

Abstract
Herein, we disclose that electrochemical stimulation induces new photocatalytic activity from a range of structurally diverse conventional photocatalysts. These studies uncover a new electron primed photoredox catalyst capable of promoting the reductive cleavage of strong C(sp2)–N and C(sp2)–O bonds even when reduction potentials hundreds of mV more negative than Li0 are required. We illustrate several examples of the synthetic utility of these deeply reducing but otherwise safe and mild catalytic conditions. Finally, we employ electrochemical current measurements to perform a reaction progress kinetic analysis. This technique reveals that the improved activity of this new system is a consequence of an enhanced catalyst stability profile.
Keywords: electron-primed photoredox catalysis, electrophotocatalysis, reductive cleavage, radical anions, kinetic analysis
Graphical Abstract

Electrochemical activation of numerous conventional photocatalysts was found to induce potent photoreductant activity. These studies resulted in the discovery of an electron-primed photoredox catalyst capable of cleaving strong aryl C–N and C–O bonds to aryl radical intermediates. Mechanistic experiments revealed that enhanced activity relative to previously developed electron-primed photoredox systems was a result of improved catalyst stability.
Introduction
Reductive activation of organic molecules through single electron transfer (SET) is a fundamental elementary step at the heart of a myriad of synthetically useful transformations.[1–4] In recent years, photoredox catalysis has emerged as a mild and chemoselective method to induce redox events.[5–10] Unfortunately, while 400 nm light possesses sufficient energy for a maximum driving force of 3.1 eV, this energy is diminished by 25–50% through vibrational relaxation, internal conversion, and intersystem crossing.[11] As a consequence, many abundant but thermodynamically stable molecules remain inert to photoredox activation.[12],[13] Indeed, in the context of reductions, alkali metals have remained reductants of unparalleled potency for over a century. These reagents continue to be used in both academic[14,15] and industrial[16] settings despite their implicit hazards, poor chemoselectivity, and inextricable chemical waste. To address this, the development of new strategies to deliver extreme reduction potentials (significantly more negative than –2 V vs. SCE) with the safety and chemoselectivity profile of photoredox catalysis is an emerging area of considerable contemporary interest.[11,17–22]
Over the past several years, numerous groups,[23–28] including ours,[26] have examined catalytic systems designed to leverage mildly reducing radical species as a new family of photocatalysts (Figure 1, top). We have dubbed these reductively activated species electron-primed photoredox catalysts to distinguish them from more conventional photocatalytic reductants. Pioneering work from König used a consecutive photoinduced electron transfer (conPET) approach to photochemically generate an electron-primed photocatalyst, albeit one that did not possess an excited state reduction potentials more negative than –2 V vs. SCE.[29] The conPET strategy requires a carefully balanced system; both catalyst oxidation states must engage in excited state intermolecular SET under a single set of reaction conditions.[19] Additionally, the byproducts of catalyst activation, which are typically reactive amine radical cations and easily reduced iminium ions, must not deactivate the catalyst or interfere in subsequent steps.[30] These fundamental challenges associated with catalyst generation and turnover have resulted in only a small collection of electron-primed photocatalytic systems being identified in the subsequent years[29,31] despite photophysical studies establishing that numerous persistent radical anions absorb visible light.[32–36]
Figure 1:

(top) established catalysts known to promote reductive SET events through electron-primed photoredox catalysis and (bottom) electrochemically driven electron-primed photoredox catalysis and a new catalyst discovered in this work derived from 4-DPAIPN. Ar = 2,6-diisopropylphenyl. R = p-OMePh. R' = 2-ethylhexyl.
We envisioned that electrochemistry would offer a flexible approach to generate electron-primed photoredox catalysts as cathodic reduction is highly tunable and divided cell electrolysis excludes interfering oxidized byproducts.[37–39] Indeed, we previously used this approach to introduce a novel electron-primed photocatalyst capable of reducing aryl chloride substrates with Ered negative than Li0.[26] Contemporaneous efforts by Lambert and Lin disclosed that 9,10-dicyanoanthracene, an electron-primed photoredox catalyst previously accessed via conPET,[31] exhibits enhanced reactivity towards aryl chloride substrates when driven electrochemically.[25] However, while these two discoveries validated the use of electrochemistry to generate potent photoreductants, both of these electrophotocatalysts remained structurally analogous to established conPET-based photocatalysts. While rapid progress has been made in net-oxidative electrophotocatalytic transformations,[40–48] electrophotocatalytic reductions remain comparatively underdeveloped.[49,50] Herein, we employ cathodic reduction to elicit new photocatalytic activity from numerous organic and inorganic structures. These studies reveal a new electron-primed photoredox catalyst that enables cleavage of strong C(sp2)–N and C(sp2)–O bonds.
Results and Discussion
The generation of aryl radical intermediates is a well-established arena to benchmark new photoreductants. Bench-stable[51–54] trialkylanilinium salts and activated phenols are readily accessible and can be reductively cleaved to aryl radical intermediates through deeply reducing direct electrolysis or alkali metal reductants, however, they remain difficult to activate under photocatalytic conditions.[55,56] Within the past year, Larionov and König illustrated that anilide and thiolate photocatalysts are capable of promoting the borylation of anilinium salts and activated phenols via photoreduction.[57,58] However, boron plays a non-innocent role in these processes and photochemical net-reductive transformations of these substrates remains limited. Reductive defunctionalization is a powerful synthetic tactic to leverage aniline and phenol activating groups in a traceless manner.[59–62] Current methods to remove these directing groups rely on harsh dissolving metal conditions[63,64] or palladium catalysis.[65,66] We envisioned that cleavage of these strong bonds was a perfect arena to explore new potent reductants given recently reported halogen-atom transfer strategies, which in some cases can circumvent deeply reducing potentials,[67] are unlikely to be amenable to the cleavage of these less polarizable heteroatoms.[68,69]
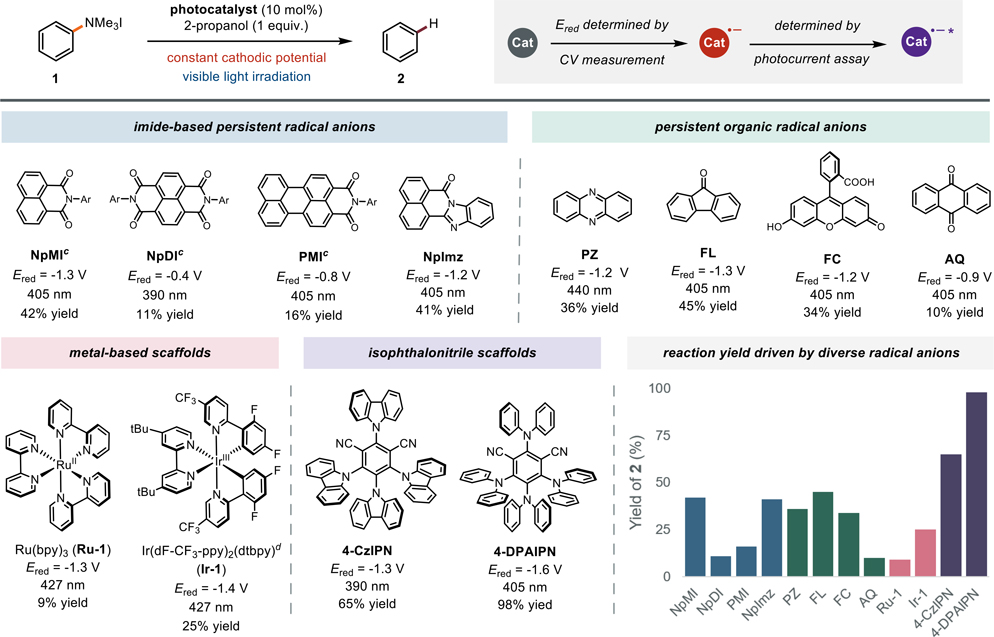
We initiated our studies with the reductive cleavage of an N,N,N-trimethyl anilinium salt, 1 (Table 1). We anticipated that the thermodynamic and kinetic challenges presented by aryl C(sp2)–N bond cleavage would expose the limitations of current electron-primed photocatalysts. We found that NpMI, the electron-primed photoredox catalyst we recently reported,[26] could cleave the C(sp2)–N bond in 42% yield under a constant cathodic potential and visible light irradiation. This result validated that an electron-primed photoredox system is capable of engaging this substrate but also highlighted the need for improved catalysts. We next evaluated a collection of structures related to the NpMI core. Electrochemistry facilitated rapid catalyst evaluation in two primary ways: (1) cyclic voltammetry studies established the minimum cathodic potential to generate the radical anion photocatalyst and (2) evaluation of wavelength dependent photocurrent established the optimal irradiation wavelength (see SI for details).[70] These studies revealed that various derivatives of NpMI including NpDI, PMI, and NpImz each provided the defunctionalized product, albeit in reduced yield relative to NpMI. Given these data, we concluded that a fundamentally different catalyst scaffold was likely necessary to efficiently promote these challenging reductions.
Table 1:
|
Reactions were conducted on a 0.2 mmol scale in DMF (0.1 M nBu4NPF6) with RVC cathode and anode. Reaction were run for 12 hours. Yields provided are of 2, determined by GC analysis.
all redox potentials reported relative to SCE.
Ar = 2,6-diisopropylphenyl.
Reaction was conducted with 1 mol % catalyst. See the supportlng information (SI) for further details.
We next targeted more structurally diverse persistent radical anion precursors that have not been explored as electron-primed photocatalysts.[36,71–74] We found that phenazine, fluorenone, and fluorescein each promote reduction of 1 in comparable yields to NpMI under appropriate electrophotocatalytic conditions. Control reactions revealed that no conversion is observed in the absence of electrolysis indicating that the photoactivity of the neutral structures is insufficient to drive defunctionalization of 1. These data suggest that electrochemical reduction can coax potent photocatalytic activity out of a much broader range of molecules than previously appreciated. Next, we recognized that nearly all photoredox catalysts, by design, undergo reversible redox events and many possess persistent radical anion congeners.[6,10,75] We questioned whether the structural features that render molecules effective as conventional photoredox catalysts would translate to the electron-primed photoredox manifold.[76,23,77] Intriguingly, we found that electrolysis at the Ered of several commonly employed photoredox catalysts Ru(bpy)3,[78] Ir(dF-CF3-ppy)2(dtbpy),[78] and 4-CzIPN[79] turned on photocatalytic activity in this challenging reduction.[80] While there is a sole report proposing photochemical activity of the reduced congener of an Ir-based photoredox catalyst,[81] these are the first data consistent with either Ru-based or isophthalonitrile structures acting as electron-primed photoredox catalysts. Given that cathodic reduction of 4-CzIPN resulted in a meaningful improvement in photochemical deamination yield, we examined other isophthalonitrile catalysts. This investigation revealed that 4-DPAIPN[82] promotes the reduction of model substrate 1 in nearly quantitative yield under electrophotocatalytic conditions. Overall, the structural diversity of the potent photocatalysts identified through these studies suggest that reductively induced photoactivity is a general phenomenon and provides a clear link between catalyst structure and reaction outcome.
We next evaluated whether 4-DPAIPN was promoting this reaction through excitation of a cathodically generated species. Under electrochemical stimulation 4-DPAIPN acts as a far more potent photoreductant than anticipated by its established redox potentials (E1/2 PC+/PC*) = −1.3 V and E1/2 (PC/PC•-) = −1.5 V vs. SCE)[82] (Figure 2). First, we conducted a series of control experiments and found that catalyst, electrolysis, and light were all required for product formation. Next, we examined whether electrochemical reductio no f4-DPAIPN was necessary to promote the defunctionalization reaction. Inspired by an elegant experiment conducted by Lambert and Lin,[25] we measured the defunctionalization yield at varied cathodic potentials. Overlaying these data with the cyclic voltammogram of 4-DPAIPN illustrates that reactivity is observed only when a sufficient potential to reduce 4-DPAIPN is applied. These data are fully consistent with cathodic catalyst reduction and subsequent excitation as necessary steps for this difficult reductive transformation.[83]
Figure 2:

Reactions were conducted on a 0.2 mmol scale in DMF (0.1 M nBu4NPF6) and run for 12 hours. aCV is of 4-DPAIPN. See Supporting Information (SI) for more details.
We next probed the scope of this catalytic C(sp2)–N cleavage process (Table 2). We found ethers (4), free alcohols (5), esters (6), and amides (7) as well as heterocycles such as piperazine (7), pyrrolidine (8), and morpholine (9) were all welltolerated. Notably, this reaction enables a molecular editing strategy wherein an N-aryl ring can be replaced by an alkyl group through an alkylation/reductive cleavage sequence (11) as both aryl and amine fragments can be recovered after C(sp2)–N reduction. Given the promising activity of this catalytic system in the cleavage of anilinium salts, we turned our attention to more difficult to reduce C(sp2)–O bonds. Phenol derivatives (e.g. triflates and phosphates) possess deep reduction potentials (typically <–2.7 V vs. SCE).[84–86] Despite the energetic demands of C(sp2)–O cleavage, phosphate ester substrates bearing a range of functional groups such as esters (13), amides (14), ethers (15), benzylic amines (16), unprotected alcohols and tertiary amines (18) as well as heterocycles such as imidazole (17) and piperazine (19) each underwent productive C(sp2)–O cleavage. While each of these reactions are conducted far below the cathodic potential required to reduce the substrate, we questioned whether deeply reducing electrolysis could recapitulate this electrophotocatalytic activity. To probe this, we carried out direct electrolysis reactions on two substrates bearing functional groups to investigate the role of the catalyst in preserving chemoselectivity. Under constant current conditions in the absence of catalyst, 16 and 19 showed significant conversion to an intractable mixture containing <20% product. By promoting reduction through a photocatalytic mediator under mild electrochemical potentials, chemoselectivity and functional group tolerance can be vastly improved compared to direct electrolysis conditions.
Table 2:
Scope of Aryl C(sp2)-N and C(sp2)-O Bond Cleavagea

|
Reactions were conducted on a 0.2 mmol scale and run for 12 h in a divided celi with RVC electrodes. Et3N (2 equiv) was added to the anodic chamber as a terminal reductant.
The counter ion is OTf unless otherwise noted.
1 used as the iodide salt.
NMR yield.
GC yield. See the SI for experimental detalls.
Phenols are electron-donating groups that enable a wide range of reactions at the arene core.[87–91] We envisioned that the scope of products accessible using these processes could be expanded through a chemoselective excision of the phenolic activating group via an electrophotocatalytic system (Figure 3). For example, this strategy allows selective formation of meta-substituted products inaccessible via direct Friedel–Crafts reactions.[92] To illustrate this strategy, we prepared a suite of meta-substituted arene products from simple precursors using a phenol-directed alkylation-defunctionalization sequence (20–23). As a direct comparison, we subjected 23 to constant current conditions in the absence of catalyst and observed high conversion with substantially diminished yield. To demonstrate the value of a phenol-directed alkylation-defunctionalization approach, we targeted the synthesis of a tricyclic resorcinol derivative that was developed as a conformationally restricted cannabinoid agonist. The route, devised by Makriyannis,[93] hinged on phenol-enabled Friedel–Crafts alkylation followed by a Li0-promoted excision of the phenol activating group. In our hands, the Friedel–Crafts process and subsequent phosphorylation proceeded smoothly to deliver tricyclic intermediate 24. Gratifyingly, electron-primed photoredox C(sp2)–O cleavage furnished intermediate 25 despite a nearly 2 V underpotential supplied at the cathode. Global demethylation furnished 26 in 18% yield over 4 steps. These data demonstrate how this new catalytic platform can directly fit into synthetic sequences and circumvent the need for more hazardous chemical reductants in the preparation of complex biologically active molecules.
Figure 3:

Phenol as a traceless directing group enabled by electron-primed photoredox catalysis. a Yields reported are for the phosphate defunctionalization step. b NMR yield. c Isolated yield, (a) 70% aq MeS03H, 70 °C, 12 h (45% yield), (b) CIP(0)(0Et)2, DABCO, MeCN, 18 h (70% yield), (c) BBr3, DCM, -78 °C, 12 h, (91% yield). Conversion of 24 to 25 was accomplished under conditions analogous to those in Table 2. See SI for experimental details.
We next questioned whether the aryl radical intermediates generated upon reductive cleavage of anlinium salts and phosphate esters could be intercepted by classic aryl radical traps. We investigated several aryl radical coupling reactions: phosphonylation, borylation, and heteroarylation (27-29). We found that both C(sp2)–N and C(sp2)–O radical precursors were amenable to each of these radical coupling reactions (Figure 4).
Figure 4:

redox neutral coupling reactions from aryl C(sp2)-N and C(sp2)-O bonds. All reactions conducted on a 0.2 mmol scale and reported as NMR yields. aNMR yield. bCatalyst loading was 2.5 mol%. See the SI for details.
Having established that 4-DPAIPN is a broadly effective electron-primed photoredox catalyst with immediate synthetic utility, we next aimed to understand the origin of the improved performance of 4-DPAIPN relative to prior electron-primed photoredox catalysts. Specifically, we questioned whether 4-DPAIPN possessed enhanced reactivity, superior catalyst stability,[94] or both. To address this question, we envisioned that electrochemical current could be employed as a non-invasive in situ rate monitoring technique to unlock tools from reaction progress kinetic analysis (RPKA).[95] This method can reveal phenomena such as catalyst decomposition and product inhibition typically invisible to classic initial rate kinetics because the analysis is conducted under typical preparative conditions. We conducted a "same excess” experiment with both our previously reported electron-primed photocatalyst, NpMI, and 4-DPAIPN to compare the extent of catalyst deactivation in each case (Figure 5). We selected aryl chloride 30 as the model substrate because both catalysts can engage this substrate under constant potential conditions and preliminary investigations indicated it exhibited a well-behaved kinetic profile.[96,97] We carried out two separate constant potential experiments for NpMI at different initial concentrations of 30 (traces a and b). When the conversion rate of 30 is plotted as a function of [30], the two curves do not overlay. This indicates either catalyst death or product inhibition.[98] Inhibition by the arene product was excluded by addition of 31, which did not restore overlay between the curves. Furthermore, NpMI exhibited an unusual kinetic profile consistent with decomposition into a new catalytically active species that subsequently decomposes. These data implicate rapid deactivation of NpMI under these conditions. In stark contrast, an analogous “same excess” experiment with 4-DPAIPN resulted in clean first order reaction profiles that nearly overlay. These data are consistent with turnover limiting photoreduction of 30 and minimal catalyst decomposition or product inhibition (see SI for details). As with NpMI, addition of 31 excluded product inhibition and suggests 4-DPAIPN decomposition occurs[99–102] but is attenuated relative to NpMI. These data indicate that the improved performance of 4-DPAIPN can be attributed to it forming a more robust electron-primed photoredox catalyst. Indeed, the initial rate of dehalogenation promoted by NpMI is faster than 4-DPAIPN but rapid decomposition of this catalyst renders it ineffective for more challenging substrates that are slower to fragment following reduction.
Figure 5:

Reactions performed on a 0.4 mmol scale.Trace a and d: [ArCI]0 = 0.08 M {standard reaction concentration), Trace b and e: [ArCI]0 = 0.052 M, Trace c and f: [ArCI]0 = 0.052 M, [ArH]0 = 0.028 M. See the SI for details
Conclusion
Overall, we have demonstrated that electrochemistry is an effective tool to explore structurally diverse electron-primed photoredox catalysts. These investigations revealed electrochemical reduction can induce potent photoreductant behavior from structurally diverse catalyst precursors. Among these, a common photoredox catalyst, 4-DPAIPN, is an exceptionally reducing electron-primed photoredox catalyst. This discovery enabled a new catalytic system to promote the reductive cleavage of diverse C(sp2)–N and C(sp2)–O bonds, which we anticipate will enable an array of synthetic sequences that previously would have mandated alkali metal reductants. Finally, we illustrated how principles from RPKA could be directly employed in electrophotocatalysis, using electrochemical current to monitor reaction rate in situ throughout a reaction. We anticipate radical anions will serve as a structurally diverse family of photoredox catalysts for challenging reductive processes and that these studies will provide a roadmap for the use of electrochemistry to both drive and interrogate such systems.
Supplementary Material
Acknowledgements
We thank Prof. Alison Wendlandt for helpful suggestions. We thank the Stahl lab for their assistance in use of spectroelectrochemical equipment. Additionally, we thank the Stahl, Weix, Yoon, and Schomaker groups for sharing their chemical inventory. Tracy Drier is acknowledged for electrochemical glassware fabrication. This work was financially supported by the Office of the Vice Chancellor for Research and Graduate Education at the University of Wisconsin–Madison with funding from the Wisconsin Alumni Research Foundation. This material is based upon work supported by the National Science Foundation under Grant No. (2047108). Acknowledgment is made to the Donors of the American Chemical Society Petroleum Research Fund for partial funding of this research (60677-DNI1). Spectroscopic instrumentation was supported by a generous gift from Paul. J. and Margaret M. Bender, NSF (CHE-1048642), and NIH (1S10 OD020022-1).
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].Ashby EC, Acc. Chem. Res. 1988, 21, 414–421. [Google Scholar]
- [2].Zhang N, Samanta SR, Rosen BM, Percec V, Chem. Rev. 2014, 114, 5848–5958. [DOI] [PubMed] [Google Scholar]
- [3].Broggi J, Terme T, Vanelle P, Angew. Chem., Int. Ed. 2014, 53, 384–413. [DOI] [PubMed] [Google Scholar]
- [4].Eberson L, in Advances in Physical Organic Chemistry (Eds.: Gold V, Bethell D), Academic Press, 1982, pp. 79–185. [Google Scholar]
- [5].Prier CK, Rankic DA, MacMillan DWC, Chem. Rev. 2013, 113, 5322–5363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Shaw MH, Twilton J, MacMillan DWC, J. Org. Chem. 2016, 81, 6898–6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Romero NA, Nicewicz DA, Chem. Rev. 2016, 116, 10075–10166. [DOI] [PubMed] [Google Scholar]
- [8].Skubi KL, Blum TR, Yoon TP, Chem. Rev. 2016, 116, 10035–10074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].DiRocco DA, Dykstra K, Krska S, Vachal P, Conway DV, Tudge M, Angew. Chem. Int. Ed. 2014, 53, 4802–4806. [DOI] [PubMed] [Google Scholar]
- [10].Narayanam JMR, Stephenson CRJ, Chem. Soc. Rev. 2010, 40, 102–113. [DOI] [PubMed] [Google Scholar]
- [11].Arias-Rotondo DM, McCusker JK, Chem. Soc. Rev. 2016, 45, 5803–5820. [DOI] [PubMed] [Google Scholar]
- [12].Kundu KK, Rakshit AK, Das MN, Electrochim. Acta 1972, 17, 1921–1937. [Google Scholar]
- [13].Roth HG, Romero NA, Nicewicz DA, Synlett 2016, 27, 714–723. [Google Scholar]
- [14].Chuang KV, Xu C, Reisman SE, Science 2016, 353, 912–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].He C, Stratton TP, Baran PS, J. Am. Chem. Soc. 2019, 141, 29–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Joshi DK, Sutton JW, Carver S, Blanchard JP, Org. Process Res. Dev. 2005, 9, 997–1002. [Google Scholar]
- [17].Speckmeier E, Fischer TG, Zeitler K, J. Am. Chem. Soc. 2018, 140, 15353–15365. [DOI] [PubMed] [Google Scholar]
- [18].Peters BK, Rodriguez KX, Reisberg SH, Beil SB, Hickey DP, Kawamata Y, Collins M, Starr J, Chen L, Udyavara S, Klunder K, Gorey TJ, Anderson SL, Neurock M, Minteer SD, Baran PS, Science 2019, 363, 838–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Glaser F, Kerzig C, Wenger OS, Angew. Chem., Int. Ed. 2020, 5, 10266–10284. [DOI] [PubMed] [Google Scholar]
- [20].Connell TU, Fraser CL, Czyz ML, Smith ZM, Hayne DJ, Doeven EH, Agugiaro J, Wilson DJD, Adcock JL, Scully AD, D. E. Gómez, Barnett NW, Polyzos A, Francis PS, J. Am. Chem. Soc. 2019, 13. [DOI] [PubMed] [Google Scholar]
- [21].Shon J-H, Kim D, Rathnayake MD, Sittel S, Weaver J, Teets TS, Chem. Sci. 2021, 12, 4069–4078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kerzig C, Guo X, Wenger OS, J. Am. Chem. Soc. 2019, 141, 2122–2127. [DOI] [PubMed] [Google Scholar]
- [23].MacKenzie IA, Wang L, Onuska NPR, Williams OF, Begam K, Moran AM, Dunietz BD, Nicewicz DA, Nature 2020, 580, 76–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Cole JP, Chen D-F, Kudisch M, Pearson RM, Lim C-H, Miyake GM, J. Am. Chem. Soc. 2020, 142, 13573–13581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kim H, Kim H, Lambert TH, Lin S, J. Am. Chem. Soc. 2020, 142, 2087–2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Cowper NGW, Chernowsky CP, Williams OP, Wickens ZK, J. Am. Chem. Soc. 2020, 142, 5, 2093–2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Caby S, Bouchet LM, Argüello JE, Rossi RA, Bardagi JI, ChemCatChem 2021, 13, 3001–3009. [Google Scholar]
- [28].Chen Y-J, Lei T. Hu H-L, Wu H-L, Zhou S, Li X-B, Chen B, Tung CH, Wu L-Z, Matter 2021, 4, 1–13. [Google Scholar]
- [29].Ghosh I, Ghosh T, Bardagi JI, König B, Science 2014, 346, 725–728.. [DOI] [PubMed] [Google Scholar]
- [30].Et3N can promote premature radical quenching through hydrogen atom transfer and back electron transfer to Et3N•+ For details see ref [7].
- [31].Neumeier M, Sampedro D, Májek M, de la Peña O’Shea VA, von Wangelin A. Jacobi, Pérez-Ruiz R, Chem. Eur. J. 2018, 24, 105–108.. [DOI] [PubMed] [Google Scholar]
- [32].Gosztola D, Niemczyk MP, Svec W, Lukas AS, Wasielewski MR, J. Phys. Chem. A 2000, 104, 6545–6551. [Google Scholar]
- [33].Fujitsuka M, Kim SS, Lu C, Tojo S, Majima T, J. Phys. Chem. B 2015, 119, 7275–7282. [DOI] [PubMed] [Google Scholar]
- [34].Nakashima H, Shida T, Nakatsuji H, J. Chem. Phys. 2012, 136, 214306. [DOI] [PubMed] [Google Scholar]
- [35].La Porte NT, Martinez JF, Chaudhuri S, Hedström S, Batista VS, Wasielewski MR, Coord. Chem. Rev. 2018, 361, 98–119. [Google Scholar]
- [36].Fox M. Anne., Chem. Rev. 1979, 79, 253–273. [Google Scholar]
- [37].Yu Y, Guo P, Zhong J-S, Yuan Y, Ye K-Y, Org. Chem. Front. 2019, 7, 131–135. [Google Scholar]
- [38].Barham JP, König B, Angew. Chem., Int. Ed. 2020, 59, 11732–11747.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Liu J, Lu L, Wood D, Lin S, ACS Cent. Sci. 2020, 6, 1317–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Huang H, Lambert TH, Angew. Chem. int. Ed. 2020, 59, 658–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Huang H, Strater ZM, Rauch M, Shee J, Sisto TJ, Nuckolls C, Lambert TH, Angew. Chem. Int. Ed. 2019, 58, 13318–13322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Huang H, Strater ZM, Lambert TH, J. Am. Chem. Soc. 2020, 142, 1698–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Huang H, Lambert TH, Angew. Chem. int. Ed. 2021,60, 11163–11167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Capaldo L, Quadri LL, Ravelli D, Angew. Chem., int. Ed. 2019, 58, 17508–17510. [DOI] [PubMed] [Google Scholar]
- [45].Yan H, Hou Z-W, Xu H-C, Angew. Chem. int. Ed. 2019, 58, 4592–4595.. [DOI] [PubMed] [Google Scholar]
- [46].Wu S, Žurauskas J, Domański M, Hitzfeld PS, Butera V, Scott DJ, Rehbein J, Kumar A, Thyrhaug E, Hauer J, Barham JP, Org. Chem. Front 2021,8, 1132–1142. [Google Scholar]
- [47].Barham and coworkers have shown that tuning of catalyst structure has a significant impact on the reactivity observed in the context of oxidative electrophotocatalysis. This observation was leveraged to improve electrophotocatalytic oxidation reactions, see ref [46].
- [48].Qiu Y, Scheremetjew A, Finger LH, Ackermann L, Chem. Eur. J, 2020, 26, 3241–3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Far fewer examples exist of electrophotochemical reductions compared to oxidative electrophotochemistry–the existing reports exclusively activate aryl chloride substrates. For details see refs: [25], [26], [27], [28].
- [50].While this manuscript was under review, an electrophotocatalytic reductive olefination system was reported. This new system relied on careful tuning of the N-Aryl substituent of napthlene imide catalysts, see: Angew. Chem. int. Ed. DOI: 10.1002/anie.202105895.
- [51].Aniline substrates can be converted into diazonium salts, which readily undergo reductive fragmentation to aryl radical intermediates. However, these salts are explosive and the conditions to generate them are highly oxidizing. For more information, see: [52], [53], [54].
- [52].Oger N, Grognec EL, Felpin F-X, Org. Chem. Front. 2015, 2, 590–614.. [Google Scholar]
- [53].Chakma P, Digby ZA, Shulman MP, Kuhn LR, Morley CN, Sparks JL, Konkolewicz D, ACS Macro Lett. 2019, 8, 95–100. [DOI] [PubMed] [Google Scholar]
- [54].Washington JB, Chem. Sci. 2021, 12, 6949–6963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Azzena U, Denurra T, Melloni G, Fenude E, Rassu G, J. Org. Chem. 1992, 57, 1444–1448. [Google Scholar]
- [56].Xu H, Yu B, Zhang H, Zhao Y, Yang Z, Xu J, Han B, Liu Z, Chem. Commun. 2015, 51, 12212–12215. [DOI] [PubMed] [Google Scholar]
- [57].Jin S, Dang Hang. T., Haug GC, He R, Nguyen VD, Nguyen VT, Arman HD, Schanze KS, Larionov OV, J. Am. Chem. Soc. 2020, 142, 1603–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Wang S, Wang H, König B, Chem, 2021, 7, 1414–1416. [Google Scholar]
- [59].Kalvet I, Deckers K, Funes-Ardoiz I, Magnin G, Sperger T, Kremer M, Schoenebeck F, Angew. Chem. Int. Ed. 2020, 59, 7721–7725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Duclos RI, Lu D, Guo J, Makriyannis A, Tetrahedron Lett. 2008, 49, 5587–5589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Manmade A, Marshall JL, Minns RA, Dalzell H, Razdan RK, J. Org. Chem. 1982, 47, 1717–1721. [Google Scholar]
- [62].Paras NA, MacMillan DWC, J. Am. Chem. Soc. 2002, 124, 7894–7895. [DOI] [PubMed] [Google Scholar]
- [63].Aviv H, Bar R, Schickler M, Amselem S, United States Patent Application: 0040110827 - High Enantiomeric Purity Dexanabinol for Pharmaceutical Compositions, A1, 20040110827. [Google Scholar]
- [64].Paras NA, Simmons B, MacMillan DWC, Tetrahedron 2009, 65, 3232–3238. [Google Scholar]
- [65].Burgett AWG, Li Q, Wei Q, Harran PG, Angew. Chem. Int. Ed. 2003, 42, 4961–4966. [DOI] [PubMed] [Google Scholar]
- [66].Kodama S, Hamashima Y, Nishide K, Node M, Angew. Chem. Int. Ed. 2004, 43, 2659–2661. [DOI] [PubMed] [Google Scholar]
- [67].Since halogen atom transfer relies on bond strength and polarizability, the vast majority of examples involve organoiodide or organobromide substrates. For examples, see: refs [68] and [69].
- [68].Constantin T, Zanini M, Regni A, Sheikh NS, Juliá F, Leonori D, Science 2020, 367, 1021–1026. [DOI] [PubMed] [Google Scholar]
- [69].Constantin T, Juliá F, Sheikh NS, Leonori D, Chem. Sci. 2020, 11, 12822–12828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Costentin C, Fortage J, Collomb M-N, J. Phys. Chem. Lett 2020, 11, 6097–6104. [DOI] [PubMed] [Google Scholar]
- [71].Fujita M, Ishida A, Majima T, Takamuku S, J. Phys. Chem. 1996, 100, 5382–5387. [Google Scholar]
- [72].Compton RG, Coles BA, Pilkington MBG, J. Chem. Soc., Faraday Trans. 1 1988, 84, 4347. [Google Scholar]
- [73].Eggins BR, Robertson PKJ, J. Chem. Soc., Faraday Trans. 199490, 2249–2256. [Google Scholar]
- [74].Fujitsuka M, Majima T, Journal of Photochemistry and Photobiology C: Photochemistry Reviews 2018, 35, 25–37. [Google Scholar]
- [75].Romero NA, Nicewicz DA, Chem. Rev. 2016, 116, 10075–10166. [DOI] [PubMed] [Google Scholar]
- [76].Given recent work from Nicewicz [23] and our group [77] that suggested that the odd electron conger of two common photoredox catalysts was photocatalytically active, we hypothesized that photochemical activity of oxidized and reduced photocatalysts might be a more general phenomenon.
- [77].Targos K, Williams OP, Wickens ZK, J. Am. Chem. Soc. 2021, 143, 4125–4132. [DOI] [PubMed] [Google Scholar]
- [78].Koike T, Akita M, Inorg. Chem. Front. 2014, 1, 562–576. [Google Scholar]
- [79].Shang T-Y, Lu L-H, Cao Z, Liu Y, He W-M, Yu B, Chem. Commun. 2019, 55, 5408–5419. [DOI] [PubMed] [Google Scholar]
- [80].We also questioned whether these catalysts could act either directly as photoreductants or electrochemically mediate the transformation (without input of light). Given that the PC/PC•− couple is more negative in each case than the PC*/PC•+ couple, we conducted control experiments with each catalyst in the dark and found none furnished defunctionalized product without irradiation.
- [81].Giedyk M, Narobe R, Weiß S, Touraud D, Kunz W, König B, Nat. Catal. 2020, 3, 40–47. [Google Scholar]
- [82].Singh PP, Srivastava V, Org. Biomol. Chem. 2021, 19, 313–321. [DOI] [PubMed] [Google Scholar]
- [83].While these data suggest reductive generation of the radical anion of 4-DPAIPN is necessary for reaction success, at this stage, we cannot exclude subsequent functionalization of the radical anion to form a secondary photoactive species, which could be either closed or open shell.
- [84].While direct electrolysis at deeply reducing potentials or C(sp2)–O homolysis via high energy UV light can rupture these bonds, these approaches remain limited. For examples, see: ref [56], [85] and [86].
- [85].Mfuh AM, Doyle JD, Chhetri B, Arman HD, Larionov OV, J. Am. Chem. Soc. 2016, 138, 2985–2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Liu W, Yang X, Gao Y, Li C-J, J. Am. Chem. Soc. 2017, 139, 8621–8627. [DOI] [PubMed] [Google Scholar]
- [87].Calloway NO, Chem. Rev. 1935, 17, 327–392. [Google Scholar]
- [88].Blicke FF, McCarty FJ, J. Org. Chem. 1959, 24, 1061–1069. [Google Scholar]
- [89].Omura Y, Taruno Y, Irisa Y, Morimoto M, Saimoto H, Shigemasa Y, Tetrahedron Lett. 2001, 42, 7273–7275. [Google Scholar]
- [90].Cuny GD, Tetrahedron Lett. 2004, 45, 5167–5170. [Google Scholar]
- [91].Huang Z, Lumb J-P, ACS Catal. 2019, 9, 1, 521–555. [Google Scholar]
- [92].Roberts RM, Friedel-Crafts Alkylation Chemistry : A Century of Discovery, Dekker M, [1984] ©1984, New York, 1984. [Google Scholar]
- [93].Lu D, Nikas SP, Han X-W, Parrish DA, Makriyannis A, Tetrahedron Lett. 2012, 53, 35, 4636–4638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Hong SH, Day MW, Grubbs RH, J. Am. Chem. Soc. 2004, 126, 7414–7415. [DOI] [PubMed] [Google Scholar]
- [95].Blackmond DG, Angew. Chem. Int. Ed. 2005, 44, 4302–4320. [DOI] [PubMed] [Google Scholar]
- [96].Altering electrolyte identity to lithium perchlorate as well as increasing electrolyte concentration significantly improved reproducibility of current traces of both 4-DPAIPN and NpMI with little to no impact on substrate conversion or product yield.
- [97].We validated that current correlated to reaction rate by comparing the reaction profiles obtained via aliquot-based GC measurements to the current readout. These data indicated that current was directly proportional to reaction rate but required a minor scalar correction factor for faradaic efficiency. See SI for details.
- [98].If catalyst is not decomposing, the rate of substrate conversion should be independent of previous catalyst turnovers. Therefore, for a well-behaved system, rate of conversion should be identical across multiple reactions at each substrate concentration. Accordingly, plotting rate vs. [substrate] for reactions with different initial substrate concentration should overlay. Lack of overlay indicates that different amounts of catalyst are active, which could be explained by either catalyst death or product inhibition. For a detailed discussion of RPKA, see ref [95].
- [99].Previous studies have found that 4-CzIPN can decompose via attack by carbon-centered radical intermediates. It is possible that 4-DPAIPN could undergo analogous decomposition mechanisms with radical intermediates. For examples, see: refs [100], [101], [102].
- [100].Grotjahn S, König B, Org. Lett. 2021, 23, 3146–3150. [DOI] [PubMed] [Google Scholar]
- [101].Donabauer K, Maity M, Berger AL, Huff GS, Crespi S, König B, Chem. Sci. 2019, 10, 5162–5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Kong D, Munch M, Qiqige Q, Cooze CJC, Rotstein BH, Lundgren RJ, J. Am. Chem. Soc. 2021, 143, 2200–2206. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.