

Abstract
Transcription factor 4 (TCF4, also known as ITF2 or E2-2) is a type I basic helix-loop-helix (bHLH) transcription factor. Autosomal dominant mutations in TCF4 cause Pitt-Hopkins syndrome (PTHS), a rare syndromic form of autism spectrum disorder (ASD). In this review, we provide an update on the progress regarding our understanding of TCF4 function at the molecular, cellular, physiological and behavioral levels with a focus on phenotypes and therapeutic interventions. We examine upstream and downstream regulatory networks associated with TCF4 and discuss a range of in vitro and in vivo data with the aims of understanding emerging TCF4-specific mechanisms relevant for disease pathophysiology. In conclusion, we provide comments about exciting future avenues of research that may provide insights into potential new therapeutic targets for PTHS.
Keywords: transcription factor 4, neurodevelopmental disorder, Pitt-Hopkins Syndrome, oligodendrocyte, bHLH
TCF4 mutations in Pitt-Hopkins Syndrome
Transcription factor 4 (TCF4) is a gene located on chromosome 18 that encodes a type I basic helix-loop-helix (bHLH) transcription factor and dimerizes with itself or other members within the same protein family. This dimer complex of E-proteins recognizes E-box (CANNTG) sequences located within promoter and enhancer regions of target genes and regulates their expression [1,2]. Autosomal dominant mutations in TCF4 are causal for a rare neurodevelopmental disorder called Pitt-Hopkins syndrome (PTHS), first described in two unrelated patients in 1978 [3]. Reliable figures for prevalence of this disorder are not available [4], however estimates range between 1 in 34,000–41,000 births [5]. PTHS predominantly arises from de novo mutations within the TCF4 gene locus, with rare instances of parental mosaicism [6,7]. Clinically, patients are characterized by intellectual disability, developmental delay, breathing abnormalities, absent or limited speech, motor delay, seizure, constipation, and facial features including wide mouth and a broad nasal base with a high bridge [5,8–11]. Seizure activity is observed in approximately 30–50% of patients [4,5,12]. In addition, brain imaging studies identify several abnormalities that vary between patients, including underdevelopment of the corpus callosum, smaller hippocampus, enlarged caudate nuclei, and cerebellar and vermis hypoplasia [9,10,13–15]. Many of these features are not fully penetrant and can complicate diagnosis. Therefore accurate diagnosis requires genetic screening to rule out other disorders that share overlapping clinical characteristics such as Rett syndrome, Angelman syndrome and Mowat-Wilson syndrome [13,14,16].
A variety of causal mutations within the TCF4 locus have been identified, and include both missense and nonsense mutations that range from single point mutations and small deletions to large truncations in the TCF4 gene. Depending on the mutation, the affected allele leads to haploinsufficient expression of TCF4 protein, or expression of a truncated/mutated TCF4 protein which is capable of acting in a dominant-negative or hypomorphic manner [17, 18]. Indeed, the majority of de novo mutations lie within the evolutionary conserved bHLH domain of TCF4 that is required for dimerization and subsequent DNA binding. Mutations in the 5’ end of the gene, that are upstream of regulatory regions and only affect expression of long isoforms, are associated with mild to moderate non-syndromic intellectual disability without the typical features of PTHS [19–21]. Attempts to correlate genotype and phenotype are reported with mixed results. One report found a positive correlation among patients harboring missense mutations with a high probability of developing seizures [5], however this correlation was not present in another study [9]. These mixed results are likely due to a combination of small sample sizes and variation in genetic background. Therefore, as more patients are identified and characterized, confidence in genotype/phenotype correlations may increase.
Molecular biology of TCF4
The human TCF4 gene spans 437 kbs on chromosome 18q21.2 and contains 41 exons. Considering both alternative splicing and alternate transcription initiation sites, so far 18 unique protein isoforms, with differing N-terminals and relatively conserved C-terminals, are identified [22]. Among these 18 N-terminally distinct types of protein isoforms, 5 are expressed only in testis (TCF4-J, TCF4-K, TCF4-L, TCF4-M and TCF4-N) [22]. Functional domain analysis identified that TCF4 contains two canonical activation domains, AD1 and AD2, a TFIID-interacting domain (AD3), a bHLH motif located near the C-terminus, and a nuclear localization sequence (NLS) [17,22,23]. The complexity of this genomic structure appears evolutionarily conserved across species. Type I bHLH protein tends to be widely expressed across diverse tissues and similarly Tcf4 is expressed in a variety of organs [22]. However, the highest level of Tcf4 expression is found in the CNS, where expression is observed in most major brain structures [22]. Tcf4 expression is also developmentally regulated, with the most abundant expression observed during fetal development [24–26].
The function of TCF4 is further complicated by the large number of alternatively spliced transcripts and the resulting protein isoforms. The functional properties of specific protein isoforms depend on which 5’ exon and internal exons are included in the translated protein. Both the in vitro subcellular localization and transcriptional control are differentially regulated based on isoform specificity. For instance, isoforms containing the NLS are found localized to the nucleus, whereas isoforms lacking the NLS require a heterodimerization partner to gain access to nucleus through a piggy-back mechanism [22]. Complexity quickly multiplies when considering how each PTHS-associated mutation may affect the function of specific protein isoforms. Two groups carried out a detailed analysis of PTHS-associated mutations and assessed the functional consequences of a variety of mutations [17,27]. They showed that missense mutations in the bHLH domain and reading frame elongating mutations damage DNA-binding and transactivation function in a manner dependent on dimer context. Elongating and missense mutations at the dimer interface of the bHLH domain destabilizes the protein, whereas missense mutations outside of the bHLH domain cause no apparent functional deficits. In vitro transcription assays using these various mutants leads to variable context-specific impairments ranging from hypomorphic to dominant-negative effects and suggests these diverse mechanisms could possibly contribute to phenotypic variability observed in PTHS patients [17,18,27].
Understanding upstream regulation of TCF4 expression and transcription is critical to developing therapeutic strategies designed to overcome TCF4 haploinsufficiency, however our current understanding of this regulation is limited. Hennig and colleagues [28] showed that pharmacological activation of WNT/β-catenin pathway in induced pluripotent stem cells (iPSCs)-derived neural progenitor cells (NPC) and neurons derived from PTHS patients led to an increase in TCF4 expression. In addition, they and others showed upstream modification of chromatin regulatory elements through inhibition of class I histone deacetylases (HDAC) also increased TCF4 expression [28,29]. Another identified upstream regulator of Tcf4 is TCF3, an E-box protein family member. Tcf3 expression precedes Tcf4 expression in the developing cortex and knockout of Tcf3 reduced TCF4 protein expression, likely through TCF3 binding to an E-box sequence within the second exon of Tcf4 [25]. Lastly, the maternally imprinted transcriptional regulator ZAC1/PLAGL1 was identified to be an upstream regulator of Tcf4 expression in a mouse neural stem cell line. ZAC1 regulates Tcf4 expression by binding the proximal promoter and first intron of a long isoform of Tcf4 (Tcf4-b) but not the promoter of the shorter isoform (Tcf4-a) [30]. Together, these studies identify upstream regulatory pathways that are capable of increasing Tcf4 expression and therefore could be suitable therapeutic targets. However, these pathways regulate a broad set of downstream genes in a context-dependent manner and may lack the specificity necessary when considering their therapeutic potential.
An additional therapeutic approach involves activation of TCF4-mediated transcription through manipulation of neuronal activity. Sepp and colleagues [31] showed that depolarization of primary cortical neurons significantly enhanced TCF4-dependent transcription. They demonstrated this regulation was triggered by calcium influx and required PKA-dependent phosphorylation of TCF4 [31]. In addition, a functional requirement for upstream regulation by calcium influx and phosphorylation of TCF4 was demonstrated in vivo, whereby TCF4 overexpression in the developing rat prefrontal cortex produced an abnormal distribution of pyramidal neurons within cortical columns. This phenotype required activity-dependent Ca2+ signaling and TCF4 phosphorylation [31,32]. The activity-dependent regulation of TCF4 function has important implications for its involvement in brain development, learning and memory, all of which are disrupted in PTHS. However, safely and effectively manipulating neuronal activity presents significant challenges as a therapeutic intervention.
As a member of the E-box protein family, Tcf4 functions as a transcriptional regulator. Identifying genes regulated by TCF4 is critical to gaining insights about disease etiology and potential therapeutic targets. Unfortunately, the E-box consensus motif, “CANNTG”, is relatively non-specific; while the complex context-specific nature of bHLH dimers precludes in silico identification of genes regulated by TCF4. ChIP-seq studies in SH-SY5Y neuroblastoma cells by two independent labs identified over 10,000 TCF4 binding sites across the genome that could be assigned to over 5500 genes [33,34]. Enrichment analysis of TCF4 target genes identified functions associated with nervous system development, neurogenesis, and ion transport. Moreover, TCF4 knockdown in the same neuroblastoma cell line led to 1204 differentially expressed genes (DEGs), and these DEGs were involved in several signaling pathways including TGF-β signaling, epithelial to mesenchymal transition, and apoptosis. In addition, knockdown of TCF4 led to differential expression of several neurodevelopmental disorder genes including UBE3A, FRM1, and MEF2C [35]. However, cellular context is an important consideration, as TCF4 knockdown in a human neural progenitor cell line led to identification of DEGs associated with cell cycle [36]. Hennig and colleagues [28] generated a stable human NPC line that overexpressed a Flag-tagged TCF4 (isoform 10) and performed ChIP-seq on differentiated neurons. Among all the binding sites observed, they noted sites near genes encoding the synaptic proteins GRIA1 and DLG2. In addition to these whole-genome ChIP-seq approaches, several ChIP-qPCR studies have identified direct targets of TCF4 binding and include Bmp7 [37], Nrxn1 [38], Gadd45g [31], Gjb2 and Plp1 [39]. Beyond these ChIP-seq studies, several groups have identified specific downstream targets of TCF4 using a variety of cell and animal models. Manipulation of TCF4 expression in these models lead to altered expression of a variety of downstream targets which include two ion channels, Scn10a (Nav1.8) and Kcnq1 (Kv7.1) [26], Wnt7b [40], Gadd45g [31], Nrxn1 [27,38], Syn and Dlg1 [41]. The role of these downstream target genes in the generation of phenotypes will be discussed in greater detail below. Together, these studies indicate TCF4 regulates a variety of neural genes that are involved in early development, differentiation, intrinsic excitability, synapses and survival. However, TCF4 is expressed in a variety of cell types in the central nervous system (see below), and therefore it will be useful to generate cell-specific ChIP-seq datasets to improve our understanding of TCF4 regulation in specific cellular contexts.
TCF4 at the cellular level
The functional significance of any gene is often associated with its spatiotemporal expression pattern and fortunately a detailed understanding of TCF4 expression pattern is emerging within specific cell types of the brain. Early RNA sequencing studies from human postmortem brain and qPCR of rat brain samples showed that TCF4 expression is elevated during fetal development and begins to subside around birth, but is still present in the adult brain [26]. These findings were then replicated in mouse, rhesus monkey, and human [24,25,42,43], suggesting an evolutionarily conserved role of TCF4 across species. More detailed studies in mouse, show Tcf4 is turned on early in brain development, around embryonic day 11.5 (E11.5) where it is highly expressed in proliferative zones and primordia of the cortex, hippocampus, cerebellum, amygdala, striatum and hypothalamus [42]. This high expression seemingly follows the early wave of neurogenesis as the migrating postmitotic neurons begin to populate cortical layer 5/6. Although its expression level begins to be downregulated after E11.5 in the ventricular zone (VZ) and subventricular zone (SVZ), TCF4 remains moderately expressed in the VZ/SVZ and can be detected in later born neurons that eventually become layer 2/3 pyramidal neurons. This type of developmental regulation of TCF4 across different proliferation/maturation stages is not present in the hippocampus, where TCF4 expression persists at a high levels both embryonically and postnatally [42], partially due to ongoing neurogenesis that continues into adulthood in the hippocampus.
Cell-type specific distribution of TCF4 was elegantly demonstrated using a Tcf4 reinstatement mouse model in which a GFP reporter is under the control of the endogenous Tcf4 promoter, thus allowing regional and cell-type specific visualization of TCF4 expression [44]. Kim and colleagues showed that TCF4 is expressed in several major cell types of the brain including excitatory and inhibitory neurons, astrocytes, and oligodendrocytes. This includes three major subclasses of all cortical inhibitory neurons, categorized by expression of PV, SOM and 5HT3aR. However, microglia which are labeled by the marker IBA1 do not appear to express TCF4. This specificity across these four major cell types was consistent across cortex and hippocampus, however in striatum PV and SOM interneuron populations expressed TCF4 but not medium spiny projection neurons or cholinergic interneurons. In the cerebellum, TCF4 is enriched in the molecular and internal granule layers but is absent in the external granule layer and Purkinje cell layers at P10 and older. Together, these expression data indicate TCF4 is critical during early brain development, but continues to have functional relevance in specific cell types throughout the lifespan. This developmental and cell type-specific expression pattern complicates the timeframe for when therapeutic approaches will be most amenable and whether specific cell-types should be targeted.
Phenotypes associated with altered TCF4 expression: Development
The peak of Tcf4 expression coincides with cortical neurogenesis, migration and neuronal maturation suggesting it plays a role in regulating these processes as shown in Figure 1. To begin to understand the functional role of TCF4 during development several groups have performed TCF4 loss-of-function (LOF) studies in the developing mouse and fruit fly brain. These studies have observed several consistent phenotypes that indicate disease causing mutations have profound effects on early brain development. In Drosophila, LOF of Daughterless (Da), a Tcf4 orthologue, disrupts neural development and leads to complete loss of peripheral neurons [45]. These various developmental defects can be rescued by overexpression of human TCF4 in the fly nervous system early in development [46]. Similarly in the mouse, Tcf4’s functional role begins early in cortical development as it regulates NPC proliferation and lineage specification of pyramidal neurons in the developing cortex [25,30,43] and hippocampus [40]. In addition, TCF4 LOF is reported to disrupt neuronal migration in the cortex [25,37] and hippocampus which may be due to abnormal radial glial fibers [40]. These early developmental phenotypes likely contribute to the abnormal distribution of cortical pyramidal cells within cortical layers and columns [25,32,43], reduced cortical thickness, hypoplasia of the hippocampus, malformations of the corpus callosum and reduced brain weight [40,43,47,48], which are concordant phenotypes observed in the PTHS patient population [9,10,13–15].
Fig. 1. Disease causing mutations in Tcf4 disrupt brain development at various developmental stages and ultimately compromise behavior.
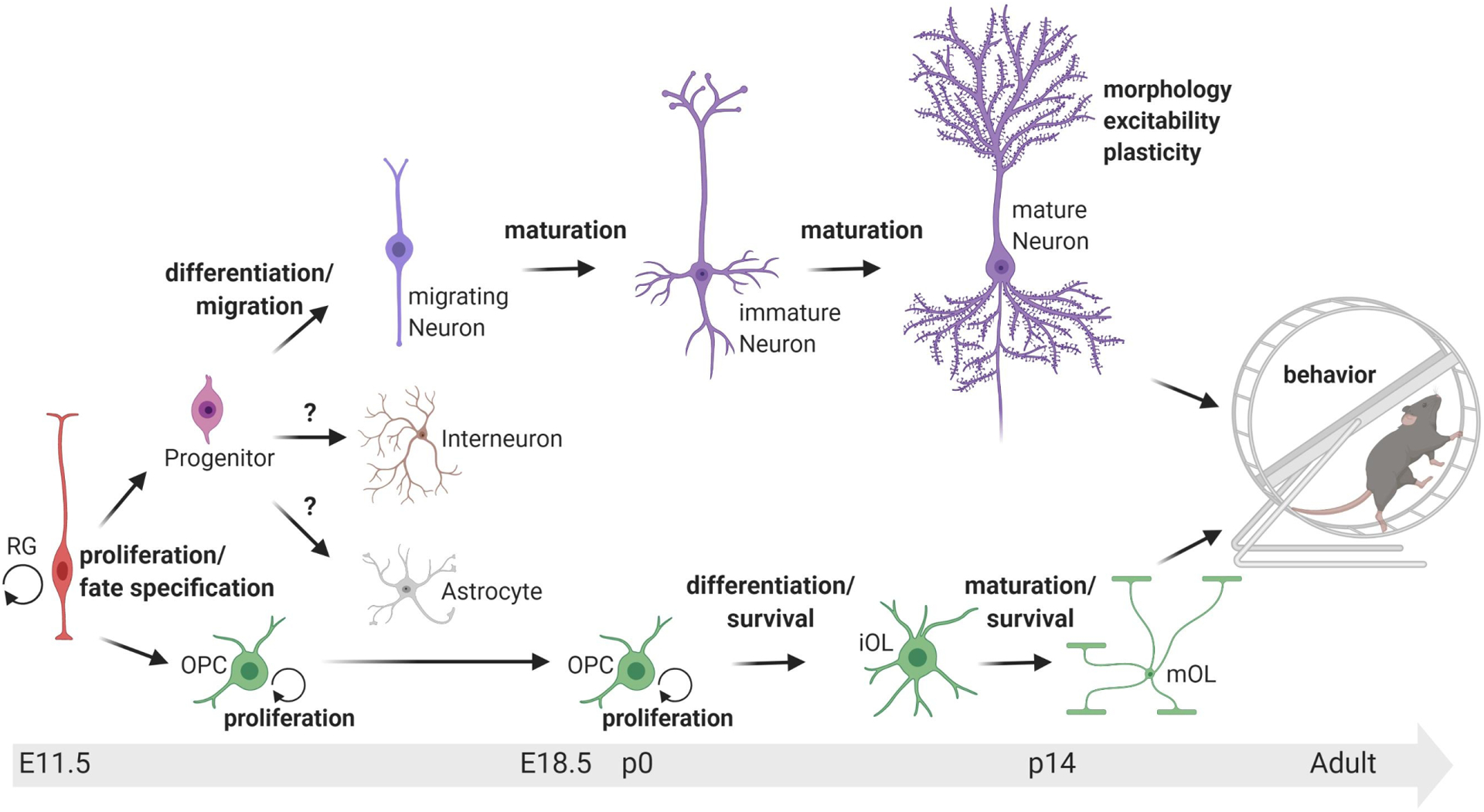
TCF4 is expressed in radial glia cells (RG), progenitor cells, excitatory neurons, inhibitory neurons, astrocytes, oligodendrocyte precursor cells (OPC), and oligodendrocytes (OLs). In the neuronal lineage, Tcf4 mutation disrupts progenitor proliferation and fate specification, migration, differentiation, maturation, morphology, excitability, and plasticity. In the oligodendrocyte lineage, Tcf4 mutation enhances OPC proliferation and reduces the density of mature OLs. TCF4 is expressed in astrocytes and interneurons, but how Tcf4 mutation in these cell types affects their function is unknown. Ultimately, these developmental phenotypes lead to behavioral deficits in PTHS mouse models. Bold type indicates biological process for which Tcf4 mutation produces a phenotype. iOL = immature oligodendrocyte, mOL = mature oligodendrocyte.
Altered early brain development due to TCF4 mutation sets the stage for abnormal neuronal and circuit development and therefore several studies have focused on the effects of TCF4 LOF in neurons. For instance, TCF4 appears to be critical for neurite development as several studies show defects in neurite length and complexity in mouse cortex and olfactory bulb due to TCF4 gain-of-function and LOF [25,38,47]. In addition, TCF4 appears to regulate the development of synapses. TCF4 LOF was shown to decrease the number of synaptophysin puncta in primary neuronal cultures [25] and increase the number of synaptic boutons in the Drosophila neuromuscular junction through regulation of Neurexin [38]. TCF4 also appears to regulate intrinsic excitability, synaptic transmission and synaptic plasticity. Embryonic knockout of Tcf4 resulted in downregulation of Kcna1 (Kv1.1), a voltage-gated potassium channel involved in a variety of human diseases [43,49]. In addition, knockdown of Tcf4 in the rat prefrontal cortex caused two peripherally enriched ion channels, Scn10a (Nav1.8) and Kcnq1 (Kv7.1) to be ectopically expressed in the brain, which resulted in abnormal membrane excitability of pyramidal neurons [26,43]. These results led to the hypothesis that inhibition of Nav1.8 may be a potential therapeutic target for PTHS. Interestingly, a high-throughput drug screen against Nav1.8 identified the FDA-approved calcium channel blocker nicardipine [50], and treatment of a PTHS mouse model with nicardipine led to rescue of a variety of behavioral deficits [51]. Lastly, in several TCF4 LOF mouse models, hippocampal long-term synaptic plasticity was exaggerated and evoked synaptic transmission showed elevated NMDA receptor function. Both of these phenotypes were consistent with deficits observed in several hippocampus-dependent behaviors [29,48].
Phenotypes associated with altered TCF4 expression: Cell-type specificity
TCF4 expression is widespread across the brain and body, yet it is heterogeneous within the same tissue, as its expression is often observed only in a subset of cell types within the tissue. For instance, in the striatum, PV-positive GABAergic neurons do express TCF4, whereas the GABAergic medium spiny neurons do not [44]. Such cell-type specific regulation indicates TCF4 is playing unique roles in specific cell types. Additional examples exist showing TCF4 is critical for the genesis and/or survival of certain cellular populations. For instance, TCF4 is an essential regulator of plasmacytoid dendritic cell (PDC) development [52]. PDCs are a distinct type of immune cell that specialize in type I interferon (IFNα) secretion in response to viral nucleic acids, and TCF4 LOF completely prevents the production of PDCs and IFNα. Moreover, this phenotype was observed in blood samples from PTHS patients, where a significant reduction in the number of PDCs and IFNα was observed [52]. In addition, TCF4 LOF was shown to disrupt development in the midbrain whereby a lack of MATH1/TCF4 heterodimers resulted in the loss of a restricted neuronal population within the pontine nucleus [53]. During dopaminergic neurogenesis, TCF4 LOF causes temporal dysregulation of developmentally relevant genes such as Th, Nxph3 and Ahd2, likely through upregulation of Ascl1, and suggests TCF4 regulates differentiation of a subset of dopaminergic neurons [54]. Lastly, two studies show TCF4 is critical for oligodendrocyte (OL) differentiation and/or survival in the spinal cord and brain [24,39]. Both groups demonstrated Tcf4 mutation led to a cell autonomous reduction in mature OLs and a concomitant reduction in myelination. Phan and colleagues further demonstrated the reduced myelination led to a functional deficit whereby a larger proportion of action potentials were observed traveling along unmyelinated axons. Furthermore, Wedel and colleagues demonstrated that defects in OL differentiation appear to involve TCF4 preferentially binding to OLIG2, a critical bHLH domain transcription factor involved in OL development. Moreover, OLIG1, a bHLH family member that is required for OL differentiation, was shown to be downregulated in Tcf4 knockout mice [43]. Together, these results indicate that TCF4 is critical in the regulation of cellular differentiation and/or survival across numerous cell types in the body and brain. However, our understanding of TCF4 expression and regulation of cell-type specific development is limited, as knowledge of TCF4 specific functions in the majority of these cell types is still lacking.
Phenotypes associated with altered TCF4 expression: Behavior
How the molecular, cellular and physiological phenotypes add up to the clinical features of PTHS is complex. One potential way forward is to determine which phenotypes underlie specific behavioral deficits in the PTHS mouse model. If rescue of cellular and/or circuit level phenotypes can normalize behavioral deficits, this improves confidence that a potential therapy could be developed for humans. Fortunately, several groups have now demonstrated consistent behavioral deficits across a variety of PTHS mouse models. Initially, Kennedy and colleagues showed that TCF4 haploinsufficient animals were hyperactive and displayed deficits in social interaction, ultrasonic vocalization, prepulse inhibition, and spatial and associative learning and memory [29]. Many of these behavioral deficits were then replicated by Thaxton and colleagues in a study that characterized 4 genetically different PTHS mouse models. Across these four mouse models, they observed hyperactivity, reduced anxiety, and deficits in spatial learning [48]. A third study has also replicated these initial behavioral deficits along with identifying additional abnormalities including abnormal nesting, increased self-grooming, and reduced grip strength [51]. It is worth noting that some of these behavioral phenotypes are also present in fruit flies. Silencing of Da was shown to impair appetitive associative learning and memory in fly larvae and locomotion in adult flies [41].
One important step to identifying therapies for PTHS is to demonstrate rescue of behavioral deficits in the PTHS mouse model. Along these lines, HDAC inhibitors, administered either with intraperitoneal injection of the broad spectrum inhibitor SAHA or via intracerebroventricular injection of antisense oligonucleotide against Hdac2, were shown to be effective at rescuing contextual and trace-cued threat recognition memory in a PTHS mouse model [29]. Similarly in fruit flies, feeding SAHA was able to rescue the negative geotaxis phenotype associated with silencing Da in female flies [41]. In addition, the dihydropyridine calcium channel inhibitor, nicardipine, was shown to be effective at rescuing a variety of behavioral deficits in the PTHS mouse model. This effect was potentially through blockade of the sodium channel Nav1.8 [51], which was found upregulated in the brain after TCF4 downregulation [26]. Finally, an exciting future direction concerns the elegantly created Tcf4 reinstatement mouse, which allows for genetic rescue of TCF4 expression in a cell type- and/or temporally-specific manner [44]. Future experiments will determine if reinstatement of TCF4 in specific cell types can lead to cellular and behavioral rescue, thus allowing for the identification of specific cell populations amenable to therapeutic targeting. Moreover, such a model may help optimize the ideal temporal timeframe of rescue for specific cell types, which will be essential for the development of therapies. If behavioral phenotypes are amenable to rescue when TCF4 is reinstated in postnatal mice, it would suggest gene therapy approaches may be beneficial to PTHS patients. However, if postnatal reinstatement of TCF4 does not rescue phenotypes, then gene therapies approaches may not be suitable unless delivered in utero, and in this case developing therapies that normalize brain function by modifying pathways downstream of TCF4 should be prioritized.
Conclusions
Over the past few decades significant progress has been made in our understanding of the etiology of PTHS and the identification of upstream and downstream targets that may greatly inform therapeutic strategies. Given its broad expression pattern across many cell types in the central nervous system, a greater understanding of its cell-type specific role is warranted. For instance, an understanding of the function of TCF4 in GABAergic interneurons is completely lacking. In early development, TCF4 is heavily expressed in regions of the basal forebrain that generate inhibitory neurons [43,47] and TCF4 is expressed postnatally in various subclasses of GABAergic interneurons [44]. Moreover, Ascl1, a known TCF4 binding partner that is a critical regulator of interneuron development, was shown to be upregulated in Tcf4 knockouts [43,55,56] and therefore could potentially impact the development and function of interneurons. An imbalance of excitation and inhibition is commonly observed in numerous neuropsychiatric disorders [57] and therefore determining if this type of imbalance is present in PTHS mouse models may shed light on new avenues for therapeutic intervention.
Two critical questions are outstanding: which phenotypes should be targeted l for greatest improvement in clinical outcomes and when are these phenotypes most amenable to rescue. Clearly from the research literature, disease causing mutations lead to profound defects in brain development, some of which may be hardwired defects that are resistant to postnatal normalization. However, postnatal brain function can be modified by targeting signaling pathways, synaptic transmitter receptors and/or ion channels and therefore normalization of brain function may be possible. Functional studies using patient derived iPSCs will be useful for determining whether cellular and physiological deficits in mouse models translate to the human context. A set of patient and parental control iPSCs are now available [58] to the field and studies using these lines may help prioritize phenotypes that when normalized may have clinical significance. In addition, studies using the Tcf4 reinstatement mouse model will be instrumental to determine which phenotypes are hardwired during brain development and which phenotypes are responsive to rescue in the postnatal period. This mouse model will also be incredibly useful to determine whether TCF4 reinstatement in specific cell-types can lead to gainful improvements. For instance, one cell type that may be capable of overcoming temporal specificity are OLs which are reduced in PTHS mouse models [24,39]. Oligodendrocyte precursor cells (OPCs) are the most abundant proliferating cell population in the adult human brain, allowing for regeneration of myelin throughout the lifespan. It will be exciting to determine if postnatal reinstatement of TCF4 within OPCs increases OL numbers and myelination and ultimately leads to functional improvements in physiology and behavior in the PTHS mouse model.
Continued research is necessary to determine which genes are upstream regulators of TCF4 expression, as modulation of these regulators could potentially overcome TCF4 haploinsufficiency. In addition, increasing our understanding of genes regulated downstream of TCF4 could also lead to the identification of additional therapeutic targets. However, it is important that these studies keep in mind the cellular and temporal context of TCF4 function, as this understanding will be necessary for identifying contextually-specific therapeutic targets. In addition, single-cell RNA sequencing (sc-RNAseq) is now routine and the TCF4 field would greatly benefit from obtaining a sc-RNAseq dataset from at least one of the PTHS mouse models. So far, there are four bulk RNAseq datasets from several different PTHS mouse models available, which have provided valuable insights about the biological functions of TCF4 [24,25,29,43]. However, given the fact that TCF4 expression is heterogeneous even within the same brain area, the resolution of bulk RNAseq is not sufficient. Thus, sc-RNAseq analysis will provide important details about DEGs within specific cell types while also identifying common pathways that are consistently dysregulated between cell types that may be more appropriate for therapeutic intervention.
Acknowledgement
We are grateful for the vision and generosity of the Lieber and Maltz families, who made this work possible. We thank the parents of the Pitt-Hopkins Research Foundation for providing both inspiration and financial support for this work. We thank Dr. Brittany Davis for her critical comments on this manuscript.
Funding Sources
This work was supported by the Lieber Institute for Brain Development, the Pitt-Hopkins Research Foundation Award, NIH R01 grant 5R01MH110487
Footnotes
Conflict of Interest Statement
The authors have no conflicts of interest to declare.
References
- 1.Massari ME, Murre C. Helix-loop-helix proteins: regulators of transcription in eucaryotic organisms. Mol Cell Biol. 2000January;20(2):429–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dennis DJ, Han S, Schuurmans C. bHLH transcription factors in neural development, disease, and reprogramming. Brain Res. 2019February15;1705:48–65. [DOI] [PubMed] [Google Scholar]
- 3.Pitt D, Hopkins I. A syndrome of mental retardation, wide mouth and intermittent overbreathing. Aust Paediatr J 1978September;14(3):182–4. [DOI] [PubMed] [Google Scholar]
- 4.de Winter CF, Baas M, Bijlsma EK, van Heukelingen J, Routledge S, Hennekam RCM. Phenotype and natural history in 101 individuals with Pitt-Hopkins syndrome through an internet questionnaire system. Orphanet J Rare Dis. 2016April12;11:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rosenfeld JA, Leppig K, Ballif BC, Thiese H, Erdie-Lalena C, Bawle E, et al. Genotype-phenotype analysis of TCF4 mutations causing Pitt-Hopkins syndrome shows increased seizure activity with missense mutations. Genet Med. 2009November;11(11):797–805. [DOI] [PubMed] [Google Scholar]
- 6.Steinbusch CVM, van Roozendaal KEP, Tserpelis D, Smeets EEJ, Kranenburg-de Koning TJ, de Waal KH, et al. Somatic mosaicism in a mother of two children with Pitt-Hopkins syndrome. Clin Genet. 2013January;83(1):73–7. [DOI] [PubMed] [Google Scholar]
- 7.Kousoulidou L, Tanteles G, Moutafi M, Sismani C, Patsalis PC, Anastasiadou V. 263.4 kb deletion within the TCF4 gene consistent with Pitt-Hopkins syndrome, inherited from a mosaic parent with normal phenotype. Eur J Med Genet. 2013June;56(6):314–8. [DOI] [PubMed] [Google Scholar]
- 8.Peippo MM, Simola KOJ, Valanne LK, Larsen AT, Kähkönen M, Auranen MP, et al. Pitt-Hopkins syndrome in two patients and further definition of the phenotype. Clin Dysmorphol. 2006April;15(2):47–54. [DOI] [PubMed] [Google Scholar]
- 9.Whalen S, Héron D, Gaillon T, Moldovan O, Rossi M, Devillard F, et al. Novel comprehensive diagnostic strategy in Pitt-Hopkins syndrome: clinical score and further delineation of the TCF4 mutational spectrum. Hum Mutat. 2012January;33(1):64–72. [DOI] [PubMed] [Google Scholar]
- 10.Marangi G, Ricciardi S, Orteschi D, Lattante S, Murdolo M, Dallapiccola B, et al. The Pitt-Hopkins syndrome: report of 16 new patients and clinical diagnostic criteria. Am J Med Genet A. 2011July;155A(7):1536–45. [DOI] [PubMed] [Google Scholar]
- 11.Sweetser DA, Elsharkawi I, Yonker L, Steeves M, Parkin K, Thibert R. Pitt-Hopkins Syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Stephens K, et al. , editors. GeneReviews®. Seattle (WA): University of Washington, Seattle; 1993. [PubMed] [Google Scholar]
- 12.Sparber P, Filatova A, Anisimova I, Markova T, Voinova V, Chuhrova A, et al. Various haploinsufficiency mechanisms in Pitt-Hopkins syndrome. Eur J Med Genet. 2020October15;104088. [DOI] [PubMed] [Google Scholar]
- 13.Zweier C, Peippo MM, Hoyer J, Sousa S, Bottani A, Clayton-Smith J, et al. Haploinsufficiency of TCF4 causes syndromal mental retardation with intermittent hyperventilation (Pitt-Hopkins syndrome). Am J Hum Genet. 2007May;80(5):994–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Amiel J, Rio M, de Pontual L, Redon R, Malan V, Boddaert N, et al. Mutations in TCF4, encoding a class I basic helix-loop-helix transcription factor, are responsible for Pitt-Hopkins syndrome, a severe epileptic encephalopathy associated with autonomic dysfunction. Am J Hum Genet. 2007May;80(5):988–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Pontual L, Mathieu Y, Golzio C, Rio M, Malan V, Boddaert N, et al. Mutational, functional, and expression studies of the TCF4 gene in Pitt-Hopkins syndrome. Hum Mutat. 2009April;30(4):669–76. [DOI] [PubMed] [Google Scholar]
- 16.Zollino M, Zweier C, Van Balkom ID, Sweetser DA, Alaimo J, Bijlsma EK, et al. Diagnosis and management in Pitt-Hopkins syndrome: First international consensus statement. Clin Genet. 2019February18;95(4):462–78. [DOI] [PubMed] [Google Scholar]
- 17.Sepp M, Pruunsild P, Timmusk T. Pitt-Hopkins syndrome-associated mutations in TCF4 lead to variable impairment of the transcription factor function ranging from hypomorphic to dominant-negative effects. Hum Mol Genet. 2012July1;21(13):2873–88. [DOI] [PubMed] [Google Scholar]
- 18.Rannals MD, Page SC, Campbell MN, Gallo RA, Mayfield B, Maher BJ. Neurodevelopmental models of transcription factor 4 deficiency converge on a common ion channel as a potential therapeutic target for Pitt Hopkins syndrome. Rare Dis. 2016August5;4(1):e1220468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kharbanda M, Kannike K, Lampe A, Berg J, Timmusk T, Sepp M. Partial deletion of TCF4 in three generation family with non-syndromic intellectual disability, without features of Pitt-Hopkins syndrome. Eur J Med Genet. 2016June;59(6–7):310–4. [DOI] [PubMed] [Google Scholar]
- 20.Maduro V, Pusey BN, Cherukuri PF, Atkins P, du Souich C, Rupps R, et al. Complex translocation disrupting TCF4 and altering TCF4 isoform expression segregates as mild autosomal dominant intellectual disability. Orphanet J Rare Dis. 2016May14;11(1):62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bedeschi MF, Marangi G, Calvello MR, Ricciardi S, Leone FPC, Baccarin M, et al. Impairment of different protein domains causes variable clinical presentation within Pitt-Hopkins syndrome and suggests intragenic molecular syndromology of TCF4. Eur J Med Genet. 2017November;60(11):565–71. [DOI] [PubMed] [Google Scholar]
- 22.Sepp M, Kannike K, Eesmaa A, Urb M, Timmusk T. Functional diversity of human basic helix-loop-helix transcription factor TCF4 isoforms generated by alternative 5’ exon usage and splicing. PLoS One. 2011July15;6(7):e22138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen W-Y, Zhang J, Geng H, Du Z, Nakadai T, Roeder RG. A TAF4 coactivator function for E proteins that involves enhanced TFIID binding. Genes Dev. 2013July15;27(14):1596–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Phan BN, Bohlen JF, Davis BA, Ye Z, Chen H-Y, Mayfield B, et al. A myelin-related transcriptomic profile is shared by Pitt-Hopkins syndrome models and human autism spectrum disorder. Nat Neurosci. 2020February3;23(3):375–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li H, Zhu Y, Morozov YM, Chen X, Page SC, Rannals MD, et al. Disruption of TCF4 regulatory networks leads to abnormal cortical development and mental disabilities. Mol Psychiatry. 2019January31;24(8):1235–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rannals MD, Hamersky GR, Page SC, Campbell MN, Briley A, Gallo RA, et al. Psychiatric risk gene transcription factor 4 regulates intrinsic excitability of prefrontal neurons via repression of scn10a and KCNQ1. Neuron. 2016April6;90(1):43–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Forrest M, Chapman RM, Doyle AM, Tinsley CL, Waite A, Blake DJ. Functional analysis of TCF4 missense mutations that cause Pitt-Hopkins syndrome. Hum Mutat. 2012December;33(12):1676–86. [DOI] [PubMed] [Google Scholar]
- 28.Hennig KM, Fass DM, Zhao W-N, Sheridan SD, Fu T, Erdin S, et al. WNT/β-Catenin Pathway and Epigenetic Mechanisms Regulate the Pitt-Hopkins Syndrome and Schizophrenia Risk Gene TCF4. Mol Neuropsychiatry. 2017July14;3(1):53–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kennedy AJ, Rahn EJ, Paulukaitis BS, Savell KE, Kordasiewicz HB, Wang J, et al. Tcf4 regulates synaptic plasticity, DNA methylation, and memory function. Cell Rep. 2016September6;16(10):2666–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schmidt-Edelkraut U, Daniel G, Hoffmann A, Spengler D. Zac1 regulates cell cycle arrest in neuronal progenitors via Tcf4. Mol Cell Biol. 2014March;34(6):1020–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sepp M, Vihma H, Nurm K, Urb M, Page SC, Roots K, et al. The intellectual disability and schizophrenia associated transcription factor TCF4 is regulated by neuronal activity and protein kinase A. J Neurosci. 2017October25;37(43):10516–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Page SC, Hamersky GR, Gallo RA, Rannals MD, Calcaterra NE, Campbell MN, et al. The schizophrenia- and autism-associated gene, transcription factor 4 regulates the columnar distribution of layer 2/3 prefrontal pyramidal neurons in an activity-dependent manner. Mol Psychiatry. 2018;23(2):304–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xia H, Jahr FM, Kim N-K, Xie L, Shabalin AA, Bryois J, et al. Building a schizophrenia genetic network: transcription factor 4 regulates genes involved in neuronal development and schizophrenia risk. Hum Mol Genet. 2018September15;27(18):3246–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Forrest MP, Hill MJ, Kavanagh DH, Tansey KE, Waite AJ, Blake DJ. The psychiatric risk gene transcription factor 4 (TCF4) regulates neurodevelopmental pathways associated with schizophrenia, autism, and intellectual disability. Schizophr Bull. 2018August20;44(5):1100–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Forrest MP, Waite AJ, Martin-Rendon E, Blake DJ. Knockdown of human TCF4 affects multiple signaling pathways involved in cell survival, epithelial to mesenchymal transition and neuronal differentiation. PLoS One. 2013August23;8(8):e73169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hill MJ, Killick R, Navarrete K, Maruszak A, McLaughlin GM, Williams BP, et al. Knockdown of the schizophrenia susceptibility gene TCF4 alters gene expression and proliferation of progenitor cells from the developing human neocortex. J Psychiatry Neurosci. 2017;42(3):181–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen T, Wu Q, Zhang Y, Lu T, Yue W, Zhang D. Tcf4 Controls Neuronal Migration of the Cerebral Cortex through Regulation of Bmp7. Front Mol Neurosci. 2016October3;9:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.D’Rozario M, Zhang T, Waddell EA, Zhang Y, Sahin C, Sharoni M, et al. Type I bHLH Proteins Daughterless and Tcf4 Restrict Neurite Branching and Synapse Formation by Repressing Neurexin in Postmitotic Neurons. Cell Rep. 2016April12;15(2):386–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wedel M, Fröb F, Elsesser O, Wittmann M-T, Lie DC, Reis A, et al. Transcription factor Tcf4 is the preferred heterodimerization partner for Olig2 in oligodendrocytes and required for differentiation. Nucleic Acids Res. 2020May21;48(9):4839–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang Y, Lu Z, Zhang Y, Cai Y, Yun D, Tang T, et al. Transcription factor 4 safeguards hippocampal dentate gyrus development by regulating neural progenitor migration. Cereb Cortex. 2020May14;30(5):3102–15. [DOI] [PubMed] [Google Scholar]
- 41.Tamberg L, Jaago M, Säälik K, Sirp A, Tuvikene J, Shubina A, et al. Daughterless, the Drosophila orthologue of TCF4, is required for associative learning and maintenance of the synaptic proteome. Dis Model Mech. 2020July30;13(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jung M, Häberle BM, Tschaikowsky T, Wittmann M-T, Balta E-A, Stadler V-C, et al. Analysis of the expression pattern of the schizophrenia-risk and intellectual disability gene TCF4 in the developing and adult brain suggests a role in development and plasticity of cortical and hippocampal neurons. Mol Autism. 2018March22;9:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mesman S, Bakker R, Smidt MP. Tcf4 is required for correct brain development during embryogenesis. Mol Cell Neurosci. 2020May28;106:103502. [DOI] [PubMed] [Google Scholar]
- 44.Kim H, Berens NC, Ochandarena NE, Philpot BD. Region and cell type distribution of TCF4 in the postnatal mouse brain. Front Neuroanat. 2020July17;14:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Caudy M, Grell EH, Dambly-Chaudière C, Ghysen A, Jan LY, Jan YN. The maternal sex determination gene daughterless has zygotic activity necessary for the formation of peripheral neurons in Drosophila. Genes Dev. 1988July;2(7):843–52. [DOI] [PubMed] [Google Scholar]
- 46.Tamberg L, Sepp M, Timmusk T, Palgi M. Introducing Pitt-Hopkins syndrome-associated mutations of TCF4 to Drosophila daughterless. Biol Open. 2015November30;4(12):1762–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schoof M, Hellwig M, Harrison L, Holdhof D, Lauffer MC, Niesen J, et al. The basic helix-loop-helix transcription factor TCF4 impacts brain architecture as well as neuronal morphology and differentiation. Eur J Neurosci. 2020June;51(11):2219–35. [DOI] [PubMed] [Google Scholar]
- 48.Thaxton C, Kloth AD, Clark EP, Moy SS, Chitwood RA, Philpot BD. Common Pathophysiology in Multiple Mouse Models of Pitt-Hopkins Syndrome. J Neurosci. 2018January24;38(4):918–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Paulhus K, Ammerman L, Glasscock E. Clinical Spectrum of KCNA1 Mutations: New Insights into Episodic Ataxia and Epilepsy Comorbidity. Int J Mol Sci. 2020April17;21(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dalecki AG, Zorn KM, Clark AM, Ekins S, Narmore WT, Tower N, et al. High-throughput screening and Bayesian machine learning for copper-dependent inhibitors of Staphylococcus aureus. Metallomics. 2019March20;11(3):696–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ekins S, Puhl AC, Davidow A. Repurposing the dihydropyridine calcium channel inhibitor nicardipine as a nav1.8 inhibitor in vivo for pitt hopkins syndrome. Pharm Res. 2020June11;37(7):127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cisse B, Caton ML, Lehner M, Maeda T, Scheu S, Locksley R, et al. Transcription factor E2-2 is an essential and specific regulator of plasmacytoid dendritic cell development. Cell. 2008October3;135(1):37–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Flora A, Garcia JJ, Thaller C, Zoghbi HY. The E-protein Tcf4 interacts with Math1 to regulate differentiation of a specific subset of neuronal progenitors. Proc Natl Acad Sci USA. 2007September25;104(39):15382–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mesman S, Wever I, Smidt MP. Tcf4 is involved in subset specification of mesodiencephalic dopaminergic neurons. Biomedicines. 2021March20;9(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Casarosa S, Fode C, Guillemot F. Mash1 regulates neurogenesis in the ventral telencephalon. Development. 1999February1;126(3):525–34. [DOI] [PubMed] [Google Scholar]
- 56.Bertrand N, Castro DS, Guillemot F. Proneural genes and the specification of neural cell types. Nat Rev Neurosci. 2002July;3(7):517–30. [DOI] [PubMed] [Google Scholar]
- 57.Sohal VS, Rubenstein JLR. Excitation-inhibition balance as a framework for investigating mechanisms in neuropsychiatric disorders. Mol Psychiatry. 2019May14;24(9):1248–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sripathy SR, Wang Y, Moses RL, Fatemi A, Batista DA, Maher BJ. Generation of 10 patient-specific induced pluripotent stem cells (iPSCs) to model Pitt-Hopkins Syndrome. Stem Cell Res. 2020September17;48:102001. [DOI] [PMC free article] [PubMed] [Google Scholar]