

Abstract
Exposure to environmental toxicants is prevalent, hazardous and linked to varied detrimental health outcomes and disease. Polychlorinated biphenyls (PCBs), a class of hazardous organic chlorines once widely used for industrial purposes, are associated with neurodegenerative disease and oxidative stress in both in vitro and in vivo models. Here, we investigated the impact of Aroclor 1254, a commercially available PCB mixture, on primary murine astrocytes to determine the response to this once ubiquitously used toxicant on the most numerous cells of the central nervous system (CNS). Astrocytes are a critical component of homeostasis throughout the CNS, including at the blood-brain barrier, where they serve as the primary defense against xenobiotics entering the CNS, and at the synapse, where they are closely coupled to neurons through several metabolic pathways. We hypothesized that PCBs cause astrocytic oxidative stress and related dysfunction including altered metabolism. We exposed primary murine cortical astrocytes to PCBs and report an increased expression of antioxidant genes (Prdx1, Gsta2, Gfap, Amigo2) in response to oxidative stress. Our data show increased ATP production and spare respiratory capacity in astrocytes exposed to 10 μM (~ 3 ppm) of PCBs. This dose also causes an increase in glucose uptake that is not seen at a higher dose (50 μM) suggesting that, at a lower dose, astrocytes are able to engage compensatory mechanisms to promote survival. Together, these data suggest that exposure to polychlorinated biphenyls impact astrocytic metabolism, which is important to consider both in the context of human health and disease and in cell culture and animal models.
Keywords: polychlorinated biphenyls, seahorse, environmental toxicant, glia, mitochondria, reactive oxygen species, homeostasis, brain, Parkinson’s disease, neurodegeneration, antioxidant
INTRODUCTION
In recent years, our understanding of the central nervous system (CNS) has expanded to recognize the important role of glial cells. This significant shift away from a predominantly neuron-centric view of brain physiology, pathology, and implicated processes therein, has led to important discoveries in areas ranging from basic science to therapeutics (Blackburn et al. 2009; Liddelow and Barres 2017). Astrocytes, a type of glial (non-neuronal) cell, are the most numerous cell type in the CNS (Han et al. 2013) and are unique in their homeostatic functions (Brown and Ransom 2007; Souza et al. 2019). Many astrocytic functions underlie proper neuronal transmission and maintenance (Bélanger et al. 2011) and, while this aspect of astrocyte biology is the subject of increasingly more research, it is clear that these cells are important for brain function in ways that exceed direct astrocyte-neuron interactions (Allaman et al. 2011). The CNS is a notably complex organ system that requires its constitutive structural, cellular and molecular machinery to work both independently and as part of complex, multi-system networks (Maffei and Fontanini 2009; Kulkarni et al. 2018). In other words, proper communication between neurons and glia as well as glia-glia communication are critical for neuronal function, health, and wellbeing. This is particularly apparent when the brain is provoked, for example during exposure to endogenous and/or exogenous stressors.
Neuronal degeneration can result from perturbation of endogenous processes (proteostasis, for example) (Dasuri et al. 2013), exposure to exogenous compounds, or a combination of internal and external factors (Bossy-Wetzel et al. 2004; Fleming 2017). For example, oxidative stress is causatively linked to neurodegeneration as a result of both endogenous and exogenous stressors (Barnham et al. 2004; Migliore and Coppedè 2009). Environmental toxicants represent a large category of exogenous human stressors, ranging from acute to chronic, life-long exposure. Organochlorines are a specific class of environmental toxicants that are known to cause oxidative stress. These chemicals are ubiquitous and, due to their high lipophilicity, chemical stability and long half-lives, particularly potent environmental toxicants (Hawker and Connell 1988; Grandjean et al. 2008). Several members of this class including polychlorinated biphenyls (PCBs), are linked to neurological dysfunction wherein neuronal oxidative stress is implicated, such as in Parkinson’s disease (PD) (Hatcher-Martin et al. 2012; Bouchard et al. 2014). PCBs are also associated with neurodevelopmental deficits (reviewed in (Pessah et al. 2019)), neuroendocrine dysfunction (Gore 2001) as well as oxidative stress and cellular dysfunction in multiple animal models (Seegal et al. 1994; Hennig et al. 1999; Lyng et al. 2007).
PCBs cause direct neuronal oxidative damage (Lee and Opanashuk 2004) but the effects on glial cells remain understudied and are the focus of the work presented herein. In order to have a more complete understanding of the impact of compounds, including PCBs as a representative oxidative stress-inducing toxicant, on the brain in general and on neurons, it is important to understand the direct effect on astrocytes.
The physiological processes of astrocytes include, among others, various aspects of metabolism and antioxidant support. The metabolism of astrocytes plays a uniquely important role in meeting the high energy demands of the brain (Bélanger et al. 2011). Astrocytic end-feet abutting the cerebral vasculature enable astrocytes to quickly take up glucose, which is then metabolized through glycolysis, the tricarboxylic acid (TCA) cycle, and oxidative phosphorylation. Astrocytes have a higher glycolytic rate than other cells, suggesting that increased glucose uptake is a mechanism by which astrocytes can maintain this metabolic profile (Itoh et al. 2003; Herrero-Mendez et al. 2009; Bittner 2010). The importance of the metabolic coupling between astrocytes and neurons is manifest in the antioxidant support astrocytes provide neurons. This relationship and the ways in which it can be perturbed is critical to our understanding of neurodegeneration and damage.
The reactivity state of astrocytes in response to injury and in neurodegenerative disease can exacerbate inflammation and neuronal dysfunction. Therefore, it is not surprising that astrocytes play an important role in PD pathogenesis (reviewed in (Booth et al. 2017)). Recent work indicates that astrocytes may have multiple functions in PD including acting as antigen presenting cells (Rostami et al. 2020) and as non-cell autonomous mediators of neurodegeneration (di Domenico et al. 2019). Furthermore, in post-mortem studies of brains from PD patients there is growing evidence that the pathogenic protein, α-synuclein accumulates in astrocytes (Wakabayashi et al. 2000; Braak et al. 2007). In PD, where the dopaminergic neurons of the substantia nigra degenerate, many of the genes associated with monogenic cases of PD are expressed by astrocytes, for example, PARK7 (DJ-1) which is associated with oxidative stress and mitochondrial function in astrocytes (Zhang et al. 2016). Interestingly, in addition to region-specific transcriptional profiles, the substantia nigra is known to contain fewer astrocytes than the ventral tegmental area, where dopaminergic cell bodies involved primarily in mesocorticolimbic systems reside (Damier et al. 1993; Nair-Roberts et al. 2008; Kostuk et al. 2019). The reactive astrocyte phenotype described by Liddelow et al (2017) and discussed above is also relevant to PD. In a recent study, blocking the ability of microglia to activate astrocytes to the A1 phenotype reduced behavioral deficits and the loss of dopamine neurons in a mouse model of sporadic PD (Yun et al. 2018). Characterization of the specific phenotype of astrocytes during PD pathogenesis is still in its infancy but the role of these cells can perhaps be best understood by a careful examination of their response(s) to specific perturbations.
Here, we utilize gene expression, oxidative stress, glucose uptake, and metabolism assays to interrogate the effects of PCBs on primary murine astrocytes to determine how this cell type, with specific roles in homeostasis, responds to this environmental toxicant as a representative oxidative stressor. We demonstrate that PCBs directly: increase astrocyte oxidative stress, promote the mRNA expression of a subset of antioxidant response element regulated genes, alter mitochondrial metabolism, and affect glucose uptake. Our work highlights the importance of understanding astrocytic responses to stressors in the maintenance of brain health.
RESULTS
Polychlorinated biphenyls induce oxidative stress in astrocytes
PCBs cause oxidative stress in several disease models and immortalized cell-lines (Hennig et al. 2002; Lyng and Seegal 2008; Zhu et al. 2009) including a dopaminergic neuronal-like cell line (MN9Ds; Lee and Opanashuk 2004; Lee et al. 2006) and cerebellar granule cells (Mariussen et al. 2002). The doses of PCBs (administered in the form of mixtures [Aroclors, for example] and individual congeners) used in both in vitro and in vivo studies have varied greatly (Ulbrich and Stahlmann 2004; Klocke and Lein 2020) making a thorough assessment of the toxicity and implicated pathways challenging. For example, stress and depletion of glutathione reserves was observed at 10 ppm Aroclor 1254 in MN9Ds (Lee and Opanashuk 2004) while cellular viability was significantly decreased in a neuroblastoma cells (SH-SY5Y) after exposure to 15.2 ppm (Cocco et al. 2015).
Taking into account the body of existing in vitro data, here we exposed primary murine astrocytes to Aroclor 1254 (1.25–50 μM) for 24h, which is equivalent to 0.4 – 16 ppm. Following Aroclor 1254-exposure, total reactive oxygen species were quantified using 2’−7’-dichlorodihydrofluoresceine diacetate (H2DCFDA), a nonfluorescent compound that is converted to a highly fluorescent compound, dichlorofluorescein (DCF), in the presence of reactive oxygen species (Fig. 1a). The PCB-exposed cells had a significantly (p < 0.05) higher level of fluorescence (10 μM: 1.75 × 106 +/− 2.48 × 104 [SEM]; 20 μM: 1.78 × 106 +/− 3.059 × 104 [SEM]; 50 μM: 1.85 × 106 +/− 4.5 × 104 [SEM]) compared to the vehicle astrocytes (1.5 × 106 +/− 1.68 × 104 [SEM]) demonstrating that exposure to a biologically relevant dose of Aroclor 1254 (10 μM or 3 ppm) results in significant oxidative stress in astrocytes.
Fig. 1. Polychlorinated biphenyls induce oxidative stress in astrocytes without impacting cellular viability.
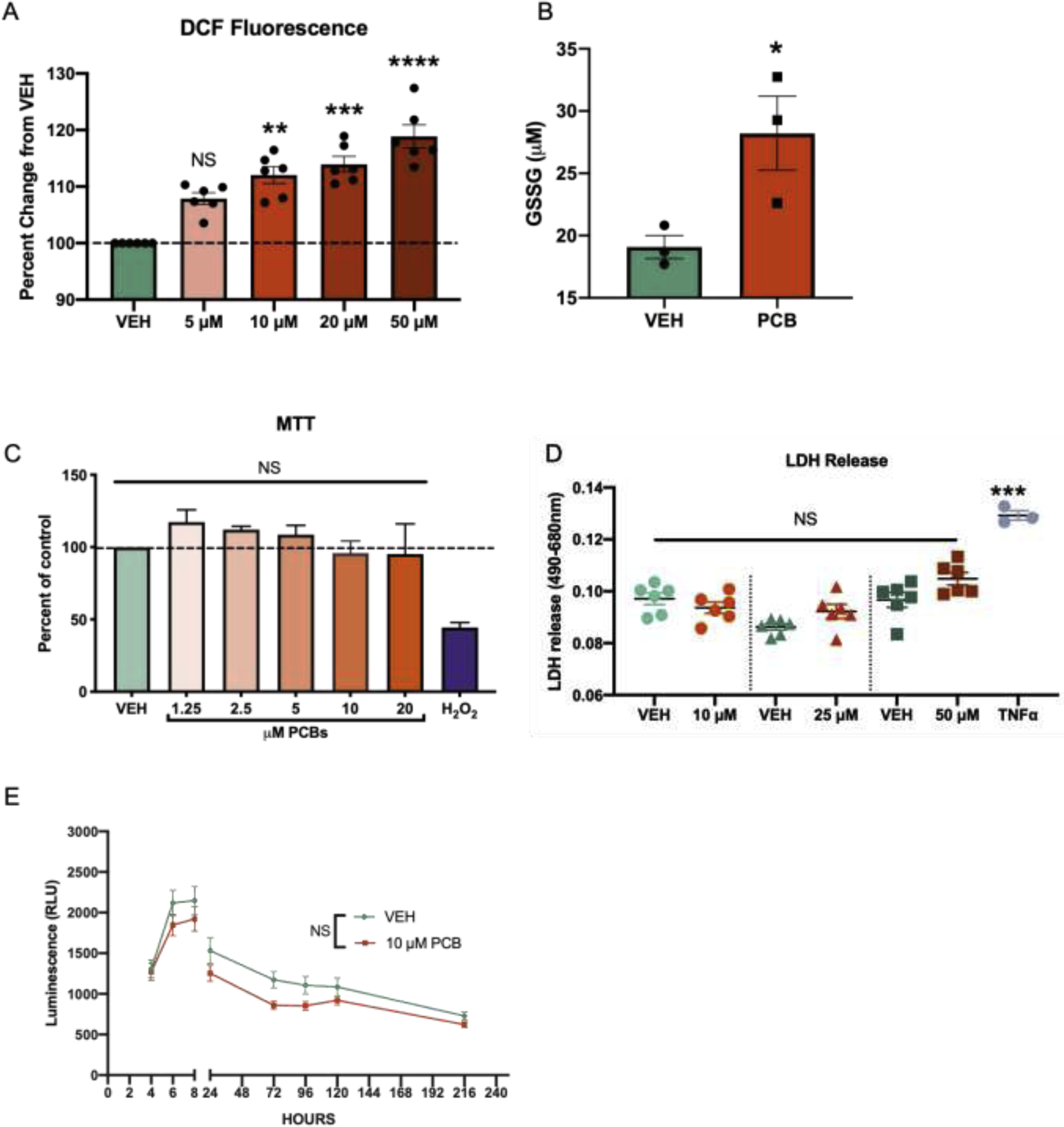
A. DCF quantification of vehicle and PCB-exposed astrocytes (24 h), n = 6, Data analyzed via one-way ANOVA**p = 0.0015, ***p = 0.0002, ****p <0.0001. B. GSSG quantification of vehicle and PCB-exposed astrocyte lysates (24 h), n = 3, p = 0.04. C. Formazan quantification with an MTT assay of PCB or methanol-exposed astrocytes (24 h) and hydrogen peroxide (100 μM) positive control, n = 6. Methanol concentration is matched v/v to highest 50 μM PCB dose. D. LDH release (24 h) including TNF-α positive control, data analyzed via one-way ANOVA ***p = 0.0015. E. RealTime-MT glo assay quantifying cellular viability over a 216 h period. Data analyzed by two-way ANOVA (time × treatment) and Šidák’s multiple comparisons test; 4-hour (p > 0.99), 6 hour (p = 0.86), 8 hour (p = 0.9), 24 hour (p = 0.78), 72 hour (p = 0.169), 96 hour (p = 0.45), 120 hour (p = 0.85) and 216 hour (p = 0.6). All data represented as mean ± SEM.
Given the increased oxidative stress in astrocytes observed with exposure to PCBs, glutathione, a tripeptide antioxidant abundant in astrocytes (Raps et al. 1989; Dringen and Hirrlinger 2003) was analyzed. In the presence of reactive oxygen species, reserves of reduced glutathione (GSH) are oxidized to form oxidized glutathione (GSSG). As such, GSSG was quantified to understand the engagement of this important cellular defense pathway. Primary murine astrocytes were exposed to PCBs (10 μM, 24 h) as described above and a GSSG detection assay was used to quantify the concentration of oxidized glutathione in PCB and vehicle-exposed astrocytes. Primary astrocytes exposed to PCBs had a statistically significant (p = 0.04) increase in the concentration of GSSG compared to vehicle-exposed cells (Fig. 1b). These results suggest that a depletion of GSH reserves may be an important avenue of response in PCB-induced toxicity and confirms that PCBs cause an increase in oxidative stress in astrocytes.
In order to determine whether cell death is increased with PCB exposure, a cell viability, MTT, assay was performed on a range of doses (1.25 – 20 μM). Hydrogen peroxide (H2O2) was included as a positive control for decreased oxidative stress-induced viability (Whittemore et al. 1995). Primary murine astrocytes were exposed to vehicle, 1.25, 2.5, 5, 10 or 20 μM of PCBs for 24 hours before the cell viability assay was performed. At doses ≤ 20 μM, there was no significant change in cellular viability (Fig. 1c). This finding was confirmed using a complementary cytotoxicity assay which quantifies lactate dehydrogenase (LDH) release following 24 hours with 10, 25 and 50 μM PCB treatment (Fig. 1d). Lactate dehydrogenase (LDH) is a cytoplasmic enzyme that is found in all cells. LDH is released into the cellular media when the plasma membrane is damaged, as is the case in necrotic and apoptotic cell death (Kumar et al. 2018). Tumor necrosis factor alpha (TNFα) was included as a positive control verifying both that the primary astrocytes are able to release LDH and that the released LDH was detectable (Park and Bowers 2010; Chan et al. 2013; Olmos and Lladó 2014). Together these data suggest that astrocytes exposed to 10 μM PCBs undergo increased oxidative stress in the absence of reduced cellular viability.
Given the chronic and persistent nature of exposure to many environmental toxicants, including PCBs, cell viability was analyzed over a longer exposure time to PCBs at a range of doses. A non-lytic continuous read cell viability assay (RealTime-Glo™ MT Cell Viability assay, Promega #G9711) was used which allowed for longer PCB exposure times at various doses (2.5, 5,10, 20 or 50 μM PCBs) for up to 216 hours. There were no significant differences between vehicle and PCB concentration at any time point (data not shown for 2.5, 5, 20 and 50 μM exposure). Comparison of the 10 μM PCB-exposed astrocytes and vehicle revealed no significant difference in cell viability at any time point throughout the assay (Fig. 1e). This data indicates that PCB-exposed astrocytes have an increased burden of ROS at 24 h but remain viable, suggesting that they are engaging cellular pathways to mitigate damage.
Upregulation of antioxidant genes in response to PCB exposure
In order to better understand which cellular pathways might be engaged in response to PCBs, and because PCBs and other organochlorines are associated with Parkinson’s disease (supplemental Fig. 2), we performed an in silico screen using the Comparative Toxicogenomics Database (CTD, data queried on Feb 17th, 2019) (Davis et al. 2021) (Fig. 2a). Three parameters were used to determine genes of interest. In order to determine functional relevance for the central nervous system, we defined one of the three parameters as genes linked to Parkinson’s disease (PD) (Hatcher-Martin et al. 2012). The second parameter was genes associated with PCB exposure. The third parameter, genes associated with antioxidant activity, was based on our findings outlined in Fig. 1 since PCB exposure caused oxidative stress in astrocytes. These in silico analyses identified 54 overlapping genes (supplementary Fig. 3). Genes of interest were identified by taking tissue and cellular expression as well as isoform redundancy into account.
Fig. 2. Upregulation of antioxidant genes in response to PCB exposure.
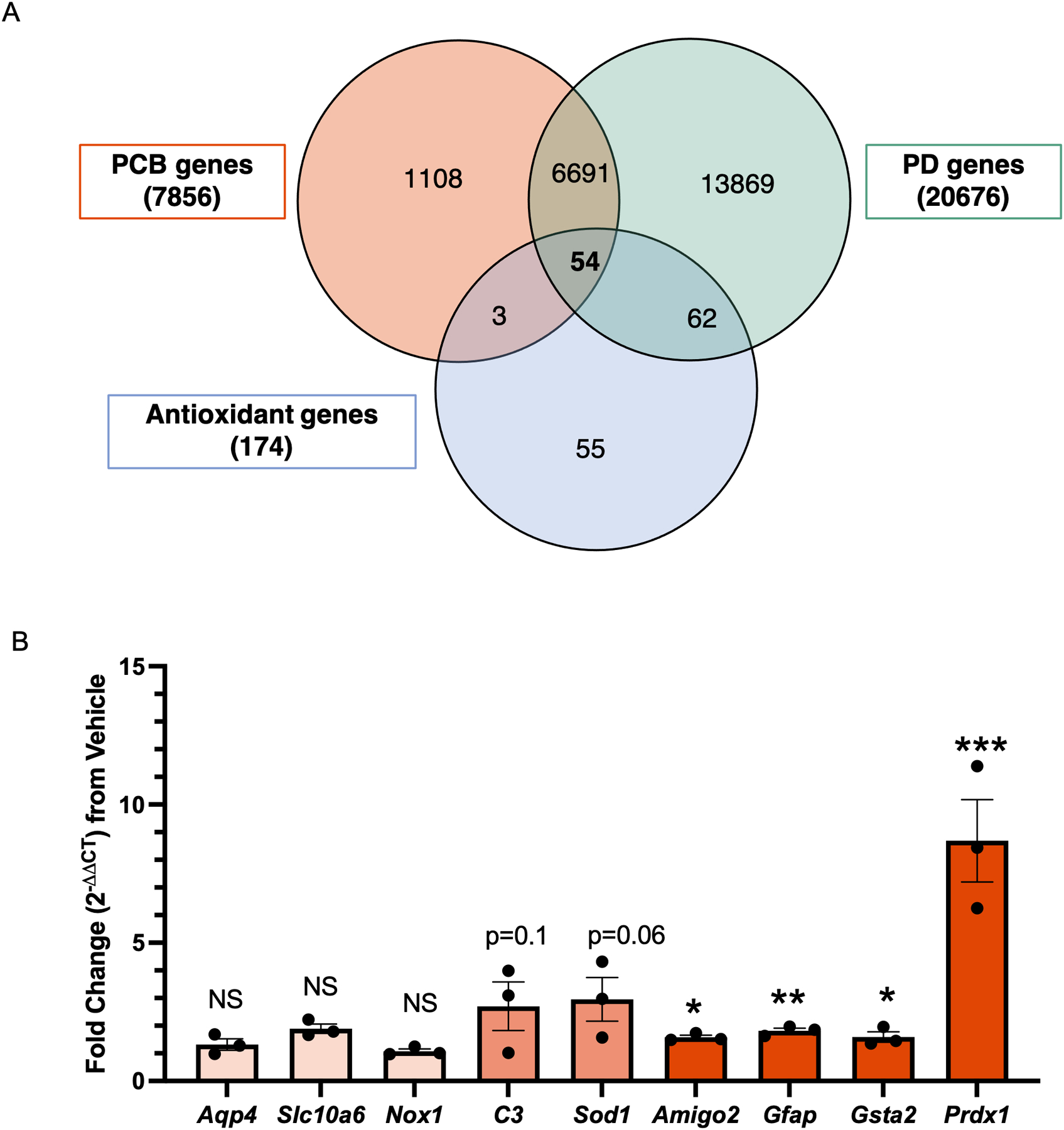
A. Comparative toxicogenomics database screen of antioxidant genes with published associations to both PCBs and Parkinson’s disease. B. qRT-PCR (mRNA), fold change of 10 μM PCB exposed astrocytes compared to vehicle (methanol) exposure. Data analyzed via Student’s t-test, represented as mean ± SEM with 3 biological replicates and was replicated in 2 to 3 independent experiments, *p = 0.02, **p = 0.008, ***p = 0.0003.
Three genes were selected (Gsta2, Prdx1 and Sod1) from this screen and we added an additional six genes that are associated with astrocyte reactivity (Aqp4, Nox1, Slc10a6, C3, Amigo2, Gfap) (Liddelow et al. 2017). All nine genes were examined using quantitative real-time reverse transcription PCR (qRT-PCR). PCB exposure of astrocytes resulted in a significant increase in antioxidant genes including glutathione-s-transferase 2 (Gsta2, 1.6 fold increase), and peroxiredoxin 1 (Prdx1, 8.6 fold increase). Of the genes relating to astrocyte reactivity, Gfap (p = 0.003, 1.8 fold increase) and Amigo2 (p = 0.02, 1.5 fold increase) showed small but significant increases following exposure to PCBs; C3 (p = 0.1, 2.7 fold increase) and Sod1 (p = 0.06, 2.9 fold increase) were trending towards increased but not statistically significant. There was no significant change in the antioxidant gene NADPH oxidase 1 (Nox1), or the astrocyte reactivity related genes, aquaporin 4 (Aqp4) and solute carrier family 10 member 6 (Slc10a6) (Fig. 2b).
Metabolic changes with PCB exposure
Given that the oxidative state of astrocytes can impact their metabolic function and homeostatic processes (reviewed in (McCann and Maguire-Zeiss 2021)), we next investigated the metabolic profile of PCB-exposed primary murine astrocytes using the Seahorse Extracellular Flux analyzer. We examined mitochondrial oxygen consumption rate (OCR) in 10 μM PCB-exposed astrocytes and found no significant differences in basal respiration, proton leak, or maximal respiration (Fig. 3a–c). Interestingly, there were significant increases in the ATP production in the 10 μM PCB-exposed astrocytes (Fig. 3d). We tested a higher dose (50 μM) and found no significant differences in OCR, basal and maximal respiration or ATP production (Fig. 3e–h). There were no significant differences in proton leak with either treatment (Fig. 3i), however there was a significant increase in the spare respiratory capacity of the 10 μM PCB-exposed astrocytes (Fig. 3j). This data suggests that at the lower (10 μM) dose, the astrocytes are engaging mechanisms to overcome metabolic dysfunction whereas this mechanism fails at the higher dose (50 μM).
Fig. 3. Metabolic changes with PCB exposure.
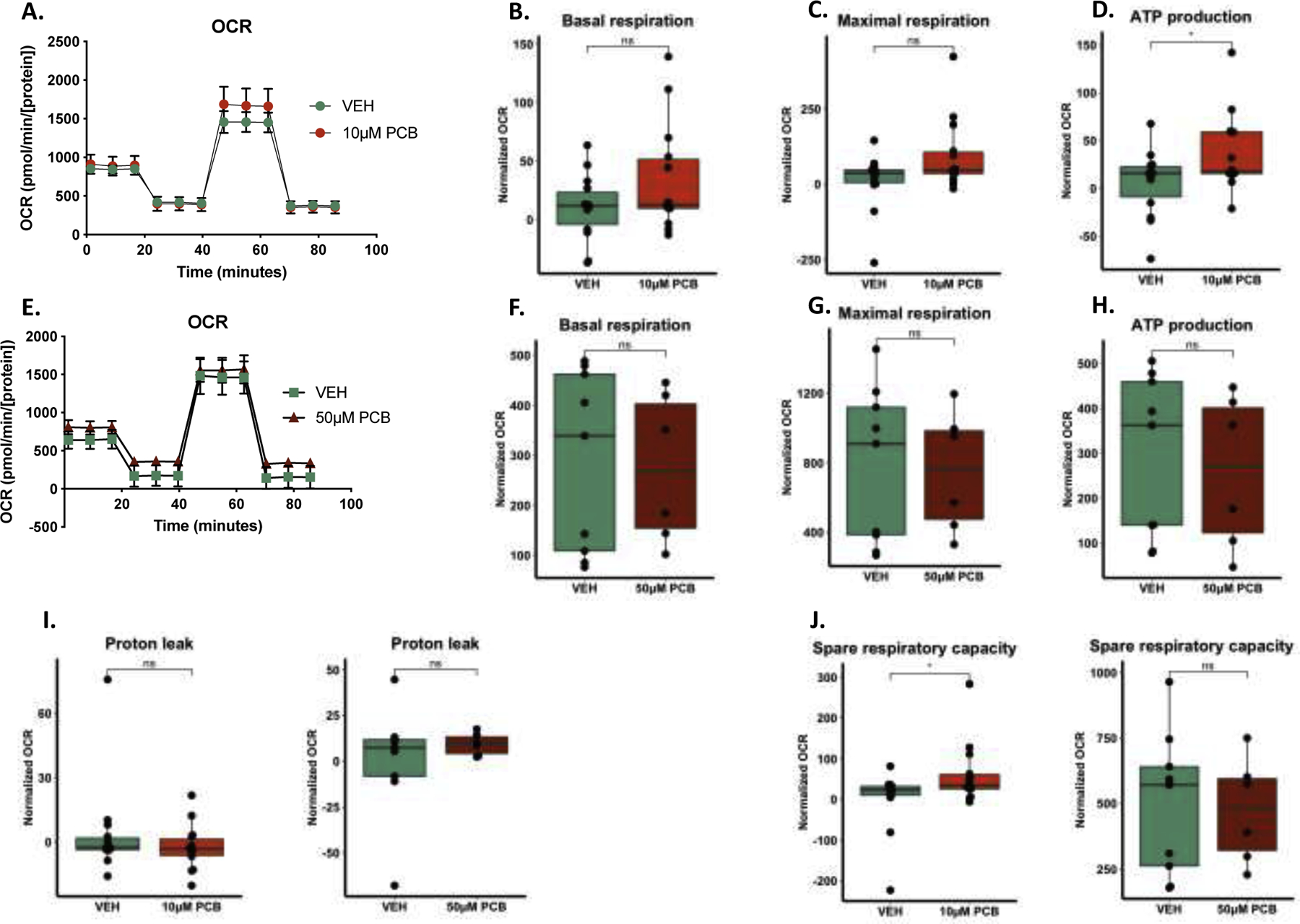
A-D. 10 μM PCB-exposed astrocytes have increased ATP production after 24 h compared to vehicle but no significant differences in basal or maximal respiration (data is normalized to cell density). E-H. 50 μM PCB exposure for 24 h does not result in significant differences in basal respiration, maximal respiration or ATP production. I. Proton leak is not impaired by either 10 or 50 μM PCB exposure. J. Spare respiratory capacity is increased in the 10 μM PCB exposed astrocytes but not the 50 μM condition. Data analyzed via generalized linear model, data represented as mean ± SEM with 3–5 biological replicates from three independent experiments.
Increased glucose uptake with low dose exposure to PCBs
Throughout the CNS, astrocytes play important roles at the synapses, where they partake in an intensive metabolic exchange with neurons that is essential for neuronal survival and at the blood-brain barrier where astrocytic endfeet ensheathe the vasculature and express GLUT1, a glucose transporter23. The metabolic processes that regulate glucose levels are critical for brain function, including synaptic transmission and global homeostasis (Allaman et al. 2011; Han et al. 2013; Chai et al. 2017; Verkhratsky et al. 2019). The ability of astrocytes to serve as sensors of metabolic changes in the microenvironment places these cells in the unique position of regulating glucose dynamics within the CNS and from the periphery into the brain (García-Cáceres et al. 2016). In order to determine whether astrocyte function is affected by PCB exposure we examined glucose uptake in astrocytes. Interestingly, 10 μM PCB-exposed astrocytes demonstrate increased glucose uptake compared with vehicle-exposed cells (Fig. 4a) whereas 50 μM PCB-exposed astrocytes do not (Fig. 4b), suggesting that in response to low-dose (10 μM) PCB exposure, astrocytes are responding to metabolic demand.
Fig. 4. Increased glucose uptake with low dose exposure to PCBs.
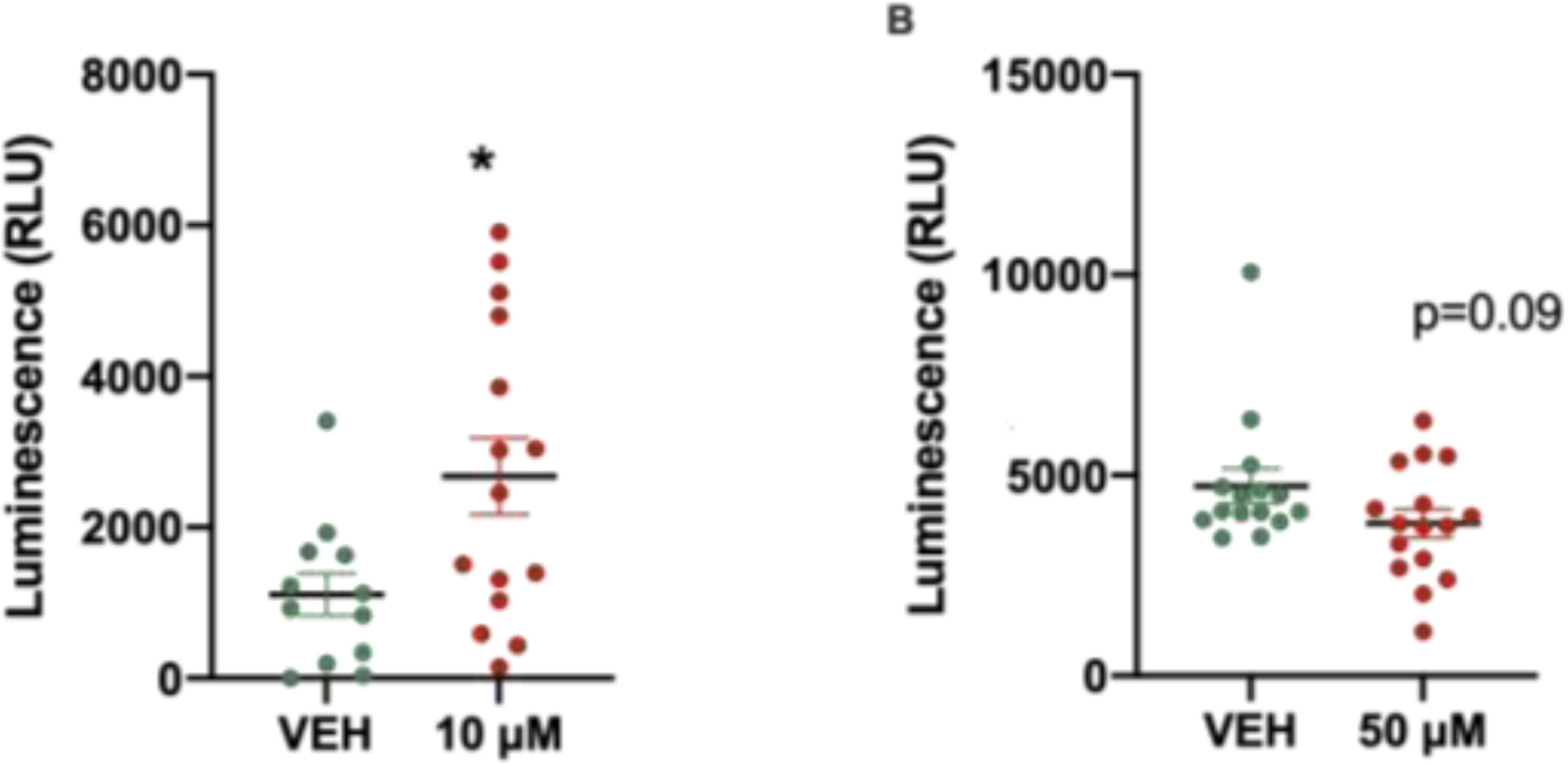
A. 10 μM PCB exposure results in increased astrocyte glucose uptake compared to vehicle. B. 50 μM exposure of astrocytes does not significantly alter glucose uptake. Data represented as mean ± SEM with 3 – 6 biological replicates from 3 independent experiments, unpaired two-tailed t-test, *p = 0.01.
DISCUSSION
Together, the work presented here shows that astrocytes exposed to PCBs are subjected to increased oxidative stress, display an antioxidant response, and are able to increase glucose uptake and ATP production. Glucose, the main substrate for energy in the brain, is metabolized by three successive processes: glycolysis, the tricarboxylic acid (TCA) cycle, and oxidative phosphorylation. This final process, oxidative phosphorylation, is the largest driver of cellular ATP production. Our data suggests that at a 10 μM exposure to PCBs, astrocytes are able to increase glucose uptake and drive ATP production to compensate for increases in reactive oxygen species burden. This mechanism is not engaged in the higher dose (50 μM) exposure, where the astrocytes do not increase glucose uptake and are unable to utilize increased energy expenditure.
This suggests that the mechanisms engaged at the lower dose are less effective at a higher dose. As indicated by increased spare respiratory capacity (Fig 3j), it is possible that the 10 μM-PCB exposed astrocytes are adapting to utilize increased oxidative phosphorylation in an attempt to meet energy and antioxidant demands. Extending these studies to determine the impact of both low and high PCB exposure on glycolysis as well as a more detailed examination of GLUT expression and function is an important future direction. Furthermore, a comparison of the expression and function of both high- and low-affinity glucose transporters in response to PCB exposure represents a next step to address the mechanism by which this shift occurs (Jouroukhin et al. 2018; Koepsell 2020). Importantly, glutathione detoxification is an ATP-consuming process and as such, it is possible that an increase in ATP is required to drive production of glutathione, the synthesis of which is increased in response to oxidative stress (Dringen et al. 2015). Addressing these mechanisms will deepen our understanding of astrocyte responses to toxicants in order to determine more completely how exposure to this toxicant alters the metabolic profile of astrocytes.
Our findings reported here are concordant with the hormesis principle of toxicology wherein stimulation is observed at low doses of exposure to chemical agents that cause toxicity at high doses, resulting in a biphasic response (Tapia 2006; Calabrese et al. 2007; Mattson 2008). Hormesis, which is related to terms such as “adaptive response” or “preconditioning” that also describe nonlinear responses, is increasingly shown to be an important consideration in the evaluation of dose-response models in biological systems (Calabrese et al. 1999, 2007; Calabrese and Baldwin 2003). While this concept as it pertains to public health decisions is controversial, and has a great potential for adverse health outcomes if applied in a regulatory context, it is clear that “nonmonotonic” dose responses are important to consider in the context of basic science and interpretation of data with an emphasis on understanding the biological machinery underpinning these responses (Thayer et al. 2005).
With regards to PCBs, hermetic effects have been shown in several models including human lung fibroblasts (Hashmi et al. 2015) and in iron sequestration in the striatum of PCB-exposed mice (Lee et al. 2012). When considering the impact of PCBs on primary neuronal cultures, where decreased cellular viability is shown to occur after a 4 h exposure to PCBs at concentrations between 30 and 100 μM in rat cortical cultures (Sánchez-Alonso et al. 2004) and there is evidence of differential outcomes between hippocampal and cortical neurons (Howard et al. 2003), it is critical to consider the impact of the attendant astrocytes in these culture systems. At higher doses (>10 μM, as we show here), it is possible that astrocytes fail to engage compensatory mechanisms to promote their own survival in addition to that of neurons. As it becomes increasingly evident that astrocytic phenotypes differ with anatomical regions, this heterogeneity may contribute to an explanation of differing neuronal vulnerabilities (Matyash and Kettenmann 2010; Xin and Bonci 2018; Matias et al. 2019).
Although much of the work regarding toxicants in the brain has focused on neurons, here it is clear that astrocytes are an important component of the brain’s response to this xenobiotic compound. Since neurons are often co-cultured with astrocytes in vitro, our work supports the need to understand individual cell-type responses (neurons and glia separately) before interpreting the neuronal-glia combined responses to environmental toxicants. This is important because astrocytic metabolism and redox status are intricately tied to that of neurons, and the brain in general. As such, this research suggests that the ability of PCBs to impair astrocyte function will have long-term, detrimental consequences for the brain.
PCBs are persistent environmental pollutants and, despite their production being banned in most countries by 1985, are continually linked to novel instances of disease and environmental disasters. These compounds are part of a larger class of organochlorines, including compounds like trichloroethylene, which is still used in dry cleaning, therefore understanding the impact that PCBs have on astrocytes can inform our understanding of other compounds. Furthermore, the predicted lipid-normalized concentration of PCB-153 (2,2′,4,4′,5,5′-hexachloro-biphenyl) is approximately 50 ng/g lipid (Quinn et al. 2011), and up to approximately 10 ng/g lipid in the brain (Mitchell 2013) and although there are many factors influencing an individuals’ life time exposure to PCBs (including geography, occupation, diet), the impact of these compounds on human health is significant. For neurodegenerative diseases, like Parkinson’s disease, where sporadic cases far outnumber familial cases, it is possible that chronic exposure to hazardous compounds over the course of an individual’s lifetime coincide with age-related vulnerabilities and cellular dysfunction resulting in disease. Importantly, the prevalence of neurodegenerative diseases is increasing (Brookmeyer et al. 2011; Kowal et al. 2013; Hebert et al. 2013) and, as individuals who were exposed to PCBs prior to regulation continue aging, this represents an impactful avenue of research.
From both experimental and epidemiological models of disease, there is compelling evidence that exposure to organochlorine environmental toxicants is associated with oxidative stress and the development of PD (Seegal et al. 1994; Lee and Opanashuk 2004; Richardson and Miller 2004; Hatcher-Martin et al. 2012). There is also evidence of increased susceptibility of dopaminergic neurons to oxidative stress a result of metabolites produced during dopamine synthesis, suggesting that oxidative stress and related cellular sequelae are important for both the development and progression of PD (Graham 1978; Jenner 2003). Importantly, as the antioxidant functions of astrocytes are critical for mounting a productive antioxidant response, so too is astrocytic oxidative stress important for the propagation of oxidative injury and PD pathogenesis (Rizor et al. 2019). In addition, in vitro studies using the C6 glioma cell line, demonstrate that PCBs disrupt the protein kinase C pathway and increases expression of neurotrophic factors, suggesting a role for astrocytes in multiple mechanisms associated with PCB exposure (Gurley et al. 2007; Adornetto et al. 2013). As astrocytic functions and processes are increasingly associated with various aspects of PD, including neuroinflammation and metabolic dysfunction, uncovering the role of these cells in disease pathogenesis will aid in our comprehensive understanding of this disorder. Furthermore, as environmental toxicants like PCBs impact astrocytic processes, considering the nexus of PCB exposure and astrocytic processes implicated in PD, as mentioned above, can inform future research directions.
In this study, we investigated a range of doses (1.25–50 μM) and used 10 μM Aroclor 1254 for the majority of our studies, as it is the lowest dose where we detected reactive oxygen species. Additionally, we sought to use a paradigm that enabled us to study the cellular response in the absence of cell death. The data discussed here show that a significant increase in OCR at 10 μM is concomitant with increased glucose uptake, indicating an increase in metabolic activity that promotes cellular survival. The 10 μM PCB-exposed astrocytes also upregulated the expression of specific genes involved in cellular detoxification (Gsta-2, Prdx-1), suggesting that a subset of Nrf2-ARE gene pathways are involved in the cellular response to PCBs in astrocytes.
The glutathione system, which is particularly robust in astrocytes (Dringen and Hirrlinger 2003), is rapidly engaged in the service of xenobiotic conjugation and detoxification. Furthermore, oxidized glutathione (GSSG) can be utilized as a proxy for the degree of oxidative stress within a cell. Here, we showed that there was a significant increase in GSSG in PCB-exposed astrocytes, as well as an increase in GSTA-2 by qRT-PCR. As astrocytes provide neurons with essential precursors for neuronal glutathione synthesis (CysGly) (Dringen and Hirrlinger 2003), the PCB-induced depletion of reduced glutathione (GSH) suggests that astrocytic ability to properly detoxify this compound is impaired, yet the astrocytes are able to survive. It is important to point out that in this study, we did not examine whether long-term low dose exposure alters mitochondrial function and metabolism and is an important focus of future studies. We also have not yet examined the broader impact of PCBs on the intact brain. The field currently lacks consensus on the appropriate in vivo dose; that work is an important next step.
As the world’s population ages, we have yet to see the full effect of PCB-toxicity on humans. Approximately 43% of the world’s population was exposed to PCBs prior to the latest date of their ban (Council of the European Union 1996). The impact of this ubiquitous toxicant is likely to be profound, therefore, ongoing research focused on the intersection of neurodegeneration and environmental toxicant exposure should take into account the important role of glial cells.
METHODS AND MATERIALS
Animals
Adult C57/Bl6J mice were bred and housed in accordance with the Georgetown University Institutional Animal Care and Use Committee and the ethical guidelines of the National Institutes of Health. Animals were housed with two females and one male per cage with food and water ad libitum.
Aroclor 1254
Aroclor 1254 (11097-69-1, AccuStandard, #C-254S-M) was diluted from a stock concentration of 1000 μg/mL in methanol. For exposure, Aroclor 1254 was diluted in serum-free astrocyte medium to the appropriate concentration as noted. Methanol was used as the vehicle (see figure legends for details).
Primary astrocyte culture
Primary astrocytes derived from C57/Bl6J mice were prepared as previously described (Daniele 2014). In brief, brains were isolated from p0–p2 mice, meninges removed and cortices microdissected. Cortices were then minced in media (HBSS supplemented with 100 μg/mL P/S [penicillin/streptomycin]), allowed to settle, 2 mL of media removed, and resuspended in complete media (Minimum Essential Medium Earle’s [MEM] supplemented with 1 mM L-glutamine, 1 mM sodium pyruvate, 0.6% v/v D(+)-Glucose, 100 μg/mL P/S, 4% v/v fetal bovine serum [FBS], and 6% v/v horse serum), centrifuged, and resuspended again. The mixed glia were dissociated by gentle trituration and cultured in T-75 flasks in a humidified incubator (37 °C, 5% CO2). At 14–21 days in vitro (DIV), astrocytes were isolated following a shaking procedure: 4 h shake on a rotary shaker (200 RPM, 37 °C), media and non-adherent cells removed, adherent astrocytes washed twice with warmed PBS and isolated following incubation in 1X trypsin/EDTA at 37 °C. Astrocytes were subsequently passaged into a T-175 flask and cultured in astrocyte medium (MEM supplemented with 100 μg/mL P/S, 1 mM L-glutamine, and 10% FBS). Before plating for experiments, T-175 flasks were shaken overnight (200 RPM, 37 °C) to remove contaminating non-astrocytic cells, the media removed, cells washed twice with warmed PBS, astrocytes isolated by trypsin digestion and plated for experiments in astrocyte medium as described below. Astrocyte cultures were periodically verified as 90–95% pure using AQP4, GFAP and DAPI immunofluorescence.
Exposure to PCBs or vehicle
Astrocytes were exposed to PCBs in the form of Aroclor 1254 (supplier and stock information above) or vehicle (methanol). The concentrations and time of exposure is noted in the figure legends. The cells were plated for the format of the experiment (cell number and well-format noted in the respective sections) and, four days later, were washed with serum-free media (MEM supplemented with 100 μg/mL P/S, 1 mM L-glutamine) and then incubated with serum-free media for 1 h. Next, the serum-free media was removed, and the appropriate treatment solution was added to the well in the following volume: 3 mL for 6-well plate (qRT-PCR), 2 mL for 12-well plate (GSH/GSSG), 300 μL for 96-well plate (H2DCFDA, MTT, LDH and glucose uptake), and 100 μL for Seahorse 96-well plate.
Reactive oxygen species quantification
Total reactive oxygen species were quantified using 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA) (Thermo Fisher #D399). The compound was resuspended in 100% ethanol to a final stock concentration of 10 mM H2DCFDA. After exposure to Aroclor 1254 (PCB, concentration noted in figure legend) or methanol (VEH, matched v/v with 50 μM PCB dose) for 24 h, astrocytes (3 × 104 cells/well) were washed in serum-free astrocyte medium and incubated with H2DCFDA at 10 μM for 30 m in the dark and read on a fluorescent microplate reader (Molecular Devices SpectraMax iD3) at Ex/Em 490/530.
Glutathione measurement
Glutathione quantification was completed using a reduced glutathione (GSH) and oxidized glutathione (GSSG) ratio detection assay (Abcam, ab138881) per the protocol described by the manufacturer. Astrocytes (2.5 × 105 cells/well) were plated on a 12-well plate and exposed to PCB or vehicle as described above. After PCB or vehicle exposure, cells were washed twice in PBS then lysed in 0.5% NP-40 in PBS. Following lysis and collection, deproteinization was completed using trichloroacetic acid and 1 M NaHCO3 was used to adjust the pH to 5. Deproteinization was confirmed by the DC Protein Assay (Bio-Rad #500011). After accounting for dilution during deproteinization, GSSG was calculated using the following formula: (total glutathione – GSH)/2.
Viability analysis
Viability analysis was performed by formazan quantification in a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (Roche, #11465007001). Primary astrocytes were grown in a 96 well plate (3 × 104 cells/well). Prior to treatment the cells were washed with serum-free astrocyte media (MEM supplemented with 100 μg/mL P/S, 1 mM L-glutamine), and then incubated in serum-free astrocyte media for 1 h. Then the serum-free media was removed and the appropriate treatment (Aroclor 1254 [concentration noted in figure], methanol [matched to highest concentration of Aroclor 1254] or hydrogen peroxide [100 μM]) for 24 hours, treatment media removed and replaced with 100 μL serum-free astrocyte media. MTT labelling reagent was added to the well at a final concentration of 0.5 mg/mL, after which the cells were incubated for 4 h in a humidified incubator (37 °C, 5% CO2), then 100 μL of solubilization solution was added to each well and the plate was incubated overnight. Formazan quantification was completed using a microplate reader at 550 nm wavelength. LDH release was analyzed to determine plasma membrane damage using Pierce LDH Cytotoxicity Assay Kit (Thermo Scientific #88953) as per the manufacturer’s instruction. Data shown is 490nm – 680nm absorbance reading. Cells were exposed to PCBs or methanol as described above and a volume-matched vehicle (methanol) group was included for each (10, 25, and 50 μM) PCB treatment group. TNF-α was included as a positive control for LDH release at a concentration of 60 ng/mL.
qRT-PCR
Following PCB or vehicle treatment for 24 h, astrocyte RNA (300 ng) was prepared using RNeasy mini kit (Qiagen #74104) and reverse transcribed in a 60 μL reaction volume using a High-Capacity RNA-to-cDNA kit (Thermo Fisher #4387406). To quantify gene expression changes, quantitative real-time reverse transcription polymerase chain reaction (qRT-PCR) to quantify gene expression changes was performed in a 384-well plate format using SsoAdvanced Universal Probes Supermix (Biorad, 1725281) and Taqman primer/probe pairs. Taqman primers/probes as follows: AQP4 (Mm00802131_m1), SLC10A6 (Mm00512730_m1), NOX1 (Mm00549170_m1), C3 (Mm01232779_m1), SOD1 (Mm01344233_g1), AMIGO2 (Mm00662105_s1), GFAP (Mm01253030_1), GSTA2 (Mm00833353_mH), PRDX1 (Mm01621996_s1), 18s rRNA (HS999999_s1). The small eukaryotic ribosomal subunit (18S) was used as an endogenous control and 18S rRNA expression level was not statistically different between vehicle and treatment conditions. ABI Prism 7900HT Sequence Detection System (Life Technologies) was used to perform qRT-PCR reactions. Data were analyzed using the 2−ΔΔCt relative quantification of gene expression method which consists, in brief, of normalizing gene of interest expression to the endogenous control (18S rRNA), followed by comparison to biological control (vehicle) and calculation of fold change utilizing 2−(ΔΔCt).
Seahorse extracellular flux analyzer experiments
Primary murine cortical astrocytes were plated at 3 × 104 cells/well in astrocyte media (MEM supplemented with 100 μg/mL P/S, 1 mM L-glutamine, and 10% FBS) and allowed to rest for 3 days. Cells were then treated in serum-free astrocyte media with Aroclor 1254 or methanol, at the concentration noted in the figure legend. The mitochondrial stress test protocol was followed as per the manufacturer’s instructions (Agilent). Briefly, the medium was replaced with DMEM supplemented with 1 mM pyruvate, 2 mM glutaMAX (Thermo Fisher #35050079) and 10 mM glucose (pH of 7.2–7.4). A sensor cartridge was equilibrated with Seahorse XF Calibrant at 37 °C in a CO2-free incubator overnight. The analysis protocol was comprised of three consecutive measurements to determine oxygen consumption rate (OCR) prior to the injection of each compound; oligomycin (final concentration 0.5 μM), FCCP (2 μM), and rotenone/antimycin (0.5 μM each). Data shown is combined across three independent experiments and was analyzed using a general linear model.
Glucose uptake
Glucose uptake was quantified using Glucose Uptake-Glo™ Assay (Promega J1341). Astrocytes were plated on a 96-well plate (30,000 cells/well) and treated as described for other assays (in brief, for 24 h following 1 h serum starvation). The cells were incubated with the 2DG6P detection reagent for 1 h at room temperature and luminescence was detected on a microplate Molecular Devices SpectraMax iD). Data shown is from three independent experiments.
Curated interaction and Gene Ontology analysis
All of the curated gene, compound and disease comparisons in the Comparative Toxicogenomics Database (CTD) are publicly available (Davis et al. 2021). The comparisons shown here were conducted with data downloaded on February 7, 2019. For analysis of gene-disease and gene-gene ontology association (Fig 2A), the following Medical Subject Heading (MeSH) IDs were used: Parkinson’s disease (D010300) and PCBs (D011078). The gene ontology (GO) name for antioxidant activity used was 16209. A dataset was created including the inferred gene associations were for D010300 (PD) and D011078 (PCBs) and the curated gene associations for antioxidant activity (GO: 16209). This dataset of genes was then analyzed using the MyVenn tool to identify overlapping genes, resulting in a list of 54 genes with known antioxidant activity and inferred associations with PD and PCBs.
Gene ontology analysis was performed on these 54 genes to identify the most enriched biological processes in Homo sapiens using the Gene Ontology Consortium’s GO enrichment analysis (http://geneontology.org) (Ashburner et al. 2000; Mi et al. 2019; Gene Ontology Consortium 2021) to determine the specific oxidant species and antioxidant processes associated with both PD and PCBs. These data were analyzed using a Fisher’s Exact test with Bonferroni correction. The p-values for the most enriched (>100 fold enrichment) processes are: removal of superoxide radicals (p = 2.50 × 10−10), cellular oxidant detoxification (p = 9.73 × 10−97), cellular response to superoxide (p = 5.47 × 10−10), response to superoxide (p = 1.53 × 10−9), cellular response to oxygen radical (p = 5.4 × 10−10), response to oxygen radical (p = 1.51 × 10−11), hydrogen peroxide catabolic process (p = 8.63 × 10−19), glutathione derivative metabolic process (p = 2.74 × 10−13), cellular detoxification (p = 1.34 × 10−94), detoxification (p = 3.69 × 10−91), and the response to lipid hydroperoxides (p = 2.55 × 10−3). To identify curated gene-disease associations, the MeSH IDs for PCBs (D011078), trichloroethylene (D014241), and dieldrin (D004026) were analyzed (Supplemental Figure 2).
Statistical analyses
Data were analyzed using unpaired t test, one-way analysis of variance or two-way analysis of variance (ANOVA) (as noted). All data are reported at mean ± SEM with significance set at p ≤ 0.05. Specific replicate values for each experiment and assay are as noted in the figure legend. Statistical analyses were conducted on biological replicates using GraphPad Prism version 9.0.1 for Mac, GraphPad Software, San Diego, California USA, www.graphpad.com and R (R Core Team 2019).
Supplementary Material
Highlights.
Astrocytes exposed to PCBs demonstrate an increase in reactive oxygen species in the absence of cell death.
Astrocytes exposed to PCBs exhibit increased expression of a subset of antioxidant response genes.
Astrocytic ATP production, spare respiratory capacity and glucose uptake are increased in response to PCB exposure.
Acknowledgements
This study was supported by funds from NIH NINDS 5T32NS041218 (M.S.M), a Georgetown University Medical Center Graduate Student Organization student research grant proposal award (M.S.M), GUMC funds (K.M.Z.) and R01NS108810 (K.C. & K.M.Z.).
Abbreviations –
- PCB
- PD
- CNS
- H2DCFDA
- DCF
- LDH
- ROS
- GSH
- GSSG
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Conflicts of Interest
The authors have no competing commercial interests or other conflicts of interest.
References
- Adornetto A, Pagliara V, Di Renzo G, Arcone R (2013) Polychlorinated biphenyls impair dibutyryl cAMP-induced astrocytic differentiation in rat C6 glial cell line. FEBS Open Bio. 10.1016/j.fob.2013.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allaman I, Bélanger M, Magistretti PJ (2011) Astrocyte-neuron metabolic relationships: for better and for worse. Trends Neurosci 34:76–87. 10.1016/j.tins.2010.12.001 [DOI] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, et al. (2000) Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25:25–9. 10.1038/75556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnham KJ, Masters CL, Bush AI (2004) Neurodegenerative diseases and oxidatives stress. Nat Rev Drug Discov 3:205–214. 10.1038/nrd1330 [DOI] [PubMed] [Google Scholar]
- Bélanger M, Ilaman I, Magistretti PJ (2011) Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab 14:724–38. 10.1016/j.cmet.2011.08.016 [DOI] [PubMed] [Google Scholar]
- Bittner CX (2010) High resolution measurement of the glycolytic rate. Front Neuroenergetics 2:1–11. 10.3389/fnene.2010.00026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackburn D, Sargsyan S, Monk PN, Shaw PJ (2009) Astrocyte function and role in motor neuron disease: A future therapeutic target? Glia 57:1251–1264. 10.1002/glia.20848 [DOI] [PubMed] [Google Scholar]
- Booth HDE, Hirst WD, Wade-Martins R (2017) The Role of Astrocyte Dysfunction in Parkinson’s Disease Pathogenesis. Trends Neurosci 40:358–370. 10.1016/j.tins.2017.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossy-Wetzel E, Schwarzenbacher R, Lipton SA (2004) Molecular pathways to neurodegeneration. Nat Med 10 Suppl:S2–9. 10.1038/nm1067 [DOI] [PubMed] [Google Scholar]
- Bouchard MF, Oulhote Y, Sagiv SK, et al. (2014) Polychlorinated biphenyl exposures and cognition in older U.S. adults: NHANES (1999–2002). Environ Health Perspect 122:73–78. 10.1289/ehp.1306532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Sastre M, Del Tredici K (2007) Development of α-synuclein immunoreactive astrocytes in the forebrain parallels stages of intraneuronal pathology in sporadic Parkinson’s disease. Acta Neuropathol 114:231–241. 10.1007/s00401-007-0244-3 [DOI] [PubMed] [Google Scholar]
- Brookmeyer R, Evans DA, Hebert L, et al. (2011) National estimates of the prevalence of Alzheimer’s disease in the United States. Alzheimers Dement 7:61–73. 10.1016/j.jalz.2010.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AM, Ransom BR (2007) Astrocyte glycogen and brain energy metabolism. Glia 55:1263–1271. 10.1002/glia.20557 [DOI] [PubMed] [Google Scholar]
- Calabrese EJ, Bachmann KA, Bailer AJ, et al. (2007) Biological stress response terminology: Integrating the concepts of adaptive response and preconditioning stress within a hormetic dose-response framework. Toxicol Appl Pharmacol 222:122–128. 10.1016/j.taap.2007.02.015 [DOI] [PubMed] [Google Scholar]
- Calabrese EJ, Baldwin LA (2003) Hormesis: The Dose-Response Revolution. Annu. Rev. Pharmacol. Toxicol 43:175–197 [DOI] [PubMed] [Google Scholar]
- Calabrese EJ, Baldwin LA, Holland CD (1999) Hormesis: A highly generalizable and reproducible phenomenon with important implications for risk assessment. Risk Anal 19:261–281. 10.1023/A:1006977728215 [DOI] [PubMed] [Google Scholar]
- Chai H, Diaz-Castro B, Shigetomi E, et al. (2017) Neural Circuit-Specialized Astrocytes: Transcriptomic, Proteomic, Morphological, and Functional Evidence. Neuron 95:531–549.e9. 10.1016/j.neuron.2017.06.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan FK-M, Moriwaki K, De Rosa MJ (2013) Detection of Necrosis by Release of Lactate Dehydrogenase Activity. In: Encyclopedic Dictionary of Genetics, Genomics and Proteomics. John Wiley & Sons, Inc., Hoboken, NJ, USA, pp 65–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocco S, Secondo A, Del Viscovo A, et al. (2015) Polychlorinated biphenyls induce mitochondrial dysfunction in SH-SY5Y neuroblastoma cells. PLoS One 10:. 10.1371/journal.pone.0129481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Council of the European Union (1996) Directive 96/59/EC. 31–35 [Google Scholar]
- Damier P, Hirsch EC, Zhang P, et al. (1993) Glutathione peroxidase, glial cells and Parkinson’s disease. Neuroscience 52:1–6. 10.1016/0306-4522(93)90175-f [DOI] [PubMed] [Google Scholar]
- Dasuri K, Zhang L, Keller JN (2013) Oxidative stress, neurodegeneration, and the balance of protein degradation and protein synthesis. Free Radic Biol Med 62:170–185. 10.1016/j.freeradbiomed.2012.09.016 [DOI] [PubMed] [Google Scholar]
- Davis AP, Grondin CJ, Johnson RJ, et al. (2021) Comparative Toxicogenomics Database (CTD): update 2021. Nucleic Acids Res 49:D1138–D1143. 10.1093/nar/gkaa891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- di Domenico A, Carola G, Calatayud C, et al. (2019) Patient-Specific iPSC-Derived Astrocytes Contribute to Non-Cell-Autonomous Neurodegeneration in Parkinson’s Disease. Stem Cell Reports 12:213–229. 10.1016/j.stemcr.2018.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dringen R, Brandmann M, Hohnholt MC, Blumrich E-M (2015) Glutathione-Dependent Detoxification Processes in Astrocytes. Neurochem Res 40:2570–82. 10.1007/s11064-014-1481-1 [DOI] [PubMed] [Google Scholar]
- Dringen R, Hirrlinger J (2003) Glutathione pathways in the brain. Biol Chem 384:505–16. 10.1515/BC.2003.059 [DOI] [PubMed] [Google Scholar]
- Fleming SM (2017) Mechanisms of Gene-Environment Interactions in Parkinson’s Disease. Curr Environ Heal reports 4:192–199. 10.1007/s40572-017-0143-2 [DOI] [PubMed] [Google Scholar]
- García-Cáceres C, Quarta C, Varela L, et al. (2016) Astrocytic Insulin Signaling Couples Brain Glucose Uptake with Nutrient Availability. Cell 166:867–880. 10.1016/j.cell.2016.07.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gene Ontology Consortium (2021) The Gene Ontology resource: enriching a GOld mine. Nucleic Acids Res 49:D325–D334. 10.1093/nar/gkaa1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gore AC (2001) Environmental toxicant effects on neuroendocrine function. Endocrine 14:235–246. 10.1385/ENDO:14:2:235 [DOI] [PubMed] [Google Scholar]
- Graham DG (1978) Oxidative pathways for catecholamines in the genesis of neuromelanin and cytotoxic quinones. Mol Pharmacol 14:633–643 [PubMed] [Google Scholar]
- Grandjean P, Budtz-Jørgensen E, Barr DB, et al. (2008) Elimination half-lives of polychlorinated biphenyl congeners in children. Environ Sci Technol 42:6991–6996. 10.1021/es800778q [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurley GH, Jelaso AM, Ide CF, Spitsbergen JM (2007) Effects of polychlorinated biphenyls (PCBs) on expression of neurotrophic factors in C6 glial cells in culture. Neurotoxicology 28:1264–71. 10.1016/j.neuro.2007.08.005 [DOI] [PubMed] [Google Scholar]
- Han X, Chen M, Wang F, et al. (2013) Forebrain engraftment by human glial progenitor cells enhances synaptic plasticity and learning in adult mice. Cell Stem Cell 12:342–353. 10.1016/j.stem.2012.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashmi MZ, Khan KY, Hu J, et al. (2015) Hormetic effects of noncoplanar PCB exposed to human lung fibroblast cells (HELF) and possible role of oxidative stress. Environ Toxicol 30:1385–1392. 10.1002/tox.22008 [DOI] [PubMed] [Google Scholar]
- Hatcher-Martin JM, Gearing M, Steenland K, et al. (2012) Association between polychlorinated biphenyls and Parkinson’s disease neuropathology. Neurotoxicology 33:1298–304. 10.1016/j.neuro.2012.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawker DW, Connell DW (1988) Octanol-Water Partition Coefficients of Polychlorinated Biphenyl Congeners. Environ Sci Technol 22:382–387. 10.1021/es00169a004 [DOI] [Google Scholar]
- Hebert LE, Weuve J, Scherr PA, Evans DA (2013) Alzheimer disease in the United States (2010–2050) estimated using the 2010 census. Neurology 80:1778–83. 10.1212/WNL.0b013e31828726f5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennig B, Hammock BD, Toborek M, et al. (2002) International Journal of Hygiene and Environmental Health PCB-induced oxidative stress in endothelial cells: modulation by nutrients [DOI] [PubMed] [Google Scholar]
- Hennig B, Slim R, Toborek M, Robertson LW (1999) Linoleic acid amplifies polychlorinated biphenyl-mediated dysfunction of endothelial cells. J Biochem Mol Toxicol 13:83–91. [DOI] [PubMed] [Google Scholar]
- Herrero-Mendez A, Almeida A, Fernández E, et al. (2009) The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C-Cdh1. Nat Cell Biol 11:747–752. 10.1038/ncb1881 [DOI] [PubMed] [Google Scholar]
- Howard AS, Fitzpatrick R, Pessah I, et al. (2003) Polychlorinated biphenyls induce caspase-dependent cell death in cultured embryonic rat hippocampal but not cortical neurons via activation of the ryanodine receptor. Toxicol Appl Pharmacol 190:72–86. 10.1016/s0041-008x(03)00156-x [DOI] [PubMed] [Google Scholar]
- Itoh Y, Esaki T, Shimoji K, et al. (2003) Dichloroacetate effects on glucose and lactate oxidation by neurons and astroglia in vitro and on glucose utilization by brain in vivo. Proc Natl Acad Sci U S A 100:4879–4884. 10.1073/pnas.0831078100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenner P (2003) Oxidative stress in Parkinson’s disease. Ann Neurol 53:S26–S38. 10.1002/ana.10483 [DOI] [PubMed] [Google Scholar]
- Jouroukhin Y, Kageyama Y, Misheneva V, et al. (2018) DISC1 regulates lactate metabolism in astrocytes: implications for psychiatric disorders. Transl Psychiatry 8:76. 10.1038/s41398-018-0123-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klocke C, Lein PJ (2020) Evidence implicating non-dioxin-like congeners as the key mediators of polychlorinated biphenyl (Pcb) developmental neurotoxicity. Int. J. Mol. Sci 21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koepsell H (2020) Glucose transporters in brain in health and disease. Pflugers Arch Eur J Physiol 472:1299–1343. 10.1007/s00424-020-02441-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostuk EW, Cai J, Iacovitti L (2019) Subregional differences in astrocytes underlie selective neurodegeneration or protection in Parkinson’s disease models in culture. Glia glia.23627. 10.1002/glia.23627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowal SL, Dall TM, Chakrabarti R, et al. (2013) The current and projected economic burden of Parkinson’s disease in the United States. Mov Disord 28:311–318. 10.1002/mds.25292 [DOI] [PubMed] [Google Scholar]
- Kulkarni A, Chen J, Maday S (2018) Neuronal autophagy and intercellular regulation of homeostasis in the brain. Curr Opin Neurobiol 51:29–36. 10.1016/j.conb.2018.02.008 [DOI] [PubMed] [Google Scholar]
- Kumar P, Nagarajan A, Uchil PD (2018) Analysis of Cell Viability by the Lactate Dehydrogenase Assay. Cold Spring Harb Protoc 2018:465–468. 10.1101/pdb.prot095497 [DOI] [PubMed] [Google Scholar]
- Lee DW, Gelein RM, Opanashuk LA (2006) Heme-oxygenase-1 promotes polychlorinated biphenyl mixture aroclor 1254-induced oxidative stress and dopaminergic cell injury. Toxicol Sci 90:159–167. 10.1093/toxsci/kfj052 [DOI] [PubMed] [Google Scholar]
- Lee DW, Notter SA, Thiruchelvam M, et al. (2012) Subchronic polychlorinated biphenyl (aroclor 1254) exposure produces oxidative damage and neuronal death of ventral midbrain dopaminergic systems. Toxicol Sci 125:496–508. 10.1093/toxsci/kfr313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DW, Opanashuk LA (2004) Polychlorinated biphenyl mixture aroclor 1254-induced oxidative stress plays a role in dopaminergic cell injury. Neurotoxicology 25:925–939. 10.1016/j.neuro.2004.05.005 [DOI] [PubMed] [Google Scholar]
- Liddelow SA, Barres BA (2017) Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 46:957–967. 10.1016/j.immuni.2017.06.006 [DOI] [PubMed] [Google Scholar]
- Liddelow SA, Guttenplan KA, Clarke LE, et al. (2017) Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541:481–487. 10.1038/nature21029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyng GD, Seegal RF (2008) Polychlorinated biphenyl-induced oxidative stress in organotypic co-cultures: Experimental dopamine depletion prevents reductions in GABA. Neurotoxicology 29:301–308. 10.1016/j.neuro.2007.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyng GD, Snyder-Keller A, Seegal RF (2007) Polychlorinated biphenyl-induced neurotoxicity in organotypic cocultures of developing rat ventral mesencephalon and striatum. Toxicol Sci 97:128–139. 10.1093/toxsci/kfm027 [DOI] [PubMed] [Google Scholar]
- Maffei A, Fontanini A (2009) Network homeostasis: a matter of coordination. Curr Opin Neurobiol 19:168–173. 10.1016/j.conb.2009.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariussen E, Myhre O, Reistad T, Fonnum F (2002) The polychlorinated biphenyl mixture Aroclor 1254 induces death of rat cerebellar granule cells: The involvement of the N-methyl-D-aspartate receptor and reactive oxygen species. Toxicol Appl Pharmacol 179:137–144. 10.1006/taap.2002.9353 [DOI] [PubMed] [Google Scholar]
- Matias I, Morgado J, Gomes FCA (2019) Astrocyte Heterogeneity: Impact to Brain Aging and Disease. Front Aging Neurosci 11:59. 10.3389/fnagi.2019.00059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP (2008) Hormesis defined. Ageing Res. Rev 7:1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matyash V, Kettenmann H (2010) Heterogeneity in astrocyte morphology and physiology. Brain Res Rev 63:2–10. 10.1016/j.brainresrev.2009.12.001 [DOI] [PubMed] [Google Scholar]
- McCann MS, Maguire-Zeiss KA (2021) Environmental toxicants in the brain: a review of astrocytic metabolic dysfunction. Environ Toxicol Pharmacol 103608. 10.1016/j.etap.2021.103608 [DOI] [PubMed] [Google Scholar]
- Mi H, Muruganujan A, Ebert D, et al. (2019) PANTHER version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res 47:D419–D426. 10.1093/nar/gky1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migliore L, Coppedè F (2009) Environmental-induced oxidative stress in neurodegenerative disorders and aging. Mutat Res - Genet Toxicol Environ Mutagen 674:73–84. 10.1016/j.mrgentox.2008.09.013 [DOI] [PubMed] [Google Scholar]
- Nair-Roberts RG, Chatelain-Badie SD, Benson E, et al. (2008) Stereological estimates of dopaminergic, GABAergic and glutamatergic neurons in the ventral tegmental area, substantia nigra and retrorubral field in the rat. Neuroscience 152:1024–1031. 10.1016/j.neuroscience.2008.01.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olmos G, Lladó J (2014) Tumor necrosis factor alpha: A link between neuroinflammation and excitotoxicity. Mediators Inflamm. 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park KM, Bowers WJ (2010) Tumor necrosis factor-alpha mediated signaling in neuronal homeostasis and dysfunction. Cell. Signal 22:977–983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pessah IN, Lein PJ, Seegal RF, Sagiv SK (2019) Neurotoxicity of polychlorinated biphenyls and related organohalogens. Acta Neuropathol. 138:363–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn CL, Wania F, Czub G, Breivik K (2011) Investigating intergenerational differences in human PCB exposure due to variable emissions and reproductive behaviors. Environ Health Perspect 119:641–646. 10.1289/ehp.1002415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2019) R: A language and environment for statistical computing. In: R Found. Stat. Comput Vienna, Austria. https://www.r-project.org/ [Google Scholar]
- Raps SP, Lai JCK, Hertz L, Cooper AJL (1989) Glutathione is present in high concentrations in cultured astrocytes but not in cultured neurons. Brain Res 493:398–401. 10.1016/0006-8993(89)91178-5 [DOI] [PubMed] [Google Scholar]
- Richardson JR, Miller GW (2004) Acute exposure to aroclor 1016 or 1260 differentially affects dopamine transporter and vesicular monoamine transporter 2 levels. Toxicol Lett 148:29–40. 10.1016/j.toxlet.2003.12.006 [DOI] [PubMed] [Google Scholar]
- Rizor A, Pajarillo E, Johnson J, et al. (2019) Astrocytic Oxidative/Nitrosative Stress Contributes to Parkinson’s Disease Pathogenesis: The Dual Role of Reactive Astrocytes. Antioxidants 8:265. 10.3390/antiox8080265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rostami J, Fotaki G, Sirois J, et al. (2020) Astrocytes have the capacity to act as antigen-presenting cells in the Parkinson’s disease brain. J Neuroinflammation 17:119. 10.1186/s12974-020-01776-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez-Alonso JA, López-Aparicio P, Recio MN, Pérez-Albarsanz MA (2004) Polychlorinated biphenyl mixtures (Aroclors) induce apoptosis via Bcl-2, Bax and caspase-3 proteins in neuronal cell cultures. Toxicol Lett 153:311–326. 10.1016/j.toxlet.2004.05.012 [DOI] [PubMed] [Google Scholar]
- Seegal RF, Bush B, Brosch KO (1994) Decreases in dopamine concentrations in adult, non-human primate brain persist following removal from polychlorinated biphenyls. Toxicology 86:71–87. 10.1016/0300-483X(94)90054-X [DOI] [PubMed] [Google Scholar]
- Souza DG, Almeida RF, Souza DO, Zimmer ER (2019) The astrocyte biochemistry. Semin. Cell Dev. Biol 95:142–150 [DOI] [PubMed] [Google Scholar]
- Tapia PC (2006) Sublethal mitochondrial stress with an attendant stoichiometric augmentation of reactive oxygen species may precipitate many of the beneficial alterations in cellular physiology produced by caloric restriction, intermittent fasting, exercise and dietary p. Med Hypotheses 66:832–843. 10.1016/j.mehy.2005.09.009 [DOI] [PubMed] [Google Scholar]
- Thayer KA, Melnick R, Burns K, et al. (2005) Fundamental flaws of hormesis for public health decisions. Environ Health Perspect 113:1271–1276. 10.1289/ehp.7811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulbrich B, Stahlmann R (2004) Developmental toxicity of polychlorinated biphenyls (PCBs): A systematic review of experimental data. Arch Toxicol 78:252–268. 10.1007/s00204-003-0519-y [DOI] [PubMed] [Google Scholar]
- Verkhratsky A, Parpura V, Vardjan N, Zorec R (2019) Physiology of astroglia. In: Advances in Experimental Medicine and Biology. Springer New York LLC, pp 45–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakabayashi K, Hayashi S, Yoshimoto M, et al. (2000) NACP alphasynuclein-positive filamentous inclusions. Acta Neuropathol 99:14–20 [DOI] [PubMed] [Google Scholar]
- Whittemore ER, Loo DT, Watt JA, Cotmans CW (1995) A detailed analysis of hydrogen peroxide-induced cell death in primary neuronal culture. Neuroscience 67:921–932. 10.1016/0306-4522(95)00108-U [DOI] [PubMed] [Google Scholar]
- Xin W, Bonci A (2018) Functional Astrocyte Heterogeneity and Implications for Their Role in Shaping Neurotransmission. Front Cell Neurosci 12:141. 10.3389/fncel.2018.00141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun SP, Kam TI, Panicker N, et al. (2018) Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat Med 24:931–938. 10.1038/s41591-018-0051-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Sloan SA, Clarke LE, et al. (2016) Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron 89:37–53. 10.1016/j.neuron.2015.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Kalen AL, Li L, et al. (2009) Polychlorinated-biphenyl-induced oxidative stress and cytotoxicity can be mitigated by antioxidants after exposure. Free Radic Biol Med 47:1762–1771. 10.1016/j.freeradbiomed.2009.09.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.