

Abstract
Neurodevelopmental disorders (NDDs) that affect cognition, social interaction, and learning, including autism spectrum disorder (ASD) and intellectual disability (ID), have a strong genetic component. Our current understanding of risk genes highlights two main groups of dysfunction: those in genes that act as chromatin modifiers and those in genes that encode for proteins localized at or near synapses. Understanding how dysfunction in these genes contributes to phenotypes observed in ASD and ID remains a major question in neuroscience. In this review, we highlight emerging evidence suggesting that dysfunction in dendrites—regions of neurons that receive synaptic input—may be key to understanding features of neuronal processing affected in these disorders. Dendritic integration plays a fundamental role in sensory processing, cognition, and conscious perception, processes hypothesized to be impaired in NDDs. Many high-confidence ASD genes function within dendrites where they control synaptic integration and dendritic excitability. Further, increasing evidence demonstrates that several ASD/ID genes, including chromatin modifiers and transcription factors, regulate the expression or scaffolding of dendritic ion channels, receptors, and synaptic proteins. Therefore, we discuss how dysfunction of subsets of NDD-associated genes in dendrites lead to defects in dendritic integration and excitability and may be one core phenotype in ASD and ID.
Keywords: neurodevelopmental disorder, epilepsy, autism spectrum disorder, dendrite, channelopathy
Introduction
Autism spectrum disorder (ASD) and intellectual disability (ID) are two of the most common neurodevelopmental disorders (NDDs), with an incidence of 1 in 54 births [1]. These NDDs have a strong genetic component, and substantial progress has been made to uncover the genetic architecture that contributes to impaired neurobiology and behavioral phenotypes. A key challenge for the field has been to translate these findings into an understanding of pathophysiology at the cellular and circuit level, with the goal of identifying key points of convergence across ASD/ID genes within the brain. Many excellent commentaries and reviews have advanced major themes of convergence in these disorders, including deficits in synaptic balance, connectivity, predictive processing, and in “top-down bottom-up” processing of stimuli relative to internal brain states [2–9]. Mechanistically, considerable attention has been paid to how NDD-associated genes affect synapse function. Here, we discuss how extending beyond synapses to the dendrites in which they reside—and understanding how alterations in dendritic structure, excitability, and integration, either driven by or driving synaptic alterations—can help link theories of ASD/ID to cellular mechanisms affected in these disorders.
In this review, we focus primarily on neocortical regions and neuronal processing within pyramidal cell dendrites. As a site for higher-order sensory processing, planning, and cognition, the neocortex has long been considered a key site for ASD/ID pathophysiology. Neocortical volume expands considerably across mammalian evolution, with parallel expansion in circuit complexity [10]. The neocortex is a multilayered structure, with each layer containing specific cell classes that have unique morphologies and synaptic connectivity. These synapses can be local, within regions that process similar information, or long-range, spanning regions to provide associative, contextual information to different regions of cortex. Pyramidal cells, which have dendrites spanning multiple layers, are uniquely positioned to integrate both local and long-range inputs, thereby acting as cellular structures that integrate disparate streams of input to help guide behavior. In particular, layer 5 pyramidal cells, which have dendrites that span all layers, have emerged as an important cell class for sensory discrimination and conscious perception [11]. Interestingly, co-expression network analysis from whole-exome sequencing data revealed multiple high-confidence ASD genes are highly expressed in layers 5 and 6 glutamatergic projection neurons, highlighting these neurons as a central locus at which ASD risk genes converge [12–16]. Thus, a better understanding of how these neurons process information may shed light on the neurobiological mechanisms disrupted in NDDs. We note, however, that pyramidal cells are just one of many cell classes that are important for ASD/ID etiology and that many of the themes discussed here are broadly applicable across brain regions, including striatum, cerebellum, and other subcortical regions [17–25].
Current understanding of pyramidal cell dendritic function
Neocortical pyramidal cells in layers 2, 3, and 5 have characteristic dendritic morphologies. Basal dendrites in proximity to the soma are studded with spines that receive input from neighboring pyramidal cells as well as partners from similar cortical regions in the contralateral hemisphere [26–28]. These pyramidal cells also have one or more apical dendrites that arborize extensively in layer 1, a layer relatively devoid of somata but rich in neurites. Synapses made in layer 1 onto the apical tufts of pyramidal cells largely arise from long-range sources, including other regions of cortex, thalamocortical recurrent loops, and sources of neuromodulatory transmitters [29–31]. Thus, neocortex circuitry is organized to that allow associative processing by making comparisons between local inputs and long-range inputs, reflecting circuit states in other brain regions (Fig. 1A). Within this framework, pyramidal cells play a central role, as their dendrites can sample both local sources in basal dendrites and long-range sources in their apical dendrites. In 1999, Larkum and colleagues provided the first empirical observations that dendrites of layer 5 pyramidal cells can perform such computations, allowing for coincidence detection between basal and apical regions [32]. In these experiments, they observed that pairing action potentials (APs) in the axon initial segment (AIS) with excitatory synaptic input into the apical tuft can lead to supralinear voltage responses in the dendrite that, in turn, result in enhanced neuronal output in the form of a high frequency AP burst (Fig. 1B). This mechanism occurred only within a narrow temporal window, thereby serving as a coincidence detector for near-simultaneous activation of these two compartments. And lastly, they showed that dendritic supralinearities can be vetoed by local inhibitory inputs. Remarkably, as discussed below, the cellular mechanisms that support each of these features have been shown to be disrupted in ASD/ID.
Figure 1: Mechanisms of synaptic integration on layer 5 pyramidal neurons dendrites in neocortex.
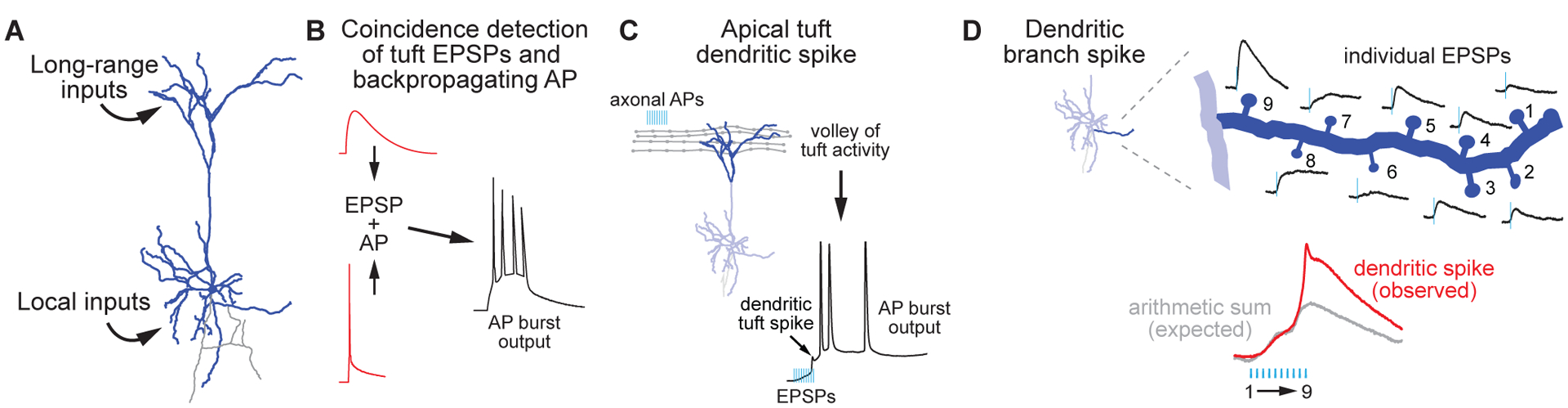
A) Approximation of synaptic inputs to neocortical layer 5 pyramidal neurons, which tend to integrate long-range synaptic inputs onto apical tuft dendritic branches and local inputs on basal dendrites.
B) Coincidence detection between apical dendritic excitatory post-synaptic potentials (EPSPs) and action potentials (APs) from basal regions generates supralinear depolarizations and axonal AP bursts.
C) High-frequency AP bursts onto apical tuft dendrites alone can evoke dendritic spikes and subsequent APs from the axon.
D) Synchronous synaptic input on dendritic branches drives individual EPSPs. When combined in a cascade of events from distal to more proximal parts of the soma, these EPSPs summate to generate events larger than their arithmetic sum (e.g., dendritic spike).
This landmark discovery brought forth more than a decade of research on cellular mechanisms present in dendrites that allow for “feature detection” in synaptic inputs. These features, which have been described in detail in excellent review articles [33–35], include: 1) the generation of “dendritic spikes”, which occur when synaptic input paired with or without AP-mediated depolarization results in regenerative supralinearities within dendritic compartments, independent of APs initiated in the axon initial segment (Fig. 1B–C) [36]; 2) detection of coincident synaptic input onto single dendritic branches, independent of the location of this branch within the overall dendritic arbor; 3) the directionality of activation of such inputs on single branches (i.e., in a cascade either toward or away from the soma; Fig. 1D) [37]; and 4) XOR (eXclusively OR) computations, where dendrites are capable of signaling the occurrence of one of two inputs (X or Y), but not both (X and Y), a process previously thought to require a multicellular network [38]. These processes often result from coordinated synaptic engagement within a small region of dendrite, which allows synaptic depolarizations to accumulate, thereby relieving voltage-dependent magnesium block of N-methyl-D-aspartate (NMDA) receptors [39]. This, in itself, promotes further local depolarization, but NMDA receptor activation is often not the sole mechanism underlying dendritic supralinarities. As these dendrites depolarize, several voltage-gated channels can be recruited, including multiple sodium (NaV) and calcium (CaV) channel proteoforms [40–42]. In addition, other channels can be inactivated, including potassium (KV) and hyperpolarization-activated cyclic nucleotide-gated (HCN) channels [43–45]. Thus, any disruption in dendritically localized channels or receptors that contribute to dendritic excitability—whether localized to the synapse itself or instead in the dendritic shaft—could interfere with pyramidal cell processing and synaptic plasticity associated with dendritic supralinearities [46,47].
Dendritic supralinearities are increasingly being recognized as critical for active perception and decision making in vivo. Using calcium imaging approaches and direct electrophysiological recording from dendrites, the apical dendritic tuft of both layer 2/3 and layer 5 pyramidal cells has been shown to be engaged during sensory perception [48–50]. These dendritic spikes are often associated with a barrage of APs that, in turn, engages thalamocortical loops thought to be critical for neocortical processing [28,51]. Causal manipulations that block the generation of dendritic spikes, often by engaging inhibitory networks that preferentially target the apical tuft, interfere with perception[52]. Furthermore, these dendritic tuft spikes appear to be critical for conscious perception, as they are some of the first subcellular events to be disrupted by anesthetics [53]. Together, these data identify a major role for pyramidal cell dendrites in higher-order neocortical function. Furthermore, they highlight the complexity and convergence of multiple processes that regulate dendritic excitability, which includes intrinsic mechanisms within the dendrite, excitatory and inhibitory synaptic input to the apical tuft, and, discussed further below, neuromodulatory pathways that alter signal processing in such regions.
Disruption in how dendrites process sensory input may fit into a mechanistic theory of ASD that proposes an altered balance in “top-down bottom-up” processing, with a bias towards bottom-up processing in ASD/ID. In terms of cognitive processes, this can be thought of as an inability to attend to specific stimuli (in the case of sensory processing), or specific streams of thought (in the case of associative, or internal state processing), due to a loss of “top-down” attentional control. Rather, “bottom-up” inputs are processed without filters, potentially resulting in sensory overload, thereby resulting in issues in identifying brain states that are relevant compared to those that instead should be ignored [54,55]. Given the prominent role of neocortical pyramidal cells in coupling associative, top-down information with local, bottom-up signals, instantiated through interactions between apical and basal arbors, it is critical to consider whether such processes are disrupted in NDDs.
Impaired dendritic excitability and synaptic integration in NDDs
Recent work to understand the brain regions and cell-types important for ASD has found that multiple genes converge onto pyramidal cells, with deep-layer pyramidal cells in prefrontal cortex of particular importance [56]. As discussed in detail above, dendrites of deep-layer pyramidal neurons play a central role in facilitating information flow throughout the neocortex by functioning as coincidence detectors to integrate synaptic inputs from both local and long-range projections. Importantly, abnormal dendritic processing and synaptic integration within these neurons has been suggested to contribute to the social, cognitive, and communication deficits typically characteristic of NDDs [57].
The largest gene discovery effort to date identified approximately 102 genes highly associated with ASD [58]. Emerging from these large-scale genetic studies are “channelopathies” or dysfunction of various ion channels as causative factors in ASD pathogenesis [59–66]. A substantial number of these genes are of the NaV, CaV, and potassium channel families, as well as HCN channels [66]. Interestingly, many of these channels are more commonly associated with dendritic function, rather than the synapse proper, and are abundantly expressed in neocortical pyramidal cell dendrites [67–71] (Fig. 2A). As such, pathogenic variants in genes that encode ion channels would likely have direct detrimental effects on dendritic excitability. Consistent with this, both rare variants and common polymorphisms are found within channelopathy-associated genes, some of the most prevalent include SCN2A, SCN1A, KCNQ3, and CACNA1E [66,72–78]. In addition to overt effects on channel-related genes, a number of high-confidence ASD genes encode proteins found directly within synapses that regulate synapse structure, function, and connectivity, including SHANK3, SYNGAP1, NLGN4 and GRIN2B [66]. Given their prominent role in supporting synaptic activity, dysfunction in these genes could further impair dendritic excitability. Lastly, a number of ASD-gene products that include chromatin modifiers, transcription factors, translation regulators, scaffolding proteins, and signaling molecules may also affect the expression, localization, and/or trafficking of ion channels, receptors, and other synaptic proteins [66]. Thus, dysfunction in these genes may indirectly disrupt dendritic function and coincidence detection. Below, we discuss how multiple high-confidence ASD genes are associated with altered dendritic structure, function, and integration in neocortical pyramidal neurons and highlight impaired dendritic excitability and coincidence detection as a central hub for ASD gene convergence.
Figure 2: Multiple high-confidence ASD/ID genes converge on neocortical excitatory synapses.

A) Several neurodevelopmental disorder (NDD)-associated genes encode for ion channels that are localized throughout the dendrites of neocortical pyramidal neurons.
B) Similarly, NDD-associated genes encode for a range of synaptic proteins essential for synaptic transmission that contribute to dendritic supralinearities.
C) “Covert” NDD-genes include the chromatin modifiers CHD2 and CHD8 and the transcription factor TBR1. They regulate the expression levels of many “overt” NDD-genes as well as additional “covert genes”, which play critical roles in synaptic integration and dendritic processing.
D) FMRP is another “covert” regulator that suppresses the translation of multiple synaptic and dendritic proteins associated with NDDs. These include various ion channels (i.e. NaV1.2 and multiple calcium channels), SHANK scaffolding proteins, SynGAP, neuroligins, and GluRN2B.
Sodium channels
In several different neuron classes, the generation and propagation of dendritic potentials is dependent on voltage-gated sodium channels [79–85]. While the importance of sodium channels in dendritic excitability has been known for some time, only recently have we begun to understand the cellular and subcellular localization patterns of different sodium channel subtypes and their distinct roles in neuronal excitability. The predominant voltage-gated sodium channel alpha subunits expressed in the adult mammalian neocortex are SCN1A, SCN2A, and SCN8A, which encode NaV1.1, NaV1.2, and NaV1.6, respectively [86,87]. Each of these genes is associated with neurodevelopmental disorders, including both ASD/ID and various forms of epilepsy. Reviews of alterations in sodium channel structure and function have highlighted how NDD-associated variants affect both biophysical properties as well as channel trafficking and modulation [88–95]. SCN3A, which encodes NaV1.3, is expressed transiently during early development, and dysfunction in this gene affects cortical folding and is associated with epileptic encephalopathy [96,97]. Whether this in turn affects dendritic processing in more mature circuits has yet to be investigated.
SCN2A has repeatedly emerged from large-scale exome-sequencing studies with some of the strongest association scores to ASD of any gene in the genome [66,98–100]. SCN2A encodes NaV1.2, which exhibits unique cell-type and domain-specific expression patterns throughout development. In the neocortex, NaV1.2 is expressed in glutamatergic pyramidal neurons along with NaV1.6, whereas NaV1.1 is the predominant sodium channel expressed in inhibitory interneurons [101–106]. Early in neocortical development, prior to 1–2 years of age in humans and postnatal day 7 in mice, NaV1.2 is the only identifiable sodium channel isoform localized to the AIS [107–109]. In mature pyramidal neurons, NaV1.2 is replaced by NaV1.6 in the distal AIS and the nodes of Ranvier [110,111]. NaV1.6 has a lower voltage threshold for activation, making these sites more susceptible to spike generation. Later in development, NaV1.2 appears to play a more dominant role in dendritic, rather than axonal, excitability throughout neocortex [112]. In hippocampus, freeze-fracture immunogold labeling has revealed that NaVs are localized exclusively to dendritic shafts, not spines [113]. This may also be the case in neocortex. Human variants in SCN2A are broadly categorized into different classes of neurodevelopmental disorders associated with increased or decreased channel function. Gain-of-function variants in SCN2A that enhance NaV1.2 activity are associated with benign infantile familial seizures (BIFS) and epileptic encephalopathy (EE) [114–116]. By contrast, loss-of-function (LoF) variants in SCN2A are found in individuals with ASD and ID and consist of either missense variants that dampen NaV1.2 function or protein truncating variants (PTVs) that result in haploinsufficiency [90,93,117]. In neocortical layer 5 pyramidal neurons, heterozygous LoF in Scn2a was shown to severely attenuate dendritic calcium transients evoked by backpropagating Aps (bAP) [112]. This deficit was associated with a range of synaptic deficits: excitatory synapses had features more commonly observed in less mature cells, including an excess of silent synapses, and bAP-mediated synaptic plasticity was impaired [112]. Furthermore, other aspects of dendritic excitability, including the generation of local dendritic spikes, may be affected by Scn2a LoF. In hippocampus, for example, synaptic plasticity can be modulated by the generation of sodium-mediated dendritic spikes [118]. While these effects are likely mediated by NaV1.6 in hippocampal neurons, NaV1.2 may be similarly critical in neocortical dendrites [119].
Loss-of-function of SCN8A, in contrast to SCN2A, is not associated with ASD but instead with seizure-free ID [120,121]. This may have to do with the different roles of these two sodium channels. In SCN2A LoF cases, dendritic excitability is impaired, but axonal excitability is intact [112]. With SCN8A LoF, axonal excitability is impaired, both within pyramidal cells and also in the majority of inputs to neocortex that also rely on NaV1.6 for axonal conduction [122]. Therefore, it is likely that SCN8A LoF results in profound synaptic impairments that ID diagnoses are most common.
Whether NaV1.6 loss also affects neocortical dendritic processing is less clear. Current compartmental models of Scn2a haploinsufficiency best recapitulate empirical data when both NaV1.2 and NaV1.6 are expressed in equal densities in the somatodendritic domain [112]. By extension, SCN8A heterozygosity should have similar effects as SCN2A heterozygosity. While heterozygous conditions have not been examined, conditional knockout of Scn8a in neocortex has been studied [123]. In these conditions, AP-evoked dendritic calcium transients were unaffected by NaV1.6 deletion; however, transients were imaged in dendritic regions relatively proximal to the soma, a region where NaV1.2 heterozygosity also had little-to-no effect [112]. Whether more distal compartments have altered electrogenesis remains unknown. Furthermore, compensation for NaV1.6 by NaV1.2 was evident in the AIS of these neurons. It is therefore possible that NaV1.6 loss was also compensated in dendrites [124].
NaV1.1 (SCN1A) is the predominant sodium channel expressed in GABAergic inhibitory interneurons [125]. SCN1A is well known for its causative link to Dravet’s Syndrome (DS) and genetic epilepsy with febrile seizures plus (GEFS+) [126–128]. Haploinsufficiency of Scn1a in mice dampens interneuron excitability, resulting in disinhibition of the cortical network and in turn, seizures, premature death, and cognitive deficits as observed in DS patients [125,129–134]. SCN1A is also associated with ASD through exome-sequencing and familial studies [66,135,136]; however, the mechanisms by which dysfunction of NaV1.1 contributes to ASD are not as well understood. Interestingly, recent work has shown that excitability deficits in parvalbumin-positive interneurons resolve by postnatal day 50 in Scn1a+/− mice, whereas deficits in vasoactive intestinal polypeptide (VIP)-expressing interneurons persist [137]. These VIP neurons form disynaptic disinhibitory circuits with pyramidal cell apical tuft dendrites through somatostatin interneurons [138]. As such, regulation of dendritic excitability may be indirectly affected in Scn1a+/− conditions. Such hypotheses are explored in detail in a companion manuscript in this issue [139].
Calcium channels
CaVs play fundamental roles in dendritic excitability and synaptic integration. Activation of CaVs promotes regenerative depolarizations which generate both linear and nonlinear dendritic spikes [140,141]. AP backpropagation into the apical dendrites of neocortical pyramidal neurons also engages CaVs, which then modulate other ion channels and receptors, stimulate various signaling cascades, and regulate gene expression [142–144]. These active dendritic processes increase the probability of AP firing, mediate dendritic neurotransmitter release, and regulate synaptic plasticity, such as spike timing-dependent plasticity (STDP) and long-term potentiation (LTP) [145,146]. Therefore, dendritic CaVs have profound effects on synaptic integration and coincidence detection in neocortical pyramidal neurons, and dysfunction of CaVs due to genetic variation would have severe effects on dendritic excitability and neocortical processing [147].
VGCCs are broadly classified into two groups based on electrophysiological properties: high voltage-activated (HVA) channels consist of L-, N-, P-/Q- and R-type calcium channels and low voltage-activated (LVA) channels include T-type channels [148]. The L-type calcium channels are encoded by CACNA1S, CACNA1C, CACNA1D, and CACNA1F and include Cav1.1, Cav1.2, Cav1.3, and Cav1.4, respectively. P/Q-, N-, and R-type calcium channels correspond to Cav2.1, Cav2.2, and Cav2.3 and are products of CACNA1A, CACNA1B, and CACNA1E [148]. Lastly, T-type channels include Cav3.1, Cav3.2, and Cav3.3 encoded by CACNA1G, CACNA1H, and CACNA1I [148]. Electrophysiological and immunohistochemical studies have shown that all calcium channel subtypes are present within dendrites [149,150]. However, the distribution patterns of each subtype is heterogeneous across brain regions, cell-types, and even different neuronal domains [151,152]. CaV1 channels are found on the soma, proximal dendritic shafts, and within spines of hippocampal and deep-layer neocortical neurons [153,154]. These channels are known to play an important role in triggering intracellular cascades related to synaptic plasticity [155–159]. In parallel, neocortical dendritic excitability can also be supported by CaV2.x and CaV3.x channel isoforms [160–162]. CaV2.2 isoforms, in particular, appear highly expressed in layer 5 pyramidal dendrites [163]. Their density is highest in proximal apical dendrites, gradually decreasing into the distal dendritic arbors.
Dysfunction of multiple CaVs may contribute to ASD [164–166]; ASD gene variants are found in loci that encode for almost all CaV alpha subunits and their associated beta subunit partners [167]. Somewhat confusingly, both gain- and loss-of-function are associated with ASD. Gain-of-function (GoF) variants were found in genes that encode for CaV1 channels including CACNA1C, CACNA1D, and CACNA1F, resulting in excess calcium influx due to impaired voltage-dependent inactivation [168–171]. By contrast, loss-of-function variants were identified in CACNA1A (CaV2.1) and CACNA1H (CaV3.2), both of which reduce voltage-dependent activation and channel conductance [172,173]. Thus, it is likely that dendritic calcium electrogenesis is altered in many of these cases. Moving forward, a central question will be to determine how neocortical dendritic impairments contribute to ASD pathophysiology in particular cases, or if CaV function in other brain regions or neuronal compartments is more important. For example, CaV2.1 channels, which are common to dendrites, are also critical for neurotransmitter release at presynaptic terminals across the brain [174–179]. Thus, dysfunction in these channels may have an indirect effect on dendritic function, altering synaptic input to dendritic regions, but also may have profound effects on axonal integration and short-term plasticity in presynaptic boutons [180–183].
HCN Channels
Hyperpolarization-activated cyclic nucleotide-gated (HCN) channels are expressed broadly in neocortical neurons. In general, HCN channels are expressed at higher density in thick-tufted pyramidal cells of the pyramidal tract rather than thin-tufted pyramidal cells that project within the telencephalon [184]. Within thick-tufted cells, HCN channels are expressed at increased density in more distal parts of the dendrite, including the apical dendritic tuft [185]. These nonselective cation channels have relatively unique voltage dependence and kinetics: they are open at hyperpolarized potentials and close slowly with depolarization. This can create conditions in which the channels contribute to resonant oscillations in membrane potential [186]. Moreover, since they are gated by cyclic nucleotides, HCN current amplitude can be regulated by second messengers. Of note, HCN channels are tightly coupled to and regulated by group 1 metabotropic glutamate receptors (mGluRs) present in the primary apical dendrite of deep-layer pyramidal cells, and depolarizations mediated by group 1 mGluRs in apical dendritic shafts are driven in part by reduced HCN function [187,188]. Thus, these channels can play a major role in regulating coupling between apical and basal compartments in neocortical dendrites.
Four HCN isoforms are expressed in the brain [189]. In neocortex, HCN1 is most heavily expressed in dendrites. HCN2 is also expressed in distal apical dendrites, but HCN3 and HCN4 are not found in neocortex [190]. Despite expression of both HCN1 and HCN2, to date, only variants in HCN1 have been shown to associate with NDDs, including epileptic encephalopathy, ASD, and ID [191]. Similar to SCN2A, variant genotype-phenotype correlations are emerging, with GoF variants more commonly associated with epilepsy and LoF more commonly associated with ASD/ID [192]. Consistent with these direct effects on neocortical HCN channels, similar reductions in HCN expression are observed with other ASD-associated genes, including mouse models of Fmr1 and Shank3 [193–196].
Potassium Channels
Potassium channels are ubiquitously expressed in pyramidal cell dendrites, helping to set resting membrane potential and the time-course of dendritic electrogenesis [197–201]. There are conflicting reports regarding potassium channel density in the apical dendrite relative to the soma, with some studies reporting low ratios [202], whereas others report high ratios [203], possibly due to differences in experimental approach. Regardless, both transient (IA) and sustained (IKD) potassium currents are observed throughout the dendrite. The precise isoforms that generate such currents have been difficult to dissect, in large part due to significant diversity in the channel isoforms that can produce similar currents. For example, IA can be produced by homotetramers of KV1.4, KV3.4, KV4.1, KV4.2, or KV4.3, or by heteromeric channels containing a mix of these subunits [204].
While IA and IKD-related genes have not been shown to be associated with ASD/ID, two other potassium channel genes have been identified: KCNQ3 and KCNMA1[66]. KCNQ3 is a member of the KV7 family of potassium channels that produces a slow outward current, with expression best characterized in axonal domains [205,206] and therefore likely falling outside the framework proposed here. KCNMA1 encodes a calcium-activated potassium current (BK) found throughout pyramidal cell arbors, including the apical tuft, where it regulates the duration of dendritic spikes [207]. Interestingly, in contrast to many other genes, ASD association stems exclusively from missense variants for both KCNQ3 and KCNMA1, with no evidence that protein truncation contributes to such conditions. This contrasts markedly with cases of epileptic encephalopathy where LoF is common, at least for KCNQ3 [208,209]. Such conditions mirror observations for the sodium channel SCN2A, where GoF and LoF largely associate with epilepsy and ASD/ID, respectively. Of note, current gene discovery methods in NDDs rely heavily on identifying protein truncations, as it is clear that such truncations impair protein function. Missense variants, by contrast, may have little to no effect on channel function. Thus, without proper electrophysiological validation, it is difficult to have confidence that such variants contribute to disease. One hint is recurrence. Missense variants in the same location, identified in patients with similar conditions, can increase confidence. This is the case for KCNQ3 [210], but fewer recurrent variants have been identified to date in other potassium channel genes. Given the opposing roles of NaV and KV channels in dendritic excitability, one could envision that ASD-associated variants would result in GoF conditions in dendritically localized KVs (i.e., missense variants). Potential discovery of such cases will require far larger genetic cohorts than currently available.
Glutamate Receptors
The overwhelming majority of synaptic inputs onto neocortical layer 5 pyramidal neurons occur on distal dendritic tufts, basal dendrites, and oblique dendrites of the main apical shaft, making these domains key sites for synaptic integration [211–215]. Within these regions, the synchronous activation of multiple glutamatergic synapses produces a local “NMDA spike”, which is a summation of regenerative events that require AMPA and NMDA-receptor activation [216,217]. NMDA spikes are high amplitude, long duration events (several hundred milliseconds) capable of depolarizing the local dendritic domain for sustained periods of time and are even able to drive somatic depolarization [218]. NMDA spikes regulate synaptic integration by modulating the dendritic environment to promote active synaptic input, spatiotemporal information processing, and to regulate long-term potentiation and depression—processes hypothesized to be impaired in ASD/ID [219–221]. In addition, excitable dendritic spines on layer 5 pyramidal neurons are necessary for regulating AP backpropagation, and changes in spine function and morphology significantly influence AP backpropagation efficacy [222] Glutamatergic synapses and dendritic spines are major contributors to synaptic integration and dendritic excitability of layer 5 pyramidal neurons. Consistent with this, impairments in glutamatergic synapses are strongly implicated in the pathophysiology of ASD and ID [223–225].
NMDA receptors are ionotropic glutamate receptors critical for fast excitatory neurotransmission and NMDA spike initiation [226]. GRIN2B, which encodes the GluN2B subunit of NMDA receptors, is one of the most strongly associated genes to ASD and hundreds of variants in GRIN2B are found in individuals with NDDs including both ASD and ID [66,99,227–229]. Large cohort studies identified numerous LoF variants in GRIN2B as causative for ASD and ID [230–232]. NMDA receptor subunits undergo highly regulated spatiotemporal expression patterns, magnifying the detrimental effects LoF variants in GRIN2B would have on normal neurodevelopment. The GluN2B subunit is highly expressed throughout the brain during prenatal development and gradually decreases during postnatal development, becoming predominantly expressed in forebrain cortical neurons where it plays an essential role in dendritic function [233]. Therefore, LoF ASD variants in GRIN2B would be expected to severely disrupt layer 5 pyramidal neuron development and synaptogenesis. Consistent with this, multiple LoF variants in GRIN2B cause significant reductions in glutamatergic transmission, with decreased excitatory postsynaptic current and impaired excitatory neuron maturation [234–237]. While human variants in GRIN2B severely disrupt glutamatergic synapse function, the effects of GRIN2B loss on NMDA spike initiation and dendritic excitability have, to our knowledge, not been investigated in neocortex. However, one could easily foresee severe disruptions in dendritic excitability that may contribute to associated disorders in activity-dependent neuronal development.
In addition to ionotropic glutamate receptors, changes in the function of group 1 metabotropic glutamate receptors have long been associated with ASD/ID [238,239]. The bulk of studies focused on their role in synaptic transmission and regulation of plasticity [240,241]. This in and of itself will have effects on dendritic integration, but there may also be more direct ways in which mGluRs can regulate neocortical dendritic excitability. These metabotropic receptors are expressed at high density in the apical dendritic shaft connecting the soma with the apical tuft. In sensory cortices, such mGluRs receive input from thalamus and result in membrane depolarization that improves cooperativity between apical and basal arbors during sensory discrimination [242,243]. This thalamic activation of mGluRs is hypothesized to act as a gate for sensory responses [244]. Thus, loss of mGluR function commonly observed in ASD/ID models may interfere with this process.
Dendritic scaffolding and maintenance proteins
The recruitment and localization of ionotropic and metabotropic glutamate receptors is essential for normal synaptic integration. Several proteins involved in AMPA and NMDA receptor (as well as ion channel) localization, spine maintenance, synaptic connectivity, and plasticity are associated with ASD and ID, including NLGN1, NLGN3, CNTNAP2, SHANK3, and SYNGAP1 (Fig. 2B) [66,245–248]. Thus, impairments in synaptic function and dendritic integration are not limited to direct deficits of dendritic receptors and ion channels discussed above, but can also result from dysfunction in scaffolding and maintenance proteins that tther those channels in place. As seen below, the majority of work on these proteins has focused on essential synapse function, and to date, “downstream” effects on dendritic excitability have yet to be tested. This is an enticing area of investigation in the future, potentially tying scaffold function in the synapse to broader dendritic deficits observed with other NDD-associated genes.
SHANKs are a family of scaffolding proteins highly enriched within the postsynaptic density (PSD) of dendritic spines, with significant associations to syndromic and idiopathic ASD as well as ID [66,249–253]. Here, they are regarded as major organizers of excitatory synapse structure and function and directly interact with numerous proteins within the PSD, including the GluR1 subunit of AMPA receptors, PSD-95-associated proteins, Homer, and other SHANK family members [253]. As seen with other cytoskeletal proteins important for spine formation and maintenance, genetic deletion of SHANK proteins reduces the levels of AMPA and NMDA receptors [254]. This, in turn, results in significant morphological and functional deficits as well as reductions in spine density in neocortical neurons of SHANK-deficient mice [255–257]. In addition, ASD variants in SHANK3 result in reduced mGluR group 1 receptor expression and disrupt mGluR-dependent synaptic plasticity [258].
ASD-associated variants are found within NLGN1, NLGN3, and NLGN4, which encode NLGN1, NLGN3, and NLGN4 members of the neuroligin family of cell adhesion molecules [259]. Neuroligins are localized within postsynaptic domains of dendritic spines and form transmembrane connections with presynaptic neurexin proteins that promote synapse development, function, and maintenance [260]. In addition, neuroligins interact with PSD-95, SHANKs, and AMPA and NMDA receptors within spines to regulate excitatory synaptic formation and transmission in hippocampal neurons [261,262]. Mouse models of Nlgn1 and Nlgn3 knockout as well as LoF missense variants display behavioral phenotypes characteristic of ASD, including social interaction deficits and repetitive behaviors [263–266], but these behaviors are likely attributed to multiple brain regions including neocortex, hippocampus, and striatum.
Genetic variants in CNTNAP2 are found in individuals with ASD [267–270]. Contactin-associated protein-like 2 (CASPR-2), product of the CNTNAP2 gene, is a member of the neurexin family. CASPR-2 is a presynaptic transmembrane protein known to control glutamatergic synapse connectivity and spine formation, dendritic arborization, and synaptic transmission [271]. Cntnap2 knockout mice demonstrate malformations of neocortical development with upper layer glutamatergic neurons mislocalized into layers 5 and 6, which would be expected to have severe effects on integration and neocortical processing [272]. Cntnap2 knockdown in mouse prefrontal cortical pyramidal neurons caused reduced excitatory synaptic transmission, abnormal network activity, and synaptic dysfunction, which resulted in behavioral phenotypes similar to those found in ASD [273,274]. Furthermore, Cntnap2 null mice have reduced numbers of parvalbumin-positive (PV+) interneurons and the remaining PV+ cells exhibited abnormal intrinsic physiological properties[275]. These findings highlight a key role for CNTNAP2 in regulating synaptic communication between both excitatory and inhibitory neurons throughout the neocortex.
SYNGAP1 is a significant risk gene for ID, ASD, and epileptic encephalopathy [66,232,276–278]. Heterozygous, de novo LoF variants in SYNGAP1 are causative in approximately 1% of individuals with ID and developmental delay, demonstrating its essential role in brain development and function [279,280]. SYNGAP1 encodes the synaptic Ras GTPase-activating protein (SynGAP), which is highly enriched in the postsynaptic density of mature neocortical and hippocampal pyramidal neurons [281]. SynGAP negatively activates the small GTP-ases Ras- and Rap-GAP to promote AMPA receptor trafficking, membrane incorporation, and synaptic long-term potentiation (LTP) and depression (LTD) [282,283]. In addition to its role in synaptic plasticity, SynGAP is fundamental for dendritic spine maturation. In neocortical neurons, SynGAP expression levels rise dramatically during postnatal development, around postnatal day 14 in mice [284]. Normal SynGAP function is critical during this developmental window as haploinsufficient Syngap1 mice display aberrant dendritic spine maturation; however, knockout of Syngap1 late in development had no significant effect on synaptic function [285,286]. Therefore, human variants that disrupt SynGAP function during critical periods of synaptogenesis would be expected to have permanent effects on excitatory integration and synaptic plasticity in developed neurons. However, a recent study by Creson et al. demonstrated that re-expression of SynGAP in adult Syngap1 heterozygous mice restored neuronal excitability deficits, memory impairments, and seizure thresholds (Creson Colgin 2019). These findings offer insight on the development of new treatment strategies that rescue expression or function of SynGAP to treat SYNGAP-associated NDDs in adult patients.
GABA Receptors
Gamma-aminobutyric acid (GABA) is the primary inhibitory neurotransmitter in the adult mammalian brain. Dysfunction of inhibitory microcircuits within the neocortex has been linked to ASD and other NDDs through a variety of mechanisms including abnormal development, migration, intrinsic properties, or connectivity [275,287,288]. Different subtypes of GABAergic interneurons synapse onto discrete domains of excitatory neurons, including layer 5 pyramidal neurons in the mPFC, where they play unique roles in modulating pyramidal cell excitability and plasticity. PV-positive interneurons target the somatodendritic domain and AIS of pyramidal neurons, whereas SOM cells primarily synapse onto the dendrites [289–291]. Martinotti SOM+ interneurons synapse onto distal apical dendrites of layer 5 pyramidal neurons, where they tightly control dendritic activity [292]. Specifically, they inhibit pyramidal cell firing during sustained periods of high activity and are also capable of effectively blocking the generation of dendritic spikes [293]. Lastly, vasoactive intestinal peptide (VIP) interneurons are found in layer 2/3 and preferentially target other neocortical interneurons including PV and SOM neurons in layer 5 [294–296]. Therefore, pathogenic variants in genes involved in GABAergic interneuron subtype function or in genes that encode GABA receptor subunits could potentially impair neocortical neuron dendritic processing. Several genes that encode GABA receptors have been implicated in neurodevelopmental disorders including ASD [297–300]. Whole-exome sequencing studies revealed GABRB2 and GABRB3, which code for the GABAA receptor beta-2 and beta-3 subunits, are among the top 102 high-confidence ASD genes [66]. Familial studies identified single nucleotide polymorphisms (SNPs) within genes that encode additional GABA receptor subunits including GABRA4 (alpha-4), GABRB1 (beta-1), and GABRB3 (beta-3) in patients with ASD [301,302]. In addition, a recent study found a 2.4 Mb duplication of 4p12 to 4p11 that consists of GABA receptor subunit gene clusters that include GABRA4, GABRA2, GABRB1, and GABRG1 in four siblings with ASD [303]. A similar duplication of this GABA receptor gene cluster has been reported in other individuals with ASD who present with a range of behavioral phenotypes [304–307]. Consistent with genetic studies, multiple GABA receptor subunits were found at significantly lower expression levels in postmortem brain of ASD individuals as compared to neurotypical controls [308,309]. While GABAergic interneurons and multiple GABA receptor subunits are involved in ASD pathophysiology, it will be important for future work to determine how GABAergic circuitry functions in tandem with glutamatergic receptors to control dendritic excitability and integration and how this is altered in ASD. In addition, pathogenic variants in several ASD-associated genes mentioned throughout this review, such as CNTNAP2, TBR1, and PTEN, result in both glutamatergic and GABergic deficits, which could further potentiate impairments in dendritic processing.
Taken together, these findings suggest that although the etiology of ASD is heterogenous, excitatory synapse dysfunction and impaired neocortical dendritic processing may be one core feature among some individuals with ASD and ID.
“Covert” mechanisms in ASD/ID that regulate dendritic impairments
Efforts to identify convergence between ASD genes have broadly separated high-confidence ASD genes into two main groups: synaptic function and transcriptional regulation. However, evidence suggests that these two groups intersect at multiple levels, perhaps leading to dysfunction of shared downstream processes [310–312]. In support of this, recent work by Jing et al. used Perturb-Seq, a novel high-throughput genetic screen that allows for single-cell resolution of the phenotypes caused by introducing an array of genetic perturbations, to demonstrate points of convergence between multiple seemingly diverse ASD/NDDs in unique cell-types and transcriptomic networks across the developing brain [313]. Here, we have discussed how dysfunction of multiple NDD-associated genes that encode for various subtypes of ion channels, receptors, and scaffolding proteins impair dendritic excitability and synaptic integration in neocortical pyramidal neurons. We term these particular genes “overt” genes, as loss or dysfunction of their protein products directly results in neocortical dendritic deficits. By contrast, an additional pool of NDD-associated genes that encode chromatin modifiers, transcriptional and translational regulators, and trafficking proteins may function in a more “covert” way to also affect dendritic integration, in part through their regulation of “overt” mechanisms discussed above. Below, we highlight emerging evidence of such interactions in select NDD-associated genes.
Fragile X syndrome is the single most common form of inherited ID and monogenic cause of ASD [314]. Fragile X syndrome is caused by the expansion of a CGG repeat in the 5’ UTR region of FMR1, which results in loss of FMR1 and, in turn, its protein product FMRP (Fragile X Mental Retardation Protein) (Fig. 2D) [315]. FMRP is an RNA binding protein that suppresses the translation of numerous mRNA targets including multiple Shanks, Scn2a, multiple calcium channel genes, Syngap1, Nlgns, and Grin2b that are essential for normal dendritic excitability [316,317]. FMRP shuttles mRNA from the nucleus throughout the cytoplasm, but it is also highly enriched within dendritic spines, where it colocalizes with many of its mRNA targets [315]. The loss of FMRP would therefore likely lead to aberrant expression of multiple proteins involved in dendritic morphology and integration. Consistent with this, neocortical neurons of individuals with Fragile X syndrome, including layer 5 neurons, have abnormally long, immature spines [318]. In addition, activation of mGluRs upregulates FMRP expression within spines. This demonstrates a functional link between mGluR stimulation and local translation in spines, a process necessary for excitatory synapse function, morphology, and plasticity [319,320]. The reciprocal relationship also exists as FMRP controls the translation of mGluR and loss of FMRP causes increased mGluR signaling, a process known as the mGluR hypothesis of fragile X syndrome [321]. Ultimately, the dendritic phenotypes observed in Fragile X syndrome patients and animal models of FMR1 loss overlap with animal models of haploinsufficiency of multiple FMRP mRNA targets (i.e. Scn2a+/− and Syngap1+/− mice), further highlighting impaired neocortical dendrites as a point of convergence between genes with the strongest association scores.
In addition to genes that regulate translation, damaging variants in genes that control transcription can also affect genes directly involved in dendritic excitability (Fig. 2C). The chromatin modifiers CHD2 and CHD8, each carrying strong ASD association, are both expressed in human deep-layer neocortical neurons, where they regulate the expression of several other genes involved in brain development [56,66,322]. Further, CHD8 and CHD2 targets include numerous ASD-linked genes, including SCN2A, GRIN2B, SHANK2, POGZ, TBR1, and ANK2 [323]. Therefore, haploinsufficiency of CHD2 and CHD8 could impair dendritic excitability and integration by affecting target genes involved in transcription, dendritic scaffolding, and excitability. Consistent with this, Chd8 knockdown in mice causes delayed neocortical neuron migration and reduced dendritic outgrowth [324].
TBR1 is another ASD-associated transcriptional regulator that controls the expression of several other genes involved in the etiology of ASD [325]. Tbr1 controls the transcription of Grin2b, Scn2a, and Ank2, genes involved in excitatory synaptic function, dendritic excitability, and protein localization in axonal structures, respectively [326]. TBR1, a T-box transcription factor, is regarded as a master regulator of cortical development as it plays a fundamental role in the differentiation and identity of deep-layer neocortical pyramidal neurons [327–329]. Tbr1 mutant mice have fewer excitatory and inhibitory synapses onto neocortical dendrites that are partially due to reduced WNT signaling [330,331]. Both excitatory and inhibitory synaptic deficits can be rescued with WNT agonists [332]. Interestingly, Tbr1 heterozygosity in layer 6 pyramidal neurons converts their dendritic arborization into layer 5-like apical dendrites, where ectopic growth of the apical dendrite extends into layer 1 instead of their typical termination with little arborization in layer 4 [333]. How these ectopic dendritic tufts impact the function of affected neocortical areas remains unclear. Interestingly, these conditions may serve as an excellent model in which to test the relative function of different deep-layer pyramidal cells, as this would create situations where layer 6 neurons may have typical basal inputs but vastly different apical inputs. One major question would be to test whether such neurons now adopt processing features more commonly associated with layer 5 cells, rather than those ascribed to layer 6 (e.g., gain control of cortical columns) [334].
PTEN encodes the protein PTEN, a phosphatase strongly associated with ASD and intellectual disability [335,336]. PTEN is highly expressed in both glutamatergic and GABAergic neurons where it acts as an inhibitor of PI3K/Akt signaling through activation of receptor tyrosine kinases (RTKs) [337–339]. Conditional knockout of Pten in neocortical glutamatergic pyramidal neurons results in aberrant dendritic and axonal arborization and somatic overgrowth, due to heightened β-catenin expression, causing macrocephaly and deficits in social behaviors [340–342]. Homozygous loss of Pten weakened excitatory synaptic transmission and plasticity in hippocampal neurons [342]. The effects of Pten loss on dendritic excitability in neocortical neurons remains unclear [342–344]. In addition, PTEN loss in mouse cortical GABAergic interneurons results in reduced somatostatin cell number shifting the ratio of PV/somatostatin interneurons, which, in turn, may lead to abnormal dendritic function and integration [345].
Here, we have discussed only a few “covert” genes that have been shown to directly regulate the expression or localization of “overt” genes in dendrites of neocortical pyramidal neurons. This is likely the tip of an iceberg that will be better revealed as deep genetic phenotyping of multiple ASD-associated models emerges in the coming years. Loss-of-function of these “covert” genes results in functional, morphological, and organizational deficits on neocortical neurons, particularly in layer 5. Of note, many of these transcriptional regulators modify expression levels of multiple “overt” genes at once, making predictions about the final effects on synaptic and dendritic function difficult without empirical tests.
CONCLUSION AND FUTURE DIRECTIONS
As mentioned at the outset, this review focused primarily on the role of dendritic integration in neocortical areas for several reasons. First, our understanding of neocortical dendritic function is relatively refined both in terms of cellular mechanisms and in vivo function, with clear models emerging for the role of dendritic integration in conscious perception and decision making. Second, neocortical pyramidal cells appear to be a point of convergence for several ASD/ID-associated genes acting both overtly and perhaps covertly to affect dendritic integration. As highlighted recently for the function of psychedelics [346], our hope is that future studies take dendritic excitability into consideration in parallel with key experiments examining molecular, cellular, systems, and behavioral consequences of ASD/ID-associated variation in such genes.
ASD and ID are brain-wide disorders, and while neocortex is thought to be important for ASD/ID etiology, it is not affected in isolation. A wealth of studies have highlighted the importance of other key circuits, including basal ganglia, amygdala, hippocampus, and cerebellum [87,347–365]. Importantly, many of the concepts discussed above, including the mechanisms supporting dendritic nonlinearities, are relevant across these structures. Thus, while we highlight neocortex here, these themes likely extend to other structures, each with their own rules governing how dendritic excitability shapes processing of relevant information.
Moving forward, several areas of investigation will be critical to better understand how dendrites are affected in ASD/ID. First, more complete genotype-phenotype correlations of genes known to regulate dendritic function will help to better identify the precise conditions in which aspects of dendritic integration are affected. For example, in both SCN2A and GRIN2B, a large number of missense variants can be associated with a range of disorders, likely due to differential effects in channel biophysics, ligand affinity, trafficking, etc. While a great deal can be done in heterologous expression systems, it can often be difficult to understand the end effects of such variation in non-native systems. Second, deep phenotyping of the neuronal proteome, perhaps focusing on “overt” dendritic excitability genes, in “covert” gene models may us to help better understand how chromatin modification and other forms of gene regulation affect the dendrite. Lastly, development of ASD/ID-relevant behavioral approaches that are amenable to simultaneous imaging, recording, and ideally, manipulation, of dendritic activity in ASD/ID relevant models will not only be key to understanding the role of dendrites in neurodevelopmental disorders, but also may shed light on components of dendritic function necessary for neurotypical processing.
Acknowledgements
We are grateful to members of the Bender Lab, including Anna Lipkin and Selin Schamiloglu, as well as Matthew McGregor, Dr. Guy Bouvier, Dr. Stephan Sanders, and Dr. John Rubenstein for discussions and comments on this review. This work was supported by the Simons Foundation Autism Research Initiative (513133, 629287) and the NIH (MH112729 and MH125978).
Footnotes
Conflict of Interest Statement
The authors have no conflicts of interest to declare.
BIBLIOGRAPHY
- 1.Maenner MJ, Shaw KA, Baio J, Washington A, Patrick M, DiRienzo M, et al. Prevalence of autism spectrum disorder among children aged 8 Years-Autism and developmental disabilities monitoring network, 11 Sites, United States, 2016. MMWR Surveillance Summaries. 2020; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Keller GB, Mrsic-Flogel TD. Predictive Processing: A Canonical Cortical Computation. Neuron. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sohal VS, Rubenstein JLR. Excitation-inhibition balance as a framework for investigating mechanisms in neuropsychiatric disorders. Molecular Psychiatry. 2019; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Polleux F, Lauder JM. Toward a developmental neurobiology of autism. Mental Retardation and Developmental Disabilities Research Reviews. 2004. [DOI] [PubMed] [Google Scholar]
- 5.Brambilla P, Hardan A, Ucelli Di Nemi S, Perez J, Soares JC, Barale F. Brain anatomy and development in autism: Review of structural MRI studies. Brain Research Bulletin. 2003. [DOI] [PubMed] [Google Scholar]
- 6.Cook J, Barbalat G, Blakemore SJ. Top-down modulation of the perception of other people in schizophrenia and autism. Frontiers in Human Neuroscience. 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nelson SB, Valakh V. Excitatory/Inhibitory Balance and Circuit Homeostasis in Autism Spectrum Disorders. Neuron. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rubenstein JLR, Merzenich MM. Model of autism: Increased ratio of excitation/inhibition in key neural systems. Genes, Brain and Behavior. 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Markram K, Markram H. The intense world theory - A unifying theory of the neurobiology of autism. Frontiers in Human Neuroscience. 2010; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hill J, Inder T, Neil J, Dierker D, Harwell J, Van Essen D. Similar patterns of cortical expansion during human development and evolution. Proceedings of the National Academy of Sciences of the United States of America. 2010; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aru J, Suzuki M, Larkum ME. Cellular Mechanisms of Conscious Processing. Trends in Cognitive Sciences. 2020. [DOI] [PubMed] [Google Scholar]
- 12.Gulsuner S, Walsh T, Watts AC, Lee MK, Thornton AM, Casadei S, et al. XSpatial and temporal mapping of de novo mutations in schizophrenia to a fetal prefrontal cortical network. Cell. 2013; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li M, Santpere G, Kawasawa YI, Evgrafov OV., Gulden FO, Pochareddy S, et al. Integrative functional genomic analysis of human brain development and neuropsychiatric risks. Science. 2018; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parikshak NN, Luo R, Zhang A, Won H, Lowe JK, Chandran V, et al. XIntegrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell. 2013; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Werling DM, Pochareddy S, Choi J, An JY, Sheppard B, Peng M, et al. Whole-Genome and RNA Sequencing Reveal Variation and Transcriptomic Coordination in the Developing Human Prefrontal Cortex. Cell Reports. 2020; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Willsey AJ, Sanders SJ, Li M, Dong S, Tebbenkamp AT, Muhle RA, et al. XCoexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell. 2013; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mei Y, Monteiro P, Zhou Y, Kim JA, Gao X, Fu Z, et al. Adult restoration of Shank3 expression rescues selective autistic-like phenotypes. Nature. 2016; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Benthall KN, Ong SL, Bateup HS. Corticostriatal Transmission Is Selectively Enhanced in Striatonigral Neurons with Postnatal Loss of Tsc1. Cell Reports. 2018; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rothwell PE, Fuccillo MV., Maxeiner S, Hayton SJ, Gokce O, Lim BK, et al. Autism-associated neuroligin-3 mutations commonly impair striatal circuits to boost repetitive behaviors. Cell. 2014; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peixoto RT, Wang W, Croney DM, Kozorovitskiy Y, Sabatini BL. Early hyperactivity and precocious maturation of corticostriatal circuits in Shank3B â ‘/â ‘ mice. Nature Neuroscience. 2016; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Langen M, Schnack HG, Nederveen H, Bos D, Lahuis BE, de Jonge MV., et al. Changes in the Developmental Trajectories of Striatum in Autism. Biological Psychiatry. 2009; [DOI] [PubMed] [Google Scholar]
- 22.Schuetze M, Park MTM, Cho IYK, Macmaster FP, Chakravarty MM, Bray SL. Morphological alterations in the thalamus, striatum, and pallidum in autism spectrum disorder. Neuropsychopharmacology. 2016; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hampson DR, Blatt GJ. Autism spectrum disorders and neuropathology of the cerebellum. Frontiers in Neuroscience. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang SSH, Kloth AD, Badura A. The Cerebellum, Sensitive Periods, and Autism. Neuron. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Manoli DS, State MW. Autism Spectrum Disorder Genetics and the Search for Pathological Mechanisms. American Journal of Psychiatry. 2021; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hooks BM, Mao T, Gutnisky DA, Yamawaki N, Svoboda K, Shepherd GMG. Organization of cortical and thalamic input to pyramidal neurons in mouse motor cortex. Journal of Neuroscience. 2013; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gökçe O, Bonhoeffer T, Scheuss V. Clusters of synaptic inputs on dendrites of layer 5 pyramidal cells in mouse visual cortex. eLife. 2016; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aru J, Suzuki M, Larkum ME. Cellular Mechanisms of Conscious Processing. Trends in Cognitive Sciences. 2020. [DOI] [PubMed] [Google Scholar]
- 29.Rubio-Garrido P, Pérez-De-Manzo F, Porrero C, Galazo MJ, Clascá F. Thalamic input to distal apical dendrites in neocortical layer 1 is massive and highly convergent. Cerebral Cortex. 2009; [DOI] [PubMed] [Google Scholar]
- 30.Anastasiades PG, Collins DP, Carter AG. Mediodorsal and ventromedial thalamus engage distinct L1 circuits in the prefrontal cortex. bioRxiv. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Genescu I, Garel S. Being superficial: a developmental viewpoint on cortical layer 1 wiring. Current Opinion in Neurobiology. 2021. [DOI] [PubMed] [Google Scholar]
- 32.Larkum ME, Zhu JJ, Sakmann B. A new cellular mechanism for coupling inputs arriving at different cortical layers. Nature. 1999; [DOI] [PubMed] [Google Scholar]
- 33.Major G, Larkum ME, Schiller J. Active properties of neocortical pyramidal neuron dendrites. Annual Review of Neuroscience. 2013. [DOI] [PubMed] [Google Scholar]
- 34.Larkum ME, Nevian T, Sandier M, Polsky A, Schiller J. Synaptic integration in tuft dendrites of layer 5 pyramidal neurons: A new unifying principle. Science. 2009; [DOI] [PubMed] [Google Scholar]
- 35.Stuart GJ, Spruston N. Dendritic integration: 60 years of progress. Nature Neuroscience. 2015. [DOI] [PubMed] [Google Scholar]
- 36.Branco T, Häusser M. The single dendritic branch as a fundamental functional unit in the nervous system. Current Opinion in Neurobiology. 2010. [DOI] [PubMed] [Google Scholar]
- 37.Branco T, Clark BA, Häusser M. Dendritic discrimination of temporal input sequences in cortical neurons. Science. 2010; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gidon A, Zolnik TA, Fidzinski P, Bolduan F, Papoutsi A, Poirazi P, et al. Dendritic action potentials and computation in human layer 2/3 cortical neurons. Science. 2020; [DOI] [PubMed] [Google Scholar]
- 39.Antic SD, Zhou WL, Moore AR, Short SM, Ikonomu KD. The decade of the dendritic NMDA spike. Journal of Neuroscience Research. 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Spratt PWE, Ben-Shalom R, Keeshen CM, Burke KJ, Clarkson RL, Sanders SJ, et al. The Autism-Associated Gene Scn2a Contributes to Dendritic Excitability and Synaptic Function in the Prefrontal Cortex. Neuron. 2019; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Golding NL, Spruston N. Dendritic sodium spikes are variable triggers of axonal action potentials in hippocampal CA1 pyramidal neurons. Neuron. 1998; [DOI] [PubMed] [Google Scholar]
- 42.Williams SR, Stuart GJ. Mechanisms and consequences of action potential burst firing in rat neocortical pyramidal neurons. Journal of Physiology. 1999; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Berger T, Senn W, Lüscher HR. Hyperpolarization-activated current Ih disconnects somatic and dendritic spike initiation zones in layer V pyramidal neurons. Journal of Neurophysiology. 2003; [DOI] [PubMed] [Google Scholar]
- 44.Harnett MT, Magee JC, Williams SR. Distribution and function of HCN channels in the apical dendritic tuft of neocortical pyramidal neurons. Journal of Neuroscience. 2015; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Harnett MT, Xu NL, Magee JC, Williams SR. Potassium channels control the interaction between active dendritic integration compartments in layer 5 cortical pyramidal neurons. Neuron. 2013; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sandler M, Shulman Y, Schiller J. A Novel Form of Local Plasticity in Tuft Dendrites of Neocortical Somatosensory Layer 5 Pyramidal Neurons. Neuron. 2016; [DOI] [PubMed] [Google Scholar]
- 47.Frick A, Magee J, Johnston D. LTP is accompanied by an enhanced local excitability of pyramidal neuron dendrites. Nature Neuroscience. 2004; [DOI] [PubMed] [Google Scholar]
- 48.Smith SL, Smith IT, Branco T, Häusser M. Dendritic spikes enhance stimulus selectivity in cortical neurons in vivo. Nature. 2013; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xu NL, Harnett MT, Williams SR, Huber D, O’Connor DH, Svoboda K, et al. Nonlinear dendritic integration of sensory and motor input during an active sensing task. Nature. 2012; [DOI] [PubMed] [Google Scholar]
- 50.Takahashi N, Oertner TG, Hegemann P, Larkum ME. Active cortical dendrites modulate perception. Science. 2016; [DOI] [PubMed] [Google Scholar]
- 51.Takahashi N, Ebner C, Sigl-Glöckner J, Moberg S, Nierwetberg S, Larkum ME. Active dendritic currents gate descending cortical outputs in perception. Nature Neuroscience. 2020; [DOI] [PubMed] [Google Scholar]
- 52.Takahashi N, Oertner TG, Hegemann P, Larkum ME. Active cortical dendrites modulate perception. Science. 2016; [DOI] [PubMed] [Google Scholar]
- 53.Suzuki M, Larkum ME. General Anesthesia Decouples Cortical Pyramidal Neurons. Cell. 2020; [DOI] [PubMed] [Google Scholar]
- 54.Pellicano E Sensory symptoms in autism: A blooming, buzzing confusion? Child Development Perspectives. 2013; [Google Scholar]
- 55.Cook J, Barbalat G, Blakemore SJ. Top-down modulation of the perception of other people in schizophrenia and autism. Frontiers in Human Neuroscience. 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Willsey AJ, Sanders SJ, Li M, Dong S, Tebbenkamp AT, Muhle RA, et al. XCoexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell. 2013; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Johnston D, Frick A, Poolos NP. Dendrites and disease. In: Dendrites. 2016. [Google Scholar]
- 58.Satterstrom FK, Kosmicki JA, Wang J, Breen MS, De Rubeis S, An JY, et al. Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell. 2020; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brager DH, Johnston D. Channelopathies and dendritic dysfunction in fragile X syndrome. Brain Research Bulletin. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kullmann DM. Neurological channelopathies. Annual Review of Neuroscience. 2010. [DOI] [PubMed] [Google Scholar]
- 61.Heron SE, Scheffer IE, Berkovic SF, Dibbens LM, Mulley JC. Channelopathies in Idiopathic Epilepsy. Neurotherapeutics. 2007; [DOI] [PubMed] [Google Scholar]
- 62.Schmunk G, Gargus JJ. Channelopathy pathogenesis in autism spectrum disorders. Frontiers in Genetics. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Spillane J, Kullmann DM, Hanna MG. Genetic neurological channelopathies: Molecular genetics and clinical phenotypes. Journal of Neurology, Neurosurgery and Psychiatry. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Marcantoni A, Calorio C, Hidisoglu E, Chiantia G, Carbone E. Cav1.2 channelopathies causing autism: new hallmarks on Timothy syndrome. Vol. 472, Pflugers Archiv European Journal of Physiology. 2020. [DOI] [PubMed] [Google Scholar]
- 65.Kruth KA, Grisolano TM, Ahern CA, Williams AJ. SCN2A channelopathies in the autism spectrum of neuropsychiatric disorders: A role for pluripotent stem cells? Vol. 11, Molecular Autism. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Satterstrom FK, Kosmicki JA, Wang J, Breen MS, de Rubeis S, An JY, et al. Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell. 2020; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shah MM, Hammond RS, Hoffman DA. Dendritic ion channel trafficking and plasticity. Trends in Neurosciences. 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vacher H, Mohapatra DP, Trimmer JS. Localization and targeting of voltage-dependent ion channels in mammalian central neurons. Physiological Reviews. 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Remy S, Beck H, Yaari Y. Plasticity of voltage-gated ion channels in pyramidal cell dendrites. Current Opinion in Neurobiology. 2010. [DOI] [PubMed] [Google Scholar]
- 70.Hausser M, Spruston N, Stuart GJ. Diversity and dynamics of dendritic signaling. Science. 2000. [DOI] [PubMed] [Google Scholar]
- 71.Trimmer JS, Rhodes KJ. Localization of voltage-gated ion channels in mammalian brain. Annual Review of Physiology. 2004. [DOI] [PubMed] [Google Scholar]
- 72.De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. 2014; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Iossifov I, O’Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sanders SJ. Next-generation sequencing in autism spectrum disorder. Cold Spring Harbor Perspectives in Medicine. 2019; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sanders SJ, He X, Willsey AJ, Ercan-Sencicek AG, Samocha KE, Cicek AE, et al. Insights into Autism Spectrum Disorder Genomic Architecture and Biology from 71 Risk Loci. Neuron. 2015; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Alonso-Gonzalez A, Rodriguez-Fontenla C, Carracedo A. De novo mutations (DNMs) in autism spectrum disorder (ASD): Pathway and network analysis. Frontiers in Genetics. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Takata A, Miyake N, Tsurusaki Y, Fukai R, Miyatake S, Koshimizu E, et al. Integrative Analyses of De Novo Mutations Provide Deeper Biological Insights into Autism Spectrum Disorder. Cell Reports. 2018; [DOI] [PubMed] [Google Scholar]
- 79.Stuart GJ, Sakmann B. Active propagation of somatic action potentials into neocortical pyramidal cell dendrites. Nature. 1994; [DOI] [PubMed] [Google Scholar]
- 80.Häusser M, Stuart G, Racca C, Sakmann B. Axonal initiation and active dendritic propagation of action potentials in substantia nigra neurons. Neuron. 1995; [DOI] [PubMed] [Google Scholar]
- 81.Vetter P, Roth A, Häusser M. Propagation of action potentials in dendrites depends on dendritic morphology. Journal of Neurophysiology. 2001; [DOI] [PubMed] [Google Scholar]
- 82.Golding NL, Kath WL, Spruston N. Dichotomy of action-potential backpropagation in CA1 pyramidal neuron dendrites. Journal of Neurophysiology. 2001; [DOI] [PubMed] [Google Scholar]
- 83.Spruston N, Schiller Y, Stuart G, Sakmann B. Activity-dependent action potential invasion and calcium influx into hippocampal CA1 dendrites. Science. 1995; [DOI] [PubMed] [Google Scholar]
- 84.Stuart G, Schiller J, Sakmann B. Action potential initiation and propagation in rat neocortical pyramidal neurons. Journal of Physiology. 1997; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Golding NL, Spruston N. Dendritic sodium spikes are variable triggers of axonal action potentials in hippocampal CA1 pyramidal neurons. Neuron. 1998; [DOI] [PubMed] [Google Scholar]
- 86.Marban E, Yamagishi T, Tomaselli GF. Structure and function of voltage-gated sodium channels. Journal of Physiology. 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wang SSH, Kloth AD, Badura A. The Cerebellum, Sensitive Periods, and Autism. Neuron. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Blanchard MG, Willemsen MH, Walker JB, Dib-Hajj SD, Waxman SG, Jongmans MCJ, et al. De novo gain-of-function and loss-of-function mutations of SCN8A in patients with intellectual disabilities and epilepsy. Journal of Medical Genetics. 2015;52(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Liu Y, Schubert J, Sonnenberg L, Helbig KL, Hoei-Hansen CE, Koko M, et al. Neuronal mechanisms of mutations in SCN8A causing epilepsy or intellectual disability. Brain. 2019;142(2). [DOI] [PubMed] [Google Scholar]
- 90.Wolff M, Johannesen KM, Hedrich UBS, Masnada S, Rubboli G, Gardella E, et al. Genetic and phenotypic heterogeneity suggest therapeutic implications in SCN2A-related disorders. Brain. 2017;140(5). [DOI] [PubMed] [Google Scholar]
- 91.Fry AE, Rees E, Thompson R, Mantripragada K, Blake P, Jones G, et al. Pathogenic copy number variants and SCN1A mutations in patients with intellectual disability and childhood-onset epilepsy. BMC Medical Genetics. 2016;17(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pablo JL, Wang C, Presby MM, Pitt GS. Polarized localization of voltage-gated Na+ channels is regulated by concerted FGF13 and FGF14 action. Proceedings of the National Academy of Sciences of the United States of America. 2016;113(19). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Meisler MH, Hill SF, Yu W. Sodium channelopathies in neurodevelopmental disorders. Vol. 22, Nature Reviews Neuroscience. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Catterall WA, Lenaeus MJ, Gamal El-Din TM. Structure and pharmacology of voltage-gated sodium and calcium channels. Vol. 60, Annual Review of Pharmacology and Toxicology. 2020. [DOI] [PubMed] [Google Scholar]
- 95.Sanders SJ, Campbell AJ, Cottrell JR, Moller RS, Wagner FF, Auldridge AL, et al. Progress in Understanding and Treating SCN2A-Mediated Disorders. Vol. 41, Trends in Neurosciences. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Smith RS, Kenny CJ, Ganesh V, Jang A, Borges-Monroy R, Partlow JN, et al. Sodium Channel SCN3A (NaV1.3) Regulation of Human Cerebral Cortical Folding and Oral Motor Development. Neuron. 2018; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zaman T, Helbig I, Božović IB, DeBrosse SD, Bergqvist AC, Wallis K, et al. Mutations in SCN3A cause early infantile epileptic encephalopathy. Annals of Neurology. 2018; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Stessman HAF, Xiong B, Coe BP, Wang T, Hoekzema K, Fenckova M, et al. Targeted sequencing identifies 91 neurodevelopmental-disorder risk genes with autism and developmental-disability biases. Nature Genetics. 2017; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Iossifov I, O’Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sanders SJ. Next-generation sequencing in autism spectrum disorder. Cold Spring Harbor Perspectives in Medicine. 2019; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ogiwara I, Miyamoto H, Morita N, Atapour N, Mazaki E, Inoue I, et al. Nav1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: A circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. Journal of Neuroscience. 2007; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hu W, Tian C, Li T, Yang M, Hou H, Shu Y. Distinct contributions of Nav1.6 and Nav1.2 in action potential initiation and backpropagation. Nature Neuroscience. 2009; [DOI] [PubMed] [Google Scholar]
- 103.Tian C, Wang K, Ke W, Guo H, Shu Y. Molecular identity of axonal sodium channels in human cortical pyramidal cells. Frontiers in Cellular Neuroscience. 2014; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Bender KJ, Trussell LO. The physiogy of the axon initial segment. Annual Review of Neuroscience. 2012. [DOI] [PubMed] [Google Scholar]
- 105.Li T, Tian C, Scalmani P, Frassoni C, Mantegazza M, Wang Y, et al. Action Potential Initiation in Neocortical Inhibitory Interneurons. PLoS Biology. 2014; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Yamagata T, Ogiwara I, Mazaki E, Yanagawa Y, Yamakawa K. Nav1.2 is expressed in caudal ganglionic eminence-derived disinhibitory interneurons: Mutually exclusive distributions of Nav1.1 and Nav1.2. Biochemical and Biophysical Research Communications. 2017; [DOI] [PubMed] [Google Scholar]
- 107.Van Wart A, Trimmer JS, Matthews G. Polarized distribution of ion channels within microdomains of the axon initial segment. Journal of Comparative Neurology. 2007; [DOI] [PubMed] [Google Scholar]
- 108.Gazina EV, Leaw BTW, Richards KL, Wimmer VC, Kim TH, Aumann TD, et al. “Neonatal” Nav1.2 reduces neuronal excitability and affects seizure susceptibility and behaviour. Human Molecular Genetics. 2015; [DOI] [PubMed] [Google Scholar]
- 109.Boiko T, Van Wart A, CaldWell JH, Levinson SR, Trimmer JS, Matthews G. Functional specialization of the axon initial segment by isoform-specific sodium channel targeting. Journal of Neuroscience. 2003; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kole MHP, Stuart GJ. Signal Processing in the Axon Initial Segment. Neuron. 2012. [DOI] [PubMed] [Google Scholar]
- 111.Bender KJ, Trussell LO. The physiogy of the axon initial segment. Annual Review of Neuroscience. 2012. [DOI] [PubMed] [Google Scholar]
- 112.Spratt PWE, Ben-Shalom R, Keeshen CM, Burke KJ, Clarkson RL, Sanders SJ, et al. The Autism-Associated Gene Scn2a Contributes to Dendritic Excitability and Synaptic Function in the Prefrontal Cortex. Neuron. 2019; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lorincz A, Nusser Z. Molecular identity of dendritic voltage-gated sodium channels. Science. 2010; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Howell KB, McMahon JM, Carvill GL, Tambunan D, Mackay MT, Rodriguez-Casero V, et al. SCN2A encephalopathy: A major cause of epilepsy of infancy with migrating focal seizures. Neurology. 2015;85(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ben-Shalom R, Keeshen CM, Berrios KN, An JY, Sanders SJ, Bender KJ. Opposing Effects on NaV1.2 Function Underlie Differences Between SCN2A Variants Observed in Individuals With Autism Spectrum Disorder or Infantile Seizures. Biological Psychiatry. 2017;82(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Liao Y, Deprez L, Maljevic S, Pitsch J, Claes L, Hristova D, et al. Molecular correlates of age-dependent seizures in an inherited neonatal-infantile epilepsy. Brain. 2010;133(5). [DOI] [PubMed] [Google Scholar]
- 117.Crawford K, Xian J, Helbig KL, Galer PD, Parthasarathy S, Lewis-Smith D, et al. Computational analysis of 10,860 phenotypic annotations in individuals with SCN2A-related disorders. Genetics in Medicine. 2021; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kim S, Kim Y, Lee SH, Ho WK. Dendritic spikes in hippocampal granule cells are necessary for long-term potentiation at the perforant path synapse. eLife. 2018; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kim Y, Hsu CL, Cembrowski MS, Mensh BD, Spruston N. Dendritic sodium spikes are required for long-term potentiation at distal synapses on hippocampal pyramidal neurons. eLife. 2015; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wagnon JL, Barker BS, Ottolini M, Park Y, Volkheimer A, Valdez P, et al. Loss-of-function variants of SCN8A in intellectual disability without seizures. Neurology: Genetics. 2017; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Makinson CD, Tanaka BS, Lamar T, Goldin AL, Escayg A. Role of the hippocampus in Nav1.6 (Scn8a) mediated seizure resistance. Neurobiology of Disease. 2014; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Makinson CD, Tanaka BS, Lamar T, Goldin AL, Escayg A. Role of the hippocampus in Nav1.6 (Scn8a) mediated seizure resistance. Neurobiology of Disease. 2014; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Katz E, Stoler O, Scheller A, Khrapunsky Y, Goebbels S, Kirchhoff F, et al. Role of sodium channel subtype in action potential generation by neocortical pyramidal neurons. Proceedings of the National Academy of Sciences of the United States of America. 2018; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Katz E, Stoler O, Scheller A, Khrapunsky Y, Goebbels S, Kirchhoff F, et al. Role of sodium channel subtype in action potential generation by neocortical pyramidal neurons. Proceedings of the National Academy of Sciences of the United States of America. 2018; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ogiwara I, Miyamoto H, Morita N, Atapour N, Mazaki E, Inoue I, et al. Nav1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: A circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. Journal of Neuroscience. 2007; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Escayg A, Goldin AL. Sodium channel SCN1A and epilepsy: Mutations and mechanisms. Epilepsia. 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lossin C A catalog of SCN1A variants. Brain and Development. 2009. [DOI] [PubMed] [Google Scholar]
- 128.Yamakawa K Mutations of Voltage-Gated Sodium Channel Genes SCN1A and SCN2A in Epilepsy, Intellectual Disability, and Autism. In: Neuronal and Synaptic Dysfunction in Autism Spectrum Disorder and Intellectual Disability. 2016. [Google Scholar]
- 129.Yu FH, Mantegazza M, Westenbroek RE, Robbins CA, Kalume F, Burton KA, et al. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nature Neuroscience. 2006; [DOI] [PubMed] [Google Scholar]
- 130.Ogiwara I, Iwasato T, Miyamoto H, Iwata R, Yamagata T, Mazaki E, et al. Nav1.1 haploinsufficiency in excitatory neurons ameliorates seizure-associated sudden death in a mouse model of dravet syndrome. Human Molecular Genetics. 2013; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Cheah CS, Yu FH, Westenbroek RE, Kalume FK, Oakley JC, Potter GB, et al. Specific deletion of NaV1.1 sodium channels in inhibitory interneurons causes seizures and premature death in a mouse model of Dravet syndrome. Proceedings of the National Academy of Sciences of the United States of America. 2012; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kalume F, Westenbroek RE, Cheah CS, Yu FH, Oakley JC, Scheuer T, et al. Sudden unexpected death in a mouse model of Dravet syndrome. Journal of Clinical Investigation. 2013; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Rubinstein M, Han S, Tai C, Westenbroek RE, Hunker A, Scheuer T, et al. Dissecting the phenotypes of Dravet syndrome by gene deletion. Brain. 2015; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Tai C, Abe Y, Westenbroek RE, Scheuer T, Catterall WA. Impaired excitability of somatostatin- and parvalbumin-expressing cortical interneurons in a mouse model of Dravet syndrome. Proceedings of the National Academy of Sciences of the United States of America. 2014; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.O’Roak BJ, Deriziotis P, Lee C, Vives L, Schwartz JJ, Girirajan S, et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nature Genetics. 2011; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Weiss LA, Escayg A, Kearney JA, Trudeau M, MacDonald BT, Mori M, et al. Sodium channels SCN1A, SCN2A and SCN3A in familial autism. Molecular Psychiatry. 2003; [DOI] [PubMed] [Google Scholar]
- 137.Favero M, Sotuyo NP, Lopez E, Kearney JA, Goldberg EM. A transient developmental window of fasts-piking interneuron dysfunction in a mouse model of dravet syndrome. Journal of Neuroscience. 2018; 138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Tremblay R, Lee S, Rudy B. GABAergic Interneurons in the Neocortex: From Cellular Properties to Circuits. Neuron. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Goff KM, Goldberg EM. A Role for Vasoactive Intestinal Peptide Interneurons in Neurodevelopmental Disorders. Developmental Neuroscience [Internet]. 2021April1;1–13. Available from: https://www.karger.com/Article/FullText/515264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Williams SR, Stuart GJ. Action potential backpropagation and somato-dendritic distribution of ion channels in thalamocortical neurons. Journal of Neuroscience. 2000; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Larkum ME, Zhu JJ, Sakmann B. A new cellular mechanism for coupling inputs arriving at different cortical layers. Nature. 1999; [DOI] [PubMed] [Google Scholar]
- 142.Catterall WA. Voltage-gated calcium channels. Cold Spring Harbor Perspectives in Biology. 2011; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Markram H, Helm PJ, Sakmann B. Dendritic calcium transients evoked by single back-propagating action potentials in rat neocortical pyramidal neurons. The Journal of Physiology. 1995; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Almog M, Korngreen A. A quantitative description of dendritic conductances and its application to dendritic excitation in layer 5 pyramidal neurons. Journal of Neuroscience. 2014; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Oakley JC, Schwindt PC, Crill WE. Dendritic calcium spikes in layer 5 pyramidal neurons amplify and limit transmission of ligand-gated dendritic current to soma. Journal of Neurophysiology. 2001; [DOI] [PubMed] [Google Scholar]
- 146.Feldman DE. The Spike-Timing Dependence of Plasticity. Neuron. 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Higley MJ, Sabatini BL. Calcium signaling in dendritic spines. Cold Spring Harbor Perspectives in Biology. 2012; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Catterall WA, Perez-Reyes E, Snutch TP, Striessnig J. International Union of Pharmacology. XLVIII. Nomenclature and structure-function relationships of voltage-gated calcium channels. Vol. 57, Pharmacological Reviews. 2005. [DOI] [PubMed] [Google Scholar]
- 149.Magee JC, Johnston D. Characterization of single voltage-gated Na+ and Ca2+ channels in apical dendrites of rat CA1 pyramidal neurons. The Journal of Physiology. 1995; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Magee JC, Johnston D. Synaptic activation of voltage-gated channels in the dendrites of hippocampal pyramidal neurons. Science. 1995; [DOI] [PubMed] [Google Scholar]
- 151.Schlick B, Flucher BE, Obermair GJ. Voltage-activated calcium channel expression profiles in mouse brain and cultured hippocampal neurons. Neuroscience. 2010; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Morton RA, Norlin MS, Vollmer CC, Valenzuela CF. Characterization of L-type voltage-gated Ca2+ channel expression and function in developing CA3 pyramidal neurons. Neuroscience. 2013; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.di Biase V, Tuluc P, Campiglio M, Obermair GJ, Heine M, Flucher BE. Surface traffic of dendritic Ca V1.2 calcium channels in hippocampal neurons. Journal of Neuroscience. 2011; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Jenkins MA, Christel CJ, Jiao Y, Abiria S, Kim KY, Usachev YM, et al. Ca2+-Dependent facilitation of cav1.3 ca2+ channels by densin and ca2+/calmodulin-dependent protein kinase II. Journal of Neuroscience. 2010; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Deisseroth K, Heist EK, Tsien RW. Translocation of calmodulin to the nucleus supports CREB phosphorylation in hippocampal neurons. Nature. 1998; [DOI] [PubMed] [Google Scholar]
- 156.Mermelstein PG, Bito H, Deisseroth K, Tsien RW. Critical dependence of cAMP response element-binding protein phosphorylation on L-type calcium channels supports a selective response to EPSPs in preference to action potentials. Journal of Neuroscience. 2000; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Bauer EP, Schafe GE, LeDoux JE. NMDA Receptors and L-Type Voltage-Gated Calcium Channels Contribute to Long-Term Potentiation and Different Components of Fear Memory Formation in the Lateral Amygdala. Journal of Neuroscience. 2002; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Grover LM, Teyler TJ. Two components of long-term potentiation induced by different patterns of afferent activation. Nature. 1990; [DOI] [PubMed] [Google Scholar]
- 159.Weisskopf MG, Bauer EP, LeDoux JE. L-type voltage-gated calcium channels mediate NMDA-independent associative long-term potentiation at thalamic input synapses to the amygdala. Journal of Neuroscience. 1999; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Leresche N, Lambert RC. T-type calcium channels in synaptic plasticity. Channels. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Catterall WA, Few AP. Calcium Channel Regulation and Presynaptic Plasticity. Neuron. 2008. [DOI] [PubMed] [Google Scholar]
- 162.Simms BA, Zamponi GW. Neuronal voltage-gated calcium channels: Structure, function, and dysfunction. Neuron. 2014. [DOI] [PubMed] [Google Scholar]
- 163.Vacher H, Mohapatra DP, Trimmer JS. Localization and targeting of voltage-dependent ion channels in mammalian central neurons. Physiological Reviews. 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Krey JF, Dolmetsch RE. Molecular mechanisms of autism: a possible role for Ca2+ signaling. Current Opinion in Neurobiology. 2007. [DOI] [PubMed] [Google Scholar]
- 165.Lu AT-H, Dai X, Martinez-Agosto JA, Cantor RM. Support for calcium channel gene defects in autism spectrum disorders. Molecular Autism. 2012; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Liao X, Liao X, Li Y. Genetic associations between voltage-gated calcium channels and autism spectrum disorder: A systematic review. Molecular Brain. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Liao X, Liao X, Li Y. Genetic associations between voltage-gated calcium channels and autism spectrum disorder: A systematic review. Molecular Brain. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, et al. CaV1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell. 2004; [DOI] [PubMed] [Google Scholar]
- 169.Pinggera A, Lieb A, Benedetti B, Lampert M, Monteleone S, Liedl KR, et al. CACNA1D de novo mutations in autism spectrum disorders activate cav1.3 l-type calcium channels. Biological Psychiatry. 2015; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Pinggera A, Mackenroth L, Rump A, Schallner J, Beleggia F, Wollnik B, et al. New gain-of-function mutation shows CACNA1D as recurrently mutated gene in autism spectrum disorders and epilepsy. Human molecular genetics. 2017; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Hemara-Wahanui A, Berjukow S, Hope CI, Dearden PK, Wu SB, Wilson-Wheeler J, et al. A CACNA1F mutation identified in an X-linked retinal disorder shifts the voltage dependence of Cav1.4 channel activation. Proceedings of the National Academy of Sciences of the United States of America. 2005; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Damaj L, Lupien-Meilleur A, Lortie A, Riou É, Ospina LH, Gagnon L, et al. CACNA1A haploinsufficiency causes cognitive impairment, autism and epileptic encephalopathy with mild cerebellar symptoms. European Journal of Human Genetics. 2015; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Splawski I, Yoo DS, Stotz SC, Cherry A, Clapham DE, Keating MT. CACNA1H mutations in autism spectrum disorders. Journal of Biological Chemistry. 2006; [DOI] [PubMed] [Google Scholar]
- 174.Indriati DW, Kamasawa N, Matsui K, Meredith AL, Watanabe M, Shigemoto R. Quantitative localization of Cav2.1 (P/Q-Type) voltage-dependent calcium channels in Purkinje cells: Somatodendritic gradient and distinct somatic Coclustering with calcium-activated potassium channels. Journal of Neuroscience. 2013;33(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Nimmrich V, Gross G. P/Q-type calcium channel modulators. Vol. 167, British Journal of Pharmacology. 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Li L, Bischofberger J, Jonas P. Differential gating and recruitment of P/Q-, N-, and R-type Ca2+ channels in hippocampal mossy fiber boutons. Journal of Neuroscience. 2007;27(49). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Poncer JC, McKinney RA, Gähwiler BH, Thompson SM. Either N- or P-type calcium channels mediate GABA release at distinct hippocampal inhibitory synapses. Neuron. 1997;18(3). [DOI] [PubMed] [Google Scholar]
- 178.Brown SP, Safo PK, Regehr WG. Endocannabinoids inhibit transmission at granule cell to Purkinje cell synapses by modulating three types of presynaptic calcium channels. Journal of Neuroscience. 2004;24(24). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Ritzau-Jost A, Delvendahl I, Rings A, Byczkowicz N, Harada H, Shigemoto R, et al. Ultrafast action potentials mediate kilohertz signaling at a central synapse. Neuron. 2014;84(1). [DOI] [PubMed] [Google Scholar]
- 180.Citri A, Malenka RC. Synaptic plasticity: Multiple forms, functions, and mechanisms. Vol. 33, Neuropsychopharmacology. 2008. [DOI] [PubMed] [Google Scholar]
- 181.Burke KJ, Bender KJ. Modulation of Ion channels in the Axon: Mechanisms and function. Vol. 13, Frontiers in Cellular Neuroscience. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Dolphin AC, Lee A. Presynaptic calcium channels: specialized control of synaptic neurotransmitter release. Vol. 21, Nature Reviews Neuroscience. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Zucker RS, Regehr WG. Short-term synaptic plasticity. Vol. 64, Annual Review of Physiology. 2002. [DOI] [PubMed] [Google Scholar]
- 184.Shah MM, Hammond RS, Hoffman DA. Dendritic ion channel trafficking and plasticity. Trends in Neurosciences. 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Brandalise F, Kalmbach BE, Mehta P, Thornton O, Johnston D, Zemelman BV., et al. Fragile X Mental Retardation Protein Bidirectionally Controls Dendritic Ih in a Cell Type-Specific Manner between Mouse Hippocampus and Prefrontal Cortex. Journal of Neuroscience. 2020; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Shah MM. Cortical HCN channels: Function, trafficking and plasticity. Journal of Physiology. 2014; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Kalmbach BE, Chitwood RA, Dembrow NC, Johnston D. Dendritic generation of mGluR-mediated slow afterdepolarization in layer 5 neurons of prefrontal cortex. Journal of Neuroscience. 2013; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Gao SH, Wen HZ, Shen LL, Zhao YD, Ruan HZ. Activation of mGluR1 contributes to neuronal hyperexcitability in the rat anterior cingulate cortex via inhibition of HCN channels. Neuropharmacology. 2016; [DOI] [PubMed] [Google Scholar]
- 189.Shah MM. Cortical HCN channels: Function, trafficking and plasticity. Journal of Physiology. 2014; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Notomi T, Shigemoto R. Immunohistochemical Localization of Ih Channel Subunits, HCN1–4, in the Rat Brain. Journal of Comparative Neurology. 2004; [DOI] [PubMed] [Google Scholar]
- 191.Nava C, Dalle C, Rastetter A, Striano P, De Kovel CGF, Nabbout R, et al. De novo mutations in HCN1 cause early infantile epileptic encephalopathy. Nature Genetics. 2014; [DOI] [PubMed] [Google Scholar]
- 192.Marini C, Porro A, Rastetter A, Dalle C, Rivolta I, Bauer D, et al. HCN1 mutation spectrum: From neonatal epileptic encephalopathy to benign generalized epilepsy and beyond. Brain. 2018; [DOI] [PubMed] [Google Scholar]
- 193.Kalmbach BE, Johnston D, Brager DH. Cell-type specific channelopathies in the prefrontal cortex of the fmr1-/y mouse model of fragile X syndrome. eNeuro. 2015; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Zhang Y, Bonnan A, Bony G, Ferezou I, Pietropaolo S, Ginger M, et al. Dendritic channelopathies contribute to neocortical and sensory hyperexcitability in Fmr1-/y mice. Nature Neuroscience. 2014; [DOI] [PubMed] [Google Scholar]
- 195.Yi F, Danko T, Botelho SC, Patzke C, Pak C, Wernig M, et al. Autism-associated SHANK3 haploinsufficiency causes Ih channelopathy in human neurons. Science. 2016; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Brandalise F, Kalmbach BE, Mehta P, Thornton O, Johnston D, Zemelman BV., et al. Fragile X Mental Retardation Protein Bidirectionally Controls Dendritic Ih in a Cell Type-Specific Manner between Mouse Hippocampus and Prefrontal Cortex. Journal of Neuroscience. 2020; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Frick A, Magee J, Koester HJ, Migliore M, Johnston D. Normalization of Ca2+ signals by small oblique dendrites of CA1 pyramidal neurons. Journal of Neuroscience. 2003; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Biró ÁA, Brémaud A, Falck J, Ruiz AJ. A-type K + channels impede supralinear summation of clustered glutamatergic inputs in layer 3 neocortical pyramidal neurons. Neuropharmacology. 2018; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Kampa BM, Letzkus JJ, Stuart GJ. Requirement of dendritic calcium spikes for induction of spiketiming-dependent synaptic plasticity. Journal of Physiology. 2006; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Hoffman DA, Magee JC, Colbert CM, Johnston D. K+ channel regulation of signal propagation in dendrites of hippocampal pyramidal neurons. Nature. 1997; [DOI] [PubMed] [Google Scholar]
- 201.Stuart GJ, Häusser M. Dendritic coincidence detection of EPSPs and action potentials. Nature Neuroscience. 2001; [DOI] [PubMed] [Google Scholar]
- 202.Schaefer AT, Helmstaedter M, Schmitt AC, Bar-Yehuda D, Almog M, Ben-Porat H, et al. Dendritic voltage-gated K+ conductance gradient in pyramidal neurones of neocortical layer 5B from rats. Journal of Physiology. 2007; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Harnett MT, Xu NL, Magee JC, Williams SR. Potassium channels control the interaction between active dendritic integration compartments in layer 5 cortical pyramidal neurons. Neuron. 2013; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Noh W, Pak S, Choi G, Yang S, Yang S. Transient potassium channels: Therapeutic targets for brain disorders. Frontiers in Cellular Neuroscience. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Pan Z, Kao T, Horvath Z, Lemos J, Sul JY, Cranstoun SD, et al. A common ankyrin-G-based mechanism retains KCNQ and Na V channels at electrically active domains of the axon. Journal of Neuroscience. 2006; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Chung HJ, Jan YN, Jan LY. Polarized axonal surface expression of neuronal KCNQ channels is mediated by multiple signals in the KCNQ2 and KCNQ3 C-terminal domains. Proceedings of the National Academy of Sciences of the United States of America. 2006; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Bock T, Stuart GJ. The impact of BK channels on cellular excitability depends on their subcellular location. Frontiers in Cellular Neuroscience. 2016; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Soh H, Pant R, LoTurco JJ, Tzingounis AV. Conditional deletions of epilepsy-associated KCNQ2 and KCNQ3 channels from cerebral cortex cause differential effects on neuronal excitability. Journal of Neuroscience. 2014; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Miceli F, Soldovieri MV, Joshi N, Weckhuysen S, Cooper EC, Taglialatela M. KCNQ3-Related Disorders. GeneReviews®. 1993. [Google Scholar]
- 210.Geisheker MR, Heymann G, Wang T, Coe BP, Turner TN, Stessman HAF, et al. Hotspots of missense mutation identify neurodevelopmental disorder genes and functional domains. Nature Neuroscience. 2017; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Markram H, Muller E, Ramaswamy S, Reimann MW, Abdellah M, Sanchez CA, et al. Reconstruction and Simulation of Neocortical Microcircuitry. Cell. 2015; [DOI] [PubMed] [Google Scholar]
- 212.Bernander O, Koch C, Douglas RJ. Amplification and linearization of distal synaptic input to cortical pyramidal cells. Journal of Neurophysiology. 1994; [DOI] [PubMed] [Google Scholar]
- 213.Spruston N Pyramidal neurons: Dendritic structure and synaptic integration. Nature Reviews Neuroscience. 2008. [DOI] [PubMed] [Google Scholar]
- 214.Antic SD, Zhou WL, Moore AR, Short SM, Ikonomu KD. The decade of the dendritic NMDA spike. Journal of Neuroscience Research. 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Hausser M, Spruston N, Stuart GJ. Diversity and dynamics of dendritic signaling. Science. 2000. [DOI] [PubMed] [Google Scholar]
- 216.Schiller J, Major G, Koester HJ, Schiller Y. NMDA spikes in basal dendrites of cortical pyramidal neurons. Nature. 2000; [DOI] [PubMed] [Google Scholar]
- 217.Lee KFH. A unique mechanism of NMDA spike initiation supports a distinct role in synaptic input integration. Journal of Neuroscience. 2012; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Rhodes P The properties and implications of NMDA spikes in neocortical pyramidal cells. Journal of Neuroscience. 2006; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Emptage N, Bliss TVP, Fine A. Single synaptic events evoke NMDA receptor-mediated release of calcium from internal stores in hippocampal dendritic spines. Neuron. 1999; [DOI] [PubMed] [Google Scholar]
- 220.Matsuzaki M, Honkura N, Ellis-Davies GCR, Kasai H. Structural basis of long-term potentiation in single dendritic spines. Nature. 2004; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Lüscher C, Nicoll RA, Malenka RC, Muller D. Synaptic plasticity and dynamic modulation of the postsynaptic membrane. Nature Neuroscience. 2000. [DOI] [PubMed] [Google Scholar]
- 222.Tsay D, Yuste R. Role of dendritic spines in action potential backpropagation: A numerical simulation study. Journal of Neurophysiology. 2002; [DOI] [PubMed] [Google Scholar]
- 223.Forrest MP, Parnell E, Penzes P. Dendritic structural plasticity and neuropsychiatric disease. Nature Reviews Neuroscience. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Zoghbi HY, Bear MF. Synaptic dysfunction in neurodevelopmental disorders associated with autism and intellectual disabilities. Cold Spring Harbor Perspectives in Biology. 2012; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Guang S, Pang N, Deng X, Yang L, He F, Wu L, et al. Synaptopathology involved in autism spectrum disorder. Frontiers in Cellular Neuroscience. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Mori H, Mishina M. Structure and function of the NMDA receptor channel. Neuropharmacology. 1995. [DOI] [PubMed] [Google Scholar]
- 227.Hu C, Chen W, Myers SJ, Yuan H, Traynelis SF. Human GRIN2B variants in neurodevelopmental disorders. Journal of Pharmacological Sciences. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Platzer K, Yuan H, Schütz H, Winschel A, Chen W, Hu C, et al. GRIN2B encephalopathy: Novel findings on phenotype, variant clustering, functional consequences and treatment aspects. Journal of Medical Genetics. 2017; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. 2014; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Kenny EM, Cormican P, Furlong S, Heron E, Kenny G, Fahey C, et al. Excess of rare novel loss-of-function variants in synaptic genes in schizophrenia and autism spectrum disorders. Molecular Psychiatry. 2014; [DOI] [PubMed] [Google Scholar]
- 231.Hu C, Chen W, Myers SJ, Yuan H, Traynelis SF. Human GRIN2B variants in neurodevelopmental disorders. Journal of Pharmacological Sciences. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.O’Roak BJ, Deriziotis P, Lee C, Vives L, Schwartz JJ, Girirajan S, et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nature Genetics. 2011; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron. 1994; [DOI] [PubMed] [Google Scholar]
- 234.Adams DR, Yuan H, Holyoak T, Arajs KH, Hakimi P, Markello TC, et al. Three rare diseases in one Sib pair: RAI1, PCK1, GRIN2B mutations associated with Smith-Magenis Syndrome, cytosolic PEPCK deficiency and NMDA receptor glutamate insensitivity. Molecular Genetics and Metabolism. 2014; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Bell S, Maussion G, Jefri M, Peng H, Theroux JF, Silveira H, et al. Disruption of GRIN2B Impairs Differentiation in Human Neurons. Stem Cell Reports. 2018; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Fedele L, Newcombe J, Topf M, Gibb A, Harvey RJ, Smart TG. Disease-associated missense mutations in GluN2B subunit alter NMDA receptor ligand binding and ion channel properties. Nature Communications. 2018; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Williams K, Russell SL, Shen YM, Molinoff PB. Developmental switch in the expression of NMDA receptors occurs in vivo and in vitro. Neuron. 1993; [DOI] [PubMed] [Google Scholar]
- 238.D’Antoni S, Spatuzza M, Bonaccorso CM, Musumeci SA, Ciranna L, Nicoletti F, et al. Dysregulation of group-I metabotropic glutamate (mGlu) receptor mediated signalling in disorders associated with Intellectual Disability and Autism. Neuroscience and Biobehavioral Reviews. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Lüscher C, Huber KM. Group 1 mGluR-Dependent Synaptic Long-Term Depression: Mechanisms and Implications for Circuitry and Disease. Neuron. 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Telias M Molecular mechanisms of synaptic dysregulation in fragile X syndrome and autism spectrum disorders. Frontiers in Molecular Neuroscience. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Lüscher C, Huber KM. Group 1 mGluR-Dependent Synaptic Long-Term Depression: Mechanisms and Implications for Circuitry and Disease. Neuron. 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Viaene AN, Petrof I, Sherman SM. Properties of the thalamic projection from the posterior medial nucleus to primary and secondary somatosensory cortices in the mouse. Proceedings of the National Academy of Sciences of the United States of America. 2011; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Kalmbach BE, Chitwood RA, Dembrow NC, Johnston D. Dendritic generation of mGluR-mediated slow afterdepolarization in layer 5 neurons of prefrontal cortex. Journal of Neuroscience. 2013; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Mease RA, Metz M, Groh A. Cortical Sensory Responses Are Enhanced by the Higher-Order Thalamus. Cell Reports. 2016; [DOI] [PubMed] [Google Scholar]
- 245.Varoqueaux F, Aramuni G, Rawson RL, Mohrmann R, Missler M, Gottmann K, et al. Neuroligins Determine Synapse Maturation and Function. Neuron. 2006; [DOI] [PubMed] [Google Scholar]
- 246.Lin YC, Frei JA, Kilander MBC, Shen W, Blatt GJ. A subset of autism-associated genes regulate the structural stability of neurons. Frontiers in Cellular Neuroscience. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Monteiro P, Feng G. SHANK proteins: Roles at the synapse and in autism spectrum disorder. Nature Reviews Neuroscience. 2017. [DOI] [PubMed] [Google Scholar]
- 248.Forrest MP, Parnell E, Penzes P. Dendritic structural plasticity and neuropsychiatric disease. Nature Reviews Neuroscience. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Leblond CS, Nava C, Polge A, Gauthier J, Huguet G, Lumbroso S, et al. Meta-analysis of SHANK Mutations in Autism Spectrum Disorders: A Gradient of Severity in Cognitive Impairments. PLoS Genetics. 2014; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Gauthier J, Spiegelman D, Piton A, Lafreniére RG, Laurent S, St-Onge J, et al. Novel de novo SHANK3 mutation in autistic patients. American Journal of Medical Genetics, Part B: Neuropsychiatric Genetics. 2009; [DOI] [PubMed] [Google Scholar]
- 251.Boccuto L, Lauri M, Sarasua SM, Skinner CD, Buccella D, Dwivedi A, et al. Prevalence of SHANK3 variants in patients with different subtypes of autism spectrum disorders. European Journal of Human Genetics. 2013; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Sala C, Vicidomini C, Bigi I, Mossa A, Verpelli C. Shank synaptic scaffold proteins: Keys to understanding the pathogenesis of autism and other synaptic disorders. Journal of Neurochemistry. 2015. [DOI] [PubMed] [Google Scholar]
- 253.Monteiro P, Feng G. SHANK proteins: Roles at the synapse and in autism spectrum disorder. Nature Reviews Neuroscience. 2017. [DOI] [PubMed] [Google Scholar]
- 254.Bozdagi O, Sakurai T, Papapetrou D, Wang X, Dickstein DL, Takahashi N, et al. Haploinsufficiency of the autism-associated Shank3 gene leads to deficits in synaptic function, social interaction, and social communication. Molecular Autism. 2010; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Jiang Y hui, Ehlers MD. Modeling Autism by SHANK Gene Mutations in Mice. Neuron. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Hung AY, Futai K, Sala C, Valtschanoff JG, Ryu J, Woodworth MA, et al. Smaller dendritic spines, weaker synaptic transmission, but enhanced spatial learning in mice lacking Shank1. Journal of Neuroscience. 2008; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Martínez-Cerdeño V Dendrite and spine modifications in autism and related neurodevelopmental disorders in patients and animal models. Developmental Neurobiology. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Lee K, Vyas Y, Garner CC, Montgomery JM. Autism-associated Shank3 mutations alter mGluR expression and mGluR-dependent but not NMDA receptor-dependent long-term depression. Synapse. 2019; [DOI] [PubMed] [Google Scholar]
- 259.Jamain S, Quach H, Betancur C, Råstam M, Colineaux C, Gillberg C, et al. Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nature Genetics. 2003; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Südhof TC. Neuroligins and neurexins link synaptic function to cognitive disease. Nature. 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Nam CI, Chen L. Postsynaptic assembly induced by neurexin-neuroligin interaction and neurotransmitter. Proceedings of the National Academy of Sciences of the United States of America. 2005; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Heine M, Groc L, Frischknecht R, Béïque JC, Lounis B, Rumbaugh G, et al. Surface mobility of postsynaptic AMPARs tunes synaptic transmission. Science. 2008; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Blundell J, Blaiss CA, Etherton MR, Espinosa F, Tabuchi K, Walz C, et al. Neuroligin-1 deletion results in impaired spatial memory and increased repetitive behavior. Journal of Neuroscience. 2010; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Nakanishi M, Nomura J, Ji X, Tamada K, Arai T, Takahashi E, et al. Functional significance of rare neuroligin 1 variants found in autism. PLoS Genetics. 2017; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Tabuchi K, Blundell J, Etherton MR, Hammer RE, Liu X, Powell CM, et al. A neuroligin-3 mutation implicated in autism increases inhibitory synaptic transmission in mice. Science. 2007; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Rothwell PE, Fuccillo MV., Maxeiner S, Hayton SJ, Gokce O, Lim BK, et al. Autism-associated neuroligin-3 mutations commonly impair striatal circuits to boost repetitive behaviors. Cell. 2014; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Alarcón M, Abrahams BS, Stone JL, Duvall JA, Perederiy JV., Bomar JM, et al. Linkage, Association, and Gene-Expression Analyses Identify CNTNAP2 as an Autism-Susceptibility Gene. American Journal of Human Genetics. 2008; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Arking DE, Cutler DJ, Brune CW, Teslovich TM, West K, Ikeda M, et al. A Common Genetic Variant in the Neurexin Superfamily Member CNTNAP2 Increases Familial Risk of Autism. American Journal of Human Genetics. 2008; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Poot M, Beyer V, Schwaab I, Damatova N, Van’T Slot R, Prothero J, et al. Disruption of CNTNAP2 and additional structural genome changes in a boy with speech delay and autism spectrum disorder. Neurogenetics. 2010; [DOI] [PubMed] [Google Scholar]
- 270.Bakkaloglu B, O’Roak BJ, Louvi A, Gupta AR, Abelson JF, Morgan TM, et al. Molecular Cytogenetic Analysis and Resequencing of Contactin Associated Protein-Like 2 in Autism Spectrum Disorders. American Journal of Human Genetics. 2008; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Anderson GR, Galfin T, Xu W, Aoto J, Malenka RC, Südhof TC. Candidate autism gene screen identifies critical role for cell-adhesion molecule CASPR2 in dendritic arborization and spine development. Proceedings of the National Academy of Sciences of the United States of America. 2012; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Peñagarikano O, Abrahams BS, Herman EI, Winden KD, Gdalyahu A, Dong H, et al. Absence of CNTNAP2 leads to epilepsy, neuronal migration abnormalities, and core autism-related deficits. Cell. 2011; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Sacai H, Sakoori K, Konno K, Nagahama K, Suzuki H, Watanabe T, et al. Autism spectrum disorder-like behavior caused by reduced excitatory synaptic transmission in pyramidal neurons of mouse prefrontal cortex. Nature Communications. 2020; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Peñagarikano O, Abrahams BS, Herman EI, Winden KD, Gdalyahu A, Dong H, et al. Absence of CNTNAP2 leads to epilepsy, neuronal migration abnormalities, and core autism-related deficits. Cell. 2011; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Vogt D, Cho KKA, Shelton SM, Paul A, Huang ZJ, Sohal VS, et al. Mouse Cntnap2 and Human CNTNAP2 ASD Alleles Cell Autonomously Regulate PV + Cortical Interneurons. Cerebral Cortex. 2018;28(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Hamdan FF, Gauthier J, Spiegelman D, Noreau A, Yang Y, Pellerin S, et al. Mutations in SYNGAP1 in Autosomal Nonsyndromic Mental Retardation . New England Journal of Medicine. 2009; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Rauch A, Wieczorek D, Graf E, Wieland T, Endele S, Schwarzmayr T, et al. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: An exome sequencing study. The Lancet. 2012; [DOI] [PubMed] [Google Scholar]
- 278.Vlaskamp DRM, Shaw BJ, Burgess R, Mei D, Montomoli M, Xie H, et al. SYNGAP1 encephalopathy: A distinctive generalized developmental and epileptic encephalopathy. Neurology. 2019; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Berryer MH, Hamdan FF, Klitten LL, Møller RS, Carmant L, Schwartzentruber J, et al. Mutations in SYNGAP1 Cause Intellectual Disability, Autism, and a Specific Form of Epilepsy by Inducing Haploinsufficiency. Human Mutation. 2013; [DOI] [PubMed] [Google Scholar]
- 280.Parker MJ, Fryer AE, Shears DJ, Lachlan KL, Mckee SA, Magee AC, et al. De novo, heterozygous, loss-of-function mutations in SYNGAP1 cause a syndromic form of intellectual disability. American Journal of Medical Genetics, Part A. 2015; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Gou G, Roca-Fernandez A, Kilinc M, Serrano E, Reig-Viader R, Araki Y, et al. SynGAP splice variants display heterogeneous spatio-temporal expression and subcellular distribution in the developing mammalian brain. Journal of Neurochemistry. 2020; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Araki Y, Hong I, Gamache TR, Ju S, Collado-Torres L, Shin JH, et al. Syngap isoforms differentially regulate synaptic plasticity and dendritic development. eLife. 2020; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Kim JH, Liao D, Lau LF, Huganir RL. SynGAP: A synaptic RasGAP that associates with the PSD-95/SAP90 protein family. Neuron. 1998; [DOI] [PubMed] [Google Scholar]
- 284.Gou G, Roca-Fernandez A, Kilinc M, Serrano E, Reig-Viader R, Araki Y, et al. SynGAP splice variants display heterogeneous spatio-temporal expression and subcellular distribution in the developing mammalian brain. Journal of Neurochemistry. 2020; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Clement JP, Ozkan ED, Aceti M, Miller CA, Rumbaugh G. SYNGAP1 links the maturation rate of excitatory synapses to the duration of critical-period synaptic plasticity. Journal of Neuroscience. 2013; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Clement JP, Aceti M, Creson TK, Ozkan ED, Shi Y, Reish NJ, et al. Pathogenic SYNGAP1 mutations impair cognitive development by disrupting maturation of dendritic spine synapses. Cell. 2012; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Di J, Li J, O’Hara B, Alberts I, Xiong L, Li J, et al. The role of GABAergic neural circuits in the pathogenesis of autism spectrum disorder. Vol. 80, International Journal of Developmental Neuroscience. 2020. [DOI] [PubMed] [Google Scholar]
- 288.Naka A, Adesnik H. Inhibitory circuits in cortical layer 5. Vol. 10, Frontiers in Neural Circuits. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Silberberg G, Markram H. Disynaptic Inhibition between Neocortical Pyramidal Cells Mediated by Martinotti Cells. Neuron. 2007;53(5). [DOI] [PubMed] [Google Scholar]
- 290.Markram H, Toledo-Rodriguez M, Wang Y, Gupta A, Silberberg G, Wu C. Interneurons of the neocortical inhibitory system. Vol. 5, Nature Reviews Neuroscience. 2004. [DOI] [PubMed] [Google Scholar]
- 291.Reyes A, Lujan R, Rozov A, Burnashev N, Somogyi P, Sakmann B. Target-cell-specific facilitation and depression in neocortical circuits. Nature Neuroscience. 1998;1(4). [DOI] [PubMed] [Google Scholar]
- 292.Riedemann T Diversity and function of somatostatin-expressing interneurons in the cerebral cortex. Vol. 20, International Journal of Molecular Sciences. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Murayama M, Pérez-Garci E, Nevian T, Bock T, Senn W, Larkum ME. Dendritic encoding of sensory stimuli controlled by deep cortical interneurons. Nature. 2009;457(7233). [DOI] [PubMed] [Google Scholar]
- 294.Goff KM, Goldberg EM. Vasoactive intestinal peptide-expressing interneurons are impaired in a mouse model of dravet syndrome. eLife. 2019;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Pfeffer CK, Xue M, He M, Huang ZJ, Scanziani M. Inhibition of inhibition in visual cortex: The logic of connections between molecularly distinct interneurons. Nature Neuroscience. 2013;16(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Dávid C, Schleicher A, Zuschratter W, Staiger JF. The innervation of parvalbumin-containing interneurons by VIP-immunopositive interneurons in the primary somatosensory cortex of the adult rat. European Journal of Neuroscience. 2007;25(8). [DOI] [PubMed] [Google Scholar]
- 297.Braat S, Kooy RF. The GABAA Receptor as a Therapeutic Target for Neurodevelopmental Disorders. Vol. 86, Neuron. 2015. [DOI] [PubMed] [Google Scholar]
- 298.Fatemi SH, Folsom TD. GABA receptor subunit distribution and FMRP-mGluR5 signaling abnormalities in the cerebellum of subjects with schizophrenia, mood disorders, and autism. Vol. 167, Schizophrenia Research. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Chen CH, Huang CC, Cheng MC, Chiu YN, Tsai WC, Wu YY, et al. Genetic analysis of GABRB3 as a candidate gene of autism spectrum disorders. Molecular Autism. 2014;5(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Kang JQ, Barnes G. A common susceptibility factor of both autism and epilepsy: Functional deficiency of GABAA receptors. Journal of Autism and Developmental Disorders. 2013;43(1). [DOI] [PubMed] [Google Scholar]
- 301.Buxbaum JD, Silverman JM, Smith CJ, Greenberg DA, Kilifarski M, Reichert J, et al. Association between a GABRB3 polymorphism and autism. Molecular Psychiatry. 2002;7(3). [DOI] [PubMed] [Google Scholar]
- 302.Ma DQ, Whitehead PL, Menold MM, Martin ER, Ashley-Koch AE, Mei H, et al. Identification of significant association and gene-gene interaction of GABA receptor subunit genes in autism. American Journal of Human Genetics. 2005;77(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Riley JD, Delahunty C, Alsadah A, Mazzola S, Astbury C. Further evidence of GABRA4 and TOP3B as autism susceptibility genes. European Journal of Medical Genetics. 2020;63(5). [DOI] [PubMed] [Google Scholar]
- 304.Polan MB, Pastore MT, Steingass K, Hashimoto S, Thrush DL, Pyatt R, et al. Neurodevelopmental disorders among individuals with duplication of 4p13 to 4p12 containing a GABA A receptor subunit gene cluster. European Journal of Human Genetics. 2014;22(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Collins AL, Ma D, Whitehead PL, Martin ER, Wright HH, Abramson RK, et al. Investigation of autism and GABA receptor subunit genes in multiple ethnic groups. Neurogenetics. 2006;7(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Sagar A, Pinto D, Najjar F, Guter SJ, Macmillan C, Cook EH. De novo unbalanced translocation (4p duplication/8p deletion) in a patient with autism, OCD, and overgrowth syndrome. American Journal of Medical Genetics, Part A. 2017;173(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Sabaratnam M, Turk J, Vroegop P. Case report: Autistic disorder and chromosomal abnormality 46, XX duplication (4) p12-p13. European Child and Adolescent Psychiatry. 2000;9(4). [DOI] [PubMed] [Google Scholar]
- 308.Fatemi SH, Reutiman TJ, Folsom TD, Rustan OG, Rooney RJ, Thuras PD. Downregulation of GABAA receptor protein subunits α6, β2, δ, ε, γ2, θ, and ρ2 in superior frontal cortex of subjects with autism. Journal of Autism and Developmental Disorders. 2014;44(8). [DOI] [PubMed] [Google Scholar]
- 309.Fatemi SH, Reutiman TJ, Folsom TD, Thuras PD. GABAA receptor downregulation in brains of subjects with autism. Journal of Autism and Developmental Disorders. 2009;39(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Sestan N, State MW. Lost in Translation: Traversing the Complex Path from Genomics to Therapeutics in Autism Spectrum Disorder. Vol. 100, Neuron. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Quesnel-Vallières M, Weatheritt RJ, Cordes SP, Blencowe BJ. Autism spectrum disorder: insights into convergent mechanisms from transcriptomics. Vol. 20, Nature Reviews Genetics. 2019. [DOI] [PubMed] [Google Scholar]
- 312.Manoli DS, State MW. Autism Spectrum Disorder Genetics and the Search for Pathological Mechanisms. American Journal of Psychiatry. 2021; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Jin X, Simmons SK, Guo A, Shetty AS, Ko M, Nguyen L, et al. In vivo Perturb-Seq reveals neuronal and glial abnormalities associated with autism risk genes. Science. 2020;370(6520). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Gatchel JR, Zoghbi HY. Diseases of unstable repeat expansion: mechanisms and common principles. Nature Reviews Genetics. 2005. [DOI] [PubMed] [Google Scholar]
- 315.Gatchel JR, Zoghbi HY. Diseases of unstable repeat expansion: mechanisms and common principles. Nature Reviews Genetics. 2005. [DOI] [PubMed] [Google Scholar]
- 316.Fernández E, Rajan N, Bagni C. The FMRP regulon: From targets to disease convergence. Frontiers in Neuroscience. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Chmielewska JJ, Kuzniewska B, Milek J, Urbanska K, Dziembowska M. Neuroligin 1, 2, and 3 Regulation at the Synapse: FMRP-Dependent Translation and Activity-Induced Proteolytic Cleavage. Molecular Neurobiology. 2019; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Hinton VJ, Brown WT, Wisniewski K, Rudelli RD. Analysis of neocortex in three males with the fragile X syndrome. American Journal of Medical Genetics. 1991; [DOI] [PubMed] [Google Scholar]
- 319.Bear MF, Huber KM, Warren ST. The mGluR theory of fragile X mental retardation. Trends in Neurosciences. 2004; [DOI] [PubMed] [Google Scholar]
- 320.Huber KM, Gallagher SM, Warren ST, Bear MF. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proceedings of the National Academy of Sciences of the United States of America. 2002; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Bear MF, Huber KM, Warren ST. The mGluR theory of fragile X mental retardation. Trends in Neurosciences. 2004; [DOI] [PubMed] [Google Scholar]
- 322.Lamar KMJ, Carvill GL. Chromatin remodeling proteins in epilepsy: Lessons from CHD2-associated epilepsy. Frontiers in Molecular Neuroscience. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Cotney J, Muhle RA, Sanders SJ, Liu L, Willsey AJ, Niu W, et al. The autism-associated chromatin modifier CHD8 regulates other autism risk genes during human neurodevelopment. Nature Communications. 2015; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Xu Q, Liu YY, Wang X, Tan GH, Li HP, Hulbert SW, et al. Autism-associated CHD8 deficiency impairs axon development and migration of cortical neurons. Molecular Autism. 2018; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Notwell JH, Heavner WE, Darbandi SF, Katzman S, McKenna WL, Ortiz-Londono CF, et al. TBR1 regulates autism risk genes in the developing neocortex. Genome Research. 2016; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Notwell JH, Heavner WE, Darbandi SF, Katzman S, McKenna WL, Ortiz-Londono CF, et al. TBR1 regulates autism risk genes in the developing neocortex. Genome Research. 2016; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Hevner RF, Shi L, Justice N, Hsueh YP, Sheng M, Smiga S, et al. Tbr1 regulates differentiation of the preplate and layer 6. Neuron. 2001; [DOI] [PubMed] [Google Scholar]
- 328.Bedogni F, Hodge RD, Elsen GE, Nelson BR, Daza RAM, Beyer RP, et al. Tbr1 regulates regional and laminar identity of postmitotic neurons in developing neocortex. Proceedings of the National Academy of Sciences of the United States of America. 2010; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.McKenna WL, Betancourt J, Larkin KA, Abrams B, Guo C, Rubenstein JLR, et al. Tbr1 and Fezf2 regulate alternate corticofugal neuronal identities during neocortical development. Journal of Neuroscience. 2011; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Fazel Darbandi S, Robinson Schwartz SE, Pai ELL, Everitt A, Turner ML, Cheyette BNR, et al. Enhancing WNT Signaling Restores Cortical Neuronal Spine Maturation and Synaptogenesis in Tbr1 Mutants. Cell Reports. 2020; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Fazel Darbandi S, Robinson Schwartz SE, Qi Q, Catta-Preta R, Pai ELL, Mandell JD, et al. Neonatal Tbr1 Dosage Controls Cortical Layer 6 Connectivity. Neuron. 2018; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Fazel Darbandi S, Robinson Schwartz SE, Pai ELL, Everitt A, Turner ML, Cheyette BNR, et al. Enhancing WNT Signaling Restores Cortical Neuronal Spine Maturation and Synaptogenesis in Tbr1 Mutants. Cell Reports. 2020; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Fazel Darbandi S, Robinson Schwartz SE, Qi Q, Catta-Preta R, Pai ELL, Mandell JD, et al. Neonatal Tbr1 Dosage Controls Cortical Layer 6 Connectivity. Neuron. 2018; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Olsen SR, Bortone DS, Adesnik H, Scanziani M. Gain control by layer six in cortical circuits of vision. Nature. 2012; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Zhou J, Parada LF. PTEN signaling in autism spectrum disorders. Vol. 22, Current Opinion in Neurobiology. 2012. [DOI] [PubMed] [Google Scholar]
- 336.Lv JW, Cheng TL, Qiu ZL, Zhou WH. Role of the PTEN signaling pathway in autism spectrum disorder. Neuroscience Bulletin. 2013;29(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Stiles BL. Phosphatase and tensin homologue deleted on chromosome 10: Extending its PTENtacles. Vol. 41, International Journal of Biochemistry and Cell Biology. 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Bourgeron T A synaptic trek to autism. Vol. 19, Current Opinion in Neurobiology. 2009. [DOI] [PubMed] [Google Scholar]
- 339.Ljungberg MC, Sunnen CN, Lugo JN, Anderson AE, D’Arcangelo G. Rapamycin suppresses seizures and neuronal hypertrophy in a mouse model of cortical dysplasia. DMM Disease Models and Mechanisms. 2009;2(7–8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Chen Y, Huang WC, Séjourné J, Clipperton-Allen AE, Page DT. Pten mutations alter brain growth trajectory and allocation of cell types through elevated β-Catenin signaling. Journal of Neuroscience. 2015;35(28). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Kwon CH, Luikart BW, Powell CM, Zhou J, Matheny SA, Zhang W, et al. Pten Regulates Neuronal Arborization and Social Interaction in Mice. Neuron. 2006;50(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Fraser MM, Bayazitov IT, Zakharenko SS, Baker SJ. Phosphatase and tensin homolog, deleted on chromosome 10 deficiency in brain causes defects in synaptic structure, transmission and plasticity, and myelination abnormalities. Neuroscience. 2008;151(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Luikart BW, Schnell E, Washburn EK, Bensen ASL, Tovar KR, Westbrook GL. Pten knockdown in vivo increases excitatory drive onto dentate granule cells. Journal of Neuroscience. 2011;31(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Williams MR, De-Spenza T, Li M, Gulledge AT, Luikart BW. Hyperactivity of newborn pten knock-out neurons results from increased excitatory synaptic drive. Journal of Neuroscience. 2015;35(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Vogt D, Cho KKA, Lee AT, Sohal VS, Rubenstein JLR. The Parvalbumin/Somatostatin Ratio Is Increased in Pten Mutant Mice and by Human PTEN ASD Alleles. Cell Reports. 2015;11(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Savalia NK, Shao LX, Kwan AC. A Dendrite-Focused Framework for Understanding the Actions of Ketamine and Psychedelics. Trends in Neurosciences. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Subramanian K, Brandenburg C, Orsati F, Soghomonian JJ, Hussman JP, Blatt GJ. Basal ganglia and autism – a translational perspective. Autism Research. 2017. [DOI] [PubMed] [Google Scholar]
- 348.Calderoni S, Bellani M, Hardan AY, Muratori F, Brambilla P. Basal ganglia and restricted and repetitive behaviours in Autism Spectrum Disorders: Current status and future perspectives. Epidemiology and Psychiatric Sciences. 2014; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Prat CS, Stocco A, Neuhaus E, Kleinhans NM. Basal ganglia impairments in autism spectrum disorder are related to abnormal signal gating to prefrontal cortex. Neuropsychologia. 2016; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Muehlmann AM, Lewis MH. Abnormal repetitive behaviours: Shared phenomenology and pathophysiology. Journal of Intellectual Disability Research. 2012; [DOI] [PubMed] [Google Scholar]
- 351.Sorrells SF, Paredes MF, Velmeshev D, Herranz-Pérez V, Sandoval K, Mayer S, et al. Immature excitatory neurons develop during adolescence in the human amygdala. Nature Communications. 2019; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Phelps EA, LeDoux JE. Contributions of the amygdala to emotion processing: From animal models to human behavior. Neuron. 2005. [DOI] [PubMed] [Google Scholar]
- 353.Schumann CM, Amaral DG. Stereological analysis of amygdala neuron number in autism. Journal of Neuroscience. 2006; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Bauman M, Kemper TL. Histoanatomic observations of the brain in early infantile autism. Neurology. 1985; [DOI] [PubMed] [Google Scholar]
- 355.Bauman ML, Kemper TL. Neuroanatomic observations of the brain in autism: A review and future directions. International Journal of Developmental Neuroscience. 2005. [DOI] [PubMed] [Google Scholar]
- 356.DeLong GR. Autism, amnesia, hippocampus, and learning. Neuroscience and Biobehavioral Reviews. 1992; [DOI] [PubMed] [Google Scholar]
- 357.Xu Q, Zuo C, Liao S, Long Y, Wang Y. Abnormal development pattern of the amygdala and hippocampus from childhood to adulthood with autism. Journal of Clinical Neuroscience. 2020; [DOI] [PubMed] [Google Scholar]
- 358.Shalom D Ben. Memory in autism: Review and synthesis. Cortex. 2003; [DOI] [PubMed] [Google Scholar]
- 359.Becker EBE, Stoodley CJ. Autism spectrum disorder and the cerebellum. In: International Review of Neurobiology. 2013. p. 1–34. [DOI] [PubMed] [Google Scholar]
- 360.Schmahmann JD. The cerebellum and cognition. Neuroscience Letters. 2019. [DOI] [PubMed] [Google Scholar]
- 361.Stoodley CJ. The Cerebellum and Neurodevelopmental Disorders. Cerebellum. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Bruchhage MMK, Bucci MP, Becker EBE. Cerebellar involvement in autism and ADHD. In: Handbook of Clinical Neurology. 2018. [DOI] [PubMed] [Google Scholar]
- 363.Hansel C Deregulation of synaptic plasticity in autism. Neuroscience Letters. 2019. [DOI] [PubMed] [Google Scholar]
- 364.Tsai PT. Autism and cerebellar dysfunction: Evidence from animal models. Seminars in Fetal and Neonatal Medicine. 2016. [DOI] [PubMed] [Google Scholar]
- 365.Schuetze M, Park MTM, Cho IYK, Macmaster FP, Chakravarty MM, Bray SL. Morphological alterations in the thalamus, striatum, and pallidum in autism spectrum disorder. Neuropsychopharmacology. 2016; [DOI] [PMC free article] [PubMed] [Google Scholar]