

Abstract
Purpose:
Glioblastoma (GBM), neoplasms derived from glia and neuro-glial progenitor cells, are the most common and lethal malignant primary brain tumors diagnosed in adults, with a median survival of 14 months. GBM tumorigenicity is often driven by genetic aberrations in receptor tyrosine kinases (RTKs), such as amplification and mutation of EGFR.
Experimental Design:
Using a Drosophila glioma model and human patient-derived GBM stem cells and xenograft models, we genetically and pharmacologically tested whether the YAP and TAZ transcription co-activators, effectors of the Hippo pathway that promote gene expression via TEAD co-factors, are key drivers of GBM tumorigenicity downstream of oncogenic EGFR signaling.
Results:
YAP and TAZ are highly expressed in EGFR-amplified/mutant human GBMs, and their knockdown in EGFR-amplified/mutant GBM cells inhibited proliferation and elicited apoptosis. Our results indicate that YAP/TAZ-TEAD directly regulate transcription of SOX2, C-MYC, and EGFR itself to create a feedforward loop to drive survival and proliferation of human GBM cells. Moreover, the benzoporphyrin derivative verteporfin, a disruptor of YAP/TAZ-TEAD mediated transcription, preferentially induced apoptosis of cultured patient-derived EGFR-amplified/mutant GBM cells, suppressed expression of YAP/TAZ transcriptional targets, including EGFR, and conferred significant survival benefit in an orthotopic xenograft GBM model. Our efforts led us to design and initiate a phase 0 clinical trial of Visudyne, an FDA-approved liposomal formulation of verteporfin, where we used intraoperative fluorescence to observe verteporfin uptake into tumor cells in GBM tumors in human patients.
Conclusions:
Together, our data suggest that verteporfin is a promising therapeutic agent for EGFR amplified and mutant GBM.
INTRODUCTION
Glioblastomas (GBMs), neoplasms composed of glial cells and their precursors, are deadly primary tumors of the central nervous system (CNS). Genomic analyses indicate that the most frequent genetic lesions in GBMs include amplification, mutation, and/or overexpression of receptor tyrosine kinases (RTKs), such as EGFR, PDGFRA, and MET, and mutations that constitutively activate the PI-3 kinase (PI3K) pathway (1). Nearly 60% of GBMs show EGFR copy gain or amplification, which is often accompanied by gain-of-function EGFR mutations (1). The most prevalent EGFR variant in GBM is EGFRvIII, in which deletion of exons 2-7 produces a constitutively active kinase that potently drives tumorigenesis (2). GBMs are incurable with current therapies, and, to date, treatments that target EGFR and PI3K signaling pathways in GBM have failed in clinical trials due to poor absorption into tumors, insufficient inhibition of their cognate targets, and/or redundancies between effectors (3). Given their aggressive nature, there is a pressing need to identify new therapeutic strategies for GBM.
To uncover factors that act in parallel or downstream of EGFR in glial tumorigenesis, we performed in vivo RNAi modifier screens in Drosophila models of EGFR-PI3K mutant gliomas followed by genetic and pharmacologic validation of human modifier orthologs in GBM models (4,5). In our Drosophila models, glial-specific co-overexpression of constitutively active human (EGFRvIII) or Drosophila EGFR (dEGFRλ) with the catalytic subunit of PI3K (dp110CAAX) drives malignant transformation of glia, which exhibit neoplastic characteristics similar to human GBM cells (4,5). Top candidates from our Drosophila screens include the core Hippo pathway serine/threonine kinases Hippo and Warts, which exacerbated EGFR-mediated glial neoplasia (5) (Supplementary Fig. S1A–B, Supplementary Table S1).
Interactions between Hippo pathway kinases and EGFR signaling in glial neoplasia are intriguing because targets of Hippo pathway kinases, the Yorkie (Yki) transcription factor and its mammalian orthologs YAP and TAZ, are effectors of RTK signaling pathways in developing stem/progenitor cells (6,7). Warts family kinases (human LATS1/2), which are regulated by Hippo-mediated phosphorylation, directly phosphorylate Yki/YAP/TAZ, leading to their inactivation through degradation to constrict stem/progenitor cell proliferation, whereas signaling through RTK pathways, such as EGFR, inactivate Hippo pathway kinases to stimulate Yki/YAP/TAZ protein accumulation, nuclear translocation, and transcriptional activation (6). In Drosophila GBM models, we observed that Yki is ectopically overexpressed in neoplastic EGFR-PI3K glia, and that Yki knockdown suppressed proliferation of neoplastic EGFR-PI3K glia (Supplementary Fig. S1A–D, Supplementary Table S1), consistent with a previous report that Yki is required downstream of EGFR signaling in Drosophila retinal glia development (8). To stimulate expression of target genes, Yki binds to several transcriptional co-activators, including the TEA domain (TEAD) family transcriptional co-activators, such as Scalloped (Sd) in Drosophila (6,7). Sd RNAi significantly reduced proliferation of neoplastic Drosophila EGFR-PI3K glia (Table S1), indicating that TEAD co-activators also promote glial neoplasia. Established TEAD-dependent Yki/YAP/TAZ transcriptional targets include genes that promote stem/progenitor cell proliferation and self-renewal that, when overexpressed, promote tumorigenesis in GBM (9–12). Based on our Drosophila data, we set out to determine if YAP and/or TAZ may drive TEAD-dependent tumorigenesis as a consequence of EGFR activity in GBM.
In this study, we followed-up on our Drosophila data, and uncovered links between EGFR-driven gliomagenesis and the YAP/TAZ pathway. One intent of our approach was to uncover ectopically upregulated EGFR effectors in GBMs that may be useful therapeutic targets. Based on our results presented below, we hypothesized that inhibition of YAP/TAZ-TEAD transcriptional activation may be a tumor-cell specific therapeutic strategy for EGFR-amplified/mutant GBM, and we tested the therapeutic potential of the YAP/TAZ-TEAD inhibitor verteporfin. Verteporfin, which is an FDA-approved drug, is a fluorescent porphyrin that shows similarities to protoporphyrin IX that is used for fluorescence-assisted intraoperative tumor mapping for GBM (13). We leveraged these properties to uncover links between EGFR-driven tumorigenesis and the YAP/TAZ pathway, and to translate our findings into the clinic to determine whether verteporfin may serve as a therapy for EGFR-driven GBMs in human patients.
MATERIALS AND METHODS
Drosophila genetics and imaging
Drosophila stocks were obtained from the Bloomington stock center and VDRC (stocks used listed in Table S1). Drosophila GBM models were previously established and crosses to create tumorous larval brains were performed as described (5). All stocks were cultured on standard corn meal molasses food at 25°C. All genotypes were established by standard genetic crossing. For immunofluorescence, aged matched larval brains were dissected with forceps, fixed in 4% paraformaldehyde, processed, stained, and imaged as previously described (4). The following antibodies were used: 8D12 mouse anti-Repo (1:10, Developmental Studies Hybridoma Bank, AB_528448) and anti-Yki (1:200, a gift from Iswar Hariharan, Department of Molecular and Cell Biology, University of California, Berkeley, CA). Secondary antibodies were conjugated to Cy3, Alexa-Flour-488, or Alexa-Flour-647 (1:100-250, Jackson Laboratories). Brains were mounted on glass slides ventral side down in vectashield and whole mount imaged on a Zeiss LSM 700 confocal system. For experiments where protein levels were compared between genotypes, all samples were prepared, subjected to immunohistochemistry, imaged, and image processed in a parallel manner side by side. 6 or more brains were stained with each Ab combination, and representative images are shown for each result. Brain phenotypes shown were highly penetrant, with approximately 75-100% of animals showing the growth phenotypes described. Images were analyzed in Zeiss Zen Software and processed in Photoshop.
TMA processing
All human tumor specimens were collected from surgical specimens donated for research with written informed consent of patients and were collected and used according to recognized ethical guidelines (Declaration of Helsinki, CIOMS, Belmont Report, GCP, Nuremberg Code) in a protocol (IRB00045732) approved by the Institutional Review Board at Emory University. Paraffin embedded human brain tumor specimens and tumor tissue microarrays with matched control tissue were prepared and sectioned using the Winship Core Pathology Laboratory. Antigen retrieval and immunohistochemical staining was performed as specified by manufacturer’s guidelines for each specific antibody. Total YAP and TAZ protein levels were detected by IHC in tumor tissue specimens and staining was evaluated based on cytoplasmic and nuclear staining levels.
The following antibodies were used for immunohistochemistry on TMAs and other paraffin sections: anti-YAP (1:200, Cell Signaling Technology, 14074, AB_2650491), anti-TAZ (1:100, Sigma Aldrich, HPA007415, AB_1080602), anti-MET (1:200, Cell Signaling Technology, 8198, AB_10858224), anti-EGFR (1:50, Cell Signaling Technology, 4267, AB_2864406), anti-SOX2 (1:250, Cell Signaling Technology, 3728, AB_2194037), anti-MYC and Ki67 (Emory Medical Labs). Results were scored in consultation with neuropathologists according to standard clinical criteria on a scale of 1 and 2 (low staining), 3 or 4 (high staining, with 4 being more uniform), and images of immunoreactivity were taken on an Olympus DP72 CCD camera.
Additional tumor genotype and phenotype results were obtained from de-identified clinical reports (CNV array data, sequencing generated as standard of care), or from research RNAseq. Tumor genotypes (eg. EGFR, MET, or PDGFRA status, PTEN, PIK3CA status) were annotated and compared to YAP/TAZ expression status.
Patient-derived gliomasphere cultures
GBM39, shared by C. David James (Department of Neurological Surgery, Northwestern University, Evanston, IL), was created from human GBMs serially xenografted. GBM301 gliomasphere (GSC) cultures were derived at UCLA and maintained in culture as described (14). HNPCs were obtained from Lonza (PT-2599). pGBM2 and pGBM6 were obtained from Michelle Monje (Department of Neurology and Neurological Sciences, Stanford University, Stanford, CA). All other gliomasphere cultures were established according to published protocols (14), using surgical specimens of adult GBMs collected under IRB protocol IRB00045732. GBMs at Emory undergo diagnostic genomic profiling and genotyping as standard of care, and this data is de-identified and provided to us through our participation in the IRB protocol IRB00045732 and Emory University Brain Tumor Sample and Information Resource (BTSIR). Cell culture was performed as previously described (5). Cells were genotyped by RNAseq and/or qPCR and immunoblots for known distinguishing markers and mutations. Cells were screened for mycoplasma using a universal mycoplasma detection kit (ATCC 30-1012K) every 2-4 months as needed and prior to all xenograft experiments.
Lentiviral shRNAs (validated pLKO shRNA vectors for YAP and TEAD4 from Sigma; validated pGIPZ shRNA vectors for TAZ from Thermo Fisher) were prepared in serum-free media and used on adherent serum-free cultured GSC lines as previously described (5). Liposomal pharmaceutical grade verteporfin (Visudyne, Bausch Health) and chemical grade verteporfin (Sigma) were used in cell culture as per published assays (15), and cells were treated in darkness so as to limit light and PDT-related effects. For verteporfin treatment assays on established neurospheres, GSC cells were dissociated with Accutase, plated at defined densities (250,000-500,000 cells per well in 6 well plates), and cultured for 24-48 hours prior to verteporfin addition. CA3 (CIL56, S8661, Selleck Chemicals) and Peptide17 (S8164, Selleck Chemicals) were used to treat GSC cultures prepared in the same manner. GSC cultures assessed for proliferation and survival using WST-1 assays, immunofluorescence, and immunoblots as previously described (5,14).
Mouse genetic model of EGFRvIII mutant GBM
All experimental procedures with mice were approved by the Institutional Animal Care and Use Committee (IACUC; protocol 2001765, 201700534). Mouse breeding and genotyping strategies are described in (16). EGFRvIII-expressing mouse neurospheres and matched control neurospheres from the same genetic background were produced in vitro from mouse neonatal neural stem cells isolated from Cdkn2a−/−; Ptenfl/fl; EGFRvIII-fl-stop-fl mice and control Cdkn2a−/−;Ptenfl/fl mice that we both bred together over 6+ generations into the C57BL/6 background (confirmed by SNP markers). Subventricular zones (SVZs) were dissected from P0-P2 pups of and dissociated into single cells with Accutase enzyme (Biolegend, 423201). Cell culture was performed as previously described (5,14). Cell cultures from individual animals, created from matched litters, were initially plated in 6 well plates and to grow neurospheres over 1-2 weeks. Neurospheres were passaged at 1-2 million cells per 10 cm plate every week. 250,000 cells were then plated in 6-well plates and treated with concentrated adenovirus Cre (Vector Biolabs) at 50-100 MOI, and then expanded for another 5-7 days before they were checked by immunoblot to confirm loss of PTEN protein expression and gain of EGFRvIII expression. Cells were cultured for up to 8 passages for in vitro experiments and for injections to form in vivo tumors. To establish tumors in vivo in 6-week old C57BL/6 mice, 200,000 cells in 5 μl in 1xDPBS were stereotactically injected per animal using a 23 Hamilton syringe at a position 2.5 mm to the right and 1 mm anterior to the bregma at a depth of 2.5-3 mm.
Immunoblot analysis
Cultured cells were collected and washed with 1xPBS and lysed in RIPA buffer containing protease and phosphatase inhibitors. The following antibodies were used for immunoblotting following the manufacturer’s recommendations: anti-YAP (1:1000, Cell Signaling, 14074), anti-YAP-S127-P (1:1000, Cell Signaling, 13008, AB_2650553), anti-TAZ (1:1000, Cell Signaling, 4883, AB_1904158), anti-TAZ (1:200, Novus, 85067, AB_11013779), anti-panTEAD (1:1000, Cell Signaling, 13295, AB_2687902), anti-TEAD1 (1:1000, Cell Signaling, 12292, AB_2797873), anti-TAZ-S89-P (1:1000, Cell Signaling, 59971, AB_2799578), anti-TEAD4 (1:500, Abcam, 58310, AB_945789), anti-EGFR (1:5000, BD, 610017, AB_2096701), anti-EGFR (1:1000, Cell Signaling, 4267), anti-SOX2 (1:1000, Cell Signaling, 14962, AB_2798664), anti-MYC (1:1000, Cell Signaling, D84C12, AB_2798629), anti-PARP (1:1000, Cell Signaling, 9542, AB_2160739), anti-LATS1 (1:1000, Cell Signaling, 3477, AB_2133513), anti-LATS2 (1:1000, Cell Signaling, 5888. AB_10835233), (anti-SOX9 (1:500, R&D Systems, AF3075, AB_2194160), anti-S100beta (1:1000, Abcam, ab41548, AB_956280), anti-PDGRα (1:1000, Cell Signaling, 3174, AB_2162345), anti-MET (1:1000, Cell Signaling, 8198, AB_10858224), anti-TRK (1:1000, Cell Signaling, 92991, AB_2800196), anti-FGFR3 (1:1000, Cell Signaling, 4574, AB_2246903), anti-Nestin (1:1000, Cell Signaling, 33475, AB_2799037), anti-CD31 (1:1000, Cell Signaling, 3528, AB_2160882), anti-CD44 (1:1000, Cell Signaling, 37259, AB_2750879), anti-GFAP (1:1000, Cell Signaling, 80788, AB_2799963), anti-IGFR2 (1:1000, Cell Signaling, 14364, AB_2798462), anti-CDK4 (1:1000, Cell Signaling, 12790, AB_2631166), anti-STAT3 (1:1000, Cell Signaling, 9139, AB_331757), anti-STAT1 (1:1000, Cell Signaling, 14994, AB_2737027), anti-RAS (1:1000, Cell Signaling, 3965, AB_2180216), anti-RAC (1:1000, Cell Signaling, 2465, AB_2176152), anti-BCL2L2 (1:1000, Cell Signaling, 2724, AB_10691557), and anti-actin (1:200, Developmental Studies Hybridoma Bank, JLA20, AB_528068). Blots were imaged by chemiluminescence on a Bio-Rad ChemiDoc imaging system, and ImageJ was used for densitometry quantification.
Quantitative PCR
RNA was isolated from GSCs using standard Trizol extraction. cDNA was generated using iScript Reverse Transcription Supermix for RT-qPCR (Bio-Rad, 1708840) with 1μg of RNA. qPCR was run using SsoAdvanced Universal SYBR Green Supermix (Bio-Rad, 1725270). Data was analyzed using delta-delta Ct method and normalized to control in each experiment to generate fold-change values.
Limiting dilution assay for neurosphere formation
Cells were dissociated and plated in a 96-well plate at concentrations varying from 1 to 1000 cells per well depending on experimental application. For drug treatment assays, cells were plated in 90 μL of media and 10 μL of media-verteporfin solution was added the following day for a total of 100 μL per well. For shRNA assays, cells were infected and harvested 48 hours post-infection and plated. Cells were observed 7 days after plating, counting wells that contained spheres and calculating neurosphere formation capacity as published (17).
Chromatin immunoprecipitation
Tiling primers were generated along the EGFR promoter referencing published reports and Encode data on predicted and known TEAD binding sites in the EGFR promoter. ChIP with antibodies to YAP (1:1000, Cell Signaling, 14074) and TEAD4 (1:500, Abcam, 58310, AB_945789) was carried out on GBM39 cells, which express YAP/TAZ and TEAD4 and harbor EGFRvIII amplicons, using SimpleCHIP kit and following manufacturer instructions (Cell Signaling Technology, 9003S). qPCR was performed on these ChIP isolates using primers for the EGFR promoter as compared to control primers for known TEAD target genes and nonspecific genomic sites.
Proteomics
Cultured GSCs were treated with verteporfin or DMSO for 6 hours, total cell pellets were collected and subjected to label-free total proteomic profiling according to published protocols (18,19). A total of 5482 different proteins were identified in our profiles. Bioinformatic analysis performed with Toppogene suite (toppgene.cchmc.org).
Xenograft models and Visudyne treatment
For orthotopic xenografts, 1x105 cells were stereotactically injected into the brains of Nod/SCID-gamma (NSG) mice using established techniques (5). Briefly, 6-week old female NSG mice were implanted with 100,000 GBM39 patient-derived xenograft glioma stem cells using a 23 Hamilton syringe and a stereotaxic frame with the coordinates 1.5 mm lateral (left) and 1 mm anterior to bregma. Cells were implanted at a depth of 2.5 mm.
For intraperitoneal (IP) drug delivery, following an engraftment period of 2 weeks, 100 mg/kg liposomal verteporfin (Visudyne) or saline was injected in the abdomen every other day over 2 weeks for a total of 8 doses as per published protocols (15). For intrathecal drug delivery, following an engraftment period of 2 weeks, micro-osmotic pumps were implanted contralaterally using the coordinates 1.1mm lateral (right) and 0.5 mm posterior to bregma (Alzet, pump model 1002, brain infusion kit 3). Micro-osmotic pumps were pre-filled with 100 μl saline (vehicle) or Visudyne (verteporfin 200 μg in 100 μl) and have a delivery rate of 0.25μl/h. Mice were monitored for neurological signs of tumor burden before sacrificing in the case of the survival endpoint experiment, or after 7 days for the verteporfin uptake experiment.
Survival was calculated with Kaplan-Meier plots. Following experimental endpoints, brains were harvested and processed for protein, mRNA, and histopathological analysis/IHC/imaging to assess tumor cell properties, gene/protein expression, and verteporfin penetration. Neuropathology was scored in collaboration with neuropathologists.
Ex vivo slice culture
For organotypic ex vivo slice cultures, xenografts were created in NSG mice, tumor-bearing brains were removed unfixed from affected animals and sliced into 200 micron thick coronal sections (Leica vibratome), and full slices are cultured in 6 well plates in serum-free neural stem/progenitor cell media. These slices, which reliably survive for 3-9 days, contain stroma, including vasculature and neuronal tissues, and tumor. Drugs are delivered to the culture media, and responses in tumor tissue are stained and imaged with confocal microscopy.
Immunofluorescence and confocal imaging
For immunofluorescence, neurospheres or brain slices were fixed with 4% paraformaldehyde, washed with 1X PBS .3% Triton X-100, and stained in 6 or 12 well plates with anti-EGFR (1:1000, BD, 610017, AB_2096701), anti-human-Vimentin (1:400, Abcam, ab8069, AB_306239), anti-Cleaved-Caspase-3 (1:100, Cell Signaling, 9664, AB_2070042), anti-SOX2 (1:400, Cell Signaling, 4900, AB_10560516), anti-MYC (1:100, Cell Signaling, 18583), anti-Nestin (1:100, Cell Signaling, 33475, AB_2799037), anti-YAP (1:200, Abcam, ab56701), anti-TAZ (1:200, BD, clone M2-616, AB_1645338), anti-Phospho-Histone-H3 (1:200, Cell Signaling, 9706, AB_331748), and/or DRAQ7 (Cell Signaling, 1:100) to stain nuclear DNA and chromosomes. Secondary antibodies were conjugated to Cy3, Alexa-Flour-488, or Alexa-Flour-647 (1:100-250, Jackson Laboratories).
For dye exclusion assays to quantify apoptosis, unfixed live neurospheres were treated with 3 μg/mL ethidium bromide to stain apoptotic cells and with 5 μg/mL acridine orange (Biotium, 70012) as a counter stain, and live neurospheres were immediately imaged. Samples were mounted on glass slides in vectashield and whole mount imaged on a Zeiss LSM 700 confocal system. For experiments where protein levels were compared between treatments, all samples were treated, fixed, subjected to immunohistochemistry, imaged, and images were processed in a parallel manner side by side. For neurospheres, over 25-50 spheres per treatment were stained with each Ab combination, and 4-6 representative spheres were imaged for each result. Images were analyzed in Zeiss Zen Software and processed in Photoshop.
To quantify ethidium-bromide-positive apoptotic cells and phospho-Histone-H3-positive proliferating cells, manual counting was performed on representative 25 μm thick Z-stacks of whole neurospheres. For calculating Cleaved-Caspase-3:Vimentin staining ratios for slice cultures, Imaris software was used on Z-stack images to determine tumor volumes and over-all pixels for each stain within the tumor volume. Statistical analyses performed using Prism.
IND exemption for Visudyne
Verteporfin is approved in combination with PDT for the treatment of macular degeneration. The use of verteporfin for treatment of GBM is experimental, and we applied for an Investigational New Drug Application (IND) exemption with the FDA, which was granted because, except for the dose, our use meets the criteria laid out in 21 CFR 312.2(b)(1). The doses are new, but because of low documented toxicity and rapid clearance of the drug, this was not likely to significantly increase risk.
Treatment plan for phase 0 study of verteporfin treatment for GBM
The clinical protocol for the phase 0 trial was prepared, reviewed, and approved according to recognized ethical guidelines (Declaration of Helsinki, CIOMS, Belmont Report, GCP, Nuremberg Code) in a protocol (IRB00082472) approved by the Institutional Review Board at Emory University. Written informed consent was obtained from participants who were scheduled to undergo surgery for suspected or known recurrent GBM. Men, women, and members of all ethnic groups were eligible for the study. Participants were excluded if they were pregnant, had a history of allergic reactions attributed to compounds of similar chemical or biologic composition to verteporfin, or had a personal or family history of porphyrias. The primary endpoint was to determine if verteporfin enters GBM cells in situ. Secondary endpoints of this study included characterization of verteporfin uptake and fluorescence in tumor cells, and assessment of acute toxicity associated with a single dose of verteporfin followed by surgery.
Each participant had a suspected GBM by history and imaging studies (MRI) and was deemed to be a surgical candidate according to standard of care and received a single dose of Visudyne before surgery. The starting dose of Visudyne was 0.15mg /kg intravenously, the dose at which Visudyne is administered for ocular disease. The higher dose was 0.3 mg/kg, which has been evaluated in human clinical trials and during development of Visudyne (20). The trial patients are listed in Table S2. Visudyne was administered on an inpatient basis as a one-time, single-dose of 0.15-0.3mg verteporfin/kg 4-6 hours prior to surgery. After verteporfin was administered, image-guided microsurgical resection of tumor was undertaken and pathologic confirmation of tumor type was made by neuropathology, as per standard of care. On encountering tumor intraoperatively, the operative microscopy and camera system (Zeiss Pentero 900) was used to illuminate the tumor bed and take photographs.
After satisfying clinical needs, a portion of resected tumor was analyzed for verteporfin concentration. Following surgery, supportive care was provided as per standard of care. Participants were followed for a week or more for toxicities following administration, and no Visudyne-related adverse events were observed.
LC-MS/MS analysis of verteporfin
Verteporfin present in tissue samples from mice and human patients was extracted using acetonitrile then analyzed by LC-MS/MS, and quantified by comparison to standards of known concentrations at the Proteomics facility of Cornell University according to published protocols (21) (http://www.biotech.cornell.edu/brc/proteomics-and-mass-spectrometry).
Statistical Analysis
IHC co-expression and correlations were calculated using chi-square test. qPCR and WST-1 p-values were calculated using 1-way or 2-way ANOVA and multiple comparison calculations using Graphpad Prism. Image quantification datasets for IHC, slice cultures, and neurosphere immunofluorescence were analyzed using unpaired t tests in Graphpad Prism. Survival curves and their significance were calculated using Log-rank (Mantel-Cox) test in Graphpad Prism. Proteomics data was analyzed by multiple t tests and volcano plots in Graphpad Prism.
Supplementary Table S3 includes a list of all abbreviations, acronyms, and symbols.
RESULTS
YAP and TAZ co-overexpression is correlated with EGFR alterations in human GBM.
The paralogous YAP and TAZ transcription factors, both human orthologs of Yki, play highly conserved roles in proliferation in normal tissue stem/progenitor cells (6). Given the tumor-cell specific overexpression of Yki and strong effect of Yki knockdown in neoplastic EGFR-PI3K Drosophila glia, we examined YAP, TAZ, and TEAD expression and function in relation to EGFR and other RTKs in human GBM tissues and models.
To determine relationships between YAP and TAZ expression and tumor genotype, we used immunohistochemistry (IHC) on representative adult human GBM tumor tissue microarrays (TMAs). YAP and TAZ expression was not detected in normal adult glia by IHC on normal brain tissue specimens (Supplementary Fig. S2A), consistent with previous studies (22). Among GBMs, 75% (21/28) showed high nuclear YAP and/or TAZ in tumor cells, and 61% (17/28) co-overexpressed YAP and TAZ (Fig. 1A). Using IHC and molecular pathology to determine RTK and PI3K status of tumors, we observed YAP/TAZ overexpression in all GBMs with PI3K pathway mutations (Table S4). We also observed YAP and/or TAZ overexpression in 88% (21/24) of GBM tumors with RTK alterations (Fig. 1A, Supplementary Table S4). In tumors with EGFR amplification or copy gain, 88% (14/16) co-overexpressed YAP and TAZ (Supplementary Table S4). 5 tumors with YAP-TAZ overexpression contained alterations in other RTKs, such as MET amplification or NTRK2 overexpression, and all of these tumors overexpressed EGFR (Table S4). Furthermore, xenografts of EGFR-amplified/mutant tumors maintained YAP/TAZ overexpression (Fig. 1A, Supplementary Fig. S2B). Therefore, YAP and TAZ co-overexpression are significantly correlated with EGFR alterations (Fig. 1A). In contrast, GBMs that showed low YAP and TAZ expression harbored IDH1 mutations (3/7), PDGFRA amplification with low EGFR expression (3/7), or TP53 mutations (1/7) (Supplementary Fig. S2A, Supplementary Table S4). Results were confirmed with immunoblots on tumor tissue from TMA specimens (Supplementary Fig. S3). Our results are consistent with previous studies that indicate GBMs frequently overexpress TAZ and that EGFR-amplified tumors overexpress YAP (22,23).
Figure 1. YAP and TAZ upregulation is associated with EGFR and RTK alterations and overexpression in GBM tumor cells.
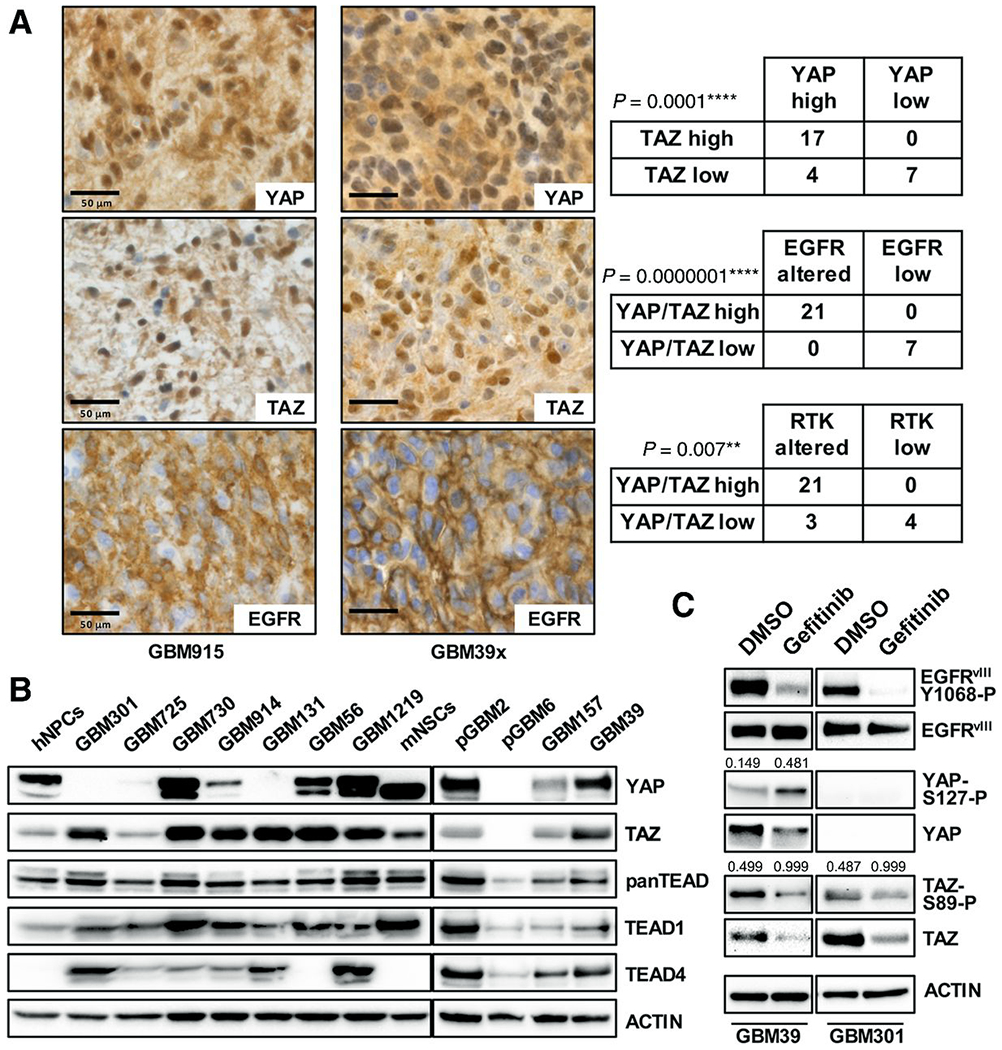
(A) Immunohistochemistry (IHC) for nuclear and cytoplasmic YAP and TAZ expression in GBM tumor cells that overexpress EGFR (upper) due to amplification and/or mutation, GBM patient tissue (left, representative case) and GBM39 xenograft (right, representative case). Statistical analysis demonstrated correlations between high YAP and TAZ expression levels with each other, with EGFR alterations (amplification, copy gain, and/or overexpression), and with other RTKs alterations (MET, NTRK2, and PDGFRA) in TMA specimens. **p≤.01; ****p≤.0001 with chi-squared test.
(B) A panel of patient-derived GBM GSC cultures (several made from tumor specimens used for IHC), showing expression of YAP, TAZ, and TEAD family members, as compared to normal human neural progenitor cells (hNPCs) and mouse neonatal neural stem cells (mNSCs). GSC RTK alterations: amplified EGFR (GBM725, GBM914, and GBM1219), amplified EGFRvIII (GBM301, pGBM2, GBM39), amplified and/or overexpressed MET (GBM730, GBM914, GBM56), and overexpressed NTRK2(GBM131), and PDGFRA overexpression and PN identity (pGBM6, GBM157) (14).
(C) YAP and TAZ protein levels and phosphorylation after treatment of EGFRvIII-mutant GBM39 and GBM301 cultures with 5 μmol/L gefitinib for 24 hours. Numbers indicate densitometry results used to determine the amount of YAP phosphorylated on S127 (YAP-S127-P) relative to total YAP protein and the amount of TAZ phosphorylated on S89 (TAZ-S89-P) relative to total TAZ protein detected. Phosphorylated EGFR levels shown as evidence of EGFR kinase inhibition by gefitinib.
Molecular classification of GBMs has provided insight into dominant transcriptional programs and mutations found in GBMs, through which subtypes named proneural (PN), mesenchymal (MES), and classical (CL) were established (24–26). This classification associated mutations in IDH1 and/or overexpression of PDGFRA with the PN subtype, alterations in NF1 and/or MET with the MES subtype, and amplification/mutation/overexpression of EGFR and expression of astrocytic cell fate markers, such as SOX9, with the CL subtype (24–26). Using a gene panel developed to distinguish PN, MES, and CL tumors(16), we performed immunoblots to subtype tumor specimens and found that tumors which consistently co-overexpress YAP and TAZ frequently show CL markers (Supplementary Fig. S3, Supplementary Table S4). In contrast, PN tumors express low YAP and TAZ as previously observed (22,23).
In our bioinformatic analysis of GBM subtypes, CL tumors consistently co-overexpress YAP and TAZ mRNA (16), linking YAP and TAZ gene expression to EGFR status. Further analysis of recent data available from The Cancer Genome Atlas (TCGA) showed that, in GBM, high EGFR mRNA expression significantly correlated with high YAP mRNA expression and that EGFR copy gain and amplification significantly correlated with TAZ mRNA overexpression and reduced LATS1 mRNA expression (Supplementary Fig. S4A–B), indicating these pathway components may be epigenetically co-regulated. We also observed reduced LATS1 and MST1 mRNA expression in higher grade gliomas, along with associations between elevated YAP and TAZ mRNA and reduced LATS1 mRNA and poor survival in GBM patients (Supplementary Fig. S4C–D) and low/no detectable LATS1/2 protein in GBM tumor tissue (Supplementary Fig. S3), consistent with prior studies postulating that downregulation of Hippo pathway components contributes to high grade disease (22,23,27). Notably, direct genetic alterations do not account for YAP/TAZ overexpression in GBM: our analysis of TCGA data show exceedingly low frequency amplification of YAP (4/1144) and TAZ(3/1144) and rare mutations in Hippo pathway components, including homodeletions in LATS1(4/1144), LATS2(5/1144), MST1(8/1144), and MST2(8/1144). Together, our data indicate that EGFR alterations, characteristic of tumors with a predominant CL signature, are significantly associated with YAP and TAZ co-overexpression in GBM.
Representative patient-derived human GBM stem-cell containing gliomasphere (GSC) cultures created from RTK-altered tumors showed strong YAP and/or TAZ expression, with all but two cultures showing TAZ overexpression and 64%(7/11) cultures showing YAP overexpression (Fig. 1C). Among GSCs without EGFR alterations, pGBM6 showed no YAP/TAZ expression and GBM157, a PN line (14), showed low YAP/TAZ expression (Fig. 1C). Normal human neural progenitor cells (hNPCs) and immortalized Ink4a/Arf−/− mouse neonatal neural stem cells (mNSCs) also expressed YAP and TAZ (Fig. 1C), but at lower levels, consistent with roles for these factors in neural progenitor/stem cells (28,29). Humans have 4 TEAD orthologs, TEAD1-4, with TEAD1 expression being common to mNSCs, hNPCs, and GSCs, and TEAD4 expression being specific to GSC lines (Fig. 1C). In GBM301 and GBM39 GSCs, which harbor extrachromosomal amplified EGFRvIII (5,30,31), treatment with EGFR inhibitor gefitinib decreased total YAP and TAZ protein levels and increased proportions of phospho-Ser127-YAP and phospho-Ser89-TAZ (Fig. 1D), which are subject to degradation (6,7), showing that EGFR catalytic activity is required for YAP and TAZ overexpression and suggesting that EGFR inhibition evoked YAP/TAZ phosphorylation and degradation (7). Together, these data indicate that YAP and TAZ upregulation is a consequence of constitutive EGFR activity in GSCs.
YAP/TAZ-TEAD are required for GBM proliferation and are direct regulators of EGFR expression in GBM.
YAP is expressed in symmetrically dividing neural stem/progenitor cells in the embryonic brain, where it stimulates proliferation and expansion of these cells during neurogenesis (28,29). In GBM, previous studies demonstrate that YAP is required for proliferation of standard serum-grown glioma cell lines, and that TAZ is required for tumorigenicity of MES GSCs (22,23). In EGFR-mutant GSC lines, we observed that YAP and TAZ knockdown by lentiviral shRNAs induced apoptosis, diminished tumor cell proliferation, and reduced expression of stem cell and astrocytic cell fate markers (Fig. 2A–E), consistent with roles in GBM tumor stem cell self-renewal.
Figure 2. YAP/TAZ-TEAD activity transcriptionally upregulates of SOX2, MYC, and EGFR expression to promote tumor stem cell identity, survival, and proliferation in GBM gliomaspheres.
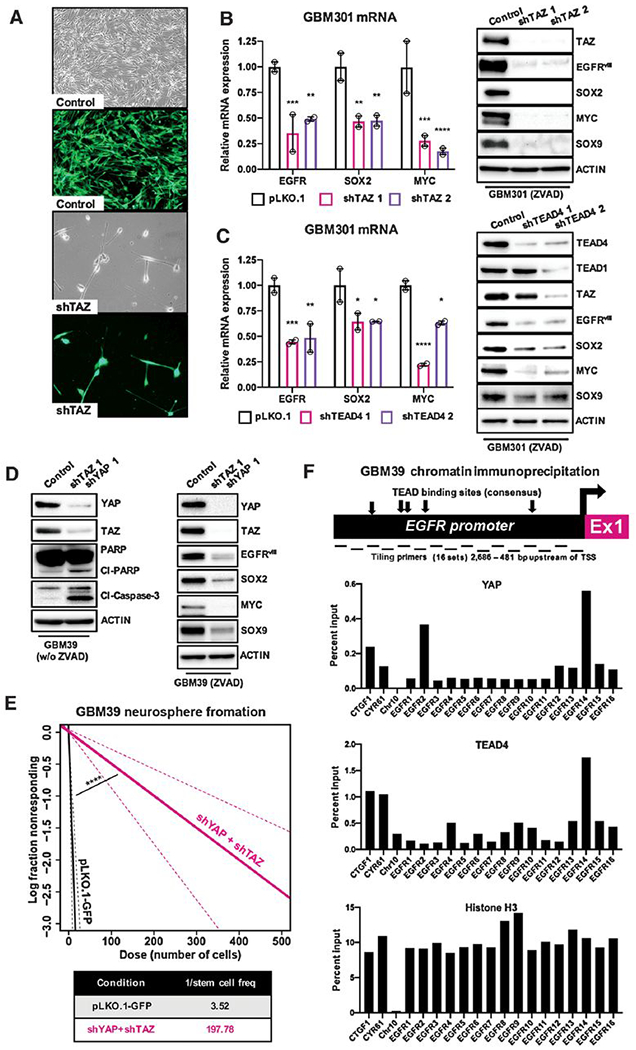
(A) Representative micrographs to show changes in cell growth and viability in GBM301 GSCs grown adherently on matrigel in serum-free conditions infected with shRNAs targeting TAZ or non-targeting GFP control lentivirus; 5 days following lentivirus infection.
(B, C) mRNA and protein expression of tumor stem cell factors SOX2, MYC, and EGFR and SOX9, an astrocytic cell-fate marker of CL GBM, in GBM301 GSCs cells treated with lentiviral shRNA knockdown of TAZ and TEAD4. Cells treated with ZVAD (20 μmol/L) to prevent apoptosis and preserve signaling pathways, and harvested 3 days post-infection. Graphs show representative experiments, and error bars show variance between experimental replicates.
(D) Cleaved-Caspase-3 and cleaved-PARP expression in GBM39 GSCs treated with lentiviral shRNA knockdown of YAP and TAZ or non-targeting GFP control lentivirus; expression of tumor stem cell factors, such as SOX2 and MYC, and the astrocytic CL GBM marker SOX9 upon YAP or TAZ knockdown in GBM39 GSCs concurrently treated with shRNAs and ZVAD (20 μmol/L). All cells harvested 3 days post-infection.
(E) Limiting dilution assays in which GBM39 GSCs were infected with shRNAs targeting YAP and TAZ or non-targeting GFP control lentivirus, plated at low density of 1-500 cells per well in 96 well plates, and observed for neurosphere formation 9 days post-infection.
(F) ChIP-qPCR from GBM39 GSCs using primer tiling for the EGFR locus indicates binding by YAP and TEAD at the EGFR promoter. Diagram depicting known and predicted TEAD consensus sites relative to qPCR tiling primers for the EGFR promoter (2,686 – 481 bp upstream from transcriptional start site). qPCR primers for known TEAD sites in CTGF and CYR61 included as positive controls; primers to a TEAD-negative region in Chromosome 10 used as negative controls. ChIP-qPCR results analyzed for percent enrichment of specific regions by primer number. H3 ChIP-qPCR results demonstrate ubiquitous H3 binding to all tiling sites within the EGFR promoter. ChIP-qPCR peaks coincide with EGFR primers 2-4 and 13-16, which overlaps with known and predicted consensus TEAD binding sites.
*p≤.05; **p≤.01; ***p≤.001; ****p≤.0001 with ANOVA multiple comparisons test.
YAP and TAZ family transcription factors bind TEADs and other transcriptional co-factors on promoters and enhancers of a broad swath of target genes in a manner dependent on cell-type and developmental stage (7). To identify YAP/TAZ-TEAD target genes that promote GBM tumorigenesis, we took a candidate-based approach. In embryonic stem (ES) cells and other stem/progenitor cells, YAP family transcription factors promote expression of TEAD-driven target genes that control stem cell identity, including SOX2 and MYC transcription factors, which themselves are master transcription factors required to promote and maintain GBM tumor stem cell identity and proliferation (9–12). In EGFR mutant GSC lines, YAP, TAZ, and TEAD4 knockdown reduced SOX2 and C-MYC mRNA and protein expression (Fig. 2B–D), although TEAD4 knockdown had a less significant effect, likely due to redundant TEAD1 (Fig. 2C). Thus, YAP/TAZ-TEAD family transcription factors may be master regulators of tumor stem cell identity downstream of EGFR in CL GBM. Consistent with regulation of CL tumor stem cell identity (24), YAP/TAZ knockdown reduced expression of SOX9 and prevented tumor stem cell self-renewal as measured by limiting dilution assay (Fig. 2B, D–E).
Reports indicate that, in other cell types, EGFR itself is also a YAP-TEAD transcriptional target (32). In GBM, EGFR and EGFR mutant variants are subject to overexpression from extrachromosomal amplicons (1), but the epigenetic mechanisms that promote EGFR overexpression in GBM are not understood. In GBM301 and GBM39, which harbor extrachromosomal amplicons encoding EGFRvIII (5,30,31), YAP, TAZ, and TEAD4 knockdown reduced EGFR mRNA and protein expression (Fig. 2B–D), suggesting that YAP/TAZ-TEAD drives EGFR overexpression in EGFR-amplified/mutant GBM. To determine if YAP/TAZ-TEAD directly regulate EGFR transcription in GBM, we generated tiled primer pairs across the EGFR promoter and performed chromatin immunoprecipitation (ChIP) analysis to assess YAP and TEAD binding to the EGFR promoter. Using YAP and TEAD4 ChIP coupled with qPCR in GBM39 cells, which express YAP, TAZ, TEAD4 and EGFRvIII amplicons, we found that YAP and TEAD4 binding is enriched at known and predicted TEAD consensus sequences in the EGFR promoter (Fig. 2F) (32,33). Known TEAD target sites for established YAP/TAZ target genes CTGF and CYR61 showed expected binding to YAP and TEAD4, while unrelated sites on chromosome 10 showed no evidence of YAP or TEAD4 binding (Fig. 2F). Together, our results indicate that, in primary-patient-derived GSCs, EGFR signaling upregulates YAP and TAZ protein levels and that YAP/TAZ-TEAD directly upregulate gene expression of factors required for tumorigenesis, such as SOX2, C-MYC, and EGFR itself, thereby creating and maintaining an EGFR-dependent YAP/TAZ feedforward loop that promotes and maintains GBM.
Pharmacologic inhibition of YAP/TAZ-TEAD inhibits GBM tumor cell tumor formation.
Studies show that YAP promotes initiation and progression of other tumor types (7), and that pharmacologic inhibition of YAP-TEAD transcriptional activation with the drug verteporfin blocks tumor cell growth in vitro in culture and in vivo in animal models (15). Verteporfin binds to the conserved TEAD interaction domain in YAP, disrupts YAP-TEAD binding, and induces YAP/TAZ protein degradation, preventing transcriptional transactivation (15,34). Because TAZ has not been previously shown to be inhibited by verteporfin, we performed co-IP experiments in GSCs and found that verteporfin blocked association between TAZ and TEAD4 (Supplementary Fig. S5A). Thus, verteporfin likely inhibits TAZ-mediated transcriptional activation of TEAD targets.
To determine the effect of verteporfin on YAP/TAZ-TEAD transcriptional activation in RTK-dependent GBM cells, we tested verteporfin on representative GSCs at dose ranges (.5-2μg/mL) shown to block YAP-TEAD-dependent growth of YAP/TAZ-dependent tumor types but be nontoxic to other cells (15,21). Verteporfin is a porphyrin and is intrinsically fluorescent (peak excitation 410/30 nm, emission 690/700 nm) (15), allowing us to visualize intracellular verteporfin absorption (Fig. 3A). Verteporfin treatment reduced protein levels and nuclear localization of YAP and TAZ in GSCs within 24 hours and inhibited growth and provoked apoptosis of YAP/TAZ-expressing EGFR-mutant GSCs in a dose-dependent manner, with IC50 in our assay at .5 μg/mL (Fig. 3B–E, Supplementary Fig. S5B–E, Fig. S6A–E). Using ZVAD to block apoptosis, we found that verteporfin reduced proliferation in YAP/TAZ-expressing EGFR-mutant GSCs (Fig. 3F). In contrast, verteporfin had weaker pro-apoptotic and anti-proliferative effects on control hNPCs, YAP/TAZ negative pGBM6, and PN GBM157 (Fig. 3E–F, Supplementary Fig. S6A–E). In limiting dilution assays for neurosphere formation, verteporfin treatment differentially prevented self-renewal of EGFR-mutant GSCs compared to representative hNPCs, YAP/TAZ negative GSCs, or PN GSCs (Fig. 3G, Supplementary Fig. S6F). To confirm our observations, we tested alternate YAZ/TAZ inhibitors, which similarly provoked apoptosis, growth arrest, and reduced expression of EGFR and stem cell markers (Fig. S7A–C).
Figure 3. Pharmacologic inhibition of YAP/TAZ transcription with verteporfin specifically inhibits stem cell identity and induces apoptosis and growth arrest in GBM gliomasphere cultures.
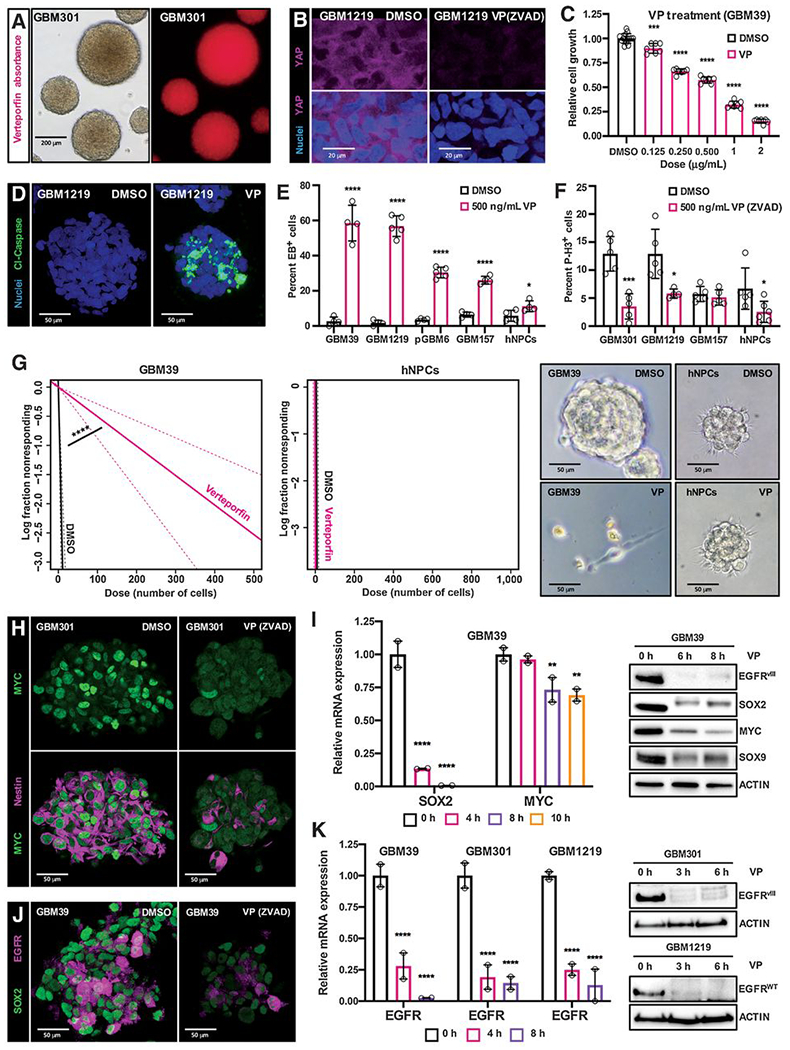
(A) Verteporfin (VP) absorption in GSC cultures in vitro, as detected via fluorescence. Cells were incubated with .5 μg/mL (.69 μmol/L) verteporfin for 12 hours.
(B) YAP protein levels and nuclear localization in YAP/TAZ-positive EGFR-mutant GSCs (GBM1219); GSCs were treated with either DMSO or .5 μg/mL verteporfin and ZVAD (20 μmol/L, to prevent apoptosis and preserve signaling pathways) for 24 hours. YAP visualized by immunofluorescence. 3 μm optical projections.
(C) WST-1 assay on EGFR-mutant GSCs (GBM39) to examine their viability and growth when treated with variable doses of verteporfin for 48 hours.
(D-F) Verteporfin toxicity in YAP/TAZ-positive EGFR-mutant GSCs compared to control cells. (D) Representative GBM1219 GSCs were treated with .5 μg/mL of verteporfin for 24 hours and Cleaved-Caspase-3 (Cl-Caspase-3) was visualized by immunofluorescence to examine apoptosis, 22 μm optical projections. (E, F) YAP/TAZ positive EGFR-mutant GSCs (GBM39, GBM1219), YAP/TAZ-negative GSCs (pGBM6), EGFR-negative PN GSCs (GBM157), and hNPCs were treated with .5 μg/mL of verteporfin for 24 hours. (E) Apoptosis visualized by ethidium bromide (EB) dye exclusion assays and (F) proliferation visualized by phospho-Histone-H3 (P-H3) immunofluorescence on cells treated with 20 μmol/L zVAD to prevent apoptosis, both quantified in 25 μm confocal z-stacks.
(G) Limiting dilution assays in which EGFR-mutant GSCs (GBM39) and hNPCs were plated at low density of 1-1000 cells per well in 96 well plates, incubated with 1 μg/mL verteporfin, and observed for neurosphere formation after 7 days.
(H-K) Tumor stem cell marker mRNA and protein levels in the indicated GSCs treated with verteporfin or DMSO as a control, as measured by qPCR, immunoblot, and immunofluorescence. 3 μm optical projections. Cells were treated with .5 μg/mL verteporfin for 24 hours for immunofluorescence and 1 μg/mL verteporfin for indicated timepoints in qPCR and immunoblots. ZVAD (20 μmol/L) was used to prevent apoptosis. Graphs show representative qPCR experiments, and error bars show range between experimental replicates.
*p≤.05; **p≤.01; ***p≤.001; ****p≤.0001 with unpaired t tests. Symbols for each bar indicate replicates per condition.
Verteporfin has also been shown to promote apoptosis of tumor cells through YAP/TAZ-TEAD independent mechanisms, including reactive oxygen radicals (ROS) generation, when used at doses 3-6-fold higher than our present study (35–38). However, even with prolonged verteporfin treatment at 3μg/mL, YAP/TAZ-negative GSCs did not undergo significant apoptosis and growth arrest (Supplementary Fig. S6D–F), indicating that YAP/TAZ expression largely dictates the pro-apoptotic and anti-proliferative effects of verteporfin on GBM cells.
We next assessed expression of direct YAP/TAZ-TEAD transcriptional targets in response to verteporfin treatment, using ZVAD to block apoptosis. We found that in EGFR-mutant/amplified GSC cultures, verteporfin reduced mRNA and protein expression of SOX2, MYC, and EGFR, indicating inhibition of YAP/TAZ target gene expression, and verteporfin reduced expression of SOX9, suggesting of loss of CL tumor stem cell identity (Fig. 3H–K). In hNPCs, verteporfin treatment reduced EGFR expression, which indicates that verteporfin affects normal autosomal EGFR gene expression, although verteporfin did not affect SOX2 or C-MYC expression (Supplementary Fig. S6C), which suggests differential regulation of these target genes in normal neural stem/progenitor cells compared to GSCs, perhaps due to dissimilar regulatory pathways. To test whether short-term verteporfin treatment affects tumor initiation by GSCs, we treated GBM39 cells with verteporfin or DMSO for 4-6 hours (before apoptosis is detected, Supplementary Fig. S8A) and intracranially implanted these cells into immunocompromised mice. We found that short-term verteporfin treatment blocked tumor initiation by GSCs (Supplementary Fig. S8A–C), perhaps due to reduced SOX2 and MYC expression.
To determine if EGFR confers sensitivity to verteporfin, we tested verteporfin on Cdkn2a−/−; Pten−/− mNSCs transformed by EGFRvIII expressed from a fl-stop-fl transgenic construct used in a mouse genetic model for EGFRvIII-driven GBM (16). The advantage of this model is that additional confounding genetic alterations are not present that may explain verteporfin response. Compared to non-transformed control Cdkn2a−/−; Pten−/− mNSCs, in EGFRvIII; Cdkn2a−/−; Pten−/− mNSCs, which overexpressed Yap and Taz in response to EGFR catalytic activity, verteporfin treatment preferentially induced apoptosis, reduced expression of Yap, Taz, Sox2, and Myc, decreased self-renewal and proliferation, and blocked tumor initiation in orthotopic implantation assays (Supplementary Fig. S9A–I), as in human GSCs. In this model verteporfin treatment did not affect transgene-driven EGFRvIII expression (Supplementary Fig. S9F), although verteporfin cooperated with EGFR inhibition to reduce viability of EGFRvIII; Cdkn2a−/−; Pten−/− mNSCs (Supplementary Fig. S9B).
To identify broader changes underlying pro-apoptotic and anti-proliferative effects of verteporfin, we subjected EGFR-amplified/mutant GSCs to total cell proteomic profiling using published protocols (18,19). Short-term 6 hour verteporfin treatment significantly reduced expression of approximately 5% of proteins detected (278/5482), including established YAP/TAZ target genes that drive gliomagenesis and stem cell identity (e.g., EGFR and SOX2) and other proteins that control cell survival (BCL2L2), proliferation and mitosis (CDK4), stem cell identity (STAT1 and STAT3), and small GTPases that transduce RTK signaling (RAS, RAC, and RAB proteins, Supplementary Table S5–S6, Supplementary Fig. S10–S11A). Bioinformatic analysis demonstrates that verteporfin significantly reduced expression of metabolic enzymes and mitochondrial regulators (IDH, TCA cycle proteins, Table S5–S6). Many proteins altered in response to verteporfin are not established YAP/TAZ-TEAD target genes, and their coordinate regulation may reflect YAP/TAZ-dependent effects on GBM cells, or, alternately, the combination of YAP/TAZ-dependent and off-target YAP/TAZ-independent effects that together cooperate to block GBM cell survival and proliferation. In support of the latter, in GBM301, TAZ knockdown decreased expression of several proteins altered by verteporfin treatment, such as CDK4 and RAC GTPases (Supplementary Fig. S11B), suggesting that their expression depends on TAZ, whereas TAZ knockdown did not affect expression of other proteins reduced by verteporfin treatment, including BCL2L2, STAT transcription factors, and RAS GTPases (Supplementary Fig. S11B), suggesting that their expression is altered by verteporfin in a YAP/TAZ-independent manner.
Verteporfin treatment decreases EGFR and SOX2 expression and slows tumor growth in orthotopic GBM xenograft models.
We hypothesized that inhibition of YAP/TAZ-TEAD transcriptional activity may be therapeutic in EGFR-amplified/mutant GBM, and we tested the efficacy of verteporfin treatments in pre-clinical ex vivo and in vivo models. Porphyrins and their precursors have been subjects of intensive development for brain tumor therapy and intraoperative brain tumor imaging because GBM and glioma cells readily absorb these molecules in vitro and in vivo in animal models and human patients (13,39,40). Yet, there are no similar studies for verteporfin absorption or therapeutic activity in GBM model systems or patients.
To determine if verteporfin could inhibit tumor growth in intact brain tissue, we first used organotypic brain slice culture assays for testing effectiveness. Brains bearing GBM39 xenografts were sliced to generate organotypic cultures grown in serum-free GSC medium which were treated with verteporfin or DMSO (control) before immunostaining and imaging by confocal microscopy (Fig. 4A). Verteporfin treatment reduced EGFR expression in GBM39 tumor cells in xenograft slices (Fig. 4B). Additionally, verteporfin induced cell death in tumor tissue, which was visualized and quantified with co-incident staining for cleaved-Caspase-3 and a human-specific Vimentin antibody to visualize tumor cells (Fig. 4C–D). These data indicate that verteporfin treatment promotes tumor cell specific death within the tumor microenvironment.
Figure 4. Verteporfin treatment induces apoptosis in GBM cells cultured in ex vivo organotypic cultures.
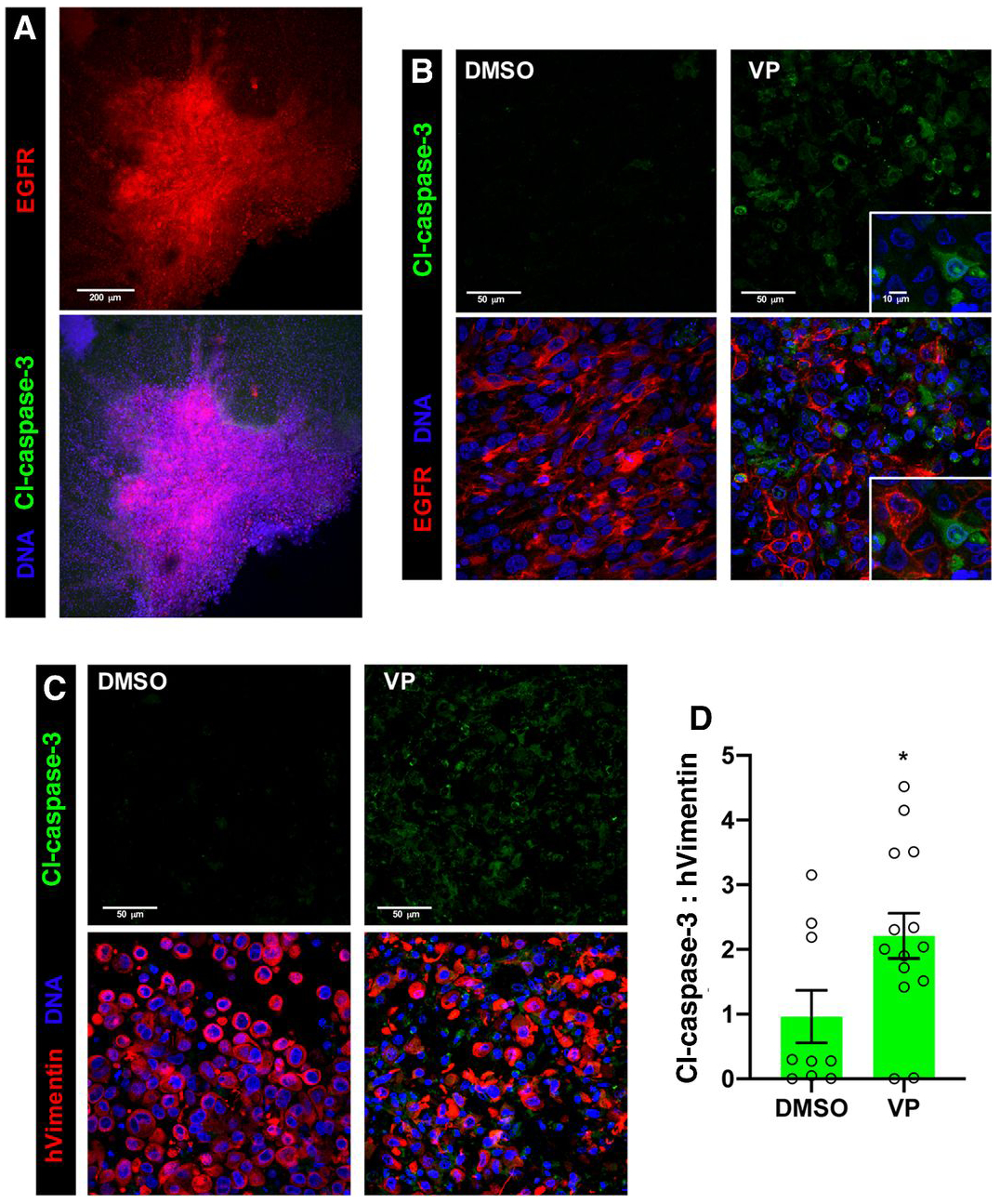
Ex vivo organotypic slice cultures created from GBM39 xenografts implanted into NSG mice, treated with DMSO (control), 1μg/mL verteporfin (VP) for 24 hours, and/or other indicated agents, and then stained and imaged with confocal microscopy. DRAQ7 DNA dye (blue) labels all cell nuclei in tumor and stroma. 3 μm optical projections.
(A) Low magnification view of a representative control slice culture showing tumor integrated into surrounding normal brain tissue, tumor cells marked by EGFR immunostaining (red); cleaved-Caspase-3 immunostaining (green) in DMSO treated tumor and normal tissue.
(B) Immunostaining for EGFR (red), with verteporfin treatment. Inset shows close-up of apoptotic cells (green, cl-Caspase-3) adjacent to EGFR-positive tumor cells.
(C) Immunostaining for cleaved-Caspase-3 (Cl-Caspase-3, green, upper panels), labeling apoptotic cells, and human Vimentin (hVimentin, red, lower panels overlay), which specifically labels human tumor cells in the mouse brain parenchyma in organotypic tumor xenograft slice cultures.
(D) Ratios for total cleaved-Caspase-3 and hVimentin immunostaining in organotypic slice cultures; values measured in 30-50 μm confocal z-stacks using Imaris. Graphs shows values from individual experimental samples from multiple experiments.
*p≤.05 with unpaired t test.
To determine if verteporfin slows tumor growth in vivo, animals bearing GBM xenografts were treated with Visudyne, a liposomal formulation of verteporfin that is FDA-approved to treat macular degeneration. In pilot experiments, mice harboring GBM xenografts received single treatments of Visudyne by intraperitoneal (IP) injection to assess verteporfin absorption in the brain. We determined that verteporfin is absorbed by tumor cells using fluorescent microscopy, penetrating at least 200 microns from major blood vessels in tumors (Supplementary Fig. S12A). In a survival experiment where we treated with IP Visudyne every other day for 2 weeks, we observed that Visudyne-treated mice lived longer than controls (Supplementary Fig. S12B), although the effect was not statistically significant, and, at the time of host death, verteporfin-treated tumors had grown as large as control tumors (Supplementary Fig. S12C).
Visudyne is formulated for intravenous (IV) administration, and IP administration does not model this well in vivo. To improve CNS uptake, we administered Visudyne via intraventricular osmotic pump to continuously deliver drug to the brain (pump validation shown in Supplementary Fig. S13A–B). As proof of concept, we used fluorescence imaging to determine if verteporfin is absorbed into tumor cells in vivo in our xenograft models. Sections from mouse brains treated with intraventricular Visudyne for 7 days showed verteporfin fluorescence at 650nm that was not present in untreated brains (Fig. 5A). Mass spectrometry quantification from animals treated with intraventricular Visudyne for 14 days showed verteporfin present at .67-1.46 ng/mg brain tissue. To test the therapeutic effects of verteporfin, GBM xenograft-bearing mice were treated with Visudyne by intraventricular osmotic pump which delivered drug continuously for 14 days or more. Visudyne treatment resulted in increased tumor necrosis, as evidenced in H&E stains in 86%(6/7) of animals examined, long-term reductions in EGFR and SOX2 expression in tumor tissue near pump implantation sites, persistent reduction of proliferation in tumor bulk, and significantly extended median survival approximately 30% longer in treated animals than control animals (Fig. 5B–D, Supplementary Fig. S14). These experiments demonstrate that verteporfin is absorbed into GBM xenograft tumors in vivo, that verteporfin treatment downregulates expression of YAP/TAZ target genes, and that verteporfin significantly slows tumor progression in xenograft models of EGFR-mutant GBM.
Figure 5. Visudyne treatment decreases tumor growth and downregulates tumor stem cell identity and EGFR expression in an orthotopic GBM xenograft model.
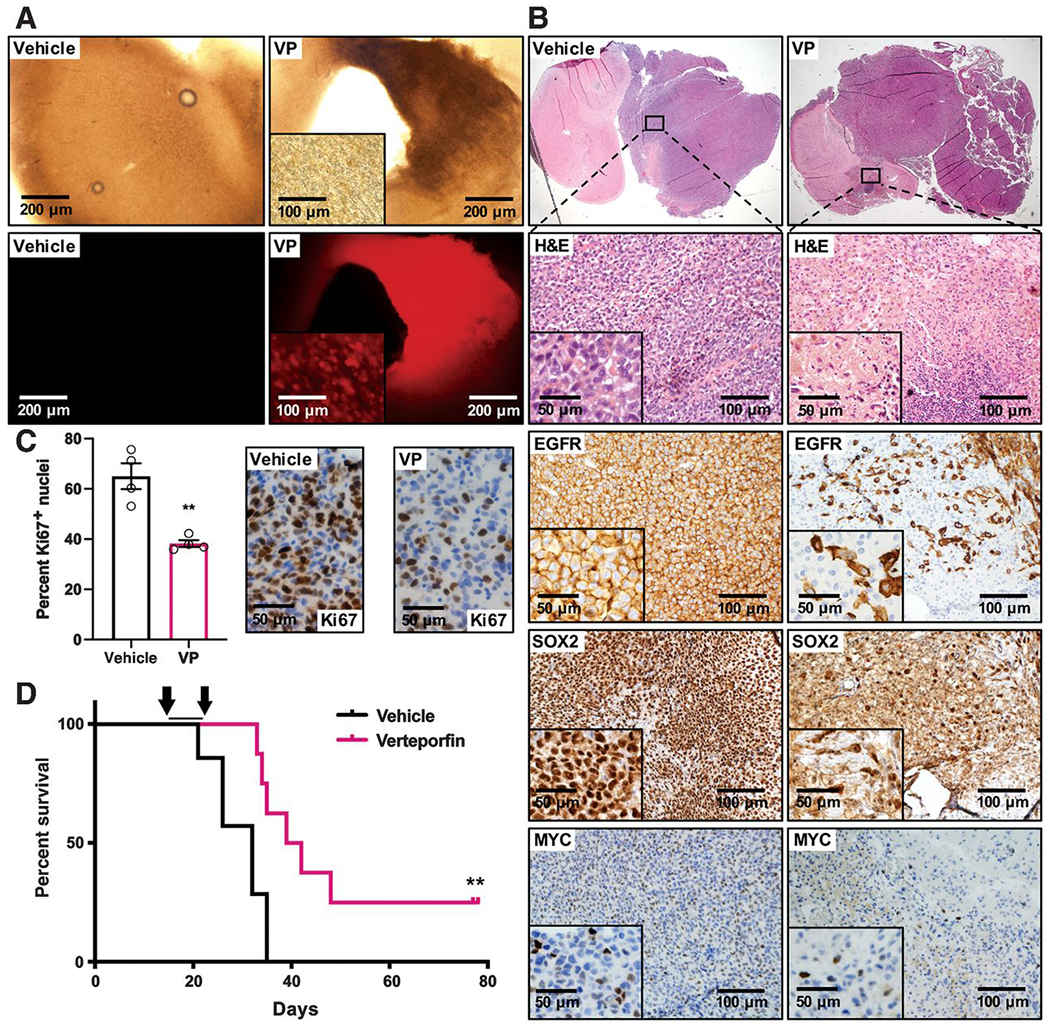
GBM39 GSCs were orthotopically implanted into 6-week-old NSG mice. Following an engraftment period of two weeks, osmotic pumps were implanted into the right ventricle to deliver vehicle (saline) or liposomal verteporfin (VP, 200 μg, Visudyne) at a constant rate over a 2-week period.
(A) Fluorescence imaging to examine verteporfin absorption; 200 μm brain slices of vehicle and verteporfin treated brains 7 days after pump implantation.
(B) H&E staining of representative brain sections from vehicle and verteporfin-treated mice. Close-up panels and insets show H&E to highlight necrosis and pyknotic nuclei and IHC for EGFR, SOX2, and MYC expression in vehicle and verteporfin-treated tumors.
(C) Ki67 IHC to assess tumor cell proliferation in tumor bulk in response to vehicle or verteporfin treatment. Graph shows Ki67-positive cells as a proportion of total cells in tumor bulk in terminal tumors from vehicle and verteporfin-treated animals, >1000 total cells counted per section per data point. **p≤.01 with unpaired two-tailed t-test.
(D) Kaplan-Meier survival curve showing survival of tumor-bearing mice treated with verteporfin (Visudyne, n=8) or control vehicle (n=7) by osmotic pump for two weeks (arrows), median survival: 32 days for vehicle, 40.5 days for verteporfin treatment. **p≤.01 with Log-rank (Mantel-Cox) test.
Phase 0 clinical trial to test Visudyne absorbance into human GBM.
To date, there are no clinical studies of verteporfin in human glioma patients. Given the strong correlation between YAP/TAZ expression and EGFR status, importance of YAP/TAZ-TEAD activity for gliomagenesis, and strong anti-tumor effects of verteporfin in vitro and in vivo, we sought to evaluate the therapeutic potential of verteporfin. For this purpose, we initiated a phase 0 clinical trial to determine if verteporfin can be absorbed by GBM tumors. Participants who were scheduled to undergo surgery for suspected or known recurrent GBM were consented for this study and entered into a dose-escalating clinical trial (Fig. 6A). We administered Visudyne intravenously (IV) at FDA-approved doses on an inpatient basis one time prior to surgery. The first patient enrolled received 0.15 mg/kg of verteporfin which was poorly visualized, so the next five patients received 0.3 mg/kg of verteporfin without side effects. Two of these patients had resections which showed mostly necrosis and/or radiation related changes with little viable tumor. The remaining four patients had WHO Grade IV GBM (Table S2).
Figure 6. Verteporfin is absorbed by human patient GBM tissue in a phase 0 clinical trial.
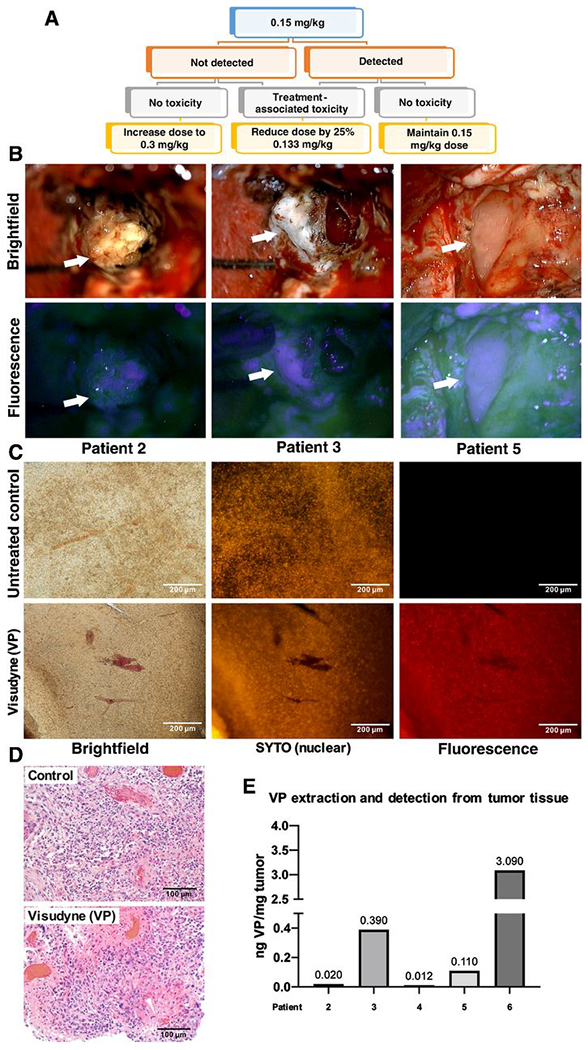
(A) Schematic of phase 0 clinical trial design. Participants received a single dose of Liposomal verteporfin (Visudyne) I.V. before surgery, at a starting of dose of 0.15 mg/kg at which verteporfin (VP) is administered to patients undergoing PDT for glaucoma. When verteporfin was not clearly detected in tumor cells at 0.15 mg/kg in the first participants, the following participants received 0.3 mg/kg.
(B) Intraoperative microscopy was accomplished with variable magnification over 2-40x to optimize visualization of tissues and structures of interest. Fluorescence was accomplished by adapting the surgical operating microscope with a filter set that modified the standard xenon light to provide fluorescence excitation in the wavelength range 390-440nm and for observation in the 600-700nm range. Arrows denote various portions of tumor under standard white light (upper) and with fluorescence (lower), respectively.
(C) Representative tumor tissue slice from a trial participant and a control patient who was not treated with verteporfin (left). Fluorescent nucleic acid counterstain (SYTO) was used to visualize cell nuclei (middle), and fluorescence using a verteporfin-specific excitation and emission filter is shown on the right.
(D) Micrographs of H&E staining on matched tissue from the tumor in (C) to visualize tumor cellularity in the region used to visualize verteporfin fluorescence.
(E) Representative tumor tissue from patients was digested, and solubilized verteporfin extracts were analyzed using mass spectrometry according to methods. Extracted verteporfin was detected at 0.012-3.090 ng/mg tumor in treated patients.
Porphyrin-related fluorescence has been clinically utilized to develop 5-aminolevulinic acid (5-ALA) and protoporphyrin IX (PPIX) imaging agents for fluorescence-assisted intraoperative microscopy for high grade glioma resection (13). Verteporfin has virtually the same excitation and emission spectra as PPIX, so we utilized intraoperative fluorescence-assisted microscopy to determine if verteporfin was visible in tumor tissue similar to PPIX. On encountering tumor during surgery, intraoperative microscopy (Zeiss Pentero 900 system) was used to illuminate the tumor bed with blue light (400-410 nm) to produce far red (620-700 nm) fluorescence in tumor cells in live patients, and photographs of tumor and adjacent tissue were taken through the microscope using a camera adapted for imaging the far-red emissions spectrum (Fig. 6B).
Unfixed resected tumor tissues from phase 0 participants were sectioned, counter-stained with nuclear markers and cell membrane markers to confirm cellularity, and imaged with fluorescence microscopy. Resected tumor tissue from Visudyne-treated patients were found to have fluorescence from verteporfin absorption into tumor cells while tumor tissue from patients who were not treated with Visudyne had no evidence of fluorescence (Fig. 6C–D). Absorbed verteporfin was extracted from tumor tissue from phase 0 participants for detection and quantification of by mass spectrometry. Our results indicated verteporfin was present in tumor tissues at varying ranges (Fig. 6E). By mass spectrometry, tissues from untreated control patients showed no signals in the same range as verteporfin, indicating that background from tissue did not contribute to our quantification of verteporfin in treated patients. However, previous studies show that verteporfin may not be fully extracted from tissues because it covalently links to proteins, (21), so we are uncertain if our measurements accurately represent verteporfin absorbed into tumors. Yet, together, our data indicate that IV administered liposomal verteporfin is effectively absorbed into GBM tumor cells in human patients.
DISCUSSION
In this study, we discovered that YAP family transcription factors and TEAD co-factors are important drivers of GBM tumorigenesis downstream of EGFR, using a combination of Drosophila models, GSC cultures, and xenograft models. The CL GBM subtype is characterized by EGFR-amplification and/or mutation, chromosome 7 gain, CDKN2A loss, and co-occurring mutations in other RTK and PI3K pathway components, and these tumors show an astrocytic transcriptional signature (1,26). In CL GBMs, we found that YAP and TAZ are co-overexpressed in response to EGFR kinase activity and are required for GBM cell proliferation, stem cell self-renewal, and tumor progression. YAP/TAZ-TEAD transcriptional targets include stem cell master regulators SOX2 and MYC, and EGFR itself, which act together with YAP/TAZ-TEAD in a tumor-promoting feedforward loop. Based on these data, we conclude that YAP and TAZ co-overexpression is common to EGFR-amplified/mutant CL GBMs, and that therapeutic strategies that target both factors may benefit patients with these tumors.
Our efforts also uncovered a promising GBM treatment, verteporfin, which is an inhibitor of YAP/TAZ-TEAD interactions (15), that was efficacious in reducing expression of EGFR and SOX2 and in extending median survival of GBM-bearing mice in orthotopic xenografts (Fig. 7). Through a phase 0 clinical trial, we demonstrate that liposomal verteporfin was effectively absorbed by GBM cells in human patients. Here we show that verteporfin disrupted TAZ-TEAD binding and reduced YAP/TAZ protein levels and nuclear localization, confirming that verteporfin is a dual targeting irreversible inactivator of YAP/TAZ proteins (34). In preliminary analyses, tumor tissue from Visudyne phase 0 participants showed low YAP/TAZ protein levels compared to a representative untreated control patient (Supplementary Fig. S15), which suggests that sufficient verteporfin may be absorbed to disrupt YAP/TAZ protein expression in vivo in humans. However, we do not have untreated control tissue for phase 0 participants for comparison, so we cannot be certain that low YAP and TAZ expression is due to verteporfin.
Figure 7. Summary Model for Verteporfin Therapeutic Intervention in Glioma.
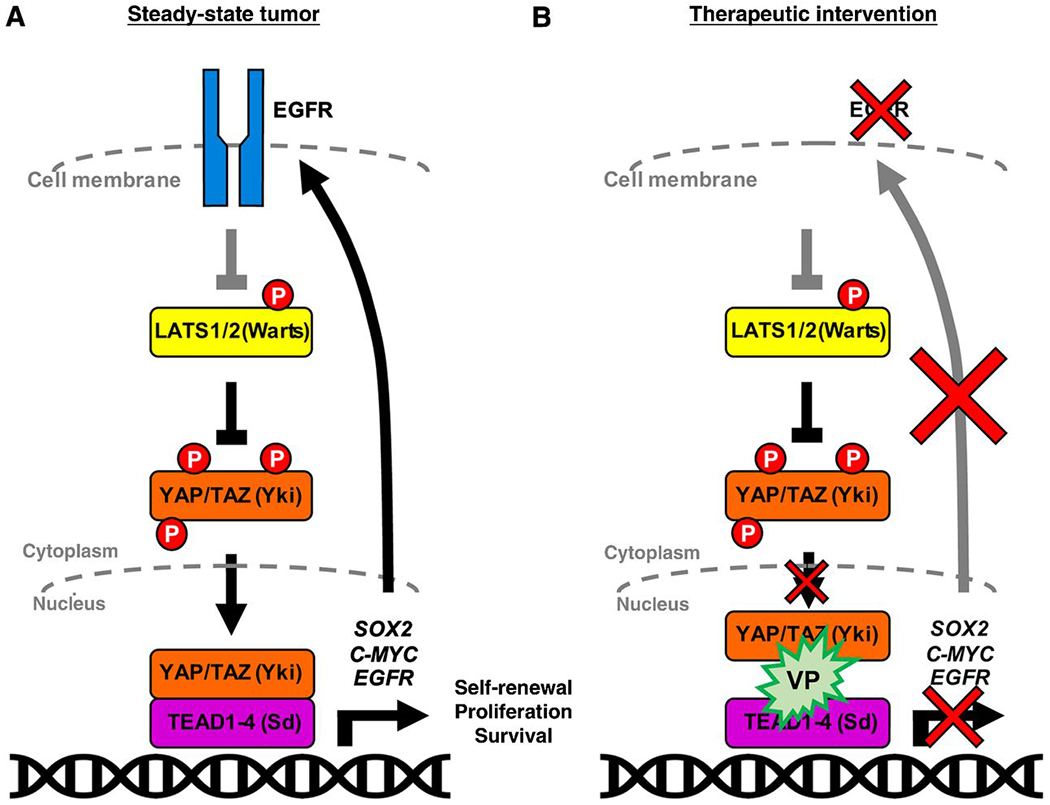
(A) We have shown that in GBM EGFR acts through YAP/TAZ, perhaps through inhibition of Hippo pathway kinases such as LATS1, to activate stem cell transcriptional regulators in combination with TEAD binding partners and transcription co-factors. YAP/TAZ thereby drive transcription of downstream targets which lead to stem cell maintenance and tumor growth and create a feed-forward loop with EGFR itself. These ultimately drive stem cell programs such as self-renewal, proliferation, and survival.
(B) Illustration representing therapeutic intervention with verteporfin, a fluorescent porphyrin derivative, which blocks the interaction between YAP/TAZ and TEAD and abrogates YAP/TAZ nuclear localization, leading to a downregulation of transcriptional targets and a consequent decreased EGFR expression and loss of stem identity and survival in GSCs.
In our Drosophila GBM model, Hippo pathway kinases Hippo, Warts, or Tao, which regulate the YAP/TAZ ortholog Yki (6,7), all counteract EGFR-driven neoplasia (5), implying EGFR signaling negatively regulates Hippo pathway kinase activity. Consistent with this observation, we found that EGFR inhibition increased the proportion of YAP and TAZ phosphorylated at LATS1/2 target sites, which promote their degradation. However, we did not observe consistent LATS protein expression in CL tumors (Supplementary Fig. S3), and therefore other effectors likely regulate YAP/TAZ phosphorylation and degradation in conjunction with EGFR. Crosstalk between Hippo-YAP/TAZ and RTK pathways, which together modulate growth, progression, and drug resistance in other tumor types, is complex (7). In other contexts, EGFR signaling bypasses Hippo kinases and upregulates YAP family transcription proteins through PI3K, MAPK, SRC, and CREB effector pathways (8,41–44). We and others have observed that CL GBMs show significant YAP and TAZ mRNA upregulation (22). Furthermore, we observed that Taz is overexpressed in our mouse model of EGFRvIII-driven GBM compared to normal neural/stem-progenitor cells, and we observed that exogenously increasing Taz mRNA levels enhanced EGFRvIII-driven tumor progression (16), implying that epigenetic mechanisms that increase YAP/TAZ overexpression promote tumor progression. In developing tissues, YAP and TAZ are differentially regulated downstream of cell adhesion signaling, GPCR signaling, and mechanochemical signaling (7), and these pathways could preferentially upregulate TAZ in CL GBM. Further study will be necessary to unravel connections between EGFR signaling and regulation of YAP and TAZ expression and function in GBM.
While functional differences between YAP and TAZ in GBM are unclear, YAP and TAZ share TEAD transcriptional targets (7), like EGFR, SOX2, and C-MYC in GBM, that, when functionally reduced, lead to loss of stem-cell-like self-renewal and reduced tumor growth (9,10). In GBM with amplified EGFR, which is typically on extrachromosomal amplicons (30,31), EGFR protein can be overexpressed at 10-20 fold or more relative to tumors without amplification (45). With such high levels in tumors, effective targeting of EGFR protein kinase activity with small molecule inhibitors in patients is highly unlikely at doses non-toxic to other tissues (3). Treatments that reduce total EGFR expression levels, such as verteporfin, may be useful for overcoming intrinsic resistance to EGFR-targeted therapies. Other established TEAD-dependent transcriptional targets genes in tumor cells include redundant RTKs, such as MET and AXL, and RTK ligands, such as IGF2, CTGF, and EGFR ligands (6,7,23,46), which promote tumorigenesis and/or resistance to EGFR-PI3K targeted therapies (3). Thus, YAP/TAZ-TEAD inhibition may impact functionally redundant RTKs and prevent resistance to therapeutics that target RTK signaling pathways in GBM. In preliminarily experiments, using slice culture assays we tested verteporfin combined with the B-RAF inhibitor dabrafenib (selected based on synergy with YAP inhibition (47), which increased apoptosis in tumor cells compared to verteporfin alone (Supplementary Fig. S16), suggesting that YAP/TAZ inhibitors may have more effective anti-tumor activity in combination with drugs that target RTK pathways.
Verteporfin also has YAP/TAZ-TEAD-independent off-target mechanisms of action that likely cooperate with YAP/TAZ-TEAD inhibition and reduced EGFR expression to contribute to anti-tumor activity. Verteporfin has been used clinically as a photosensitizer in photodynamic therapy (PDT) for neo-vascular macular degeneration, where it is activated by laser light to focally generate ROS to eliminate the abnormal blood vessels. In previous studies, ROS generation in response to high dose verteporfin is toxic to standard glioma cell lines by YAP-independent means (37,38). Additional non-PDT-mediated mechanisms of verteporfin toxicity include induction of autophagy pathways and proteotoxicity (35,36). Using proteomics, we uncovered alterations in verteporfin-treated EGFR mutant GSCs that likely result from on-target YAP/TAZ-dependent and off-target YAP/TAZ-independent mechanisms (Supplementary Table S4–S5, Supplementary Fig. S10–S11), which may both contribute to therapeutic efficacy. For example, we found that verteporfin reduced STAT3 levels in a YAP/TAZ independent manner, similar to previous studies (35). Furthermore, our analysis indicates that verteporfin treatment reduced proteins involved in the TCA cycle and oxidative phosphorylation in mitochondria (Supplementary Table S4–S5), many of which are involved in biosynthesis and metabolism essential to tumor cell growth. Previous studies show that verteporfin accumulates in mitochondria such that alterations in mitochondrial function may also be a YAP/TAZ independent effect of verteporfin (37,38). However, off-target YAP/TAZ and EGFR independent effects of verteporfin do not reduce its potential as a therapeutic agent for GBM. The off-target effects of cancer therapeutics often contribute as much to their clinical efficacy as their on-target effects (48), and further research on the on-target and off-target combinatorial effects of verteporfin will be important to the development of verteporfin and additional therapies that target YAP/TAZ-TEAD.
Verteporfin is a porphyrin derivative, and porphyrins related to verteporfin cross the blood-brain barrier and accumulate in the brain (39,40). Previous research established that porphyrins are readily absorbed by GBM cells both in vitro and in vivo in animal models and human patients (13,39,40). Our study is the first to confirm with fluorescence microscopy and mass spectrometry that verteporfin, delivered in the FDA-approved Visudyne liposomal formulation, accumulates in GBM cells in animal models and human patients. Visudyne is designed to optimize delivery to neovasculature by IV injection, which is difficult to deliver repeatedly in mouse models. We tested two different modes of delivery in mouse models, and we observed a survival benefit by administering Visudyne directly into the CSF via intraventricular osmotic pump but did not observe a significant survival benefit by administering Visudyne by IP injection. Our data indicate that enhanced CNS delivery improves verteporfin efficacy. We speculate that reformulation optimized for CNS absorption will improve verteporfin efficacy in brain tumors. Towards this goal, other groups have developed nanoparticle delivery mechanisms for verteporfin using animal models of glioma (49,50).
Our experiments have uncovered a therapeutically relevant dependency on YAP/TAZ-TEAD activity in EGFR-amplified/mutant GBM, demonstrated that these tumors display a clinically relevant therapeutic vulnerability to pharmacologic treatment with the YAP/TAZ-TEAD inhibitor and porphyrin verteporfin, and confirmed verteporfin absorption into GBM tissues in a phase 0 clinical study of human patients. Our results may spur the development of verteporfin as a new treatment for GBMs and other gliomas.
Supplementary Material
STATEMENT OF TRANSLATIONAL RELEVANCE.
There is an urgent unmet clinical need for new and effective treatments for glioblastomas and related high-grade gliomas. This project has uncovered a therapeutically relevant dependency on YAP and TAZ transcriptional activators in human GBMs that are driven by EGFR amplification/mutation, demonstrated that these tumors display a therapeutic vulnerability to pharmacologic treatment with the YAP/TAZ inhibitor and porphyrin derivative verteporfin, and confirmed verteporfin absorption into GBM tissues in a phase 0 clinical study of human patients. Our results establish a basis for the development of verteporfin and/or other YAP/TAZ inhibitors as possible treatments for EGFR driven glioblastomas.
ACKNOWLEDGEMENTS
We thank Zhihong Chen, Kai Nie, Dolores Hambardzumyan, and Colleen Mosley for technical assistance, Anh N. Tran for R programming assistance, William Read for assistance with the Phase 0 trial protocol, and the Winship Cancer Institute’s Cancer Animal Models Core for assistance with mouse breeding.
Funding:
This work was supported by grants from the American Brain Tumor Association, the Rally Foundation for Childhood Cancer Research, the Winship Cancer Institute, and NINDS to RDR, an R25 training supplement from NINDS and a postdoctoral fellowship from the Neurosurgery Research and Education Foundation to KV, a T32 training award in Translational Neurology from NINDS to NHB, and a postdoctoral fellowship from the Rally Foundation for Childhood Cancer Research to SYO.
Footnotes
Conflict of Interest: The authors have no conflicts of interest regarding this manuscript.
REFERENCES
- 1.Brennan CW, Verhaak RGW, McKenna A, Campos B, Noushmehr H, Salama SR, et al. The Somatic Genomic Landscape of Glioblastoma. Cell 2013;155(2): 462–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nishikawa R, Ji XD, Harmon RC, Lazar CS, Gill GN, Cavenee WK, et al. A mutant epidermal growth factor receptor common in human glioma confers enhanced tumorigenicity. Proc Natl Acad Sci U S A 1994;91(16):7727–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shergalis A, Bankhead A 3rd, Luesakul U, Muangsin N, Neamati N. Current Challenges and Opportunities in Treating Glioblastoma. Pharmacol Rev 2018;70(3):412–45 doi 10.1124/pr.117.014944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Read RD, Cavenee WK, Furnari FB, Thomas JB. A drosophila model for EGFR-Ras and PI3K-dependent human glioma. PLoS Genet 2009;5(2):e1000374 doi 10.1371/journal.pgen.1000374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Read RD, Fenton TR, Gomez GG, Wykosky J, Vandenberg SR, Babic I, et al. A kinome-wide RNAi screen in Drosophila Glia reveals that the RIO kinases mediate cell proliferation and survival through TORC2-Akt signaling in glioblastoma. PLoS Genet 2013;9(2):e1003253 doi 10.1371/journal.pgen.1003253PGENETICS-D-12-01408 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Misra JR, Irvine KD. The Hippo Signaling Network and Its Biological Functions. Annu Rev Genet 2018;52:65–87 doi 10.1146/annurev-genet-120417-031621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zheng Y, Pan D. The Hippo Signaling Pathway in Development and Disease. Dev Cell 2019;50(3):264–82 doi 10.1016/j.devcel.2019.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reddy BV, Irvine KD. Regulation of Hippo signaling by EGFR-MAPK signaling through Ajuba family proteins. Dev Cell 2013;24(5):459–71 doi 10.1016/j.devcel.2013.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang J, Wang H, Li Z, Wu Q, Lathia JD, McLendon RE, et al. c-Myc is required for maintenance of glioma cancer stem cells. PLoS One 2008;3(11):e3769 doi 10.1371/journal.pone.0003769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gangemi RM, Griffero F, Marubbi D, Perera M, Capra MC, Malatesta P, et al. SOX2 silencing in glioblastoma tumor-initiating cells causes stop of proliferation and loss of tumorigenicity. Stem Cells 2009;27(1):40–8 doi 10.1634/stemcells.2008-0493. [DOI] [PubMed] [Google Scholar]
- 11.Lian I, Kim J, Okazawa H, Zhao J, Zhao B, Yu J, et al. The role of YAP transcription coactivator in regulating stem cell self-renewal and differentiation. Genes Dev 2010;24(11):1106–18 doi 10.1101/gad.1903310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Choi W, Kim J, Park J, Lee DH, Hwang D, Kim JH, et al. YAP/TAZ Initiates Gastric Tumorigenesis via Upregulation of MYC. Cancer Res 2018;78(12):3306–20 doi 10.1158/0008-5472.CAN-17-3487. [DOI] [PubMed] [Google Scholar]
- 13.Stummer W, Pichlmeier U, Meinel T, Wiestler OD, Zanella F, Reulen HJ, et al. Fluorescence-guided surgery with 5-aminolevulinic acid for resection of malignant glioma: a randomised controlled multicentre phase III trial. The Lancet Oncology 2006;7(5):392–401 doi 10.1016/S1470-2045(06)70665-9. [DOI] [PubMed] [Google Scholar]
- 14.Laks DR, Masterman-Smith M, Visnyei K, Angenieux B, Orozco NM, Foran I, et al. Neurosphere formation is an independent predictor of clinical outcome in malignant glioma. Stem Cells 2009;27(4):980–7 doi 10.1002/stem.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu-Chittenden Y, Huang B, Shim JS, Chen Q, Lee SJ, Anders RA, et al. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev 2012;26(12):1300–5 doi 10.1101/gad.192856.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen Z, Herting CJ, Ross JL, Gabanic B, Puigdelloses Vallcorba M, Szulzewsky F, et al. Genetic driver mutations introduced in identical cell-of-origin in murine glioblastoma reveal distinct immune landscapes but similar response to checkpoint blockade. Glia 2020. doi 10.1002/glia.23883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu Y, Smyth GK. ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods 2009;347(1-2):70–8 doi 10.1016/j.jim.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 18.Seyfried NT, Dammer EB, Swarup V, Nandakumar D, Duong DM, Yin L, et al. A Multi-network Approach Identifies Protein-Specific Co-expression in Asymptomatic and Symptomatic Alzheimer’s Disease. Cell Syst 2017;4(1):60–72 e4 doi 10.1016/j.cels.2016.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wingo TS, Duong DM, Zhou M, Dammer EB, Wu H, Cutler DJ, et al. Integrating Next-Generation Genomic Sequencing and Mass Spectrometry To Estimate Allele-Specific Protein Abundance in Human Brain. J Proteome Res 2017;16(9):3336–47 doi 10.1021/acs.jproteome.7b00324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huggett MT, Jermyn M, Gillams A, Illing R, Mosse S, Novelli M, et al. Phase I/II study of verteporfin photodynamic therapy in locally advanced pancreatic cancer. Br J Cancer 2014;110(7):1698–704 doi 10.1038/bjc.2014.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bang LG, Dasari VR, Kim D, Gogoi RP. Differential gene expression induced by Verteporfin in endometrial cancer cells. Sci Rep 2019;9(1):3839 doi 10.1038/s41598-019-40495-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Orr BA, Bai H, Odia Y, Jain D, Anders RA, Eberhart CG. Yes-associated protein 1 is widely expressed in human brain tumors and promotes glioblastoma growth. J Neuropathol Exp Neurol 2011;70(7):568–77 doi 10.1097/NEN.0b013e31821ff8d8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bhat KP, Salazar KL, Balasubramaniyan V, Wani K, Heathcock L, Hollingsworth F, et al. The transcriptional coactivator TAZ regulates mesenchymal differentiation in malignant glioma. Genes Dev 2011;25(24):2594–609 doi 25/24/2594 [pii] 10.1101/gad.176800.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Neftel C, Laffy J, Filbin MG, Hara T, Shore ME, Rahme GJ, et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019;178(4):835–49 e21 doi 10.1016/j.cell.2019.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010;17(1):98–110 doi S1535-6108(09)00432-2 [pii] 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brennan C, Momota H, Hambardzumyan D, Ozawa T, Tandon A, Pedraza A, et al. Glioblastoma subclasses can be defined by activity among signal transduction pathways and associated genomic alterations. PLoS One 2009;4(11):e7752 doi 10.1371/journal.pone.0007752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang H, Geng D, Gao J, Qi Y, Shi Y, Wang Y, et al. Expression and significance of Hippo/YAP signaling in glioma progression. Tumour Biol 2016;37:15665–15676 doi 10.1007/s13277-016-5318-1. [DOI] [PubMed] [Google Scholar]
- 28.Cao X, Pfaff SL, Gage FH. YAP regulates neural progenitor cell number via the TEA domain transcription factor. Genes Dev 2008;22(23):3320–34 doi 10.1101/gad.1726608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lavado A, Park JY, Pare J, Finkelstein D, Pan H, Xu B, et al. The Hippo Pathway Prevents YAP/TAZ-Driven Hypertranscription and Controls Neural Progenitor Number. Dev Cell 2018;47(5):576–91 e8 doi 10.1016/j.devcel.2018.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koga T, Li B, Figueroa JM, Ren B, Chen CC, Carter BS, et al. Mapping of genomic EGFRvIII deletions in glioblastoma: insight into rearrangement mechanisms and biomarker development. Neuro Oncol 2018;20(10):1310–20 doi 10.1093/neuonc/noy058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nathanson DA, Gini B, Mottahedeh J, Visnyei K, Koga T, Gomez G, et al. Targeted therapy resistance mediated by dynamic regulation of extrachromosomal mutant EGFR DNA. Science 2014;343(6166):72–6 doi 10.1126/science.1241328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Song S, Honjo S, Jin J, Chang SS, Scott AW, Chen Q, et al. The Hippo Coactivator YAP1 Mediates EGFR Overexpression and Confers Chemoresistance in Esophageal Cancer. Clin Cancer Res 2015;21(11):2580–90 doi 10.1158/1078-0432.CCR-14-2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stein C, Bardet AF, Roma G, Bergling S, Clay I, Ruchti A, et al. YAP1 Exerts Its Transcriptional Control via TEAD-Mediated Activation of Enhancers. PLoS Genet 2015;11(8):e1005465 doi 10.1371/journal.pgen.1005465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gibault F, Bailly F, Corvaisier M, Coevoet M, Huet G, Melnyk P, et al. Molecular Features of the YAP Inhibitor Verteporfin: Synthesis of Hexasubstituted Dipyrrins as Potential Inhibitors of YAP/TAZ, the Downstream Effectors of the Hippo Pathway. ChemMedChem 2017;12(12):954–61 doi 10.1002/cmdc.201700063. [DOI] [PubMed] [Google Scholar]
- 35.Zhang H, Ramakrishnan SK, Triner D, Centofanti B, Maitra D, Gyorffy B, et al. Tumor-selective proteotoxicity of verteporfin inhibits colon cancer progression independently of YAP1. Sci Signal 2015;8(397):ra98 doi 10.1126/scisignal.aac5418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Donohue E, Tovey A, Vogl AW, Arns S, Sternberg E, Young RN, et al. Inhibition of autophagosome formation by the benzoporphyrin derivative verteporfin. J Biol Chem 2011;286(9):7290–300 doi 10.1074/jbc.M110.139915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eales KL, Wilkinson EA, Cruickshank G, Tucker JHR, Tennant DA. Verteporfin selectively kills hypoxic glioma cells through iron-binding and increased production of reactive oxygen species. Sci Rep 2018;8(1):14358 doi 10.1038/s41598-018-32727-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peng TI, Chang CJ, Guo MJ, Wang YH, Yu JS, Wu HY, et al. Mitochondrion-targeted photosensitizer enhances the photodynamic effect-induced mitochondrial dysfunction and apoptosis. Ann N Y Acad Sci 2005;1042:419–28 doi 10.1196/annals.1338.035. [DOI] [PubMed] [Google Scholar]
- 39.Hill JS, Kahl SB, Kaye AH, Stylli SS, Koo MS, Gonzales MF, et al. Selective tumor uptake of a boronated porphyrin in an animal model of cerebral glioma. Proc Natl Acad Sci U S A 1992;89(5):1785–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hill JS, Kaye AH, Sawyer WH, Morstyn G, Megison PD, Stylli SS. Selective uptake of hematoporphyrin derivative into human cerebral glioma. Neurosurgery 1990;26(2):248–54. [DOI] [PubMed] [Google Scholar]
- 41.Azad T, Nouri K, Janse van Rensburg HJ, Maritan SM, Wu L, Hao Y, et al. A gain-of-functional screen identifies the Hippo pathway as a central mediator of receptor tyrosine kinases during tumorigenesis. Oncogene 2020;39(2):334–55 doi 10.1038/s41388-019-0988-y. [DOI] [PubMed] [Google Scholar]
- 42.Xia H, Dai X, Yu H, Zhou S, Fan Z, Wei G, et al. EGFR-PI3K-PDK1 pathway regulates YAP signaling in hepatocellular carcinoma: the mechanism and its implications in targeted therapy. Cell Death Dis 2018;9(3):269 doi 10.1038/s41419-018-0302-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen P, Zhao D, Li J, Liang X, Li J, Chang A, et al. Symbiotic Macrophage-Glioma Cell Interactions Reveal Synthetic Lethality in PTEN-Null Glioma. Cancer Cell 2019;35(6):868–84 e6 doi 10.1016/j.ccell.2019.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen J, Harris RC. Interaction of the EGF Receptor and the Hippo Pathway in the Diabetic Kidney. J Am Soc Nephrol 2016;27(6):1689–700 doi 10.1681/ASN.2015040415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schulte A, Gunther HS, Martens T, Zapf S, Riethdorf S, Wulfing C, et al. Glioblastoma stem-like cell lines with either maintenance or loss of high-level EGFR amplification, generated via modulation of ligand concentration. Clin Cancer Res 2012;18(7):1901–13 doi 10.1158/1078-0432.CCR-11-3084. [DOI] [PubMed] [Google Scholar]
- 46.Al-Moujahed A, Brodowska K, Stryjewski TP, Efstathiou NE, Vasilikos I, Cichy J, et al. Verteporfin inhibits growth of human glioma in vitro without light activation. Sci Rep 2017;7(1):7602 doi 10.1038/s41598-017-07632-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lin L, Sabnis AJ, Chan E, Olivas V, Cade L, Pazarentzos E, et al. The Hippo effector YAP promotes resistance to RAF- and MEK-targeted cancer therapies. Nat Genet 2015;47(3):250–6 doi 10.1038/ng.3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lin A, Giuliano CJ, Palladino A, John KM, Abramowicz C, Yuan ML, et al. Off-target toxicity is a common mechanism of action of cancer drugs undergoing clinical trials. Science translational medicine 2019;11(509) doi 10.1126/scitranslmed.aaw8412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pellosi DS, Paula LB, de Melo MT, Tedesco AC. Targeted and Synergic Glioblastoma Treatment: Multifunctional Nanoparticles Delivering Verteporfin as Adjuvant Therapy for Temozolomide Chemotherapy. Mol Pharm 2019;16(3):1009–24 doi 10.1021/acs.molpharmaceut.8b01001. [DOI] [PubMed] [Google Scholar]
- 50.Shah SR, Kim J, Schiapparelli P, Vazquez-Ramos CA, Martinez-Gutierrez JC, Ruiz-Valls A, et al. Verteporfin-Loaded Polymeric Microparticles for Intratumoral Treatment of Brain Cancer. Mol Pharm 2019;16(4):1433–43 doi 10.1021/acs.molpharmaceut.8b00959. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.