

Abstract
The seven members of the A3 family of cytidine deaminases (A3A to A3H) share a conserved catalytic activity that converts cytidines in single-stranded (ss) DNA into uridines, thereby inducing mutations. After their initial identification as cell-intrinsic defenses against HIV and other retroviruses, A3s were also found to impair many additional viruses. Moreover, some of the A3 proteins (A3A, A3B and A3H haplotype I) are dysregulated in cancer cells, thereby causing chromosomal mutations that can be selected to fuel progression of malignancy. Viral mechanisms that increase transcription of A3 genes or induce proteasomal degradation of A3 proteins have been characterized. However, only a few underlying biological mechanisms regulating levels of A3s in uninfected cells have been described. Here, we characterize that the von Hippel–Lindau tumor suppressor (pVHL), via its CRLpVHL, induces degradation of all 7 A3 proteins. Two independent lines of evidence supported the conclusion that the multi-protein CRLpVHL complex is necessary for A3 degradation. CRLpVHL more effectively induced degradation of nuclear, pro-cancer A3 (A3B) than the cytoplasmic, anti-retroviral A3 (A3G). These results identify specific cellular factors that regulate A3s post-translationally.
Introduction
Evidence indicates that the 7 members of the apolipoprotein B mRNA-editing enzyme catalytic polypeptide-like 3 (APOBEC3; A3) family of cellular cytidine deaminases (A3A, -B, -C, D, F, G, and -H) have been positively selected throughout primate evolution by their interplay with endogenous retroelements and exogenous pathogens.1-5 This conserved deaminase catalytic function results in different A3s introducing mutations in both exogenous and endogenous single-stranded DNA (ssDNA) substrates.6 Variation in levels of A3s in different human cell types is associated with differences in pathogenesis of human immunodeficiency virus (HIV) infection and cancer, albeit in opposite directions.
Studies have proven that cytoplasmic A3D, A3F, A3G, and A3H can be encapsidated in virions budding from HIV-producing cells and that this decreases infectivity of those virions.7-15 Levels of those A3s in the cytoplasm that enable this incorporation into virions are higher in some T lymphocyte and myeloid cell differentiation states.16, 17 HIV infectivity is decreased more if virions are produced from specific types of T lymphocytes with relatively up-regulated cytoplasmic A3s, in comparison to those produced from T lymphocytes with lower levels of cytoplasmic A3s.16, 18 Virion A3, after entry into the next target cell, causes hypermutations in the ssDNA intermediate of the HIV genome produced during reverse transcription that can restrict virus production from that target cell.10, 11, 13, 19-21 Furthermore, A3-mediated mutations have also been implicated in the editing of a broad range of other viral genomes.5 In some such cases, A3 proteins have been shown to also restrict replication of these other viruses.22
On the other hand, nuclear-localized A3A, A3B, and A3H haplotype I (A3H I) are a source of chromosomal mutations in cancer cells that has been associated with accelerated progression in a range of cancers.23-31 In particular, A3B signature, clustered hypermutations (kataegis) are evident in many cancer types and not in non-malignant cells.23-27 These chromosomal hypermutations caused by A3B are a source of genetic diversity in cancer cells that can confer a selective advantage in proliferation, metastasis, and/or drug resistance to subpopulations of cells in a tumor.27-32. More recently, A3A has also been documented to be a source of mutations in multiple cancers.33-36
Distinct pathways of transcriptional regulation have been characterized to date for A3A, A3B, and A3G.37 Virus-cell interactions are characterized for the cancer-associated human papillomavirus (HPV) and polyomavirus that can inactivate specific components of the E2F transcription factor network to de-repress A3B transcription.38, 39 HPV-16 E6 protein also upregulates A3B transcription.40-42 Other mechanisms of transcriptional upregulation have been reported in cancers, including non-viral cancers.43-47 However, the relatively low magnitude of correlation noted between A3B mRNA levels and the degree of APOBEC3 signature chromosomal mutation in cells from breast and other cancers suggests that post-transcriptional mechanisms may also contribute to regulation.23, 24, 48 Modulation of nuclear A3 levels by mechanisms other than transcription has indeed been recently reported for specific Epstein-Barr virus and HPV proteins.49, 50 HPV-16 E7 protein has been reported to stabilize levels of the A3A protein post-translationally by inhibiting its Cullin 2-mediated degradation, although the lack of evidence in that report for an interaction between E7 and A3A indicates that the underlying mechanism requires further characterization.50
It is also well characterized how a retroviral gene product induces Cullin-RING E3 ubiquitin ligase (CRL)-mediated degradation of cytoplasmic A3 proteins.13, 51-56 HIV-1, HIV-2 and simian immunodeficiency viruses (SIVs), along with some other retroviruses, encode related virion infectivity factors (Vifs) that function as substrate recognition receptors to recruit different cytoplasmic A3s (particularly A3F, G, and H) to a specific CRL complex.57, 58 This CRLVif includes Elongin (ELO) B/C, Cullin (CUL) 5, RING-box protein (Rbx) 2 and the co-factors core binding factor-β as well as the Ariadne RING-in-between-RING (RBR) E3 ubiquitin protein ligase (ARIH2).59-63 This complex polyubiquitinates the cytoplasmic A3 proteins, directing them to the proteasome for degradation prior to the assembly and budding of retroviral particles.13, 64-66 Degradation of cytoplasmic A3s by HIV-1 Vif enables the evasion of A3-mediated restriction. Treatment with proteasome inhibitors effectively negates this A3-depleting activity of HIV-1 Vif that promotes HIV infectivity.67 While A3B is resistant to HIV-1 Vif 68, the Vif produced by a specific strain of SIV, SIVmaC239, has demonstrated the ability to degrade human A3B.69-72
Since cellular processes can regulate homeostasis of many proteins, we studied if there was a specific mechanism for post-translational degradation of A3 cytidine deaminases. We report here that the tumor suppressor von-Hippel Lindau protein (pVHL), and its CRL, mediate degradation of each member of the A3 family. This identification of a functional parallel with retroviruses represents a critical first step to enabling additional insights into cancer biology and future development of therapeutics to specifically modulate cellular levels of different A3 proteins.
Materials and Methods
Cell Lines, Culture Conditions, Proteasomal Inhibitors and siRNA.
All the cell lines used in this study were obtained from ATCC and maintained at 37 °C and 5% CO2. HEK293T and MCF7 cells were cultivated in DMEM (containing 4.5 g/liter glucose, L-glutamine, and sodium pyruvate) medium plus 10% fetal bovine serum, 50 IU/ml penicillin, and 50 μg/ml streptomycin. H1299 and T47D cells were cultivated in RPMI medium plus 10% fetal bovine serum, 50 IU/ml penicillin, and 50 μg/ml streptomycin. A549 cells were maintained in F12K, plus 10% fetal bovine serum, 50 IU/ml penicillin, and 50 μg/ml streptomycin. HTB41 cells were maintained in McCoy's 5a Medium Modified plus 10% fetal bovine serum, 50 IU/ml penicillin, and 50 μg/ml streptomycin.
The proteasomal inhibitors, MG132 and Lactacystin were purchased from Santa Cruz Biotechnology and Epoxomicin was purchased from Cayman Chemical. All the inhibitors were prepared according to manufacturers’ protocols. DMSO was used as a control in these experiments since the proteasome inhibitors are dissolved in DMSO.
To downregulate endogenous pVHL gene expression, siRNAs were purchased from Dharmacon. For specific pVHL down regulation, ON-TARGETplus VHL siRNAs (J-003936-11, J-003936-12, termed siVHL 1 and siVHL 2) were used and siRNA ON-TARGETplus Non-Targeting Control Pool (D-001810-10, termed Contro/siNT) was used as a treatment control. Transfection reagents for the siRNA were also purchased from Dharmacon. VHL-targeting shRNA was obtained from Dr. Wuhan Xiao from the Chinese Academy of Sciences in Wuhan, China 73.
Plasmids
Tagged A3 and pVHL expression vectors were recurrently used in this study. APOBEC3A-myc DNA was kindly provide by Dr. Jinwoo Ahn from University of Pittsburg 74. We obtained APOBEC3B-HA DNA through the NIH AIDS Reagent Program (catalog #: 11090), where it was originally provided Dr. Bryan R. Cullen 68. The APOBEC3D-V5-His expression DNA used was also obtained through the NIH AIDS Reagent Program (catalog #: 11433), provided by Dr. Yong-Hui Zheng 15. APOBEC3C-HA, APOBEC3F-HA and APOBEC3G-HA vectors were described previously 8, 9. APOBEC3H-HA haplotypes I and II were kindly provided by Drs. Marcel Ooms and Viviana Simon from Icahn School of Medicine at Mount Sinai 14. HA-VHL and mutant HA-VHLC162F expression DNAs were kindly provided by Dr. Michael Ohh from University of Toronto 75. ARIH1 and HIF1α expression DNA was purchased from Addgene 76, 77 The Flag-tagged pVHL construct was obtained from Dr. Wuhan Xiao from the Chinese Academy of Sciences in Wuhan, China 73.
Immunoblotting
HEK293T cells were plated at a density of 8 × 105 cells/well in a 6-well culture plate 24 h prior to transfection with individual plasmids as indicated in individual figure legends. Linear polyethyleneimine (PEI; 25 kDa; Polysciences, Inc.) was used, as described 78. Twenty-four hours after transfection, cells were lysed in a 1% NP-40 based lysis buffer with the addition of a protease inhibitor cocktail (Millipore Sigma) and subsequently sonicated to release nuclear-localized proteins followed by centrifugation at 10,000 × g for 10 min at 4 °C. Protein concentrations of the cell lysates were quantified using a BCA protein assay (Thermo Fisher Scientific) and normalized. Equal amounts of proteins were denatured using a 10% DTT and 4X LDS sample buffer (Thermo Fisher Scientific), boiled for 7 minutes at 92°C, and analyzed by electrophoresis through a 4-12% Bolt Bis-Tris electrophoresis gel (Thermo Fisher Scientific). After electrophoresis, separated proteins were transferred to a PVDF membranes (Thermo Fisher Scientific) and processed for western blot analysis using protein-specific antibodies; anti-HA (Millipore Sigma), anti-FLAG (Millipore Sigma), anti-MYC (Millipore Sigma), anti-V5 (Thermo Fisher Scientific), anti-A3B (in-house generated), anti-VHL (Santa Cruz Biotechnology), anti-HIF1α (Santa Cruz Biotechnology), anti-Lamin A/C (Santa Cruz Biotechnology), and anti-GAPDH (Millipore Sigma). Fluorescent-labelled secondary antibodies were used in conjunction with the Odyssey CLx imaging system (Li-Cor) to detect protein signal.
Protein Band Quantification
Following signal detection, protein bands were quantified using the ImageJ software available through the National Institutes of Health. Image files produced by the Odyssey CLx imaging system were converted into 32-bit, black-and-white (black bands/white background) files that were then quantified using the gel analysis feature and measuring the area under the curve for peaks that represented the target protein. Quantified bands for A3s and pVHL were normalized based on their corresponding Lamin C/GAPDH values. To calculate the relative A3B/pVHL signal intensities, the normalized band values were then divided by control values for the non-targeting siRNA or DMSO for the knockdown and proteasome inhibition experiments, respectively. To obtain the degradation ratio in co-transfected samples in 293T cells, the band quantification values for A3 proteins co-transfected with either WT pVHL or C162F were divided by the values from control samples that were co-transfected with an empty pcDNA control vector. For the statistical analysis of A3B vs A3G degradation ratios, a paired Wilcoxon test was performed on 11 repeats of these experiments, wherein the pairing was between samples that were cultured, collected, and processed on the same date.
RT-qPCR Analysis
Cellular RNA was purified from cell pellets using the RNeasy kit (Qiagen) recommended by the manufacture and used as a template for the production of cDNA using the SuperScript III first-strand synthesis system (Thermo Fisher Scientific). Quantitative PCR was then performed using the primer sequences for A3B and TBP as designed by Refsland et al. 79 and for VHL as described by Lu et al. 80 (Table 1). Quantitative PCR was performed using a SYBR Green stain (Thermo Fisher Scientific) and analyzed using a QuantStudio 6 Flex Real-Time PCR system (Thermo Fisher Scientific). Gene normalization was performed using TATA box-binding protein (TBP).
Immunoprecipitation
HEK293T cells were plated at a density of 8 × 106 cells/plate in a 10 cm culture plate 24 h prior to transfection with individual plasmids as indicated in individual figure legends using PEI transfection method. Twenty-four hours after transfection, cell lysates were processed as described above. Immunoprecipitation experiments were performed using antibody-conjugated agarose beads for anti-HA (Thermo Fisher Scientific), anti-FLAG (Millipore Sigma), and anti-MYC (Millipore Sigma) as indicated in individual figures. Between 500-1000 μg of clarified protein lysate was incubated overnight with the beads, which were then washed five times with wash buffer and remaining buffer was removed using a 27-gauge needle. The beads were then resuspended in 2X DTT-based sample buffer and boiled for 3 minutes before being analyzed using immunoblotting as described above.
Proteomics and Data Analysis
HEK293T cells were transfected with an empty pcDNA3.1 vector, or with a pcDNA3.1 plasmids expressing HA-tagged A3B using PEI transfection method. Samples were treated with Epoxomicin to increase protein-protein interactions by inhibiting proteasomal degradation of cellular proteins. Cell lysates were collected and processed as described above in immunoblotting section. The resulting cell lysates were then incubated separately with agarose-conjugated beads that were anti-HA (Thermo Fisher Scientific) as well as isotype matched agarose-conjugated mouse IgG beads (Millipore Sigma). These were incubated overnight at 4°C, washed and denatured as described above. Samples were run on SDS-PAGE gel and a gel band was subject for in-gel digestion. The gel band was washed in 100 mM Ammonium Bicarbonate (AmBic)/Acetonitrile (ACN) and reduced with 10 mM dithiothreitol at 50°C for 30 minutes. Cysteines were alkylated with 100 mM iodoacetamide in the dark for 30 minutes at room temperature. The gel band was washed in 100mM AmBic/ACN prior to adding 600 ng trypsin for an overnight incubation at 37 °C. Supernatant containing peptides was saved into a new tube. The gel was then washed at room temperature for 10 minutes with gentle shaking in 50% ACN/5% FA, and supernatant was saved to peptide solution. Wash step was repeated each by 80% ACN/5% FA, and 100% ACN, and all supernatants were saved in peptide solution and then subjected to the speedvac dry. After lyophilization, peptides were reconstituted with 5% ACN/0.1% FA in water and injected onto a trap column (150 μm ID X 3cm in-house packed with ReproSil C18, 3 μm) coupled with an analytical column (75 μm ID X 10.5 cm, PicoChip column packed with ReproSil C18, 3 μm) (New Objectives, Inc.). Samples were separated using a linear gradient of solvent A (0.1% formic acid in water) and solvent B (0.1% formic acid in ACN) over 120 minutes using a Dionex UltiMate 3000 Rapid Separation nanoLC (Thermo Fisher Scientific). MS data were obtained on an Orbitrap Elite Mass Spectrometer (Thermo Fisher Scientific). Data were searched using Mascot (Matrix Science) v.2.5.1 against the Swiss-Prot Human database (2019) and results were reported at 1% FDR in Scaffold v.4.8.4 (Proteome Software). Only proteins in which the anti-HA pulldown on both samples yielded at least twice the total spectrum count compared to the mouse IgG control samples were considered. For our own analysis, a protein was considered to be exclusively bound to A3B if the total spectrum count was four times greater in the A3B expressing samples compared to the empty pcDNA3.1 control sample.
Results
Proteasome inhibition increases levels of endogenous APOBEC3B
To begin investigating potential post-translational regulation of A3s, we treated two human cell lines, MCF7 (derived from a breast cancer) and H1299 (derived from a lung cancer) with proteasome inhibitors. These cell lines were selected because it is well established that biopsied breast and lung cancer cells display high levels of A3-mediated cytidine deaminase mutations in vivo.23, 24, 81, 82
MCF7 and H1299 cells were treated with proteasome inhibitors MG132, Lactacystin and Epoxomicin. The effect of proteasome inhibition on endogenous levels of A3B protein was subsequently analyzed through immunoblotting (Figure 1A). Levels of endogenous A3B protein increased upon treatment with each of the proteasome inhibitors in H1299 cells. In the MCF7 cells, MG132 and Epoxomicin treatments resulted in increased A3B; Lactacystin-induced inhibition did not, indicating variable effects of these inhibitors in different cell types. Though Epoxomicin and MG132 both inhibit the 20S proteolytic core of the 26S proteasome, this inhibition by MG132 is reversible; moreover, MG132 is not entirely specific against the proteasome as it also inhibits the action of calpain proteases and lysosomal cathepsins.83, 84 Treatment with the irreversible inhibitor, Epoxomicin, had the greatest and most consistent effect on levels of A3B protein, while the more reversible MG132 treatment did not have a reliably reproducible effect in repeated experiments (not shown).
Figure 1. Proteasome inhibition increases levels of endogenous APOBEC3B.

Immunoblotting was done for endogenous A3B in cells cultured in 6-well plates and treated overnight (18 hours) with proteasome inhibitors. Cells were lysed, and then protein amounts were normalized, denatured, and separated via electrophoresis. (A) H1299 and MCF7 cells were treated with a panel of proteasome inhibitors; MG132 (5 μM), Lactacystin (5 μM) and Epoxomicin (500 nM). (B) A3B protein (immunoblot in left panel and its quantitation in middle panel) and mRNA levels (relative to TBP, right panel) were measured by immunoblotting and RT-qPCR respectively from duplicate 6-well cultures of A549 lung cancer cells (n=3), (C) The same analyses were done for MCF7 breast cancer cells (n=3), and (D) HTB-41 head/neck cancer cells (n=4). For all panels, values are means ± SD error bars. For each cell line, protein level quantifications were normalized to the DMSO control for each replicate.
Since A3B-induced hypermutations have also been observed in head/neck cancers, we next studied the head/neck cancer cell line, HTB-41. Both HTB-41 and another non-small cell lung cancer (NSCLC) cell line, A549, were treated with Epoxomicin. Epoxomicin-induced proteasomal inhibition of these cells consistently resulted in 2- to 3-fold increases in A3B protein levels compared to control treated cells (Figures 1B-D and S1).
Next, we considered whether the specific, potent, and irreversible inhibition of the 26S proteasome by Epoxomicin might affect transcriptional mechanisms of A3B regulation. Reverse transcriptase quantitative PCR (RT-qPCR) measured A3B mRNA levels in the absence and presence of Epoxomicin treatment (Figure 1B-D). We did not observe any instances of increased A3B mRNA following proteasome inhibitor treatment in any of the cell lines tested; in fact, decreased A3B mRNA was consistently observed, as has also been reported by others.44 This is consistent with the increased levels of A3B protein observed in these cell lines not being indirectly due to transcriptional activation.
Proteomic analysis identifies A3B-pVHL interaction
The observed consistent increase in levels of endogenous A3B protein with irreversible proteasome inhibition suggests a cellular regulatory mechanism involving proteasomal degradation. We next sought to identify cellular protein(s) interacting with A3B through proteomic analysis. To do this, 293T cells were transfected with either an empty control vector or HA-tagged A3B expression plasmids in the presence of Epoxomicin to minimize A3B degradation. Cell lysates were immunoprecipitated using HA-specific antibody-conjugated agarose beads. Through an in-gel digestion followed by mass spectrometry, protein database analysis, and the removal of non-specific peptides, we identified 39 potential candidate proteins that were specifically bound to HA-tagged A3B, including the pVHL tumor suppressor protein (Figure 2). The most extensively characterized cellular function of pVHL is as a substrate receptor that recruits the HIF1α transcription factor to a CRL (CRLpVHL). CRLpVHL polyubiquitinates HIF1α, which directs it for subsequent degradation through the 26S proteasome.85-89 Additional factors associated with ubiquitin-mediated proteasomal degradation were found to interact with A3B, including PSMC5 – a gene encoding a component of the 19S regulatory subunit of the 26S proteasome that recognizes polyubiquitinated substrates – and UBA52, a ubiquitin encoding gene.90, 91 The identification of these proteins prompted further study of a putative functional role of pVHL in A3B degradation.
Figure 2. Proteomic analysis identifies A3B-pVHL interaction.
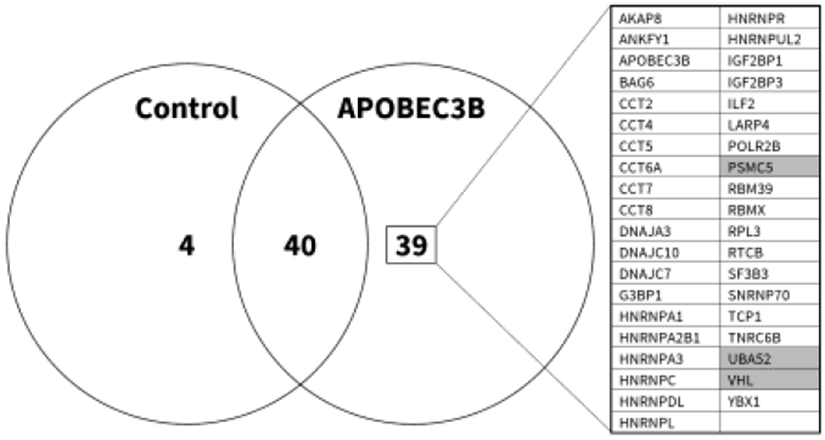
8 million 293T cells were cultured in 10 cm plates, and transfected on the following day with 10 μg of an HA-tagged A3B expression plasmid, or an empty control vector, in the presence of Epoxomicin. Lysates were subsequently incubated with HA- or IgG- conjugated agarose beads in order to affinity-purify A3B and identify non-specific protein-bead interactions, respectively. Proteins were denatured and separated on SDS-PAGE gels. Mass spectrometry analysis identified 39 interacting proteins, as shown, with shading indicating those that were related to ubiquitination and/or the proteasome.
Down-regulation of pVHL increases endogenous A3B protein levels
To test whether pVHL regulates cellular levels of endogenous A3B, we treated MCF7 and H1299 cells with short interfering RNAs (siRNAs) targeting VHL mRNA, or with a non-targeting control siRNA. Two days post-transfection with siRNAs, cell lysates from both MCF7 and H1299 were analyzed by immunoblotting for endogenous levels of A3B and pVHL (Figures 3A-B and S2A). We observed that VHL knockdown led to a consistent increase in A3B protein levels in both MCF7 and H1299 cells. Furthermore, short hairpin RNAs (shRNAs) against VHL yielded a similar result in MCF7 cells (Figure S2B).
Figure 3. Down-regulation of pVHL increases endogenous A3B protein levels.

MCF7 cells were cultured in 10 cm plates and, one day later, were treated with non-targeting siRNA (siNT) and siRNAs targeting VHL (siVHL 1, 2) in order to knockdown endogenous pVHL. (A, B) Endogenous A3B and pVHL protein levels assessed by immunoblot (n=3) (A) and bands quantified and normalized to the siNT control (B). (C) RT-qPCR analysis of samples from (A) quantifying A3B and VHL mRNA, normalized to TBP. For all panels, values are means ± SD error bars.
Given that the principal substrate of pVHL, HIF1α, is a transcription factor that activates the expression of a myriad of metabolic genes, we also tested whether a knockdown of VHL could be promoting HIF1α-induced transcriptional activation that could explain the increased levels of A3B protein observed. To test this, RT-qPCR quantification of A3B and VHL mRNA in MCF7 samples treated with VHL-specific siRNA was performed. We observed no significant increase in A3B mRNA following siRNA knockdown of VHL (Figure 3C). These results implicate pVHL as a post-translational regulator of A3B protein.
Ectopic expression of pVHL decrease levels of A3 proteins
We next studied whether pVHL can regulate other A3 proteins. 293T cells co-transfected with pVHL and individual A3-expressing plasmids were lysed and analyzed by immunoblotting one day post-transfection. We consistently observed a decrease in every A3 protein tested following the transfection of pVHL (Figure 4A). Importantly, we also demonstrated that our pVHL expression construct was capable of inducing the degradation of the canonical substrate HIF1α (Figure S3). When the co-transfection of A3B, -F, and -G expression vectors, each with the pVHL-expressing plasmid, was done in the presence of cycloheximide to inhibit translation, the level of each of these three A3s was also reduced, again consistent with post-translational regulation (Figure S4). These results demonstrated that pVHL degrades A3 substrates regardless of their localization or size, consistent with a conserved, post-translational regulatory mechanism across the A3 protein family.
Figure 4. Ectopic expression of pVHL decrease levels of A3 proteins.
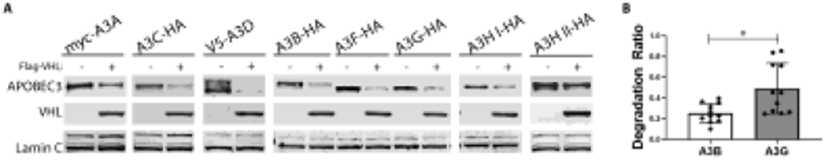
800,000 293T cells were seeded on 6-well plates one day prior to co-transfection with plasmids expressing the indicated tagged A3 proteins (500 ng) along with N-terminal Flag-tagged pVHL (1000 ng). Cells were collected 24 hrs post-transfection, lysed, and then separated on 4-12% Bis-Tris gradient gels. A3 and pVHL signals were normalized to the Lamin C band signal. (A) Representative immunoblots of ectopically expressed A3 proteins with co-transfected empty control and Flag-VHL plasmids. (B) Multiple replicate samples of A3B and A3G that were run simultaneously from the experiment illustrated in (A) were quantified and normalized to the Lamin C band signal to yield a degradation ratio of normalized A3B to A3G signals (n=11), values are means ± SD error bars. A Wilcoxon matched-pairs signed rank test determined P value of 0.0322 for comparison of degradation rations of A3B to A3G.
Given the extensive literature implicating nuclear-localized A3B in promoting chromosomal mutagenesis in cancer cells, and the well-established anti-HIV properties of cytoplasmic A3G, our initial focus was on studying A3B and A3G. During the repeated experiments (11 replicates) using immunoblotting after co-transfection of pVHL with either A3B or A3G, we observed that A3B was more sensitive to pVHL-mediated degradation compared to A3G. The protein bands for A3B and A3G were each quantified and normalized to Lamin C signal quantity in each respective experiment. A ratio that measured the lamin-normalized level of each of these two A3s following transfection with pVHL-containing versus empty control vector showed significantly greater degradation of A3B than A3G (P=0.032, Figure 4B).
CRLpVHL formation is necessary for A3 degradation
A genetic approach was employed to determine if the CRLpVHL ubiquitin ligase complex was necessary to degrade A3 substrates, as occurs with CRLVif in HIV- and SIV- infected cells. The naturally occurring and well-characterized C162F mutant pVHL has been repeatedly shown to be unable to bind Elongin C (ELOC). As a result, a functional CRL complex is not formed when C162F pVHL is present, leading to failure of C162F pVHL to induce degradation of HIF1α.92-96
To determine whether pVHL-ELOC binding and the formation of a functional CRLpVHL complex was necessary to induce A3 degradation, we transfected either tagged wild-type (WT) pVHL or C162F pVHL- expression plasmids into 293T cells along with each of the A3 proteinexpressing plasmids. One day post-transfection, cell lysates were collected and analyzed by immunoblotting. WT pVHL consistently induced degradation of A3s, but C162F pVHL expression had no appreciable effect on A3 protein levels (Figure 5A). Quantification of multiple immunoblots of each experiment confirmed the decreases in A3 protein levels following WT pVHL expression and this was not seen with expression of C162F pVHL (Figure 5B-E).
Figure 5. CRLpVHL formation is necessary for A3 degradation.
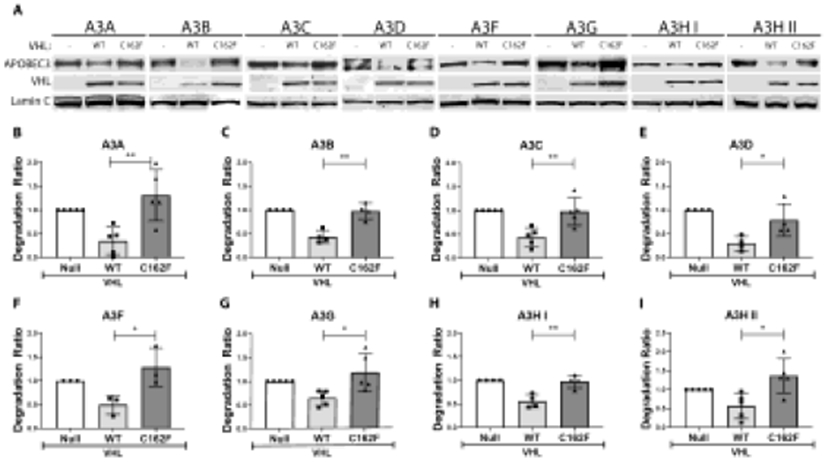
293T cells were cultured in 6-well plates of 800,000 cells per well, and transfected with various A3-expression constructs (500 ng) along with a plasmid expressing either no protein (empty vector or null control), WT pVHL, or C162F pVHL (1000 ng of each) the next day. (A) Representative western blots of each A3 protein expressed along with the null control, WT pVHL, and C162F pVHL are shown. (B-I) Quantification of the different A3 bands following multiple repeats of these experiments are shown. For all panels, values are means of a ratio (“A3 degradation ratio”) of A3 protein signal following the expression of each pVHL normalized to the null control with each point representing an independent replicate. Error bars are ± SD. Significance of difference between each degradation ratio, comparing change from A3 band quantitation in the null control following WT pVHL versus C162F pVHL expression, was analyzed by an unpaired t-test. Significant differences are indicated as either *P < 0.05 or **P < 0.01.
Additionally, we transfected increasing amounts of either wild type or C162F pVHL expression plasmids along with a constant amount of individual A3 expression plasmids. There was a consistent concentration-dependent relationship between increased WT pVHL expression and decreasing A3 protein levels, whereas increasing expression of C162F pVHL demonstrated no such decrease in A3 levels (Figure S5). Cumulatively, these observations strongly suggest that pVHL-ELOC binding and the formation of a functional CRLVHL complex are necessary for pVHL-mediated A3 degradation.
Post-translational modifications of pVHL are not required for A3 degradation
Post-translational modification of certain lysine residues of pVHL influences intracellular localization and activity. Lysines are modified at residue 159 (by neddylation); at residue 171 (by both SUMOlyation and ubiquitination); and at residue 196 (by ubiquitination).75, 97-99 Singlesite arginine mutants that prevent those modifications at each of these residues (K159R, K171R, K196R) were studied in order to determine the necessity of post-translational modification at each of these lysines of pVHL for A3 down-regulation (Figure 6). Mutants lacking each single one of these post-translational modifications retained the ability to degrade A3s, although further work is suggested by these data (as described in the Discussion below).
Figure 6. Post-translational modifications of pVHL are not required for A3 degradation.
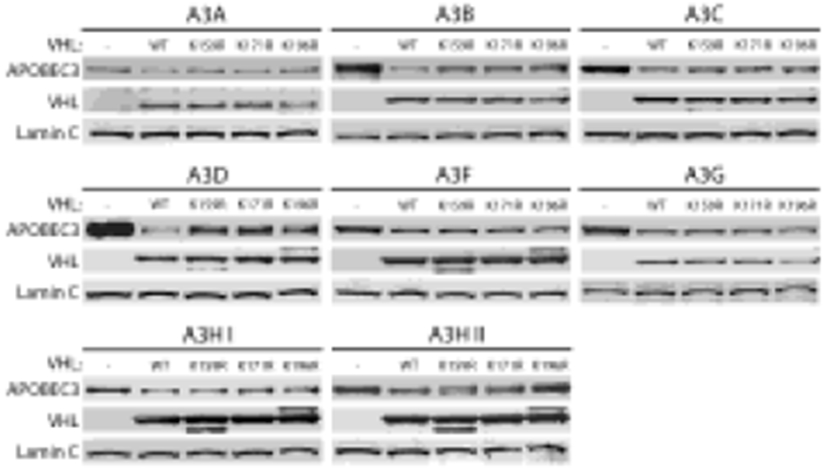
6-well plates were cultured with 800,000 293T cells/well one day before co-transfection with one of the tagged A3 proteins and either an empty control vector, Flag-tagged WT pVHL, or Flag-tagged K159R, K171R, or K196R pVHL. Representative immunoblots are shown.
A3 proteins interact with pVHL
The role of substrate receptor proteins, like pVHL and HIV-1 Vif, for CRLs is twofold: firstly, to bind the target substrate, and secondly to effectively interact with the CRL complex. In doing so, substrate receptors recruit targeted proteins to the CRL ubiquitin ligase machinery that then mediates the ubiquitination of the substrate. Therefore, the established mechanism of degradation employed by HIV-1 Vif (for cytoplasmic A3s) and pVHL (for HIF1α) necessitates their interaction with the substrate .
To investigate whether pVHL binds to the A3 proteins, 293T cells were co-transfected with pVHL and individual A3s. One day post-transfection, cell lysates were collected, and pulldown experiments were carried out using anti-HA, anti-MYC, and anti-FLAG conjugated agarose beads followed by immunoblotting. The immunoblotting results demonstrated that A3A, A3B, A3C, A3F, A3G, A3H haplotype I, and A3H haplotype II each interact with pVHL (Figure 7A, B). These results corroborated the finding from our proteomic approach that identified an interaction between endogenous pVHL and A3B and extended documentation of such interactions to other A3 family members, except one. A3D was not studied here because an expression plasmid with a tag allowing immunoprecipitations, as done for the other A3s, was not able to be obtained or constructed.
Figure 7. A3 proteins interact with pVHL.
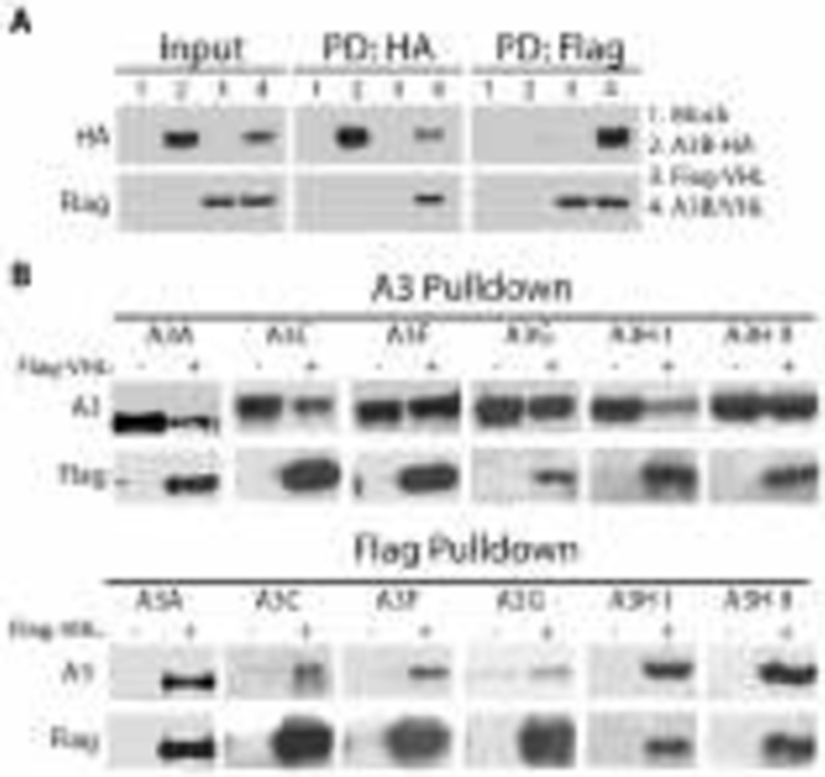
8 million 293T cells were cultured in 10 cm plates for one day prior to transfection with tagged A3-proteins (10 μg) and Flag-tagged pVHL (10 μg). Complexes of pVHL with each A3 were affinity purified with anti-HA antibodies, except for A3A, which is Myc-tagged and was thusly purified with anti-Myc antibody. In addition, anti-Flag antibody was used to isolate A3-pVHL complexes in a complementary manner. (A) Immunoblot of the input and affinity purified tagged A3B and pVHL following pulldown with anti-HA and -Flag conjugated beads, respectively. (B) Immunoblots of expanded affinity pulldown experiments for additional members of the APOBEC3 family (A3A, -C, -F, -G, -H I, H II).
ARIH1 potentiates pVHL-mediated A3 degradation
Recent studies have found that members of the Ariadne family of RING-in-between-RING (RBR) E3 ligases (ARIH) interact with CRLs in order to efficiently prime initial monoubiquitination of a substrate, which catalyzes subsequent polyubiquitination.100-102 Earlier data showed that the ARIH family member ARIH1 interacted with Cullin 2-based CRLs to initiate pVHL-mediated degradation of HIF1α.103 Therefore, we investigated whether ARIH1 is involved in pVHL-mediated A3 regulation. Each tagged A3 protein was transfected at a constant concentration along with pVHL and ARIH1 individually, as well as in combination with each other in a triple-transfection. Addition of ARIH1 to pVHL did not have a discernable impact on pVHL-mediated degradation of A3D, A3H haplotype I and A3H haplotype II; intensity of immunoblot bands of these 3 A3 proteins appeared unaffected by the addition of ARIH1 to pVHL (Figure 8D, G, H, comparing lanes 2 and 4). However, the addition of ARIH1 to pVHL decreased intensity on immunoblot of A3B, A3C, A3F, and A3G relative to only pVHL coexpression with an A3 (Figure 8A, B, C, E, F, comparing lanes 2 and 4). This evidence of increased degradation of several A3s by adding over-expression of ARIH1 to that of pVHL is consistent with our other data implicating a role for the CRLpVHL complex in A3 regulation, although further work is also suggested as described in the Discussion below.
Figure 8. Addition of ARIH1 increases pVHL-mediated degradation of several A3s.
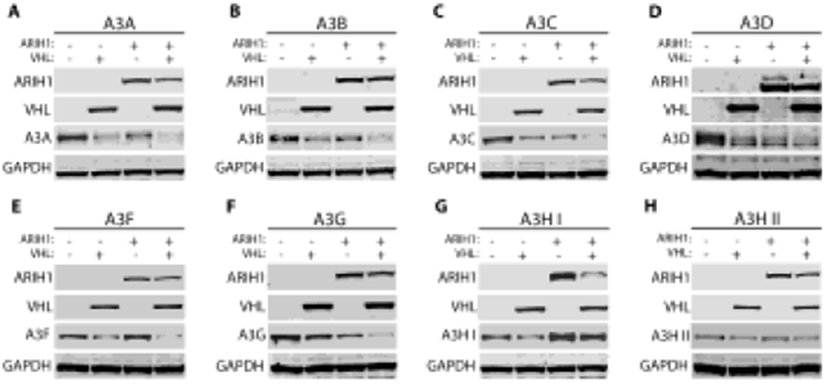
293T cells were cultured in 6-well plates with 800,000 cells/well one day before co-transfection with an A3 expression plasmid (500 ng) and either an empty control vector, a plasmid expressing HA-tagged pVHL (500 ng), a plasmid expressing Flag-tagged ARIH1 (500 ng), or both the pVHL and ARIH1-expression plasmids (500 ng of each). In order to compare A3 protein levels upon expression of pVHL, ARIH1, or both, immunoblot experiments were performed. (A-H) Representative immunoblots of these experiments are shown. To evaluate if adding ARIH1 increases pVHL effect on each A3, the relevant comparison in each panel is the A3 band intensity in lane 2 with only pVHL over-expressed, relative to the A3 band intensity in lane 4 with both ARIH1 and pVHL over-expressed.
Discussion
This report identifies a post-translational regulatory mechanism that degrades A3A, A3B, A3C, A3D, A3F, A3G, A3H I, and A3H II through the activity of CRLpVHL and the proteasome. Results from two independent methods show that the tumor suppressor protein pVHL interacts with A3 proteins either directly or indirectly to mediate their degradation. Proteomic analysis identified endogenous pVHL as interacting with A3B (Figure 2) and co-immunoprecipitation experiments showed that each A3 interacted with pVHL (Figure 7). Genetic knockdown of pVHL increased endogenous A3B protein (Figures 3 and S2), and over-expression of pVHL decreased levels of each A3 protein (Figure 4). Two independent lines of evidence supported the conclusion that the multi-protein CRLpVHL complex87, 94 is necessary for A3 degradation (Figures 5 and 8). This cellular mechanism functionally parallels how retrovirus-encoded Vif proteins deplete infected cells of cytoplasmic A3s by recruiting them to CRLVif.
Although differential cellular levels of the A3 cytidine deaminases play pathogenic roles in cancer progression as well as defense against multiple exogenous viruses and retrotransposons, there has been only limited characterization to date of how they are regulated in uninfected cells. Transcriptional upregulation of A3B expression has been reported in some non-viral cancers.43-47– However, a comparison of relative levels of A3B RNA and APOBEC signature mutations between two breast cancer cell lines exemplifies some of the data suggesting that there may be regulatory mechanisms at an additional level beyond transcription. APOBEC3 signature mutations were 50% of the BT-474 breast cancer line’s total chromosomal mutations, with A3B RNA being 0.59-fold change relative to the normalizing TBP RNA level (Supplementary Table 3 in 48). In contrast, fewer, 22%, of total mutations were APOBEC signature mutations in the HCC202 breast cancer cell line, despite higher A3B RNA levels at almost 2-fold change relative to the normalizing TBP RNA (Supplementary Table 3 in 48). A mechanism of post-translational degradation of A3B could explain this discordance between A3B mRNA levels and A3 signature mutation frequencies.
Results here indicated that the binding of ELOC to pVHL, which is essential for forming the functional, multi-protein CRLpVHL complex87, 94, is necessary for A3 degradation. It has been well characterized that pVHL must bind to ELOC to form the full CRLpVHL and induce degradation of its canonical substrate, HIF1α, as mutations in the BC-box of pVHL render the protein incapable of degrading HIF1α.95, 96 Exogenous expression of one such BC-box mutant that decreases HIF1α degradation, C162F pVHL, did not degrade A3s, in contrast to WT pVHL (Figure 5).
An additional factor in the CRLpVHL complex is ARIH1, which monoubiquinates a substrate to ‘prime’ subsequent polyubiquitination of the substrate.101, 104 Previous studies demonstrated that when ARIH1 is downregulated, the activity of CRLpVHL is decreased such that the substrate HIF1α is less well degraded.103 Similar to the effect on HIF1α, the over-expression of ARIH1 along with pVHL appeared to enhance the degradation of some A3s (A3A, A3B, A3C, A3F, and A3G), relative to the degradation induced by pVHL over-expression alone. The results for ARIH1 effects on A3A, A3B, A3C, A3F, and A3G here add to the C162F pVHL data in supporting a role of the CRLpVHL complex in regulating many A3s. However, the lack of discernable change in immunoblot band intensity with adding ARIH1 to pVHL over-expression here for A3D, A3H haplotype I and A3H haplotype II suggests additional characterization of ARIH1 in future work to more thoroughly evaluate if there is indeed lesser impact of ARIH1 on the pVHL effect on some A3s.
We also found that mutants unable to be post-translationally modified at specific, single pVHL lysine residues implicated in pVHL functions separate from its role in a CRL and in its cellular localization (K159R, K171R, K196R)75, 97-99 could still degrade A3s. However, the immunoblots suggested these mutants may degrade A3B, A3C, A3D, A3HI and II slightly less well than the other A3s (Figure 6). This suggests further study of the role of post-translational modifications of pVHL at those residues in optimizing degradation of specific A3s.
A role for proteasomal degradation was also shown in this work, consistent with CRLpVHL activity on A3s directing them to the proteasome. Proteasome inhibition by MG132 was reported previously to rescue A3G from HIV-1 Vif-mediated degradation.52, 105 However, MG132 did not impact uninfected cell levels of A3G in earlier work.56, 69, 105 The observation here that MG132, a reversible inhibitor of the proteosome, did not reproducibly cause increases in A3B may help explain those earlier results. In our study, we also treated cancer cell lines with the more specific and irreversible proteasome inhibitor, Epoxomicin, as well as MG132. Epoxomicin consistently increased levels of endogenous A3B protein. These increases in A3B protein seen with Epoxomicin were not attributable to enhanced transcription here; indeed, decreased A3B RNA was consistently observed after Epoxomicin treatment. Decreased endogenous A3B RNA has also been reported after MG132 treatment of the MCF10A breast cancer cell line and attributed to decreased degradation of p100 diminishing PKC-NFκB-driven transcription from the A3B promoter.44
Cumulatively, results from these diverse experiments lead us to posit that pVHL functions as a substrate receptor for the CRLpVHL complex. In this role, pVHL binds A3s and recruits them – through an interaction with Elongin C – to the CRLpVHL complex for polyubiquitination. Polyubiqitination then targets A3s for degradation via the proteasome.
This pathway of CRLpVHL-mediated degradation was shown here to be conserved among A3s regardless of their localization in the nucleus (A3B), cytoplasm (A3D, A3F, A3G, A3H II), or both (A3A, A3C, A3H I). However, results also indicate a greater sensitivity of A3B to CRLpVHL mediated degradation relative to A3G. This raises additional potential mechanisms by which loss of pVHL function can affect cancer progression via A3B that can be addressed by future research. It may also inform strategies to study if reversing A3B up-regulation may slow cancer progression. Current efforts to attempt to reverse A3B effects on cancer involve inhibition of its deaminase activity or its sequestration in perinuclear bodies49, 106, 107; either strategy may also affect other A3s and potentially impair defenses against exogenous retroviruses and endogenous retroelements in non-malignant cells. Results here suggest further work may open opportunities to develop interventions that preferentially increase CRLpVHL activity to enhance degradation of A3B in cancer cells, while minimizing loss of immune cell-intrinsic defenses against viruses provided by A3G. This is particularly pertinent to cancers lacking a viral etiology that are more frequent and aggressive in HIV patients, even during antiretroviral therapy. These findings may also inform therapeutic strategies, given that targeting the ubiquitin-proteasome system is already being successfully exploited via two different treatment modalities – inhibiting and hijacking the proteasome. Multiple proteosome inhibitors have proven efficacious in cancer therapy, and more are in development.108, 109 PROteolysis TArgeting Chimera (PROTAC) technology redirects the ubiquitin-proteasome system, inducing polyubiquitination and proteasomal degradation of a specific target protein.110 A PROTAC therapeutic is now in a phase I clinical trial.111 Results here suggest potential to study if inhibiting this pathway of A3 degradation may have antiviral effects and/or if reversing CRLpVHL mechanisms of A3B upregulation may decrease its mutational burden and slow cancer progression.
Limitations of this study include a reliance on the 293T cell line and co-transfections of A3 proteins along with pVHL. It is likely that the level of expression of these proteins in 293T cells is greater than their physiological levels in primary cells. However, co-transfection into 293T cells was a useful model system to study whether this regulatory pathway was conserved amongst different A3 family members. We also confirmed effects on endogenous A3B in cancer cell lines. Future studies into this mechanism will undoubtedly need to utilize more physiologically relevant cells, in particular primary immune cells in which A3s play a pivotal role in HIV restriction and biopsy-derived cancer cells.
In summary, this study identifies a CRLpVHL-dependent mechanism for physiological regulation of the entire A3 family of proteins, DNA-editing enzymes whose diverse roles in critical cellular processes have recently been increasingly appreciated. This includes evidence of a role for A3s in many different virus infections, as well as many cancers.5, 24 Moreover, selectivity of CRLpVHL activity against some A3 family members (e.g.; greater activity against A3B than A3G) was found. Further characterization of these A3 regulatory mechanisms can advance understanding of cancer progression. There is also potential for more work on these A3 family-wide regulatory mechanisms, and how they differ for individual A3 family members. Such studies would add insights that can help develop strategies targeting the ubiquitin-proteosome system in order to selectively minimize cancer-promoting chromosomal mutagenesis or promote antiviral activity.
Background
APOBEC3 (A3) cytidine deaminases induce mutations in single-stranded DNA substrates, in some cases, fatally hypermutating virus DNA and, in others, generating selectable mutations in chromosomal DNA of cancer cells. Some mechanisms by which virus-encoded factors modulate levels of A3 proteins are known. Little has been reported about how A3s are regulated in uninfected cells.
Supplementary Material
Translational Significance.
The characterization here of a post-translational mechanism of regulation of A3 protein levels in uninfected cells has implications for biological and translational research on antiviral defenses, retroelement mobility, and cancer progression.
Acknowledgments:
We thank Hannah Hudson, Nina Calantone, and Harry E. Taylor for their continued support and feedback throughout the course of these experiments. We would additionally like to thank Dr. Goo and the Northwestern University Proteomics Core.
Funding:
This work was supported by P01 AI 131346 (G.K.S., I.C., C.S., R.T.D.), a supplement to the Robert H. Lurie Comprehensive Cancer Center (P30 CA 060553-24S3) (C.S.), the U.S. Army Medical Research and Material Command (W81XWH-15-1-0105) (M.R.), a Chicago Baseball Charities Cancer Charities Award (G.K.S.), and the Northwestern Medicine Catalyst Fund (R.T.D.). We also acknowledge core resources, a Developmental Core pilot project award (C.S.), and consultations from the Third Coast Center for AIDS Research (CFAR), an NIH funded center (P30 AI117943).
Abbreviations
- A3
Human apolipoprotein B mRNA-editing enzyme catalytic polypeptide-like 3
- pVHL
Von Hippel-Lindau tumor suppressor protein
- CRL
Cullin-RING E3 Ubiquitin Ligase
- HIV
Human immunodeficiency virus
- SIV
Simian immunodeficiency virus
- Vif
Virion infectivity factor
- siRNA
short interfering RNAs
- HIF1α
Hypoxia-inducible factor 1-alpha
- RT-qPCR
Reverse transcription quantitative polymerase chain reaction
- ELOC
Elongin C
- CUL
Cullin
- ARIH1
Ariadne RING-in-between-RING E3 ubiquitin protein ligase 1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests: The authors have read the journal's policy on disclosure of potential conflicts of interest and declare no competing interests.
References
- 1.Ito J, Gifford RJ, and Sato K, Retroviruses drive the rapid evolution of mammalian APOBEC3 genes. Proc Natl Acad Sci U S A. 2020; 117: p. 610–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anwar F, Davenport MP, and Ebrahimi D, Footprint of APOBEC3 on the genome of human retroelements. J Virol. 2013; 87: p. 8195–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Conticello SG, Thomas CJ, Petersen-Mahrt SK, and Neuberger MS, Evolution of the AID/APOBEC family of polynucleotide (deoxy)cytidine deaminases. Mol Biol Evol. 2005; 22: p. 367–77. [DOI] [PubMed] [Google Scholar]
- 4.Sawyer SL, Emerman M, and Malik HS, Ancient adaptive evolution of the primate antiviral DNA-editing enzyme APOBEC3G. PLoS Biol. 2004; 2: p. E275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Poulain F, Lejeune N, Willemart K, and Gillet NA, Footprint of the host restriction factors APOBEC3 on the genome of human viruses. PLoS Pathog. 2020; 16: p. e1008718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Refsland EW and Harris RS, The APOBEC3 family of retroelement restriction factors. Curr Top Microbiol Immunol. 2013; 371: p. 1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hultquist JF, Lengyel JA, Refsland EW, et al. , Human and rhesus APOBEC3D, APOBEC3F, APOBEC3G, and APOBEC3H demonstrate a conserved capacity to restrict Vif-deficient HIV-1. J Virol. 2011; 85: p. 11220–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Donahue JP, Levinson RT, Sheehan JH, et al. , Genetic analysis of the localization of APOBEC3F to human immunodeficiency virus type 1 virion cores. J Virol. 2015; 89: p. 2415–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Song C, Sutton L, Johnson ME, D'Aquila RT, and Donahue JP, Signals in APOBEC3F N-terminal and C-terminal deaminase domains each contribute to encapsidation in HIV-1 virions and are both required for HIV-1 restriction. J Biol Chem. 2012; 287: p. 16965–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bishop KN, Holmes RK, Sheehy AM, Davidson NO, Cho SJ, and Malim MH, Cytidine deamination of retroviral DNA by diverse APOBEC proteins. Curr Biol. 2004; 14: p. 1392–6. [DOI] [PubMed] [Google Scholar]
- 11.Harris RS, Bishop KN, Sheehy AM, et al. , DNA deamination mediates innate immunity to retroviral infection. Cell. 2003; 113: p. 803–9. [DOI] [PubMed] [Google Scholar]
- 12.Holmes RK, Koning FA, Bishop KN, and Malim MH, APOBEC3F can inhibit the accumulation of HIV-1 reverse transcription products in the absence of hypermutation. Comparisons with APOBEC3G. J Biol Chem. 2007; 282: p. 2587–95. [DOI] [PubMed] [Google Scholar]
- 13.Sheehy AM, Gaddis NC, Choi JD, and Malim MH, Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature. 2002; 418: p. 646–50. [DOI] [PubMed] [Google Scholar]
- 14.Ooms M, Brayton B, Letko M, et al. , HIV-1 Vif adaptation to human APOBEC3H haplotypes. Cell Host Microbe. 2013; 14: p. 411–21. [DOI] [PubMed] [Google Scholar]
- 15.Dang Y, Wang X, Esselman WJ, and Zheng YH, Identification of APOBEC3DE as another antiretroviral factor from the human APOBEC family. J Virol. 2006; 80: p. 10522–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vetter ML, Johnson ME, Antons AK, Unutmaz D, and D'Aquila RT, Differences in APOBEC3G expression in CD4+ T helper lymphocyte subtypes modulate HIV-1 infectivity. PLoS Pathog. 2009; 5: p. e1000292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peng G, Greenwell-Wild T, Nares S, et al. , Myeloid differentiation and susceptibility to HIV-1 are linked to APOBEC3 expression. Blood. 2007; 110: p. 393–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.De Pasquale M, Kourteva Y, Allos T, and D'Aquila RT, Lower HIV provirus levels are associated with more APOBEC3G protein in blood resting memory CD4+ T lymphocytes of controllers in vivo. PLoS One. 2013; 8: p. e76002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang H, Yang B, Pomerantz RJ, Zhang C, Arunachalam SC, and Gao L, The cytidine deaminase CEM15 induces hypermutation in newly synthesized HIV-1 DNA. Nature. 2003; 424: p. 94–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gillick K, Pollpeter D, Phalora P, Kim EY, Wolinsky SM, and Malim MH, Suppression of HIV-1 infection by APOBEC3 proteins in primary human CD4(+) T cells is associated with inhibition of processive reverse transcription as well as excessive cytidine deamination. J Virol. 2013; 87: p. 1508–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mbisa JL, Barr R, Thomas JA, et al. , Human immunodeficiency virus type 1 cDNAs produced in the presence of APOBEC3G exhibit defects in plus-strand DNA transfer and integration. J Virol. 2007; 81: p. 7099–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Warren CJ, Xu T, Guo K, et al. , APOBEC3A functions as a restriction factor of human papillomavirus. J Virol. 2015; 89: p. 688–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burns MB, Lackey L, Carpenter MA, et al. , APOBEC3B is an enzymatic source of mutation in breast cancer. Nature. 2013; 494: p. 366–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Burns MB, Temiz NA, and Harris RS, Evidence for APOBEC3B mutagenesis in multiple human cancers. Nat Genet. 2013; 45: p. 977–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leonard B, Hart SN, Burns MB, et al. , APOBEC3B upregulation and genomic mutation patterns in serous ovarian carcinoma. Cancer Res. 2013; 73: p. 7222–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Taylor BJ, Nik-Zainal S, Wu YL, et al. , DNA deaminases induce break-associated mutation showers with implication of APOBEC3B and 3A in breast cancer kataegis. Elife. 2013; 2: p. e00534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zou J, Wang C, Ma X, Wang E, and Peng G, APOBEC3B, a molecular driver of mutagenesis in human cancers. Cell Biosci. 2017; 7: p. 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Harris RS, Molecular mechanism and clinical impact of APOBEC3B-catalyzed mutagenesis in breast cancer. Breast Cancer Res. 2015; 17: p. 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Law EK, Sieuwerts AM, LaPara K, et al. , The DNA cytosine deaminase APOBEC3B promotes tamoxifen resistance in ER-positive breast cancer. Sci Adv. 2016; 2: p. e1601737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sieuwerts AM, Willis S, Burns MB, et al. , Elevated APOBEC3B correlates with poor outcomes for estrogen-receptor-positive breast cancers. Horm Cancer. 2014; 5: p. 405–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu L, Chang Y, An H, Zhu Y, Yang Y, and Xu J, High APOBEC3B expression is a predictor of recurrence in patients with low-risk clear cell renal cell carcinoma. Urol Oncol. 2015; 33: p. 340 e1–8. [DOI] [PubMed] [Google Scholar]
- 32.Swanton C, McGranahan N, Starrett GJ, and Harris RS, APOBEC Enzymes: Mutagenic Fuel for Cancer Evolution and Heterogeneity. Cancer Discov. 2015; 5: p. 704–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cortez LM, Brown AL, Dennis MA, et al. , APOBEC3A is a prominent cytidine deaminase in breast cancer. PLoS Genet. 2019; 15: p. e1008545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim YS, Sun S, Yoon JS, Ko YH, Won HS, and Kim JS, Clinical implications of APOBEC3A and 3B expression in patients with breast cancer. PLoS One. 2020; 15: p. e0230261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Buisson R, Langenbucher A, Bowen D, et al. , Passenger hotspot mutations in cancer driven by APOBEC3A and mesoscale genomic features. Science. 2019; 364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Law EK, Levin-Klein R, Jarvis MC, et al. , APOBEC3A catalyzes mutation and drives carcinogenesis in vivo. J Exp Med. 2020; 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Covino DA, Gauzzi MC, and Fantuzzi L, Understanding the regulation of APOBEC3 expression: Current evidence and much to learn. J Leukoc Biol. 2018; 103: p. 433–444. [DOI] [PubMed] [Google Scholar]
- 38.Verhalen B, Starrett GJ, Harris RS, and Jiang M, Functional Upregulation of the DNA Cytosine Deaminase APOBEC3B by Polyomaviruses. J Virol. 2016; 90: p. 6379–6386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roelofs PA, Goh CY, Chua BH, et al. , Characterization of the mechanism by which the RB/E2F pathway controls expression of the cancer genomic DNA deaminase APOBEC3B. Elife. 2020; 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mori S, Takeuchi T, Ishii Y, and Kukimoto I, Identification of APOBEC3B promoter elements responsible for activation by human papillomavirus type 16 E6. Biochem Biophys Res Commun. 2015; 460: p. 555–60. [DOI] [PubMed] [Google Scholar]
- 41.Mori S, Takeuchi T, Ishii Y, et al. , Human Papillomavirus 16 E6 Upregulates APOBEC3B via the TEAD Transcription Factor. J Virol. 2017; 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vieira VC, Leonard B, White EA, et al. , Human papillomavirus E6 triggers upregulation of the antiviral and cancer genomic DNA deaminase APOBEC3B. mBio. 2014; 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Middlebrooks CD, Banday AR, Matsuda K, et al. , Association of germline variants in the APOBEC3 region with cancer risk and enrichment with APOBEC-signature mutations in tumors. Nat Genet. 2016; 48: p. 1330–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Leonard B, McCann JL, Starrett GJ, et al. , The PKC/NF-kappaB signaling pathway induces APOBEC3B expression in multiple human cancers. Cancer Res. 2015; 75: p. 4538–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Maruyama W, Shirakawa K, Matsui H, et al. , Classical NF-kappaB pathway is responsible for APOBEC3B expression in cancer cells. Biochem Biophys Res Commun. 2016; 478: p. 1466–71. [DOI] [PubMed] [Google Scholar]
- 46.Chou WC, Chen WT, Hsiung CN, et al. , B-Myb Induces APOBEC3B Expression Leading to Somatic Mutation in Multiple Cancers. Sci Rep. 2017; 7: p. 44089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lin L, Holmes B, Shen MW, et al. , Comprehensive Mapping of Key Regulatory Networks that Drive Oncogene Expression. Cell Rep. 2020; 33: p. 108426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jarvis MC, Ebrahimi D, Temiz NA, and Harris RS, Mutation Signatures Including APOBEC in Cancer Cell Lines. JNCI Cancer Spectr. 2018; 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cheng AZ, Yockteng-Melgar J, Jarvis MC, et al. , Epstein-Barr virus BORF2 inhibits cellular APOBEC3B to preserve viral genome integrity. Nat Microbiol. 2019; 4: p. 78–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Westrich JA, Warren CJ, Klausner MJ, et al. , Human Papillomavirus 16 E7 Stabilizes APOBEC3A Protein by Inhibiting Cullin 2-Dependent Protein Degradation. J Virol. 2018; 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yu X, Yu Y, Liu B, et al. , Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science. 2003; 302: p. 1056–60. [DOI] [PubMed] [Google Scholar]
- 52.Mehle A, Strack B, Ancuta P, Zhang C, McPike M, and Gabuzda D, Vif overcomes the innate antiviral activity of APOBEC3G by promoting its degradation in the ubiquitin-proteasome pathway. J Biol Chem. 2004; 279: p. 7792–8. [DOI] [PubMed] [Google Scholar]
- 53.Kobayashi M, Takaori-Kondo A, Miyauchi Y, Iwai K, and Uchiyama T, Ubiquitination of APOBEC3G by an HIV-1 Vif-Cullin5-Elongin B-Elongin C complex is essential for Vif function. J Biol Chem. 2005; 280: p. 18573–8. [DOI] [PubMed] [Google Scholar]
- 54.Donahue JP, Vetter ML, Mukhtar NA, and D'Aquila RT, The HIV-1 Vif PPLP motif is necessary for human APOBEC3G binding and degradation. Virology. 2008; 377: p. 49–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Desimmie BA, Delviks-Frankenberrry KA, Burdick RC, Qi D, Izumi T, and Pathak VK, Multiple APOBEC3 restriction factors for HIV-1 and one Vif to rule them all. J Mol Biol. 2014; 426: p. 1220–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Conticello SG, Harris RS, and Neuberger MS, The Vif protein of HIV triggers degradation of the human antiretroviral DNA deaminase APOBEC3G. Curr Biol. 2003; 13: p. 2009–13. [DOI] [PubMed] [Google Scholar]
- 57.Russell RA, Smith J, Barr R, Bhattacharyya D, and Pathak VK, Distinct domains within APOBEC3G and APOBEC3F interact with separate regions of human immunodeficiency virus type 1 Vif. J Virol. 2009; 83: p. 1992–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Russell RA and Pathak VK, Identification of two distinct human immunodeficiency virus type 1 Vif determinants critical for interactions with human APOBEC3G and APOBEC3F. J Virol. 2007; 81: p. 8201–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wiegand HL, Doehle BP, Bogerd HP, and Cullen BR, A second human antiretroviral factor, APOBEC3F, is suppressed by the HIV-1 and HIV-2 Vif proteins. EMBO J. 2004; 23: p. 2451–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jager S, Kim DY, Hultquist JF, et al. , Vif hijacks CBF-beta to degrade APOBEC3G and promote HIV-1 infection. Nature. 2011; 481: p. 371–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Guo Y, Dong L, Qiu X, et al. , Structural basis for hijacking CBF-beta and CUL5 E3 ligase complex by HIV-1 Vif. Nature. 2014; 505: p. 229–33. [DOI] [PubMed] [Google Scholar]
- 62.Huttenhain R, Xu J, Burton LA, et al. , ARIH2 Is a Vif-Dependent Regulator of CUL5-Mediated APOBEC3G Degradation in HIV Infection. Cell Host Microbe. 2019; 26: p. 86–99 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Desimmie BA, Smith JL, Matsuo H, Hu WS, and Pathak VK, Identification of a tripartite interaction between the N-terminus of HIV-1 Vif and CBFbeta that is critical for Vif function. Retrovirology. 2017; 14: p. 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mariani R, Chen D, Schrofelbauer B, et al. , Species-specific exclusion of APOBEC3G from HIV-1 virions by Vif. Cell. 2003; 114: p. 21–31. [DOI] [PubMed] [Google Scholar]
- 65.Marin M, Rose KM, Kozak SL, and Kabat D, HIV-1 Vif protein binds the editing enzyme APOBEC3G and induces its degradation. Nat Med. 2003; 9: p. 1398–403. [DOI] [PubMed] [Google Scholar]
- 66.Stopak K, de Noronha C, Yonemoto W, and Greene WC, HIV-1 Vif blocks the antiviral activity of APOBEC3G by impairing both its translation and intracellular stability. Mol Cell. 2003; 12: p. 591–601. [DOI] [PubMed] [Google Scholar]
- 67.Wichroski MJ, Ichiyama K, and Rana TM, Analysis of HIV-1 viral infectivity factor-mediated proteasome-dependent depletion of APOBEC3G: correlating function and subcellular localization. J Biol Chem. 2005; 280: p. 8387–96. [DOI] [PubMed] [Google Scholar]
- 68.Doehle BP, Schafer A, and Cullen BR, Human APOBEC3B is a potent inhibitor of HIV-1 infectivity and is resistant to HIV-1 Vif. Virology. 2005; 339: p. 281–8. [DOI] [PubMed] [Google Scholar]
- 69.Land AM, Wang J, Law EK, et al. , Degradation of the cancer genomic DNA deaminase APOBEC3B by SIV Vif. Oncotarget. 2015; 6: p. 39969–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Molan AM, Hanson HM, Chweya CM, et al. , APOBEC3B lysine residues are dispensable for DNA cytosine deamination, HIV-1 restriction, and nuclear localization. Virology. 2017; 511: p. 74–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang J, Shaban NM, Land AM, Brown WL, and Harris RS, Simian Immunodeficiency Virus Vif and Human APOBEC3B Interactions Resemble Those between HIV-1 Vif and Human APOBEC3G. J Virol. 2018; 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gaur R and Strebel K, Insights into the dual activity of SIVmac239 Vif against human and African green monkey APOBEC3G. PLoS One. 2012; 7: p. e48850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Xiao W, Ai J, Habermacher G, et al. , U19/Eaf2 binds to and stabilizes von hippel-lindau protein. Cancer Res. 2009; 69: p. 2599–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Taura M, Song E, Ho YC, and Iwasaki A, Apobec3A maintains HIV-1 latency through recruitment of epigenetic silencing machinery to the long terminal repeat. Proc Natl Acad Sci U S A. 2019; 116: p. 2282–2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Stickle NH, Chung J, Klco JM, Hill RP, Kaelin WG Jr., and Ohh M, pVHL modification by NEDD8 is required for fibronectin matrix assembly and suppression of tumor development. Mol Cell Biol. 2004; 24: p. 3251–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Okumura F, Zou W, and Zhang DE, ISG15 modification of the eIF4E cognate 4EHP enhances cap structure-binding activity of 4EHP. Genes Dev. 2007; 21: p. 255–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kondo K, Klco J, Nakamura E, Lechpammer M, and Kaelin WG Jr., Inhibition of HIF is necessary for tumor suppression by the von Hippel-Lindau protein. Cancer Cell. 2002; 1: p. 237–46. [DOI] [PubMed] [Google Scholar]
- 78.Zhu B, Xiao Y, Yeager M, et al. , Mutations in the HPV16 genome induced by APOBEC3 are associated with viral clearance. Nat Commun. 2020; 11: p. 886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Refsland EW, Stenglein MD, Shindo K, Albin JS, Brown WL, and Harris RS, Quantitative profiling of the full APOBEC3 mRNA repertoire in lymphocytes and tissues: implications for HIV-1 restriction. Nucleic Acids Res. 2010; 38: p. 4274–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lu YY, Zhu WJ, and Xie BG, Von Hippel-Lindau gene expression in human endometrium during menstrual cycle. Mol Med Rep. 2014; 9: p. 1355–8. [DOI] [PubMed] [Google Scholar]
- 81.Tokunaga E, Yamashita N, Tanaka K, et al. , Expression of APOBEC3B mRNA in Primary Breast Cancer of Japanese Women. PLoS One. 2016; 11: p. e0168090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yan S, He F, Gao B, et al. , Increased APOBEC3B Predicts Worse Outcomes in Lung Cancer: A Comprehensive Retrospective Study. J Cancer. 2016; 7: p. 618–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Downey SL, Florea BI, Overkleeft HS, and Kisselev AF, Use of proteasome inhibitors. Curr Protoc Immunol. 2015; 109: p. 9 10 1–9 10 8. [DOI] [PubMed] [Google Scholar]
- 84.Kisselev AF, van der Linden WA, and Overkleeft HS, Proteasome inhibitors: an expanding army attacking a unique target. Chem Biol. 2012; 19: p. 99–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ivan M, Kondo K, Yang H, et al. , HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001; 292: p. 464–8. [DOI] [PubMed] [Google Scholar]
- 86.Nguyen HC, Yang H, Fribourgh JL, Wolfe LS, and Xiong Y, Insights into Cullin-RING E3 ubiquitin ligase recruitment: structure of the VHL-EloBC-Cul2 complex. Structure. 2015; 23: p. 441–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Stebbins CE, Kaelin WG Jr., and Pavletich NP, Structure of the VHL-ElonginC-ElonginB complex: implications for VHL tumor suppressor function. Science. 1999; 284: p. 455–61. [DOI] [PubMed] [Google Scholar]
- 88.Gossage L, Eisen T, and Maher ER, VHL, the story of a tumour suppressor gene. Nat Rev Cancer. 2015; 15: p. 55–64. [DOI] [PubMed] [Google Scholar]
- 89.Kaelin WG Jr., The VHL Tumor Suppressor Gene: Insights into Oxygen Sensing and Cancer. Trans Am Clin Climatol Assoc. 2017; 128: p. 298–307. [PMC free article] [PubMed] [Google Scholar]
- 90.Bard JAM, Goodall EA, Greene ER, Jonsson E, Dong KC, and Martin A, Structure and Function of the 26S Proteasome. Annu Rev Biochem. 2018; 87: p. 697–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Komander D and Rape M, The ubiquitin code. Annu Rev Biochem. 2012; 81: p. 203–29. [DOI] [PubMed] [Google Scholar]
- 92.Ohh M, Park CW, Ivan M, et al. , Ubiquitination of hypoxia-inducible factor requires direct binding to the beta-domain of the von Hippel-Lindau protein. Nat Cell Biol. 2000; 2: p. 423–7. [DOI] [PubMed] [Google Scholar]
- 93.Tanimoto K, Makino Y, Pereira T, and Poellinger L, Mechanism of regulation of the hypoxia-inducible factor-1 alpha by the von Hippel-Lindau tumor suppressor protein. EMBO J. 2000; 19: p. 4298–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lonergan KM, Iliopoulos O, Ohh M, et al. , Regulation of hypoxia-inducible mRNAs by the von Hippel-Lindau tumor suppressor protein requires binding to complexes containing elongins B/C and Cul2. Mol Cell Biol. 1998; 18: p. 732–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kibel A, Iliopoulos O, DeCaprio JA, and Kaelin WG Jr., Binding of the von Hippel-Lindau tumor suppressor protein to Elongin B and C. Science. 1995; 269: p. 1444–6. [DOI] [PubMed] [Google Scholar]
- 96.Knauth K, Cartwright E, Freund S, Bycroft M, and Buchberger A, VHL mutations linked to type 2C von Hippel-Lindau disease cause extensive structural perturbations in pVHL. J Biol Chem. 2009; 284: p. 10514–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Russell RC and Ohh M, NEDD8 acts as a 'molecular switch' defining the functional selectivity of VHL. EMBO Rep. 2008; 9: p. 486–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cai Q and Robertson ES, Ubiquitin/SUMO modification regulates VHL protein stability and nucleocytoplasmic localization. PLoS One. 2010; 5. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 99.Cai Q, Verma SC, Kumar P, Ma M, and Robertson ES, Hypoxia inactivates the VHL tumor suppressor through PIASy-mediated SUMO modification. PLoS One. 2010; 5: p. e9720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Duda DM, Olszewski JL, Schuermann JP, et al. , Structure of HHARI, a RING-IBR-RING ubiquitin ligase: autoinhibition of an Ariadne-family E3 and insights into ligation mechanism. Structure. 2013; 21: p. 1030–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yuan L, Lv Z, Atkison JH, and Olsen SK, Structural insights into the mechanism and E2 specificity of the RBR E3 ubiquitin ligase HHARI. Nat Commun. 2017; 8: p. 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kelsall IR, Duda DM, Olszewski JL, et al. , TRIAD1 and HHARI bind to and are activated by distinct neddylated Cullin-RING ligase complexes. EMBO J. 2013; 32: p. 2848–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Scott DC, Rhee DY, Duda DM, et al. , Two Distinct Types of E3 Ligases Work in Unison to Regulate Substrate Ubiquitylation. Cell. 2016; 166: p. 1198–1214 e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kleiger G and Deshaies R, Tag Team Ubiquitin Ligases. Cell. 2016; 166: p. 1080–1081. [DOI] [PubMed] [Google Scholar]
- 105.Liu B, Yu X, Luo K, Yu Y, and Yu XF, Influence of primate lentiviral Vif and proteasome inhibitors on human immunodeficiency virus type 1 virion packaging of APOBEC3G. J Virol. 2004; 78: p. 2072–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Malim MH and Pollpeter D, APOBEC restriction goes nuclear. Nat Microbiol. 2019; 4: p. 6–7. [DOI] [PubMed] [Google Scholar]
- 107.Li M, Shandilya SM, Carpenter MA, et al. , First-in-class small molecule inhibitors of the single-strand DNA cytosine deaminase APOBEC3G. ACS Chem Biol. 2012; 7: p. 506–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Manasanch EE and Orlowski RZ, Proteasome inhibitors in cancer therapy. Nat Rev Clin Oncol. 2017; 14: p. 417–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Weathington NM and Mallampalli RK, Emerging therapies targeting the ubiquitin proteasome system in cancer. J Clin Invest. 2014; 124: p. 6–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Nalawansha DA and Crews CM, PROTACs: An Emerging Therapeutic Modality in Precision Medicine. Cell Chem Biol. 2020; 27: p. 998–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mullard A, First targeted protein degrader hits the clinic. Nat Rev Drug Discov. 2019. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.