

Abstract
Antimicrobial peptides (AMPs) produced by multi-cellular organisms as their immune system’s defence against microbes are actively considered as natural alternatives to conventional antibiotics. Although substantial progress has been achieved in studying the AMPs, the microscopic mechanisms of their functioning remain not well understood. Here, we develop a new theoretical framework to investigate how the AMPs are able to efficiently neutralize bacteria. In our minimal theoretical model, the most relevant processes, AMPs entering into and the following inhibition of the single bacterial cell, are described stochastically. Using complementary master equations approaches, all relevant features of bacteria clearance dynamics by AMPs, such as the probability of inhibition and the mean times before the clearance, are explicitly evaluated. It is found that both processes, entering and inhibition, are equally important for the efficient functioning of AMPs. Our theoretical method naturally explains a wide spectrum of efficiencies of existing AMPs and their heterogeneity at the single-cell level. Theoretical calculations are also consistent with existing single-cell measurements. Thus, the presented theoretical approach clarifies some microscopic aspects of the action of AMPs on bacteria.
Keywords: antimicrobial peptides, probability of inhibition, mean inhibition times, single-cell heterogeneity, stochastic processes, bacterial clearance
1. Introduction
Antimicrobial peptides (AMPs), which are also called host defence peptides, are essential elements of the innate immune systems in multi-cellular organisms [1]. They generally provide the first line of defence against harmful bacteria without being toxic to the host organisms [1–4]. Moreover, AMPs support their hosts by exhibiting a variety of critically important biological properties such as anti-viral, anti-fungal, anti-cancer and anti-inflammatory activities [4–7]. Due to their antimicrobial actions, AMPs are frequently compared to conventional antibiotics that play a central role in modern medicine. However, there are two main differences between these classes of active molecules. First, unlike the typical antibiotics that are composed of single organic compounds, AMPs are made of (mostly positively charged) short sequences of amino acids [3]. Second, despite the fact that bacteria in multi-cellular organisms have encountered antimicrobial peptides for millions of years, acquisition of resistance by a bacterial strain against AMPs is much weaker [1,3,4,8,9]. These unique characteristics of AMPs stimulated significant efforts in exploring them as alternative therapeutic approaches against pathogenic bacteria and other types of infection [10]. In addition, new applications of AMPs in food production, agriculture and medicine have been also intensively debated [4,11].
It is widely believed that AMPs kill bacteria via one of two main routes [12,13]. AMPs might associate with the bacterial cell membrane that leads to the disruption of major processes and causes the formation of pores, allowing for the leakage of essential ions and nutrients and eventually killing the bacterium [2,13]. Alternatively, absorption of AMPs to the membrane can open new pathways for the peptides to move further inside and to act on various intracellular targets [3,4]. Recent experiments found that in some systems a large number of peptides (approx. 106–107) is needed to completely saturate the bacterial membrane and to trigger disruptive effects on bacteria [1,14]. This corresponds to AMPs operating at millimolar/micromolar concentrations. However, there are also AMPs that can kill bacteria even at much smaller nanomolar concentrations [1]. Such a wide spectrum of activities has been attributed to a variety of possible mechanisms of inhibition [1–4,12]. However, the microscopic details of AMPs' action on bacteria remain mostly unexplained.
A new direction in exploring the mechanisms of AMPs opened a recent quantitative study that investigated the dynamics of AMPs' actions by integrating single-cell and population level experiments [15]. Fast absorption and unexpected retention of LL37 antimicrobial peptides in E. coli bacteria have been observed, which led to a complex heterogeneous behaviour in the system. The cell growth was inhibited in one group of cells, while the growing sub-population survived due to the sequestration of AMPs by the non-growing fraction of bacterial cells. One of the striking observations of this study was a heterogeneous growth inhibition even at the single-cell level [15]. But the most important result of this study was a quantitative description of the bacterial inhibition dynamics by AMPs.
The importance of AMPs in protecting living organisms from various infections and microbes stimulated multiple theoretical investigations [16,17]. These studies, however, concentrated mostly on the structural aspects of entering the bacterial cell membranes and on the molecular models of pore formation. Since most AMPs are relatively short peptide chains, atomistic and coarse-grained molecular dynamics simulations have been actively used for clarifying the mechanisms of bacterial cell membrane disruption and for developing new potential drugs based on AMPs [16–18]. However, the dynamic aspects of bacterial inhibition have not been addressed. In addition, current theoretical studies have not considered the effect of stochasticity of underlying biochemical and biophysical processes. At the same time, it has been shown recently that the stochasticity plays an important role in bacterial clearance dynamics by conventional antibiotics [19,20]. In particular, these investigations led to a new definition of minimal inhibitory concentration (MIC) corrected by stochastic effects. It was suggested that even low concentrations (sub-MIC) could potentially lead to successful clearance of the bacteria [20].
In this paper, we present a new theoretical framework to analyse the bacterial clearance dynamics by AMPs. To take into account the random nature of underlying processes, a discrete-state stochastic approach of bacterial inhibition is developed. In our minimal theoretical model, normal bacterial cell growth, AMPs entering into the cell and stopping the growth are viewed as independent random processes. Using various master equations approaches [20–23], the inhibition dynamics is explicitly evaluated. Our results suggest that both entering and inhibition are equally important for the efficient functioning of AMPs. Theoretical analysis, which is consistent with available experimental observations for AMP LL37 in E. coli bacteria, also provides plausible explanations for a large range of AMP activities and for inhibition heterogeneity at the single-cell level.
2. Model
To understand the microscopic mechanisms of inhibition, let us consider a single bacterial cell surrounded initially (at t = 0) by N AMPs as illustrated in figure 1. At typical cellular conditions, this number is quite large, N ∼ 106–107 [14]. The cell can grow and divide into two new cells with a rate λ, while the AMPs can enter and attach to the bacterium with a rate constant a (figure 1). The overall entrance rate is proportional to the number of AMPs outside of the bacterium. The peptides already in the cell can lead to the growth inhibition with a rate constant k: see figure 1. Here, we also assume that the inhibition is an additive process, i.e. the rate of killing the bacterium is proportional to the number of AMPs already inside the cell. We assume that the cell growth rate λ is independent of the number of attached peptides because it is normally controlled by other external factors such as the availability of nutrients, temperature, osmotic pressure and many others [24]. The AMP entrance rate into bacterium depends on the amino acid sequence, the membrane lipid composition, details of peptide–membrane interactions and on the AMP concentration outside of the bacteria [25]. The inhibition rate reflects the biochemical and biophysical processes of membrane disruption that are still not well understood [2,3]. Note that this is a minimal description of a very complex bacterial clearance by AMPs that takes into account only the most relevant processes.
Figure 1.
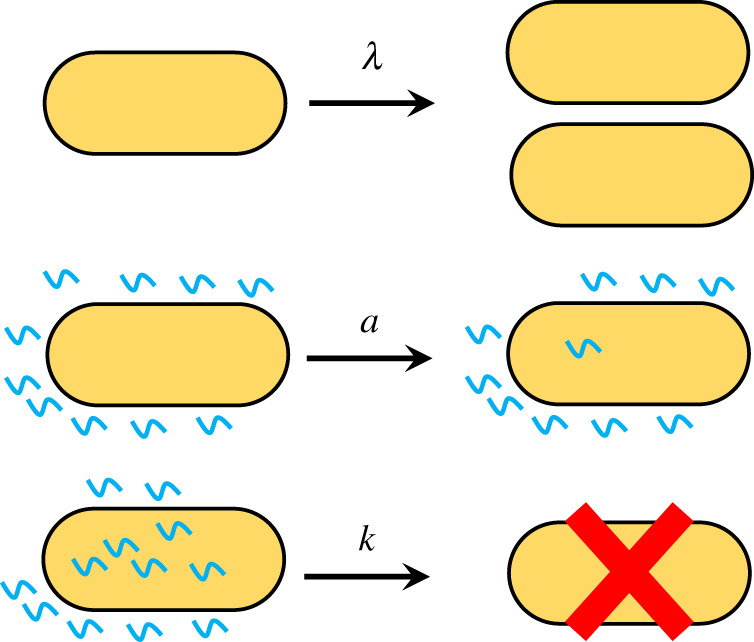
Three fundamental processes that are taking place during the interaction between AMPs and a bacterial cell: growth and division with a rate λ, peptides entering the cell with a rate a and bacterial inhibition with a rate k.
To obtain a comprehensive dynamic description of the system in figure 1, we use two complementary approaches that are based on master equations exact calculations. We will start with a first-passage probabilities method that has been successfully applied for investigations of various phenomena in chemistry, physics and biology [21,23]. It concentrates on analysing the dynamics of first arrival to specific states of the system. The second approach explores conventional forward master equations to evaluate the stationary probabilities of different discrete states in the system [23].
2.1. First-passage probabilities analysis
To analyse the system using the first-passage probabilities method, it is convenient to describe the dynamics using a kinetic scheme as presented in figure 2. The states are labelled by a variable n that describes the number of AMPs already inside the bacterium. From the state n (0 < n < N), the system can have three outcomes: the cell can grow and divide with the rate λ, one more peptide can enter the bacterium with the overall rate (N − n)a, or the cell growth can be inhibited with the overall rate nk. In the state n = 0, there are no AMPs inside the bacterium and for this reason only the growth (with the rate λ) or the peptide entering the cell (with the rate Na) are possible. In the state n = N, all originally available peptides are already in the cell and only the growth (with the rate λ) and inhibition (with the rate Nk) can happen: see figure 2.
Figure 2.

A kinetic scheme for the first-passage probabilities analysis of the bacterial inhibition by AMPs. Each discrete state in the system is labelled as n (n = 0, 1, …, N) where n corresponds to the number of already absorbed AMP molecules. Upper (green) arrows describe the uninhibited growth and cell division, lower (red) arrows describe the transitions to the inhibition, and the horizontal (black) arrows correspond to the sequential entrance of AMPs into the bacterium. Lower red dashed box corresponds to the fully inhibited state of the bacterium. Upper green dashed box corresponds to the growth state of the bacterium.
The dynamics in the system can be described by defining a function Fn(t), which is a probability density function for the cell to be inhibited at time t given that there are n AMPs inside the cell (0 ≤ n ≤ N) at t = 0. The temporal evolution of this function is governed by following backward master equations [23]:
| 2.1 |
for n = N, and
| 2.2 |
for 0 ≤ n < N. Here, we also used an additional function Fd(t) that corresponds to the probability to be inhibited at time t for the first time starting already from the inhibited state (figure 2). Clearly, the following boundary condition, Fd(t) = δ(t), must be satisfied. The physical meaning of this result is that the bacterial clearance is immediately accomplished if the cell is initially found in the inhibited state.
Introducing the Laplace transforms of the first-passage functions, , allows us to obtain full exact solutions for backward master equations (see electronic supplementary material, appendix A.1). Importantly, the first-passage probability density functions contain a comprehensive description of the bacterial clearance process. Specifically, yields the probability of bacterial inhibition given that initially n AMP molecules were contained the cell. It can be shown that the inhibition probability is given by (see electronic supplementary material, appendix A.1)
| 2.3 |
To mimic the experimental observations [15], we are interested in that describes the bacterial clearance probability starting from the state without any peptides inside the bacterium. It can be shown that this probability can be rewritten as
| 2.4 |
where parameters pj and qi are given by
| 2.5 |
These parameters can be easily understood. The mean residence time at the state j is given by 1/[λ + jk + (N − j)a], then pj is the probability of adding the additional peptide in the state j because the corresponding rate is (N − j)a. Similarly, qi is the probability of inhibition in the state i because the corresponding rate is ik. These arguments give a clear physical meaning for the expression in equation (2.4). The overall inhibition probability is the sum of inhibition probabilities from each state i ≥ 1. The factor gives the probability for the system to reach the state i without bacteria being cleared, and qi gives the probability of inhibition exactly in the state i.
To understand better the bacterial inhibition dynamics, it is interesting to consider two limiting situations. When the entrance rate is much faster than other transitions (a ≫ k, λ), the system is preferentially found in the state n = N. This is because from equation (2.5) one can estimate that pj ≃ 1, qj ≃ 0 for 0 ≤ j < N, and qN = Nk/(λ + Nk). In this limit, the probability of bacterial clearance is given by
| 2.6 |
In the opposite limit of very fast inhibition (k ≫ a, λ), the system is mostly found in the state n = 0 because even one peptide inside the bacterium can lead to the inhibition. In this case, we have from equation (2.5) pj ≃ 0, qj ≃ 1 for 0 < j ≤ N, and p0 = Na/(λ + Na). The probability of bacterial clearance then is given by
| 2.7 |
The results of our explicit calculations for the probability of bacterial clearance are presented in figure 3. Increasing the entrance and inhibition rates, as expected, leads to higher probabilities of killing the bacterium. But the important result here is that both processes (the entrance and the inhibition) are equally important for the overall inhibition. One can see this from the following arguments. For any specific value of the bacterial clearance probability , from equations (2.6) and (2.7) one might conclude that for and/or for it is not possible to achieve the inhibition probability larger than . In other words, for any peptide entrance rate below the critical value a* it is not possible to reach the inhibition probability above for any possible values of the killing rate k. Similarly, for any inhibition rate below the critical value k* it is not possible to reach the inhibition probability above for any possible values of the entrance rate a. One could also see the important role of the number of AMPs around the bacterium, N. Increasing N lowers these critical values, i.e. it is easier to kill the bacteria with more available AMPs.
Figure 3.
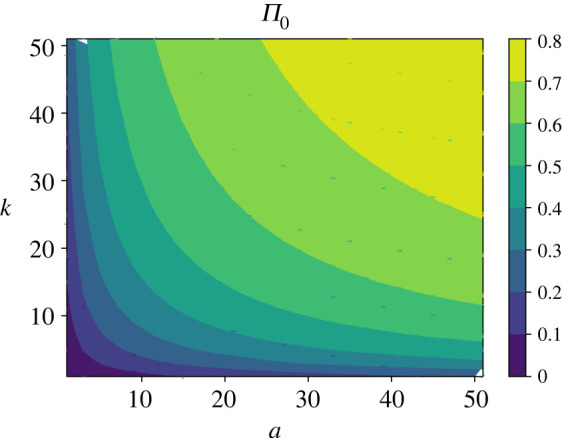
Analytical evaluation of the inhibition probability for a wide range of entrance and inhibition rates a and k (expressed in units of λ/N). For calculations, parameters N = 100 and λ = 3/60 min−1 have been used.
Another important characteristic of the bacterial clearance dynamics by AMPs is a mean inhibition time. This time scale corresponds to the average time before the bacteria will stop growing after being exposed to AMPs. This property is crucial for developing new AMP-based therapies and for evaluating the bacterial tolerance and resistance [26,27]. In the language of the first-passage probabilities method, the time Tn corresponds to the mean first-passage time to reach the inhibition state starting initially in the state n (figure 2). Using the probability density functions Fn(t), one can write
| 2.8 |
We are mostly interested in the mean inhibition times starting from the state n = 0 when there are no AMPs in the bacterium, T0. As shown in electronic supplementary material, appendix A.1, it is given by
| 2.9 |
where τj = 1/(λ + jk + (N − j)a) is the residence time for the system to be found in the state j. The physical meaning of equation (2.9) is the following. The total mean inhibition time is a sum of N contributions corresponding to the bacterial cell killing from the specific state n = 1, 2, …, N. Each such term is a product of several components. The probability of inhibition at the state n = i is given by , while the second multiplier is the sum of the residence times along this specific inhibition pathway that ends in the state n = i . At the end, everything is divided by the total probability of inhibition, , to account for those events that do not lead to the bacterial clearance (growth and division with the rate λ).
The expression for the mean inhibition time simplifies in the limiting situations. When the peptide entrance rates are very fast (a ≫ k, λ), the system is mostly found in the state n = N. The residence times in all states except the state n = N are very short, τi → 0 for i < N, while τN = 1/(λ + Nk). Then using equation (2.6) we obtain in this limit
| 2.10 |
In the opposite limit of very fast inhibition rates (a ≫ k, λ), the system will not be able to proceed beyond the state n = 1, and we have using equation (2.7),
| 2.11 |
The results of calculations for the mean inhibition times are presented in figure 4. Similarly to the inhibition probability, one can see that both entrance and killing rates are important in order to achieve fast bacterial clearance. For any desired mean inhibition time T*, for entrance and killing rates smaller than 1/N((1/T*) − λ) it is not possible to achieve the bacterial inhibition by AMPs at times shorter than T*. Comparing with figure 3, one could also conclude that, as expected, the bacterial clearance probabilities and the mean inhibition times correlate with each other: the larger is , the shorter is T0, i.e. if the probability of inhibition is large it will happen fast.
Figure 4.
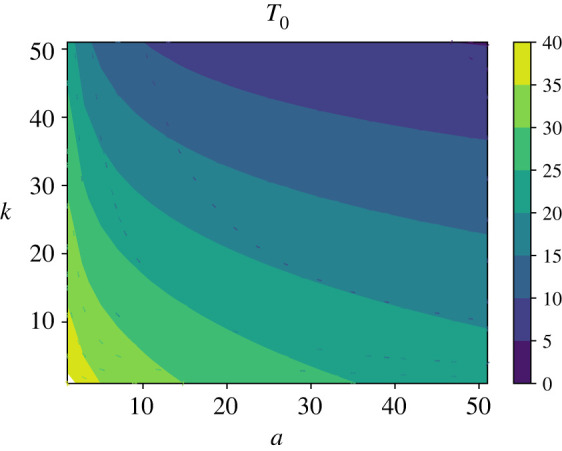
Analytical evaluation of the mean inhibition time T0 for a wide range of entrance and inhibition rates a and k (expressed in units of λ/N). For calculations, parameters N = 100 and λ = 3/60 min−1 were used.
Our theoretical method also allows us to estimate the degree of fluctuations in the inhibition dynamics by AMPs. It can be done by evaluating the noise, which might be defined as a cell-to-cell variation in the inhibition times. More specifically, we explicitly calculate the normalized variance of inhibition times:
| 2.12 |
where the expression for second moment is given in electronic supplementary material, appendix A.1. Based on these calculations, one could argue that the level of noise in the system weakly depends on both entrance and killing rates.
2.2. Forward master equations approach
There is an alternative method of analysing the dynamic properties of the AMP bacterial inhibition by using forward master equations approach. To do so, it is convenient to employ a kinetic scheme shown in figure 5. Since we are interested in the inhibition dynamics starting from the situation when there are no AMPs in the bacterium yet, as soon as the system goes to the growth (green transitions) or to the inhibition (red transitions) we reset it back to the state n = 0. This explains why all growth and inhibition transitions return to the state n = 0: see figure 5. Now, one can define a function Pn(t) as the probability to find the system in the state n (which also corresponds to n AMPs inside the bacterium) at time t. Temporal evolution of these functions is governed by a set of conventional forward master equations:
| 2.13 |
for n = 0,
| 2.14 |
for 1 ≤ n ≤ N. In addition, there is a normalization condition, .
Figure 5.

Schematic of the model for the calculation of distribution of number of peptides absorbed by cell.
At long times (t → ∞), the system reaches the stationary state where dPn(t)/dt = 0, and this allows us to solve explicitly the forward master equations as explained in electronic supplementary material, appendix A.2:
| 2.15 |
for n = 0, while
| 2.16 |
for 1 ≤ n ≤ N − 1, and
| 2.17 |
for n = N. It can be easily shown for a special simple case N = 1,
| 2.18 |
It is convenient also to rewrite equation (2.16) in the following form:
| 2.19 |
where coefficients γn are given by
| 2.20 |
To understand the behaviour of the stationary distribution Pn as a function of n, we can now evaluate using equation (2.19) the following ratio:
| 2.21 |
This suggests that Pn would decrease monotonically (Pn+1/Pn < 1) as a function of n for a < λ + (n + 1)k. This situation corresponds to the plots presented in figure 6b,c. However, a more complex behaviour is possible for a > λ + (n + 1)k when one could have a maximum in the distribution (figure 6d) or Pn might be even increasing with n (figure 6e).
Figure 6.

(a) Analytical evaluation of the Fano factor for the number of AMPs inside the bacterial cell for a wide range of entrance and inhibition rates a and k (expressed in units of λ/N). Probability distributions for (b) k = 10λ/N and a = λ/N; (c) for k = λ/N and a = 10λ/N; (d) for k = λ/N and a = 150λ/N; and (e) for k = λ/N and a = 500λ/N. For calculations, parameters N = 100 and λ = 3/60 min−1 were used.
Knowing the stationary probabilities for all states, one can estimate the dynamic properties in the system. The total inhibition flux is given by
| 2.22 |
while the total flux in the growth direction is given by
| 2.23 |
The forward and backward master equations approaches provide a complementary description of the bacterial clearance dynamics, and the connections between these approaches can be easily established. For example, the inhibition probability calculated explicitly in the first-passage approach can be also estimated using the forward master equations as
| 2.24 |
It can be shown explicitly that this expression fully agrees with the one obtained using the first-passage approach: see equation (2.4). The relations between the mean inhibition times and inhibition fluxes are more complex, but they can be provided using the approach discussed in [28].
The functions Pn not only give the stationary probabilities of different states in the system, but they also provide a stationary distribution of the number of AMPs inside the bacterial cell. One can estimate then the average number of absorbed AMPs needed to clear the bacterial infection:
| 2.25 |
But at the same time, one can rewrite equation (2.22) using the average number of absorbed AMPs, yielding
| 2.26 |
This means that the overall inhibition flux, as expected, is proportional the average number peptides inside the bacterium.
Our theoretical method also allows us to analyse the heterogeneity in inhibition dynamics at the single-cell level by quantifying the variations in the number of absorbed AMPs. For this purpose, we define a dimensionless parameter, known as a Fano factor, which is given by the ratio between the variance and the mean of the number of the absorbed peptides:
| 2.27 |
where the second moment in the distribution is equal to
| 2.28 |
The Fano factor is a convenient measure of heterogeneity because it correlates with the degree of fluctuations in the number of AMPs inside the bacterium.
The results of our explicit calculations for the distributions of absorbed peptides and the Fano factor are presented in figure 6. The analysis of the Fano factor (figure 6a) shows interesting trends. While increasing the inhibition rate k lowers the Fano factor, for larger entrance rates a the Fano factor is increasing. This can be explained by analysing the distributions of absorbed AMPs inside the bacterium. One can see that when the inhibition rate is much faster than the entrance rate the distribution is narrow because the bacterial cell stops growing before the large number of AMPs can enter inside (figure 6b). This leads to the smaller fluctuations in the number of peptides inside the bacterial cell. The situation is different for the faster entrance rates (a ≫ k) when the distribution is wide because many AMPs can enter the cell before it can be inhibited: see figure 6c,d. In this case, the fluctuations are much larger because the inhibition can happen for any number of absorbed peptides. Importantly, these arguments suggest that the Fano factor can be used as a quantitative measure of cellular heterogeneity in the inhibition dynamics of AMPs that can also provide information on the microscopic mechanisms. High heterogeneity would correspond to the systems where the entrance rates are fast, while lower heterogeneity would describe peptides with fast killing rates.
2.3. Comparison with experiments
To test our theoretical model, it is important to compare its predictions with experimental results. However, there is a limited amount of quantitative information on inhibition dynamics by AMPs at the single-cell level. A recent study provides such data by demonstrating the absorption and retention of LL37 AMPs in E. coli bacteria [15]. The rapid translocation of AMPs followed by the inhibition of bacterial cell growth was monitored using fluorescent time-lapse microscopy that allowed one to simultaneously visualize the growth of bacterial cells and the absorbance of AMPs.
While the growth rate for E.coli bacteria at the conditions used in these experiments is more or less is known, λ ≃ 0.03 min−1 [15], we need to estimate the entrance rate a and the inhibition rate k. The following arguments can be presented to approximately evaluate these parameters from experimental data [15]. In experiments, the concentration of AMPs used in single-cell measurements was c = 10 μM. Then the volume around the single cell can be roughly estimated as approximately 100 μm3, and there are N ∼ 106 peptides per cell in this volume. Experiments suggest that at these conditions in approximately 10 min all AMPs go inside the bacterium. This gives us the estimate of the parameter a ∼ 0.1 min−1. It seems that the growth does not stop for approximately 10–20 min, allowing us also to approximate k ≃ 10−7 min−1. Since the entrance rate a is much faster than other transitions in the system, we can use equations (2.6) and (2.10) to estimate the probability of inhibition and the mean inhibition time:
| 2.29 |
Note that although our estimates of the transition rates in the system are quite crude, our predictions for the mean inhibition times are consistent with experimental observations [15]. This gives a support to our theoretical model.
These calculations also suggest that our theoretical framework might naturally explain a very wide range of concentrations at which different AMPs are functioning. We can argue that those peptides that operate in the millimolar/micromolar range probably have faster entrance rates, a > k, so that a significant number of peptides is needed to enter the bacterium in order for the inhibition to be achieved. At the same time, AMPs working in the nanomolar regime probably have faster inhibition rates (k > a) since even a relatively small number of absorbed peptides can stop the growth of the bacterial cell. It seems that LL37 peptides are operating in the a > k regime that corresponds to larger heterogeneity in the system.
3. Summary and conclusion
We developed a new theoretical framework for analysing the bacterial clearance dynamics by AMPs at the single-cell level. This is done by taking into account the most relevant stochastic processes, such as the bacterial growth, AMP entrance and inhibition, as random stochastic processes. Using two complementary master equations approaches, the dynamic properties of bacterial inhibition are explicitly evaluated. It is found that both the entrance and the killing processes are equally important to support the effective action of AMPs. The probability of inhibition correlates with the speed of bacterial clearance. It is also shown that our theoretical predictions are consistent with available experimental data. In addition, it is suggested that the proposed theoretical approach might explain the wide spectrum of activities of various AMPs that operate at different concentration ranges. Furthermore, we presented a specific quantitative measure of heterogeneity and explained that larger heterogeneity is associated with faster entrance rates, while the faster inhibition rates do lead to a smaller heterogeneity.
Since the development of new AMP-based therapeutics is critically important for multiple medical applications, our theoretical results might help in designing new AMPs. We suggest that one should concentrate not only on improving the killing abilities of peptides when they are already bound to bacteria but also on the way for AMPs to enter faster. In addition, by tuning the entrance and the killing rates one might control the heterogeneity in the action of AMPs that is also important for applications. It will be interesting to test in experiments this combined strategy.
Although the presented theoretical framework is able to provide a comprehensive description for the inhibition dynamics of AMPs on bacteria, it should be emphasized that the model is very simplified and several important aspects of the system need to be explored more. Clearly, the processes of entering into the cell and the inhibition involve multiple biochemical and biophysical transformations and cannot be well approximated as single one-step stochastic transitions. In addition, it is important to consider various bacterial strategies of resistance to AMPs [29–31]. This includes the sequestration by bacterial surface structures [31], the alteration of membrane charges and/or fluidity, and the degradation and removal of AMPs by efflux pumps. Also bacteria may exhibit collective tolerance effects through a variety of different mechanisms, including the membrane-displayed proteases that degrade AMPs directly [32]. It will be interesting to extend and generalize our theoretical approach in these important directions [26,30,32,33].
Acknowledgements
We thank the anonymous reviewers for useful comments and suggestions.
Data accessibility
Additional data are available in the electronic supplementary material.
Authors' contributions
H.T. and A.B.K designed the research; H.T. and T.N.N. performed the research; H.T. and A.B.K wrote the manuscript.
Competing interests
We declare that we have no competing interests.
Funding
The work was supported by the Welch Foundation (C-1559), by the NSF (CHE-1953453 and MCB-1941106), and by the Center for Theoretical Biological Physics sponsored by the NSF (PHY-2019745).
References
- 1.Zasloff M. 2002Antimicrobial peptides of multicellular organisms. Nature 415, 389-395. ( 10.1038/415389a) [DOI] [PubMed] [Google Scholar]
- 2.Brogden KA. 2005Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 3, 238-250. ( 10.1038/nrmicro1098) [DOI] [PubMed] [Google Scholar]
- 3.Mahlapuu M, Håkansson J, Ringstad L, Björn C. 2016Antimicrobial peptides: an emerging category of therapeutic agents. Front. Cell. Infect. Microbiol. 6, 194. ( 10.3389/fcimb.2016.00194) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huan Y, Kong Q, Mou H, Yi H. 2020Antimicrobial peptides: classification, design, application and research progress in multiple fields. Front. Microbiol. 11, 2559. ( 10.3389/fmicb.2020.582779) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Divyashree M, Mani MK, Reddy D, Kumavath R, Ghosh P, Azevedo V, Barh D. 2020Clinical applications of antimicrobial peptides (AMPS): where do we stand now? Protein Pept. Lett. 27, 120-134. ( 10.2174/0929866526666190925152957) [DOI] [PubMed] [Google Scholar]
- 6.Tornesello AL, Borrelli A, Buonaguro L, Buonaguro FM, Tornesello ML. 2020Antimicrobial peptides as anticancer agents: functional properties and biological activities. Molecules 25, 2850. ( 10.3390/molecules25122850) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nizet Vet al.2001Innate antimicrobial peptide protects the skin from invasive bacterial infection. Nature 414, 454-457. ( 10.1038/35106587) [DOI] [PubMed] [Google Scholar]
- 8.Yu G, Baeder DY, Regoes RR, Rolff J. 2018Predicting drug resistance evolution: insights from antimicrobial peptides and antibiotics. Proc. R. Soc. B 285, 20172687. ( 10.1098/rspb.2017.2687) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fjell CD, Hiss JA, Hancock REW, Schneider G. 2012Designing antimicrobial peptides: form follows function. Nat. Rev. Drug Discovery 11, 37-51. ( 10.1038/nrd3591) [DOI] [PubMed] [Google Scholar]
- 10.Hancock REW, Sahl H-G. 2006Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 24, 1551-1557. ( 10.1038/nbt1267) [DOI] [PubMed] [Google Scholar]
- 11.Fox JL. 2013Antimicrobial peptides stage a comeback. Nat. Biotechnol. 31, 379-382. ( 10.1038/nbt.2572) [DOI] [PubMed] [Google Scholar]
- 12.Benfield AH, Henriques ST. 2020Mode-of-action of antimicrobial peptides: membrane disruption vs. intracellular mechanisms. Front. Med. Technol. 2, 20. ( 10.3389/fmedt.2020.610997) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peters BM, Shirtliff ME, Jabra-Rizk MA. 2010Antimicrobial peptides: primeval molecules or future drugs? PLoS Pathog. 6, e1001067. ( 10.1371/journal.ppat.1001067) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roversi D, Luca V, Aureli S, Park Y, Mangoni ML, Stella L. 2014How many antimicrobial peptide molecules kill a bacterium? The case of PMAP-23. ACS Chem. Biol. 9, 2003-2007. ( 10.1021/cb500426r) [DOI] [PubMed] [Google Scholar]
- 15.Snoussi M, Talledo JP, Del Rosario N-A, Mohammadi S, Ha B-Y, Košmrlj A, Taheri-Araghi S. 2018Heterogeneous absorption of antimicrobial peptide ll37 in Escherichia coli cells enhances population survivability. Elife 7, e38174. ( 10.7554/eLife.38174) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li J, Koh J-J, Liu S, Lakshminarayanan R, Verma CS, Beuerman RW. 2017Membrane active antimicrobial peptides: translating mechanistic insights to design. Front. Neurosci. 11, 73. ( 10.3389/fnins.2017.00073) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fox SJ, Li J, Tan YS, Nguyen MN, Pal A, Ouaray Z, Yadahalli S, Kannan S. 2016The multifaceted roles of molecular dynamics simulations in drug discovery. Curr. Pharm. Des 22, 3585-3600. ( 10.2174/1381612822666160425120507) [DOI] [PubMed] [Google Scholar]
- 18.Wang Y, Chen CH, Hu D, Ulmschneider MB, Ulmschneider JP. 2016Spontaneous formation of structurally diverse membrane channel architectures from a single antimicrobial peptide. Nat. Commun. 7, 13535. ( 10.1038/ncomms13535) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Coates J, Park BR, Le D, Şimşek E, Chaudhry W, Kim M. 2018Antibiotic-induced population fluctuations and stochastic clearance of bacteria. Elife 7, e32976. ( 10.7554/eLife.32976) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Teimouri H, Kolomeisky AB. 2019Theoretical investigation of stochastic clearance of bacteria: first-passage analysis. J. R. Soc. Interface 16, 20180765. ( 10.1098/rsif.2018.0765) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Redner S. 2001A guide to first-passage processes. Cambridge, UK: Cambridge University Press. [Google Scholar]
- 22.Van Kampen NG. 1992Stochastic processes in physics and chemistry, vol. 1. Amsterdam, The Netherlands: Elsevier. [Google Scholar]
- 23.Kolomeisky AB. 2015Motor proteins and molecular motors. Boca Raton, FL: CRC Press. [Google Scholar]
- 24.Rochman N, Si F, Sun SX. 2016To grow is not enough: impact of noise on cell environmental response and fitness. Integr. Biol. 8, 1030-1039. ( 10.1039/C6IB00119J) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shai Y. 2002From innate immunity to de-novo designed antimicrobial peptides. Curr. Pharm. Des 8, 715-725. ( 10.2174/1381612023395367) [DOI] [PubMed] [Google Scholar]
- 26.Wu F, Tan C. 2019Dead bacterial absorption of antimicrobial peptides underlies collective tolerance. J. R. Soc. Interface 16, 20180701. ( 10.1098/rsif.2018.0701) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brauner A, Fridman O, Gefen O, Balaban NQ. 2016Distinguishing between resistance, tolerance and persistence to antibiotic treatment. Nat. Rev. Microbiol. 14, 320-330. ( 10.1038/nrmicro.2016.34) [DOI] [PubMed] [Google Scholar]
- 28.Bel G, Zilman A, Kolomeisky AB. 2020Different time scales in dynamic systems with multiple outcomes. J. Chem. Phys. 153, 054107. ( 10.1063/5.0018558) [DOI] [PubMed] [Google Scholar]
- 29.Joo H-S, Fu C-I, Otto M. 2016Bacterial strategies of resistance to antimicrobial peptides. Phil. Trans. R. Soc. B 371, 20150292. ( 10.1098/rstb.2015.0292) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rodríguez-Rojas A, Baeder DY, Johnston P, Regoes RR, Rolff J. 2021Bacteria primed by antimicrobial peptides develop tolerance and persist. PLoS Pathog. 17, e1009443. ( 10.1371/journal.ppat.1009443) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Abdi M, Mirkalantari S, Amirmozafari N. 2019Bacterial resistance to antimicrobial peptides. J. Pept. Sci. 25, e3210. ( 10.1002/psc.3210) [DOI] [PubMed] [Google Scholar]
- 32.Schmidtchen A, Frick I-M, Andersson E, Tapper H, Björck L. 2002Proteinases of common pathogenic bacteria degrade and inactivate the antibacterial peptide ll-37. Mol. Microbiol. 46, 157-168. ( 10.1046/j.1365-2958.2002.03146.x) [DOI] [PubMed] [Google Scholar]
- 33.Sieprawska-Lupa Met al.2004Degradation of human antimicrobial peptide ll-37 by Staphylococcus aureus-derived proteinases. Antimicrob. Agents Chemother. 48, 4673-4679. ( 10.1128/AAC.48.12.4673-4679.2004) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Additional data are available in the electronic supplementary material.