

Abstract
Purpose:
Up to 25% of patients diagnosed as idiopathic Parkinson’s disease (IPD) have an atypical parkinsonian syndrome (APS). We had previously validated an automated image-based algorithm to discriminate between IPD, multiple system atrophy (MSA), and progressive supranuclear palsy (PSP). While the algorithm was accurate with respect to the final clinical diagnosis after long term expert follow-up, its relationship to the initial referral diagnosis and to the neuropathological gold standard is not known.
Methods:
Patients with an uncertain diagnosis of parkinsonism were referred for [18F]-fluorodeoxyglucose (FDG) PET to classify patients as IPD or as APS based on the automated algorithm. Patients were followed by a movement disorder specialist and subsequently underwent neuropathological examination. The image-based classification was compared to the neuropathological diagnosis in 15 patients with parkinsonism.
Results:
At the time of referral to PET, the clinical impression was only 66.7% accurate. The algorithm correctly identified 80% of the cases as IPD or APS (p=0.02) and 87.5% of the APS cases as MSA or PSP (p=0.03). The final clinical diagnosis was 93.3% accurate (p<0.001), but needed several years of expert follow-up.
Conclusion:
The image-based classifications agreed well with autopsy and can help to improve diagnostic accuracy during the period of clinical uncertainty.
Keywords: parkinsonism, FDG PET, differential diagnosis, neuropathology, network analysis
Introduction
The clinical diagnosis of idiopathic Parkinson’s disease (IPD) is inaccurate as 25% of patients classified as having Parkinson’s turn out to have an atypical parkinsonian syndrome (APS) [1]. Multiple system atrophy (MSA) and progressive supranuclear palsy (PSP) account for over 90% of APS cases, which can be difficult to distinguish from IPD at early disease stages [2–5]. Indeed, in a study [6] comparing neuropathological diagnosis with the initial clinical diagnosis made in untreated or ambiguously responsive parkinsonian subjects, the positive predictive value (PPV) of a PD diagnosis by a movement disorders specialist was only 26%; in early-stage patients who responded to therapy, the PPV was only 53% [6].
Metabolic imaging can improve the diagnostic accuracy in distinguishing PD from APS using observer-dependent and observer-independent methods [7–11]. We developed an automated, image-based algorithm that classifies patients based on the expression of disease-related metabolic networks at the individual subject level [12,13]. Specifically, we used 18F-fluorodeoxyglucose (FDG) PET to differentiate between IPD, MSA, and PSP based on their respective pattern expression values [12]. Several subsequent studies showed that the resulting classifications agree closely with the clinical diagnosis made by movement disorders experts three to four years after imaging blind to the results of the automated analysis. The PPV of an IPD diagnosis ranged from 92 to 98% [12,14–16].
Here, we asked how the accuracy of automated imaging classification compared to the neuropathological gold standard, and how it compared to the clinical impression at the time of referral for imaging.
Methods
Study design
We evaluated cases of uncertain diagnosis of parkinsonism that had undergone both FDG PET imaging and eventual neuropathological assessment (Figure 1). All patients were referred to the Center for Parkinson’s Disease and Other Movement Disorders at Columbia University Irving Medical Center (CUIMC) in New York by an outpatient physician/neurologist to seek the opinion of a center specializing in movement disorders. At CUIMC, all patients underwent detailed clinical assessments by a movement disorders specialist (S.F., S.A.O.). Only patients with an uncertain diagnosis were referred to The Feinstein Institutes for Medical Research (Manhasset, NY) for FDG PET imaging [12].
Fig. 1. Study design.
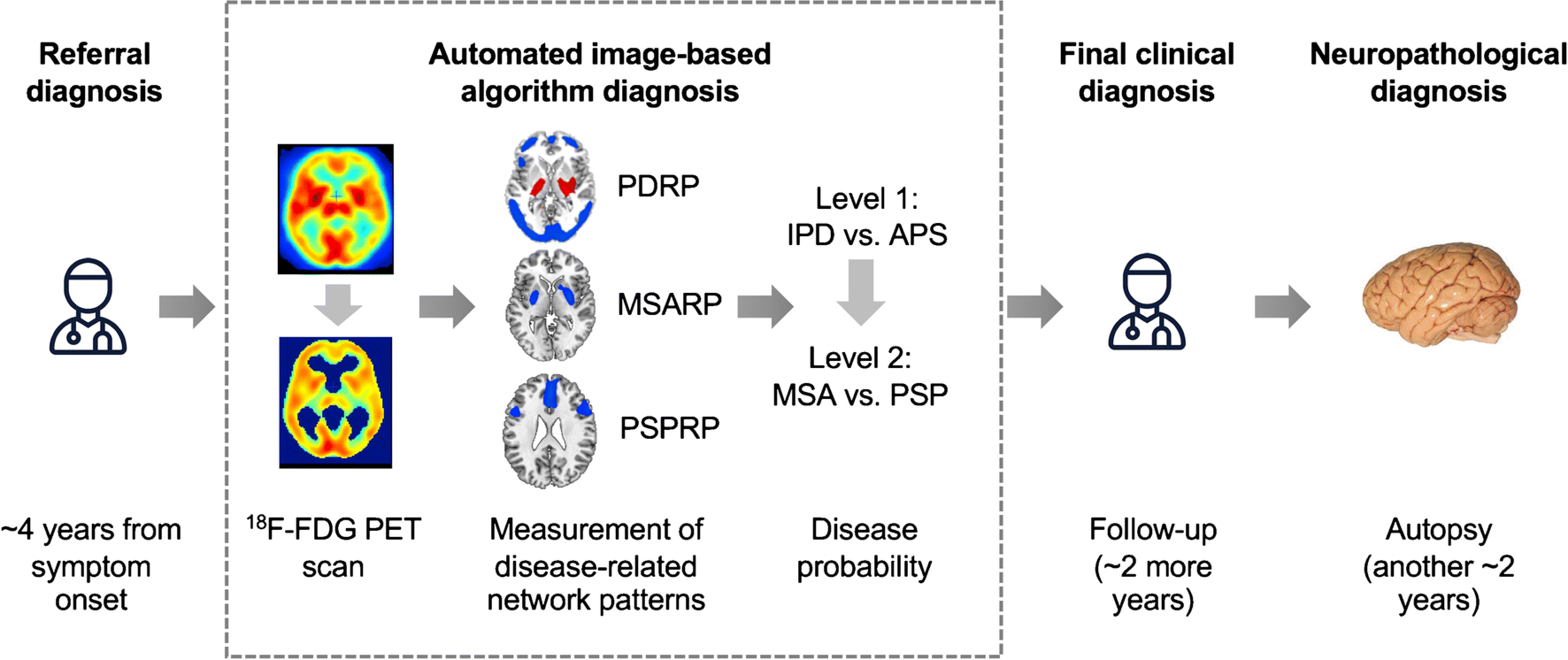
Patients with parkinsonism and uncertain clinical diagnosis were referred by a movement disorder specialist for brain imaging with FDG PET an average of 4 years from symptom onset. The first step (Level 1) determines whether the diagnosis is likely IPD or an atypical parkinsonian syndrome (APS); if the algorithm predicts an APS, Level 2 evaluates the diagnosis of progressive supranuclear palsy, and multiple system atrophy. At either Level, the pattern expression may be insufficiently developed for the algorithm to render a diagnosis, leading to an indeterminate (IND) diagnosis. The probability of each disease was calculated for every case (see Methods and Table 2). Each patient was followed clinically by a specialist who was blind to the results of the imaging algorithm. For every case, the referral diagnosis, the algorithm’s diagnosis, and the final clinical diagnosis were compared to the neuropathology, determined an average of two years after the final clinical diagnosis. [Abbreviations: 18F-FDG PET=fluorodeoxyglucose positron emission tomography; PSPRP=progressive supranuclear palsy-related pattern; MSARP=multiple system atrophy-related pattern; PDRP=Parkinson’s disease-related pattern]
The inclusion criteria for this study were as follows: 1) uncertain clinical diagnosis, i.e., a differential of two or more possible conditions or a non-specific diagnosis of parkinsonism between January 1, 1996 and January 1, 2016; and 2) no evidence of secondary cause for parkinsonism based on structural imaging. At the time of the referral to FDG PET, the movement disorders specialist recorded a referral diagnosis representing the clinical impression, i.e., the most likely of the various diagnostic possibilities. After imaging, scans were classified according to an automated image-based algorithm [12] performed by an analyst (C.C.T.), blind to patient identity, clinical diagnosis, and autopsy findings. The algorithm provided an image-based diagnosis of IPD, MSA, PSP, or indeterminate (IND) based on the pattern expression values (subject scores) computed for each patient. A final clinical diagnosis was subsequently made following at least two follow-up visits by a movement disorders specialist (S.F.), who was blind to the results of the imaging algorithm. The final clinical diagnosis of each patient was made in agreement with consensus criteria for IPD [17], MSA [18], and PSP [19]. In each case, the autopsy diagnosis was determined by a neuropathologist (J.-P.V.) at the New York Brain Bank (NYBB), who likewise was blind to the image-based diagnosis.
Study sample
206 parkinsonian patients were referred for FDG PET because of an initially uncertain diagnosis, and followed by movement disorders specialists at CUIMC to receive a final clinical diagnosis. Of these, 20 (9.7%) had postmortem examination at NYBB. Because the algorithm was validated only for the differential diagnosis of IPD, MSA, and PSP [14,15], we included patients confirmed at autopsy to have one of these disorders. Thus, four patients with other pathological diagnoses (three with corticobasal syndrome (CBS); one with an unclassified tauopathy) were excluded. Additionally, one patient had structural MRI at the time of PET, which disclosed severe atrophy, which can confound the computation of subject scores and invalidate the algorithm [14]. Thus, the image-based classification was compared to the neuropathological diagnosis in 15 patients with parkinsonism. Descriptive information from a subset of these patients have been reported previously as highlighted in Supplementary Table 1 [12].
At the end of the study, the imaging classifications and the clinical and autopsy findings were separately submitted to an independent evaluator (D.K.G.) at a third site. He opened the blind and merged the results into a final database that was shared with the other sites and used for statistical analysis.
FDG PET
All subjects were scanned using the General Electric Advance tomograph (Milwaukee, WI) at the Feinstein Institutes/Northwell Health as described previously [20]. In brief, all subjects fasted overnight and antiparkinsonian medication was withheld at least 12 hours before the scan. Scans from each subject were realigned and spatially normalized to a standard Montreal Neurological Institute (MNI)-based PET brain template and smoothed with an isotropic Gaussian kernel (10 mm) in all directions to improve the signal-to-noise ratio. Image processing was performed using Statistical Parametric Mapping (SPM5) software (Wellcome Centre for Human Neuroimaging, London, UK).
Automated pattern-based classification
Expression values (subject scores) for each of the three previously validated disease-related metabolic covariance patterns (PDRP, MSARP, and PSPRP) were computed on a prospective individual-scan basis [21] using existing software (ScAnVp, freely available at http://www.feinsteinneuroscience.org/). Subject scores for each disease pattern were standardized (z-scored) with respect to corresponding values from healthy volunteer subjects used in the original study [12].
Using logistic regression analysis, we used the subject scores from each patient to compute the probability of each disease category in that individual (see Figure 1). Based on these probabilities, we classified each of the subjects according to a two-level procedure as described previously [12]. At Level 1, each subject was classified as IPD or APS by comparing the subject’s probabilities to the cutoff probabilities for IPD (0.81) and APS (0.79) determined in the original study. Patients who had a higher probability than the cutoff value for IPD were classified as IPD, whereas those with a higher probability than the cutoff value for APS were classified as APS. At Level 2, subjects classified at Level 1 as APS were further subclassified as MSA or PSP using the previously reported cutoff probabilities for MSA (0.74) and PSP (0.55) [12]. Subjects with probabilities lower than the cutoff values were classified as “indeterminate” (IND) at each level. The pattern-based classification algorithm employed in this analysis was identical to that used in the original cohort [12] and in the subsequent validation studies [14–16].
Neuropathology
All patients underwent postmortem examination and received a histopathological diagnosis by a neuropathologist (J.-P.V.) according to established criteria [19,22–24]. Half-brains were placed in formalin and cut; tissue was processed according to published protocols [25]. Briefly, 7 μm thick sections were stained with Luxol fast blue counterstained with hematoxylin and eosin for general survey. Selected sections were stained using the Bielschowsky method to evaluate axons, neuritic plaques, and neurofibrillary and glial tangles; AT8 antibodies against hyperphosphorylated tau; and antibodies against β-amyloid, α-synuclein, ubiquitin and TDP43.
Statistical analysis
For statistical analysis, we calculated the proportions of correctly classified cases for the imaging diagnosis, the referral diagnosis and the final clinical diagnosis each in comparison with the pathological diagnosis. The binomial test was used to test the hypothesis that the proportion of correctly classified cases was different from a chance guess, i.e., a probability of 0.50, and was considered significant for p<0.05, two-tailed. All statistical analyses were performed in SAS Studio.
Results
Sample characteristics
The 15 patients in the study had a postmortem diagnosis of IPD (n=4), MSA (n=6), or PSP (n=5) (Supplementary Table 1). Thus, the majority of cases (73.3%) had an atypical form of parkinsonism at autopsy. This is not unexpected, given that autopsies are seldom performed except in the most challenging cases. The mean age of the patients at the time of imaging was 66.3 (range 44–82) years and the mean symptom duration was 4.1 (range 2–9) years at the time of PET (Supplementary Table 2). The mean time interval between PET and autopsy was 3.5 (range 0.4–5.8) years. The final clinical diagnosis was reached after a mean 1.9 (range 0.3–4.7) years after imaging. The mean time from final clinical diagnosis to autopsy was 1.7 (range 0.1–5.0) years.
Clinical referral diagnosis
At the time of the referral for PET, nine of the 15 cases were thought likely to have IPD based on the leading diagnostic impression, whereas the other six were thought to have either PSP or MSA as the most likely possibility. Relative to postmortem, the referral diagnoses were 66.7% correct, which did not differ significantly from chance (Z=1.29; p=0.19, Table 1). All five initial misdiagnoses were APS patients thought incorrectly to have IPD. However, of the six cases with an initial clinical impression of MSA or PSP, all diagnoses were subsequently confirmed at autopsy.
Table 1.
Concordance of clinical and imaging diagnosis with postmortem
| Neuropathological diagnosis | Proportion of correct clinical diagnoses | p-value | ||||
|---|---|---|---|---|---|---|
|
| ||||||
| CLINICAL | Referral diagnosis | IPD | MSA | PSP | ||
| IPD | 4 | 4 | 1 | |||
| MSA | 0 | 2 | 0 | 10/15 (66.7%) | p=0.19 | |
| PSP | 0 | 0 | 4 | |||
|
| ||||||
| Final diagnosis | IPD | MSA | PSP | |||
| IPD | 4 | 0 | 0 | |||
| MSA | 0 | 5 | 0 | 14/15 (93.3%) | p<0.001 | |
| PSP | 0 | 0 | 5 | |||
| CBS | 0 | 1 | 0 | |||
|
| ||||||
| IMAGING | Level-I (IPD vs. APS) | IPD | APS | |||
| IPD | 4 | 2 | ||||
| APS | 0 | 8 | 12/15 (80.0%) | p=0.02 | ||
| IND | 0 | 1 | ||||
|
| ||||||
| Level-II (MSA vs. PSP) | MSA | PSP | ||||
| MSA | 4 | 0 | ||||
| PSP | 0 | 3 | 7/8 (87.5%) | p=0.03 | ||
| IND | 0 | 1 | ||||
Automated image-based diagnosis
The automated algorithm produced one of three possible results: a correct diagnosis (with reference to autopsy), a misdiagnosis, or an “indeterminate” classification reflecting classification probabilities beneath the prespecified cut points. For comparison of diagnostic accuracy in this study, we considered indeterminate (IND) as an incorrect diagnosis.
Based on the neuropathological diagnosis, the automated algorithm correctly classified 80.0% of the cases at Level 1 (IPD vs APS), which was greater than chance (Z=2.32, p=0.02, Table 2). Of the 8 cases classified as APS at Level 1, the algorithm correctly classified 87.5% as MSA or PSP at Level 2, which was also greater than chance (Z=2.12, p=0.03; Table 1). Two representative cases are shown in Figure 2.
Fig. 2. Cases of autopsy-confirmed Parkinson’s disease (PD) and progressive supranuclear palsy (PSP).
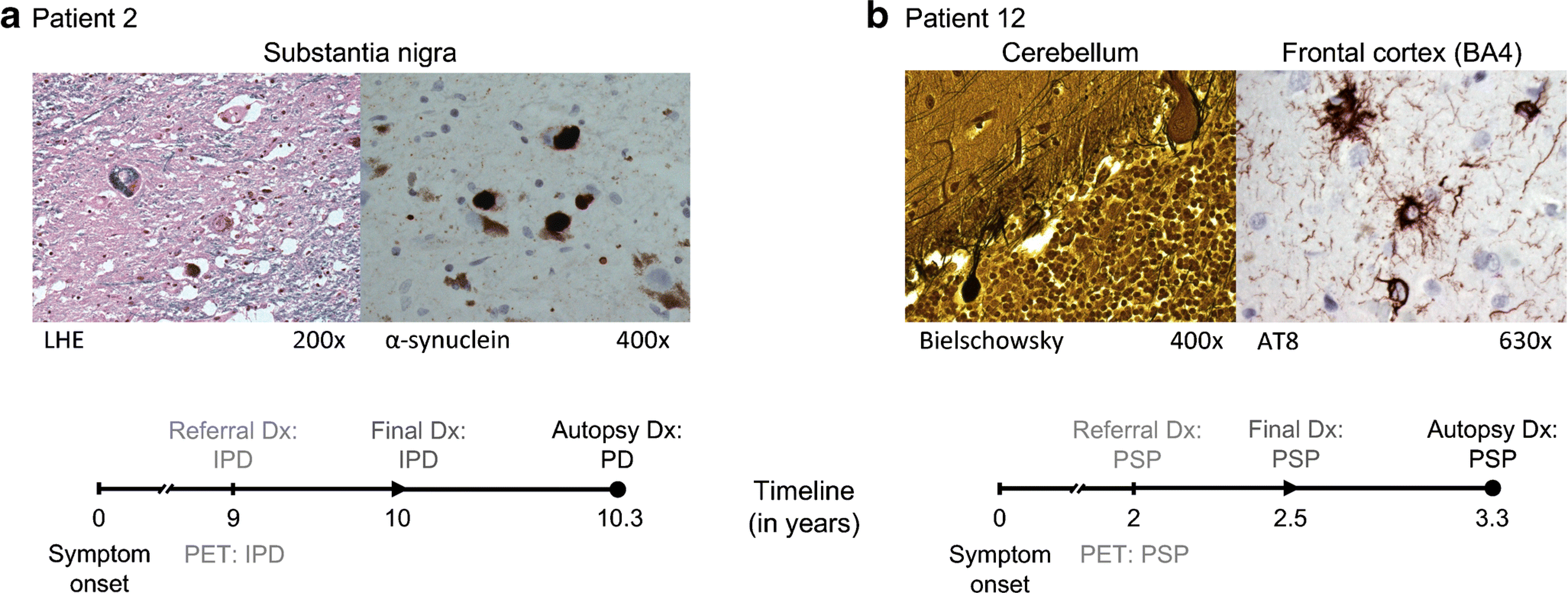
(a) Patient 2 (57-year old male) had an uncertain clinical diagnosis of idiopathic Parkinson’s disease (IPD) at the time of PET referral, roughly 9 years after symptom onset. The automated imaging algorithm classified the patient as IPD (99% likelihood). One year after imaging, a final clinical diagnosis of IPD was reached, which was confirmed on autopsy 4 months later. The neuropathological examination demonstrated Lewy body-containing neurons and severe neuronal loss in the pars compacta of the substantia nigra (LHE, 200×; left). Lewy body-containing neurons were labeled with α-synuclein (400×; right). [LHE=Luxol fast blue hematoxylin and eosin]. (b) At the time of PET referral, Patient 12 (69 years old, male, symptom duration 2 years) had an uncertain clinical diagnosis with PSP as the leading possibility. The automated imaging algorithm classified the patient as PSP (83.3% likelihood). Six months after imaging, a final clinical diagnosis of PSP was made, which was confirmed on autopsy 10 months later. The histopathological examination showed neuronal loss in the globus pallidus, substantia nigra, red nucleus, subthalamic nucleus, pons, medulla oblongata, and cerebellum. The cerebellar cortex displayed loss of Purkinje cells and presence of torpedoes (Bielschowsky, 400×; left). AT8-labeled cells including tufted astrocytes and glial cytoplasmatic inclusions were found in the paracentral cortex (630×; right), superior parietal lobe, and prefrontal cortex.
The automated algorithm misclassified two patients with APS as IPD. In both instances, IPD was also the leading referral diagnosis, but Case 9 was eventually found to have MSA at autopsy, and Case 15 to have PSP. The algorithm classified two cases as IND (Cases 10 and 13). At the time of PET referral, the leading clinical impression of Case 10 was IPD, but the final clinical diagnosis was MSA four years later, which was consistent with autopsy four months later. Because neuronal loss and gliosis of the putamen and globus pallidus were heavily lateralized on the left hemisphere, the algorithm was unable to determine whether this metabolic asymmetry was due to a reduction on the left or relative increase on the right and so rendered a classification of IND at Level 1.
Final clinical diagnosis
Based on the neuropathology, the final clinical diagnosis was correct in 14/15 (93.3%) cases. This was markedly different than chance (Z=3.36; p=0.0008, Table 1). A single case with MSA (mixed type) at autopsy (Figure 3) was incorrectly classified by the algorithm as IPD and by the expert clinician as CBD.
Fig. 3. A challenging case of pathology-proven multiple system atrophy (MSA).
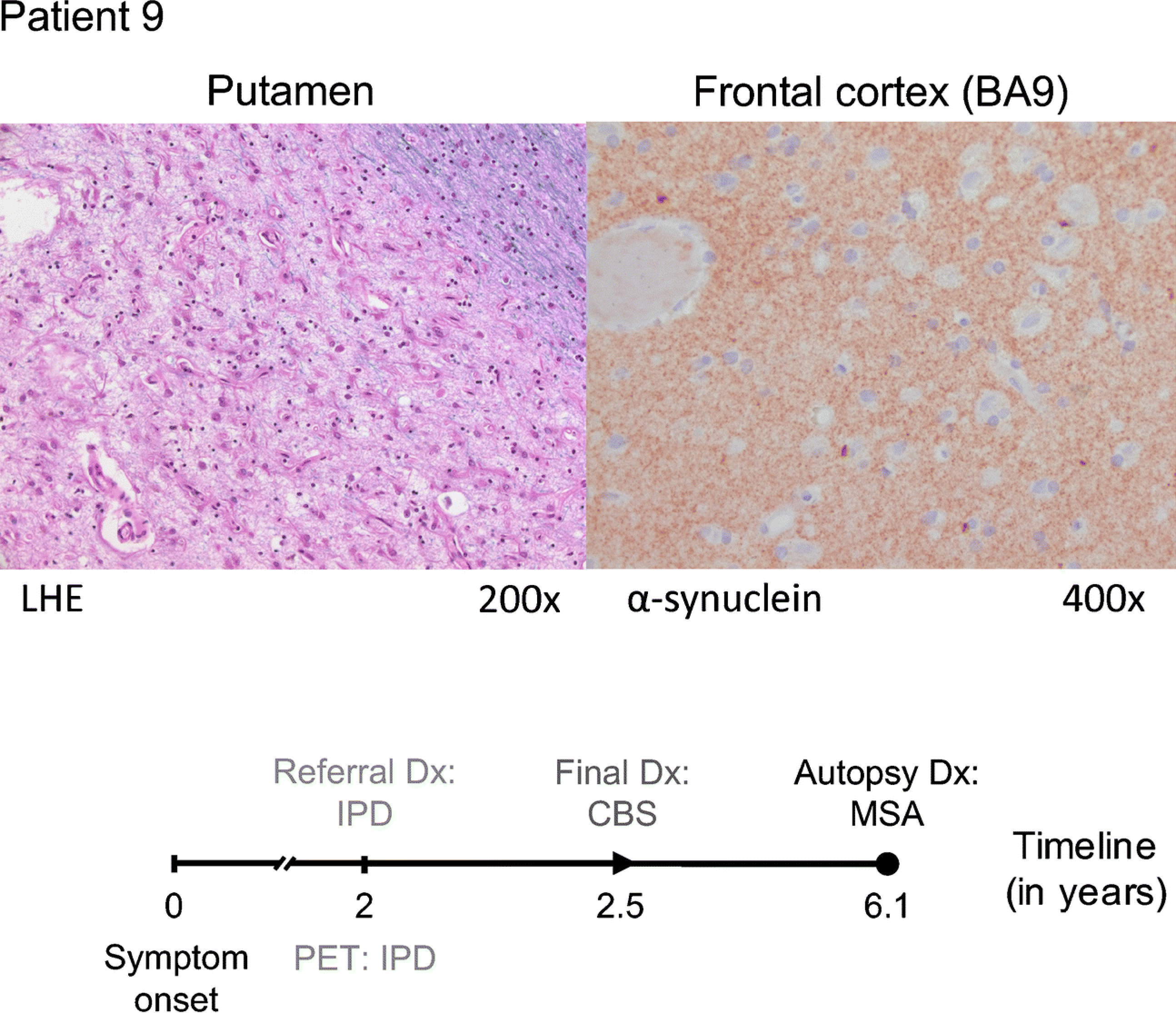
Patient 9 (64-year old female, symptom duration 2 years) was referred for FDG PET because of an uncertain clinical diagnosis with a suspicion of IPD or CBS. The automated imaging algorithm classified the patient as IPD (83.9% likelihood). After 6 months of additional clinical follow-up, the clinical diagnosis was revised to CBS. Autopsy performed four years later revealed changes consistent with MSA: severe neuronal loss and reactive gliosis in the putamen (LHE, 200×; left), pons, substantia nigra, and cerebellum. α-synuclein-labeled glial cytoplasmic inclusions were found in the frontal cortex (α-synuclein antibody, 400×; right), the striatum, the pons, amygdala, and medulla oblongata.
Discussion
In this small sample of 15 diagnostically challenging cases of parkinsonism, the clinical referral diagnosis, rendered after a mean disease duration of 4.1 years, correctly predicted the autopsy result in 67% of cases (Figure 4). Metabolic imaging in conjunction with the pattern-based algorithm was accurate in 80% (Level 1) and 88% (Level 2) of the cases. The final clinical diagnosis, reached after an average of two additional years of clinical follow-up, was even more accurate (93%). Although the sample size is a limitation of the current study, these statistics are congruent with previous studies showing close agreement between PET-based classification and the final clinical diagnosis made 3–4 years later, where the algorithm distinguished IPD from APS with 94% specificity and achieved 90% specificity for MSA and 94% for PSP [12,14–16]. The data also accord with comprehensive studies showing a close relationship between the final diagnosis of the expert clinician and autopsy [1,26]. APS clearly still presented a challenge to the expert clinician as evident in the Hughes et al. study [26], which did not incorporate brain imaging: the final clinical diagnosis, achieved after a mean disease duration of 5.3 years, was correct for 85.7% of the MSA cases and 80% of the PSP cases.
Fig. 4. Comparison of the concordance of clinical and PET diagnoses with autopsy.
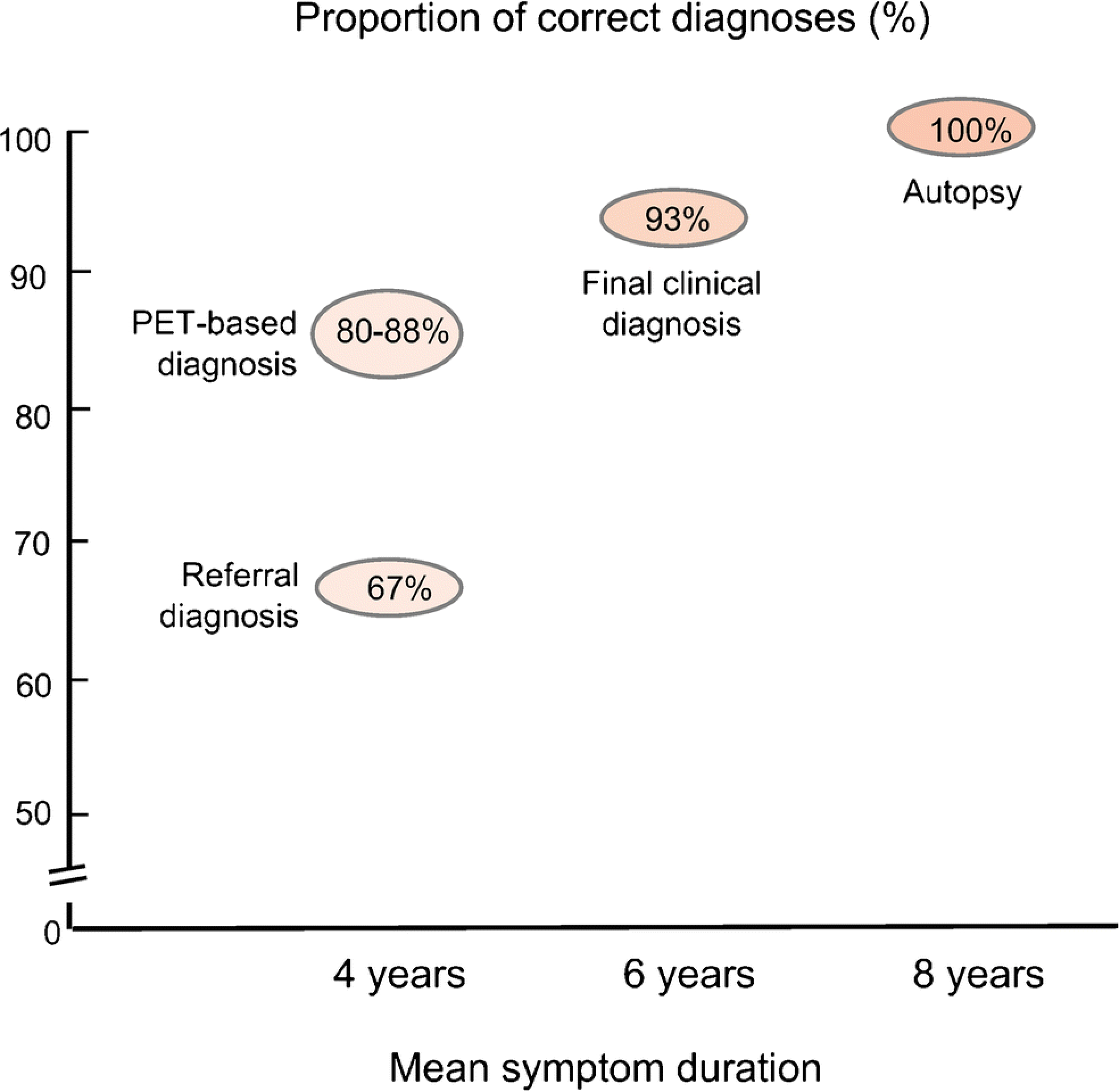
At the time of PET referral, in patients with uncertain clinical diagnosis (mean symptom duration 4 years), the referral diagnosis agreed with autopsy only 67% of the time. Automated classification based on contemporaneous PET imaging increased diagnostic accuracy to 80% for Level 1 and 87.5% for Level 2. The final clinical diagnosis, reached by the expert clinician after an average of 2 years of follow-up, was 93% concordant with autopsy findings obtained approximately 2 years later.
The low likelihood of accurate clinical diagnosis in patients with disease duration <5 years is particularly concerning, and it has not improved appreciably over the past few decades [1,6]. In the current study, the referral diagnosis, which was made by a movement disorders specialist an average of 4 years after symptom onset, was correct in only 67% of patients. Indeed, of nine cases initially thought to have IPD, five were found to have APS at postmortem examination. By contrast, all initial diagnostic impressions of APS were correct, despite the clinical uncertainty that prompted the request for imaging to begin with.
Two of the four discordant classifications of the imaging algorithm were attributed to indeterminate readouts, i.e., where the classification probability did not exceed the requisite cut point for diagnosis. In such cases, the algorithm provides a degree of doubt that is preferable to an incorrect classification [12]. That notwithstanding, only two of the misdiagnoses made by the algorithm reflected true errors. Case 9 was the most challenging (Figure 3). This patient had MSA, but was misclassified as IPD by both the initial clinical assessment and the algorithm; the final clinical diagnosis of CBS was also incorrect. It is possible that at the time of referral, the site of neurodegeneration in this patient was more nigral than striatal. This would accord with a more IPD-like clinical presentation, which includes levodopa responsiveness [3,27]. With regard to the algorithm, all three expression scores were quite low, with absolute values ≤0.6. While the case was categorized as IPD, the probability was only 83.9%, marginally exceeding the cut point (81.0%) for this diagnosis. In all likelihood, the initial classification of IPD would not have been sustained over time as neurodegeneration progressed to involve the striatum. Overall, this case was the only low expression case (pattern expression scores of all three pattern ≤ 1.0) in this study. The PPV for discriminating IPD from atypical parkinsonism was 96–98% in previous studies [12,15,28]. In cases with low expression, the PPV was reduced to 80% for IPD [12], which should be considered when making the final diagnosis in clinical practice.
Case 15 has a primary diagnosis of PSP with coexisting Alzheimer’s disease (AD) pathology, and was misclassified as IPD by the algorithm. The AD-related covariance pattern (ADRP) is characterized by relative metabolic decreases in the hippocampus, parahippocampal gyrus, and parietal and temporal association regions [29–31]. This topography includes a circumscribed area of modest overlap with the PDRP, which has minimal effect on PDRP expression in PD datasets [31]. Thus, the presence of incipient AD pathology with slight PDRP elevations would be unlikely to bias algorithmic classifications of patients as IPD, as seen in Cases 3 and 4. The PSPRP, on the other hand, has little topographic overlap with PDRP. Indeed, in a recent study, PSP co-pathologies, the most common being AD, had minimal clinical impact on the disease [32]. Despite the topographic and clinical independence of the two pathologies, it is possible that a patient with PSP-AD dual pathology will exhibit metabolic decreases in parietal cortex due to AD rather than PSP. As noted above, a localized change of this sort can cause a spurious, albeit modest, increase in PDRP expression and lead to a misclassification as IPD, as seen in Case 15.
Nonetheless, at the time of PET referral, the automated image-based classification was substantially more accurate than the contemporaneous clinical diagnosis. Specifically, three of the five patients who initially were thought (incorrectly) to have IPD were correctly classified as APS by the algorithm. As targeted proteinopathy-specific therapies are developed, confirming a clinical diagnosis of one or the other major forms of APS will become relevant [33].
Conclusion
This study confirms the advantages of clinical expertise combined with longitudinal patient observation in difficult-to-diagnose cases. Unfortunately, many patients will not have access to such expertise and few are willing to wait several years for an accurate diagnosis and a realistic prognosis. In such circumstances, reliable biomarkers will help reduce diagnostic uncertainty early in the disease course. So far, the image-based algorithm has the highest specificity and predictive value of available network biomarkers in differentiating PD, MSA, and PSP [7,12–15,34]. The advantage of metabolic imaging is that it can provide a wealth of information regarding rates of disease progression and treatment responses [13,34,35]. Perhaps more importantly, accurate early diagnosis is critical for efficient clinical trial design. Many disease-modification trials in PD focus on recent onset, drug-naïve PD patients—the very group for whom early diagnosis is least accurate [6]. Using the algorithm to screen for potential APS patients “masquerading” as IPD may enhance the likelihood of detecting a therapeutic effect. This attribute is particularly relevant in Phase 2 clinical trials in which sample sizes are smaller and significant results can be obscured by just a few misdiagnoses.
Supplementary Material
Acknowledgments
The authors thank Vicky Brandt and Dr. Martin Niethammer for helpful discussions and suggestions. We also thank Rosie Persaud for her invaluable assistance with coordinating data collection.
Funding
Aspects of this work were supported by the National Institute of Neurological Disorders and Stroke (P50 NS 071675 [Morris K. Udall Center of Excellence for Parkinson’s Disease Research at The Feinstein Institute for Medical Research] to D.E.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the National Institute of Neurological Disorders and Stroke. K.A.S. is supported by the Leopoldina Fellowship Program of the German National Academy of Sciences Leopoldina (LDS 2016-08) and the Postdoctoral Fellowship Grant No. PF-FBS-1929 from the Parkinson’s Foundation.
Conflicts of interest
K.L.P. has received clinical trial grants from Sanofi and AstraZeneca (unrelated to manuscript) and consulting fees from Allergan and Curasen (unrelated to manuscript). D.E. serves on the scientific advisory boards of and has received fees from The Michael J. Fox Foundation for Parkinson’s Research and Ovid Therapeutics (unrelated to manuscript); receives consulting fees from MeiraGTx (unrelated to manuscript); has received grants from NIH (NINDS, NIAID) (unrelated to manuscript); and is the coinventor of patents re: Markers for use in screening patients for nervous system dysfunction and a method and apparatus for using same, without financial gain. All other authors disclose no relevant conflict of interest.
Footnotes
Ethics approval
Ethical permission for this study was obtained from the Institutional Review Board of the participating institutions (Columbia University and Northwell Health).
Consent to participate
A waiver of consent was granted for this study.
Availability of data and material
Deidentified data will be made available on reasonable request from interested investigators for the purpose of replicating results.
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of a an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
References
- 1.Rizzo G, Copetti M, Arcuti S, Martino D, Fontana A, Logroscino G. Accuracy of clinical diagnosis of Parkinson disease. Neurology. 2016;86:566–76. [DOI] [PubMed] [Google Scholar]
- 2.Fahn S, Jankovic J. Principles and Practice of Movement Disorders. Philadelphia, PA: Churchill Livingstone Elsevier; 2007. [Google Scholar]
- 3.Krismer F, Wenning GK. Multiple system atrophy: insights into a rare and debilitating movement disorder. Nat Rev Neurol. 2017;13:232–43. [DOI] [PubMed] [Google Scholar]
- 4.Obeso JA, Stamelou M, Goetz CG, Poewe W, Lang AE, Weintraub D, et al. Past, present, and future of Parkinson’s disease: A special essay on the 200th Anniversary of the Shaking Palsy. Mov Disord. 2017;32:1264–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rus T, Tomše P, Jensterle L, Ležaić L, Stokin CL, Popović M, et al. Atypical clinical presentation of pathologically proven Parkinson’s disease: The role of Parkinson’s disease related metabolic pattern. Park Relat Disord. 2020;78:1–3. [DOI] [PubMed] [Google Scholar]
- 6.Adler CH, Beach TG, Hentz JG, Shill HA, Caviness JN, Driver-Dunckley E, et al. Low clinical diagnostic accuracy of early vs advanced Parkinson disease: Clinicopathologic study. Neurology. 2014;83:406–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meyer PT, Frings L, Rücker G, Hellwig S. 18F-FDG PET in Parkinsonism: Differential diagnosis and evaluation of cognitive impairment. J Nucl Med. 2017;58:1888–98. [DOI] [PubMed] [Google Scholar]
- 8.Meles SK, Teune LK, de Jong BM, Dierckx RA, Leenders KL. Metabolic Imaging in Parkinson Disease. J Nucl Med. 2017;58:23–8. [DOI] [PubMed] [Google Scholar]
- 9.Strafella AP, Bohnen NI, Perlmutter JS, Eidelberg D, Pavese N, Van Eimeren T, et al. Molecular imaging to track Parkinson’s disease and atypical parkinsonisms: New imaging frontiers. Mov Disord. 2017;32:181–92. [DOI] [PubMed] [Google Scholar]
- 10.Caminiti SP, Alongi P, Majno L, Volontè MA, Cerami C, Gianolli L, et al. Evaluation of an optimized [18F]fluoro-deoxy-glucose positron emission tomography voxel-wise method to early support differential diagnosis in atypical Parkinsonian disorders. Eur J Neurol. 2017;24:687–e26. [DOI] [PubMed] [Google Scholar]
- 11.Antonini A, Kazumata K, Feigin A, Mandel F, Dhawan V, Margouleff C, et al. Differential Diagnosis of Parkinsonism With [ 18F ] Fluorodeoxyglucose and PET. 1998;13:268–74. [DOI] [PubMed] [Google Scholar]
- 12.Tang CC, Poston KL, Eckert T, Feigin A, Frucht S, Gudesblatt M, et al. Differential diagnosis of parkinsonism: a metabolic imaging study using pattern analysis. Lancet Neurol. 2010;9:149–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schindlbeck KA, Eidelberg D. Network imaging biomarkers: insights and clinical applications in Parkinson’s disease. Lancet Neurol. 2018;17:629–40. [DOI] [PubMed] [Google Scholar]
- 14.Tripathi M, Tang CC, Feigin A, De Lucia I, Nazem A, Dhawan V, et al. Automated Differential Diagnosis of Early Parkinsonism Using Metabolic Brain Networks: A Validation Study. J Nucl Med. 2016;57:60–6. [DOI] [PubMed] [Google Scholar]
- 15.Rus T, Tomše P, Jensterle L, Grmek M, Pirtošek Z, Eidelberg D, et al. Differential diagnosis of parkinsonian syndromes: a comparison of clinical and automated - metabolic brain patterns’ based approach. Eur J Nucl Med Mol Imaging. 2020;47:2901–10. [DOI] [PubMed] [Google Scholar]
- 16.Markaki I, Tang CC, Lilja Lindström M, Eidelberg D, Svenningsson P, Savitcheva I. Automated Metabolic Pattern Analysis in the Differential Diagnosis of Parkinsonism in a Swedish Cohort [abstract]. Mov Disord. 2019;34. [Google Scholar]
- 17.Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry. 1992;55:181–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gilman S, Wenning GK, Low PA, Brooks DJ, Mathias CJ, Trojanowski JQ, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology. 2008;71:670–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hauw JJ, Daniel SE, Dickson D, Horoupian DS, Jellinger K, Lantos PL, et al. Preliminary NINDS neuropathologic criteria for Steele-Richardson-Olszewski syndrome (progressive supranuclear palsy). Neurology. 1994;44:2015–9. [DOI] [PubMed] [Google Scholar]
- 20.Ma Y, Tang C, Spetsieris PG, Dhawan V, Eidelberg D. Abnormal metabolic network activity in Parkinson’s disease: test-retest reproducibility. J Cereb Blood Flow Metab. 2007;27:597–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Spetsieris PG, Eidelberg D. Scaled subprofile modeling of resting state imaging data in Parkinson’s disease: Methodological issues. Neuroimage. 2011;54:2899–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Papp MI, Lantos PL. The distribution of oligodendroglial inclusions in multiple system atrophy and its relevance to clinical symptomatology. Brain. 1994;117:235–43. [DOI] [PubMed] [Google Scholar]
- 23.Litvan I, Hauw JJ, Bartko JJ, Lantos PL, Daniel SE, Horoupian DS, et al. Validity and reliability of the preliminary NINDS neuropathologic criteria for progressive supranuclear palsy and related disorders. J Neuropathol Exp Neurol. 1996;55:97–105. [DOI] [PubMed] [Google Scholar]
- 24.Dickson DW, Lin WL, Liu WK, Yen SH. Multiple system atrophy: A sporadic synucleinopathy. Brain Pathol. 1999;9:721–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vonsattel JPG, del Amaya MP, Keller CE. Twenty-first century brain banking. Processing brains for research: The Columbia University methods. Acta Neuropathol. 2008;115:509–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hughes AJ, Daniel SE, Ben-Shlomo Y, Lees AJ. The accuracy of diagnosis of parkinsonian syndromes in a specialist movement disorder service. Brain. 2002;125:861–70. [DOI] [PubMed] [Google Scholar]
- 27.Poston KL, Tang CC, Eckert T, Dhawan V, Frucht S, Vonsattel JP, et al. Network correlates of disease severity in multiple system atrophy. Neurology. 2012;78:1237–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tripathi M, Tang CC, Feigin A, De Lucia I, Nazem A, Dhawan V, et al. Automated Differential Diagnosis of Early Parkinsonism Using Metabolic Brain Networks: A Validation Study. J Nucl Med. 2016;57:60–6. [DOI] [PubMed] [Google Scholar]
- 29.Habeck C, Foster NL, Perneczky R, Kurz A, Alexopoulos P, Koeppe RA, et al. Multivariate and univariate neuroimaging biomarkers of Alzheimer’s disease. Neuroimage. 2008;40:1503–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Teune LK, Renken RJ, Mudali D, De Jong BM, Dierckx RA, Roerdink JBTM, et al. Validation of parkinsonian disease-related metabolic brain patterns. Mov Disord. 2013;28:547–51. [DOI] [PubMed] [Google Scholar]
- 31.Mattis PJ, Niethammer M, Sako W, Tang CC, Nazem A, Gordon ML, et al. Distinct brain networks underlie cognitive dysfunction in Parkinson and Alzheimer diseases. Neurology. 2016;87:1925–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jecmenica Lukic M, Kurz C, Respondek G, Grau-Rivera O, Compta Y, Gelpi E, et al. Copathology in Progressive Supranuclear Palsy: Does It Matter? Mov Disord. 2020;35:984–93. [DOI] [PubMed] [Google Scholar]
- 33.Giagkou N, Stamelou M. Emerging drugs for progressive supranuclear palsy. Expert Opin Emerg Drugs. 2019;24:83–92. [DOI] [PubMed] [Google Scholar]
- 34.Woo C-W, Chang LJ, Lindquist MA, Wager TD. Building better biomarkers: brain models in translational neuroimaging. Nat Neurosci. 2017;20:365–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Niethammer M, Tang CC, Vo A, Nguyen N, Spetsieris P, Dhawan V, et al. Gene therapy reduces Parkinson’s disease symptoms by reorganizing functional brain connectivity. Sci Transl Med. United States; 2018;10:aau0713. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.