

Abstract
GABAergic inhibitory interneurons of the cerebral cortex expressing vasoactive intestinal peptide (VIP-INs) are rapidly emerging as important regulators of network dynamics and normal circuit development. Several recent studies have also identified VIP-IN dysfunction in models of genetically-determined neurodevelopmental disorders (NDDs). In this article, we review the known circuit functions of VIP-INs and how they may relate to accumulating evidence implicating VIP-INs in the mechanisms of prominent NDDs. We highlight recurring VIP-IN mediated circuit motifs that are shared across cerebral cortical areas, and how VIP-IN activity can shape sensory input, development, and behavior. Ultimately, we extract a set of themes that inform our understanding of how VIP-INs influence pathogenesis of NDDs. Using publicly available single cell RNA sequencing (scRNA-seq) data from the Allen Institute, we also identify several underexplored disease-associated genes that are highly expressed in VIP-INs. We survey these genes and their shared related disease phenotypes that may broadly implicate VIP-INs in autism spectrum disorder (ASD) and intellectual disability (ID) rather than epileptic encephalopathy. Finally, we conclude with a discussion of the relevance of cell type-specific investigations and therapeutics in the age of genomic diagnosis and precision medicine.
Keywords: VIP, Autism, Epilepsy, Interneurons
Introduction
While comprising only a fraction of the total cells in the brain, interneurons (INs) have garnered significant attention due to their complex and dynamic roles in shaping network activity and behavior. Furthermore, many neurological disorders may involve impaired IN function [1]. IN dysfunction is often framed in the context of excitation/inhibition (E/I) imbalance, a useful simplification. However, INs are embedded within myriad complex repeating circuit motifs in the cerebral cortex, including feedforward, feedback, and disinhibitory interconnectivity [2]. As a result, the three largest subclasses of INs, which express parvalbumin, somatostatin, or vasocactive intestinal peptide (PV, SST, and VIP-INs) [3], take part in diverse circuit functions, from regulation of neuromodulatory influences on neocortex to spike timing and synchronization. INs also serve as mediators of normal circuit development by regulating critical period plasticity and shaping the formation of sensory maps [1]. Due to such functions, there is strong interest in identifying the role of IN subclasses in genetically-determined neurodevelopmental disorders (NDDs) that arise from dysfunctional neural circuits. VIP-INs are less studied than the other main IN subclasses, but their unique network functions could position them as a critically important locus of pathology in NDDs. Specific contributions of VIP-INs in NDDs are just beginning to be explored.
Here, we examine the circuit impact of VIP-INs, extracting recurrent themes to suggest how VIP-IN dysfunction might contribute to NDDs (Fig. 1). We summarize early evidence for VIP-IN involvement in specific NDDs such as Dravet syndrome and Rett syndrome, and more broadly across epilepsy and autism spectrum disorder (ASD). Then, using single cell RNA sequencing (scRNA-seq) data available from the Allen Institute [4], we highlight several known disease genes that exhibit cell-type specific expression in VIP-INs and may be important for VIP-IN function while also implicating VIP-IN dysfunction in NDD pathogenesis. Finally, we conclude by discussing the future potential for identification of cell-type specific dysfunction in driving forward the development of novel therapeutic approaches for NDDs.
Fig. 1. Circuit Functions of VIP-INs in NDDs.
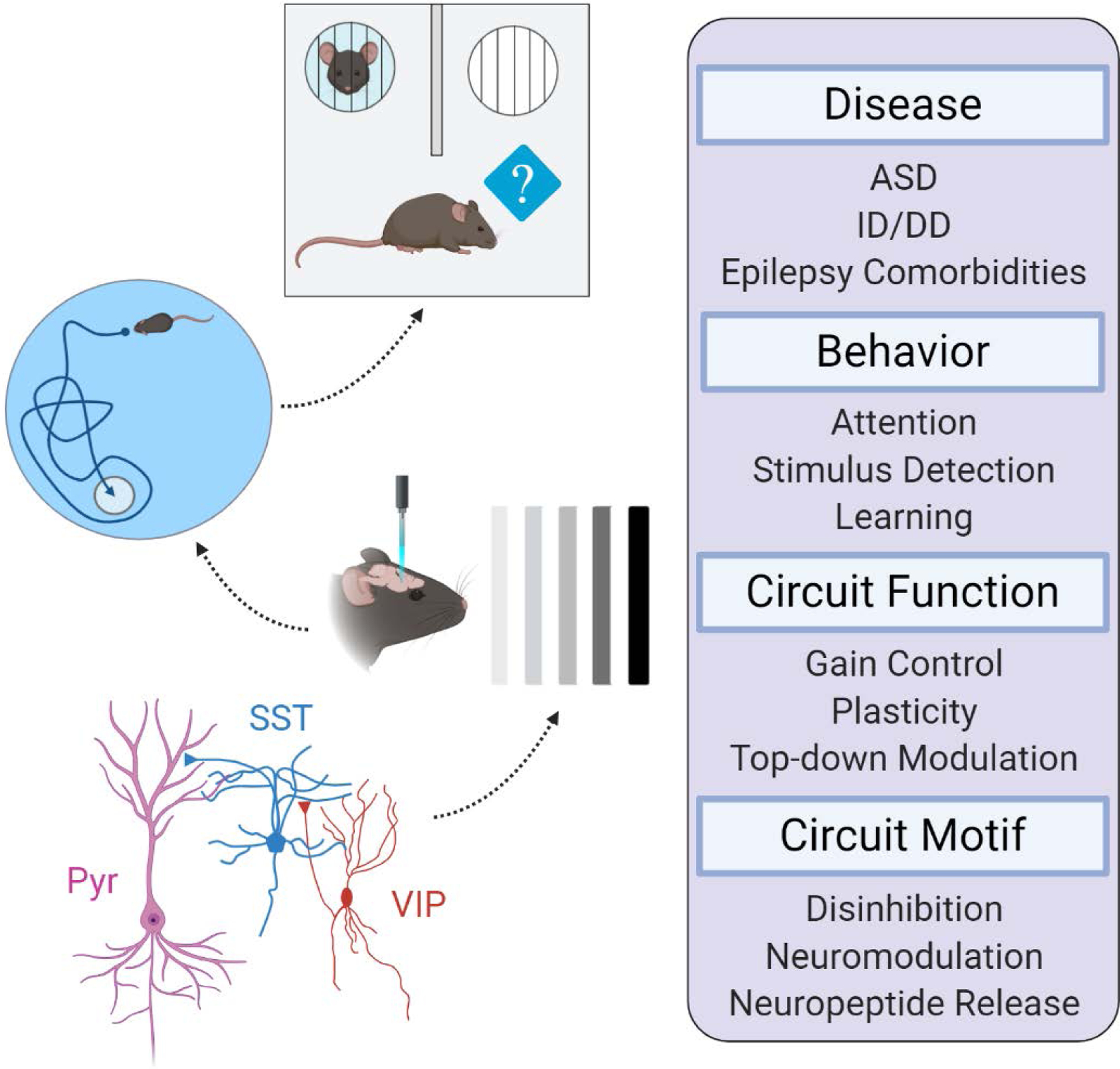
Diagrammatic representation of VIP-IN regulated circuits and their impact on biology and disease. VIP-INs are components of several repetitive circuit motifs in the cerebral cortex, including a prominent disinhibitory circuit that is conserved across many brain areas. The activity of VIP-INs not only influences network dynamics, but also the development of normal neural circuits. This shapes behaviours in several important ways, including learning, memory, and attention. Finally, impaired VIP-IN function caused by mutations in certain disease genes may cause abnormal circuit function and behaviour that underlies NDD endophenotypes like autism spectrum disorder (ASD), intellectual disability (ID), and developmental delay (DD). Figure created with BioRender.com.
Circuit functions of VIP-INs
The existence of VIP-expressing as well as IN-targeting cells in neocortex and hippocampus has been known for 30–40 years [5–9]. Comprising only 10% of cerebral cortex INs [3], understanding of VIP-IN function has been accelerated by the availability of genetic tools to target discrete IN subpopulations [10–12]. Although it is the case that all INs target other INs to some extent, several studies using VIP-Cre mice to record, image, and manipulate VIP-INs indicate that they preferentially inhibit other INs, increase activity of local pyramidal neurons, and are functionally disinhibitory [13–16]. There is considerable heterogeneity within VIP-INs including non-overlapping populations marked by the expression of cholecystokinin and calretinin, as well as a subset of VIP-INs that corelease acetylcholine [12,17–23]. There is some evidence that VIP itself can shape cerebral bloodflow and neuronal excitability, but this has not been specifically tied to VIP-IN activity [24–26]. It remains largely unknown how this within-type diversity relates to VIP-IN circuit function.
Despite unresolved aspects of diversity within the VIP-IN subclass, there remain several core features of circuits containing VIP-INs that are remarkably well conserved across brain regions [27]. For example, VIP-INs are uniformly recruited by cholinergic neuromodulation [7,15,28–30], and preferentially target SST-INs [13–15,31–34]. Taken together, this means that VIP-INs disinhibit the dendrite-targeted GABAergic input provided by SST-INs in brain states characterized by increased cholinergic tone. These occur during active behaviors like running and whisking in rodents, and are also marked by enhanced hippocampal theta rhythm [15,30,35–40]. VIP-INs express nicotinic [7,18,21,29] and muscarinic [41] acetylcholine receptors, as well as receptors for other neuromodulators including the ionotropic serotonin receptor 5-HT3R [28,42]. Beyond direct depolarization by acetylcholine, a diverse repertoire of metabotropic receptors shapes the intrinsic excitability of VIP-INs through regulating ionic conductances such as M-type potassium current [41] and T-type calcium current [28]. VIP-INs also integrate long-range cortico-cortical input [14,43]. Overall, this positions VIP-INs as a key component of top-down neuromodulation in the neocortex, defining the cellular and circuit basis for how VIP-INs shape cortical processing.
VIP-INs shape response to sensory inputs
VIP-INs play a particularly important role in shaping neocortical response to sensory inputs. The canonical VIP-IN disinhibitory circuit is found in essentially all primary sensory areas in rodents. In visual cortex (V1), VIP-IN recruitment during active behaviors increases the gain of layer 2/3 pyramidal neurons without affecting their orientation selectivity [15,30]. Correspondingly, optogenetic stimulation of VIP-INs during a visual contrast detection task improves performance, while coactivating either SST or PV-INs reduces the ability of the mouse to detect lower contrasts [44]. Long range projections from the cingulate cortex activate this circuit, providing another mechanism for top down modulation of early visual processing analogous to the frontal eye fields in primates [43]. This pathway drives narrow bands of disinhibition within visual columns and enhances both visual responses and performance on a discrimination task.
VIP-INs in auditory cortex (A1) are strongly activated by cholinergic stimulation [29], but the exact relationship between locomotion and VIP-IN-mediated disinhibition may be different than in V1 [45]. Nevertheless, direct VIP-IN activation disinhibits a subset of tone-responsive neurons, resulting in an increase in the gain of the corresponding tuning curves [16]. Cortico-cortical motor signals arising from whisking act in parallel to ascending cholinergic neuromodulation to drive VIP-IN activity in barrel cortex (S1) [14]. Interestingly, whisking causes inhibition of a subset of Martinotti-type SST-INs via VIP-INs, yet other SST-INs are actually recruited by muscarinic acetylcholine receptor activation [31]. VIP-INs are clearly able to increase the gain of sensory responses in primary sensory areas, but there may be meaningful region-specific differences in what is otherwise a highly conserved circuit motif.
While top down disinhibition through VIP-INs enhances the gain of sensory signals on a trial-by-trial basis, VIP-INs also promote plasticity and themselves exhibit plastic responses, which could influence developmental processes. Indeed, theoretical work suggests that disinhibition may be a mechanism for associative learning in the neocortex [46]. Supporting this hypothesis, inactivation, or activation, of VIP-INs in V1 can impair or enhance plasticity after monocular deprivation, respectively [47]. While the response of V1 VIP-INs to simple visual stimuli like drifting gratings is variable [48,49], VIP-INs are suppressed by familiar natural images but strongly activated by novel images during a discrimination task [50]. Moreover, VIP-INs show strong ramping activity during the omission of familiar images in the same task, suggesting that VIP-IN activity may enhance the cortical representation of salient sensory events such as the presentation of novel stimuli or the absence of expected stimuli. In A1, VIP-INs are recruited by signals reinforcing both reward and avoidance [16], further suggesting that VIP-INs are involved in encoding the salience of input features across sensory neocortex.
VIP-INs influence attention and performance across multiple learning paradigms
In addition to shaping sensory processing, VIP-INs influence learning, attention, and plasticity in distributed brain regions. VIP-INs in the basolateral amygdala (BLA) are recruited by aversive foot shocks in a fear conditioning paradigm, and VIP-IN activity is enhanced when the shock is unexpected, or is stronger than expected [34]. Furthermore, blocking VIP-IN activity during the unconditioned foot shock stimulus reduces projection neuron activity and prevents normal fear conditioning. In area CA1 of the hippocampus, VIP-INs are also modulated by locomotion [37,38], but respond strongly to rewards during spatial learning tasks [37]. Blocking VIP-IN activity abolishes the remapping of place cells in CA1 around the location of a rewarding stimulus and negatively impacts the performance of mice learning the task, while direct activation of VIP-INs concurrent with the reward improves performance.
Finally, direct activation of VIP-INs in the prefrontal cortex (PFC) improves performance on a delayed forced choice task, but activation of PV, SST, or pyramidal neurons during the task all decrease performance [51]. Some VIP-INs also show ramping activity during the delay between the cue and test response, and their ability to increase both correct hits and correct rejections argues that they improve attention to stimulus presentation during this working memory task. Similarly, VIP-INs in the PFC are recruited when a mouse is in the open arm of an elevated plus maze, and inhibiting VIP-IN activity increases exploration of the open arm [52]. Thus, VIP-INs in PFC, amygdala, and hippocampus contribute to distinct learning modalities and respond to a variety salient signals of both positive and negative valence.
VIP-INs are required for normal circuit development
Overall, VIP-INs have emerged as important components of the cortical circuits underlying attention and arousal, particularly by attaching salience to features of sensory input. Through these functions, VIP-IN activity also has a profound impact on multiple mechanisms of cortical plasticity and forms of learning. This positions VIP-INs as a potentially critical yet nearly unexplored cellular locus of circuit dysfunction in NDDs. The work described above defines roles for VIP-INs in cerebral cortical circuit operations, but it is now known that proper VIP-IN function is also required for normal development of these same circuits. Conditional VIP-IN deletion of ErBb4, a receptor important for normal migration and synapse formation, results in profound network abnormalities in V1 [53]. This includes disruption of VIP-IN functions such as state transitions and gain modulation during locomotion, but this perturbation also affects basic network features including LFP-spike phase locking and orientation selectivity of layer II/III pyramidal neurons. Interestingly, pyramidal neuron visual responses in the conditional ErBb4 knockout mice closely resemble WT responses at post-natal day (P) 15–18, but these responses progressively degrade as the mice age, further supporting a dynamic intersection between VIP-IN function in established circuits and influence over developmental processes. Critically, these circuit-level deficits cause impaired performance during a contrast discrimination task, suggesting that VIP-IN dysfunction gives rise to pathalogical brain-state regulation that may lead to abnormal behavior.
VIP-IN dysfunction is implicated in two prominent NDDs
Several recent studies directly link impaired VIP-IN function to NDDs through the use of genetic mouse models of prominent human syndromes. Rett syndrome is a early-onset NDD characterized by seizures, intellectual disability (ID), features of ASD, and repetitive stereotypical hand movements [54,55]. Most cases are found in girls and are caused by missense variants or deletions in the gene encoding methyl-CpG binding protein 2 (MECP2) located on the X chromosome. Hemizygous male mice lacking a copy of Mecp2 show many features consistent with Rett syndrome [56,57], and, while Mecp2 is expressed in essentially all neurons, conditional deletion of Mecp2 in cerebral-cortical INs replicates many of the core features of the global model [58]. Interestingly, mice also show electrophysiological and behavioral deficits when deletion of Mecp2 is further restricted to VIP-INs [58]. In particular, cortical state transitions at the onset of locomotion are diminished, and, while there is no increase in seizures or mortality, the mice show abnormal patterns of social behavior including a failure to display preference for a novel mouse versus an empty chamber. This phenotype is characteristic of many mouse models of ASD [59,60], and it is notable that the features of ASD can be dissociated from the seizure phenotype in this Rett model by selective manipulation of VIP-INs. PV or SST-IN specific deletions of Mecp2 did not cause the same social defecits. While the primarily disinhibitory role of VIP-INs may not relate to seizure succeptability in Rett syndrome, disruption of VIP-IN circuits may underlie some aspects of ASD and/or ID.
VIP-INs may also play an important role in Dravet Syndrome, a severe NDD and epileptic encephalopathy that is characterized by epilepsy, ASD, moderate to severe ID, and a high risk of sudden unexpected death (SUDEP) [61–63]. Cases are caused by pathogenic heterozygous loss of function variants in SCN1A [64], the gene encoding the voltage gated sodium channel α subunit Nav1.1, which is expressed in nearly all INs and is an important determinant of intrinsic excitability of these cells. While VIP-INs were initially thought to only express Nav1.2 [65], Nav1.1 is expressed at both the mRNA [4,18,21] and protein level [41]. PV, SST, and VIP-INs from multiple cortical regions exhibit impaired action potential generation and repetitive firing in Scn1a+/− mice, although these abnormalities display different developmental time courses [41,66–70]. For instance, neocortical PV-INs are most dysfunctional around P16-P21, corresponding to the onset of Nav1.1 expression when the mice are most prone to seizures and SUDEP, but regain normal excitability by young adulthood (P50+) [66]. CA1 horizontal stratum-oriens INs (most likely expressing SST) also show a narrow window of severe impairment around P21–24 and partially regain their function in adult mice [70]. In contrast, VIP-INs are equally impaired in juvenile and young adult Scn1a+/− mice [41] andadult Scn1a+/− mice continue to show social deficits shared by many ASD models [71]. While it is not clear how and why some IN subpopulations recover function in Scn1a+/− mice – perhaps through homeostatic upregulation of other Nav1.X channel subunits – the persistent VIP-IN dysfunction could partially underlie the durable social and cognitive deficits seen in Scn1a+/− mice. Overall, these findings closely mirror what is known about the human disease; seizure frequency typically decreases with age yet ID and ASD are durable and longstanding [72].
VIP-INs may be involved in broader neuropsychiatric disorders
Dravet syndrome and Rett syndrome are interesting early examples of VIP-IN dysfunction in NDDs. However, there is increasing consensus that many neuropsychiatric disorders have ‘developemental’ origins, including prominent clinical entities like major depression, bipolar disorder, schizophrenia, and substance abuse/addiction [73]. In mice, expression of an α5 nicotinic receptor (CHRNA5) human variant associated with nicotine abuse drastically decreases spontaneous VIP-IN activity in vivo with a corresponding increase in SST-IN activity [74]. This also causes decreased PFC network activity, and thus may underlie the hypofrontality seen in patients with this gene variant. Interestingly, Chrna5 variant mice also have social deficits – a recurring theme in all three of the above examples. At a mechanistic level, some VIP-INs in the anterior cingulate cortex respond directly to social stimuli [75], but it remains unknown how VIP-INs influence social behavior. This represents exciting new territory for basic research into VIP-IN function. The shared features of these results compared to the Dravet and Rett syndrome models point to a broader role for VIP-INs in NDDs, with particular reference to features of ASD and ID.
Searching for diseases genes important for VIP-IN function
The brief but compelling information summarized above will hopefully lead to investigation of VIP-IN function in a host of disease models in the coming years, but is it possible to predict which NDDs are more likely to involve VIP-IN dysfunction? Cutting edge tools and open source big data approaches championed by the Allen Instutue and others may allow us to do just that. Single cell RNA sequencing (scRNA-seq) provides quantitative gene expression data on thousands of genes per cell, and is scalable up to millions of cells given the appropriate resources. The Allen Institute has several large scale scRNA-seq data-sets available for public use [4,18,20], including the most recent set of data for over 1.1 million neurons from 20 different mouse brain regions using a combination of Smart-Seq and 10X genomics [4]. This massive amount of high dimensional quantitative data is well-positioned for identification of cell types, particularly when correlated with morphological and electrophysiological data [20,76–78]. However, such data is also useful for hypothesis generation when there is limited direct data available for use [41,77]. We analyzed this scRNA-seq dataset (available at https://portal.brain-map.org/atlases-and-data/rnaseq) to probe the expression of genes associated with NDDs in VIP-INs.
We selected a group of disease genes used for clinical diagnosis of NDDs that are found on standard academic and commercially available NextGen sequencing panels (CHOP Epilepsy Panel v1.0 and a commercial ASD Panel). These two panels include approximately 250 unique genes (140 on the ASD panel, 76 on the Epilepsy panel, 25 shared between the two panels) with murine homologues found in the Allen Brain data. The most common genetic causes of NDDs found in children, including both Dravet (SCN1A) and Rett syndrome (MECP2), are covered by this selection of genes. We first separated the scRNA-seq data based on clustering results from the Allen Institute [4] and focused on gene expression in VIP, SST, and PV-INs compared to all other INs (which include the Lamp5, Scng, and Meis2 subclasses) as well as all glutamatergic (Glu) neurons. We then calculated the trimmed mean expression (25%–75% of Log2(CPM)) of each gene from the above panels and ranked them based on relative expression in VIP-INs by z-scoring the expression across groups (Fig. 2 A–C). Many genes from both panels were expressed at lower levels in VIP-INs (Fig. 2D) perhaps because, on average, fewer total reads and genes are recovered from VIP-INs in this dataset compared to other cell types [4].This might particularly affect detection of transcripts with low overall expression levels (see Fig. 2D, bottom left quadrant). However, there was a subset of genes that were highly expressed in VIP-INs when compared with all INs and/or all Glu neurons (Fig. 2, Table 1).
Fig. 2. Relative expression of NDD related disease genes in VIP-INs.
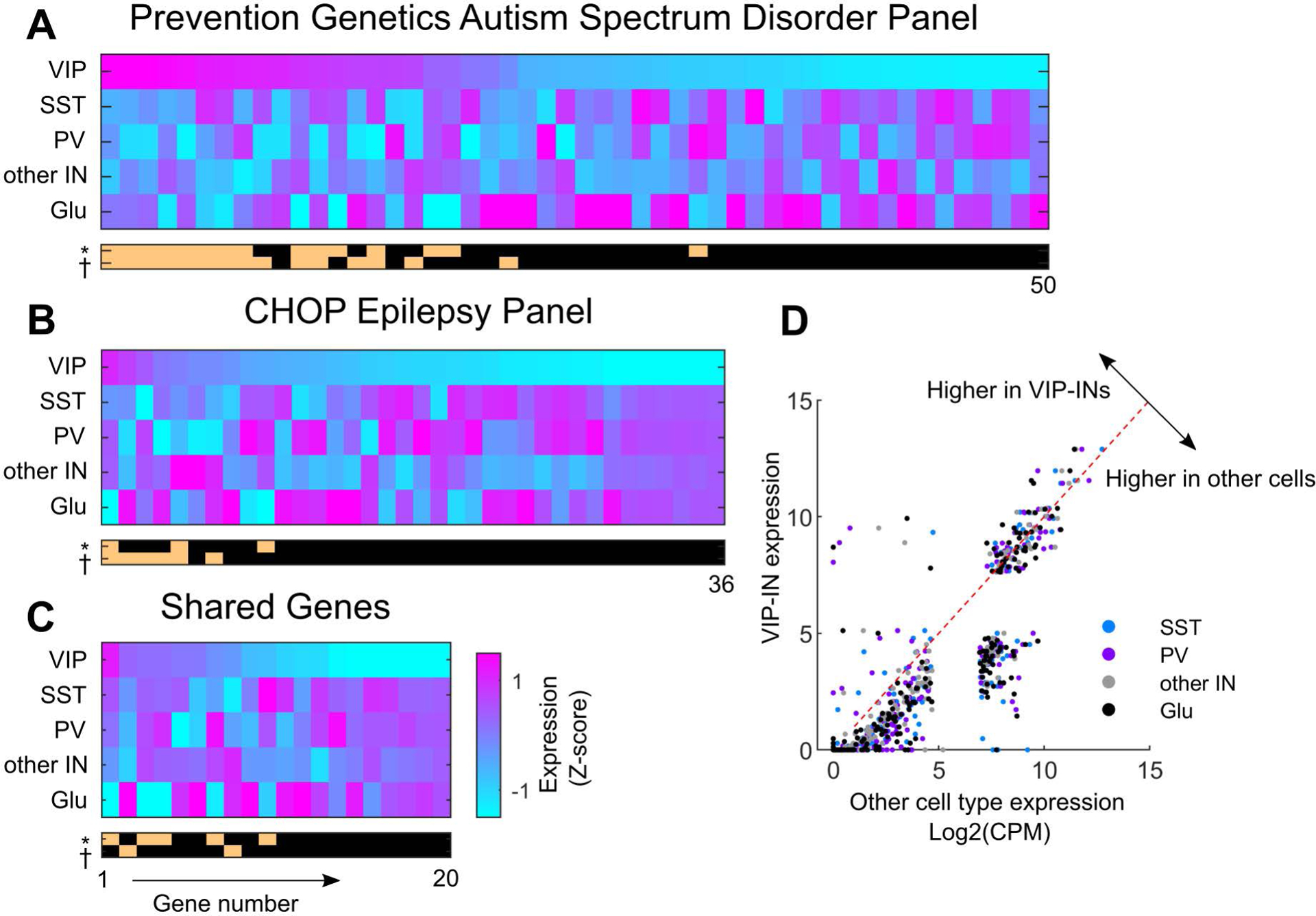
A) Heatmap showing the expression of genes in a standard NextGen sequencing panel for ASD calculated as a z-score of the trimmed mean LOG2(CPM) across the 5 indicated cell types. “Other IN” includes all INs not in the three main subclasses (VIP, SST, and PV). Genes are sorted based on relative expression in VIP-INs. Below, tan bars on a black background represent expression of each gene as significantly higher in VIP-INs compared to either all Glutamatergic cells (*) or all other INs (†, including PV, SST, and other IN). See Table 1 for raw expression values and statistical test information. B) As in Fig. 2A, but for genes only found on the CHOP Epilepsy v1.0 Panel. C) The same as Fig. 2A–B, but showing genes shared between the two panels. For clarity in Fig. 2A–C, genes with no expression in VIP-INs are omitted, including >20 genes with a mean value of 0 in all cell types, and only the first 50 out of 93 genes from the ASD panel are shown. D) A scatter plot of the trimmed mean LOG2(CPM) expression of all 241 genes, where VIP-IN expression is on the Y axis and is plotted against the other indicated cell types on the X axis. Note that equal expression in VIP-INs compared to another subclass would cause the given data point to fall on the indicated diagonal line. Most genes, particularly those that are low expressing, are expressed higher in all other cell types than in VIP-INs. The sparse distribution of points on the top-left of the diagonal line show genes that are enriched in VIP-INs.
Table 1.
Summary scRNA-seq gene expression data from the Allen Institute cell types database. Values are reported as trimmed means (25%–75%) of LOG2(CPM). Cell types were assigned based on the ‘subclass’ labels provided by the Allen Institute. ‘Glu’ includes all glutamatergic cells. Genes are ordered based on relative expression in VIP-INs (as shown in Figure 2). Significant differences between VIP-INs and all Glu (*) or all other INs combined (†) were determined by bootstrapping the difference in means 50,000 times using 10% of each sample population and a Bonferroni corrected significance cut-off. NB, some individual IN subclasses have higher expression of a given gene even if VIP-INs had higher expression than the combined IN pool. The top genes from each panel are shown up to and including all significantly different genes.
| Cell Type Specific Expression Of Neurodevelopmental Disorder Genes | |||||
|---|---|---|---|---|---|
| ASD Panel | |||||
| Gene | VIP | SST | PV | other IN | Glu |
| Nbea | 10,2 * † | 8,9 | 9,0 | 8,7 | 9,3 |
| Auts2 | 10,2 * † | 9,3 | 9,1 | 9,5 | 9,6 |
| Ank3 | 10,3 * † | 9,9 | 9,7 | 9,9 | 10,0 |
| Zbtb20 | 8,9 * † | 7,7 | 8,0 | 8,3 | 7,3 |
| Nrxn1 | 12,0 * † | 10,5 | 9,7 | 10,8 | 11,2 |
| Nrxn3 | 12,9 * † | 12,7 | 11,8 | 11,5 | 11,4 |
| Cntn6 | 5,1 * † | 4,3 | 3,0 | 1,4 | 0,5 |
| Setbp1 | 8,4 * † | 7,7 | 8,3 | 7,4 | 7,9 |
| Myt1l | 9,3 † | 9,1 | 8,6 | 8,7 | 9,1 |
| Son | 8,1 | 7,8 | 7,8 | 8,0 | 8,0 |
| Tcf4 | 11,4 * † | 10,8 | 10,9 | 11,2 | 9,5 |
| Dscam | 9,5 * † | 8,7 | 0,8 | 2,2 | 8,6 |
| Rere | 8,3 * | 8,3 | 8,2 | 8,2 | 7,8 |
| Cttnbp2 | 8,5 † | 8,0 | 7,7 | 8,1 | 8,7 |
| Cacna2d3 | 8,9 * † | 9,2 | 0,3 | 3,4 | 8,3 |
| Tnrc6b | 7,7 | 7,5 | 7,7 | 7,6 | 7,5 |
| Nfix | 8,0 † | 0,0 | 0,0 | 8,8 | 8,1 |
| Grip1 | 9,9 * | 10,4 | 10,2 | 10,6 | 3,5 |
| Cntnap2 | 11,5 * | 11,5 | 12,1 | 11,6 | 9,4 |
| Cacna1c | 7,9 | 8,2 | 7,3 | 7,9 | 8,0 |
| Tanc2 | 8,5 | 8,4 | 8,4 | 8,3 | 9,0 |
| Shank2 | 3,3 † | 2,4 | 1,8 | 3,5 | 7,4 |
| Epilepsy Panel | |||||
| Zeb2 | 9,6 * † | 8,8 | 9,3 | 9,3 | 8,0 |
| Ryr3 | 2,4 † | 1,2 | 0,0 | 0,5 | 3,2 |
| Gabra1 | 9,3 † | 4,7 | 9,8 | 9,4 | 8,7 |
| Scn3a | 2,6 † | 2,7 | 0,4 | 2,8 | 4,0 |
| Chrna4 | 0,3 * † | 0,2 | 0,0 | 0,9 | 0,0 |
| Gnao1 | 8,7 | 8,8 | 8,2 | 9,3 | 8,7 |
| Lgi1 | 4,8 † | 3,8 | 2,2 | 7,6 | 7,3 |
| Prickle1 | 7,9 | 7,5 | 8,0 | 7,9 | 9,1 |
| Atp1a3 | 9,1 | 9,4 | 9,7 | 9,2 | 8,8 |
| Kcnc1 | 5,0 * | 7,8 | 9,5 | 8,1 | 2,1 |
| Shared Genes | |||||
| Ube3a | 9,3 * | 9,2 | 9,1 | 9,0 | 8,8 |
| Grin2b | 10,4 † | 10,2 | 10,1 | 10,1 | 10,6 |
| Slc6a1 | 8,7 * | 9,3 | 10,3 | 10,5 | 0,0 |
| Scn1a | 7,8 * | 7,9 | 9,0 | 8,0 | 4,6 |
| Scn2a | 8,7 | 8,8 | 7,7 | 8,8 | 9,1 |
| Gabrb3 | 8,5 | 8,2 | 8,1 | 8,6 | 9,1 |
| Hcn1 | 8,6 * | 8,4 | 10,0 | 9,4 | 7,6 |
| Cdkl5 | 4,2 † | 1,5 | 4,0 | 7,9 | 7,8 |
| Cask | 3,6 | 4,6 | 3,6 | 4,2 | 7,1 |
| Arx | 0,5 * | 7,1 | 4,2 | 1,8 | 0,0 |
| … | |||||
| Mecp2 | 2,8 | 4,3 | 4,0 | 4,1 | 3,6 |
There were several interesting genes that emerged from this analysis that could aid the investigation into the role of VIP-INs in NDDs as well as inform our understanding of the biology underlying VIP-IN function. On the ASD Panel, NRXN1 and NRXN3 were the two most highly expressed genes in VIP-INs, and were both in the top 10 in relative expression compared to other cell types (Table 1). Neurexins (NRXN1-3) are a familiy of synaptic adhesion molecules that are critical for synapse and circuit formation during development, and are implicated in many neurospyschiatric disorders including ASD [79,80]. Their expression is uniquely regulated on a cell type and regional basis at the transcriptional, translational, and post translational level [81], yet nothing is known about how they influence VIP-IN mediated circuits. Mutations in the transcription factor TCF4 cause Pitt-Hopkins syndrome, a neurodevelopmental disorder defined by ASD with variable but typically severe developmental delay and ID [82]. TCF4 itself regulates the expression of both NRXN1 and CNTNAP2 [83], which are both enriched in VIP-INs, and pathogenic variants in NRXN1 and CNTNAP2 give rise to a Pitt-Hopkins-like syndromes [84]. However, little is known on the cell type-specific activity of any of these genes.
As an interesting aside, the neurexins, TCF4, and CNTNAP2 are also associated with neurospychiatric disorders like schizophrenia through genome wide association studies (GWAS), as are several other ASD-associated genes expressed in VIP-INs, including NBEA and CACNA1C (associated with Timothy Syndrome) [85). CHRNA5A, used in the model of nicotine addiction described above [74], is also associated with schizophrenia through GWAS [86]. While beyond the scope of this review, a similar analysis of genes related to various neuropopsychiatric disorders could provide new avenues to study the impact of VIP-INs in this class of pathology. It will be interesting to see if there are further examples of VIP-INs mediating shared characteristics between monogenetic NDDs present early in life, and complex diseases arising later in life involving oligo- or polygenic inheritance.
Many disorders present with an overlap of ASD and epilepsy in addition to Rett syndrome and Dravet syndrome. Angelman Syndrome is caused by functional silencing of the maternally-imprinted gene UBE3A (through deletion, mutation, or aberrant methylation) [87]. Interestingly, restricting deletion of Ube3a to INs enhances the abnormal EEG and seizure phenotype found in the global or Glu specific murine model [88], although relatively similar expression across IN subclasses suggests involvement of multiple cell types (Table 1). Mecp2 is present although not enriched in VIP-INs in this scRNA-seq data, but decreased Mecp2 activity can result in lower levels of Ube3a and Gabrb3 [89], both of which are found at higher levels in VIP-INs. As expected, Scn1a is highly expressed in VIP-INs compared to Glu but is similarly expressed compared to other IN subclasses.
Finally, for the Epilepsy panel genes, there were only two that were enriched in VIP-INs compared to both Glu and other INs, highlighting our suggestion that VIP-IN dysfunction may be more closely linked to ASD and ID than epilepsy. Mutations in ZEB2 cause Mowat-Wilson syndrome which is characterized by moderate to severe intelectual disability, microcephaly, a high rate of Hirschprung’s disease, as well as epilepsy (in 70–75% of cases), but is not a monogenic epilepsy syndrome per se [90,91]. While expressed at slightly higher levels in adult VIP-INs, ZEB2 is also prominently expressed accross the brain during early development which is more likely to account for the many developmental defects seen in this disorder. The second example, CHRNA4, encodes an α subunit of the nicotinic acetycholine receptor that is sparsely expressed in the brain compared to CHRNA2 and 3, similar to CHRNA5 [92]. Mutations in CHRNA4 cause a subset of autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE), a relatively pure epilepsy syndrome with some motor and cognitive comorbidities [93]. Interestingly, in two mouse models of ADNFLE expressing human Chrna4a variants, a seizure phenotype is paradoxically paired with a massive increase in inhibitory post synaptic current frequency recorded from cortical pyramidal neurons after bath application of nicotine [94]. Low dose picrotoxin is able to reduce the abnormal EEG phenotype in these mice, which is counterintuitive and suggests pathologically increased inhibition. In another study, direct activation of α4-containing nicotinic acetylcholine receptors also resulted in disinhibition in neocortical layer 5 [95]. While these results are not tied directly to VIP-IN function, they are consistent with the hypothesis that Chrna4 may be expressed on VIP-INs [7], its loss resulting in paradoxically increased inhibition in response to nicotinic stimulation. These studies highlight the complex and varied etiology of seizures, and hint at how increased inhibition could theoretically underlie epilepsy syndromes. Future studies could investigate the role of VIP-INs in this alternative framework.
For a given NDD, it is possible that the causative disease gene is expressed widely across IN or Glu subclasses (as in Ube3a and Mecp2 above). In other disorders, the causative gene may be preferentially expressed in a specific subclass or subclasses such that the disease predominantly affects a narrow population of cells. While expression of the panel genes accross cell types was correlated (Fig. 2D), the relative sparsity of genes enriched in VIP-INs suggests that there are at least a handful of diseases that follow this second pattern. We used dimensionality reduction to visualize the expression of 241 NDD-associated genes across IN subclasses (Fig. 3A). Surprisingly, the original IN subclasses identified by the Allen Institute were easily extracted from the TSNE1 and TSNE2 dimensions derived from this limited dataset using a simple branching classifier, while the same separation was not possible using random subsets of 250 genes (Fig. 3B). Therefore, the information contained within this selection of NDD-associated transcripts is sufficient to distiguish IN subclasses from one another. We suggest that differential disease gene expression across IN subclasses may manifest as distinct disease phenotypes (e.g., epilepsy, features of ASD, ADHD, etc) associated with each subclass. This highlights the potential of investigating the role of disease genes using cell type-specific genetic tools.
Fig. 3. Separation of IN subclasses based on NDD diagnostic panel genes.

A) TSNE plot generated from the first 50 principal components of the 241 unique genes between the ASD and epilepsy panel using the default settings in MATLAB 2019b. Individual cells are coloured based on the cell subclasses identified by the Allen Brain Institute, indicated in the legend. B) The same done for a random selection of 250 genes. The subtypes are more separable when the ASD/Epilepsy panel gene data is used. C) Receiver operating characteristic (ROC) curves demonstrating the effectiveness of a binary decision tree classifier fitted to the TSNE1 and TSNE2 dimensions in A and B at recovering the original subclasses labelled by the Allen Institute. Each ROC curve is created using the posterior probabilities from the trained decision tree classifier model. For each subclass, the coloured line represents a model based on the ASD/Epilepsy gene panels, and the grey lines represent 5 different selections of 250 random genes. The ‘X’ marks the optimal ROC point for each model. In all cases, the ASD/Panel genes outperform random selections.
Points of convergence between experimental models of ASD
A general theme emerging from the scRNA-seq data discussed above is that disease-related genes enriched in VIP-INs are more highly associated with ASD/ID relative to developmental epilepsy syndromes. Disorders that have prominent features of both epilepsy and ASD/ID may be the result of mixed dysfunction of multiple cell subclasses. This is perhaps best exemplified by the fact that lesions of PV-INs (genetic or otherwise) tend to produce epilepsy in mice [68,96], while lesions restricted to VIP-INs do not produce epilepsy and instead affect attention, learning, and social behavior [47,53,58]. We have discussed the evidence supporting a role for VIP-INs in the pathogenesis of NDDs with features of ASD; however, it is useful to consider this within the broader context of pathological development and function of circuit elements related to ASD, particularly in relation to the role of disinhibition in dendritic integration. For instance, SCN2A is also an ASD-associated gene expressed in INs but more prominently in glutamatergic neurons (Table 1). Unlike the DS model, Scn2a+/− mice exhibit normal PV and SST-IN excitability, yet have early impairement in layer 5b principal neuron excitability (P4) and striking deficiencies in dendritic action potential backpropogation later in development (P27+) [97]. This critically intereferes with spike timing dependent plasticity, and is also correlated with more immature dendritic morphology and a disrupted AMPA-NMDA receptor balance. These results fit well with findings from other ASD models showing abnormalities in synaptic function, either directly involving synaptic proteins or through gene transcription [98,99]. In addition, dendritic inhibition provided by SST-INs has precise and compartmentalized control over dendritic integration [100–102]. And via inhibition of SST-INs, VIP-INs are poised to influence processes such as action potential back propogation, spike timing dependent plasticity, and synaptic integration, including during developmental critical periods [47,53]. Thus, VIP-IN dysfunction may converge on this common conceptual framework for ASD-associated genes related to synaptic plasticity and dendritic excitability.
Conclusion
Towards cell type-specific therapies
We hope that the evidence so far highlights the need to investigate cell-type specific function in NDDs. But why not instead focus on targeting and correcting disease-associated genetic lesions at a global level? Part of the answer to this question lies in the heterogeneity of cell type-specific circuit functions that also change over the course of development. Taking Dravet syndrome as a case study, SCN1A was identified as the causitive gene twenty years ago [64], but the prognosis for children with Dravet syndrome remains bleak. Seizure control in Dravet Syndrome is poor, and SUDEP remains a terrifying spectre for parents. However, even in those patients who attain seizure control, ID and features of ASD remain, and the vast majority are dependent on others for care [72,103,104]. Aspects of this complex pathology may arise from dysfunction of distinct subclasses of INs that varies over time, suggesting that targeted interventions given at precise developmental timepoints may be necessary to address the distinct aspects of the disease.
Identifying a way to selectively target and manipulate INs could provide an avenue to develop new therapies for Dravet Syndrome. In contrast, global upregulation of SCN1A could theoretically be harmful, as patients with gain of function variants of SCN1A suffer from Familial Hemiplegic Migraine Type 3 [105,106] and rare SCN1A variants identified in an early-onset form of Dravet syndrome have been shown to act via gain of ion channel function [107,108]. VIP-INs remain dysfunctional in young adult Scn1a+/− mice, while PV-INs follow a different developmental trajectory; hence, SCN1A upregulation in discrete IN subclasses would allow for treatments informed by these developmental trajectories targeted to IN subclass-specific disease features. A potential solution to achieving this lies in the SCN1A gene itself. There are different enhancer elements within the SCN1A locus that drive expression in distinct cell subclasses, including E2 (which drives expression in PV-INs) and E6 (which is limited to VIP-INs) [109]. Packaging the E2 enhancer element into a virus creates a tool that selectively drives expression of a desired payload in PV-INs. Moreover, this approach appears to be equally effective in human neurons from cultured brain slices. Ultimately, cell type-specific technology like this could be paired with a gene therepy tool, such as the recently developed catalytically dead Cas9 (dCas9) system that is able to boost Nav1.1 expression and enhance the intrinsic excitability of cultured INs when targetted to the proximal promoter region of the Scn1a locus [110]. Hypothetically, targeting Scn1a-dCas9 or another gene therepy specifically to VIP-INs with the E6 promoter could improve their function without off-target effects and serve as a way to both investigate and address the underlying VIP-IN mediated component of Dravet Syndrome pathology. Perhaps, this approach could be broadly adapted as a therapy for ASD or syndromic NDDs, particularly once we know how mutations in more of the NDD genes identified above affect VIP-IN function.
Novel precision therepeutics that lack subclass specific targeting could still be effective, but ultimately will be limited by the complex and intertwined developmental trajectories of INs. For instance, a single early (P2) dose of an antisense oligonucleotide (ASO) targeting the ‘poison exon’ [111] in Scn1a suppresses a nonproductive alternative splicing event and boosts expression of the remaining copy of Scn1a, dramatically suppressing seizures and prevented SUDEP in Scn1a+/− mice [112]. However, the effect of treatment later in development (P14) is reduced, and treatment after the appearance of spontaneous seizures (P18) – a likely requirement in human patients – is unknown. Futher complicating matters, treatment with an ASO targetting Scn8a also prevents seizures and SUDEP in Scn1a+/− mice [113], perhaps indicating that disease symptoms during the chronic phase of Dravet Syndrome may involve homeostatic or pathological upregulation of Nav1.X channel subunits. Treatment boosting Scn1a expression in PV-INs later in life, after normalization of intrisic excitability, might be less effective at controlling seizures. However, treatment at a later developmental timepoint might address the durable defecit in VIP-INs while having limited unintended consequences. As this approach enters human trials (MONARCH Study; monarchstudy.com), it will be critical to determine whether ASOs are capable of not only early seizure control, but stable seizure control and SUDEP risk reduction as well as an improved neurodevelopmental trajectory without adverse effects.
As the story of VIP-INs unfolds, accumulating evidence suports the involvement of VIP-IN dysfunction in the pathogenesis of NDDs. Cutting-edge approaches in human and mouse genetics, human induced pluripotent stem cell technology, single cell omics, cell type-specific electrophysiology, opto/chemogenetic manipulation, and in vivo imaging in model systems will eventually improve our understanding of VIP-INs in disease contexts. Ultimately, gene-targeted cell type-specific therapies are on the horizon, informed by continued mechanistic investigation into the basic biological function of disease genes and the circuit roles of defined classes of cells including VIP-INs.
Acknowledgements
We would like to thank the Allen Institute for supporting open science.
Funding Sources
This work was supported by NIH NINDS F31 NS111803 to K.M.G. and NIH NINDS K08 NS097633 and R01 NS110869 and a Career Award for Medical Scientists from the Burroughs Wellcome Fund to E.M.G.
Footnotes
Conflict of Interest Statement
The authors have no conflicts of interest to declare.
References
- 1.Hattori R, Kuchibhotla KV, Froemke RC, Komiyama T Functions and dysfunctions of neocortical inhibitory neuron subtypes. Nat Neurosci. 2017;20(9):1199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kepecs A, Fishell G. Interneuron Cell Types: Fit to form and formed to fit. Nature. 2014;505(7483):318–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rudy B, Fishell G, Lee S, Hjerling-Leffler J. Three groups of interneurons account for nearly 100% of neocortical GABAergic neurons. Dev Neurobiol. 2011January1;71(1):45–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yao Z, Nguyen TN, van Velthoven CTJ, Goldy J, Sedeno-Cortes AE, Baftizadeh F, et al. A taxonomy of transcriptomic cell types across the isocortex and hippocampal formation. bioRxiv. 2020January1;2020.03.30.015214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Acsády L, Görcs TJ, Freund TF. Different populations of vasoactive intestinal polypeptide-immunoreactive interneurons are specialized to control pyramidal cells or interneurons in the hippocampus. Neuroscience. 1996;73(2):317–34. [DOI] [PubMed] [Google Scholar]
- 6.Porter JT, Cauli B, Staiger JF, Lambolez B, Rossier J, Audinat E. Properties of bipolar VIPergic interneurons and their excitation by pyramidal neurons in the rat neocortex. Eur J Neurosci. 1998December;10(12):3617–28. [DOI] [PubMed] [Google Scholar]
- 7.Porter JT, Cauli B, Tsuzuki K, Lambolez B, Rossier J, Audinat E. Selective excitation of subtypes of neocortical interneurons by nicotinic receptors. J Neurosci. 1999July;19(13):5228–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hájos N, Acsády L, Freund TF. Target selectivity and neurochemical characteristics of VIP-immunoreactive interneurons in the rat dentate gyrus. Eur J Neurosci. 1996;8(7):1415–31. [DOI] [PubMed] [Google Scholar]
- 9.Dávid C, Schleicher A, Zuschratter W, Staiger JF. The innervation of parvalbumin-containing interneurons by VIP-immunopositive interneurons in the primary somatosensory cortex of the adult rat. Eur J Neurosci. 2007;25(8):2329–40. [DOI] [PubMed] [Google Scholar]
- 10.Taniguchi H, He M, Wu P, Kim S, Paik R, Sugino K, et al. A Resource of Cre Driver Lines for Genetic Targeting of GABAergic Neurons in Cerebral Cortex. Neuron. 2011September;71(6):995–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dimidschstein J, Chen Q, Tremblay R, Rogers SL, Saldi G-A, Guo L, et al. A viral strategy for targeting and manipulating interneurons across vertebrate species. Nat Neurosci. 2016December31;19(12):1743–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.He M, Tucciarone J, Lee S, Nigro MJ, Kim Y, Levine JM, et al. Strategies and Tools for Combinatorial Targeting of GABAergic Neurons in Mouse Cerebral Cortex. Neuron. 2016;92(2):555. [DOI] [PubMed] [Google Scholar]
- 13.Pfeffer CK, Xue M, He M, Huang ZJ, Scanziani M. Inhibition of inhibition in visual cortex: the logic of connections between molecularly distinct interneurons. Nat Neurosci. 2013August30;16(8):1068–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee S, Kruglikov I, Huang ZJ, Fishell G, Rudy B. A disinhibitory circuit mediates motor integration in the somatosensory cortex. Nat Neurosci. 2013November6;16(11):1662–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fu Y, Tucciarone JM, Espinosa JS, Sheng N, Daniel P, Nicoll RA, et al. A cortical circuit for gain control by behavioral state. Cell. 2014;156(6):1139–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pi H-J, Hangya B, Kvitsiani D, Sanders JI, Huang ZJ, Kepecs A. Cortical interneurons that specialize in disinhibitory control. Nature. 2013November6;503(7477):521–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Prönneke A, Scheuer B, Wagener RJ, Möck M, Witte M, Staiger JF. Characterizing VIP neurons in the barrel cortex of VIPcre/tdTomato mice reveals layer-specific differences. Cereb Cortex. 2015;25(12):4854–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tasic B, Menon V, Nguyen TN, Kim TK, Jarsky T, Yao Z, et al. Adult mouse cortical cell taxonomy revealed by single cell transcriptomics. Nat Neurosci. 2016;19(2):335–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gouwens NW, Sorensen SA, Berg J, Lee C, Jarsky T, Ting J, et al. Classification of electrophysiological and morphological neuron types in the mouse visual cortex. Nat Neurosci. 2019;22(7):1182–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gouwens NW, Sorensen SA, Baftizadeh F, Budzillo A, Lee BR, Jarsky T, et al. Toward an Integrated Classification of Cell Types: Morphoelectric and Transcriptomic Characterization of Individual GABAergic Cortical Neurons. bioRxiv. 2020; [Google Scholar]
- 21.Paul A, Crow M, Raudales R, He M, Gillis J, Huang ZJ. Transcriptional Architecture of Synaptic Communication Delineates GABAergic Neuron Identity. Cell. 2017;171(3):522–539.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Granger AJ, Wang W, Robertson K, El-Rifai M, Zanello AF, Bistrong K, et al. Cortical ChAT+ neurons cotransmit acetylcholine and GABA in a target-and brain-region-specific manner. Elife. 2020;9:1–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Obermayer J, Luchicchi A, Heistek TS, de Kloet SF, Terra H, Bruinsma B, et al. Prefrontal cortical ChAT-VIP interneurons provide local excitation by cholinergic synaptic transmission and control attention. Nat Commun. 2019November;10(1):461723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cunha-Reis D, Caulino-Rocha A. VIP Modulation of Hippocampal Synaptic Plasticity: A Role for VIP Receptors as Therapeutic Targets in Cognitive Decline and Mesial Temporal Lobe Epilepsy. Front Cell Neurosci. 2020;14(June):1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cunha-Reis D, Ribeiro JA, Sebastião AM. VIP enhances synaptic transmission to hippocampal CA1 pyramidal cells through activation of both VPAC1 and VPAC2 receptors. Brain Res. 2005;1049(1):52–60. [DOI] [PubMed] [Google Scholar]
- 26.Yang K, Lei G, Jackson MF, MacDonald JF. The involvement of PACAP/VIP system in the synaptic transmission in the hippocampus. J Mol Neurosci. 2010;42(3):319–26. [DOI] [PubMed] [Google Scholar]
- 27.Guet-McCreight A, Skinner FK, Topolnik L. Common Principles in Functional Organization of VIP/Calretinin Cell-Driven Disinhibitory Circuits Across Cortical Areas. Front Neural Circuits. 2020;14(June):1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Prönneke A, Witte M, Möck M, Staiger JF. Neuromodulation leads to a burst-tonic switch in a subset of VIP neurons in mouse primary somatosensory (Barrel) cortex. Cereb Cortex. 2020;30(2):488–504. [DOI] [PubMed] [Google Scholar]
- 29.Askew CE, Lopez AJ, Wood MA, Metherate R. Nicotine excites VIP interneurons to disinhibit pyramidal neurons in auditory cortex. Synapse. 2019;(May):e22116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reimer J, Froudarakis E, Cadwell CR, Yatsenko D, Denfield GH, Tolias AS. Pupil Fluctuations Track Fast Switching of Cortical States during Quiet Wakefulness. Neuron. 2014;84(2):355–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Muñoz W, Tremblay R, Levenstein D, Rudy B. Layer-specific modulation of neocortical dendritic inhibition during active wakefulness. Science. 2017March3;355(6328):954–9. [DOI] [PubMed] [Google Scholar]
- 32.Walker F, Möck M, Feyerabend M, Guy J, Wagener RJ, Schubert D, et al. Parvalbumin-and vasoactive intestinal polypeptide-expressing neocortical interneurons impose differential inhibition on Martinotti cells. Nat Commun. 2016;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tyan L, Chamberland S, Magnin E, Camire O, Francavilla R, David LS, et al. Dendritic Inhibition Provided by Interneuron-Specific Cells Controls the Firing Rate and Timing of the Hippocampal Feedback Inhibitory Circuitry. J Neurosci. 2014;34(13):4534–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Krabbe S, Paradiso E, d’Aquin S, Bitterman Y, Courtin J, Xu C, et al. Adaptive disinhibitory gating by VIP interneurons permits associative learning. Nat Neurosci. 2019;22(11):1834–43. [DOI] [PubMed] [Google Scholar]
- 35.McGinley MJ, Vinck M, Reimer J, Batista-Brito R, Zagha E, Cadwell CR, et al. Waking State: Rapid Variations Modulate Neural and Behavioral Responses. Neuron. 2015;87(6):1143–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jackson J, Ayzenshtat I, Karnani MM, Yuste R. VIP+ interneurons control neocortical activity across brain states. J Neurophysiol. 2016;115(6):3008–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Turi GF, Li WK, Chavlis S, Pandi I, O’Hare J, Priestley JB, et al. Vasoactive Intestinal Polypeptide-Expressing Interneurons in the Hippocampus Support Goal-Oriented Spatial Learning. Neuron. 2019January;101(6):1150–1165.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Luo X, Guet-Mccreight A, Villette V, Francavilla R, Marino B, Chamberland S, et al. Synaptic Mechanisms Underlying the Network State-Dependent Recruitment of VIP-Expressing Interneurons in the CA1 Hippocampus. Cereb Cortex. 2020;30(6):3667–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Khan AG, Poort J, Chadwick A, Blot A, Sahani M, Mrsic-flogel TD, et al. selectivity and interactions of GABAergic interneuron classes in visual cortex. Nat Neurosci. 2018;21(June). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gasselin C, Hohl B, Vernet A, Crochet S, Petersen CCH. Cell-type-specific nicotinic input disinhibits mouse barrel cortex during active sensing. Neuron. 2021;1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goff KM, Goldberg EM. Vasoactive intestinal peptide-expressing interneurons are impaired in a mouse model of dravet syndrome. Elife. 2019;8:1–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee S, Hjerling-leffler J, Zagha E, Fishell G. The largest group of superficial neocortical GABAergic interneurons expresses ionotropic serotonin receptors. J Neurosci. 2010;30(50):16796–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang S, Xu M, Kamigaki T, Do JPH, Chang W-C, Jenvay S, et al. Long-Range and Local Circuits for Top-Down Modulation of Visual Cortical Processing. Science. 2014;345(6197):660–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cone JJ, Scantlen MD, Histed MH, Maunsell JHR. Different inhibitory interneuron cell classes make distinct contributions to visual contrast perception. eNeuro. 2019;6(1):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bigelow J, Morrill RJ, Dekloe J, Hasenstaub AR. Movement and VIP interneuron activation differentially modulate encoding in mouse auditory cortex. eNeuro. 2019;6(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wilmes KA, Clopath C. Inhibitory microcircuits for top-down plasticity of sensory representations. Nat Commun. 2019;10(1):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fu Y, Kaneko M, Tang Y, Alvarez-Buylla A, Stryker MP. A cortical disinhibitory circuit for enhancing adult plasticity. Elife. 2015;2015(4):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.de Vries SEJ, Lecoq JA, Buice MA, Groblewski PA, Ocker GK, Oliver M, et al. A large-scale standardized physiological survey reveals functional organization of the mouse visual cortex. Nat Neurosci. 2020January16;23(1):138–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Millman DJ, Ocker GK, Caldejon S, Kato I, Larkin JD, Lee EK, et al. VIP interneurons in mouse primary visual cortex selectively enhance responses to weak but specific stimuli. Elife. 2020;9:1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Garrett M, Manavi S, Roll K, Ollerenshaw DR, Groblewski PA, Ponvert ND, et al. Experience shapes activity dynamics and stimulus coding of VIP inhibitory cells. Elife. 2020;9:1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kamigaki T, Dan Y. Delay activity of specific prefrontal interneuron subtypes modulates memory-guided behavior. Nat Neurosci. 2017April24;20(6):854–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee AT, Cunniff MM, See JZ, Wilke SA, Luongo FJ, Ellwood IT, et al. VIP Interneurons Contribute to Avoidance Behavior by Regulating Information Flow across Hippocampal-Prefrontal Networks. Neuron. 2019June;102(6):1223–1234.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Batista-Brito R, Vinck M, Ferguson KA, Chang JT, Laubender D, Lur G, et al. Developmental Dysfunction of VIP Interneurons Impairs Cortical Circuits. Neuron. 2017;95(4):884–895.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chahrour M, Zoghbi HY. The Story of Rett Syndrome: From Clinic to Neurobiology. Neuron. 2007;56(3):422–37. [DOI] [PubMed] [Google Scholar]
- 55.Hagberg B, Aicardi J, Dias K, Ramos O. A progressive syndrome of autism, dementia, ataxia, and loss of purposeful hand use in girls: Rett’s syndrome: Report of 35 cases. Ann Neurol. 1983October;14(4):471–9. [DOI] [PubMed] [Google Scholar]
- 56.Guy J, Hendrich B, Holmes M, Martin JE, Bird A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat Genet. 2001March;27(3):322–6. [DOI] [PubMed] [Google Scholar]
- 57.Chen RZ, Akbarian S, Tudor M, Jaenisch R. Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat Genet. 2001March;27(3):327–31. [DOI] [PubMed] [Google Scholar]
- 58.Mossner JM, Batista-Brito R, Pant R, Cardin JA. Developmental loss of MeCP2 from VIP interneurons impairs cortical function and behavior. Elife. 2020April28;9:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tatsukawa T, Ogiwara I, Mazaki E, Shimohata A, Yamakawa K. Impairments in social novelty recognition and spatial memory in mice with conditional deletion of Scn1a in parvalbumin-expressing cells. Neurobiol Dis. 2018April;112:24–34. [DOI] [PubMed] [Google Scholar]
- 60.Antoine MW, Langberg T, Schnepel P, Feldman DE. Increased Excitation-Inhibition Ratio Stabilizes Synapse and Circuit Excitability in Four Autism Mouse Models. Neuron. 2019;101(4):648–661.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Villas N, Meskis MA, Goodliffe S. Dravet syndrome: Characteristics, comorbidities, and caregiver concerns. Epilepsy Behav. 2017September;74:81–6. [DOI] [PubMed] [Google Scholar]
- 62.Li B-M, Liu X-R, Yi Y-H, Deng Y-H, Su T, Zou X, et al. Autism in Dravet syndrome: Prevalence, features, and relationship to the clinical characteristics of epilepsy and mental retardation. Epilepsy Behav. 2011July;21(3):291–5. [DOI] [PubMed] [Google Scholar]
- 63.DRAVET C Dravet syndrome history. Dev Med Child Neurol. 2011April;53(s2):1–6. [DOI] [PubMed] [Google Scholar]
- 64.Claes L, Del-Favero J, Ceulemans B, Lagae L, Van Broeckhoven C, De Jonghe P. De Novo Mutations in the Sodium-Channel Gene SCN1A Cause Severe Myoclonic Epilepsy of Infancy. Am J Hum Genet. 2001;68(6):1327–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yamagata T, Ogiwara I, Mazaki E, Yanagawa Y, Yamakawa K. Nav1.2 is expressed in caudal ganglionic eminence-derived disinhibitory interneurons: Mutually exclusive distributions of Nav1.1 and Nav1.2. Biochem Biophys Res Commun. 2017;491(4):1070–6. [DOI] [PubMed] [Google Scholar]
- 66.Favero M, Sotuyo NP, Lopez E, Kearney JA, Goldberg EM. A Transient Developmental Window of FastSpiking Interneuron Dysfunction in a Mouse Model of Dravet Syndrome. J Neurosci. 2018;38(36):7912–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.De Stasi AM, Farisello P, Marcon I, Cavallari S, Forli A, Vecchia D, et al. Unaltered Network Activity and Interneuronal Firing during Spontaneous Cortical Dynamics in Vivo in a Mouse Model of Severe Myoclonic Epilepsy of Infancy. Cereb Cortex. 2016;26(4):1778–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rubinstein M, Han S, Tai C, Westenbroek RE, Hunker A, Scheuer T, et al. Dissecting the phenotypes of Dravet syndrome by gene deletion. Brain. 2015;138(8):2219–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tai C, Abe Y, Westenbroek RE, Scheuer T, Catterall WA. Impaired excitability of somatostatin- and parvalbumin-expressing cortical interneurons in a mouse model of Dravet syndrome. PNAS. 2014;111(30):E3139––48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Almog Y, Fadila S, Brusel M, Mavashov A, Anderson K, Rubinstein M. Developmental alterations in firing properties of hippocampal CA1 inhibitory and excitatory neurons in a mouse model of Dravet syndrome. Neurobiol Dis. 2021;148:105209. [DOI] [PubMed] [Google Scholar]
- 71.Han S, Tai C, Westenbroek RE, Yu FH, Cheah CS, Potter GB, et al. Autistic-like behaviour in Scn1a +/− mice and rescue by enhanced GABA-mediated neurotransmission. Nature. 2012September22;489(7416):385–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Genton P, Velizarova R, Dravet C. Dravet syndrome: The long-term outcome. Epilepsia. 2011;52(SUPPL. 2):44–9. [DOI] [PubMed] [Google Scholar]
- 73.Owen MJ, O’Donovan MC. Schizophrenia and the neurodevelopmental continuum:evidence from genomics. World Psychiatry. 2017October;16(3):227–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Koukouli F, Rooy M, Tziotis D, Sailor KA, O’neill HC, Levenga J, et al. Nicotine reverses hypofrontality in animal models of addiction and schizophrenia HHS Public Access Author manuscript. Nat Med. 2017;23(3):347–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Johnson C, Kretsge LN, Yen WW, Sriram B, Jimenez JC, Jinadasa TJ, et al. Distinct VIP interneurons in the cingulate cortex encode anxiogenic and social stimuli. bioRxiv. 2020; [Google Scholar]
- 76.Gouwens NW, Berg J, Feng D, Sorensen SA, Zeng H, Hawrylycz MJ, et al. Systematic generation of biophysically detailed models for diverse cortical neuron types. Nat Commun. 2018;9(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yuste R, Hawrylycz M, Aalling N, Aguilar-Valles A, Arendt D, Arnedillo RA, et al. A community-based transcriptomics classification and nomenclature of neocortical cell types. Nat Neurosci. 2020; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Scala F, Kobak D, Bernabucci M, Bernaerts Y, Cadwell CR, Castro JR, et al. Phenotypic variation of transcriptomic cell types in mouse motor cortex. Nature. 2020November12; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kim HG, Kishikawa S, Higgins AW, Seong IS, Donovan DJ, Shen Y, et al. Disruption of Neurexin 1 Associated with Autism Spectrum Disorder. Am J Hum Genet. 2008;82(1):199–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Vaags AK, Lionel AC, Sato D, Goodenberger M, Stein QP, Curran S, et al. Rare deletions at the neurexin 3 locus in autism spectrum disorder. Am J Hum Genet. 2012;90(1):133–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fuccillo MV, Földy C, Gökce Ö, Rothwell PE, Sun GL, Malenka RC, et al. Single-Cell mRNA Profiling Reveals Cell-Type-Specific Expression of Neurexin Isoforms. Neuron. 2015July;87(2):326–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sweatt JD. Pitt-Hopkins Syndrome: Intellectual disability due to loss of TCF4-regulated gene transcription. Exp Mol Med. 2013;45(4):1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Forrest M, Chapman RM, Doyle AM, Tinsley CL, Waite A, Blake DJ. Functional analysis of TCF4 missense mutations that cause Pitt-Hopkins syndrome. Hum Mutat. 2012December;33(12):1676–86. [DOI] [PubMed] [Google Scholar]
- 84.Zweier C, de Jong EK, Zweier M, Orrico A, Ousager LB, Collins AL, et al. CNTNAP2 and NRXN1 Are Mutated in Autosomal-Recessive Pitt-Hopkins-like Mental Retardation and Determine the Level of a Common Synaptic Protein in Drosophila. Am J Hum Genet. 2009November;85(5):655–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ripke S, Sanders AR, Kendler KS, Levinson DF, Sklar P, Holmans PA, et al. Genome-wide association study identifies five new schizophrenia loci. Nat Genet. 2011;43(10):969–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014July24;511(7510):421–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mabb AM, Judson MC, Zylka MJ, Philpot BD. Angelman syndrome: insights into genomic imprinting and neurodevelopmental phenotypes. Trends Neurosci. 2011June;34(6):293–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Judson MCC, Wallace MLL, Sidorov MSS, Burette ACC, Gu B, van Woerden GMM, et al. GABAergic Neuron-Specific Loss of Ube3a Causes Angelman Syndrome-Like EEG Abnormalities and Enhances Seizure Susceptibility. Neuron. 2016;90(1):56–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Samaco RC, Hogart A, Lasalle JM. Epigenetic overlab in autism-spectrum neurodevelopmental disorders: MECP2 deficiency causes reduced expression of UBE3A and GABRB3. 2005;14(4):483–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cordelli DM, Pellicciari A, Kiriazopulos D, Franzoni E, Garavelli L. Epilepsy in Mowat-Wilson syndrome: Is it a matter of GABA? Epilepsia. 2013July;54(7):1331–2. [DOI] [PubMed] [Google Scholar]
- 91.Cordelli DM, Garavelli L, Savasta S, Guerra A, Pellicciari A, Giordano L, et al. Epilepsy in Mowat-Wilson syndrome: Delineation of the electroclinical phenotype. Am J Med Genet Part A. 2013February;161(2):273–84. [DOI] [PubMed] [Google Scholar]
- 92.Dani JA, Bertrand D. Nicotinic acetylcholine receptors and nicotinic cholinergic mechanisms of the central nervous system. Annu Rev Pharmacol Toxicol. 2007;47:699–729. [DOI] [PubMed] [Google Scholar]
- 93.Motamedi GK, Lesser RP. Autosomal dominant nocturnal frontal lobe epilepsy. Adv Neurol. 2002;89:463–73. [PubMed] [Google Scholar]
- 94.Klaassen A, Glykys J, Maguire J, Labarca C, Mody I, Boulter J. Seizures and enhanced cortical GABAergic inhibition in two mouse models of human autosomal dominant nocturnal frontal lobe epilepsy. PNAS. 2006;103(50):19152–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Aracri P, Meneghini S, Coatti A, Amadeo A, Becchetti A. α4β2(*) nicotinic receptors stimulate GABA release onto fast-spiking cells in layer V of mouse prefrontal (Fr2) cortex. Neuroscience. 2016/10/26.2017January6;340:48–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Jiang X, Lachance M, Rossignol E. Involvement of cortical fast-spiking parvalbumin-positive basket cells in epilepsy. Prog Brain Res. 2016;226:81–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Spratt PWE, Ben-Shalom R, Keeshen CM, Burke KJ, Clarkson RL, Sanders SJ, et al. The Autism-Associated Gene Scn2a Contributes to Dendritic Excitability and Synaptic Function in the Prefrontal Cortex. Neuron. 2019;103(4):673–685.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. 2014;515(7526):209–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sanders SJ. First glimpses of the neurobiology of autism spectrum disorder. Curr Opin Genet Dev. 2015;33:80–92. [DOI] [PubMed] [Google Scholar]
- 100.Bloss EB, Cembrowski MS, Karsh B, Colonell J, Fetter RD, Spruston N. Structured Dendritic Inhibition Supports Branch-Selective Integration in CA1 Pyramidal Cells. Neuron. 2016March;89(5):1016–30. [DOI] [PubMed] [Google Scholar]
- 101.Chiu CQ, Lur G, Morse TM, Carnevale NT, Ellis-Davies GCR, Higley MJ. Compartmentalization of GABAergic inhibition by dendritic spines. Science. 2013May;340(6133):759–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Murayama M, Pérez-Garci E, Nevian T, Bock T, Senn W, Larkum ME. Dendritic encoding of sensory stimuli controlled by deep cortical interneurons. Nature. 2009February;457(7233):1137–41. [DOI] [PubMed] [Google Scholar]
- 103.Berkvens JJL, Veugen I, Veendrick-Meekes MJBM, Snoeijen-Schouwenaars FM, Schelhaas HJ, Willemsen MH, et al. Autism and behavior in adult patients with Dravet syndrome (DS). Epilepsy Behav. 2015June;47:11–6. [DOI] [PubMed] [Google Scholar]
- 104.Takayama R, Fujiwara T, Shigematsu H, Imai K, Takahashi Y, Yamakawa K, et al. Long-term course of Dravet syndrome: A study from an epilepsy center in Japan. Epilepsia. 2014;55(4):528–38. [DOI] [PubMed] [Google Scholar]
- 105.Cestèle S, Scalmani P, Rusconi R, Terragni B, Franceschetti S, Mantegazza M. Self-limited hyperexcitability: functional effect of a familial hemiplegic migraine mutation of the Nav1.1 (SCN1A) Na+ channel. J Neurosci. 2008July;28(29):7273–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kahlig KM, Rhodes TH, Pusch M, Freilinger T, Pereira-Monteiro JM, Ferrari MD, et al. Divergent sodium channel defects in familial hemiplegic migraine. PNAS. 2008July;105(28):9799–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Berecki G, Bryson A, Terhag J, Maljevic S, Gazina EV, Hill SL, et al. SCN1A gain of function in early infantile encephalopathy. Ann Neurol. 2019April;85(4):514–25. [DOI] [PubMed] [Google Scholar]
- 108.Sadleir LG, Mountier EI, Gill D, Davis S, Joshi C, DeVile C, et al. Not all SCN1A epileptic encephalopathies are Dravet syndrome: Early profound Thr226Met phenotype. Neurology. 2017September;89(10):1035–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Vormstein-Schneider D, Lin JD, Pelkey KA, Chittajallu R, Guo B, Arias-Garcia MA, et al. Viral manipulation of functionally distinct interneurons in mice, non-human primates and humans. Nat Neurosci. 2020;23(December). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Colasante G, Lignani G, Brusco S, Di Berardino C, Carpenter J, Giannelli S, et al. dCas9-Based Scn1a Gene Activation Restores Inhibitory Interneuron Excitability and Attenuates Seizures in Dravet Syndrome Mice. Mol Ther. 2019;28(1):1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Carvill GL, Engel KL, Ramamurthy A, Cochran JN, Roovers J, Stamberger H, et al. Aberrant Inclusion of a Poison Exon Causes Dravet Syndrome and Related SCN1A-Associated Genetic Epilepsies. Am J Hum Genet. 2018;103(6):1022–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Han Z, Chen C, Christiansen A, Ji S, Lin Q, Anumonwo C, et al. Antisense oligonucleotides increase Scn1a expression and reduce seizures and SUDEP incidence in a mouse model of Dravet syndrome. Sci Transl Med. 2020;12(558). [DOI] [PubMed] [Google Scholar]
- 113.Lenk GM, Jafar-Nejad P, Hill SF, Huffman LD, Smolen CE, Wagnon JL, et al. Scn8a Antisense Oligonucleotide Is Protective in Mouse Models of SCN8A Encephalopathy and Dravet Syndrome. Ann Neurol. 2020;87(3):339–46. [DOI] [PMC free article] [PubMed] [Google Scholar]