

Abstract
With the increasing use of ketamine as an off-label treatment for depression and the recent FDA approval of (S)-ketamine for treatment-resistant depression, there is an increased need to understand the long-term safety profile of chronic ketamine administration. Of particular concern is the neurotoxicity previously observed in rat models following acute exposure to high doses of ketamine, broadly referred to as 'Olney's lesions'. This type of toxicity presents as abnormal neuronal cellular vacuolization, followed by neuronal death and has been associated with ketamine's inhibition of the N-methyl-D-aspartate receptor (NMDAR). In this study, a pharmacological and neuropathological analysis of ketamine, the potent NMDAR antagonist MK-801, and the ketamine metabolite (2R,6R)-hydroxynorketamine [(2R,6R)-HNK)] in rats is described following both single dose and repeat dose drug exposures. Ketamine dosing was studied up to 20 mg/kg intravenously for the single-dose neuropathology study and up to 60 mg/kg intraperitoneally for the multiple-dose neuropathology study. MK-801 dosing was studied up to 0.8 mg/kg subcutaneously for both the single and multiple-dose neuropathology studies, while (2R,6R)-HNK dosing was studied up to 160 mg/kg intravenously in both studies. These studies confirm dose-dependent induction of 'Olney's lesions' following both single dose and repeat dosing of MK-801. Ketamine exposure, while showing common behavioral effects, did not induce wide-spread Olney's lesions. Treatment with (2R,6R)-HNK did not produce behavioral effects, toxicity or any evidence of Olney's lesion formation. Based on these results, future NMDAR-antagonist neurotoxicity studies should strongly consider taking pharmacokinetics more thoroughly into account.
Keywords: Neurotoxicology, NMDAR antagonists, Olney Lesions, Ketamine, Pharmacokinetics, MK-801, neuropathology
1. Introduction
Ketamine, an N-methyl-D-aspartate receptor (NMDAR) antagonist, has emerged as an effective therapy for treatment-resistant depression (Zarate 2006, Newport 2015, Wilkinson 2017). Its use as an acute and maintenance treatment is rapidly increasing for both depression and other indications such as bipolar disorder and posttraumatic stress disorder (Wilkinson 2017). The increasing use of ketamine (i.e. racemic ketamine: a mixture of (R)-ketamine and (S)-ketamine) and the recent FDA approval of intranasal (S)-ketamine to treat major depressive disorder (MDD) has fueled concerns around these drugs'acute and long-term toxicities (Strong 2018). In addition, the use ketamine for depression has been administered through a variety of routes, including intravenous, intranasal, intramuscular, and subcutaneous, with different outcomes from each route of administration (Loo 2016). Despite its value in treating depression, ketamine has several detrimental side effects, including its abuse liability, dissociation properties, and potential to induce cognitive deficits (Strong 2018). One potential neurological risk derived from the use of NMDAR antagonists is the development of a specific type of NMDAR antagonist neurotoxicity, also known as 'Olney's lesions' (Olney 1989). Originally identified by Olney and coworkers, this toxicity results from the administration of moderate to high doses of NMDAR antagonists, including PCP, MK-801, ketamine, and tiletamine and is described as vacuolization in the neurons of the retrosplenial cortex and posterior cingulate gyrus in rats (Olney 1989, Olney 1991). Vacuoles are heterogeneous in size (3 to 15 μM in diameter) and located within the cytoplasmic compartment of the neurons, which appear to originate from the saccules of the endoplasmic reticulum (Onley 1989). Following short-term administration, lesion formation is reversible, however long-term treatment with NMDAR antagonists can induce irreversible neuronal necrosis of affected brain regions (Olney 1989, Olney 1991). Such damage is revealed through hematoxylin and eosin staining which exposes necrotic neurons with brightly eosinophilic, severely shrunken cytoplasm and pykonotic or karyorrectic nuclei (Fix 1993). By four days after dosing with MK-801 the necrotic neurons are often surrounded by reactive microglial cells (Fix 1993). Later experiments identified increased HSP70 and HSP72 levels as biomarkers that correlate with NMDAR antagonism (Fix 1993, Sharp 1991, Sharp 1992, Tian 2018). Pronounced strain (Fix 1995), age and sex (Auer 1996) differences have been identified in the development of this neurotoxicity, with older rats and female rats being more susceptible (Auer 1996). A human study on subjects with a history of ketamine abuse also showed neural degeneration (Wang 2013), reinforcing the potential for neurotoxicity in individuals who repeatedly use NMDAR antagonists. Vacuole formation is difficult to confirm in humans as neuronal vacuolization is transient and likely difficult to observe in post-mortem human brains. Given the difficulty in confirming the presence of Olney's lesions in human subjects, iťs imperative that we expand upon preclinical models in hopes of better assessing the neuronal toxicity of this highly useful class of drugs (Green 2009). The shift from short duration, single-use ketamine administration for anesthesia to repeated, subanesthetic use of ketamine to treat depression, pain, and other indications intensifies this need.
For this study, we focused on the pharmacology, behavioral effects, and neurotoxicity of ketamine in rats. We employed a multiple dosing paradigm to better mimic the human antidepressant administration regime and assessed the effects on neurotoxicity. We included MK-801, a potent NMDAR antagonist which has been associated with neurotoxicity, as a positive control. For MK-801, the (+)-MK 801 enantiomer was utilized as it yields the lesions much more readily than its enantiomer. We also included the ketamine metabolite (2R,6R)-hydroxynorketamine [(2R,6R)-HNK] in this study. Substantial preclinical evidence shows that (2R,6R)-HNK possesses antidepressant-like activity in mouse models without appreciable activity as an NMDAR antagonist at pharmacologically-relevant concentrations (Zanos 2016, Zanos 2018, Fukumoto 2019, Lumsden 2019, Riggs 2020). However, there are some inconsistencies in the reported effects of (2R,6R)-HNK in tests predictive of antidepressant effectiveness (Highland 2021). Each agent used in this study was subjected to extensive pharmacokinetic analysis in hopes of better understanding how drug exposure relates to Olney’s lesion formation.
2. Materials and Methods
2.1. Study Design
The experiments were designed as three separate studies: a single-dose, pharmacokinetic study; a single-dose neuropathology study; and a multiple-dose neuropathology study. The pharmacokinetic study was designed to understand the differences in compound exposure levels for each agent with special consideration given to the differences in route of administration and established differences based on animal sex. It was performed in Sprague-Dawley rats in order to better correspond to the strain most often used in the literature (Olney 1989, Olney 1991, Jevtovic-Todorovic 2000, Jevtovic-Todorovic 2001). The single-dose neuropathology study was designed to examine the formation of Olney’s lesions or necrotic neurons via neuropathology at rapid and delayed time points (nominally referred to as 3 or 72 hours postdose; actual necropsy/perfusion ranged from 3 to 3.33 or 72 ± 1 hour postdose, respectively). The multiple-dose neuropathology study was designed to dose the animals twice a week for two weeks, and examine the animals for the formation of Olney’s lesions of necrotic neurons via neuropathology at three time points: 3 hours after the first dose; 3 hours after the last dose; and 72 hours after the last dose. Time points were selected on the basis of the current literature, which suggests that the reversible neuronal vacuolization is most robust at 3–4 hours after dose administration, while the neuronal necrosis is most evident at 3–4 days post dosing (Olney 1989, Olney 1991, Fix 1993). The neuropathology studies were performed in Han Wistar rats in order to align with a concurrent toxicology program (data not included). All experimental treatments were administered unblinded. Animals were assigned to the treatment groups randomly.
2.2. Animals
Food and water were available ad libitum. For the neuropathology studies, Han Wistar rats (12–14 weeks of age at time of dosing, Envigo, RMS, Inc) were acclimated to the test facility for 7–18 days. Pharmacokinetic studies used 36 Sprague Dawley rats (11–15 weeks of age, 18 male, 18 female, Envigo RMS Inc.), with 3 male and 3 female animals per group which were acclimated to the test facility for 2–5 days. Neuropathology studies used 858 total animals (286 Male, 572 female, 12–14 weeks old at initial of dosing, including replacement animals) with 20 female animals per group for the single dose study and 40 female animals (30 neuropathology and 10 satellite) per group in the multiple dose study. Additionally, for the negative control group and (2R,6R)-HNK dosing cohorts 20 male animals per group were used for the single dose study and 40 male animals (30 neuropathology and 10 satellite) per group for the multiple dose study. Animals were group-housed (up to three animals/sex/cage) in polycarbonate cages. Animals were monitored for abnormal clinical observations, including changes in behavior, at least three times daily during inlife. In addition, on days of dose administration, animals were monitored at least 10, 30, and 60 minutes post dose. Animals administered MK-801 were continuously monitored for at least 2 hours following dose administration. Body weights were recorded and monitored. All procedures were performed in facilities fully accredited by the AAALAC, were in compliance with applicable animal welfare acts and were approved by the appropriate Institutional Animal Care and Use Committee (IACUC).
2.3. Compounds, Formulation, and Dosing
(2R,6R)-HNK was utilized as (2R,6R)-hydroxynorketamine hydrochloride (National Center for Advancing Translational Sciences). Ketamine was utilized as ketamine hydrochloride (Tocris Biosciences or Sigma Aldrich). MK-801 was utilized as (+)-MK 801 maleate (Tocris Biosciences). For pharmacologic studies, compounds and controls were dissolved in normal saline (0.9% w/v) until they formed a clear or colorless solution. (2R,6R)-HNK and ketamine were formulated at 2 mg/ml final concentration. MK-801 was formulated at 0.1 mg/ml final concentration. The purpose of MK-801 was to serve as a positive control for histological staining (rather than for the test articles) to ensure that lesions, when present, could be detected. Compounds were dosed using intravenous (IV), intraperitoneal (IP) or subcutaneous (SC) routes of administration as described, with SC injection in the scapular region and with ketamine IV injection via the tail vein as a slow bolus over 60 seconds.
For the neuropathology studies, (2R,6R)-HNK was formulated in a 75 mM sodium phosphate buffer, pH 7 to avoid injection of a drug substance outside of safe pH ranges. (2R,6R)-HNK was administered as an IV infusion via tail vein over 20 minutes. In the multiple dose study, (2R,6R)-HNK was administered IV via tail-vein infusion over 20 minutes on days 1, 4, 11, and 15. Ketamine was formulated in 75 mM sodium phosphate buffer, pH 7 and was administered as an IV slow bolus (approximately 60–90 seconds) via the tail vein in the single-dose study. In the multiple dose study, ketamine was administered as an IP injection on days 1, 4, 11, and 15. MK-801 was formulated in sterile water for injection, and was administered as a SC injection near the scapular region, with injections on day 1, 4, 7, and 15 for the multiple dose study. The negative control was a 75 mM sodium phosphate buffer, pH 7, prepared in sterile 0.45% (w/v) saline. This negative control was administered as an IV infusion via the tail vein over 20 minutes, with injections on day 1, 4, 7, and 15 for the multiple dose study.
2.4. Sample Collection and Analysis
Pharmacokinetic analyses were performed from blood collected via jugular vein-cannula by syringe and transferred into tubes containing K2ETDA. Samples were collected prior to dosing, and at 0.083, 0.25, 0.5, 1, 2, 4, 8, 12, and 24 hours after dosing. Blood was centrifuged within 1 hour of collection, and the resulting plasma was placed in 96 well tubes, and stored at − 70 °C to – 80 °C. Plasma samples were analyzed by liquid chromatography-mass spectrometry (LC-MS/MS) for (2R,6R)-HNK, ketamine, or MK-801 concentrations as appropriate.
2.5. Neuropathology
For single dose studies, animals were euthanized either 3 hours after dosing or 72 hours after dosing. For multiple dose studies, animals were euthanized at 3 hours after dosing on day 1, 3 hours after dosing on day 15, or 72 hours after dosing on day 15. For the multiple dose studies, animals were fasted overnight prior to euthanasia. Animals were anesthetized with sodium pentobarbital, exsanguinated via upper body perfusion with heparinized sodium nitrate saline, followed by 4% paraformaldehyde fixative. The brain was removed, divided into 7 sections as previously described as the recommended practices for sampling and processing the nervous system (Rao 2011; Bolon 2013), and paraffin embedded within 72 hours after fixation. Slides were made of the brain at a thickness of 5 μM. Slides were stained using hematoxylin and eosin, fluorojade-B, or immunostained using Iba-1 (Abcam cat # Ab5076) and GFAP (Life technologies cat # 003218). Histopathological examination included a qualitative evaluation for vacuolization (H&E) and necrosis (H&E and Fluoro Jade B). Relative changes in immunolabeling for Iba-1 (ionized Ca-binding protein) and GFAP (glial fibrillary acidic protein) were also based on subjective (increase or decrease) comparisons to the control group.
2.6. Data and Statistical Analysis
Pharmacokinetic calculations were performed using Microsoft Excel (Microsoft, Redmond, WA) and PKSolver (Zhang 2010). Non-compartmental analysis was applied to determine the following parameters: C0 (Back-extrapolated concentration at time 0); Cmax (Maximum observed concentration); Tmax (time of maximum observed concentration); AUC0-t (Area under the concentration-time curve from hour 0 to the last measurable concentration, estimated by the linear trapezoidal rule); AUC0-inf (Area under the concentration time curve from hour 0 to infinity, calculated as AUC0-inf = AUC0-t + Ct/λz where Ct is the last measurable concentration and λz is the elimination rate constant estimated using log-linear regression during the terminal elimination phase); t1/2 (Elimination half-life determined by ln(2)/λz); CL (Clearance, calculated as Dose / AUC0-inf), Vss (Volume of distribution at steady-state, calculated as CL * MRT0-inf. MRT0-inf is defined as the mean residence time for the drug.) CL/F (apparent total clearance after non-intravenous administration); and Vz/F (Apparent volume of distribution during the terminal phase after non-intravenous administration). Table 1 reports selected parameters, as mean values (N=3 unless otherwise noted). Other abbreviations as follows: M (males); F (females); IV (intravenous); IP (intraperitoneal); SC (subcutaneous).
Table 1:
Dosing and pharmacokinetic parameters upon administration of (2R,6R)-HNK, Ketamine, or MK-801
| Group | Compound | Dose (mg/kg) | N and Sex | Route of administration | Cmax (ng/mL) | AUC(0-t) (ng *h / mL) | t1/2 (h) | Cl (ml/h/kg) | Vss (mL/kg) |
|---|---|---|---|---|---|---|---|---|---|
| 1a | (2R,6R)-HNK | 10 | 3, M | IVa | 5050 | 3480 | 4.3b | 2620b | 15700b |
| 1b | (2R,6R)-HNK | 10 | 3, F | IVa | 5840 | 4520 | 4.7 | 2320 | 6000 |
| 2a | Ketamine | 10 | 3, M | IVc | 3250 | 1440 | 0.96 | 6920 | 2940 |
| 2b | Ketamine | 10 | 3, F | IVc | 3347 | 1940 | 1.78 | 5180 | 5230 |
| 3a | Ketamine | 10 | 3, M | SC | 1790 | 1590 | 1.09 | 4230d | 9980e |
| 3b | Ketamine | 10 | 3, F | SC | 1550 | 2090 | 1.06 | 4780d | 7270e |
| 4a | MK-801 | 0.5 | 3, M | IV | 108 | 101 | 1.54 | 4430 | 6570 |
| 4b | MK-801 | 0.5 | 3, F | IV | 96 | 180 | 2.96 | 1700 | 7140 |
| 5a | MK-801 | 0.5 | 3, M | IP | 17.5 | 43.8 | 1.77 | 11400d | 27500e |
| 5b | MK-801 | 0.5 | 3, F | IP | 29.3 | 94.5 | 5.2 | 2130d | 15910e |
| 6a | MK-801 | 0.5 | 3, M | SC | 40.4 | 110 | 1.78 | 4350d | 11200e |
| 6b | MK-801 | 0.5 | 3, F | SC | 55.3 | 513 | 7.67 | 860d | 9550e |
Pharmacokinetic abbreviations are defined in the methods
Administered as an IV infusion over 20 minutes.
N=2
Administered as a slow push over 60 seconds.
Represents CL/F.
Represents Vz/F
Pharmacokinetic data was used to generate pharmacokinetic charts (Figure 1) utilizing GraphPad Prism 8 to show the 0 – 4 or 0 – 24 hour time window as appropriate, with mean values and standard deviations displayed. Statistical significance was calculated using the unpaired t-test (GraphPad Prism 8) for comparing AUC values for male and female groups (Figure S1). One * indicates p < 0.05, ** indicates p < 0.01, *** indicates p < 0.001. For statistical significance in the neuropathological evaluation, the Fisher’s exact test was utilized.
Figure 1:
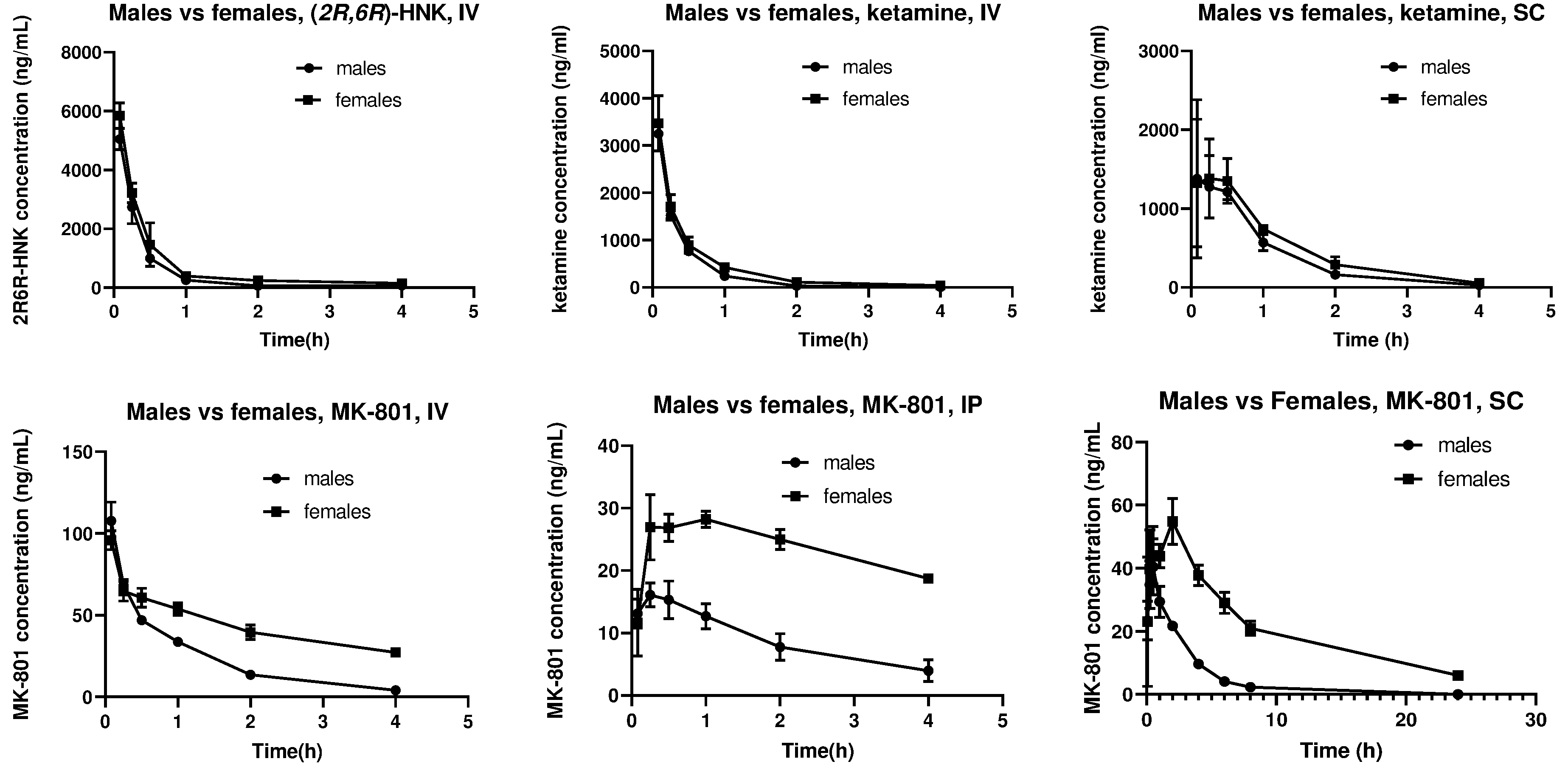
Sex differences in the drug exposure of (2R,6R)-HNK (10 mg/kg), ketamine (10 mg/kg), or MK-801 (0.5 mg/kg), when administered via various routes. IV: Intravenous; IP: intraperitoneal; SC: subcutaneous. Data are mean +/- standard deviation (N=3).
3. Results
3.1. Behavioral effects of (2R,6R)-HNK, Ketamine, and MK-801 in pharmacokinetic studies
(2R,6R)-HNK, ketamine, and MK-801 were administered to different groups of animals, with each group comprised of 3 female and 3 male animals (Table 1). (2R,6R)-HNK and ketamine were administered at a dose of 10 mg/kg, while MK-801 was administered at a dose of 0.5 mg/kg. Administration of (2R,6R)-HNK was not associated with hyperactivity or ataxia in either male or female animals. IV administration of ketamine generated hyperactivity in both male and female animals, which resolved within 1 hour. SC administration of ketamine at the same dose had no behavioral effects. IV and IP MK-801 administration led to ataxia in male animals which resolved by 6 to 7 hours after administration. Female animals given the same MK-801 dose became severely ataxic shortly after administration and became non-responsive by 4 to 6 hours. According to IACUC guidelines, all female animals were euthanized at 5.5 to 6 hours due to lack of improvement. SC dosing of MK-801 also caused ataxia in both male and female animals. In the male animals, the ataxia began to resolve by 4 hours after administration. Ataxia persisted in the female groups past 8 hours but resolved by 24 hours after administration.
3.2. Pharmacokinetic analysis of (2R,6R)-HNK, Ketamine, and MK-801
In both male and female groups, plasma levels of (2R,6R)-HNK (10 mg/kg) showed rapid drug elimination, with concentrations dropping from over 5000 ng/ml to under 500 ng/ml in less than an hour (Figure 1, Table 1). No significant differences in drug exposure were found in male versus female animals (Figure S1).
The pharmacokinetic parameters of ketamine (10 mg/kg) were assessed using both IV and SC administration. IV administration resulted in a higher Cmax in both sexes compared to SC administration (Table 1, Figure 1). However, there was no significant difference in the AUC values between the SC and IV ketamine administration (p = 0.19). Female animals demonstrated significantly higher levels of ketamine compared to male animals following both SC and IV administration (Table 1, Figure S1).
The pharmacokinetic parameters of MK-801 (0.5 mg/kg) were assessed using three different routes of administration (IV, IP, and SC). With all three routes of administration, levels of MK-801 were significantly higher in female animals compared to male animals (Table 1, Figure S1). This difference was most pronounced in the SC dosing, where female animals demonstrated an AUC level almost 5-fold above those found in male animals.
3.3. Single Dose Neuropathology Study of (2R,6R)-HNK, Ketamine, and MK-801
The posterior cingulate and retrosplenial cortices, regions both known to be affected by NMDAR antagonist neurotoxicity (Olney 1989, Horvath 1997), were assessed 3 hours and 72 hours after a single dose of ketamine, (2R,6R)-HNK, or MK-801. IV injection of ketamine at doses higher than 20 mg/kg led to a high rate of mortality in female animals. Thus, the maximum dose used to assess acute neurotoxicity was 20 mg/kg of ketamine given by IV administration. At this dose, acute ketamine showed no neuronal vacuolization or necrosis, and Iba-1 immunostaining showed no evidence of microglial activation in female or male animals.
SC administration of MK-801 (0.5 mg/kg) resulted in neuronal vacuolization in the retrosplenial cortex of all rats by 3 hours after dosing, (Fig. 2A–C, Fig. S2, Table 2). Vacuolization was observed in the posterior cingulate gyrus in 50% of the animals at the higher MK-801 dose. By 72 hours after dosing, neuronal necrosis was present in 8 of the 9 animals within the retrosplenial cortex as resolved by hematoxylin and eosin staining (H&E) and by fluorojade-B staining (Fig. 2D–I, Table 2, see figure S3 for an example of the necrosis). Iba-1 labelling revealed microglial activation in 8 of the 9 rats at the higher MK-801 dosing (Fig. 2J–L, Table 2).
Figure 2:
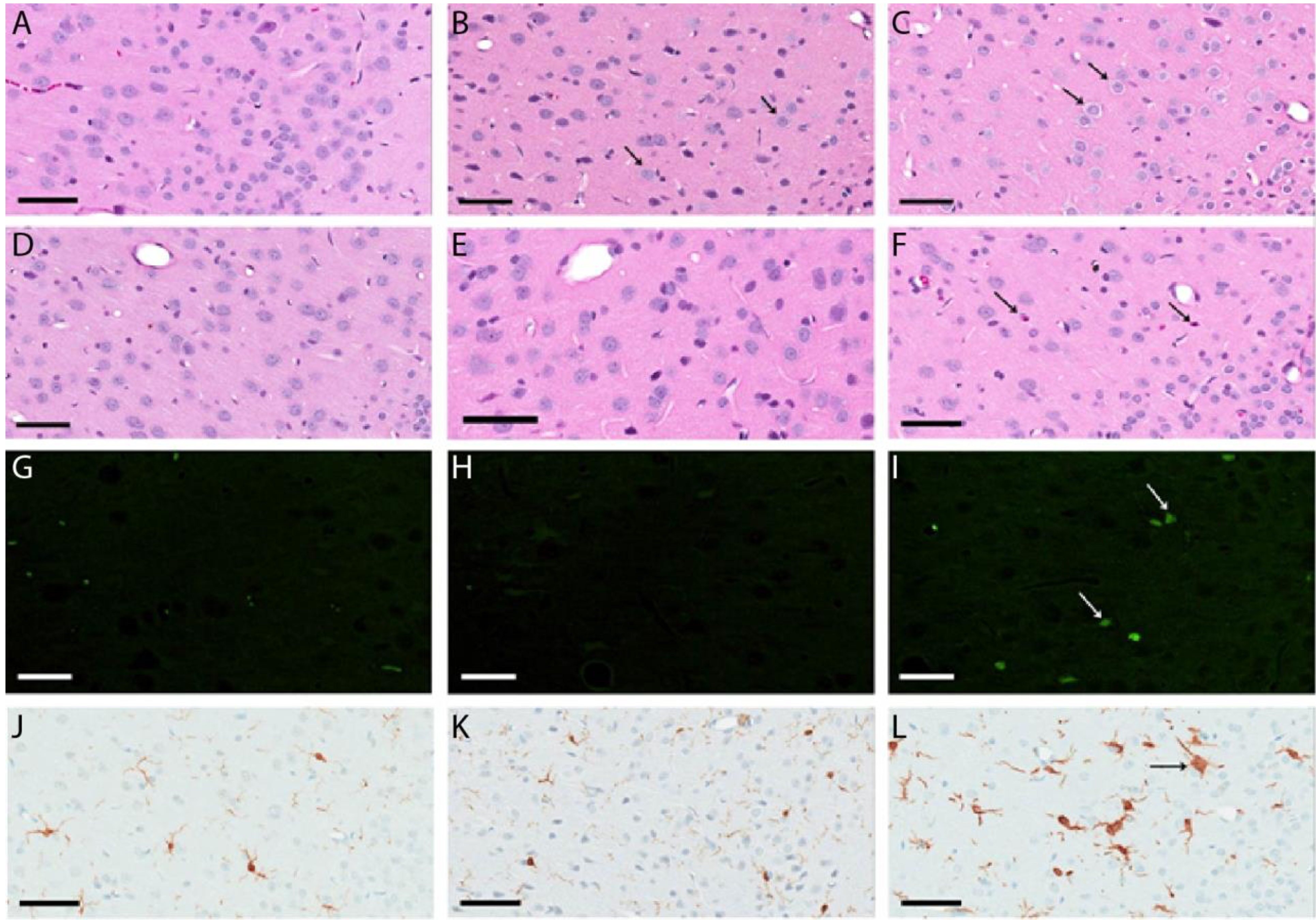
Representative neuropathology images after single dosing of saline or MK-801 in the retrosplenial cortex A) H&E staining, saline dose, 3 hour time point. B) H&E staining, 0.5 mg/kg MK-801, 3 hour time point. The small arrow designates vacuolization in the cells. C) H&E staining, 0.8 mg/kg MK-801, 3 hour time point. Note the more pronounced vacuolization. D) H&E staining, saline dose, terminal euthanization. E) H&E staining, 0.5 mg/kg MK-801, terminal euthanization. F) H&E staining, 0.8 mg/kg MK-801, terminal euthanization. Note the necrosis pointed by the arrows. G) Fluorojade staining, saline dose, terminal euthanization. H) Fluorojade staining, 0.5 mg/kg MK-801, terminal euthanization. I) Fluorojade staining, 0.8 mg/kg MK-801, terminal euthanization. Note the necrotic neurons observed by fluorojade staining J) Iba-1 staining, saline dose, terminal euthanization. K) IBA1 staining, 0.5 mg/kg MK-801, terminal euthanization. L) Iba-1 staining, 0.8 mg/kg MK-801, terminal euthanization. Note the increase in Iba-1 staining. Bar length is 0.06 mm
Table 2:
Single Dose neuropathology results
| Compound | Dose (mg/kg) | Route of administration | % animals with vacuolization | % animals with necrosis | % animals with increased Iba-1 labelling | % animals with increased GFAP labelling | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| 3 hr | 72 h | 3 h | 72 h | 3 h | 72 h | 3 h | 72 h | |||
| Negative control | 0 | IVa | 0% | 0% | 0% | 0% | ||||
| (2R,6R)-HNK | 12 | IVa | 0% | 0% | 0% | 0% | ||||
| (2R,6R)-HNK | 80 | IVa | 0% | 0% | 0% | 0% | ||||
| (2R,6R)-HNK | 120 | IVa | 0% | 0% | 0% | 0% | ||||
| (2R,6R)-HNK | 160 | IVa | 0% | 0% | 0% | 0% | ||||
| Ketamine | 3 | IVb | 0% | 0% | 0% | 0% | ||||
| Ketamine | 10 | IVb | 0% | 0% | 0% | 0% | ||||
| Ketamine | 20 | IVb | 0% | 0% | 0% | 0% | ||||
| MK-801 | 0.5 | SCc | 100% | 0% | 0% | 0% | 0% | 0% | 0% | |
| MK-801e | 0.8 | SCc | 100% | 0% | 0% | 88% | 0% | 88% | 0% | |
Ten animals/sex/group per timepoint per sex were utilized. The negative control and (2R,6R)-HNK groups utilized both male and female animals. The ketamine andMK-801 groups utilized only female animals.
Administered as an intravenous infusion over 20 minutes.
Administered as a slow intravenous injection over 60 seconds.
Subcutaneous dosing.
One of the 10 animals in the 72 hour cohort died before the end of the study and was excluded
No neuropathological changes were observed in any animals after IV injection of (2R,6R)-HNK, even at doses up to 160 mg/kg (Table 2).
3.4. Multiple Dose Neuropathology study of (2R,6R)-HNK, Ketamine, and MK-801
We next performed multiple dose neuropathology studies in rats as a model of repeat-dose ketamine administration in humans. For this study, animals were dosed twice a week for two weeks. Neuropathological changes were examined in the posterior cingulate and retrosplenial cortices at 3 hours after the first dose, 3 hours after the last dose (15 days; 363 h), and 72 hours after the last dose (432 h). For this study, the ketamine route of administration was changed to IP dosing, which allowed for higher dosing levels without increasing acute lethality.
No neurotoxicity was observed at any dose or time point after (2R,6R)-HNK dosing, up to 160 mg/kg IV, or after ketamine dosing, up to 60 mg/kg IP (Table 3). The level of neuronal vacuolization at the 3-hour time point following MK-801 administration was less pronounced relative to the single dose study. However, after multiple doses, the neuronal necrosis observed with MK-801 was more pronounced in both the number of animals affected at the 0.5 mg/kg dose (0% of animals with necrosis up to 60% of animals with necrosis, p = 0.01, Table 2, Table 3) and the relative severity of neuronal necrosis.
Table 3:
Multiple Dose Neuropathology Results
| Compound | Dose mg/kg | Route of administration | % animals with vacuolization | % animals with necrosis | % animals with increased Iba-1 labelling | % animals with increased GFAP staining | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 3h | 363 h | 432 h | 3 h | 363 h | 432 h | 3h | 363 h | 432 h | 3h | 363 h | 432 h | |||
| Negative control | NA | IVa | 0% | 0% | 0% | 0% | ||||||||
| (2R,6R)-HNK | 12 | IVa | 0% | 0% | 0% | 0% | ||||||||
| (2R,6R)-HNK | 80 | IVa | 0% | 0% | 0% | 0% | ||||||||
| (2R,6R)-HNK | 160 | IVa | 0% | 0% | 0% | 0% | ||||||||
| Ketamine | 50 | IPb | 0% | 0% | 0% | 0% | ||||||||
| Ketamine | 60 | IPb | 0% | 0% | 0% | 0% | ||||||||
| MK-801 | 0.5 | SCc | 0% | 0% | 0% | 0% | 60% | 40% | 0% | 70% | 10% | 0% | 80% | 80% |
| MK-801 | 0.8 | SCc | 30% | 0% | 0% | 0% | 100% | 90% | 0% | 90% | 40% | 0% | 100% | 100% |
Ten animals/sex/group per timepoint per sex were utilized. The negative control and (2R,6R)-HNK groups utilized both male and female animals. The ketamine andMK-801 groups utilized only female animals.363 hours represents Day 15, + 3 hours. 432 hours represents Day 15 + 72 hours
Administered as an intravenous infusion over 20 minutes.
Intraperitoneal injection.
Subcutaneous dosing.
Analysis of Iba-1 immunostaining showed activation of microglia in animals in both MK-801 groups following repeat-dose administration. GFAP immunostaining showed astrogliosis in both MK-801 dosing groups, which was not observed in the single dose study (Fig. S4, S5, Table 3). In addition, necrotic neurons were identified in the dentate gyrus of the hippocampus in one animal in the MK-801 0.8 mg/kg dosing group. Thus, multiple doses of MK-801 had greater neurotoxicity compared to single dose administration.
4. Discussion
This study provided an analysis of the neurotoxicity associated with both acute and multiple dose administration of (2R,6R)-HNK, ketamine, and MK-801 in rats, alongside detailed pharmacokinetic analysis, in order to more thoroughly understand the exact doses and exposure required for NMDAR antagonist-induced neurotoxicity. Dose ranges and different routes of administration were explored in order to elevate dosing to a highest achievable exposure of each drug as a means to demonstrate potential neurotoxicity outcomes and reflect clinical routes of administration (Loo 2016). In this study, only MK-801 treatment induced the formation of Olney’s lesions and neurotoxicity. Notably, and consistent with published literature (Fix 1995) a stark difference in the MK-801 pharmacokinetics was observed between male and female animals, with females having dramatically increased exposures (Fig. S1) and correspondingly more severe behavioral effects.
MK-801 administration resulted in dose-dependent Olney’s lesion formation and neuronal necrosis. In the initial pharmacokinetic experiments, MK-801 treated animals had an extended period of ataxia following IV, IP, or SC administration. These behavioral changes were so severe in female animals that animal welfare protocols required that the experiments be terminated after 6 hours in the IV and IP administrations in females. It has been previously reported that female rats are far more sensitive to MK-801 than male rats, with higher rates of Olney’s lesion formation and neurotoxicity given the same dosing level of MK-801 (Auer 1996). Our pharmacokinetics experiments demonstrate a profound sex difference in MK-801 exposure, especially with SC administration where the AUC in female animals was nearly five times that of the male animals, providing an explanation for the differential behavioral effects.
In contrast to MK-801, in the present study ketamine did not induce any detectable neurotoxicity at any of the doses or and routes of administration examined. The observation that ketamine does not induce Olney's lesions is a divergence from previous studies including the original observations reported by Olney and coworkers (Olney 1989, Olney 1991). The incongruity between our results and other published studies may be related to differences in route of administration, dose and strain. Olney's original reports primarily utilized a subcutaneous injection of ketamine in female Sprague-Dawley rats (Olney 1989, Olney 1991). By contrast, our neuropathology study utilized female Han Wistar rats, with either intraperitoneal or intravenous routes of administration. Another comparative study, which did find lesion formation, used doses in excess of 40 mg/kg and intraperitoneal drug injection with rats (Jevtovic-Todorovic 2001). Another study reported that intraperitoneal doses of 100 mg/kg in female rats were necessary to induce HSP-70 formation, a biomarker for NMDAR antagonist neurotoxicity (Tian 2018). In contrast, in male rats an intraperitoneal dose of 50 mg/kg was necessary to induce HSP-70 formation (Tomitaka 1996). Given the observation by Olney that female rats were more susceptible to Olney lesion induction, the exact correlation between HSP-70 induction and Olney lesion formation remains of interest (Olney 1989, Jevtovic-Todorovic 2000). Intrathecal injections of (S)-ketamine (0.7 mg/kg, once a day for 7 days) resulted in significant neurotoxicity in rabbits (Vranken 2006). In a human case report, after repeated intrathecal injection of (S)-ketamine (20–50 mg), neurological damage to the spine was observed postmortem in an adult patient (Vranken 2005). While the observed subarachnoid hemorrhage was attributed to the trauma of the catheter insertion, the authors found it unlikely that the observed spinal cord lesions were due to injection trauma (Vranken 2005). To the best of our knowledge, there are no reports of neurotoxicity associated with IV ketamine administration in rodent models. As IV administration is the most common route of administration in humans, we endeavored to use this route of administration in this study. However, our studies were limited by unacceptable toxicity in female animals when using IV dosing of ketamine at/above 20 mg/kg. Using the IP route in the repeated dose study allowed ketamine dose levels to be increased to 60 mg/kg. However, within both the acute and chronic setting, the high-dose ketamine administrations (20 mg/kg IV and 60 mg/kg IP, respectively) did not generate Olney's lesion or neuronal necrosis. This suggests that IV or IP-administered ketamine does not achieve the necessary and/or sustained concentration needed to induce Olney's lesions without also causing experiment-ending adverse events in Han Wistar rats.
Similarly, the ketamine metabolite (2R,6R)-HNK showed no neurotoxicity at any dose, even with repeat, high dose administration (160 mg/kg, IV). Some studies have demonstrated (2R,6R)-HNK can act as a partial NMDAR antagonist at high concentrations (50 μM) (Suzuki 2017). However, our results are consistent with prior studies that show no effects of (2R,6R)-HNK on NMDAR activity at pharmacologically-relevant concentrations (Zanos 2016, Lumsden 2019) and minimal to no adverse side effects at much higher concentrations (Highland 2021).
From a pharmacological perspective, the mechanism of NMDAR antagonist-induced neurotoxicity is not completely understood. There is evidence that high doses of NMDAR antagonists result in disinhibition of excitatory inputs to certain cholinergic neurons (Low 2004). This loss of inhibitory control leads to excess cholinergic inputs to cortical glutamatergic neurons in the posterior cingulate (PC) and retrosplenial cortex (RC) in rats, leading to vacuolization and ultimately cell death within these cortical neurons (Low 2004). Notably, while rats and mice demonstrate the most severe neurotoxicity in the PC and RC, other species demonstrate more severe NMDAR antagonist-induced neurotoxicity in alternate locations: guinea pigs within the frontopariental neocortex (Raboisson 1987); mice (Bender 2010; Wozniak 1996); and canines within the piriform cortex (Low 2004).
There is evidence that NMDAR antagonist-induced neurotoxicity is driven by the length of time the NMDAR is inhibited rather than the maximal concentration of the NMDAR antagonist (Bender 2010b). Ketamine is rapidly eliminated in rats when dosed IV (Figure 1, Table 1), and to a lesser extent IP, with levels dropping near to the lower limit of quantitation within two hours after IV administration and 3 hours for IP administration (Saland 2018). However, when dosed subcutaneously, that length of time is extended to four hours (Table 1, Figure 1). Interestingly Olney observed that female rats were more susceptible to ketamine-induced Olney's lesion formation when compared to male rats (Jevtovic-Todorovic, 2001). Both our data (Figure S1) and Saland's data (Saland 2018) demonstrate that ketamine has a significantly higher AUC in female rats compared to male rats. This is suggestive that the increased susceptibility for Olney lesion induction in female rats is due to ketamine’s pharmacokinetic differences between the sexes.
Simultaneous and delayed (up to 10 hours) exposure to DNQX, a competitive antagonist at AMPA and kainate receptors, protected neurons from MK-801-induced neuronal necrosis (Bender, 2010b). However, a 24-hour window between MK-801 and DNQX injections had no protective effects. When the neurotoxicity of the NMDAR antagonist nitrous oxide was tested, its administration over 12 hours was required to induce neuronal necrosis (Jevtovic-Todorovic 2001). Treatment with nitrous oxide for 8 hours or less resulted in reversible vacuole formation, but minimal to no neuronal necrosis. Further experiments coupling pharmacokinetics with clinically utilized NMDAR antagonists are needed to better understand this enigmatic form of neurotoxicity.
5. Conclusions
Here we present an in-depth pharmacokinetic analysis of ketamine, (2R,6R)-HNK, and MK-801 as a means to better understand the NMDAR-antagonist-related neurotoxicity of each agent using different routes of administration and schedules. (2R,6R)-HNK demonstrated no adverse behavioral effects at the highest doses and no observed neurotoxicity. Ketamine administration reproduced well-established behavioral effects and high-dose lethality, however, previously reported NMDAR-antagonist neurotoxicity was not confirmed. MK-801 administration produced significant behavioral effects and dose-dependent neurotoxicity. Unlike ketamine and (2R,6R)-HNK, MK-801 also possessed stark sex-based differences in exposure which likely accounts for the increased sensitivity of female rats to MK-801 toxicity. This study confirms the safety of (2R,6R)-HNK over a broad dose range (up to 160 mg/kg) with no evidence of neurotoxicity. These findings indicate that ketamine-induced Olney'slesion-type neurotoxicity may be dependent on the route of administration in acute and repeat-dose paradigms, as well as the relative pharmacokinetic exposure of ketamine. Future studies investigating NMDAR antagonist neurotoxicity should strongly consider taking sex and pharmacokinetics into account.
Supplementary Material
Highlights.
MK-801 demonstrates a pronounced sex-based disparity in pharmacokinetics in rats
MK-801 induces dose-dependent Olney’s lesion formation in Han Wistar rats
Intravenous ketamine does not produce Olney’s lesions in Wistar rats up to 20 mg/kg
(2R,6R)-HNK does not demonstrate any neurotoxicology at doses up to 160 mg/kg
Acknowledgements:
This work was supported by the Division of Preclinical Innovation, National Center for Advancing Translational Research and the Center for Cancer Research, National Cancer Institute and the Intramural Research Programs of the National Institute of Mental Health and the National Institute of Aging. This article reflects the views of the author and should not be construed to represent FDA's views or policies. The authors would like to thank Steve Van Adestine (Covance Laboratories, Inc.) for assisting with image capture.
Footnotes
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- Auer RN "Effect of Age and Sex on N-Methyl-D-Aspartate Agonist Induced Neuronal Necrosis in Rats". Stroke, 1996, 27, 473–746 [DOI] [PubMed] [Google Scholar]
- Bender C; de Olmos S; Beuno A; de Olmos J, Lorenzo A.; "Comparative analyses of the neurodegeneration induced by the non-competitive NMDA-receptor-antagonist drug MK801 in mice and rats" Neurotoxicol. Teratol. 2010a, 32, 542–550. [DOI] [PubMed] [Google Scholar]
- Bender C; Rassetto M; de Olmos JS; de Olmon S; Lorenzo A; "Involvement of AMP A/Kainateexcitotoxicity in MK801-induced neuronal death in the retrosplenial cortex." Neuroscience, 2010b, 169, 720–732. [DOI] [PubMed] [Google Scholar]
- Bolon B; Garman RH; Pardo ID; Jensen K; Sills RC; Roulouis A; Radovsky A; Bradley A; Andrews-Jones L; Butt M; Gumprecht L; "STP Position Paper: Recommended practices for Sampling and Processing the Nervous System (Brain, Spinal Cord Nerve, and Eye) during Nonclinical General Toxicity Studies." Toxicologic Pathology, 2013, 41, 1028–1048. [DOI] [PubMed] [Google Scholar]
- Fix AS.; Horn JW.; Wightman KA.; Johnson CA.; Long GG.; Starts RW.; Farber N.; Wozniak DF.; Olney JW. "Neuronal vacuolization and necrosis induced by the noncompetitive N-methyl-Daspartate (NMDA) antagonist MK(+)801 (dizocilpine maleate): a light and electron microscopic evaluation of the rat retrosplenial cortex.” Exp Neurol. 1993,123, 204–215. [DOI] [PubMed] [Google Scholar]
- Fix AS; Wozniak DW; Truex LL; McEwen M; Miller JP; Olney JW "Quantitative analysis of factors influencing neuronal necrosis induced by MK-801 in the rat posterior cingulate/retrosplenial cortex” Brain Res. 1995, 696, 194–204. [DOI] [PubMed] [Google Scholar]
- Fukumoto K; Fogaca MV; Liu RJ; Duman C; Kato T; Li XY; Duman RS; "Activitydependent brain-derived neurotrophicfactor signaling is required for the antidepressant actions of (2R,6R)-hydroxynorketmaine.” Proc. Natl. Acad. Sci. USA 2019, 116, 297–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green SM; Cote CJ "Ketamine and Neurotoxicity: Clinical Perspectives and Implications for Emergency Medicine” Ann. of Emerg. Med. 2009, 54, 181–190. [DOI] [PubMed] [Google Scholar]
- Highland JN; Zanos P; Riggs LM; Georgiou P; Clark SM; Moms PJ; Moaddel R; Thomas CJ; Zarate CA Jr.; Pereira EFR; Gould TD. "Hydroxynorketamines: Pharmacology and Potential Therapeutic Applications” Pharmacol. Rev. 2021, 73, 763–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath ZC.; Czopf J.; Buzsaki G. "MK-801-induced neuronal damage in rats” Brain Res. 1997, 753, 181–195. [DOI] [PubMed] [Google Scholar]
- Jevtovic-Todorovic V; Benshoff N; Olney JW "Ketamine potentiates cerebrocortical damage induced by the common anaesthetic agent nitrous oxide in adult rats" Br. J. Pharmacol. 2000, 130, 1692–1698.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jevtovic-Todorovic V; Wozniak DF, Benshoff ND.; Olney JW. "A comparative evaluation of the neurotoxic properties of ketamine and nitrous oxide" Brain Res. 2001, 895, 264–267. [DOI] [PubMed] [Google Scholar]
- Loo C.; Galvez V.; O'Keefe E.; Mitchell PB.; Hadzi-Pavlovic D.; Leyden J.; Harper S.; Somogyi AA, Lai R.; Weickert CS.; Glue P. "Placebo-controlled pilot trial testing dose titration and intravenous, intramuscular and subcutaneous routes for ketamine in depression" Acta Psychiatr. Scand. 2016, 134, 48–56. [DOI] [PubMed] [Google Scholar]
- Low SJ; Roland CL "Review of NMD A antagonist-induced neurotoxicity and implications for clinical development." Int. J. Clin. Pharmacol. Ther. 2004, 42, 1–14. [DOI] [PubMed] [Google Scholar]
- Lumsden EW; Troppoli TA; Myers SJ; Zanos P; Aracava Y; Kehr J; Lovett J; Kim S; Wang F-H.; Schmidt.; Jenne CE.; Yuan P.; Morris PJ.; Thomas CJ.; Zarate CA Jr.; Moaddel R.; Traynelis SF.; Pereira EFR.; Thompson SM.; Albuquerque EX.; Gould TD "Antidepressant-relevant concentrations of the ketamine metabolite (2R,6R)-hydroxynorketamine do not block NMDA receptor Function." Proc. Nat. Acad. Sci. 2019, 116, 5160–5169. [DOI] [PMC free article] [PubMed] [Google Scholar]; Newport DJ; Carpenter LL; McDonald WM; Potash JB.; Tohen M.; Nemeroff CB.; APA Council of Research Task Force on Novel Biomarkers and Treatments. "Ketamine and other NMDA antagonists: Early clinical trials and possible mechanisms in depression." Am. J. Psychiatry 2015; 172, 950–966. [DOI] [PubMed] [Google Scholar]
- Olney JW; Labruyere J; Price MT "Pathological changes induces in cerebrocortical neurons by phencyclidine and related drugs" Science, 1989, 244, 1360–1362. [DOI] [PubMed] [Google Scholar]
- Olney JW; Labruyere J; Wang G; Wozniak DF; Price MT; Sesma MA "NMDA Antagonist Neurotoxicity: Mechanism and Prevention" Science, 1991, 254, 1515–1518. [DOI] [PubMed] [Google Scholar]
- Rao DB; Little PB; Malarkey DE; Herbert RA; Sills RC "Histopathological evaluation of the nervous system in National Toxicology Program rodent studies: a modified approach/’ Toxicol. Pathol. 2011, 39, 463–470. [DOI] [PubMed] [Google Scholar]
- Riggs LM; Aracava Y; Zanos P; Fischell J; Albuquerque EX.; Pereira EFR.; Thompson SM.; Gould TD.; "(2R,6R)-hydroxynorkctaminc rapidly potentiates hippocampal glutamatergic transmission through a synapse-specific presynaptic mechanism.” Neuropsychopharmacology, 2020, 45, 426–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raboisson P; Flood K; Lehmann A; Beige O-G; "MK-801 neurotoxicity in the guinea pig cerebral cortex: Susceptibility and regional differences compared with the rat.” J. Neurosci. Res. 1998, 49, 364–371.. [PubMed] [Google Scholar]
- Saland SK; Kabbaj M "Sex Differences in the Pharmacokinetics of Low-dose Ketamine in Plasma and Brain of Male and Female Rats” J. Pharmacol. Exp. Ther. 2018, 367, 393–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp FR; Butman M; Wang S; Koistinaho J; Graham SH; Sagar SM; Noble L; Berger P; Longo FM; "Haloperidol Prevents Induction of the hsp70 Heat Shock Gene in Neutons Injured by Phencyclidine (PCP), MK801, and Ketamine.” J. Neurosci. Res. 1992, 33, 605–616. [DOI] [PubMed] [Google Scholar]
- Sharp FR; Jasper P; Hall J; Nobel L, Sagar SM. "MK-801 and ketamine induce heat shock protein HSP72 in injured neurons in the posterior and retrosplenial cortex" Ann. Neurol. 1991, 30, 801–809. [DOI] [PubMed] [Google Scholar]
- Strong CE; Kabbaj K "On the safety of repeated ketamine infusions for the treatment of depression: Effects of sex and developmental periods.” Neurobiol. Stress. 2018, 9, 166–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K; Nosyreva E; Hunt KW; Kavalali ET; Monteggia LM "The ketamine metabolite hydroxynorketamine impacts downstream signaling via NMDA receptor inhibition” Nature, 2017, 546, E1–E3. [DOI] [PubMed] [Google Scholar]
- Tian Z, Dong C; Fujita A; Fugita Y; Hashimoto K; "Expression of heat shock protein HSP-70 in the retrosplenial cortex of rat brain after administration of (R,S)-ketamine and (S)-ketamine but not (R)ketamine." Pharmacol. Biochem. Behav. 2018, 172, 17–21. [DOI] [PubMed] [Google Scholar]
- Tomitaka S; Hashimoto K; Narita N; Sakamoto A; Minabe Y; Tamura A "Memantine induces heat shock protein HSP70 in the posterior cingulate cortex, retrosplenial cortex and dentate gyrus of rat brain" Brain Res. 1996, 740, 1–5. [DOI] [PubMed] [Google Scholar]
- Vranken JH; Troost D; de Haan P; Pennings FA; van der Vegt MH; Dijkgraaf MG; Hollmann MW "Severe toxic damage to the rabbit spinal cord after intrathecal administration of preservative-free S(+)-ketamine" Anesthesiology, 2006, 105, 813–818. [DOI] [PubMed] [Google Scholar]
- Vranken JH; Troost D; Wegener JT; Kruis MR; van der Vegt MH "Neuropathological findings after continuous intrathecal administration of S(+)-ketamine for the management of neuropathic cancer pain." Pain. 2005, 117, 231–235. [DOI] [PubMed] [Google Scholar]
- Wang C; Zheng D; Xu J; Lam W; Yew DT; "Brain Damages in ketamine addicts as revealed by magnetic resonance imaging" Front. Neuroanat. 2013, 7, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson ST.; Toprak M.; Turner MS.; Levine SP.; Katz RB.; Sanacora G. "A Survey of the Clinical, Off-Label Use of Ketamine as a Treatment for Psychiatric Disorders" Am. J. Psychiatry. 2017, 174, 695–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozniak DF; Brosnan-Watters G; Nardi A; McEwen M; Corso TD; Olney JW; Fix AS; "MK-801 neurotoxicity in male mice: histologic effects and chronic impairment in spatial learning" Brain Res. 1996, 707, 165–179. [DOI] [PubMed] [Google Scholar]
- Zanos P; Moaddel R; Morris PJ; Riggs LM; Highland JN; Georgiou P; Pereira EFR.; Albuquerque EX.; Thomas CJ.; Zarate CA Jr.; Gould TD. "Ketamine and ketamine metabolite pharmacology: insights into therapeutic mechanisms” Pharmacol. Rev, 2018, 70, 621–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanos P; Moaddel R; Morris PJ; Georgiou P; Fischell J; Elmer GI; Alkondon M; Yuan P; Pribut HJ; Singh NS; Dossou KS; Fang Y; Fluang XP; Mayo CL; Wainer IW; Albuquerque EX; Thompson SM; Thomas CJ; Zarate CA Jr.; Gould TD.; "NMDAR inhibition-independent antidepressant actions of ketamine metabolites'’ Nature, 2016, 533, 481–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarate CA Jr; Singh JB; Carlson PJ; Brutsche NE; Ameli R; Luckenbaugh DA; Charney DS; Manji HK "A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression" Arch. Gen. Psychiatry, 2006, 63, 856–864. [DOI] [PubMed] [Google Scholar]
- Zhang Y; Huo M; Zhou J; Xie S; "PKSovlcr: An add-in program for pharmacokinetic and pharmacodynamic data analysis in Microsoft Excel" Comput. Methods Programs Biomed. 2010, 99, 306–314.. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.