

Abstract
Calcific aortic valve disease (CAVD) is a common heart disease that contributes to increased cardiovascular morbidity and mortality. There is a lack of effective pharmaceutical therapy because its mechanisms are not yet fully known. Ginkgo biloba extract (EGB761) is reported to alleviate vascular calcification. However, whether EGB761 protects against aortic valve calcification, a disease whose pathogenesis shares many similarities with vascular calcification, and potential molecular mechanisms remain unknown. In this study, porcine aortic valve interstitial cells (pAVIC) calcification was induced by warfarin with or without the presence of EGB761. Immunostaining was performed to establish and characterize the pAVIC phenotype. Calcium deposition and calcium content were examined by Alizarin Red S staining and an intracellular calcium content assay. Alkaline phosphatase (ALP) activity was detected by the p-nitrophenyl phosphate method. The expression levels of bone morphogenetic protein-2 (BMP2), Runt-related transcription factor 2 (Runx2), homeobox protein MSX-2 (Msx2) and phosphorylated (p)-Smad1/5 were detected by reverse transcription-quantitative PCR and western blot analysis. Consistent with these in vitro data, we also confirmed the suppression of in vivo calcification by EGB761 in warfarin-induced C57/Bl6 mice. The results indicated that both pAVICs and aortic valves tissue of mice stimulated with warfarin showed increased calcium deposition and expression of osteogenic markers (ALP, BMP2, Msx2 and Runx2), and promoted p-Smad1/5 translocation from the cytoplasm to the nucleus. The addition of EGB761 significantly inhibited p-Smad1/5 translocation from the cytoplasm to the nucleus, thus suppressing calcification. In conclusion, EGB761 could ameliorate warfarin-induced aortic valve calcification through the inhibition of the BMP2-medicated Smad1/5/Runx2 signaling pathway.
Keywords: Ginkgo biloba extract, aortic valve, calcification, bone morphogenetic protein-2/Smad1/5/ Runt-related transcription factor 2 signaling, warfarin
Introduction
Calcific aortic valve disease (CAVD) is a prevalent heart disease that contributes to increased cardiovascular mortality in almost 30% of adults >65 years and 40-50% of those >75 years (1,2). The development of this disease is thought to be initiated by endothelial injury, lipid deposition, inflammatory cell infiltration, extracellular matrix deposition and neovascularization in the valve, and is followed by a propagation phase, where several factors promote osteogenesis and calcification (3). These processes are also contributing factors to atherosclerosis. Although there are similar pathophysiological processes between them, only ~50% of patients with CAVD are accompanied by clinically significant atherosclerotic characteristics (4). To date, there is no effective medical therapy to treat or even delay the progress of CAVD.
Warfarin is an antagonist of vitamin K and plays an important anticoagulative role by inhibiting coagulation factors. It is the most important oral anticoagulant in the synthesis of numerous coagulation factors as well as in the formation of circulating anticoagulants. Warfarin is associated with severe aortic valve and vascular calcification in hemodialysis and diabetic patients and is considered to be an irritant factor for calcium phosphate deposition in aortic valves (5–7); however, its mechanism is not fully understood (8). One characteristic of the pro-osteogenic mechanism of warfarin involves the inhibition of protein carboxylation, including of the matrix Gla protein, which can prevent calcification by directly inhibiting the formation of bone morphogenetic proteins (BMPs)2 and 4 (8–10). Our previous research demonstrated that 10 μM warfarin and 1.6 mM inorganic phosphate led to accelerated calcification of porcine aortic valve interstitial cells (AVICs) (11). Moreover, warfarin was also involved in the development of aortic valve calcification in C57/Bl6 mice as previously reported (11).
Ginkgo biloba extract (EGB761) is a standardized extract consisting of two major ingredients with specific pharmacological profiles: 24% flavonoids (including quercetin, kaempferol and isorhamnetin) and 6.0% terpene trilactones (including ginkgolides A, B, C, J and bilobalide) (12). Numerous studies in animal models and humans have verified the protective effects of EGB761 on cardiovascular disease, including its well-known antioxidant property of scavenging superoxides, free radicals and nitric oxide (NO), and antiplatelet effects to reduce inflammation and atherosclerotic plaque formation (13–15). Furthermore, the protective effects of EGB761 on vascular functions have shown that it could modulate the synthesis of vasoactive substances including NO and endothelin (ET) (14).
Recent several studies have examined the effects of EGB761 on vascular calcification (16,17); the results demonstrated that EGB761 significantly reduced calcium deposition in rat aortic vascular smooth muscle cells, thereby alleviating vascular calcification. However, it is unclear whether EGB761 could reduce calcium deposition in AVICs to relieve calcification and osteogenic differentiation, and its specific mechanism remains to be elucidated. The present study hypothesized that EGB761 may be beneficial for the treatment of aortic valve calcification and osteogenesis in vivo and in vitro and aimed to investigate the potential molecular mechanisms of EGB761 treatment in cultured warfarin-induced calcification AVICs.
Materials and methods
All experimental procedures were approved by the Experimental Animal Care and Use Committee of the University of Michigan State and Nanjing Medical University, and conducted in accordance with the Guide for the Care and Use of Laboratory Animals (NIH publication No. 85-23, revised 1996).
Drugs and reagents.
Standard EGB761 was purchased from Dr. Willmar Schwabe GmbH & Co. KG. High glucose DMEM (HDMEM; 4.5 g/l) and FBS were purchased from Gibco (Thermo Fisher Scientific, Inc.). Warfarin (cat. no. PHR1435) and Alizarin Red S (cat. no. A5533) were purchased from Sigma-Aldrich (Merck KGaA). Von Kossa (cat. no. ab150687) were purchased from Abcam. BMP2 antibody (cat. no. NBP1-19751) was purchased from Novus Biologicals LLC. Anti-Runx2 (cat. no. 12556) and anti-GAPDH (cat. no. 5174) antibodies were purchased from Cell Signaling Technology, Inc. Anti-phosphorylated (p)-Smad1/5 (cat. no. ab80255) and anti-Msx2 (cat. no. ab227720) antibodies were purchased from Abcam. Human recombinant BMP2 (rhBMP-2; cat. no. B3555) was purchased from Cyagen Biosciences, Inc.
pAVIC cell culture and characterization.
pAVICs were routinely isolated and cultured as described previously, with minor modifications (18). Dissociated pAVICs were cultured in HDMEM supplemented with 10% FBS, 100 U/ml penicillin-streptomycin and 2 mM L-glutamine. Adherent pAVICs were passaged at 80% confluence. Cells between passages 3 and 5 were used in the present study. To establish and characterize pAVICs, the cells were identified by immunostaining with AVIC markers, such as anti-vimentin (1:900, cat. no. 2258, Sigma-Aldrich) and anti-a-smooth muscle actin (α-SMA; 1: 200, cat. no. 7817, Abcam) antibodies.
pAVIC cell treatment.
The treatment protocol for warfarin induction was performed as previously described (11). Once adherent pAVICs were at 80% confluence, cells were treated as follows according to different groups: i) HDMEM supplemented with 10% FBS and 100 U/ml penicillin-streptomycin [NC (negative control) group]; ii) HDMEM supplemented with 10% FBS, 100 U/ml penicillin-streptomycin, 1.6 mM inorganic phosphate (Pi) and 10 μM warfarin (warfarin group); and iii) EGB761(concentration 0.1, 0.3, 0.5 and 0.7 mg/ml), 1.6 mM Pi and 10 μM warfarin (EGB761 group). To investigate the role of the BMP-Smad signaling pathway in mediating warfarin-induced pAVIC calcification, cells were treated with 100 ng/ml rhBMP2 (19) in the presence of EGB761, 1.6 mM Pi and 10 μM warfarin (EGB761+rhBMP2 group). Different media was added into the culture media for a further 4-day incubation period and changed every 2 days.
Animal grouping.
To establish the effect of EGB761 on warfarin induced aortic valve calcification in vivo, male C57/Bl6 mice, aged 8 weeks old, were rendered aortic valve calcification as warfarin group by subcutaneous injection of 250 mg/kg/d warfarin and 30 mg/kg/d vitamin K1 every day for 4 weeks as previously reported (11). The mice that received saline water served as control group. The Intraperitoneal injection with variable dosages of EGB761 was administered simultaneously with warfarin and vitamin K1 once a day.
C57/Bl6 mice were randomly divided into four groups: i) saline water, [NC (negative control) group)]; ii) 250 mg/kg/d warfarin and 30 mg/kg/d vitamin K1 (warfarin group); iii) EGB761 (75 mg/kg/d and 125 mg/kg/d), 250 mg/kg/d warfarin and 30 mg/kg/d vitamin K1 (EGB761 group). All the mice were sacrificed by isoflurane, and selected tissues were fixed in 4% paraformaldehyde and embedded in paraffin or frozen at −80°C for later studies.
Cytotoxicity assay.
Cell Counting Kit-8 (CCK-8; Dojindo Molecular Technologies, Inc.) was used according to the manufacturer’s instructions. Cells were analyzed in quintuplicate in 96-well plates. The pAVICs calcification model was established by culture in medium containing warfarin, and the cells were treated with warfarin and/or supplemented with 0.1, 0.3, 0.5 or 0.7 mg/ml EGB761. The optical density (OD) value of each well was measured at a wavelength of 450 nm and the relative cell viability was determined as ODdrug/ODnormal.
Alizarin Red staining.
Calcified cells were measured by Alizarin Red staining. Briefly, cells were carefully washed with Ca2+-free PBS, and fixed in 4% paraformaldehyde for 10 min and in 95% ethanol for 30 min at room temperature. Subsequently, cells were stained with 2% Alizarin Red solution (pH 4.2) for 1-5 min to visualize orange-red mineralized nodules. The plates were photographed under an inverted microscope.
Von kossa staining.
Paraffin sections were routinely dewaxed to water. Incubate slide in 5% silver nitrate solution for 30-60 min with exposure to ultraviolet light or incandescent. Rinse in distilled water and incubate slide in 5% sodium thiosulfate solution for 2-3 minutes. Incubate slide in nuclear fast red solution for 5 minutes, then rinse with distilled water. Dehydrate very quickly in fresh Absolute Alcohol. Clear and mount in synthetic resin. After dehydration and baking, seal the piece with synthetic resin and put it in the oven overnight. After drying, it can be photographed under optical microscope.
Image Analysis.
Image Pro Plus Pathological Image analysis software (Media Cybernetics, Inc. Rockville, MD, USA) was used to calculate the calcification area and total area of each visual field, and the percentage of calcification area = (calcification area/total valve area) ×100%.
Intracellular calcium levels.
In order to quantify calcium levels, cells were washed with PBS and decalcified with 0.6 mM HCl at 4°C for 24 h. Subsequently, intracellular calcium levels were measured with a commercial colorimetric kit (Randox Laboratories, Ltd.) following the manufacturer’s instructions and corrected for total protein concentration with a BCA Protein assay (Pierce; Thermo Fisher Scientific, Inc.). Absorbances were read at a wavelength of 570 nm (calcium) and 562 nm (protein) using a microplate reader (BioTek Instruments, Inc.).
Alkaline phosphatase (ALP) activity assay.
ALP activity was detected using a QuantiChrom™ Alkaline Phosphatase Assay kit (Bioassay systems) according to the manufacturer’s protocol. ALP activity was normalized based on the cellular protein content.
Reverse transcription-quantitative PCR (RT-qPCR).
The CellAmp™ Direct TB Green™ RT-qPCR kit (Takara Bio, Inc.) was used to perform qPCR without the need for RNA extraction from aortic valve tissues of mice and pAVICs. RT was performed and gene expression was detected on an ABI QuantStudio 7 Flex Real-Time PCR system (Applied Biosystems; Thermo Fisher Scientific, Inc.). The primers for mouse or porcine target genes were listed in Table 1. Relative gene expression data was normalized against GAPDH via the 2−ΔΔCq method.
Table 1.
Primer sequences used in quantitative real-time PCR
| Gene name | Primer sequences (5′—3′) | |
|---|---|---|
| Porcine | Forward | GGACGAGGCAAGAGTTTCAC |
| Runx2 | Reverse | GTGGATTAAAAGGACTTGGTGC |
| Porcine | Forward | GGAGCTAGCACT GAGCGAC |
| BMP2 | Reverse | CGAAGTGAGGAGCCCAAGTT |
| Porcine | Forward | CTGGTGAAGCCCTTCGAGAC |
| Msx2 | Reverse | AGGGCTCATATGTCTTGGCG |
| Porcine | Forward | GTCGGAGTGAACGGATTTGGC |
| GAPDH | Reverse | CTTGCCGTGGGTGGAATCAT |
| Mouse | Forward | AGAGTCAGATTACAGATCCCAGG |
| Runx2 | Reverse | AGGAGGGGTAAGACTGGTCATA |
| Mouse | Forward | CCGCCGCCCAGACATA |
| Msx2 | Reverse | CTTCCGGTTGGTCTTGTGTTTC |
| Mouse | Forward | AGGTCGGTGTGAACGGATTTG |
| GAPDH | Reverse | TGTAGACCATGTAGTTGAGGTCA |
Western blotting.
Equal amounts of protein were extracted with the NE-PER Nuclear and Cytoplasmic Extraction reagent (Pierce; Thermo Fisher Scientific, Inc.), separated by 10-15% SDS-PAGE and electrotransferred to a PVDF membrane (EMD Millipore). The membranes were subsequently blocked with 5% non-fat milk in TBS and incubated overnight at 4°C with antibodies for Runx2 (1:1,000), BMP2 (1:2,300), Msx2 (1:1,000), p-Smad1/5 (1:1,000) and GAPDH (1:1,000). Subsequently, the membranes were incubated with goat anti-rabbit IgG (H+L) Alexa Fluor 700 (1:20,000; cat. no. A-21038; Invitrogen; Thermo Fisher Scientific, Inc.) for 1 h at room temperature. Fluorescent detection was performed using the Odyssey® CLX imaging system (LI-COR Biosciences).
Statistical analysis.
All results were analyzed using IBM 24.0 software (IBM Corp.). Continuous variables are expressed as the mean ± SD. Comparisons of parameters among different groups were performed using one-way ANOVA followed by Bonferroni’s multiple comparison test or paired t-test. P<0.05 was considered to indicate a statistically significant difference.
Results
Establishment and characterization of pAVICs.
Immunofluorescence staining of AVIC markers (α-SMA and vimentin) was performed to establish and characterize pAVICs, since these are a highly plastic population of resident cells with the ability to differentiate into a variety of other types, such as myofibroblasts or osteoblasts. Cells showed positive immunofluorescence staining for α-SMA (Figure. 1A) and vimentin (Figure. 1B). Cells were negative for non-specific fluorescence staining (Figure. 1C).
Figure 1.
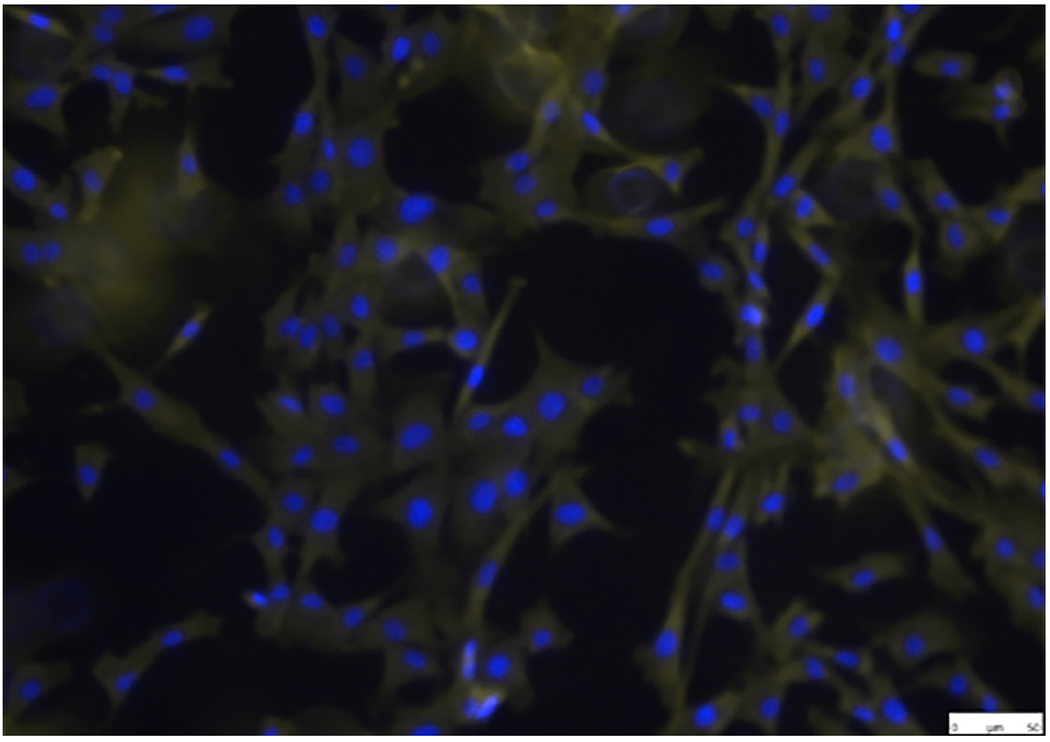
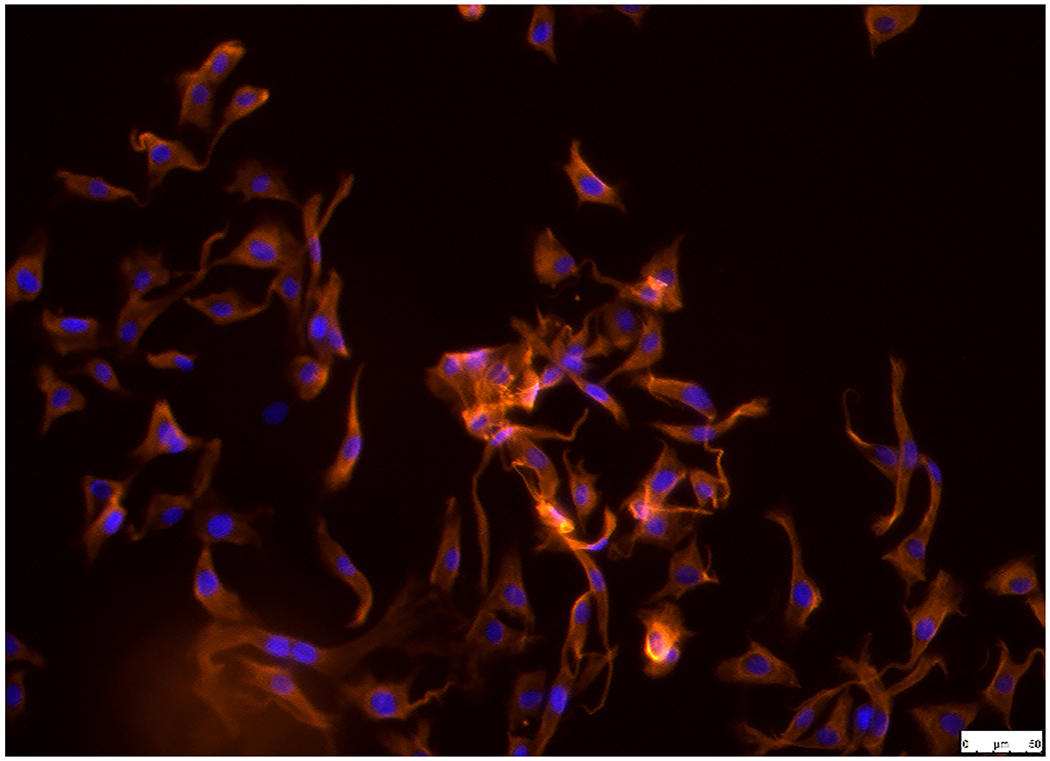
Establishment and characterization of pAVICs. Cells were positive for (A) α-SMA and (B) vimentin in pAVICs. (C) BSA (1%) was used as a negative control in order to exclude non-specific fluorescence staining. Scale bar, 50 μm. α-SMA, a-smooth muscle actin; pAVICs, porcine aortic valve interstitial cells.
EGB761 maintains pAVIC viability under warfarin treatment.
A CCK-8 kit was used to evaluate the cytotoxic effects of EGB761 on pAVIC viability under warfarin treatment. The results showed that the cell viability was decreased upon EGB761 treatment in a dose-dependent manner, where 0.1 and 0.3 mg/ml EGB761 were the most optimal and safe concentrations (Figure. 2).
Figure 2.
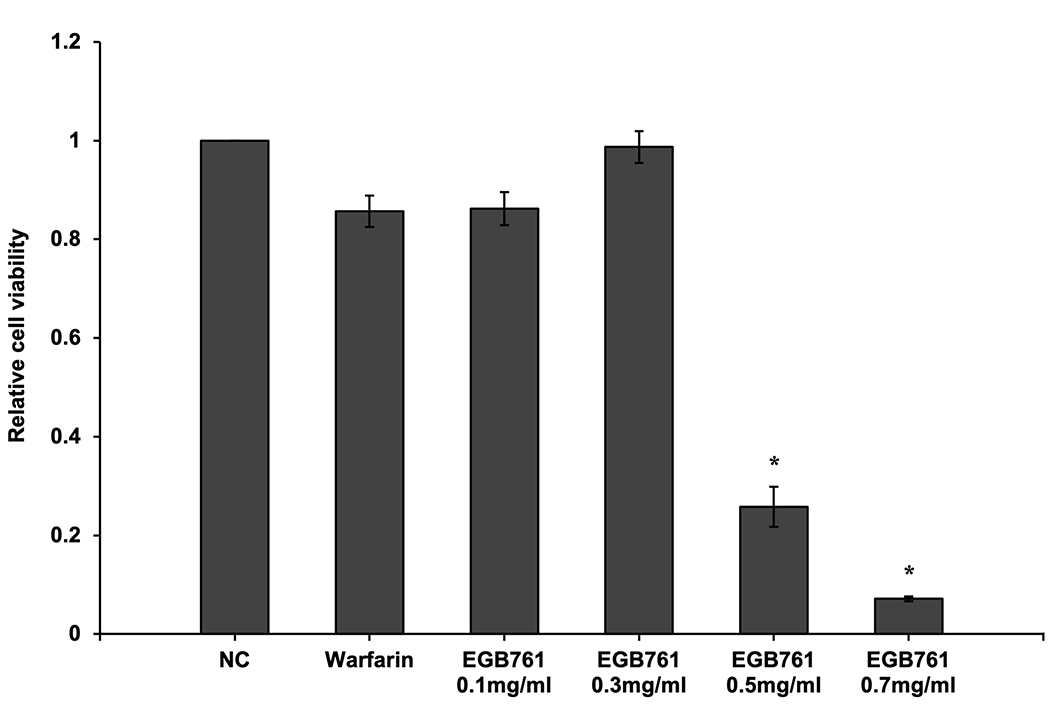
Effects of EGB761 on pAVIC viability under warfarin treatment. pAVICs were treated with warfarin and/or EGB761 at different concentrations (0.1, 0.3, 0.5 or 0.7 mg/ml). Cellular viability was detected by a Cell Counting Kit-8 kit and defined as the percentage of the NC group. n=6. *P<0.05 vs. warfarin group. pAVICs, porcine aortic valve interstitial cells; NC, negative control.
EGB761 inhibits warfarin-induced aortic valve calcification both in vitro and in vivo.
A preliminary study was conducted to determine the effects of EGB761 on warfarin-induced pAVIC calcification. Alizarin Red staining and intracellular calcium content were performed to observe calcium deposition in pAVICs. The warfarin group showed obvious orange-red mineralized nodules and increased intracellular calcium content compared with the NC group (610.01±35.48 vs. 120.0±18.47 ¼g/mg protein; P<0.05). In addition, EGB761 (0.3 mg/ml) presented an anti-calcification effect that decreased the number of orange-red mineralized nodules and intracellular calcium content (313.36±22.88 vs. 610.01±35.48 μg/mg protein; P<0.05) when compared with 0.1 mg/ml EGB761 (596.68±22.85 vs. 610.01±35.48 μg/mg protein; P>0.05). Therefore, 0.3 mg/ml EGB761 was chosen for subsequent experiments (Figure. 3A and 3B).
Figure 3.
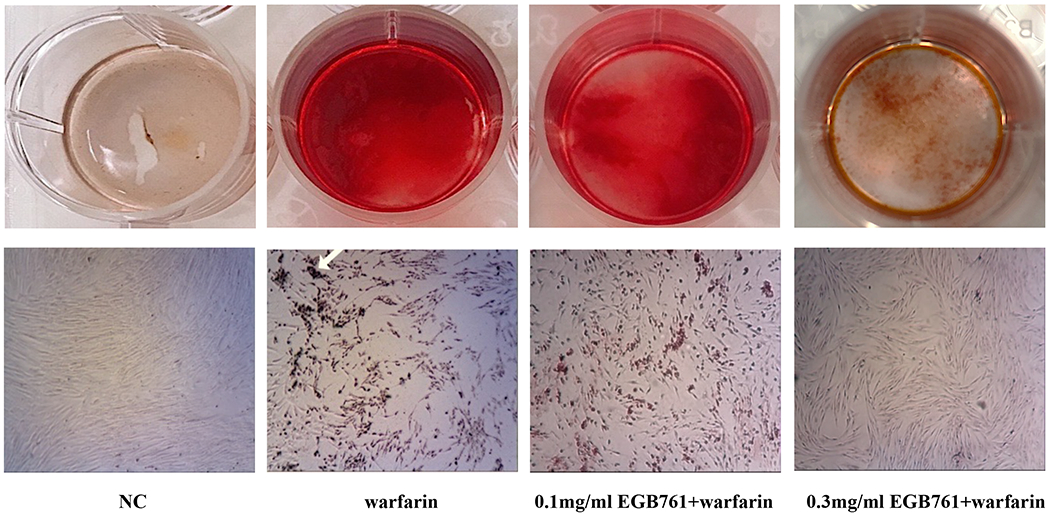
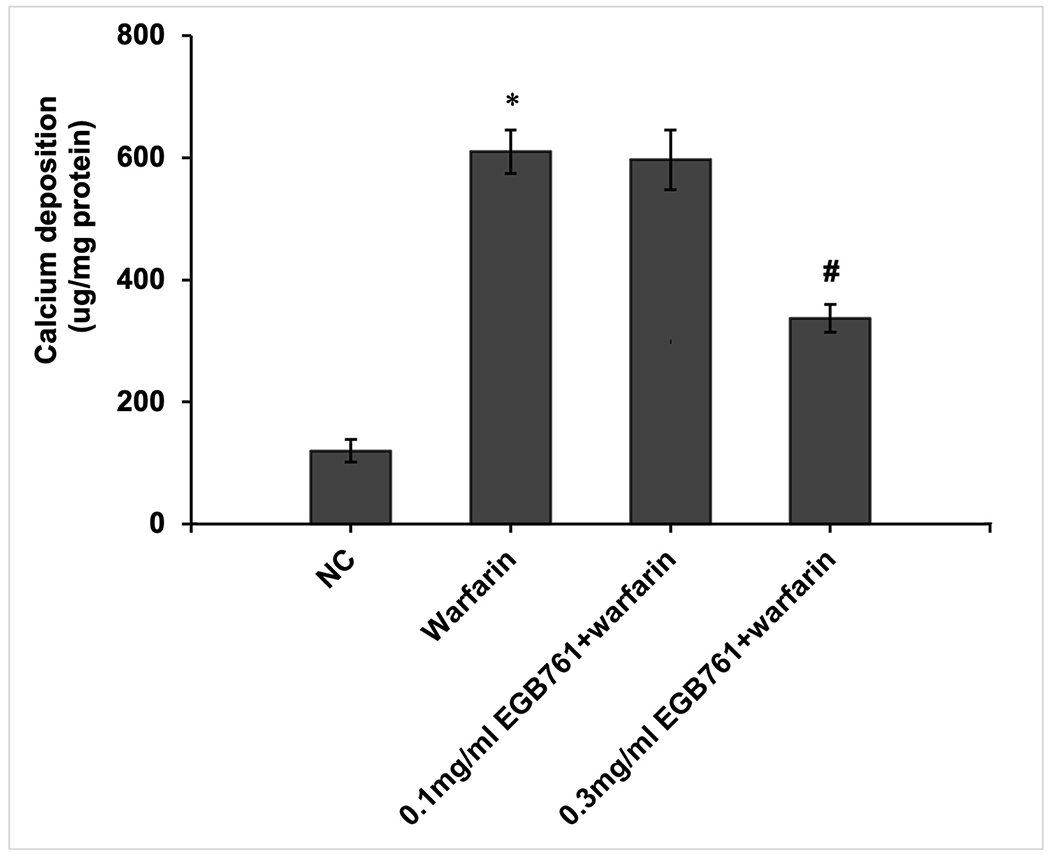
EGB761 inhibits warfarin-induced calcium deposition in pAVICs. (A) Representative Alizarin Red staining of plates (top) and microscopic images (x40 magnification; bottom). Orange-red mineralized nodules indicate matrix calcium deposition. (B) Intracellular calcium contents corrected for protein concentration. n=6. *P<0.05 vs. NC group; #P<0.05 vs. warfarin group. pAVICs, porcine aortic valve interstitial cells; NC, negative control.
We next evaluated whether EGB761 ameliorated warfarin-induced valve leaflet calcification in vivo. We observed significantly calcium deposits in aortic valve leaflets in the mice after warfarin treatment via von kossa staining, which was obviously attenuated in high-dosage of EGB761 treatment (125 mg/kg/day), but the difference was not statistically significant in low-dosage of EGB761 treatment (75 mg/kg/day) (Figure. 4A). Similarly, the percentage of calcified valve area of mice treated with warfarin was significantly increased as compared to the NC group (19.8±6.7 vs. 0.0±0.0; P<0.05). 125 mg/kg/day EGB761 also attenuated the calcified valve area (9.2±3.2 vs. 19.8±6.7; P<0.05). No significant difference of calcified valve area was observed between low-dosage of EGB761 treatment and warfarin group (17.8±5.9 vs. 19.8±6.7; P>0.05) (Figure. 4B).
Figure 4.
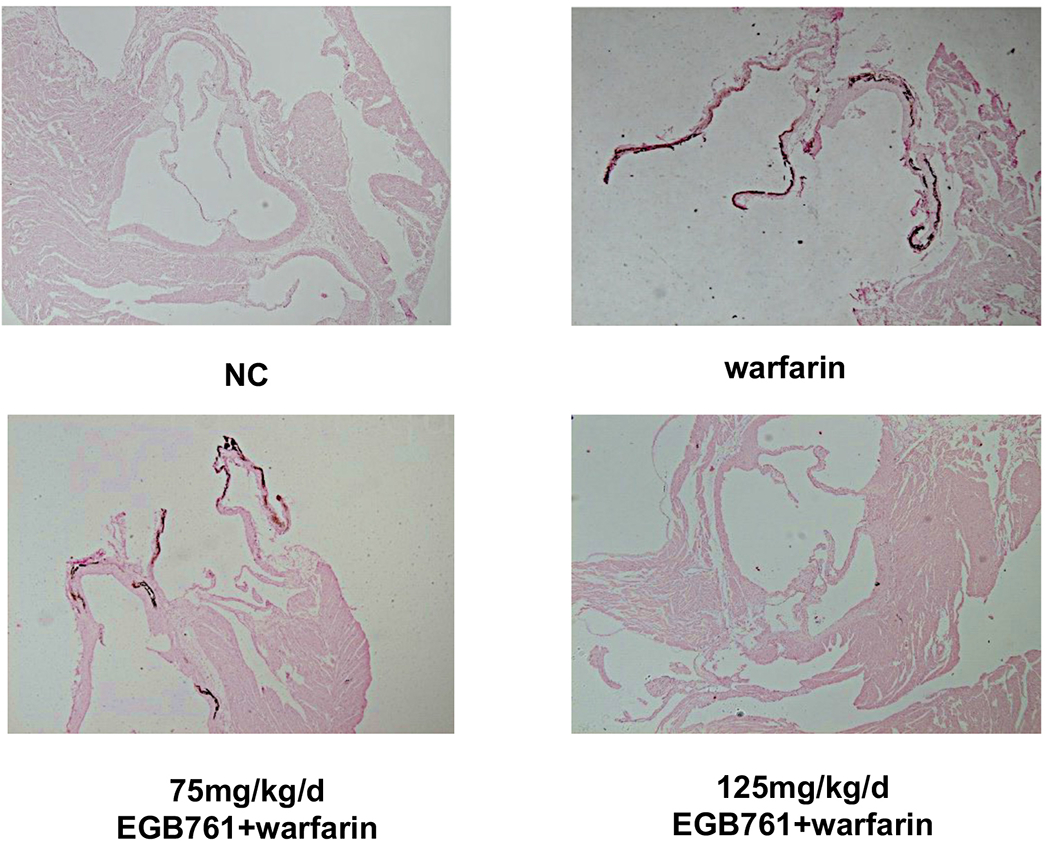
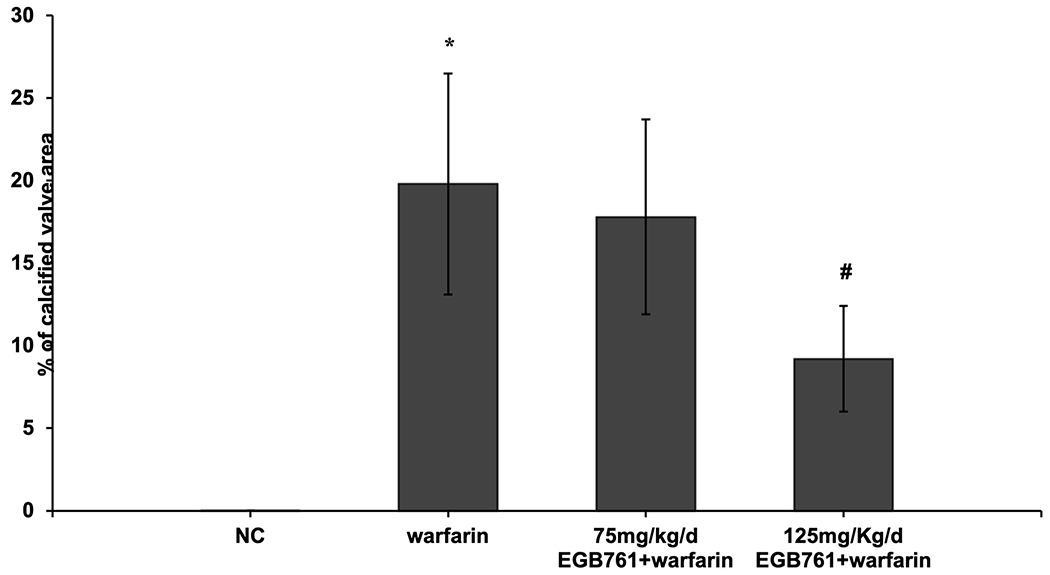
EGB761 attenuates warfarin-induced aortic valve calcification in mice. (A) Representative images of calcium deposits in aortic valve leaflet (von kossa) in mice of different groups (x40 magnification). (B) The percentage of aortic valve area occupied by calcium deposits in the mice treated with warfarin as compared to different dosage of EGB761. n=14 per group. *P<0.05 vs. NC group; #P<0.05 vs. warfarin group. NC, negative control.
EGB761 suppresses warfarin-induced osteoblastic differentiation both in vitro and in vivo.
ALP activity is recognized as an early differentiation marker of osteogenic cells. Other specific osteogenic markers include Runx2 and Msx2. To further investigate whether the inhibitory effects of EGB761 on pAVIC calcification was associated with osteogenic differentiation, ALP activity and the expression of Runx2 and Msx2 were detected. A significant increase in ALP activity was observed in cells cultured in medium with warfarin (5.998±0.682 vs. 2.873±0.277 U/mg protein; P<0.05), and this increase was significantly inhibited by 0.3 mg/ml EGB761 treatment (3.680±0.286 vs. 5.998±0.682 U/mg protein; P<0.05; Figure. 5A). The present study further investigated the expressions of Runx2 and Msx2 mRNA and protein. The results showed that the mRNA expression of Runx2 and Msx2 was increased in the warfarin group, and the aforementioned increase was significantly reversed by 0.3 mg/ml EGB761 treatment (Figure. 5B and C). Runx2 and Msx2 protein expression was also significantly upregulated by warfarin, while this expression was suppressed by 0.3 mg/ml EGB761 (Figure. 5D).
Figure 5.
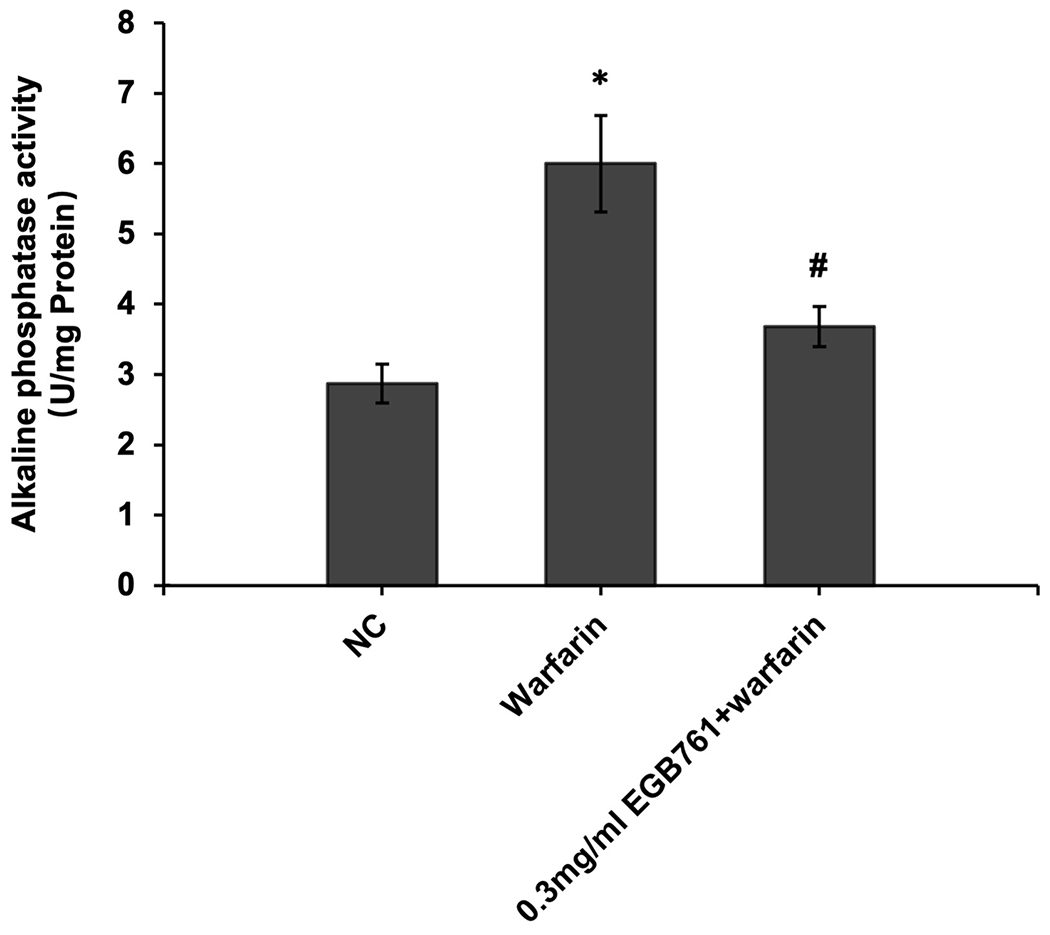
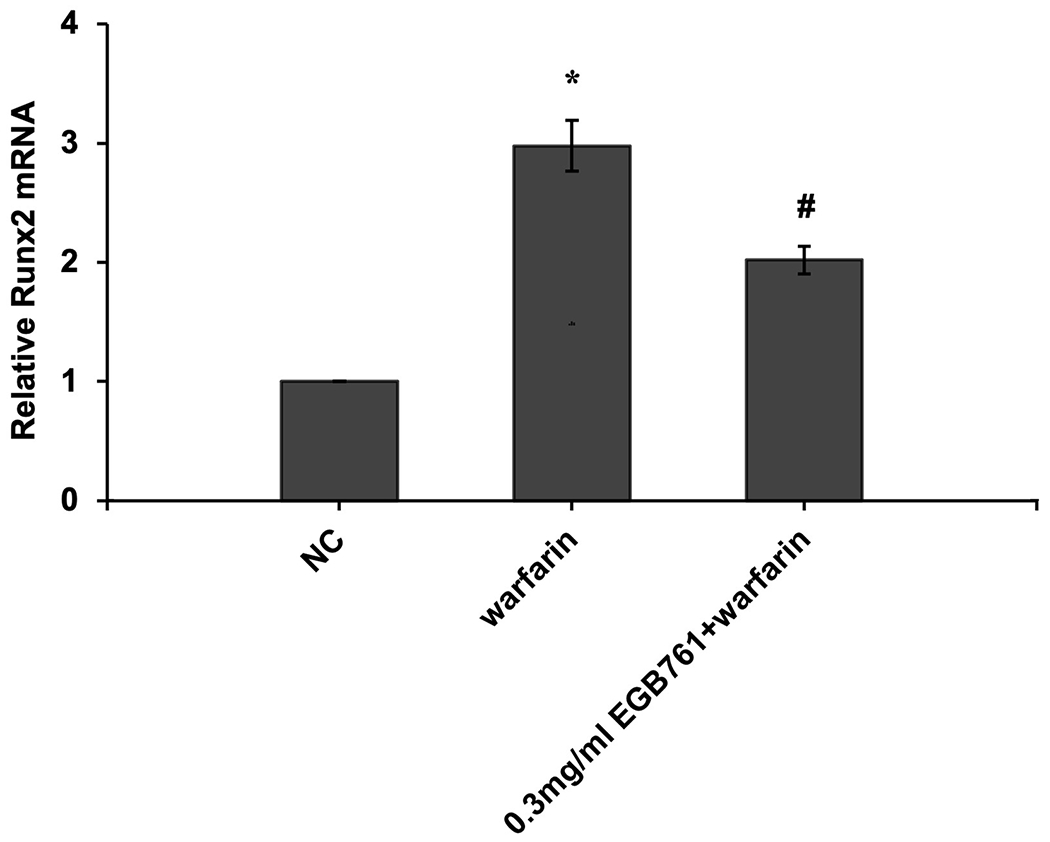
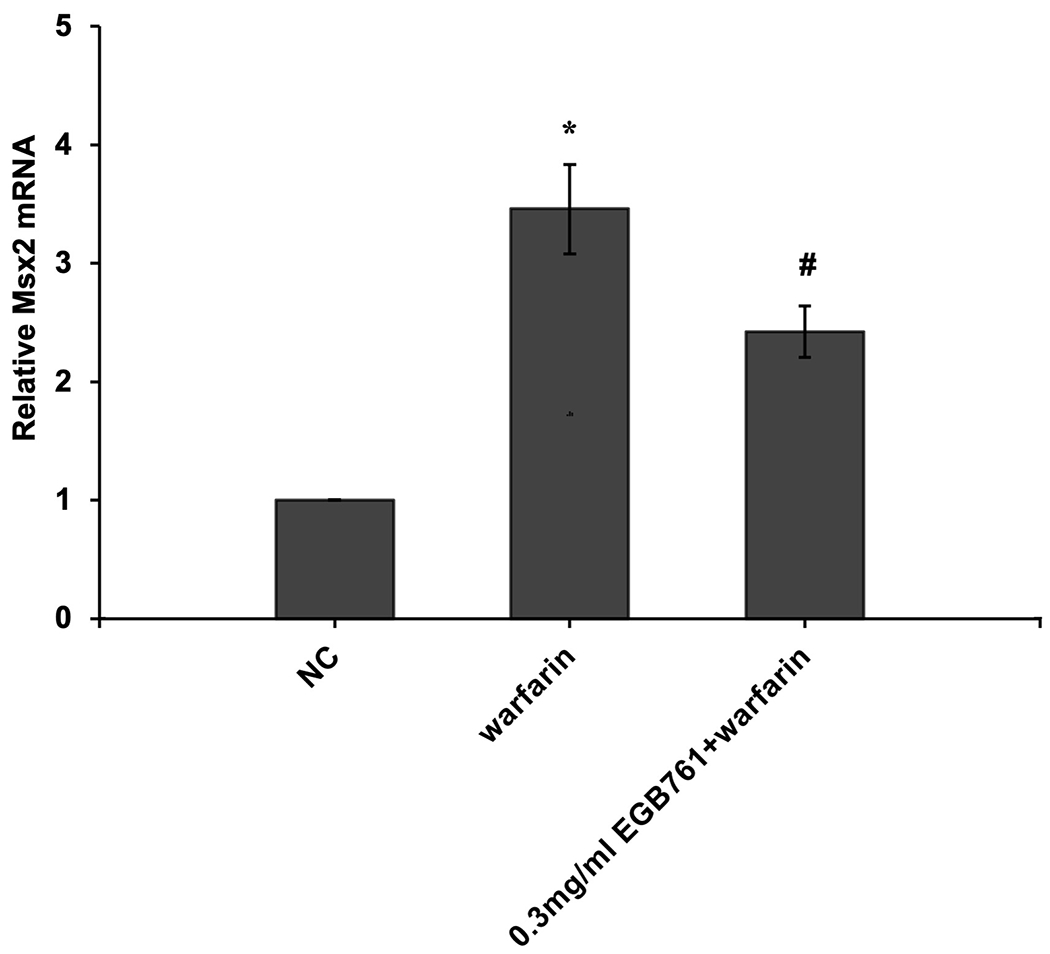
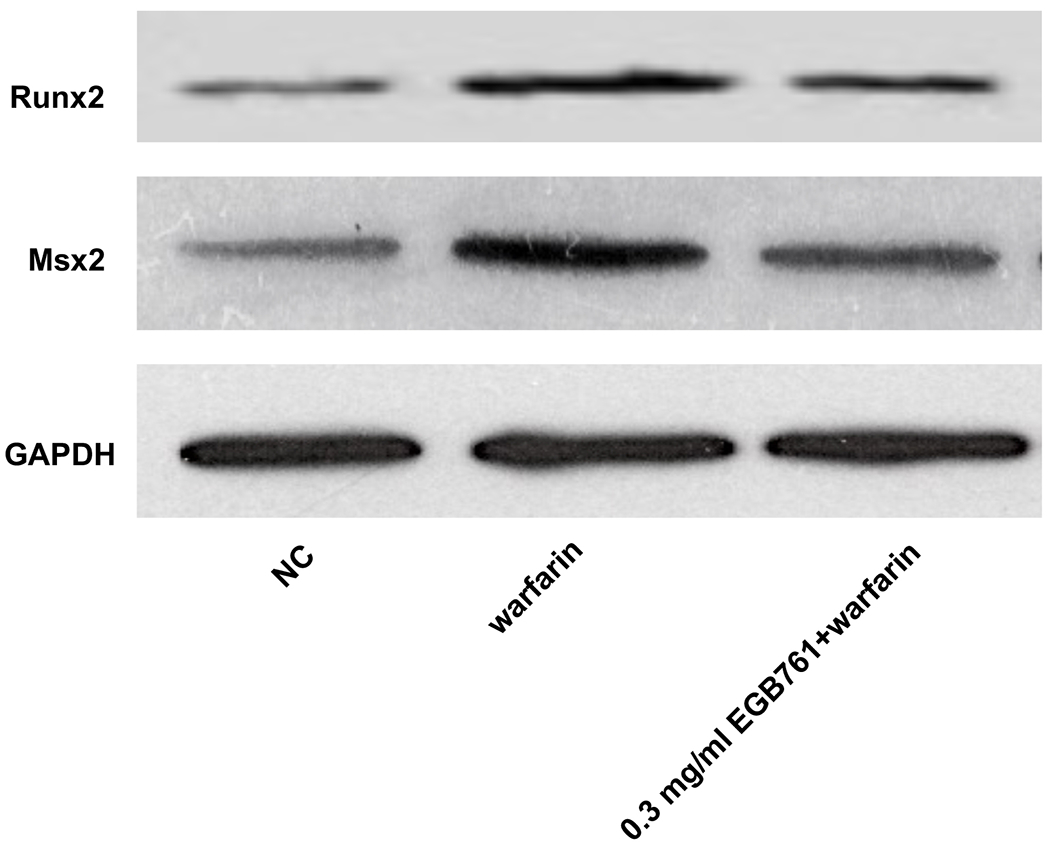
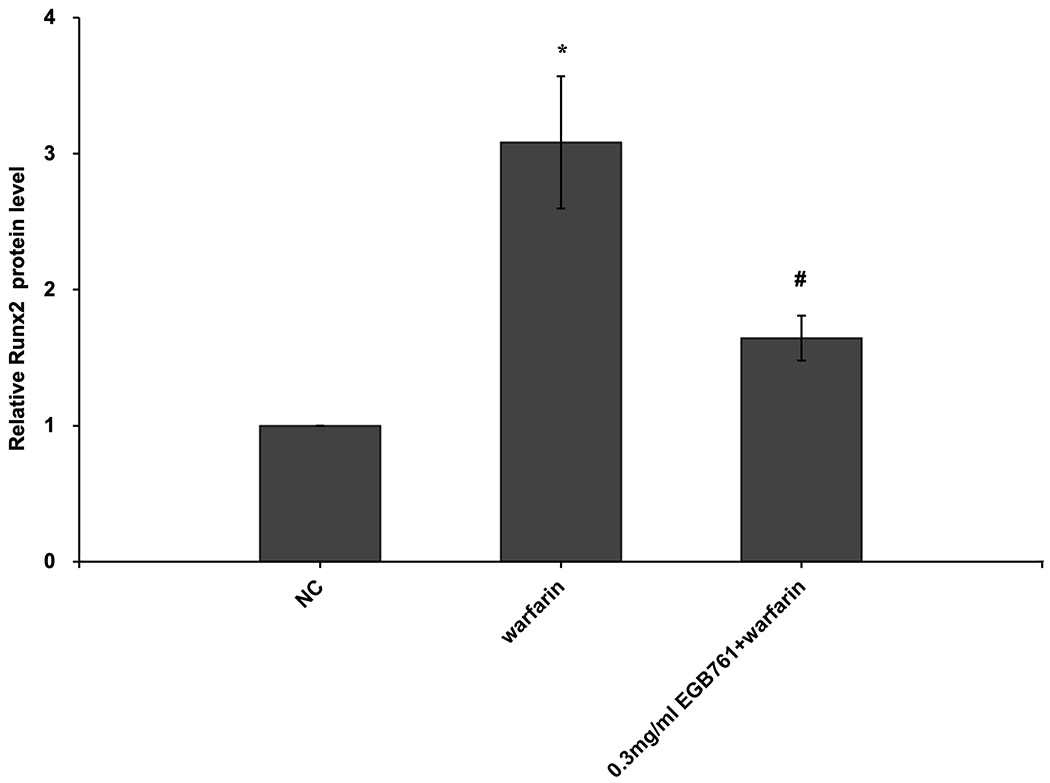
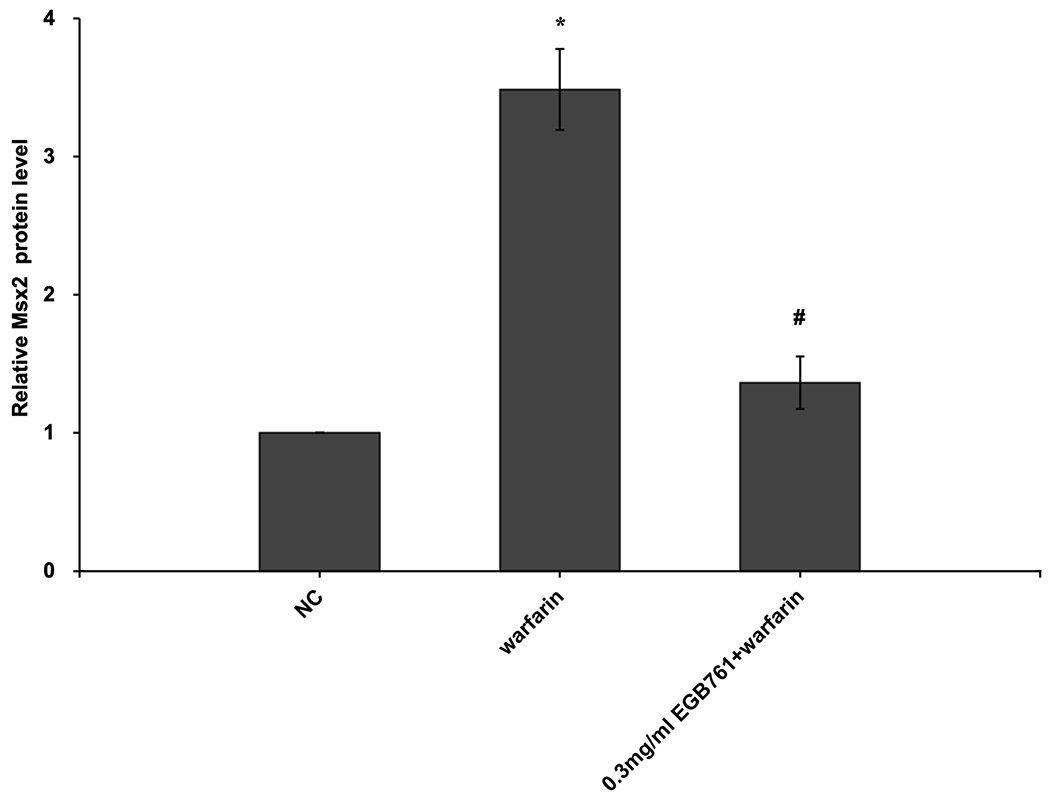
EGB761 suppresses warfarin-induced osteogenic marker expression in pAVICs. (A) ALP activity was measured and normalized to protein content. (B and C) mRNA expression of Runx2 and Msx2 in pAVICs was detected using reverse transcription-quantitative PCR. (D) Representative immunoblots of Runx2 and Msx2 expression. n=6. *P <0.05 vs. NC group; #P<0.05 vs. warfarin group. pAVICs, porcine aortic valve interstitial cells; NC, negative control; ALP, alkaline phosphatase; Runx2, Runt-related transcription factor 2; Msx2, homeobox protein MSX-2.
Moreover, as observed in vitro, 125 mg/kg/d EGB761 significantly downregulated expression of aortic valve Runx2 and Msx2 gene expression compared to warfarin group in vivo (Figure. 6A and B).
Figure 6.
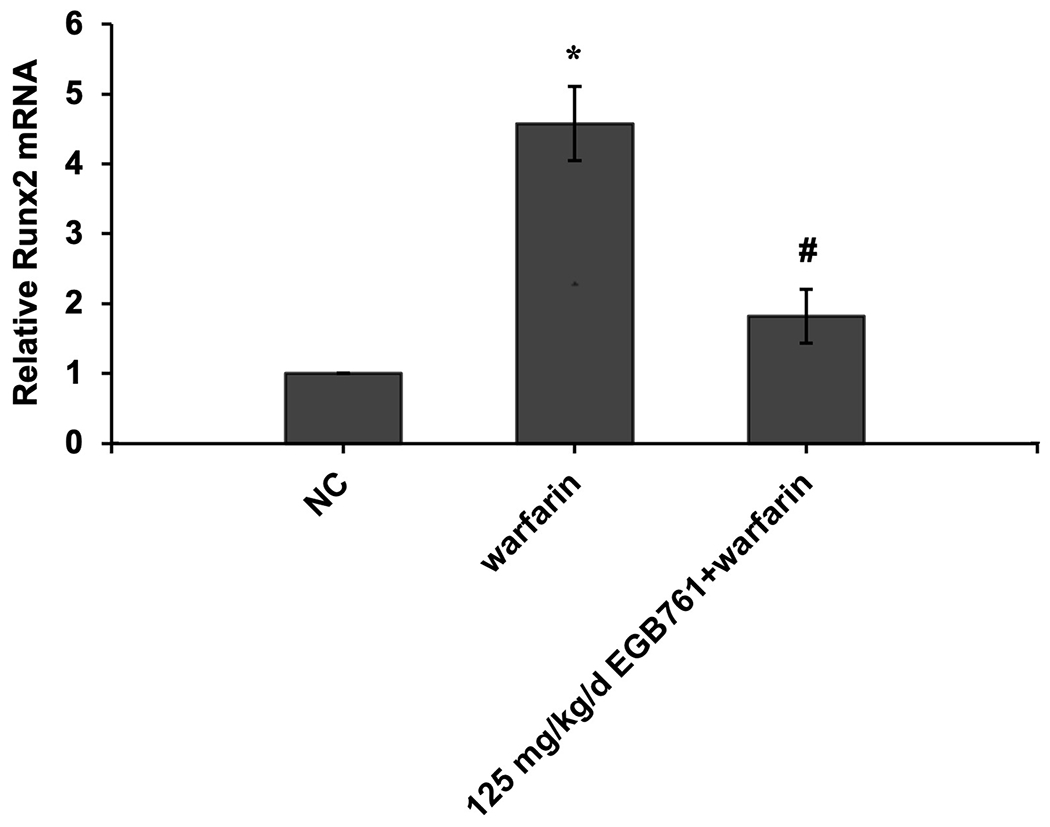
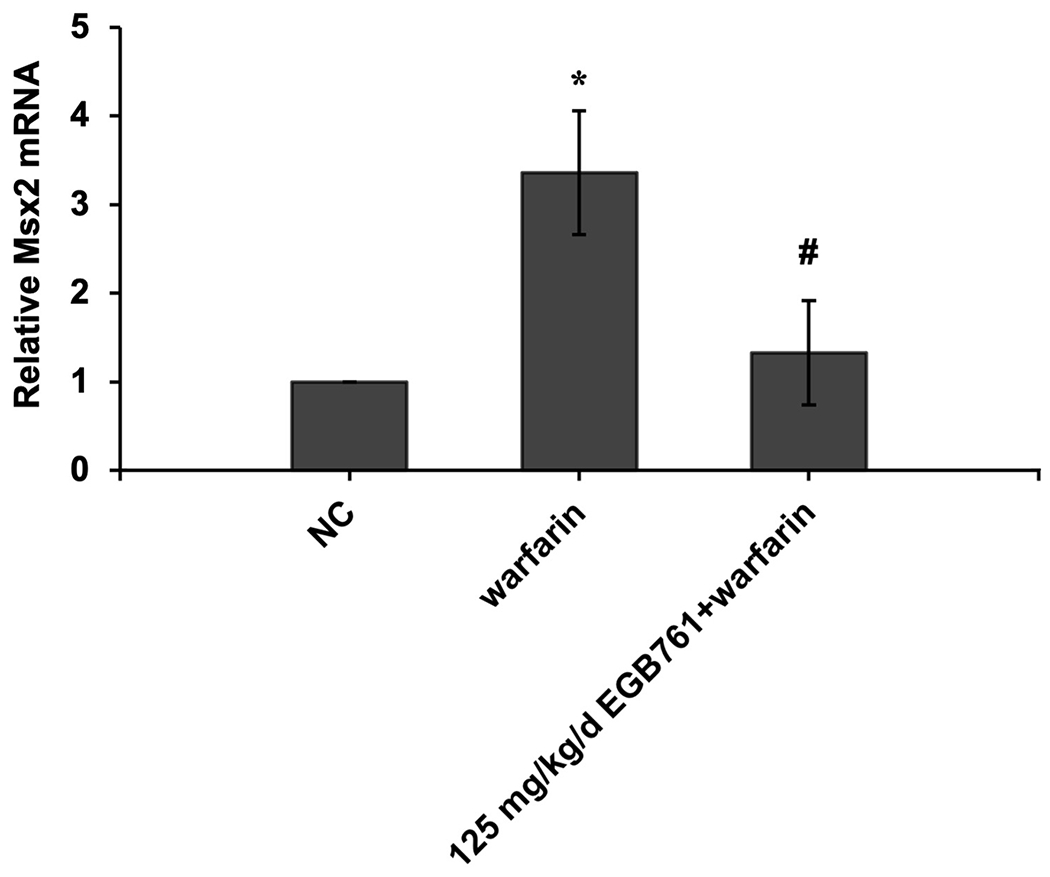
EGB761 suppresses warfarin-induced aortic valve osteogenic marker expression in mice. (A) mRNA expression of Runx2 was detected using reverse transcription-quantitative PCR. (B) mRNA expression of Msx2 was detected using reverse transcription-quantitative PCR. n=14 per group. *P<0.05 vs. NC group; #P<0.05 vs. warfarin group. NC, negative control; Runx2, Runt-related transcription factor 2; Msx2, homeobox protein MSX-2.
EGB761 alleviates pAVIC calcification via the BMP2/Smad1/5/Runx2 signaling pathway.
In order to further determine the potential molecular mechanisms of EGB761 repressing warfarin-induced pAVIC calcification, the levels of BMP2 and its downstream target, p-Smad1/5, were evaluated by RT-qPCR and western blotting. The mRNA and protein expression of BMP2 was significantly increased when pAVICs were treated with warfarin, while this increase was suppressed by EGB761 treatment (Figure. 7A and B). Once BMP2 binds to its receptor, Smad1/5 phosphorylation will be activated, in which p-Smad1/5 may translocate from the cytoplasm to the nucleus to upregulate the expression of osteogenic biomarkers. p-Smad1/5 was detected in the nucleus and cytoplasm. The results demonstrated that the levels of p-Smad1/5 in the nucleus were markedly upregulated in pAVICs stimulated with warfarin compared with the NC group, while EGB761 reversed this increase in expression. Meanwhile, the levels of p-Smad1/5 in the cytoplasm were markedly downregulated in pAVICs stimulated with warfarin, while their levels in the EGB761 group were obviously increased (Figure. 7C).
Figure 7.
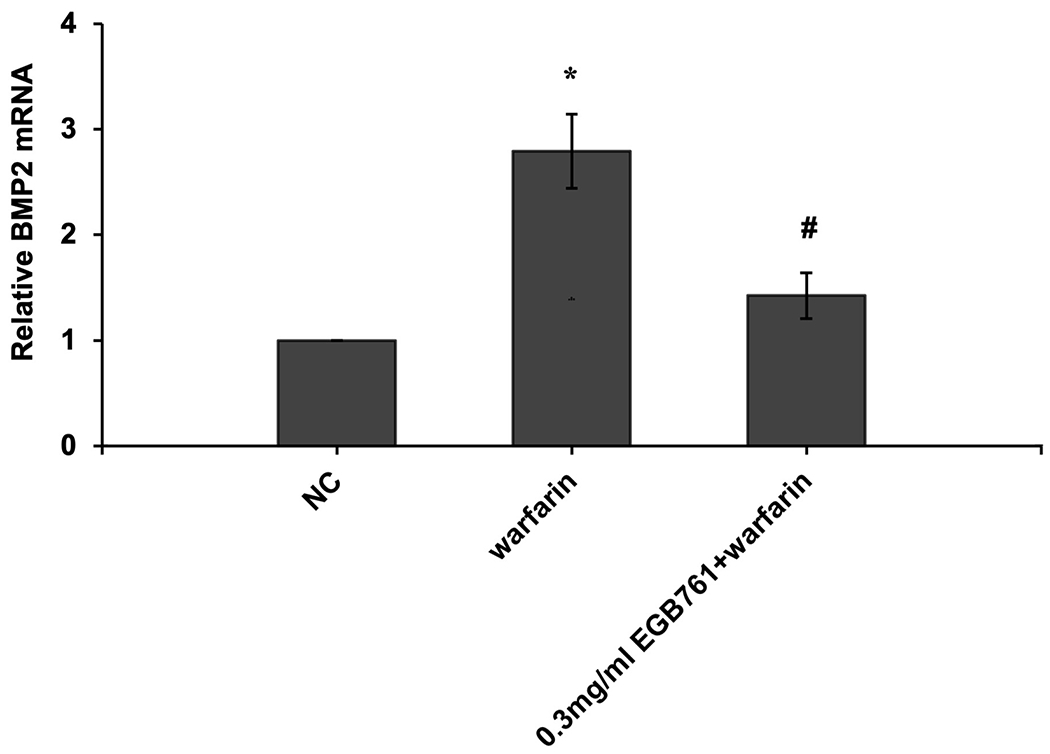
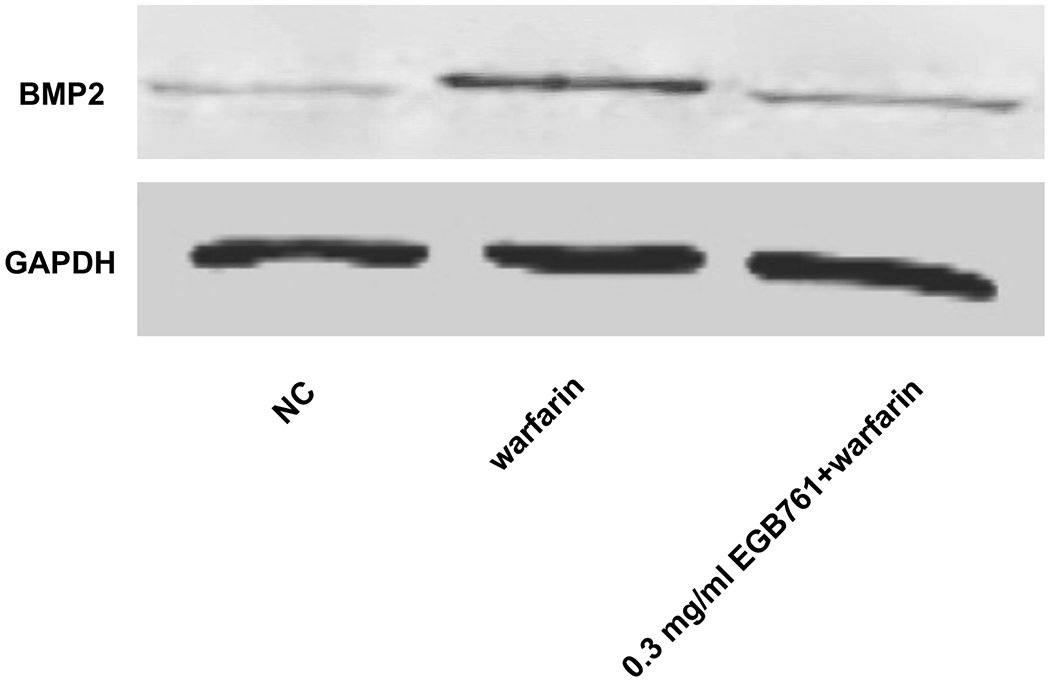
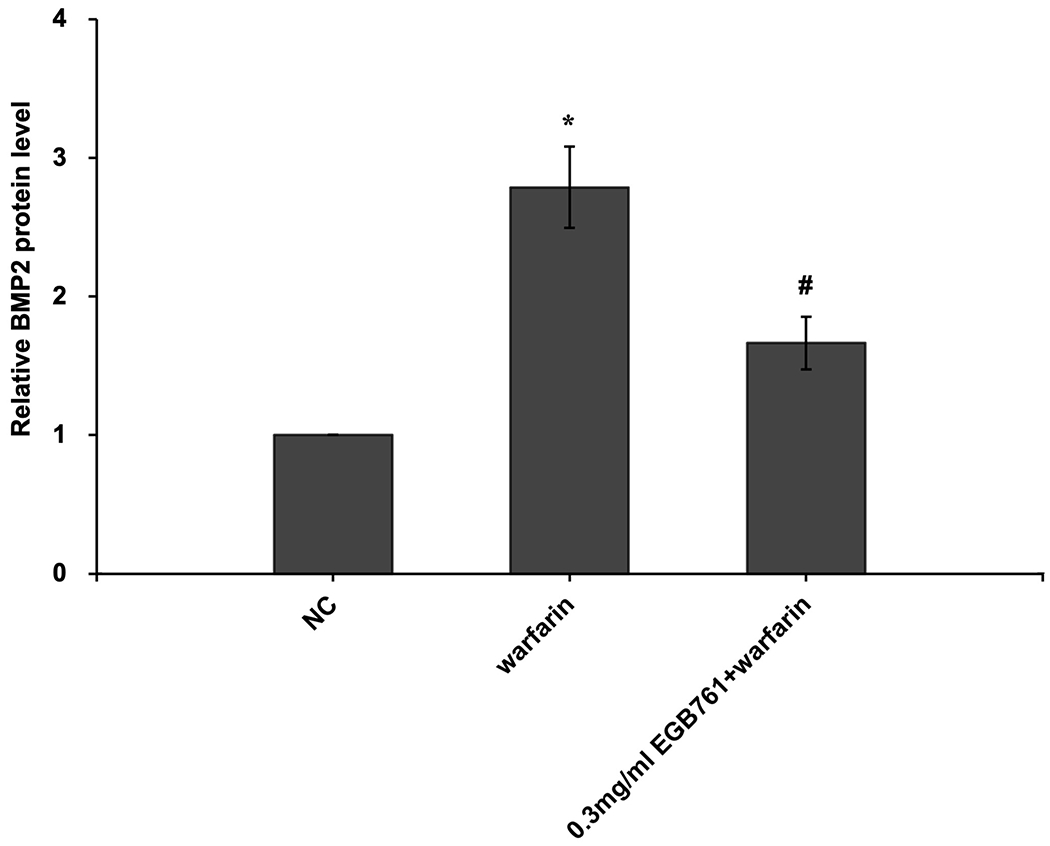
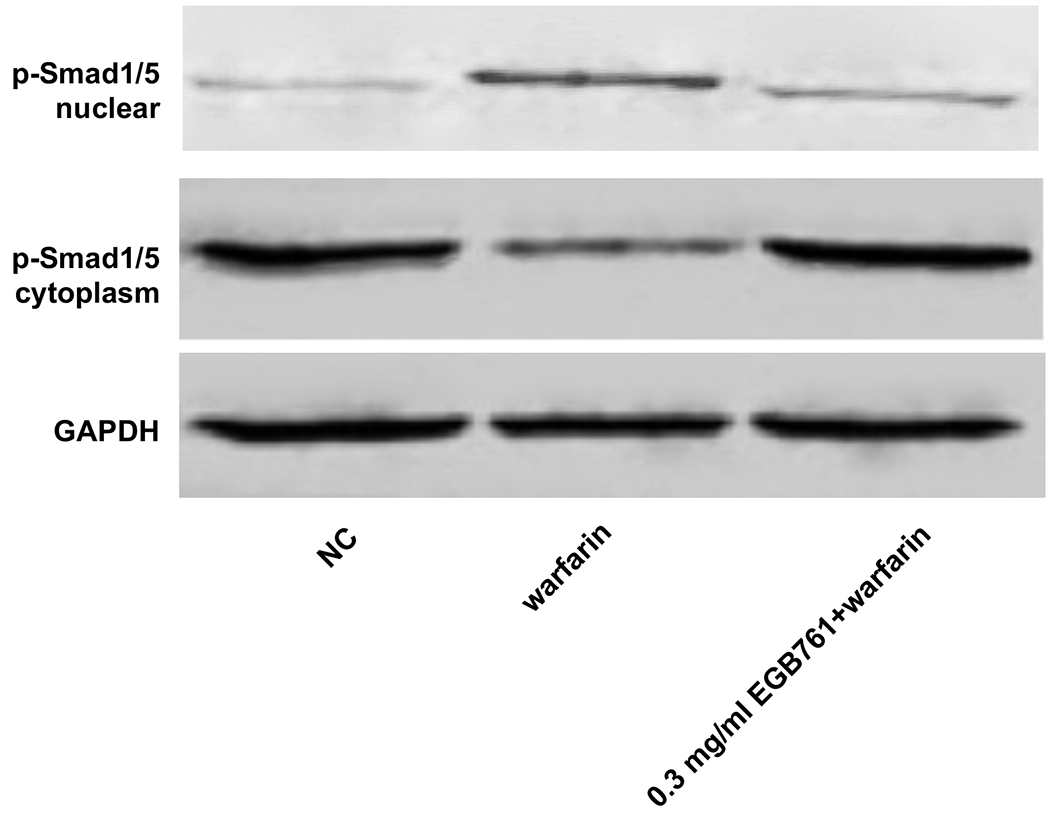
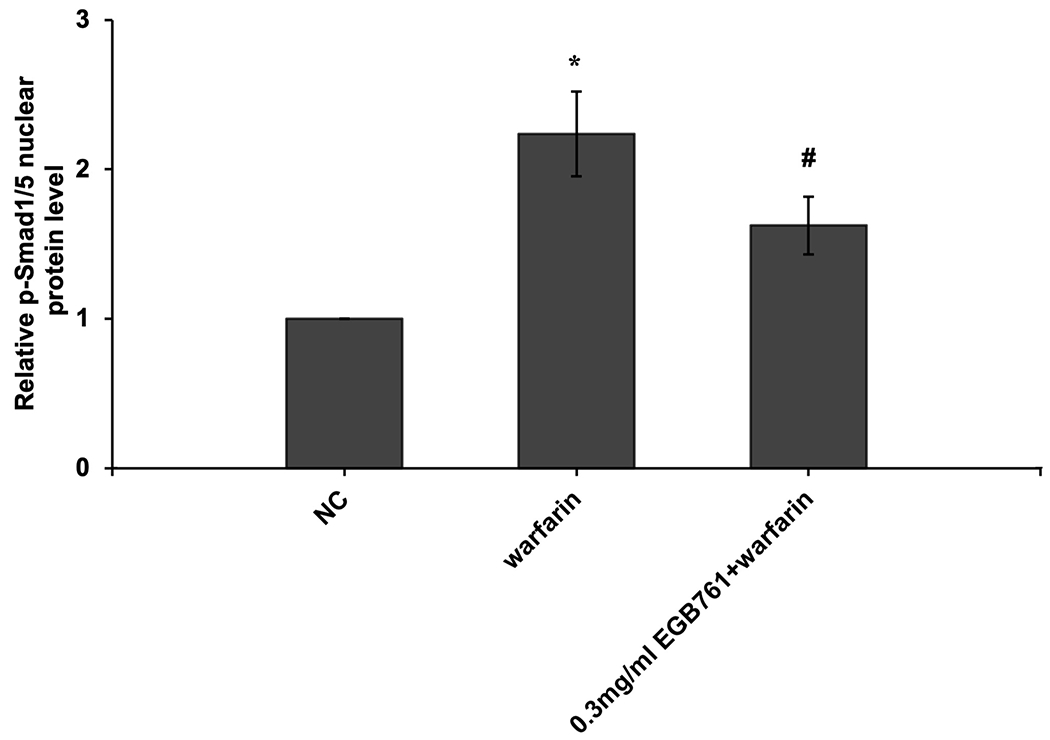
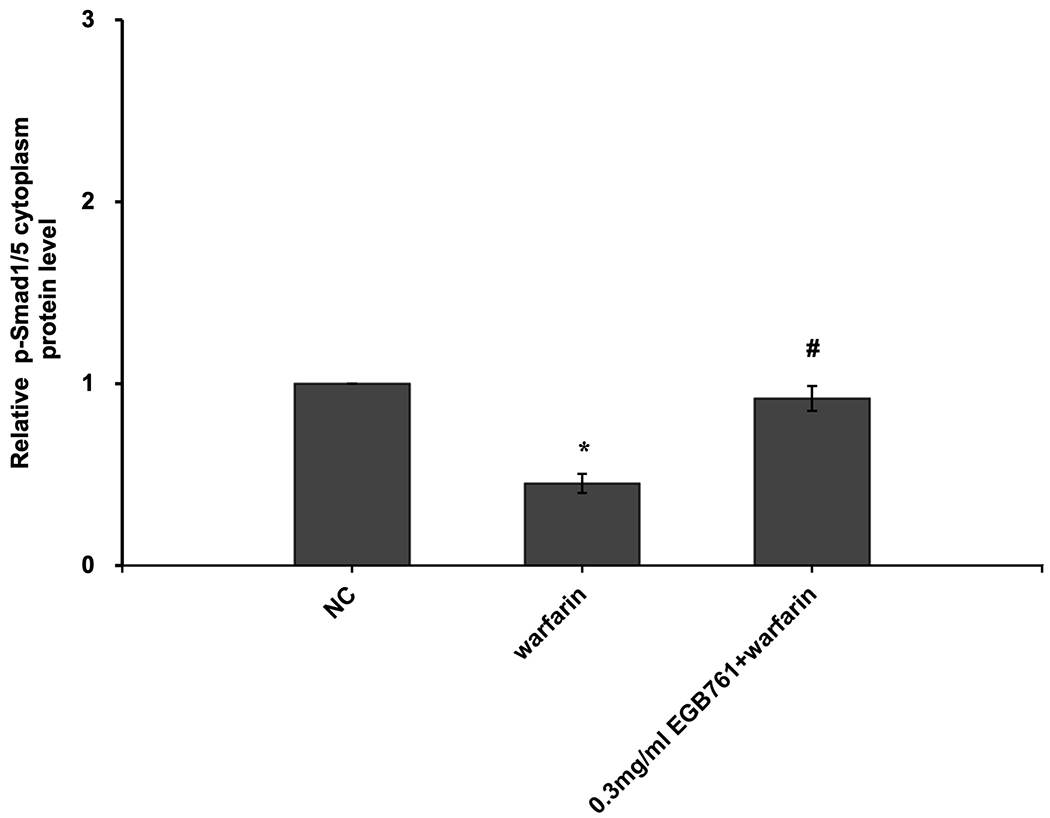
EGB761 blocks pAVIC calcification by inhibiting the BMP2/Smad1/5/Runx2 signaling pathway. (A) BMP-2 mRNA expression was detected by reverse transcription-quantitative PCR. (B) Representative immunoblot of BMP2 staining. (C) Representative immunoblot of p-Smad1/5 expression in the nucleus and cytoplasm. n=6. *P <0.05 vs. NC group; #P<0.05 vs. warfarin group. pAVICs, porcine aortic valve interstitial cells; NC, negative control; p, phosphorylated; BMP2, bone morphogenetic protein 2.
BMP2 attenuates the effects of EGB761 on inhibiting warfarin-induced pAVIC calcification.
To confirm whether the BMP-Smad signaling pathway was involved in mediating warfarin-induced pAVIC calcification, rhBMP2 was used to neutralize the effects of EGB761 on inhibiting warfarin-induced pAVIC calcification. rhBMP2 abrogated the inhibitory effects of EGB761 on calcium deposition and calcium content in pAVICs incubated with warfarin (Figure. 8A and B). In addition, rhBMP2 upregulated ALP activity and the expression of osteogenic biomarkers Runx2 and Msx2 in pAVICs treated with EGB761 (Figure. 8C–E). rhBMP2 also reversed the decrease in Runx2 protein expression by EGB761, while the increase in p-Smad1/5 levels in the cytoplasm by EGB761 was significantly inhibited by rhBMP2 (Figure. 8F).
Figure 8.
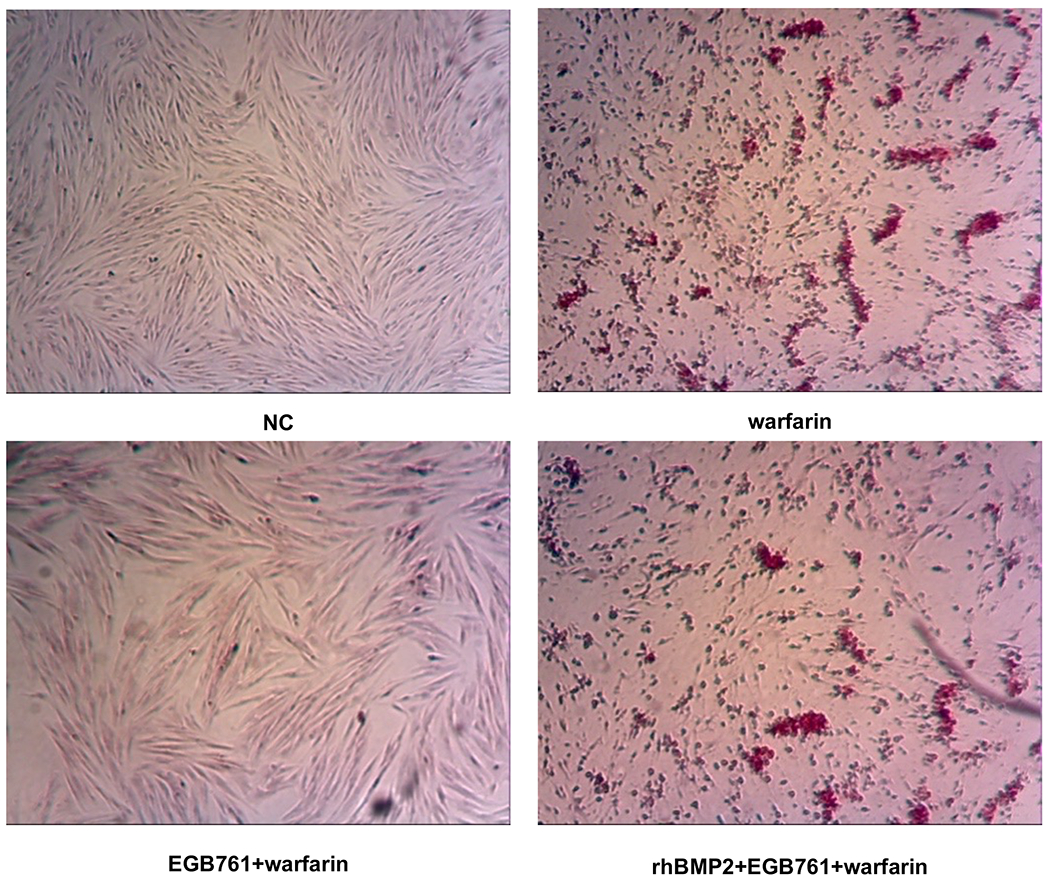
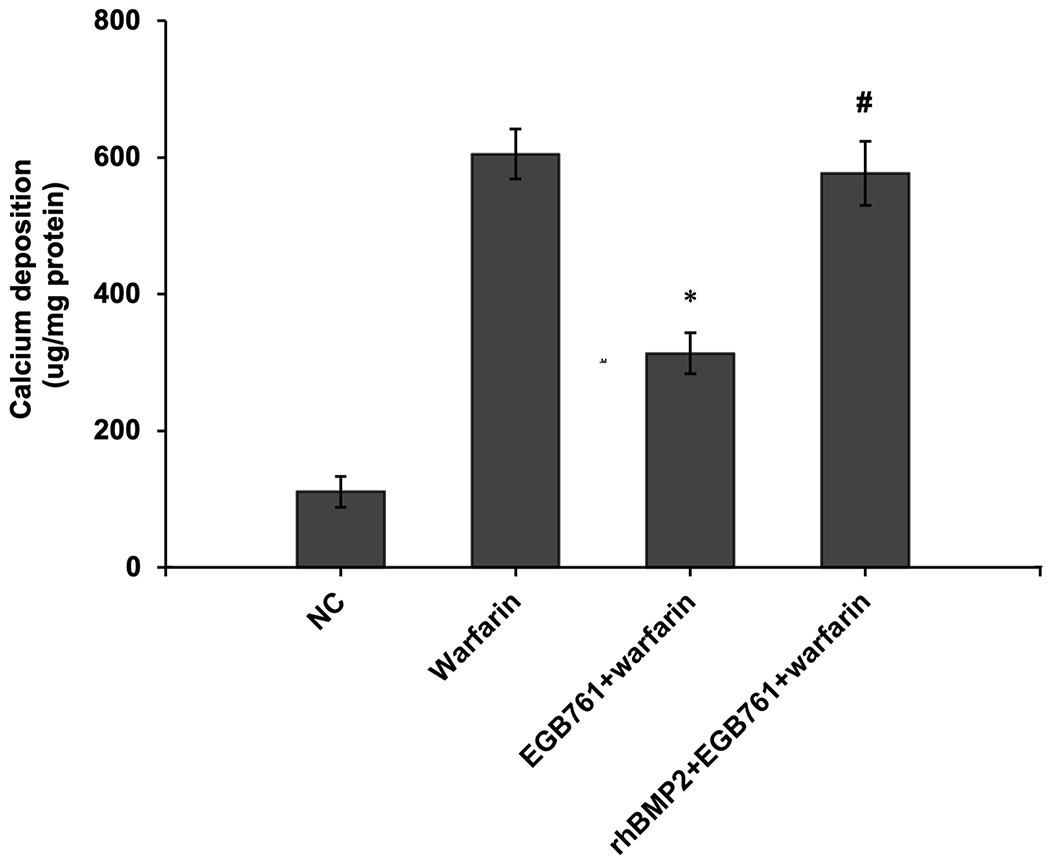

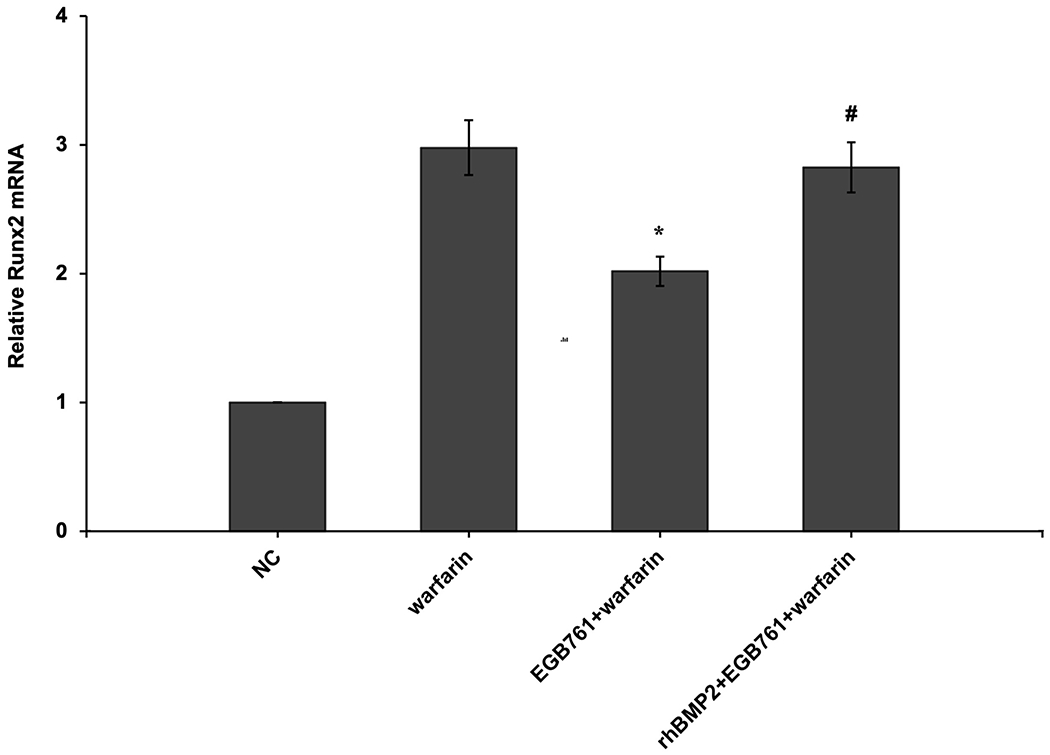
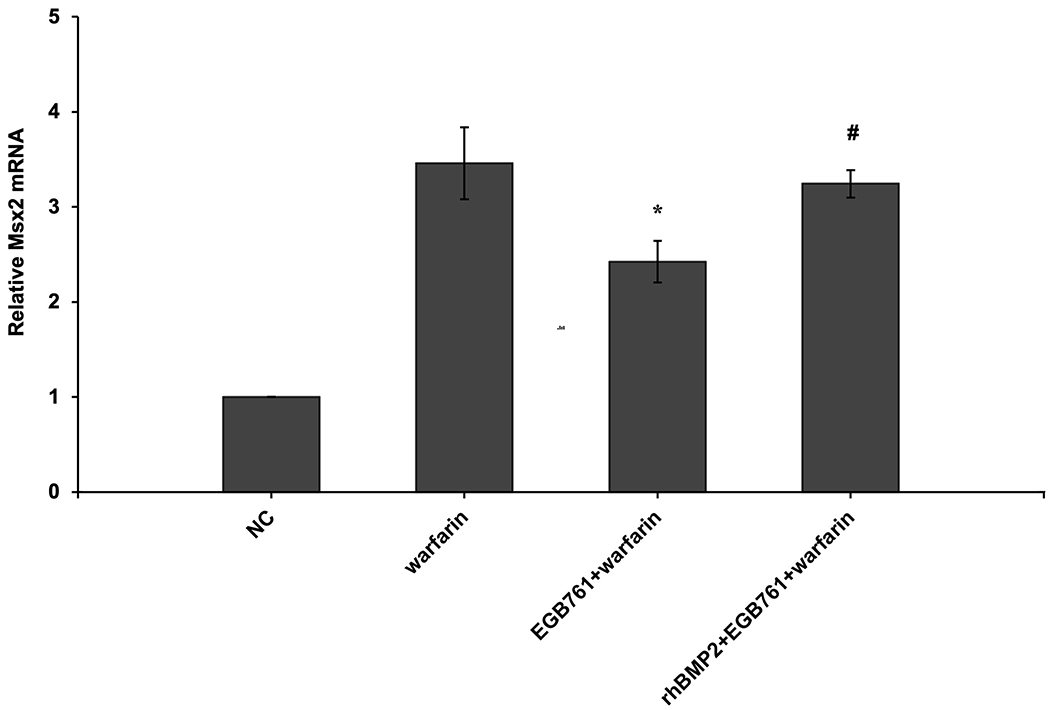
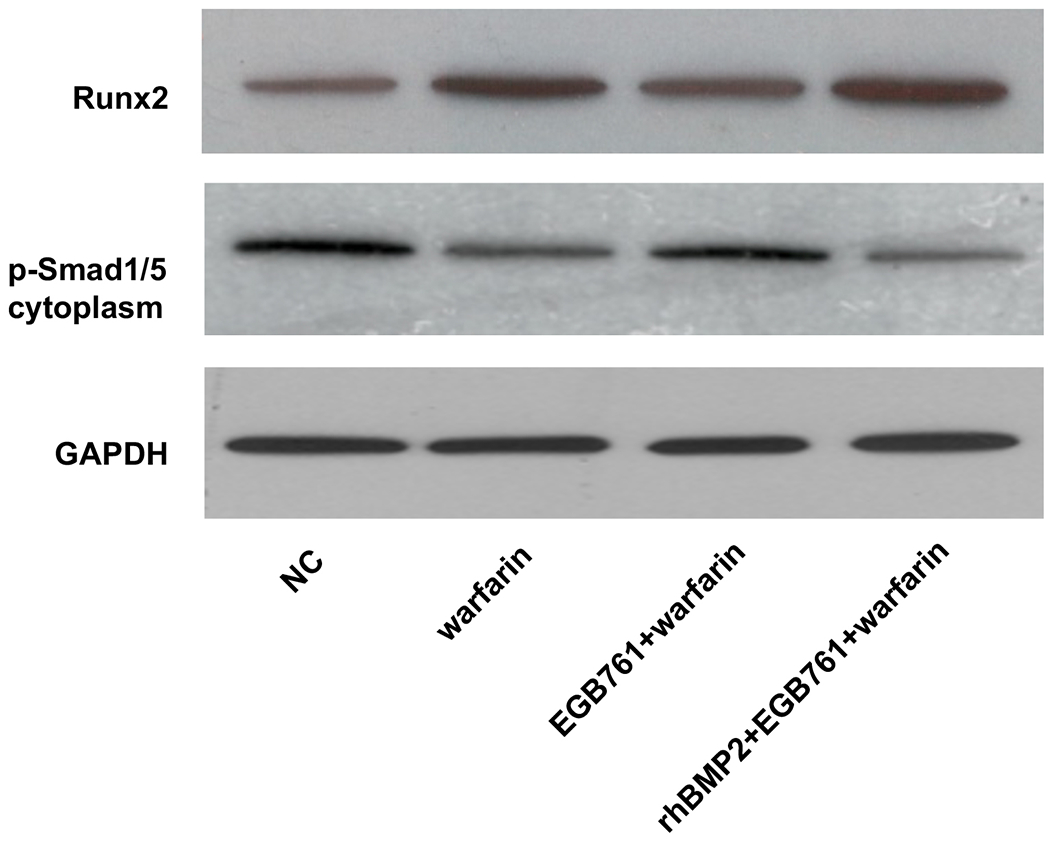
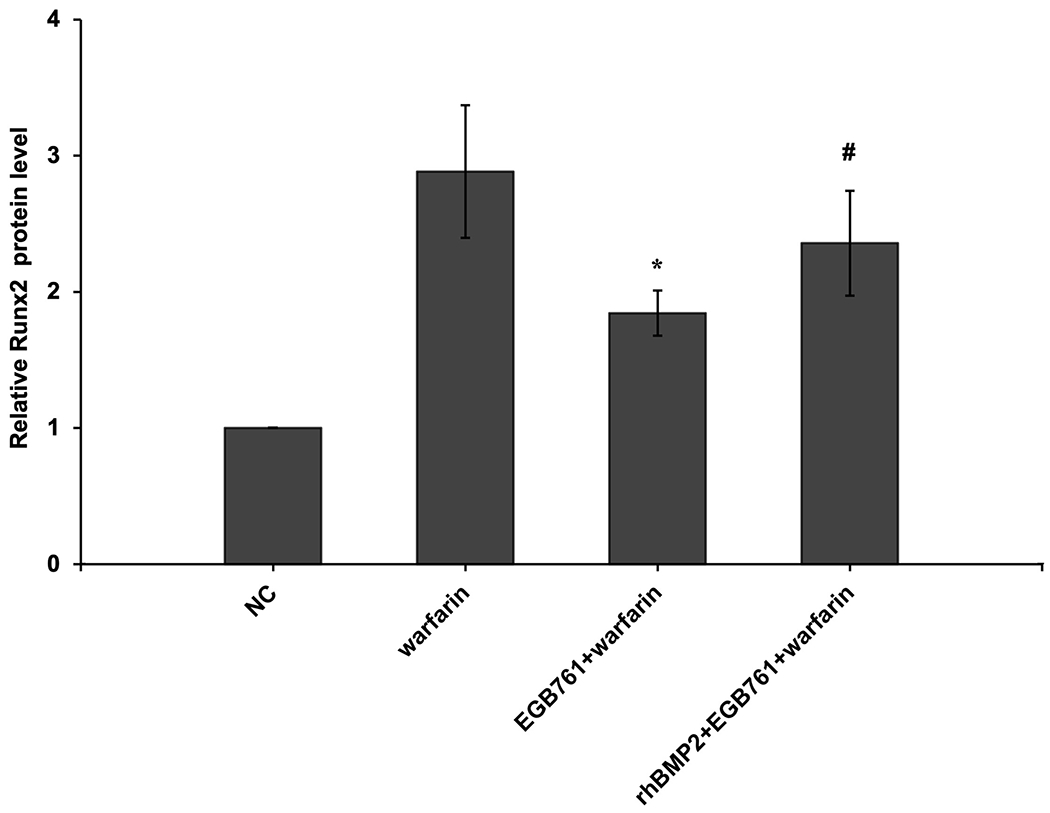
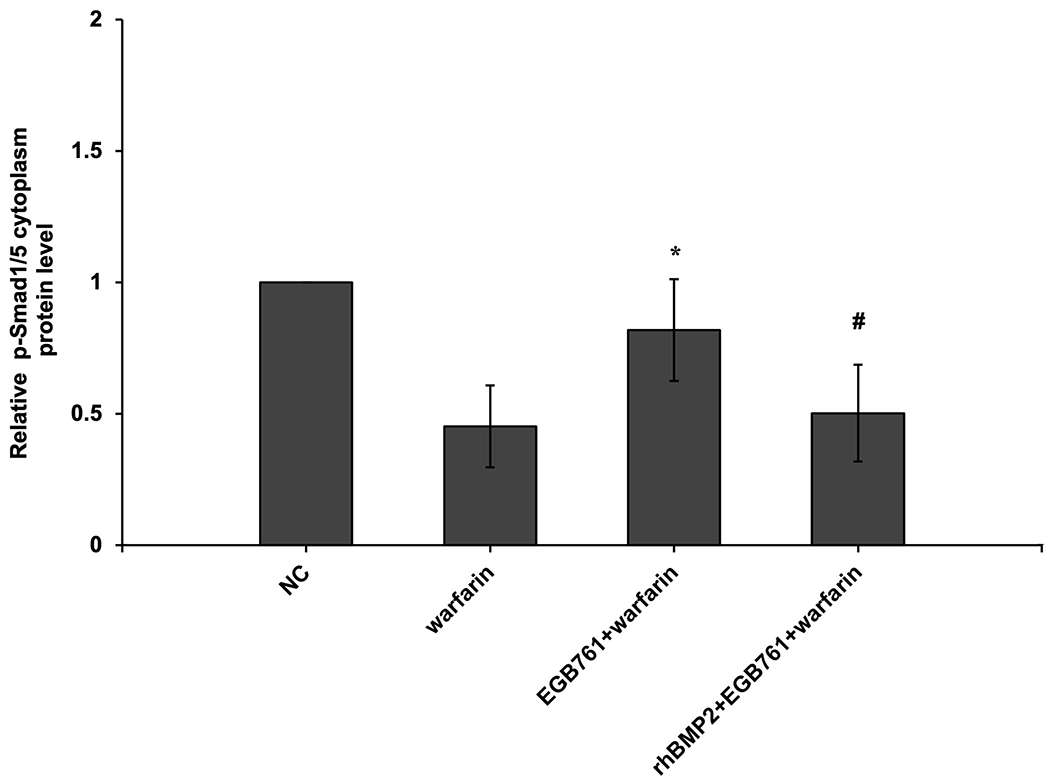
BMP2 attenuates the effects of EGB761 on inhibiting warfarin-induced pAVIC calcification. (A) Microscopy images (x40 magnification; bottom). Orange-red mineralized nodules indicate matrix calcium deposition. (B) Intracellular calcium levels corrected for protein concentration. (C) ALP activity was measured and normalized to protein content. (D and E) mRNA expression levels of Runx2 and Msx2 were detected by reverse transcription-quantitative PCR. (F) Representative immunoblot of Runx2 and p-Smad1/5 expression in the cytoplasm. n=6. *P<0.05 vs. warfarin group; #P<0.05 vs. EGB761 group. pAVICs, porcine aortic valve interstitial cells; NC, negative control; p, phosphorylated; BMP2, bone morphogenetic protein 2; ALP, alkaline phosphatase; Runx2, Runt-related transcription factor 2; Msx2, homeobox protein MSX-2.
Discussion
Calcification is ubiquitous in the progression of various diseases, including atherosclerosis, diabetes, hypertension, chronic kidney disease and dyslipidemia (20). Valve calcification is the degeneration and fibrosis of valve tissue. The main pathological features are valve leaflet thickening, matrix remodeling and calcium deposition. This ultimately leads to valve stenosis and/or insufficiency, which seriously affects the normal function of the valve (21,22). At present, CAVD is no longer considered to be a passive process of valve degradation with age and calcium accumulation, but an actively-regulated progressive disease (23). Increasing evidence from human and animal studies suggested that CAVD is similar to the osteogenic differentiation process, which is strictly regulated by numerous signaling pathways (24,25). Osteogenic differentiation is considered as a critical step in the process of valve calcification. Therefore, the identification of mechanisms associated with osteoblast differentiation may be a key regulatory point to prevent or delay valve calcification.
EGB761 has received attention due to its well-known pharmacological effects in the modern medicine treatment of cardiovascular disease. Several studies reported that the antioxidant and antiplatelet role of EGB761 has been observed in a number of animal disease models and clinical trials, including atherosclerosis (26), glucose and lipid metabolism (14), cerebral ischemia (27), and Alzheimer’s disease (28). Recent studies have shown the protective effects of EGB761 on the calcification of vascular smooth muscle cells in vitro. Li et al (16) found that EGB761 significantly reduced calcium deposition and inhibited osteogenic differentiation in rat aortic vascular smooth muscle cells. Wang et al (17) additionally reported that EGB761 prevented β-glycerophosphate-induced vascular smooth muscle cell calcification by inhibiting the Wnt/β-Catenin signaling pathway. Therefore, it may be used as a potential method for the clinical prevention and treatment of calcification.
In the present study, pAVICs incubated with therapeutic concentrations (10 μM) of warfarin combined with low dose (1.6 mM) Pi was used as an artificially inducible AVIC calcification model and to further investigate potential anticalcification and osteogenic differentiation effects of EGB761 and its underlying molecular mechanisms.
Our previous research highlighted that 10 μM warfarin rapidly induced calcification in vitro (11). The data of the present study established that mineralized nodules and calcium content were increased in the presence of 10 μM warfarin and 1.6 mM Pi, which was consistent with the findings of previous reports investigating vascular calcification (8). Tsang et al (1) also confirmed that 2.7 mM calcium potently induced AVIC calcification, whereas Pi treatment alone had no effect, regardless of the concentration. Upon addition of EGB761, mineralized nodules and intracellular calcium content significantly decreased, which may indicate that EGB761 significantly inhibited warfarin-induced AVIC calcification and calcium deposition.
In order to further investigate the inhibitory effects of EGB761 on AVIC osteogenic differentiation, ALP activity (an important early osteogenic biomarker) and Runx2 and Msx2 expression (biomarkers related to the phenotypic transition of bone formation) were evaluated. The results revealed that ALP activity and expression of osteogenic differentiation markers in AVICs were significantly increased. However, EGB761 reversed these effects and thus, alleviated osteogenic differentiation in AVICs. Consistent with these in vitro data, we also confirmed that administration of EGB761 significantly ameliorated aortic valve calcification in warfarin-treated mice, as evidenced by decreased calcium deposits and the expressions of osteogenic differentiation markers (Runx2 and Msx2) in the aortic valve.
Notch, BMP/Smad and Wnt/β-catenin signaling pathways have been identified to be associated with AVIC osteogenesis and calcification (29–32). BMP/Smad is critical for warfarin-induced AVIC osteogenesis and calcification (33). BMPs are reported to be superfamily members of TGF-β and affect bone formation. BMP2 is a key factor that promotes bone formation and osteogenic differentiation in calcified valves (34–37). In a biglycan-induced calcification model, BMP2 expression was upregulated in human AVICs (19). In addition, BMP2 increased inflammatory mediator expression to stimulate osteoblast-like phenotype transformation of AVICs (38). Yang et al (39) showed calcified nodule formation in AVICs stimulated with BMP2. Therefore, BMP2 signaling is expected to be a therapeutic target for valve calcification in the future. The present data indicated that when AVICs were stimulated with 10 μM warfarin, the protein levels of BMP2 significantly increased. Meanwhile, this was reduced upon EGB761 treatment compared with the warfarin group (P<0.05). The results suggested that EGB761 alleviated warfarin-induced AVIC calcification and osteogenic differentiation by inhibiting BMP2 expression.
Several studies demonstrated that BMP2 initiates signaling via binding to type-I and type II BMP receptors (BMPR-I and BMPR-II, respectively). BMPR-I is phosphorylated by BMPR-II kinase, which in turn activates receptor-regulated Smad. p-Smad, especially Smad1/5/8, eventually translocates into the nucleus to modulate the transcription of target genes, such as Runx2 and Msx2 (40–42). Western blot analysis further showed that the levels of p-Smad1/5 in the cytoplasm were markedly downregulated when induced by warfarin, and upregulated upon treatment with EGB761. The present study hypothesized that EGB761 can suppress warfarin-induced pAVIC osteogenic differentiation by inhibiting Smad1/5 phosphorylation to prevent its translocation from the cytoplasm to the nucleus.
To further verify the role of the BMP2/ Smad1/5/Runx2 signaling pathway on the inhibitory effects of EGB761 on AVIC calcification, AVICs were treated with EGB761 in the presence of rhBMP2. Consistent with our previous data, it was found that the addition of rhBMP2 to EGB761 and warfarin-treated AVICs reversed the inhibitory effects of EGB761 on warfarin-induced AVIC calcification. Calcium deposition and calcium content, which were inhibited by EGB761 in the AVICs, were strengthened upon the addition of rhBMP2. The expression of several osteoblastic transformation genes (Runx2, Msx2 and ALP), which were decreased by EGB761, were also upregulated by rhBMP2. In addition, rhBMP2 also significantly decreased the levels of p-Smad1/5 in the cytoplasm, which were upregulated by EGB761. These results indicated that rhBMP2 counteracted the role of EGB761 in preventing the translocation of p-Smad1/5 from the cytoplasm to the nucleus. The aforementioned results demonstrated that EGB761 inhibited AVIC calcification and osteogenic differentiation by suppressing the BMP2/Smad1/5/Runx2 signaling pathway.
Conclusion
Taken together, the present study demonstrated for the first time that EGB761 can alleviate warfarin-induced AVIC calcification by inhibiting BMP2 expression and repressing the BMP2/Smad1/5/Runx2 signaling pathway, thus protecting against warfarin-induced AVIC calcification. The present findings indicated that EGB761, as a therapeutic agent, may be used to prevent or treat aortic valve calcification diseases.
Funding
The present study was supported by the Intramural Research Program of the NINDS at NIH, the Extramural Research Program of the NIBIB (grant no. R00EB016753) and the National Natural Science Foundation of China (grant no. 81900349).
Footnotes
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Ethics approval and consent to participate
All experimental procedures were approved by the Experimental Animal Care and Use Committee of the University of Michigan State and Nanjing Medical University, and conducted in accordance with the Guide for the Care and Use of Laboratory Animals (NIH publication No. 85-23, revised 1996).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Conflict of Interest Disclosure
The authors have no conflicts of interest to declare.
References
- 1.Tsang HG, Cui L, Farquharson C, Corcoran BM, Summers KM, Macrae VE. Exploiting novel valve interstitial cell lines to study calcific aortic valve disease. Mol Med Rep. 2018. February;17(2):2100–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lindman BR, Clavel MA, Mathieu P, Iung B, Lancellotti P, Otto CM, Pibarot P. Calcific aortic stenosis. Nat Rev Dis Primers 2. 2016March3; 2:16006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pawade TA, Newby DE, Dweck MR. Calcification in Aortic Stenosis: The Skeleton Key. J Am Coll Cardiol. 2015August4;66(5):561–77. [DOI] [PubMed] [Google Scholar]
- 4.Mohler ER, Gannon F, Reynolds C, Zimmerman R, Keane MG, Kaplan FS. Bone formation and inflammation in cardiac valves. Circulation. 2001March20;103(11):1522–8. [DOI] [PubMed] [Google Scholar]
- 5.Lerner RG, Aronow WS, Sekhri A, Palaniswamy C, Ahn C, Singh T,Sandhu R, McClung JA. Warfarin use and the risk of valvular calcification. J Thromb Haemost. 2009. December;7(12):2023–7. [DOI] [PubMed] [Google Scholar]
- 6.Rennenberg RJ, van Varik BJ, Schurgers LJ, Hamulyak K, Ten Cate H, Leiner T, Vermeer C, de Leeuw PW, Kroon AA. Chronic coumarin treatment is associated with increased extracoronary arterial calcification in humans. Blood. 2010. June 17;115(24):5121–3. [DOI] [PubMed] [Google Scholar]
- 7.Holden RM, Sanfilippo AS, Hopman WM, Zimmerman D, Garland JS, Morton AR. Warfarin and aortic valve calcification in hemodialysis patients. J Nephrol. 2007;20(4):417–22. [PubMed] [Google Scholar]
- 8.Beazley KE, Eghtesad S, Nurminskaya MV. Quercetin attenuates warfarin-induced vascular calcification in vitro independently from matrix Gla protein. J Biol Chem. 2013January25;288(4):2632–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zebboudj AF, Imura M, Bostrom K. Matrix GLA protein, a regulatory protein for bone morphogenetic protein-2. J Biol Chem. 2002. February 8;277(6):4388–94. [DOI] [PubMed] [Google Scholar]
- 10.Beazley KE, Deasey S, Lima F, Nurminskaya MV. Transglutaminase 2-mediated activation of beta-catenin signaling has a critical role in warfarin-induced vascular calcification. Arterioscler Thromb Vasc Biol. 2012January;32(1):123–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gao L, Ji Y, Lu Y, Qiu M, Shen YJ, Wang YQ, Kong XQ, Shao YF, Sheng YH, Sun W. Low-level overexpression of p53 promotes warfarin-induced calcification of porcine aortic valve interstitial cells by activating Slug gene transcription. J Biol Chem. 2018March9;293(10):3780–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yeh YC, Liu TJ, Wang LC, Lee HW, Ting CT, Lee WL, Hung CJ, Wang KY, Lai HC. A standardized extract of Ginkgo biloba suppresses doxorubicin-induced oxidative stress and p53-mediated mitochondrial apoptosis in rat testes. Br J Pharmacol. 2009. January;156(1):48–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gautam J, Kushwaha P, Swarnkar G, Khedgikar V, Nagar GK, Singh D, Singh V, Jain M, Barthwal M, Trivedil R. EGb 761 promotes osteoblastogenesis, lowers bone marrow adipogenesis and atherosclerotic plaque formation. Phytomedicine. 2012. September 15;19(12):1134–42. [DOI] [PubMed] [Google Scholar]
- 14.Tian JF, Liu Y, Chen KJ. Ginkgo biloba Extract in Vascular Protection: Molecular Mechanisms and Clinical Applications. Curr Vasc Pharmacol. 2017;15(6):532–48. [DOI] [PubMed] [Google Scholar]
- 15.Shen J, Wang J, Zhao B, Hou J, Gao T, Xin W. Effects of EGb 761 on nitric oxide and oxygen free radicals, myocardial damage and arrhythmia in ischemia-reperfusion injury in vivo. Biochim Biophys Acta. 1998. April 28;1406(3):228–36. [DOI] [PubMed] [Google Scholar]
- 16.Li EG, Tian J, Xu ZH. Effects of Gingko biloba extract (EGb 761) on vascular smooth muscle cell calcification induced by beta-glycerophosphate. Ren Fail. 2016;38(4):552–7. [DOI] [PubMed] [Google Scholar]
- 17.Wang J, Qiu XB, Xu TH, Sheng ZT, Yao L. Sclerostin/Receptor Related Protein 4 and Ginkgo Biloba Extract Alleviates beta-Glycerophosphate-Induced Vascular Smooth Muscle Cell Calcification By Inhibiting Wnt/beta-Catenin Pathway. Blood Purif. 2019;47 Suppl 1(Suppl 1):17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Taylor PM, Allen SP, Yacoub MH. Phenotypic and functional characterization of interstitial cells from human heart valves, pericardium and skin. J Heart Valve Dis. 2000January;9(1):150–8. [PubMed] [Google Scholar]
- 19.Song R, Fullerton DA, Ao L, Zheng D, Zhao KS, Meng XZ. BMP-2 and TGF-beta1 mediate biglycan-induced pro-osteogenic reprogramming in aortic valve interstitial cells. J Mol Med (Berl). 2015April;93(4):403–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yutzey KE, Demer LL, Body SC, Huggins GS, Towler DA, Giachelli CM, Hofmann-Bowman MA, Mortlock DP, Rogers MB, Sadeghi MM, Aikawa E. Calcific aortic valve disease: a consensus summary from the Alliance of Investigators on Calcific Aortic Valve Disease. Arterioscler Thromb Vasc Biol. 2014. November;34(11):2387–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Towler DA. Molecular and cellular aspects of calcific aortic valve disease. Circ Res. 2013July5;113(2):198–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Steiner I, Kasparová P, Kohout A, Dominik J. Bone formation in cardiac valves: a histopathological study of 128 cases. Virchows Arch. 2007. June;450(6):653–7. [DOI] [PubMed] [Google Scholar]
- 23.Galeone A, Brunetti G, Oranger A, Greco G, Di Benedetto A, Mori G, Colucci S, Zallone A, Paparella D, Granol M. Aortic valvular interstitial cells apoptosis and calcification are mediated by TNF-related apoptosis-inducing ligand. Int J Cardiol. 2013November15;169(4):296–304. [DOI] [PubMed] [Google Scholar]
- 24.Rajamannan NM, Evans FJ, Aikawa E, Grande-Allen KJ, Demer LL, Heistad DD, Simmons CA, Masters KS, Mathieu P, O'Brien KD, Schoen FJ, Towler DA, Yoganathan AP, Otto CM. Calcific aortic valve disease: not simply a degenerative process: A review and agenda for research from the National Heart and Lung and Blood Institute Aortic Stenosis Working Group. Executive summary: Calcific aortic valve disease-2011 update. Circulation. 2011. October 18;124(16):1783–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jian B, Narula N, Li QY, Mohler ER, Levy RJ. Progression of aortic valve stenosis: TGF-beta1 is present in calcified aortic valve cusps and promotes aortic valve interstitial cell calcification via apoptosis. Ann Thorac Surg. 2003February;75(2):457–65; discussion 465-6. [DOI] [PubMed] [Google Scholar]
- 26.Liu XY, Zhao GX, Yan Y, Bao L, Chen BD, Qi RM. Ginkgolide B reduces atherogenesis and vascular inflammation in ApoE(−/−) mice. PLoS One. 2012;7(5):e36237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu LL, Hu ZY, Shen JJ, McQuillan PM. Effects of Ginkgo biloba extract on cerebral oxygen and glucose metabolism in elderly patients with pre-existing cerebral ischemia. Complement Ther Med. 2015. April;23(2):220–5. [DOI] [PubMed] [Google Scholar]
- 28.EGb 761: ginkgo biloba extract, Ginkor. Drugs R D. 2003;4(3):188–93. [DOI] [PubMed] [Google Scholar]
- 29.Ankeny RF, Thourani VH, Weiss D, Vega JD, Taylor WR, Nerem RM, Jo H. Preferential activation of SMAD1/5/8 on the fibrosa endothelium in calcified human aortic valves--association with low BMP antagonists and SMAD6. PLoS One. 2011;6(6):e20969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xie CH, Shen YN, Hu WL, Chen ZP, Li YS. Angiotensin II promotes an osteoblast-like phenotype in porcine aortic valve myofibroblasts. Aging Clin Exp Res. 2016April;28(2):181–7. [DOI] [PubMed] [Google Scholar]
- 31.Gao X, Zhang LW, Gu GJ, Wu PH, Jin S, Hu WL, Zhan CY, Li J, Li YS. The effect of oxLDL on aortic valve calcification via the Wnt/ beta-catenin signaling pathway: an important molecular mechanism. J Heart Valve Dis. 2015. March;24(2):190–6. [PubMed] [Google Scholar]
- 32.Hofmann JJ, Briot A, Enciso J, Zovein AC, Ren SX, Zhang ZW, Radtke F, Simons M, Wang YB, Iruela-Arispe ML. Endothelial deletion of murine Jag1 leads to valve calcification and congenital heart defects associated with Alagille syndrome. Development. 2012December1;139(23):4449–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu XH, Xu ZY. Osteogenesis in calcified aortic valve disease: From histopathological observation towards molecular understanding. Prog Biophys Mol Biol. 2016. November;122(2):156–161. [DOI] [PubMed] [Google Scholar]
- 34.Bostrom KI, Rajamannan NM, Towler DA. The regulation of valvular and vascular sclerosis by osteogenic morphogens. Circ Res. 2011. August 19;109(5):564–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Al-Aly Z, Shao JS, Lai CF, Huang E, Cai J, Behrmann A, Cheng SL, Towler DA. Aortic Msx2-Wnt calcification cascade is regulated by TNF-alpha-dependent signals in diabetic Ldlr−/− mice. Arterioscler Thromb Vasc Biol. 2007. December;27(12):2589–96. [DOI] [PubMed] [Google Scholar]
- 36.Shimizu T, Tanaka T, Iso T, Matsui H, Ooyama Y, Kawai-Kowase K, Arai M, Kurabayashi M. Notch signaling pathway enhances bone morphogenetic protein 2 (BMP2) responsiveness of Msx2 gene to induce osteogenic differentiation and mineralization of vascular smooth muscle cells. J Biol Chem. 2011May27;286(21):19138–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kaden JJ, Bickelhaupt S, Grobholz R, Vahl CF, Hagl S, Brueckmann M, Haase KK, Dempfle CE, Borggrefe M. Expression of bone sialoprotein and bone morphogenetic protein-2 in calcific aortic stenosis. J Heart Valve Dis. 2004July;13(4):560–6. [PubMed] [Google Scholar]
- 38.Nakagawa Y, Ikeda K, Akakabe Y, Koide M, Uraoka M, Yutaka KT, Kurimoto-Nakano R, Takahashi T, Matoba S, Yamada H, Okigaki M, Matsubara H. Paracrine osteogenic signals via bone morphogenetic protein-2 accelerate the atherosclerotic intimal calcification in vivo. Arterioscler Thromb Vasc Biol. 2010. October;30(10):1908–15. [DOI] [PubMed] [Google Scholar]
- 39.Yang XP, Fullerton DA, Su X, Ao LH, Cleveland JC, Meng XZ. Pro-osteogenic phenotype of human aortic valve interstitial cells is associated with higher levels of Toll-like receptors 2 and 4 and enhanced expression of bone morphogenetic protein 2. J Am Coll Cardiol. 2009February10;53(6):491–500. [DOI] [PubMed] [Google Scholar]
- 40.Shi YG, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003June13;113(6):685–700. [DOI] [PubMed] [Google Scholar]
- 41.Attisano L, Lee-Hoeflich ST. The Smads. Genome Biol. 2001;2(8): REVIEWS3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zwijsen A, Verschueren K, Huylebroeck D. New intracellular components of bone morphogenetic protein/Smad signaling cascades. FEBS Lett. 2003. July 3;546(1):133–9. [DOI] [PubMed] [Google Scholar]