregulation of inhibitors of calcification (53, 54).
A number of genetic diseases with complex phenotypes, including MAC, originate from inactivating mutations of genes participating in the adenosine triphosphate (ATP) metabolic pathway and shed light on the initiating steps in MAC pathogenesis. ATP is released from cells under a number of metabolic stresses and insults with ATP degradation products generally assisting cells and tissues to resist injury and to maintain homeostasis (55). It has been assumed that mutations in ectonucleotide pyrophosphatase/phosphodiesterase 1 (ENPP1) and ATP-binding cassette sub-family C member 6 (ABCC6) drive MAC via the reduction in extracellular pyrophosphate, a key endogenous inhibitor of ectopic mineralization. Deficiency of ABCC6 is considered pathogenetically linked to the development of disseminated calcifications in pseudoxantoma elasticum (56). ENPP1 breaks down ATP to AMP and pyrophosphate and Ecto-5′-nucleotidase (CD73) converts extracellular AMP into phosphate and adenosine, which can signal via the four adenosine receptors. Lack of adenosine signaling promotes AC due to CD73 deficiency in affected patients (20).a
Lack of CD73-mediated adenosine signaling causes upregulation of tissue non-specific alkaline phosphatase (TNAP); a key component of ectopic mineralization (57). The lack of CD73-mediated adenosine signaling led to increased levels of the transcription factor FOXO1, which binds the TNAP promoter and induces TNAP transcription (58). Importantly, this mechanism was also found in popliteal arteries collected from non-CD73 deficient patients presenting with MAC. This observation suggests that the mechanisms underlying CD73 deficiency – loss of adenosine signaling and upregulation of FOXO1 activity – may also have a role in a common MAC variety. Future studies should explore how CD73 and adenosine receptor protein levels change with age, and whether their induced expression and activation may protect against MAC.
Disruption of the ECM contributes to the development of MAC. This is appreciated in aneurysms, whereby calcification and disorganized and fragmented aortic elastic lamina lead to dilatation and tortuosity (59). Connective tissue disorders, such as Marfan syndrome and several types of Loeys-Dietz syndrome, also exhibit calcification in tortuous vessels localized along damaged elastic lamina, strikingly similar to vessel pathology of MAC observed in genetic diseases (60, 61). These genetic aneurysmal diseases share increased activity of the transforming growth factor β signaling pathway and it has been proposed that the calcification seen in these vessels is subsequent to the aberrant remodeling of the vessel wall (62–65). Indeed, it is well known that cells are able to sense the stiffness of their environment and that vice versa the environment itself has may induce switches in cell phenotypes (66). Stiffening of ECM transduced to VSMCs may trigger signaling events via the collagen binding discoidin domain receptor 1, which was found to promote osteogenic differentiation and calcification (67). Exploration of the significance of epigenetic imprinting stemming from the developmental origin of individual vascular beds and their propensity to calcify the medial layer provides an interesting example for prospective comprehensive disease modelling (68).
Another mechanism driving MAC pathogenesis is related to DNA damage response pathways. Upon DNA damage, the ataxia teleangiectasia mutated protein phosphorylates gH2AX, which marks the DNA lesion and triggers DNA repair proteins (69). One of these response proteins are poly[ADP-ribose]polymerases (PARPs) which trigger events such as senescence or cell death, but the product of PARPs, poly[ADP-ribose] (PAR) itself can promote the MAC by two means: the activation of osteogenic gene expression and also the amalgamation and packaging of PAR in MV that are delivered to the EM, where PAR and calcium and phosphate ions nucleate (70, 71).
The role of MVs in mineralization was first noticed in cartilage (72) but it is now appreciated to contribute to both MAC and IAC. In addition to delivering packets of the mineral building blocks of hydroxyapatite, MVs contain lipids, metabolites, microRNAs and proteins that promote the osteogenic switch of cells (73). While MVs play a role in atherosclerosis and MAC, the similarities and differences of their biogenesis, accumulation, and cargo as not been fully explored.
In T2DM patients suffering from sympathetic fibers damage, and those with sympathetic denervation following sympathectomy, MAC is a common finding; however the mechanism causing calcification in such patients remains to be clarified (74, 75).
In the settings of CKD the potential role of microbiome dysbiosis - derived uremic proteins in AC pathogenesis has been proposed (76); yet the proof of concept and confirmation in clinical studies in MAC are lacking.
Genetic considerations
For rare Mendelian disorders the rare variants with large effect size (mutations) directly cause the disease. In contrast, AC associated with the common clinical conditions such as CKD and DM appear to have a more complex etiology with multiple low frequency or common variants with small effect size interacting with non-genetic factors. For example, in patients with ABI >1.3 suggestive of PAD, a meta-analysis of genome-wide association studies (GWAS) from 21 population-based cohorts identified a single locus on chromosome 9 of genome-wide significance (77). In a study of the UK Biobank participants on whom arterial stiffness was measured, the arterial stiffness index was the subject of two GWAS analyses. These studies revealed four significant loci (near TEX41, FOXO1, C1orf21, and MRVI1), with gene-based analysis implying COL4A2 (78). Thus, although these studies provide preliminary evidence of a genetic contribution to AC phenotypes, studies with specific well-defined cohorts of patients with MAC are lacking.
Histopathology
MAC affects predominantly muscular arteries. In large arteries early stages are characterized by fine granulations of the CaP precipitates located within the media in the proximity of the internal elastic membrane, later progressing in size and distribution. In advanced stages large precipitates forming sheaths replacing large areas of the healthy media and fragmentation of the internal elastic membrane are seen (7). In some cases osteogenic transformation and invasions of the intima can be seen (18). The stages and differences between MAC and IAC have been summarized in figure 2 and in the corresponding table 1.
Figure 2. Histology of medial (MAC) and intimal (atherosclerosis) calcifications.
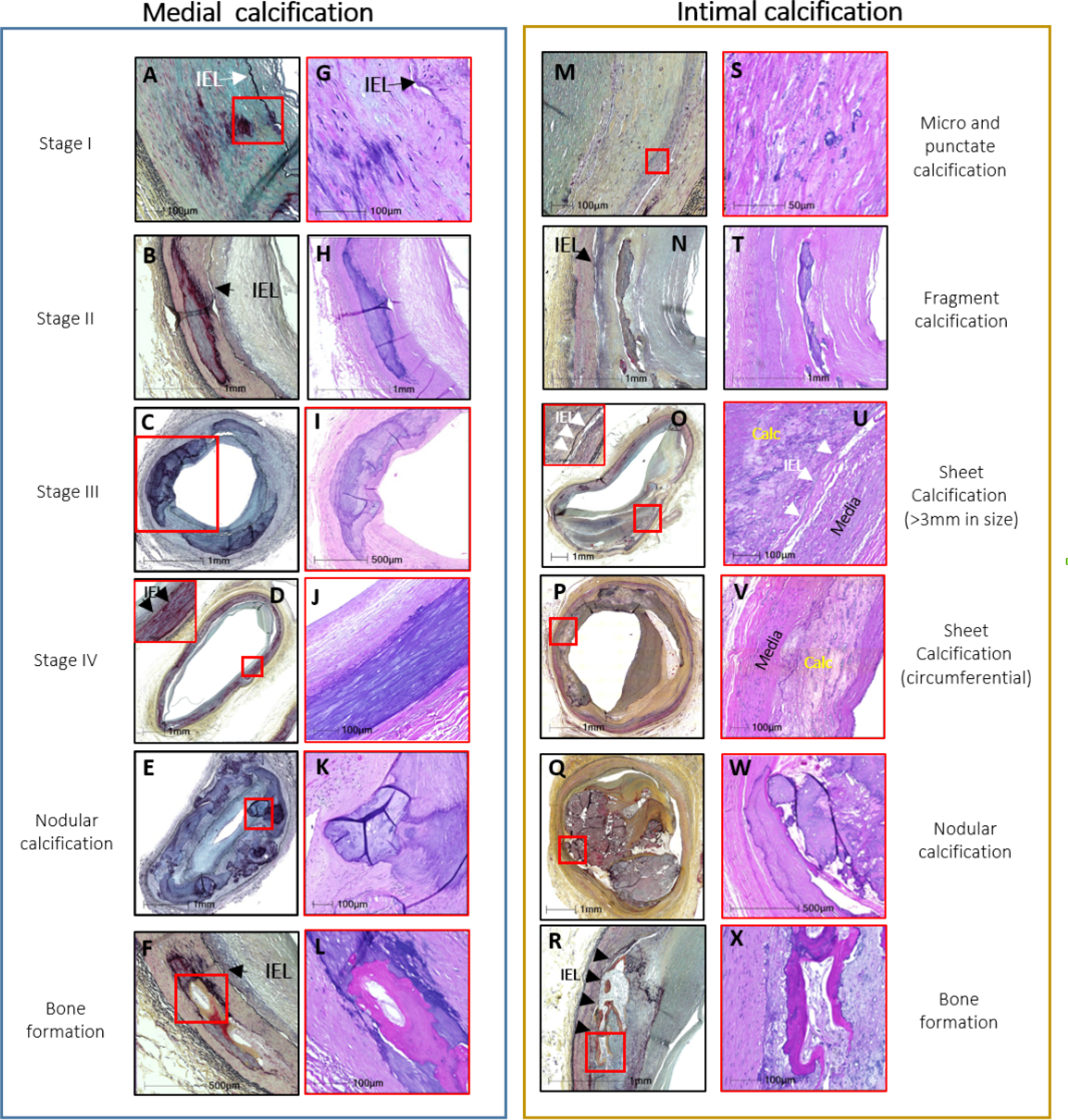
MAC and the intimal atherosclerotic calcifications are shown. The legends to each medial and intimal calcification pattern have been summarized in table 1. The histologic sections shown in the left and right columns of the medial and intimal calcifications were stained with Movat pentachrome (MP) (A-F and M-R) and hematoxylin and eosin (H&E) (G-L and S-X), respectively. The red boxes in the MP sections indicate areas of magnifications shown in the H&E sections and in stage IV (medial) and sheet (intimal) MP sections. IEL – internal elastic lamina (black arrows, black and white arrowheads). Modified and reproduced with permission from doi: 10.1016/j.jcmg.2018.08.039 and doi: 10.1016/j.ejvs.2020.08.037.
Table 1.
Stages of progression. Comparison between MAC and intimal calcifications.
| Medial Arterial Calcification | Intimal Calcification | ||
|---|---|---|---|
| I | Calcification of the internal elastic membrane with or without extension into the media (A and G). | Microcalcification (includes micro and punctate) is identified by calcium particles ranging from >0.5 μm and <1mm in diameter (M and S). | Micro and punctate calcification |
| II | Calcifications coalescence and becomes confluent (varying in size from 1 to 3 mm), forming fragments of calcification (B and H). | Small calcification is often accompanied by inflammation, areas of microcalcification coalescence forming fragments of calcification that are >1 mm but <3mm in diameter (N and T). | Fragment calcification |
| III | Calcification length > 3 mm and/or extends to involve >90° of the circumference (C and I). | Calcification of the intima >3mm or >90° (O and U). Calcification can extend and become circumferential (P and V). | Sheet Calcification |
| IV | Calcifications of the media, spanning the entire circumference (D and J). | ||
| Nodular calcification | Nodular calcification is rarely seen in medial wall which is composed of nodules of calcification often accompanied by fibrin (E and K). | Nodular calcification is composed of nodules of calcification often accompanied by fibrin with a fibrous cap (Q and W). | Nodular calcification |
| Bone formation | Bone formation may be observed in fragmented and areas of sheet calcification (F and L). Bone formation and rarely cartilaginous metaplasia may be seen in late-stage most frequently in stages III and IV, but rarely also in stage II. | Bone formation can be observed within the regions of calcification (R and X). | Bone formation |
Modified and reproduced with permission from doi: 10.1016/j.jcmg.2018.08.039; doi: 10.1016/j.ejvs.2020.08.037.
It is not known how far distal on the arterial tree MAC can spread. Some studies suggest that in small arteries numerous changes including severe intimal hyperplasia and thrombotic occlusions (7, 79, 80) along with CaP deposits (81) may occur. In amputated limbs of CLTI patients a strong correlation was found between MAC of the foot arteries and metatarsal artery obstruction. The most common obstructive lesion was a combination of intimal thickening, advanced MAC, and thrombosis that was striking “for the relatively small size of the metatarsal arteries” (82).
Hemodynamic Impairments
MAC causes arterial wall stiffening which is associated with fundamental changes in central hemodynamics: impaired Windkessel function, increase in left ventricular afterload, decrease in coronary artery perfusion and increased blood flow pulsatility (1, 83, 84). The limited studies of hemodynamics in peripheral arteries of patients with MAC revealed marked impairments suggestive of peripheral shunting (85).
Based on our preliminary observations in patients with CLTI, following vasodilatation advanced MAC appears to cause an accentuated fall in mean and diastolic arterial pressures. These data appears to suggest a decrease in the driving force due to the arterial wall rigidity and pathological peripheral vasodilatation (Figure 3) (Lanzer et al. unpublished data). These early data seem to corroborate the clinical observations in patient with CLTI due to MAC resulting in severe limb ischemia (7).
Figure 3. MAC and hemodynamics.
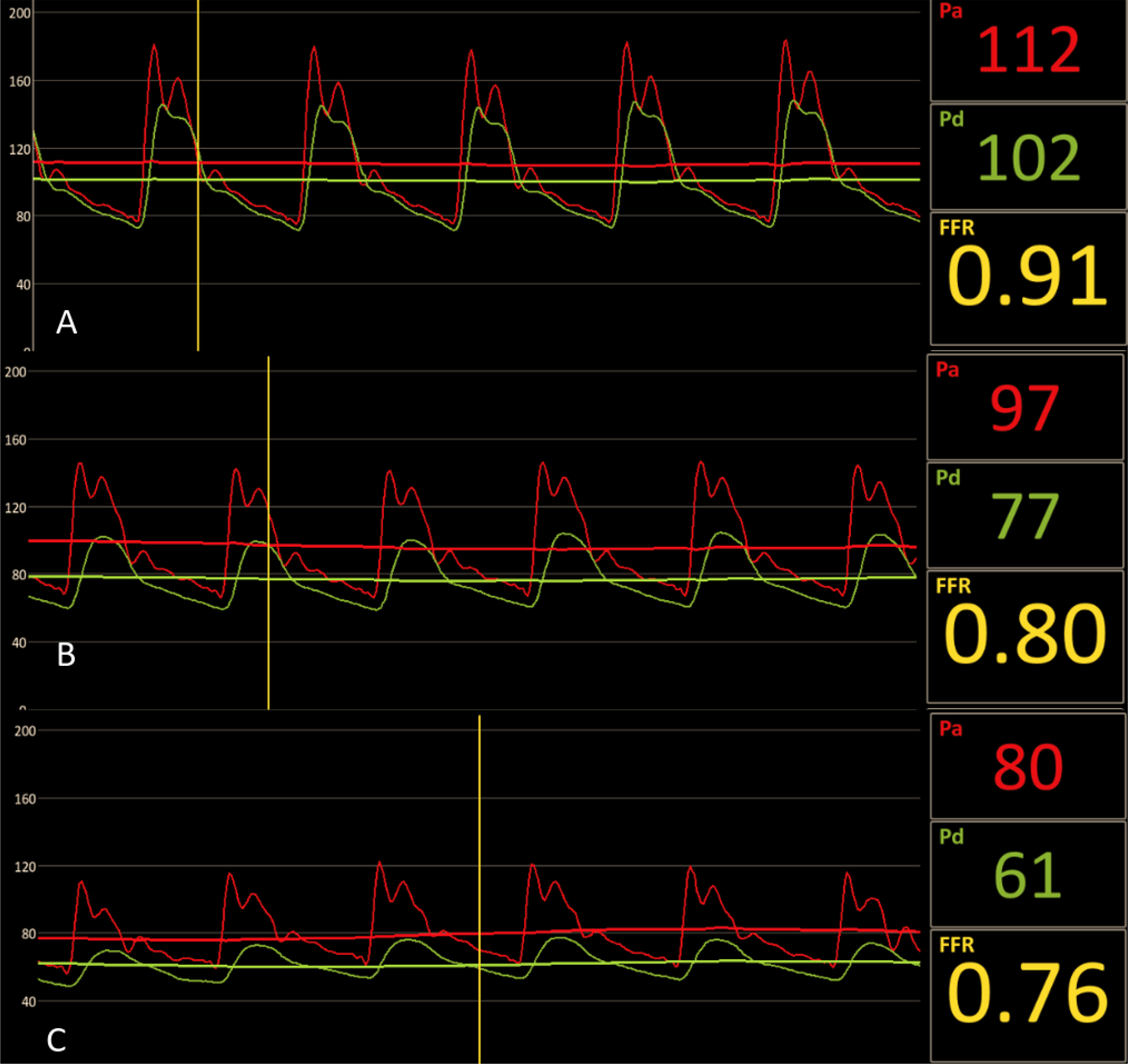
Male with diabetes mellitus and gangrene of the right middle finger and X-ray angiography demonstrating advanced MAC without evidence of stenotic lesions of the right upper extremity arteries. (A) High fidelity blood pressure recordings at the level of the mid brachial (Pa) and distal ulnar (Pd) arteries demonstrated Pd/Pa-ratio of 0.91 at baseline, (B) Pd/Pa-ratio of 0.80 following intravenous (140 μg/kg/min systemic infusion) and intra-arterial (150 μg) bolus administration of adenosine and (C) Pd/Pa-ratio of 0.76 following intra-arterial nitroglycerin (300 μg). The disproportional decrease in the distal diastolic pressures following vasodilatation appears to suggest marked microvascular dysfunction. Hemodynamically significant lesion proximal to the target territory has been excluded by X-ray angiography.
Interactions with atherosclerosis
The exact nature of interactions between MAC and atherosclerosis appears foremost biomechanical in nature. In healthy media, when atheroma develops, the luminal area is initially maintained by circumferential expansion (86). It is thought that this compensatory stage lasts until the vessel’s cross-sectional area expands by about 40%. Beyond 40%, inward remodeling occurs, leading to a decreasing lumen area. However, in the presence of MAC, the compensatory stage may be prematurely disrupted. MAC may encourage the inward phase of remodeling to start earlier, giving rise to an acceleration of luminal stenosis related to the progression of atherosclerosis. How the enhanced inward remodeling occurs physiologically remains to be fully characterized but one hypothesis suggests a competition in stiffness between the intima and media (87). If the stiffness of the media is much greater than that of the intima, the media acts as a stiff band surrounding a relatively compliant intima. With further growth the intima is forced to encroach into the lumen, leading to a reduction in blood flow. A stiffer media could also interfere with the vessel’s ability to stretch and recoil, disrupting the normal development of pressure gradients. A long-term disruption in hemodynamics may also promote an earlier onset of disease, since low endothelial shear stress is thought to be atherogenic (88).
While IAC typically occurs in advanced atherosclerotic lesions often promoted by localizing biomechanical factors (89), MAC occurs early in the course of the disease along elastic lamellae. Thus, while IAC of atherosclerosis occur in advanced disease threatening the patients with focal or multifocal plaque evolution, rupture and thromboembolic complications MAC occurs early in the disease’s course, at least in peripheral arteries and it is the main risk factor for progressive shutdown of circulation due to small artery disease.
Diagnostic evaluations
MAC is most commonly seen in the arteries of the lower and upper extremities but has been reported in all major vascular territories (90), temporal (91), facial (92) and mammary (93) arteries. Diagnosis of MAC is frequently incidental in the course of other diagnostic evaluations or a result of systematic screening in patients with PAD. In PAD, ABI has been the preferred first-line method to screen for MAC; however the validity to determine MAC was questioned (29). Recently a simple MAC score was proposed based on a planar X-ray of the foot in two projections. Looking at five vascular sites and evaluating the length of the “pipe-steam” patients can be divided in three MAC levels: absent, moderate and severe (94).
Multiple imaging modalities allowing differentiation between the intimal and medial layers of the arterial walls are suitable for MAC detection. In peripheral arteries MAC and IAC can be clearly differentiated by transcutaneous ultrasound imaging. MAC is recognized on longitudinal views by echogenic abluminal bands and typically smooth endothelial interfaces (Figure 4) while IAC appear granular and spotty (95). On conventional X-ray radiography, MAC radiopaque “pipe-steam” type shadows are typical in contrast to the irregular discrete “plaque-like” IAC (96); if combined with angiography, smooth endothelial interfaces can be appreciated (Figure 5). Computed tomography (CT) exquisitely depicts peripheral and coronary artery calcifications as high density signals, yet due to its limited spatial resolution MAC and IAC cannot be differentiated on conventional images. A distinction between these two types of calcification based on differences in patterns has been proposed on thin CT slice images (97). Compared to other imaging modalities, CT may provide a whole - body estimates of AC burden as shown in figure 6.
Figure 4. MAC in ultrasound.
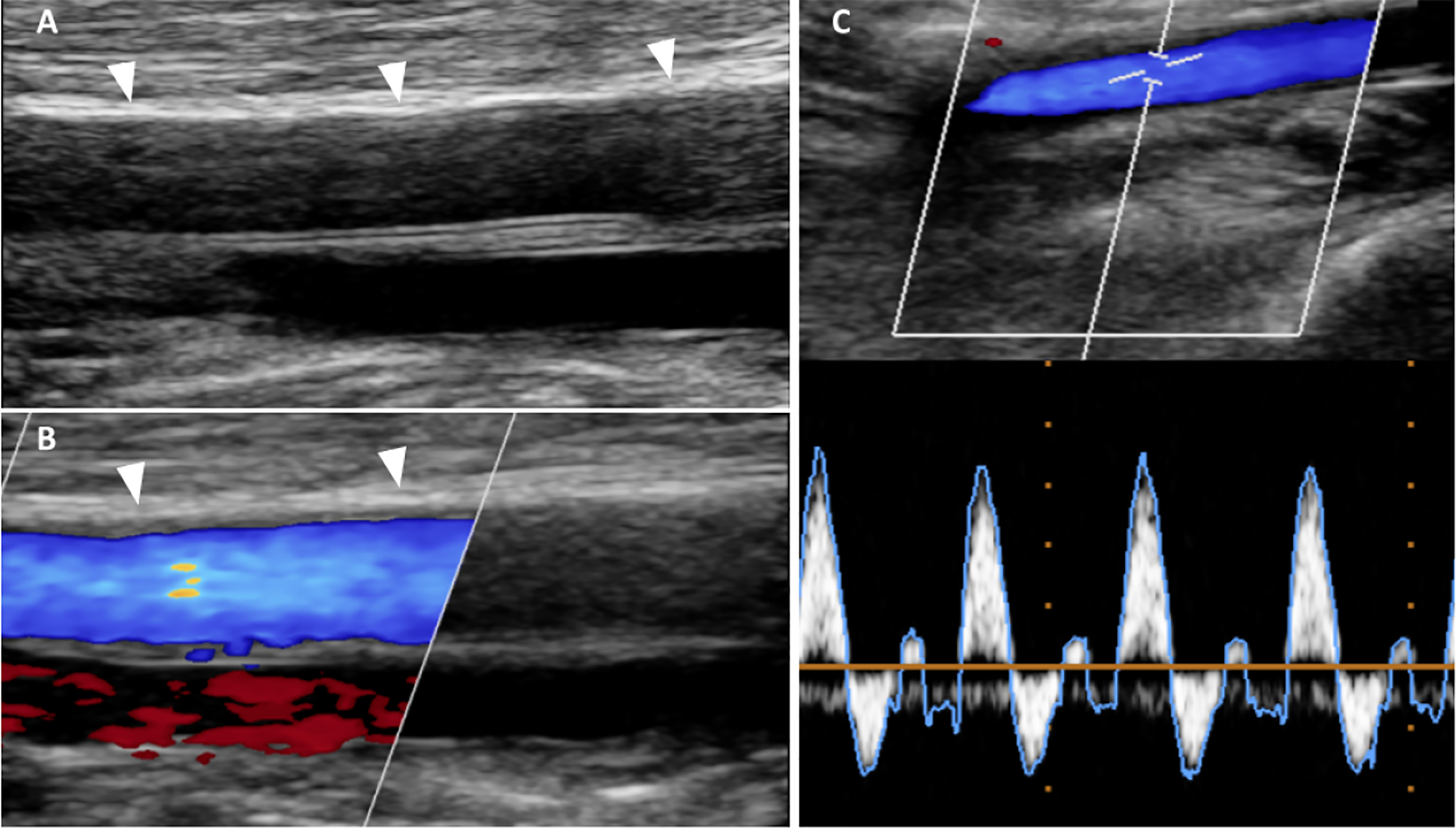
Female with chronic kidney disease and MAC demonstrated by ultrasound. (A) B-mode ultrasound longitudinal image of the left superficial femoral artery (SFA) revealed on the near end arterial wall linear homogeneous echogenic band pathognomonic for MAC; note the smooth endothelial interface of the SFA; white arrowheads denote diffuse medial calcifications.(B) Color coded Doppler (CCD) image of A demonstrates normal arterial flow signal (blue color); red color corresponds to the accompanying vein; white arrowheads as in A. (C) CCD and pulse wave Doppler (PWD) images show a normal triphasic flow pattern indicating a normal artery.
Figure 5. MAC in native X-ray and X-ray angiographic images.
(A, B) Male with diabetes mellitus and MAC demonstrated by X-ray imaging. (A) native X-ray image of the right brachial artery demonstrating the typical “rail-road track” pattern of calcification parallel to the humerus, white arrowheads at the edge of calcifications (B) X-ray angiographic image of A showing the smooth endothelial interface following contrast media filling, heavy calcification are recognized as white stripes along the lumen (white arrowhead). (C, D) Male with cardiac arrhythmias undergoing coronary angiography. (C) High resolution native X-ray image of the right coronary artery reveals disseminated pattern of calcifications (two white arrowheads on the left) suggestive of MAC seen in A; the image appears partly duplicated due the rapid motion of the cardiac base (white arrowhead on the right). (D) X-ray coronary angiographic image of C showing the smooth endothelial interface demarcated by homogeneous contrast media filling (black arrow).
Figure 6. MAC in Computed Tomography.
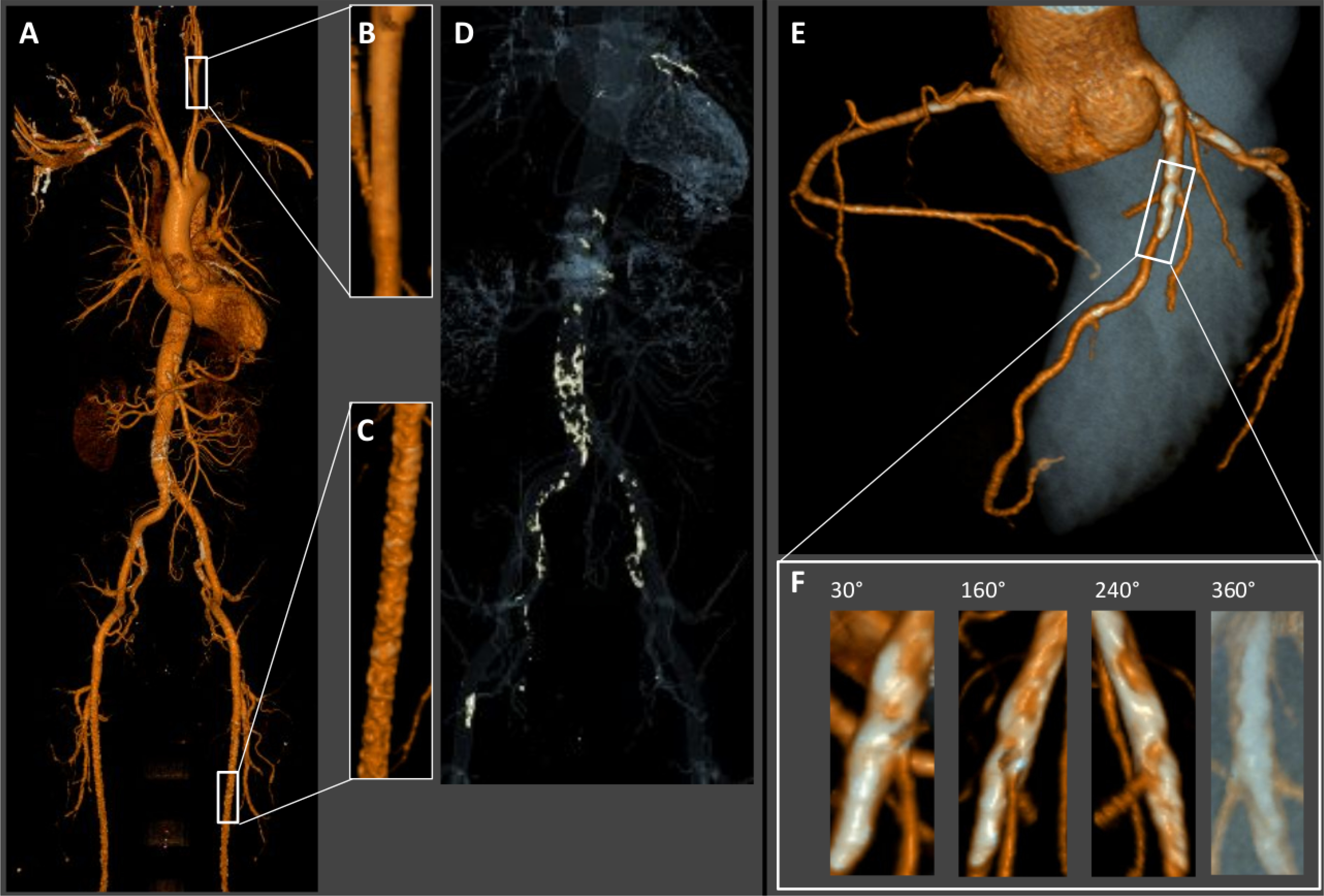
3D CT reconstruction surface rendering in a male with MAC. (A) Whole body scan reveals calcifications (white spots) within the abdominal aorta, pelvic and leg arteries. (B) Enlarged section of the internal carotid artery from A appears relatively free from calcifications. (C) Enlarged section of the superficial femoral artery with heavy diffuse calcification typical for MAC. (D) Calcium windowing highlights the calcifications of the vasculature. (E) Coronary CT- angiography shows extensive calcifications of the proximal left anterior descending (LAD) coronary artery and lesser calcifications of the proximal right and left circumflex coronary arteries. (F) Enlarged section of the proximal LAD shows heavy confluent, partly circumferential calcifications.
Coronary artery calcifications can be reliably visualized by CT and the coronary calcium scores have been used for detection and prognostication of coronary artery disease for more than two decades (98). However, to allow distinction between MAC and IAC either IVUS, or even more optimally, optical coherence tomography (OCT) with axial resolution of approximately 100μ and 10μ, respectively, are needed (99). Using OCT the artery wall layers containing calcifications are clearly visualized (Figure 7). However, due to their invasive nature both techniques are applied only if clinically indicated in selected patients.
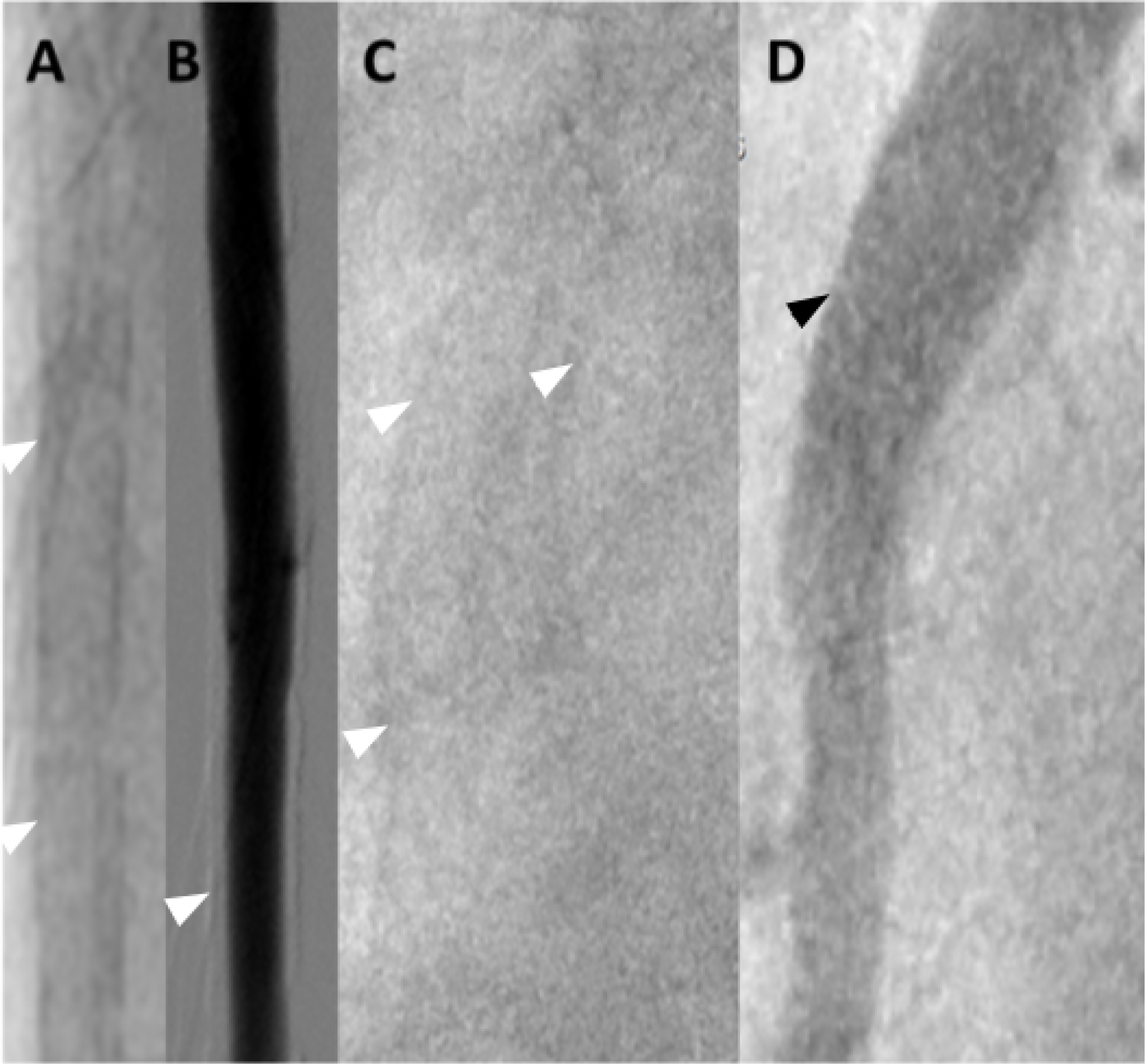
Figure 7. MAC in Optical Coherence Tomography.
On OCT MAC appears as regions with homogeneous low signal intensity and sharply demarcated border zones. (A-C) Cross-sections of the coronary arteries. (A) MAC spans more than a half of the circumference (12 to 19 o’clock); (B) Extensive, partly eccentric MAC (13 to 15 o’clock); (C) Highly eccentric MAC (10 to 12 o’clock). Note the smooth and regular intima in all three images. Courtesy and Copyright, Abbott, Inc.
The typical laboratory test and biomarkers for MAC have been summarized in the Supplement B.
Therapy
Pharmacological therapy
Slowing of MAC progression in patients with CKD has been demonstrated with the use of phosphate binders and calcimimetic agents (100, 101). An extensive body of literature on patients receiving long-term dialysis suggests that the benefit of non-calcium based phosphate binders is not limited to phosphate reduction but it extends to reduction in the calcium load that patients receive from calcium-based binders and an effective control of PTH (102). Indeed, PTH was shown to have direct pro-calcifying effects in animal experiments (103). Additionally, these binders enhanced bone mineral density while slowing AC progression (104).
Other experimental therapeutic approaches included vitamin K supplementation, pyrophosphate, tenapanor and acetazolamide. The Valkyrie study demonstrated a nominal but non-significant slower progression of calcifications in patients on hemodialysis with atrial fibrillation randomized to rivaroxaban with or without vitamin K supplementation (105).
Tenapanor, an inhibitor of the sodium-hydrogen isoform 3 protein regulating paracellular absorption of sodium and phosphate in the intestine, has demonstrated several beneficial effects such as reduction in sodium and phosphorus absorption, reduction in serum levels of phosphorus and FGF-23, and reduced heart mass in rats with chemically induced CKD. Additionally, it significantly decreased ectopic aortic calcification (106). In two human trials of hemodialysis patients, tenapanor showed a clear dose-dependent ability to reduce serum phosphate levels although the trials did not measure the effect of this drug on AC (107).
Pyrophosphate inhibits calcium and phosphate deposition in bone and soft tissue. Bisphosphonates are analogues of pyrophosphate and animal studies in the 1970’s suggested their inhibitory activity on AC. Early forms of this drug (etidronate) have successfully halted the extensive calcification of newborns with GACI allowing their survival (108), and currently a clinical trial is underway to test whether this therapy may also be useful in patients with CD73 deficiency (109). Randomized control trials in pre-dialysis and dialysis populations failed to reaffirm this hypothesis (110) and appeared to promote osteomalacia (111). Etidronate, a bisphosphonate with a mechanism of action similar to that of pyrophosphate, did slow peripheral AC in a recent trial (112).
Aldosterone expands the pro-calcifying activity of hyperphosphatemia and facilitates osteoblastic transformation of VSMCs in vitro (113), while spironolactone inhibited calcification in a Klotho-deficient mice model (114). Since patients with CKD often suffer from hyperphosphatemia and concomitant hyperaldosteronism, human trials with mineralocorticoid antagonists are currently under way.
The anti-calcifying activity of magnesium have been demonstrated in two small human trials where magnesium supplementations slowed the progression of coronary artery calcium in pre-dialysis patients (115) and a decreased propensity to develop AC in hemodialysis (116).
The most interesting recent development in treatment of AC involves a direct inhibitor of HAP, the final step in all calcification processes. SNF472, myo-inositol hexaphosphate (117) was shown to significantly slow progression of aortic valve and coronary artery calcification in patients with end-stage CKD (118).
Whether any of the modest effects described above will translate into meaningful clinical benefits in patients with MAC remains to be determined (119). Besides pharmacological interventions, renal transplantation seems to slow progression of MAC but does not cause regression of calcification (120). While CaP overload represents a valid target in patients with CKD, in disorders with preserved systemic CaP homeostasis with the exception of pseudoxantoma elasticum no therapy targets have been established.
Interventional therapy
AC is associated with increased risk of complications during surgical and percutaneous coronary and peripheral revascularization procedures (14–16). Numerous surgical and endovascular strategies were designed to improve outcomes. For example, in endovascular procedures non-compliant, scoring and cutting balloons, excimer laser ablation, rotational and orbital atherectomy and more recently intravascular lithotripsy have been employed to cope with coronary and peripheral artery calcifications (121–123). Although these procedures do not remove calcium, they fracture the superficial and/or deep calcifications and by modifying the lesions they improve stent placement and provide access to distal target sites. Figure 9 reviews histology of calcified lesions treated with endovascular procedures.
Figure 9. Calcified plaques and endovascular interventions (histology).
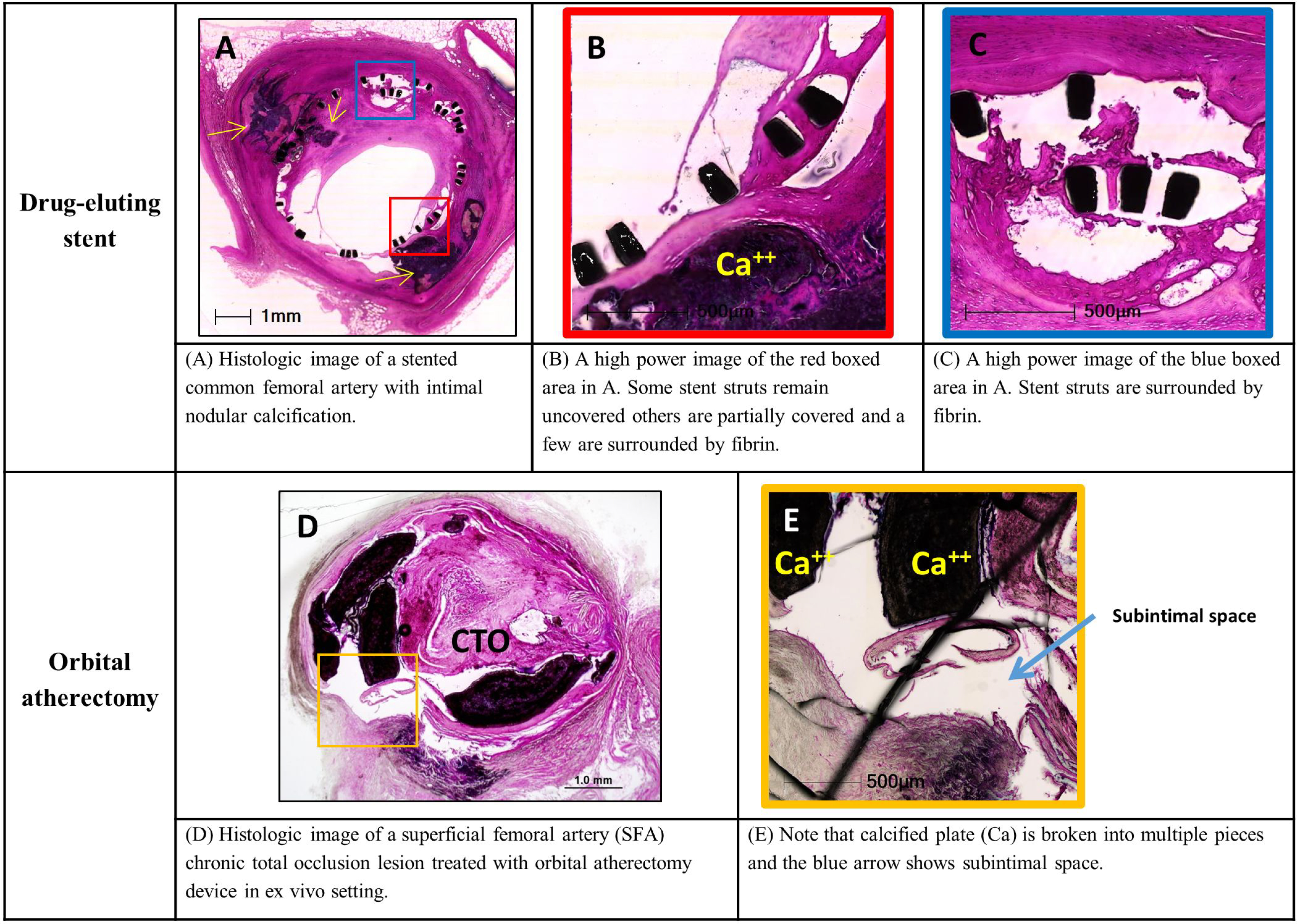
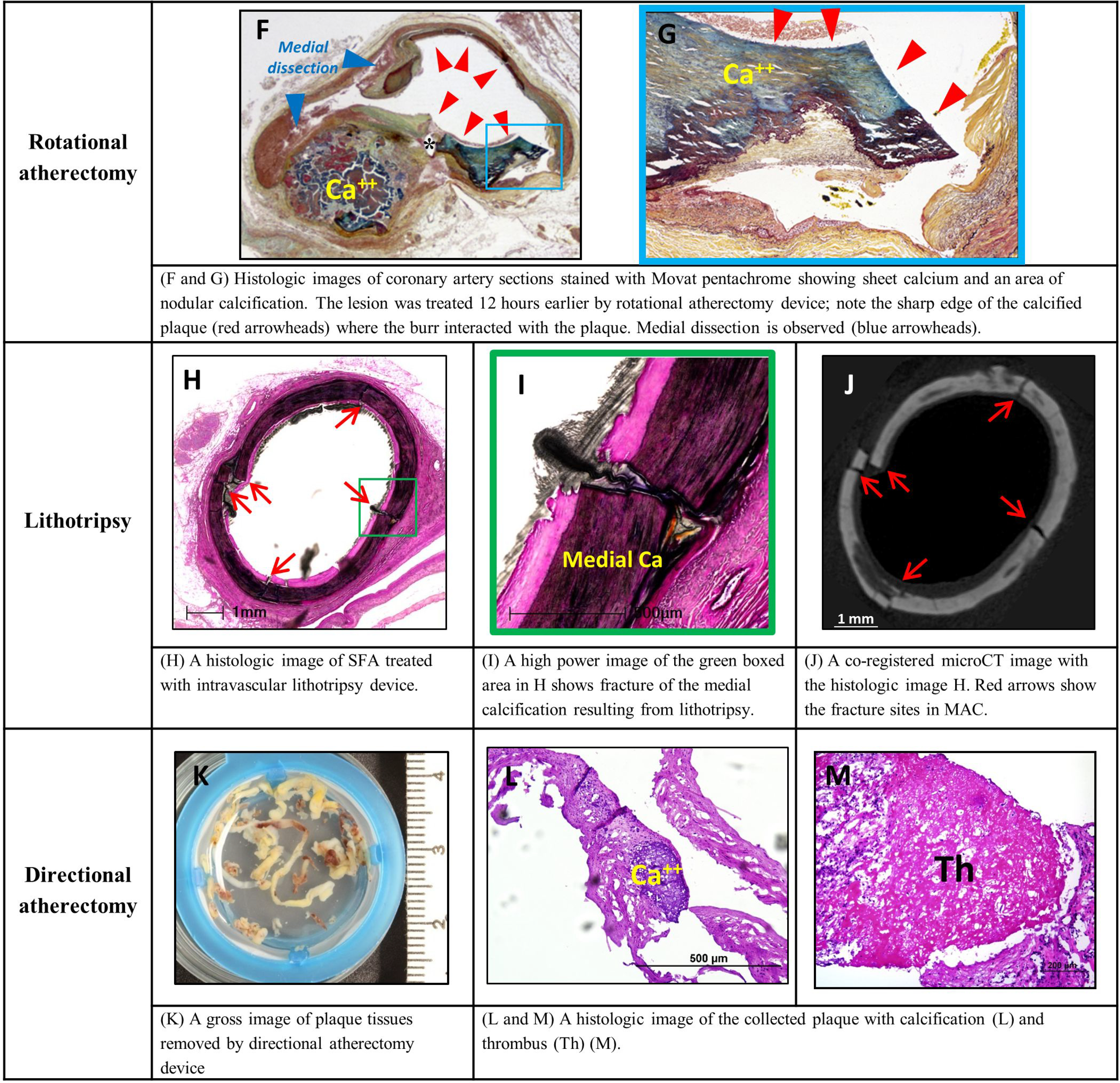
Examples of histological findings of intimal and medial calcifications following endovascular interventions. Upper panels: common femoral artery and superficial femoral artery (SFA) following drug eluting stent placement and orbital atherectomy device in ex vivo setting, respectively. Lower panels: coronary artery treated by rotational atherectomy device, SFA treated with intravascular lithotripsy device and plaque debris obtained from directional atherectomy device. Images A to D, H, I, L, M are stained with hematoxylin and eosin (H&E). Images E to G are stained with Movat pentachrome. Ca – calcifications; CTO – chronic total occlusion. Modified and reproduced with permission from DOI: 10.1016/j.jcin.2019.10.060. Images F and G: modified and reproduced with permission from doi: 10.1016/0002-8703(95)90384-4.
Future perspectives
Experimental data and plausibility considerations
Currently AC pathogenesis has been studied employing a variety of in vitro and in vivo experimental models using numerous experimental protocols replicating some but not all of the AC features encountered in human diseases (s. Supplement A). Albeit providing important insights to date, they have not uncovered novel and targetable molecular machinery that maybe used to design pharmacological prevention and therapy to treat AC.
Due to the extreme sensitivity of the CaP precipitation processes in regard to the ionic strength of Ca and P ions modified by promoters and inhibitors of mineralization, aberrations of in vivo conditions in experimental protocols bear the risk of factitious results and errors in interpretations (27) (s. Supplement C). To avoid these inconsistencies, experimental designs conforming to the laws of thermodynamics governing the CaP precipitation must be observed and first principles thinking should be applied (s. Supplement D). Identification of the triggers of CaP nucleation along with the potentially reversible stage of the amorphous phosphate formation, and the irreversible point of no return following HAP crystallization, will be critical to define the targets of pharmacological therapy (27). Replication or direct observation of the in vivo conditions, conserving the principles of thermodynamics and focusing on the medial layer, will allow insights into the pathobiology of MAC, and holds the potential for developing causal treatments.
Genetics
Genetic analysis provides an important direction of research to unravel MAC molecular pathogenesis. Genetic analyses of MAC will in the future depend on the availability of whole exome sequencing and the identification of appropriate patient cohorts e.g., multi-generational families with affected members, to help identify monogenic disorders (124, 125). The genes identified in monogenic disorders may also be relevant for polygenic disease. However, robust characterization of genetic variants contributing to polygenic forms of MAC requires the establishment of large cohorts in whom secondary causes of AC have been excluded as outlined below.
Genetic variants that promote the development of MAC as a function of comorbidities such as DM and CKD are not easily identified via familial studies. Thus, discovery of genetic variants influencing MAC risk should follow several related paths. First, to discover low frequency variants of high effect size, whole exome sequencing (WES) or whole genome sequencing (WGS) should be undertaken using large and diverse populations. Both of these techniques are limited by the ability to easily parse variants into benign or pathogenic categories (126).
Second, GWAS could be undertaken to look for common variants of small effect size that could nevertheless yield important insights into the biology of AC. For example, a GWAS of >9,400 individuals has identified an association with the histone deacetylase 9 (HDAC9) gene and atherosclerotic aortic calcification. This study showed HDAC9 to increase expression of RUNX2, which promotes the differentiation of VSMCs into cells with an osteogenic phenotype (127). Moreover, HDAC9 overexpression was shown to increase calcification in cultured VSMCs of mice, whereas deficiency of HDAC9 decreased calcification (127). Thus, while HDAC9 was demonstrated as a potential therapeutic target for atherosclerotic AC, comparable data on MAC are not available. A major limitation of GWAS is the need for well-phenotyped cohorts with a sample size in the thousands. As mentioned above, discerning between IAC and MAC is not often determined routinely. Furthermore, associations should be replicated in an independent cohort, again necessitating the collection of thousands more cases and controls. It is therefore clear that a large, international, multidisciplinary team would be required to gather sufficient cases, analyze the genetic data, and conduct in silico and laboratory studies to discover causal variants and identify candidate genes for MAC.
Third, is the need for functional validation of GWAS discoveries. The International Knockout Mouse Consortium (IKMC), whose objective is to establish knockout mouse lines for each of the protein-coding genes (128) may provide an opportunity to functionally validated candidate genes identified from GWAS and WES/WGS studies. Systematic phenotyping for AC in these knockout mouse lines could yield novel genes involved in VSMC function and the pathogenesis of MAC. However it is established that mice are not good models for human MAC (41). The challenge lies in establishing non-invasive in vivo imaging modalities for the high throughput detection and quantification of AC in mice. 18F-sodium fluoride positron emission tomography (18F-NaF PET) has potential as a sensitive in vivo imaging technique (129). If greater resolution is required, ex vivo microCT analysis could also be considered to detect microcalcification (57).
In addition to MAC presenting as co-morbidity, primary systemic MAC characterized by the absence of predisposing factors has been reported (8). In the absence of known risk factors for MAC the possibility of inherited genetic causes is likely, thereby providing an opportunity to search for additional monogenic causes of MAC in these patients. The availability of blood samples and vascular tissues from patients with primary MAC would greatly simplify research protocols providing valuable opportunities to identify the causes of MAC in such patients.
Furthermore, the striking differences in the topographic distribution of MAC between individual vascular beds with strong predisposition for the arteries of the extremities appears to coincide with the differences in the embryological origins of VSMCs (130) potentially pointing towards an epigenetic predisposition worthy of being explored.
Omics
While applications of omics’ approaches to VC are still in their infancy the research into MAC and IAC will benefit from a variety of studies employing different omics, including proteomics (131), metabolomics (132) and most recently single cell transcriptomics (133).
The in depth characterization of calcifying blood vessels or cells could yield important insights into the evolving disease processes, mainly by providing an unbiased tissue profiles. As depicted in Figure 10 omics-based tissue profiles can have different resolutions.
Figure 10. MAC and Omics.
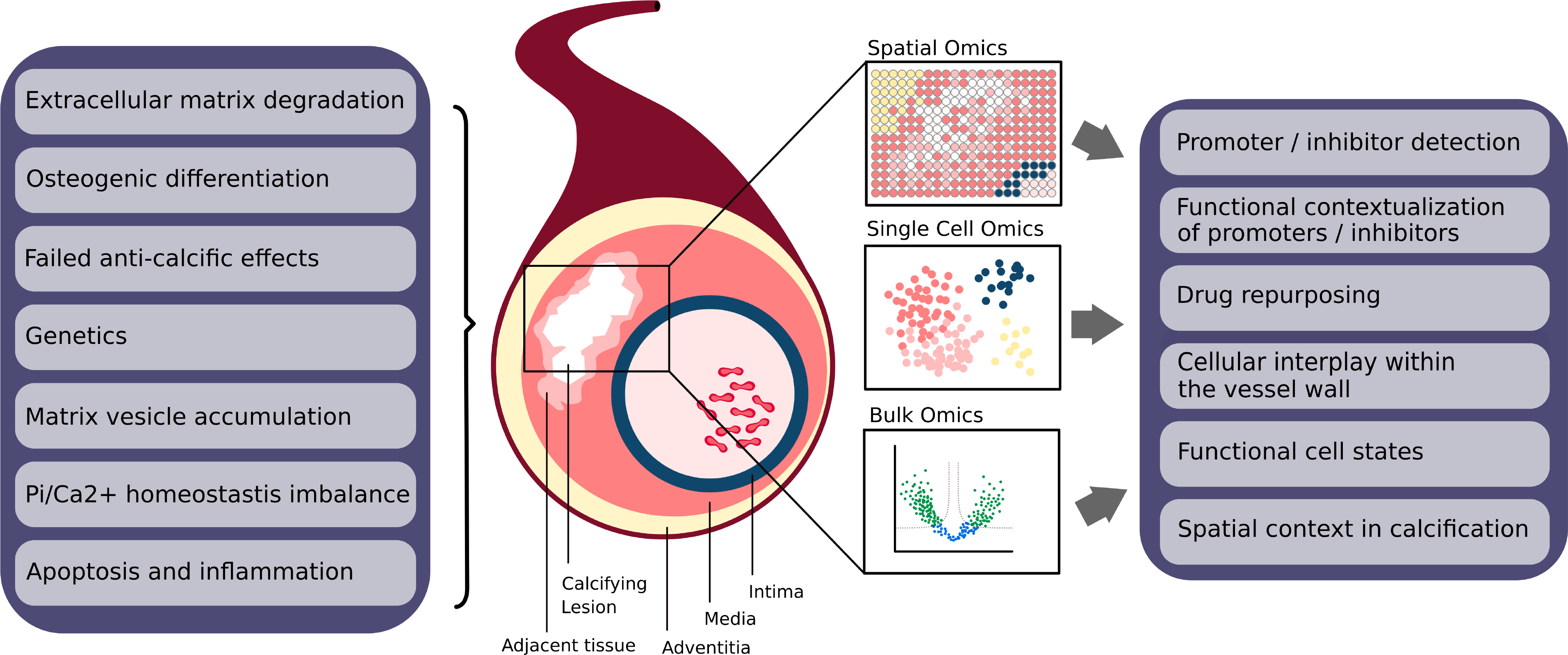
Overview of pathophysiology of MAC and the proposed Omics approaches to study MAC. (Left box) Different proposed pathophysiologic principles suggested triggering and promoting the vascular calcifications including MAC. (Middle section) Calcified sites in the vasculature can be studied with omics at different degrees of resolutions. Bulk omics (bottom) provide a gross tissue profiles. Single cell omics (middle) provide profiles of individual cells. Spatial omics (top) yields profiles of regions represented as the 2D sections of the tissues. (Right box) The proposed approaches have not been systematically applied to study MAC but hold promise to address numerous aspects of calcifications processes providing tangible insights and hypotheses for molecular pathogenesis of MAC.
To date, a few important bulk-omics-based studies on AC have been published. Long-noncoding RNAs (LncRNA) are gene regulatory transcripts longer than 200 bases that have been associated with major cardiovascular pathologies including heart failure (134) and atherosclerosis (135). Using an in vitro model, key regulatory LncRNA were detected via transcriptomics in calcifying rat VSMC (136), and the identified candidates were assayed based on prior knowledge. In this experimental set-up, LncRNA Lrrc75a-as1 was confirmed to have a mechanistic impact on in vitro calcification. A similar pipeline was applied by Schanstra et al. (137) who analysed the proteomic difference between human arterial tissue samples in patients with early and late stages of a broad spectrum of cardiovascular diseases. To identify potentially beneficial drugs, unbiased drug signatures (Connectivity-MAP) (138) were applied to match the proteomic disease signature, assuming that the drug could reverse the pathogenic phenotype. Using this approach cytosolic phospholipase A2 inhibitor was predicted to prevent AC, which was subsequently confirmed experimentally in vivo.
Regarding the evaluation of complex biological systems and the integration molecular cascades via network analysis, Song et al. assembled a network from protein-protein-interactions (PPIs) in VSMCs (139). Known proteins involved in AC were used as seed genes and mapped onto the resulting network. This was repeated for other endophenotypes such as fibrosis and inflammation to evaluate their relationship with the AC endophenotype via the PPI network. The network was further used for drug repurposing by assessing the network distance between drug targets and the calcification module. Three candidates, including the mTOR inhibitor Everolimus, were experimentally validated to reduce VSMC calcification.
Single cell omics allows analysis across the complexity spectrum down to a single cell. This appears to be of particular value to research on AC as it has enabled insights into vessel wall biology (133) and especially into cell type and cell state composition in atherosclerotic plaques (140, 141). The medial layer of arteries contains mainly VSMC, fibroblasts, pericytes and transitional cell types termed myofibroblasts (142). Single-cell technologies are likely to provide important insights into the cellular heterogeneity of VSMC and transitional cells’ states in their functional evolution along with their calcifying potential.
AC exhibit a strong topographic aspect, they begin locally restricted to specific loci of the vasculature and also locate within specific layers of the vessel wall; in MAC calcification sites occur predominantly in the arteries of the extremities and here, within the media as the characteristic single target. Thus, spatial aspects of pathogenesis are expected to be highly influential and should be addressed with proper sampling strategies, as demonstrated for calcific aortic valve disease (143) and by employing spatially resolved omics. These novel technologies assess molecular profiles while retaining their spatial information. While novel technologies are under development, spatial transcriptomics are the most advanced at present (144, 145). The blood vessels, small and well-structured organs, constitute excellent study subjects for these technologies. Here, the functional analysis of calcifying sites could substantially improve our understanding of how and why calcification occurs at sites while others are spared.
While omics have the potential to reshape research on AC, important limitations should be considered. The major ones include reproducibility batch effects, cost and coverage. Single cell and spatially resolved omics and the computational methods employed for data analysis are still in early stages of development and mechanistic insight from omics studies relying on biological assumptions may not always reflect biological reality. Nevertheless, omics holds a great promise providing data driven hypotheses requiring validation experiments, allowing causal conclusions, and as such, need to be systematically employed to support biochemical, genetic and experimental approaches to understand AC (24) and specifically MAC.
The definition of the MAC molecular biology will ultimately need to be addressed by interdisciplinary teams with complementary expertise (24, 146, 147).
The Central Illustration summarizes the key points pertaining to the pathophysiology and clinical manifestation of MAC.
Central Illustration. MAC: A Systemic Vascular Disorder Devastating Peripheral Circulation.
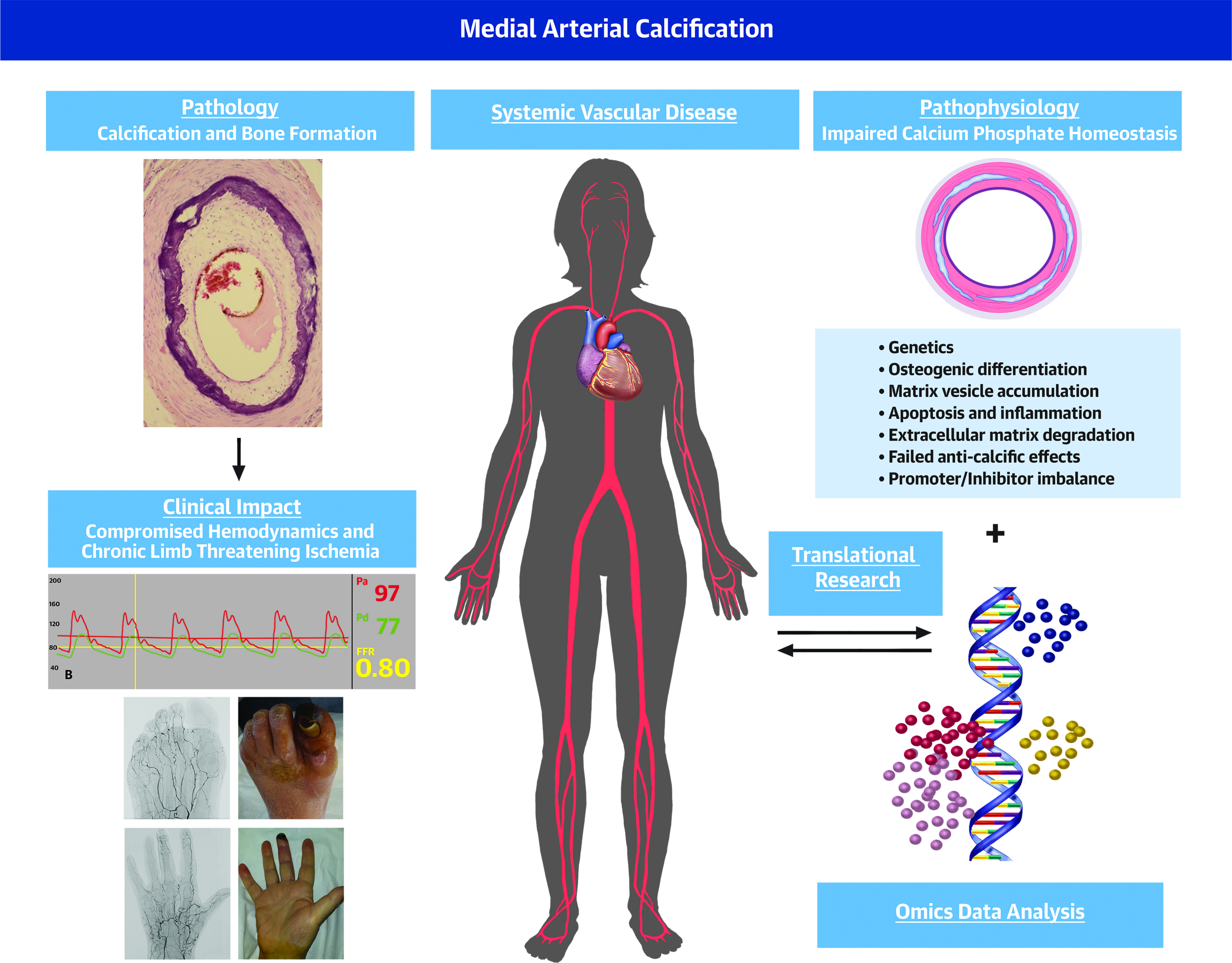
. In health calcium phosphate (CaP) homeostasis is maintained and crystallization prevented. In MAC CaP homeostasis brakes down resulting in progressive mineralization and in more advanced stages, bone formation within the medial layer. A number of pathogenetic principles including smooth muscle cells osteogenic differentiation, apoptosis, inflammation, molecular defects of matrisome have been reported to regulate the calcification process (left panels). MAC impairs hemodynamics often causing chronic limb threatening ischemia (right panels). Omics approaches holds distinct promise to define MAC molecular pathogenesis and design treatments (central panels).
Summary
MAC is a chronic systemic vascular disorder distinct from atherosclerosis, frequently, but not always associated with DM, CKD, and aging. It is part of a number of complex phenotypes observed in numerous less common diseases. The hallmarks of MAC include disseminated and progressive precipitation of CaP within the medial layer, compromise of hemodynamics, prolonged clinically silent course and CLTI. The pathogenesis of MAC is unknown. The current state of knowledge concerning MAC was reviewed and directions of future research were briefly outlined.
Supplementary Material
Figure 8. MAC in limb arteries.
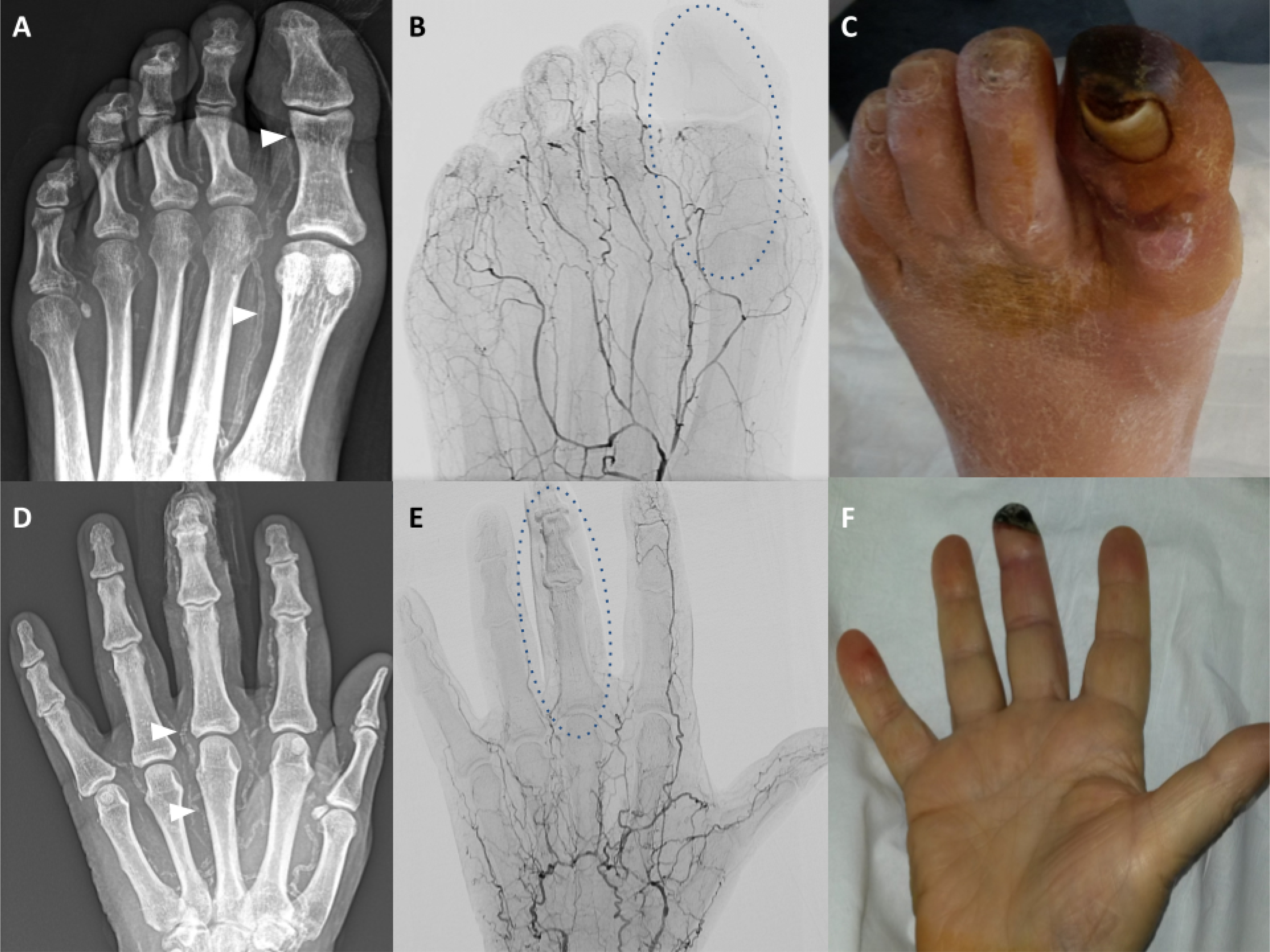
Male with the gangrene of the great toe left. (A) Native X-ray image of the left foot; heavy calcifications of the metatarsal and toe arteries, particularly of the first metatarsal and great toe arteries are shown (white arrowheads). (B) Angiographic X-ray image corresponding to A; shown is the extensive destruction of the small arteries; paucity of the distal arterial supply is highlighted in the context of the great toe (oval shaped broken line). (C) Photograph of the left foot corresponding to A; shown is extensive necrosis of the left great toe. Male with the gangrene of the mid-finger left. (D) Native X-ray image of the left hand; heavy calcifications of the metacarpal and finger arteries are shown (white arrowheads). (E) Angiographic X-ray image corresponding to A; shown are multiple occlusions of the metacarpal and finger arteries and virtual absence of arterial blood flow to the fingers (exemplified by the mid-finger, oval shaped broken line). (F) Photograph of the left hand corresponding to A; shown is distal necrosis of the mid-finger left.
Highlights.
Medial arterial calcification is a systemic vascular disorder distinct from atherosclerosis resulting in progressive calcification of the medial layer of the arterial wall.
Medial arterial calcification is frequently associated with diabetes mellitus, chronic kidney disease, and aging, and can result in severe limb ischemia.
Genetic and proteomic investigations are needed to clarify the pathogenesis of medial arterial calcification and develop specific therapeutic approaches.
Acknowledgments:
PL acknowledges fruitful discussions on coronary calcifications with Dr. Gary Mintz, NYC during the late phases of drafting the manuscript.
Funding statement
RVT is supported by: a Wellcome Trust Senior Investigator Award (RVT); National Institute for Health Research (NIHR) Senior Investigator Award (RVT); and NIHR Oxford Biomedical Research Centre Programme. CSH is supported by the National Institutes of Health HL142932 and HL117917. DF is supported by the NIHR Oxford Biomedical Research Centre. MS is supported by Charité 3R and the Bundesministerium für Bildung und Forschung. JDL and JSR are supported by Informatics for Life funded by the Klaus Tschira Foundation. JSR has received funding from GSK and Sanofi and expects consultant fees from Travere Therapeutics. AM acknowledges financial support from the Spanish Ministry of Science, Innovation, and Universities [Grant no: PGC2018_095795_B_I00].
Abbreviations
- AC
arterial calcification
- IAC
intimal arterial calcification
- MAC
medial arterial calcification
- VC
vascular calcification
Footnotes
Disclosures: The authors disclose no conflicts of interests
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Reesink KD, Spronck B. Constitutive interpretation of arterial stiffness in clinical studies: A methodological review. Am J Physiol Heart Circ Physiol 2019;316:H693–H709. [DOI] [PubMed] [Google Scholar]
- 2.Weber T, Chirinos JA. Pulsatile arterial haemodynamics in heart failure. Eur Heart J 2018;39:3847–3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chirinos JA, Segers P, Hughes T, Townsend R. Large-artery stiffness in health and Disease; JACC State-of-the-Art review. J Am Coll Cardiol 2019;74:1237–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mowafy KA, Soliman M, Hammoda AM, Soliman RM. Bilateral lower limb disabling claudication in a young man: A case of Mönckeberg’s arteriosclerosis. Vasc Endovasc Rev https://www.verjournal.com/articles/Disabling-Claudication-Monckebergs-Arteriosclerosis (June 6, 2020). [Google Scholar]
- 5.Pisani I, De Troia A, Allegri L, Corradi D, Vaglio A. Malignant Mönckeberg medial calcific sclerosis. Intern Emerg Med 2018;13:615–617. [DOI] [PubMed] [Google Scholar]
- 6.Lanzer P, Boehm M, Sorribas V, et al. Medial vascular calcification revisited: review and perspectives. Eur Heart J 2014;35:1515–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mustapha JA, Diaz-Sandoval LJ, Saab F. Infrapopliteal calcification patterns in critical limb ischemia: diagnostic, pathologic and therapeutic implications in the search for the endovascular holy grail. J Cardiovasc Surg (Torino) 2017;58:383–401. [DOI] [PubMed] [Google Scholar]
- 8.Lanzer P Mediakalzinose Mönckeberg. Z Kardiol 1998;87:586–593. [DOI] [PubMed] [Google Scholar]
- 9.Niskanen L, Siitonen O, Suhonen M, Uusitupa MI. Medial artery calcification predicts cardiovascular mortality in patients with NIDDM. Diab Care 1994;17:1252–6. [DOI] [PubMed] [Google Scholar]
- 10.Lehto S, Niskanen L, Suhonen M, Rönnemaa T, Laakso M. Medial artery calcification. A neglected harbinger of cardiovascular complications in non-insulin-dependent diabetes mellitus. Arterioscler Thromb Vasc Biol 1996;16:978–83. [DOI] [PubMed] [Google Scholar]
- 11.London GM, Guérin AP, Marchais SJ, Métivier F, Pannier B, Adda H. Arterial media calcification in end-stage renal disease: impact on all-cause and cardiovascular mortality. Nephrol Dial Transplant 2003;18:1731–40. [DOI] [PubMed] [Google Scholar]
- 12.Nelson AJ, Raggi P, Wolf M, Gold AM, Chertow GM, Roe MT. Targeting vascular calcification in chronic kidney disease. JACC Basic Transl Sci 2020; 5: 398–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pescatore LA, Gamarra LF, Lieberman M. Multifaceted mechanisms of vascular calcification in aging. Arterioscler Thromb Vasc Biol 2019;39:1307–1316. [DOI] [PubMed] [Google Scholar]
- 14.Généreux P, Madhavan MV, Mintz GS et al. Ischemic outcomes after coronary intervention of calcified vessels in acute coronary syndromes. Pooled analysis from the HORIZONS-AMI (Harmonizing Outcomes with Revascularization and Stents in Acute Myocardial Infarction) and ACUITY (Acute Catheterization and Urgent Intervention Triage Strategy) TRIALS. J Am Coll Cardiol 2014;63:1845–54. [DOI] [PubMed] [Google Scholar]
- 15.Bourantas CV, Zhang Y-J, Garg S et al. Prognostic implications of severe coronary calcification in patients undergoing coronary artery bypass surgery: an analysis of the SYNTAX study. Catheter Cardiovasc Interv 2015;85:199–206. [DOI] [PubMed] [Google Scholar]
- 16.Misare BD, Pomposelli FB Jr, Gibbons GW, Campbell DR, Freeman DV, LoGerfo FW. Infrapopliteal bypasses to severely calcified, unclampable outflow arteries: two-year results. J Vasc Surg 1996;24:6–15. [DOI] [PubMed] [Google Scholar]
- 17.Villa-Bellosta R, Egido J. Phosphate, pyrophosphate, and vascular calcification: a question of balance. Eur Heart J 2017;38;1801–1804. [DOI] [PubMed] [Google Scholar]
- 18.Deneke T, Langner K, Grewe PH, Harrer E, Müller KM. Ossification in Atherosclerotic Carotid Arteries. Z Kardiol 2001;90Suppl 3:106–15. [DOI] [PubMed] [Google Scholar]
- 19.Boström K, Watson KE, Horn S, Wortham C, Herman IM, Demer LL. Bone morphogenetic protein expression in human atherosclerotic lesions. J Clin Invest 1993;91:1800–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.St Hilaire C, Ziegler SG, Markello TC et al. Nt5e mutations and arterial calcifications. N Engl J Med 2011;364:432–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rutsch F, Ruf N, Vaingankar S, et al. Mutations in ENPP1 are associated with ‘idiopathic’ infantile arterial calcification. Nat Genet 2003;34:379–81. [DOI] [PubMed] [Google Scholar]
- 22.Wang C, Li Y, Shi L, et al. Mutations in slc20a2 link familial idiopathic basal ganglia calcification with phosphate homeostasis. Nat Genet 2012;44:254–256. [DOI] [PubMed] [Google Scholar]
- 23.Lebwohl M, Halperin M, Phelps RG. Occult pseudoxanthoma elasticum in patients with premature cardiovascular disease. N Engl J Med 1993;329:1237–1239. [DOI] [PubMed] [Google Scholar]
- 24.Rogers MA, Aikawa E. Cardiovascular calcification: artificial intelligence and big data accelerate mechanistic discovery. Nat Rev Cardiol 2019;16:261–274. [DOI] [PubMed] [Google Scholar]
- 25.Schantl AE, Ivarsson ME, Leroux J-C. Investigational pharmacological treatments for vascular calcification. Adv Ther 2019;2: 10.1002/adtp.201800094 (June 6, 2020). [DOI] [Google Scholar]
- 26.Herrmann J, Babic M, Tolle M, van der Giet M, Schuchardt M. Research models for studying vascular calcification. Int J Mol Sci 2020; 21 (6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Millan A, Lanzer P, Sorribas V. The thermodynamics of medial vascular calcification. Front Cell Develop Biol 9:633465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Krüger K Epidemiologie der peripheren arteriellen Verschlusskrankheit in Deutschland; Was ist gesichert und was ist offen? Hämostaseol 2006;26:193–196. [PubMed] [Google Scholar]
- 29.Hoek AG, Sabine R Zwakenberg SR, Petra JM Elders PJM. An elevated ankle-brachial index is not a valid proxy for peripheral medial arterial calcification. Atherosclerosis 2021;323:13–19. [DOI] [PubMed] [Google Scholar]
- 30.Narula N, Dannenberg AJ, Olin JW et al. Pathology of peripheral artery disease in patients with critical limb ischemia. J Am Coll Cardiol 2018;72:2152–63. [DOI] [PubMed] [Google Scholar]
- 31.O’Neill WC, Han KH, Schneider TM, Hennigar RA. Prevalence of nonatheromatous lesions in peripheral artery disease. Arterioscler Thromb Vasc Biol 2015;35:439–447. [DOI] [PubMed] [Google Scholar]
- 32.Mintz GS, Popma JJ, Pichard AD et al. Patterns of calcification in coronary artery disease. A statistical analysis of intravascular ultrasound and coronary angiography in 1155 lesions. Circulation 1995;91:1959–65. [DOI] [PubMed] [Google Scholar]
- 33.Abramovitz Y, Jilaihawi H, Chakravarty T, Mack MJ, Makkar RR. Porcelain aorta; a comprehensive review. Circulation 2020;131:827–836. [DOI] [PubMed] [Google Scholar]
- 34.de Jong PA, Hellings WE, Takx RAP, Isgun I, van Herwaarden JA, Mali WPTM. Computed tomography of aortic wall calcifciations in aortic dissection patients. PLoS One 20148;9(7):e102036.doi: 10.1371/journal.pone.0102036.eCollection 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Everhart JE, Pettitt DJ, Knowler WC, Rose FA, Bennett PH. Medial arterial calcification and its association with mortality and complications of diabetes. Diabetologia 1988;31:16–23. [DOI] [PubMed] [Google Scholar]
- 36.Smith DC, Bilmen GJ, Iqbal S, Robey S, Pereira M. Medial artery calcification as an indicator of diabetic peripheral vascular disease. Foot Ankle Int 2008;29:185–190. [DOI] [PubMed] [Google Scholar]
- 37.Losurdo F, Ferraresi R, Ucci A, Zanetti A, Clerici G, Zambon A. Association of infrapopliteal medial arterial calcification with lower-limb amputations in high-risk patients: A systematic review and meta-analysis. Vasc Med 2020;December29; doi: 10.1177/1358863X20979738. [DOI] [PubMed] [Google Scholar]
- 38.Schurgers LJ, Teunissen KJ, Knapen MH et al. , Novel conformation-specific antibodies against matrix gamma-carboxyglutamic acid (Gla) protein: undercarboxylated matrix Gla protein as marker for vascular calcification. Arterioscler Thromb Vasc Biol 2005; 25:1629–33. [DOI] [PubMed] [Google Scholar]
- 39.Dalmeijer GW, van der Schouw YT, Magdeleyns EJ, et al. Matrix Gla protein species and risk of cardiovascular events in type 2 diabetic patients. Diabet Care 2013;36:3766–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barrett H, O’Keeffe M, Kavanagh E, Walsh M, O’Connor EM. Is Matrix Gla Protein associated with vascular calcification? A Systematic review. Nutrients 2018March27;10(4):415. doi: 10.3390/nu10040415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Joolharzadeh P, St Hilaire C. CD73 (Cluster of Differentiation 73) and the differences between mice and humans. Arterioscler Thromb Vasc Biol 2019; 39:339–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nelson AJ, Raggi P, Wolf M, Gold AM, Chertow GM, Roe MT. Targeting Vascular Calcification in Chronic Kidney Disease. JACC Basic Transl Sci 2020; 5:398–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang C, Li Y, Shi L, et al. Mutations in SLC20A2 link familial idiopathic basal ganglia calcification with phosphate homeostasis. Nat Genet 2012; 44:254–6. [DOI] [PubMed] [Google Scholar]
- 44.Bon N, Couasnay G, Bourgine A, et al. Phosphate (Pi)-regulated heterodimerization of the high-affinity sodium-dependent Pi transporters PiT1/Slc20a1 and PiT2/Slc20a2 underlies extracellular Pi sensing independently of Pi uptake. J Biol Chem 2018; 293:2102–2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Voelkl J, Lang F, Eckardt K-U,;et al. Signaling pathways involved in vascular smooth muscle cell calcification during hyperphosphatemia. Cell Mol Life Sci 2019;76:2077–2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chavkin NW, Chia JJ, Crouthamel MH, Giachelli CM. Phosphate uptake-independent signaling functions of the type III sodium-dependent phosphate transporter, PiT-1, in vascular smooth muscle cells. Exp Cell Res 2015; 333:39–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yamada S, Leaf EM, Chia JJ, Cox TC, Speer MY, Giachelli CM. PiT-2, a type III sodium-dependent phosphate transporter, protects against vascular calcification in mice with chronic kidney disease fed a high-phosphate diet. Kidney Int 2018; 94:716–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hortells L, Sosa C, Guillén N, Lucea S, Millán A, Sorribas V. Identifying early pathogenic events during vascular calcification in uremic rats. Kidney Int 2017;92:1384–1394. [DOI] [PubMed] [Google Scholar]
- 49.Komaba H, Fukagawa M. Phosphate-a poison for humans? Kidney Int 2016;90:753–63. [DOI] [PubMed] [Google Scholar]
- 50.Villa-Bellosta R, Millan A, Sorribas V. Role of calcium-phosphate deposition in vascular smooth muscle cell calcification. Am J Physiol Cell Physiol 2011;300:C210–C220. [DOI] [PubMed] [Google Scholar]
- 51.Alam M, Kirton JP,Wilkinson FL, et al. Calcification is associated with loss of functional calcium-sensing receptor in vascular smooth muscle cells. Cardiovasc Res 2009;81:260–8. [DOI] [PubMed] [Google Scholar]
- 52.Mary A, Objois T,Brazier M, et al. Decreased monocyte calcium sensing receptor expression in patients with chronic kidney disease is associated with impaired monocyte ability to reduce vascular calcification. Kidney Int 2021;99:1382–1391. [DOI] [PubMed] [Google Scholar]
- 53.Louvet L, Büchel J, Steppan S, Passlick-Deetjen J, Massy ZA. Magnesium prevents phosphate-induced calcification in human aortic vascular smooth muscle cells. Nephrol Dial Transplant 2013;28:869–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xu J, Bai Y, Jin J, Zhang J, Zhang S, Cui L, Zhang H. Magnesium modulates the expression levels of calcification-associated factors to inhibit calcification in a time-dependent manner. Exp Ther Med 2015;9:1028–1034. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 55.Newby AC. Adenosine and the concept of ‘retaliatory metabolites’. Trends in Biochemical Sciences 1984; 9:42–44. [Google Scholar]
- 56.Li Q, van de Wetering K, Uitto J. Pseudoxanthoma elasticum as a paradigm of heritable ectopic mineralization disorders: pathomechanisms and treatment development. Am J Pathol. 2019;189:216–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jin H, St Hilaire C, Huang Y, et al. Increased activity of TNAP compensates for reduced adenosine production and promotes ectopic calcification in the genetic disease ACDC. Sci Signal 2016; 9 (458):ra121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Moorhead WJ 3rd, Chu CC, Cuevas RA, et al. Dysregulation of FOXO1 (Forkhead Box O1 Protein) drives calcification in arterial calcification due to deficiency of CD73 and is present in peripheral artery disease. Arterioscler Thromb Vasc Biol 2020; 40:1680–1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schlatmann TJ, Becker AE. Pathogenesis of dissecting aneurysm of aorta. Comparative histopathologic study of significance of medial changes. Am J Cardiol 1977; 39:21–6. [DOI] [PubMed] [Google Scholar]
- 60.Wanga S, Hibender S, Ridwan Y, et al. Aortic microcalcification is associated with elastin fragmentation in Marfan syndrome. J Pathol 2017; 243:294–306. [DOI] [PubMed] [Google Scholar]
- 61.Markello TC, Pak LK, St Hilaire C et al. Vascular pathology of medial arterial calcifications in NT5E deficiency: implications for the role of adenosine in pseudoxanthoma elasticum. Mol Genet Metab 2011; 103:44–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Canadas V, Vilacosta I, Bruna I, Fuster V. Marfan syndrome. Part 1: pathophysiology and diagnosis. Nat Rev Cardiol 2010; 7:256–65. [DOI] [PubMed] [Google Scholar]
- 63.Neptune ER, Frischmeyer PA, Arking DE, et al. Dysregulation of TGF-beta activation contributes to pathogenesis in Marfan syndrome. Nat Genet 2003; 33:407–11. [DOI] [PubMed] [Google Scholar]
- 64.Gallo EM, Loch DC, Habashi JP, et al. Angiotensin II-dependent TGF-beta signaling contributes to Loeys-Dietz syndrome vascular pathogenesis. J Clin Invest 2014; 124:448–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Callewaert BL, Willaert A, Kerstjens-Frederikse WS, et al. Arterial tortuosity syndrome: clinical and molecular findings in 12 newly identified families. Hum Mutat 2008; 29:150–8. [DOI] [PubMed] [Google Scholar]
- 66.Jaalouk DE, Lammerding J. Mechanotransduction gone awry. Nat Rev Mol Cell Biol 2009; 10:63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ngai D, Lino M, Rothenberg KE, Simmons CA, Fernandez-Gonzalez R, Bendeck MP. DDR1 (Discoidin Domain Receptor-1)-RhoA (Ras Homolog Family Member A) Axis senses matrix stiffness to promote vascular calcification. Arterioscler Thromb Vasc Biol 2020; 40:1763–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hou Y-C, C-L, Yuan T-H, M-T, Chao C-T, Lu K-C. The Epigenetic landscape of vascular calcification: An integrative perspective. Int J Mol Sci 2020, 21(3), 980; 10.3390/ijms21030980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature 2009;461(7267):1071–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang C, Xu W, Jie An J et al. Poly(ADP-ribose) polymerase 1 accelerates vascular calcification by upregulating Runx2. Nat Commun 2019;10(1):1203. doi: 10.1038/s41467-019-09174-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ngai D, Lino M, Rothenberg KE, Simmons CA, Fernandez-Gonzalez R, Bendeck MP. DDR1 (Discoidin Domain Receptor-1)-RhoA (Ras Homolog Family Member A) axis senses matrix stiffness to promote vascular calcification. Arterioscler Thromb Vasc Biol 2020;40:1763–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Anderson HC. Electron microscopic studies of induced cartilage development and calcification. J Cell Biol 1967;35:81–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Blaser MC,Aikawa E, Roles and Regulation of Extracellular Vesicles in Cardiovascular Mineral Metabolism. Front Cardiovasc Med 201821;5:187. doi: 10.3389/fcvm.2018.00187.eCollection 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Edmonds ME, Morrison N, Laws JW, Watkins PJ. Medial arterial calcification and diabetic neuropathy. BMJ 1982;284:928–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bourron O, Aubert CE, Liabeuf S, Cluzel P, Lajat-Kiss F, Dadon M, Hartemann A. Below-Knee Arterial Calcification in Type 2 Diabetes: Association With Receptor Activator of Nuclear Factor κB Ligand, Osteoprotegerin, and Neuropathy. The Journal of Clinical Endocrinology & Metabolism, 2014;99:4250–4258. [DOI] [PubMed] [Google Scholar]
- 76.Filipska I,Winiarska A, Knysak M, Stompór T. Contribution of gut microbiota-derived uremic toxins to the cardiovascular system mineralization. Toxins (Basel) 2021April10;13(4):274. doi: 10.3390/toxins13040274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Murabito JM, White CC, Kavousi M, et al. Association Between Chromosome 9p21 Variants and the Ankle-Brachial Index identified by a meta-analysis of 21 Genome-Wide Association studies. Circulation Cardiovasc Genet 2012;5:100–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zekavat SM, Aragam K, Emdin C, Khera AV, Klarin D, Zhao H, Natarajan P. Genetic Association of finger photoplethysmography-derived arterial stiffness index with blood pressure and coronary artery disease. Arterioscler Thromb Vasc Biol. 2019;39:1253–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.O’Neill WC, Han KH, Schneider TM, Hennigar RA. Prevalence of nonatheromatous lesions in peripheral arterial disease. Arterioscler Thromb Vasc Biol 2015;35:439–47. [DOI] [PubMed] [Google Scholar]
- 80.Goldenberg S, Alex M, Joshi RM, Blumenthal HAT. Nonatheromatous peripheral vascular disease of the lower extremity in diabetes mellitus. Diabetes 1959;8:261–73. [DOI] [PubMed] [Google Scholar]
- 81.Johnson RC, Leopold JA, Loscalzo J. Vascular Calcification; Circ Res 2006;99:1044–1059. [DOI] [PubMed] [Google Scholar]
- 82.Lee SJ, In-Kyu Lee I-K, Jae-Han Jeon J-H. Vascular calcification; new insights into its mechanism. Int J Mol Sci 2020April13;21(8):2685. doi: 10.3390/ijms21082685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.McEniery CM, McDonnell BJ, So A, et al. Aortic calcification is associated with aortic stiffness and isolated systolic hypertension in healthy individuals. Hypertension 2009;53:524–31. [DOI] [PubMed] [Google Scholar]
- 84.Mitchell GF. Aortic stiffness, pressure and flow pulsatility, and target organ damage. Appl Physiol 2018;125: 1871–1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Edmonds ME,Roberts VC,Watkins PJ. Blood flow in the diabetic neuropathic foot. Diabetologia 1982;22:9–15. [DOI] [PubMed] [Google Scholar]
- 86.Glagov S, Weisenberg E, Zarins CK, Stankunavicius R, Kolettis GJ; Compensatory enlargement of human atherosclerotic coronary arteries. N Engl J Med.1987;316:1371–5. [DOI] [PubMed] [Google Scholar]
- 87.Fok P-W, Lanzer P. Media sclerosis drives and localizes atherosclerosis in peripheral arteries. PLoS One 2018 doi: 10.1371/journal.pone.0205599. eCollection 2018 (June 6, 2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chatzizisis YS, Coskun AU, Jonas M, Edelman ER, Feldman CL, Stone PH. Role of endothelial shear stress in the natural history of coronary atherosclerosis and vascular remodeling. J Am Coll Cardiol 2007;49;2379–93. [DOI] [PubMed] [Google Scholar]
- 89.Stary HC, Chandler AB, Dinsmore RE, et al. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. Circulation 1995;92:1355–74. [DOI] [PubMed] [Google Scholar]
- 90.Lachman AS, Spray TL, Kerwin DM, Shugoll GI, Robert WC. Medial calcinosis of Mönckeberg. Am J Med 1977;63:615–622. [DOI] [PubMed] [Google Scholar]
- 91.Castillo BV, Torczynski E, Edward DP. Mönckeberg’s sclerosis in a temporal artery biopsy specimen. Br J Ophthalmol 1999;83:1091–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lee S-J, Choe YS, Lee JC, Park BC, Lee WJ, Kim DW. Two cases of Mönckeberg’s medial sclerosis on the face. Ann Dermatol (Soul) 2007;19:31–34. [Google Scholar]
- 93.Kim HJ, Greenerg JS, Javitte MC. Breast calcification due to Mönckeberg medial calcific sclerosis. Radiographics 1999;19:1401–3. [DOI] [PubMed] [Google Scholar]
- 94.Ferraresi R, Ucci A, Pizzuto A, et al. A novel scoring system for small artery disease and medial arterial calcification is strongly associated with major adverse events in patients with chronic limb-threatening ischemia. J Endovasc Ther Off J Int Soc Endovasc Spec 2020:1526602820966309. doi: 10.1177/1526602820966309. [DOI] [PubMed] [Google Scholar]
- 95.Liu KH, Chu WC, Kong AP, et al. US assessment of medial arterial calcification: a sensitive marker of diabetes-related microvascular and macrovascular complications. Radiology 2012;265:294–302. [DOI] [PubMed] [Google Scholar]
- 96.Lidbom A Arteriosclerosis and arterial thrombosis in the lower limb: a roentgenological study. Acta Radiol Suppl 1950;80:1–80. [PubMed] [Google Scholar]
- 97.Konijin LCD, van Overhagen H, Takx RAP, de Jong PA, Veger HTC, Mali WPThM. CT calcification patterns of peripheral arteries in patients without known peripheral arterial disease. Eur J Radiol 2020July;128:108973.doi: 10.1016/j.ejrad.2020.108973. [DOI] [PubMed] [Google Scholar]
- 98.Greenland P, Blaha MJ, Budoff MJ, Erbel R. Watson KE. Coronary Calcium Score and Cardiovascular Risk; JACC State-of-the-Art Review. J Am Coll Cardiol.2018,72434–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wang X, Matsumura M, Mintz GS et al. , In Vivo calcium detection by comparing optical coherence tomography, intravascular ultrasound, and angiography. JACC Cardiovasc Imag 2017;10:869–879. [DOI] [PubMed] [Google Scholar]
- 100.Chen N-C, Hsu C-Y, Chen C-L. The strategy to prevent and regress the vascular calcification in dialysis patients. BioMed Research Internat 2017, 10.1155/2017/903519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Raggi P, Chertow GM, Torres PU, et al. The ADVANCE study: a randomized study to evaluate the effects of cinacalcet plus low-dose vitamin D on vascular calcification in patients on hemodialysis. Nephrol Dial Transplant 2011;26:1327–1339. [DOI] [PubMed] [Google Scholar]
- 102.Jamal SA, Vandermeer B, Raggi P, et al. Effect of calcium-based versus non-calcium-based phosphate binders on mortality in patients with chronic kidney disease: an updated systematic review and meta-analysis. Lancet 2013;382(9900):1268–77. [DOI] [PubMed] [Google Scholar]
- 103.Neves KR, Graciolli FG, Reis LM, et al. Vascular calcification: contribution of parathyroid hormone in renal failure. Kidney Int 2007;71:1262–70. [DOI] [PubMed] [Google Scholar]
- 104.Raggi P, James G, Burke SK, et al. Decrease in thoracic vertebral bone attenuation with calcium-based phosphate binders in hemodialysis. J Bone Miner Res 2005;20:764–72. [DOI] [PubMed] [Google Scholar]
- 105.De Vriese AS, Caluwé R, Pyfferoen L, et al. Multicenter Randomized Controlled Trial of Vitamin K antagonist replacement by rivaroxaban with or without vitamin K2 in hemodialysis patients with atrial fibrillation: the Valkyrie Study. JASN 2020;1;186–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Leadbetter MR, Kozuka L, Kohler K et al. Gastrointestinal inhibition of sodium-hydrogen exchanger 3 reduces phosphorus absorption and protects against vascular calcification in CKD. J Am Soc Nephrol 2015;26:1138–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Block GA, Rosenbaum DP, Yan A, Chertow GM. Efficacy and safety of tenapanor in patients with hyperphosphatemia receiving maintenance hemodialysis: A Randomized phase 3 trial. J Am Soc Nephrol 2019;30:641–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Rutsch F, Böyer P, Nitschke Y et al. ,. Hypophosphatemia, hyperphosphaturia, and bisphosphonate treatment are associated with survival beyond infancy in generalized arterial calcification of infancy. Circ Cardiovasc Genet 2008;1:133–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Etidronate for Arterial Calcifications Due to Deficiency in CD73 (ACDC). https://clinicaltrials.gov/ct2/show/NCT01585402.
- 110.O’Neill C, Lomashvili KA. Recent progress in the treatment of vascular calcification. Kidney Intern 2010;78,1232–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Nitta K, Ogawa T. Vascular Calcification in End-Stage Renal Disease Patients. IN: Nitta K (ed): Chronic Kidney Diseases - Recent Advances in Clinical and Basic Research. Contrib Nephrol Basel: Karger, 2015, 185; 156–167. [DOI] [PubMed] [Google Scholar]
- 112.Kranenburg G, de Jong PA, Bartstra JW et al. , Etidronate for prevention of ectopic mineralization in patients with pseudoxanthoma elasticum. J Am Coll Cardiol. 2018;71:1117–1126. [DOI] [PubMed] [Google Scholar]
- 113.Jaffe IZ, Tintut Y, Newfell BG, Demer LL, Mendelsohn ME. Mineralocorticoid receptor activation promotes vascular cell calcification. Arterioscler Thromb Vasc Biol 2007;27:799–805. [DOI] [PubMed] [Google Scholar]
- 114.Lang F, Ritz E, Voelkl J, Alesutan I. Vascular calcification--is aldosterone a culprit? Nephrol Dial Transplant 2013;28:1080–4. [DOI] [PubMed] [Google Scholar]
- 115.Sakaguchi Y, Hamano T, Obi Y, et al. A Randomized Trial of Magnesium Oxide and Oral Carbon Adsorbent for Coronary Artery Calcification in Predialysis CKD. J Am Soc Nephrol 2019;30:1073–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Bressendorff I, Hansen D, Schou M, Pasch A, Brandi L. The Effect of increasing dialysate magnesium on serum calcification propensity in subjects with end stage kidney disease: A Randomized, controlled clinical trial. Clin J Am Soc Nephrol 2018;13:1373–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Grases F, Costa-Bauza A. Key Aspects of Myo-inositol hexaphosphate (phytate) and pathological calcifications. Molecules 2019,24, 4434; doi: 10.3390/molecules24244434109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Raggi P, Bellasi A, Bushinsky D, et al. Slowing progression of cardiovascular calcification with SNF472 in patients on hemodialysis: Results of a randomized phase 2b study. Circulation 2020;141:728–739. [DOI] [PubMed] [Google Scholar]
- 119.Hedayati SS. A novel treatment for vascular calcification in patients with dialysis-dependent chronic kidney disease: Are we there yet? Circulation 2020;141:740–742. [DOI] [PubMed] [Google Scholar]
- 120.Alappan HR. Vasanth P, Manzoor S, O’Neill WC. Vascular calcification slows but does not regress after kidney transplantation. KIReports. October08, 2020. DOI: 10.1016/j.ekir.2020.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Mintz GS. Intravascular imaging of coronary calcification and its clinical implications. JACC Cardiovasc Imag 2015;8:461–471. [DOI] [PubMed] [Google Scholar]
- 122.Madhavan MV, Tarigopula M, Mintz GS, Maehara A, Stone GW, Généreux P. Coronary artery calcification: pathogenesis and prognostic implications. J Am Coll Cardiol 2014;63:1703–14. [DOI] [PubMed] [Google Scholar]
- 123.Dini CS, Tomberli B,Mattesini A, et al. Intravascular lithotripsy for calcific coronary and peripheral artery stenoses. EuroIntervention 2019;15:714–721. [DOI] [PubMed] [Google Scholar]
- 124.Schanstra JP, Luong TTD, Makridakis M, et al. Systems biology identifies cytosolic PLA2 as a target in vascular calcification treatment. JCI Insight 2019May16;4(10):e125638. doi: 10.1172/jci.insight.125638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Stranneheim H, Wedell A. Exome and genome sequencing: a revolution for the discovery and diagnosis of monogenic disorders. J Intern Med 2016;279:3–15. [DOI] [PubMed] [Google Scholar]
- 126.Taylor JC, et al. Factors influencing success of clinical genome sequencing across a broad spectrum of disorders. Nat Genet 2015;47:717–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Malhotra R, Mauer AC, Lino Cardenas CL et al. HDAC9 is implicated in atherosclerotic aortic calcification and affects vascular smooth muscle cell phenotype. Nat Genet 2019; 51:1580–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.International Mouse Knockout Consortium; Collins FS, Rossant J, Wurst W. A mouse for all reasons. Cell 2007;128:9–13. [DOI] [PubMed] [Google Scholar]
- 129.Rucher G, Cameliere L, Fendri J, et al. Molecular imaging of endothelial activation and mineralization in a mouse model of accelerated atherosclerosis. EJNMMI Res. 2019;9:80. doi: 10.1186/s13550-019-0550-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Sinha S, Santoro MM. New models to study vascular mural cell embryonic origin: implications in vascular diseases. Cardiovasc Res 2018; 114:481–491. [DOI] [PubMed] [Google Scholar]
- 131.Herrington DM, Mao C, Parker SJ, et al. Proteomic architecture of human coronary and aortic atherosclerosis. Circulation 2018;137:2741–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Chen X, Liu L, Palacios G, et al. Plasma metabolomics reveals biomarkers of the atherosclerosis. J Sep Sci 2010;33:2776–83. [DOI] [PubMed] [Google Scholar]
- 133.Williams JW, Winkels H, Durant CP, Zaitsev K, Ghosheh Y, Ley K. Single cell RNA sequencing in atherosclerosis research. Circ Res 2020;126:1112–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Gomes CPC, Schroen B, Kuster GM, et al. EU-CardioRNA COST Action (CA17129). Regulatory rnas in heart failure. Circulation 2020;141:313–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Zhang Z, Salisbury D, Sallam T. Long noncoding rnas in atherosclerosis: JACC review topic of the week. J Am Coll Cardiol 2018;72:2380–2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Jeong G, Kwon D-H, Shin S, et al. Long noncoding RNAs in vascular smooth muscle cells regulate vascular calcification. Sci Rep 2019; 9:5848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Schanstra JP, Luong TT, Makridakis M, et al. Systems biology identifies cytosolic PLA2 as a target in vascular calcification treatment. JCI Insight 2019May16;4(10). pii: 125638. doi: 10.1172/jci.insight.125638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lamb J, Crawford ED, Peck D, et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science 2006; 313:1929–1935. [DOI] [PubMed] [Google Scholar]
- 139.Song JS, Wang RS, Leopold JA, Loscalzo J. Network determinants of cardiovascular calcification and repositioned drug treatments [published online ahead of print, 2020 Jul 8]. FASEB J 2020;34(8):11087–11100. doi: 10.1096/fj.202001062R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Fernandez DM, Rahman AH, Fernandez NF, et al. Single-cell immune landscape of human atherosclerotic plaques. Nat Med 2019;25:1576–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Depuydt MAC, Prange KHM, Slenders L, et al. Microanatomy of the Human Atherosclerotic Plaque by Single-Cell Transcriptomics. Circ Res. 2020November6;127(11):1437–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Liu M, Gomez D. Smooth muscle cell phenotypic diversity. Arterioscler Thromb Vasc Biol. 2019;39:1715–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Schlotter F, Halu A, Goto S, et al. Spatiotemporal Multi-Omics mapping generates a molecular atlas of the aortic valve and reveals networks driving disease. Circulation 2018;138:377–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Zhang M, Sheffield T, Zhan X, et al. Spatial molecular profiling: platforms, applications and analysis tools. Brief Bioinform 2021May20;22(3):bbaa145. doi: 10.1093/bib/bbaa145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Asp M, Bergenstråhle J, Lundeberg J. Spatially resolved transcriptomes-next generation tools for tissue exploration. Bioessays 2020;e1900221. [DOI] [PubMed] [Google Scholar]
- 146.Epple M, Lanzer P. How much interdisciplinarity is required to understand vascular calcifications? Formulation of four basic principles of vascular calcification. Z Kardiol 2001;90Suppl 3:2–5. [DOI] [PubMed] [Google Scholar]
- 147.Gourgas O, Marulanda J, Zhang P, Murshed M, Cerruti M. Multidisciplinary approach to understand medial arterial calcification. Arterioscl Thromb Vasc Biol 2018;38:363–372. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.