

Abstract
Imbalanced neuronal excitability homeostasis is commonly observed in patients with fragile X syndrome (FXS) and the animal model of FXS, the Fmr1 KO. While alterations of neuronal intrinsic excitability and synaptic activity at the steady state in FXS have been suggested to contribute to such a deficit and ultimately the increased susceptibility to seizures in FXS, it remains largely unclear whether and how the homeostatic response of neuronal excitability following extrinsic challenges is disrupted in FXS. Our previous work has shown that the acute response following induction of endoplasmic reticulum (ER) stress can reduce neural activity and seizure susceptibility. Because many signaling pathways associated with ER stress response are mediated by Fmr1, we asked whether acute ER stress–induced reduction of neural activity and seizure susceptibility are altered in FXS. Our results first revealed that acute ER stress can trigger a protein synthesis–dependent prevention of neural network synchronization in vitro and a reduction of susceptibility to kainic acid–induced seizures in vivo in wild-type but not in Fmr1 KO mice. Mechanistically, we found that acute ER stress–induced activation of murine double minute-2 (Mdm2), ubiquitination of p53, and the subsequent transient protein synthesis are all impaired in Fmr1 KO neurons. Employing a p53 inhibitor, Pifithrin-α, to mimic p53 inactivation, we were able to blunt the increase in neural network synchronization and reduce the seizure susceptibility in Fmr1 KO mice following ER stress induction. In summary, our data revealed a novel cellular defect in Fmr1 KO mice and suggest that an impaired response to common extrinsic challenges may contribute to imbalanced neuronal excitability homeostasis in FXS.
Keywords: FXS, FMRP, hyperexcitability, ER stress, Seizures
INTRODUCTION
The importance and dysregulation of neuronal excitability homeostasis has been described in different autism spectrum disorders (Amaral et al., 2008; Levitt et al., 2004), and multiple mechanisms underlying homeostatic control in neurons involve autism-linked genes (Beique et al., 2011; Gao et al., 2010; Hu et al., 2010; Korb et al., 2013; Shepherd et al., 2006; Tian et al., 2010; Zhong et al., 2012), including Fmr1 (Soden and Chen, 2010). Fmr1 is absent in the most common cause of mental retardation and autism, FXS. Fmr1 encodes fragile X mental retardation protein (FMRP), an RNA-binding protein that regulates translation of its bound RNAs (~4% of brain mRNAs) (Ashley et al., 1993). In the absence of FMRP, translation of the bound RNAs is misregulated, leading to FXS. FXS patients and the mouse model of FXS, the Fmr1 KO, both show elevated neuronal and circuit excitability (Gross et al., 2011; Iacoangeli and Tiedge, 2013; Repicky and Broadie, 2009), with seizure diagnosed in 15% of male individuals and 6–8% of female individuals (Berry-Kravis et al., 2010). The hyperexcitability also affects sensory perception (Lovelace et al., 2016; Rotschafer and Razak, 2014; Zhang et al., 2014) and potentially contributes to learning deficits in FXS (Gonçalves et al., 2013). While multiple mechanisms have been described to explain such hyperexcitability in FXS (Gibson et al., 2008; Martin et al., 2014; Michalon et al., 2012; Pfeiffer et al., 2010; Ronesi et al., 2012; Ronesi and Huber, 2008; Tsai et al., 2012a; Vislay et al., 2013), our current knowledge has been mainly limited to intrinsic properties and synaptic activity of Fmr1 KO neurons at the steady states. Whether and how Fmr1 KO neurons and mice respond to extrinsic challenges to homeostatically modulate their excitability remains largely unclear.
The cellular response to ER stress comprises a set of evolutionarily conserved mechanisms that help the cell adapt to and remove disturbances. When attempts to cope with the disturbances fail or when the disturbances last for an extended period of time, the ER stress response can trigger cell degeneration and death. Although ER stress has been observed in and suggested to contribute to cell death in both health and disease, its effects on neuronal excitability was unclear until our recent study showing that ER stress response can trigger a reduction of seizure susceptibility in mice (Liu et al., 2019). In the same study, we also revealed that the effect was elicited by ubiquitination of tumor suppressor p53 mediated by ubiquitin E3 ligase murine double minute-2 (Mdm2), which ultimately led to transient protein synthesis under ER stress. Because Fmr1 has been linked to many signaling pathways associated with ER stress response (Coyne et al., 2015; Di Marco et al., 2021; Taha et al., 2021; Utami et al., 2020), we asked whether ER stress–induced reduction of neural activity and seizure activity are altered in Fmr1 KO mice, and if yes, whether we can correct them.
To answer these questions, we present data in this study to show that ER stress, through a protein synthesis–dependent mechanism, can prevent an increase in neural network synchronization in vitro and reduce susceptibility to seizures in vivo in wild-type but not in Fmr1 KO mice. Mechanistically, we found that Mdm2 is abnormally inactivated while ubiquitination of p53 is impaired in Fmr1 KO neurons upon induction of ER stress. By using a p53 inhibitor, Pifithrin-α, the prevention of neural network synchronization and reduction of seizure susceptibility in Fmr1 KO mice can both be restored upon the induction of ER stress. Because ER stress can be induced following many physiological and also pathological stimuli, including reduced mitochondrial respiration and aberrant translational control that were previously seen in FXS (Guo et al., 2016; Shen et al., 2019), our data suggest that an impaired response to common cellular stress challenges may contribute to imbalanced neuronal excitability homeostasis in FXS.
MATERIALS AND METHODS
Mice
The Fmr1 KO, Mdm2flox and Emx1-Cre mice in C57BL/6J background were obtained from The Jackson Laboratory. Trio breeding was conducted throughout the study. Both male and female mice were used to prepare mixed sex cultures. All animal procedures were performed in accordance with our institutional animal care committee’s regulations. For genotyping, the primers used to detect Mdm2 loxP allele are: 5’- TGT GGA GAA ACA GTT ACT TC -3’, and 5’- GAA ACT CGG GAC TCC AAA CA -3’. The primers used to differentiate Wild type and Emx1 alleles are 5’-CGG TCT GGC AGT AAA AAC TAT C-3’ (Emx1-Cre), 5’-GTG AAA CAG CAT TGC TGT CAC TT-3’ (Emx1-Cre); 5’-AAG GTG TGG TTC CAG AAT CG-3’ (Wild type), and 5’-CTC TCC ACC AGA AGG CTG AG-3’ (Wild type).
Reagents
Dimethyl sulfoxide (DMSO) and MG132 were from Fisher Scientific. DMSO was used as a vehicle in this study. Kainic acid was from Alomone Labs. Saline was from Hanna Pharmaceutical Supply Co., Inc. Pifithrin and Thapsigargin were from AdipoGen. Cycloheximide was from Sigma. The antibodies used in this study were purchased from Santa Cruz Biotechnology (anti-Mdm2), GenScript Corporation (anti-Gapdh), and Cell Signaling (anti-p-Mdm2, anti-p53 and anti-ubiquitin). HRP-conjugated secondary antibodies were from Jackson Immunoresearch and Cell Signaling.
Primary neuron cultures
Primary neuron cultures were made from mice aged at p0–p1 and maintained in Neural Basal A medium (Invitrogen) supplemented with B27 supplement (Invitrogen), GlutaMax (final concentration at 2 mM; Invitrogen), and Cytosine β-D-arabinofuranoside (AraC, final concentration at 2 μM; Sigma). The culture medium was changed 50% on DIV 2 and every 3–4 days thereafter until the experiments on DIV 14.
MEA recording
The multi-electrode array recordings were conducted using an Axion Muse 64-channel system in single-well MEAs (M64-GL1-30Pt200, Axion Biosystems) inside a humidified incubator with 5% CO2 at 37°C. Field potentials (voltage) at each electrode relative to the ground electrode were recorded with a sampling rate of 25 kHz. After 30 min of recording the baseline (before treatment), drug(s) or vehicles indicated in each experiment were added, and the MEA dish was put back into the incubator for one hour before another 30 min of recording (after treatment). To eliminate changes in network activity caused by physical movement of the MEA, only the last 15 min of each recording were used in data analyses (Jewett et al., 2016; Zhu et al., 2017). AxIS software (Axion Biosystems) was used for the extraction of spikes (i.e. action potentials) from the raw electrical signal. After filtering, a threshold of ±6 standard deviations was independently set for each channel; activity exceeding this threshold was counted as a spike. Synchrony index was computed through AxIS software, based on a published algorithm (Eggermont, 2006), by taking the cross-correlation between any two spike trains, removing the portions of the cross-correlogram that are contributed by the auto-correlations of each spike train, and reducing the distribution to a single metric. For all the drug treatment comparisons, to minimize the influence caused by variability between cultures, the recording from each MEA dish after treatment was only compared to the baseline recording from that same culture.
Immunoprecipitation and western blotting
For immunoprecipitation (IP), cell lysates were obtained by sonicating pelleted cells in a IP buffer (50 mM Tris, pH 7.4, 120 mM NaCl, 0.5% Nonidet P-40). Eighty μg of total protein lysates was incubated for one hour at 4 °C with 0.5 μg primary antibodies. Protein A/G agarose beads (Santa Cruz Biotechnology) were added for another hour followed by three 10-min washings with IP buffer. For western blotting, after SDS-PAGE, the gel was transferred onto a polyvinylidene fluoride (PVDF) membrane (Santa Cruz Biotechnology). After blocking with 1% Bovine Serum Albumin in TBST buffer (20 mM Tris pH 7.5, 150 mM NaCl, 0.1% Tween-20), the membrane was incubated with primary antibody overnight at 4 °C, followed by three 10-min washings with TBST buffer. The membrane was then incubated with an HRP-conjugated secondary antibody for 1 hour at room temperature, followed by another three 10-min washings. Finally, the membrane was developed with an ECL Chemiluminescent Susbtrate (Pierce).
Seizure induction and scoring
Male mice at age 3-weeks old were intraperitoneally injected with kainic acid, prepared in saline solution at doses of 30 mg/kg. The total injection volume was kept close to 0.15 ml. After injection, mice were closely observed in real time for 1 hour. The intensity of seizures was assessed by Racine’s scoring system (Racine, 1972). To clearly determine seizure activity, only stage 4 (rearing and falling) and stage 5 (tonic-clonic activity) were considered positive for seizures, as previously performed (Jewett et al., 2016; Keith et al., 2012; Zhu et al., 2017).
Whole-cell patch-clamp recording
Dissociated cortical neurons, visually identified using an upright microscope (Olympus, BX51WI), were used for recordings. Recording pipettes, pulled from glass capillaries with an outer diameter of 1.5 mm on micropipette puller (P-97; Sutter Instruments), had a resistance of 4–6 MΩ when filled with internal solution containing (in mM): 130 K-gluconate, 6 KCl, 3 NaCl, 10 HEPES, 0.2 EGTA, 4 Mg-ATP, 0.4 Na-GTP, 14 Tris-phosphocreatine (pH 7.25, 285 mOsm. All recordings were carried out at room temperature (23–25°C) in external solution containing (in mM): 119 NaCl, 2.5 KCl, 4 CaCl2, 4 MgCl2, 1 NaH2PO4, 26 NaHCO3 and 11 D-Glucose, saturated with 95% O2/5% CO2 (pH 7.4, 310 mOsm), and were performed in the presence of fast synaptic transmission blockers: CNQX (20 μM), DL-APV (200 μM) and PTX (100 μM). For current-clamp, neurons were held at −60 mV. Action potential firing rates measured upon delivering constant current pulses of 500 ms in the range 0–200 pA, and the number of action potentials was averaged from 3 to 5 individual sweeps for current intensity. Neurons were omitted if the resting membrane potential was >−50 mV, if no action potentials were discharged. No series resistance compensation was used. The action potential firing rate at each current pulse was calculated as a reciprocal of the interspike intervals, measured as the time between the first and second action potential peaks (i.e. 1/interspike interval). Whole-cell current-clamp recordings were made using a Multiclamp 700B amplifier (Molecular Devices). Records were filtered at 2 kHz and digitized at 10 kHz. Data was acquired and analyzed with a Digidata 1550B interface (Molecular Devices) and the pClamp suite of software (version 10.6; Molecular Devices). Analysis of recordings was performed using Clampfit software (version 10.6; Molecular Devices).
Statistical Analysis
Statistical methods to determine significance along with sample numbers were indicated in each figure legend. In brief, ANOVA with post-hoc Tukey HSD (Honest Significant Differences) test was used for multiple comparisons between treatments or genotypes. Student’s t-test was used for analyzing spontaneous neuronal activity and paired western blotting results as indicated in each figure legend. Each “n” indicates an independent culture. Differences are considered significant at the level of p < 0.05.
RESULTS
Acute ER stress–induced reduction of seizure activity is impaired in Fmr1 KO mice
Our previous work has demonstrated that acute ER stress can reduce seizure susceptibility in mice (Liu et al., 2019). To begin determining whether such an effect can also be seen in the mouse model of FXS, the Fmr1 KO mice, we employed a kainic acid–induced seizure model in wild-type and Fmr1 KO mice, both in C57BL/6 background. To induce a preconditioning to ER stress, we intraperitoneally injected mice with saline or Thapsigargin (Tg), an ER stress inducer that acts through inhibition of Ca2+ ATPase in ER, at 2 mg/kg for three hours. Our previous work has validated the successful induction of ER stress in the brain through this approach (Eagleman et al., 2020; Liu et al., 2019). Three hours after the injections of saline or Tg, the mice received a second injection with kainic acid at 30 mg/kg. This relatively high dosage of kainic acid was used based on previous studies from others and our showing higher resistance to kainic acid–induced seizures in mice in C57BL/6 background (Ferraro et al., 2001; Liu et al., 2019). Immediately following the second injections, seizure behavior was closely monitored and scored. As shown (Fig. 1A), the wild-type mice that received Tg showed a significantly enhanced latency to the stage 4 seizures (rearing and falling) and a delayed onset of stage 5 seizures (tonic-clonic seizures), as compared to the mice receiving only saline. When we scored the seizures in Fmr1 KO mice (Fig. 1B), we observed no significant difference in the latency to the stage 4 seizures and surprisingly even a slightly quicker onset of stage 5 seizures. These results indicate that acute ER stress–induced reduction of seizure susceptibility is impaired in Fmr1 KO mice.
Figure 1. Acute ER stress reduces seizure severity in wild-type but not Fmr1 KO mice.
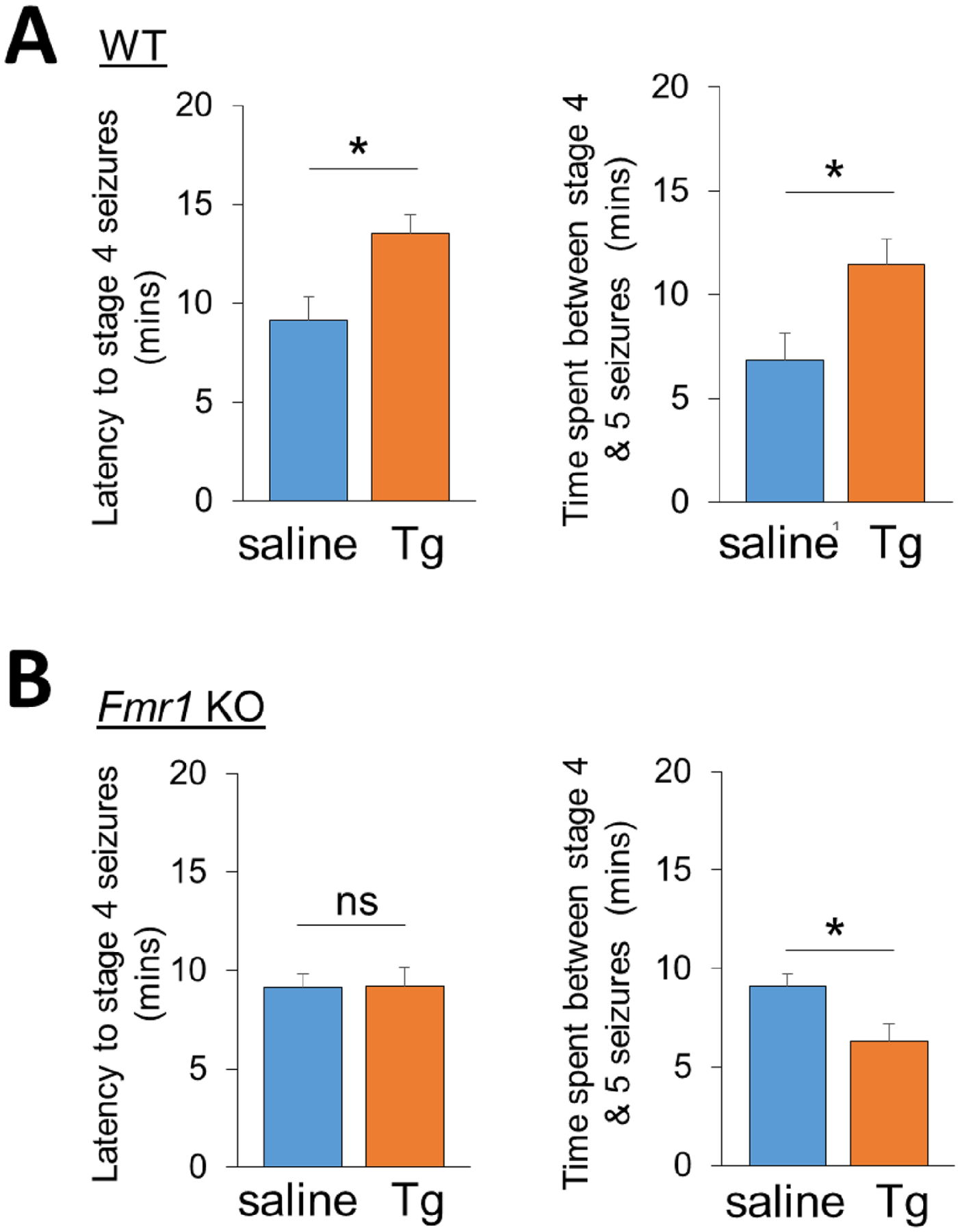
(A, B) Quantification of latency to stage 4 seizures and the time spent between stage 4 to 5 seizures from 3-weeks old wild-type (A) or Fmr1 KO (B) mice intraperitoneally injected with saline or Thapsigargin (Tg; 2 mg/kg) for 3 hours followed by injections with KA (30 mg/kg). n = 8–10 mice per group. Student’s t-test was used. Data are represented as mean ± SEM with *P<0.05, ns: non-significant.
Acute ER stress does not alter neuronal intrinsic excitability
To study the mechanism by which ER stress differentially modulates seizure activity in wild-type and Fmr1 KO mice, we asked whether ER stress is able to modulate neuronal intrinsic excitability, which is known to be critical to overall brain excitability. To this end, we performed a whole-cell patch-clamp recording in dissociated wild-type or Fmr1 KO cortical neurons treated with vehicle (DMSO) or Tg (1 μM) for 1 hour, the dosage and duration that we used previously (Liu et al., 2019), starting at days-in-vitro (DIV) 13–14. We employed a current-clamp recording to measure the action potential firing rate after delivering constant somatic current pulses for durations of 500 ms in the range of 0–200 pA (Lee and Chung, 2014; Lee et al., 2015). As shown (Figs. 2A and 2B), we did not observe any significant differences in the firing rates of action potentials between vehicle- and Tg-treated groups in either wild-type or Fmr1 KO neurons. These data suggest that, although previous studies have reported basally altered intrinsic properties and excitability in Fmr1 KO neurons, these cells do not exhibit different responses in the intrinsic excitability to acute ER stress induction.
Figure 2. Acute ER stress does not alter the neuronal intrinsic excitability.
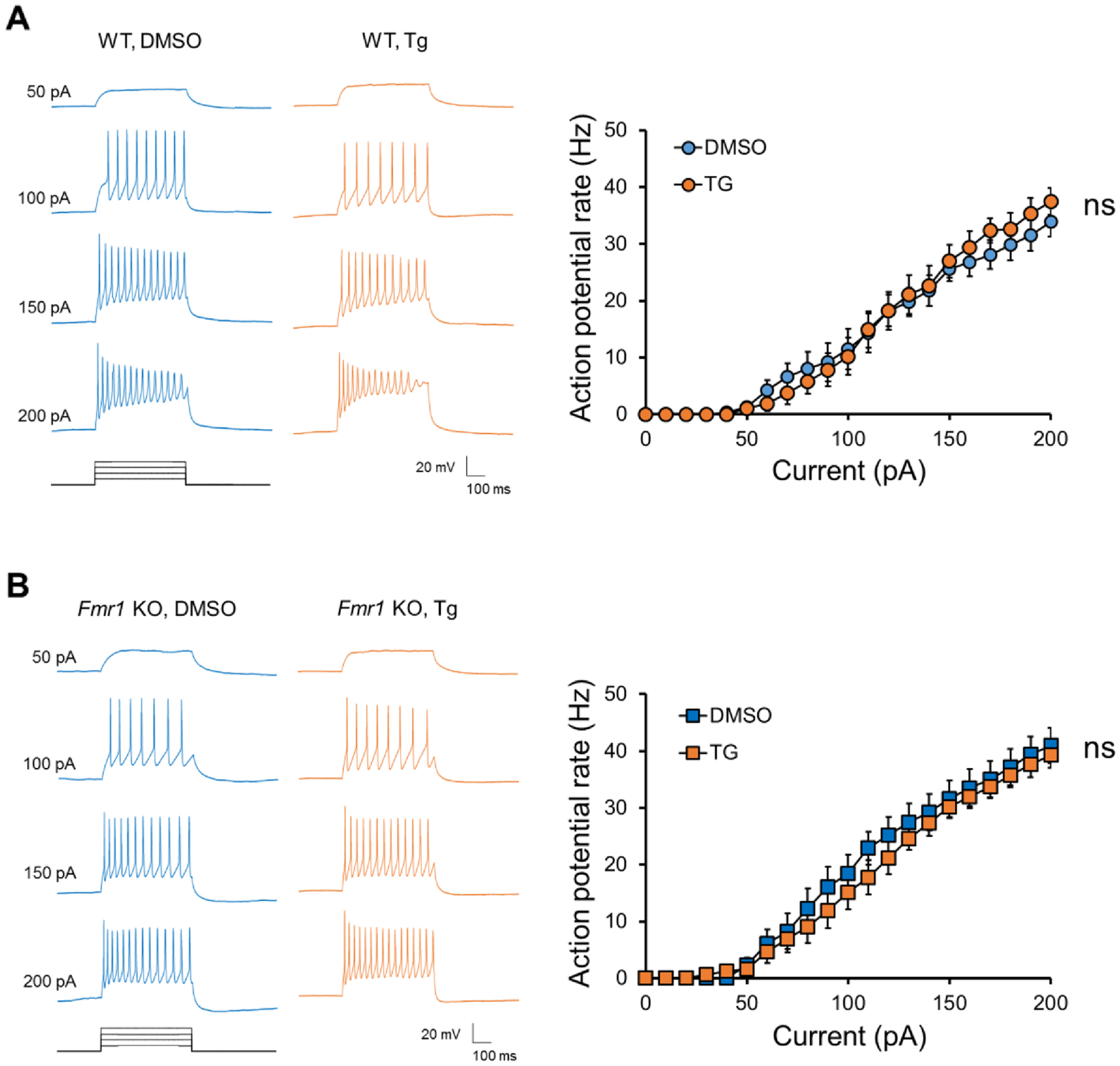
Left: Representative traces of action potentials induced by 50, 100, 150, and 200 pA from wild-type (A) or Fmr1 KO (B) neurons treated with DMSO or Thapsigargin (1 μM) for 1 hour. Right: Average action potential firing rates (Hz) evoked by 0–200 pA injection from wild-type (A) or Fmr1 KO (B) neurons treated with DMSO or Thapsigargin. n = 9–12 neurons per treatment group. The data were analyzed using a one-way ANOVA. Data are represented as mean ± SEM with ns: non-significant.
Acute ER stress induces protein synthesis- and Fmr1-dependent modulation of neural network synchronization
Since acute ER stress does not affect the neuronal excitability at the single-cell level, we asked whether ER stress can affect the synchronicity of neuronal firing at the populational level, a known factor in contributing to seizure susceptibility (Bui et al., 2015; Di Cristo et al., 2018). To answer this question, we employed a multi-electrode array (MEA) system to record the extracellular spontaneous spikes (action potentials) of primary mouse cortical neuron cultures, prepared from wild-type or Fmr1 KO mice, at DIV 13–14. To study the synchronicity of neuronal firing, we measured the cross-electrode synchronization in cultures treated with vehicle (DMSO) or Tg (1 μM) for 1 hour. As shown (Fig. 3), while Tg did not significantly alter the synchronization in wild-type cultures, it significantly elevated the synchronization in Fmr1 KO cultures. This may explain the elevated seizure susceptibility in Fmr1 KO mice following preconditioning of ER stress (Fig. 1B). Noticeably, treatment of a protein synthesis inhibitor cycloheximide (60 μM) caused a significant elevation of the synchronization in wild-type cultures while no further elevation of synchronization was seen in Fmr1 KO cultures. These results suggest that Fmr1 functions to prevent the increase of neural network synchronization through a protein synthesis–dependent manner, following acute induction of ER stress.
Figure 3. A protein translation-dependent maintenance of neural network synchronization following acute ER stress induction in wild-type but not Fmr1 KO neurons.
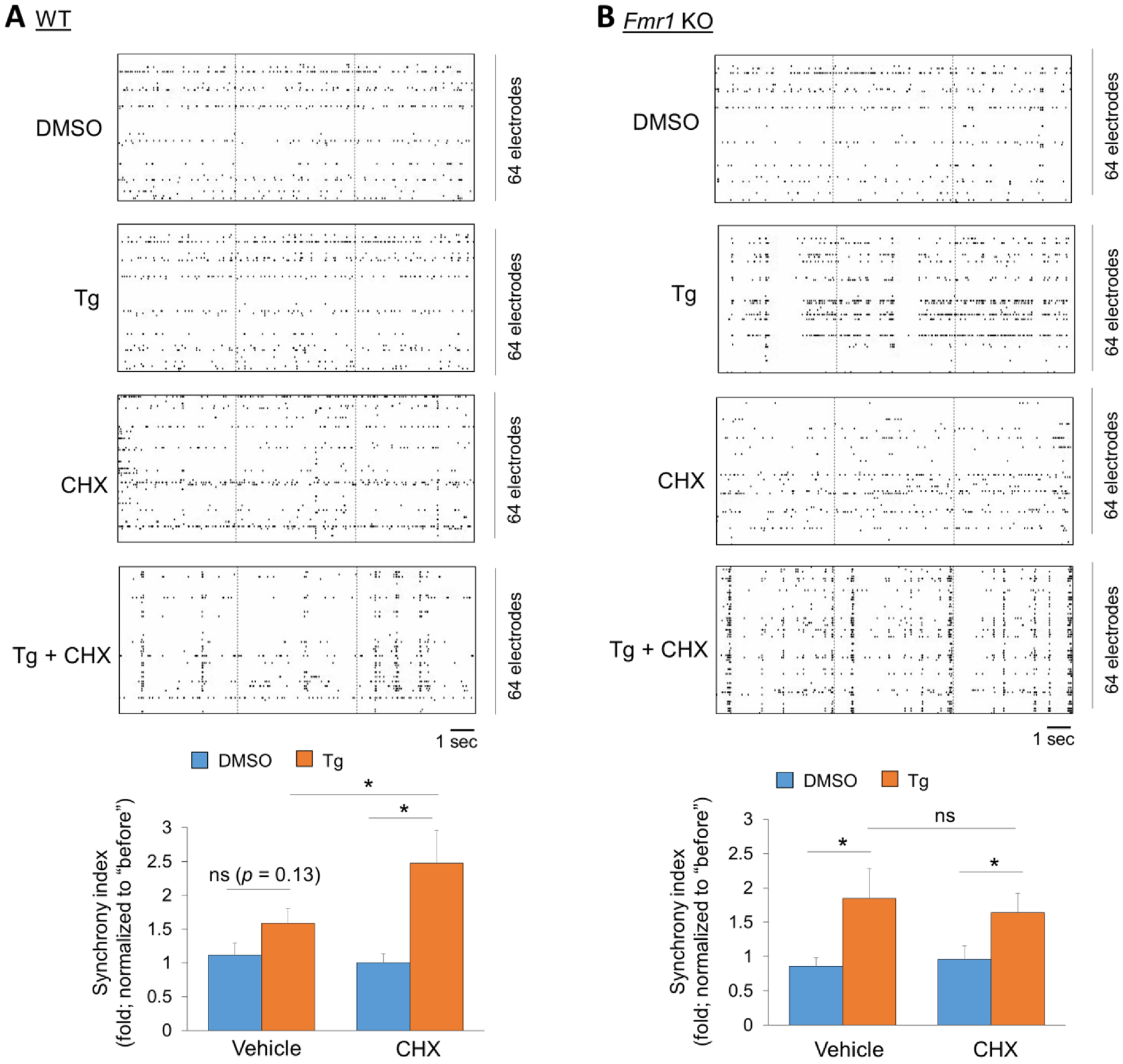
(A, B) Top: Representative raster plots of spontaneous spikes from wild-type (A) and Fmr1 KO (B) cortical neuron cultures treated with vehicle (DMSO), Thapsigargin (Tg, 1 μM), cycloheximide (CHX, 60 μM) or CHX+Tg, for 1 h at DIV14. Bottom: Quantification of synchrony index by comparing ‘after treatment’ to ‘before treatment’ of the same cultures (n = 8–11 independent cultures). A two-way ANOVA with Tukey test were used. Data are represented as mean ± SEM with *P<0.05, ns: non-significant.
Acute ER stress induces Fmr1-dependent activation of Mdm2 to maintain neural network synchronization
Because our previous work has shown that acute ER stress–induced reduction of seizure response is regulated by the ubiquitin E3 ligase Mdm2, we asked whether Mdm2 is differentially regulated upon acute ER stress induction in wild-type and Fmr1 KO cultures. As shown (Fig. 4A), treatment of Tg for 1 hour led to a significantly elevation of Mdm2 in wild-type cultures. Despite no elevated activation of Mdm2 (the levels of p-Mdm2 to total Mdm2 is similar between vehicle- and Tg-treatment groups), due to the elevated Mdm2, the total amount of phosphorylated Mdm2 (p-Mdm2 to Gapdh) is elevated in wild-type cultures treated with Tg. In Fmr1 KO cultures (Fig. 4B), however, despite an elevation of total Mdm2 as seen in wild-type cultures, Mdm2 is abnormally dephosphorylated, leading to a slight reduction of total amount of phosphorylated Mdm2. We conclude from these data that acute ER stress induction elevates the levels of active, phosphorylated Mdm2 in wild-type cultures but not in Fmr1 KO cultures.
Figure 4. Acute ER stress-induced activation of Mdm2 is impaired in Fmr1 KO neurons.
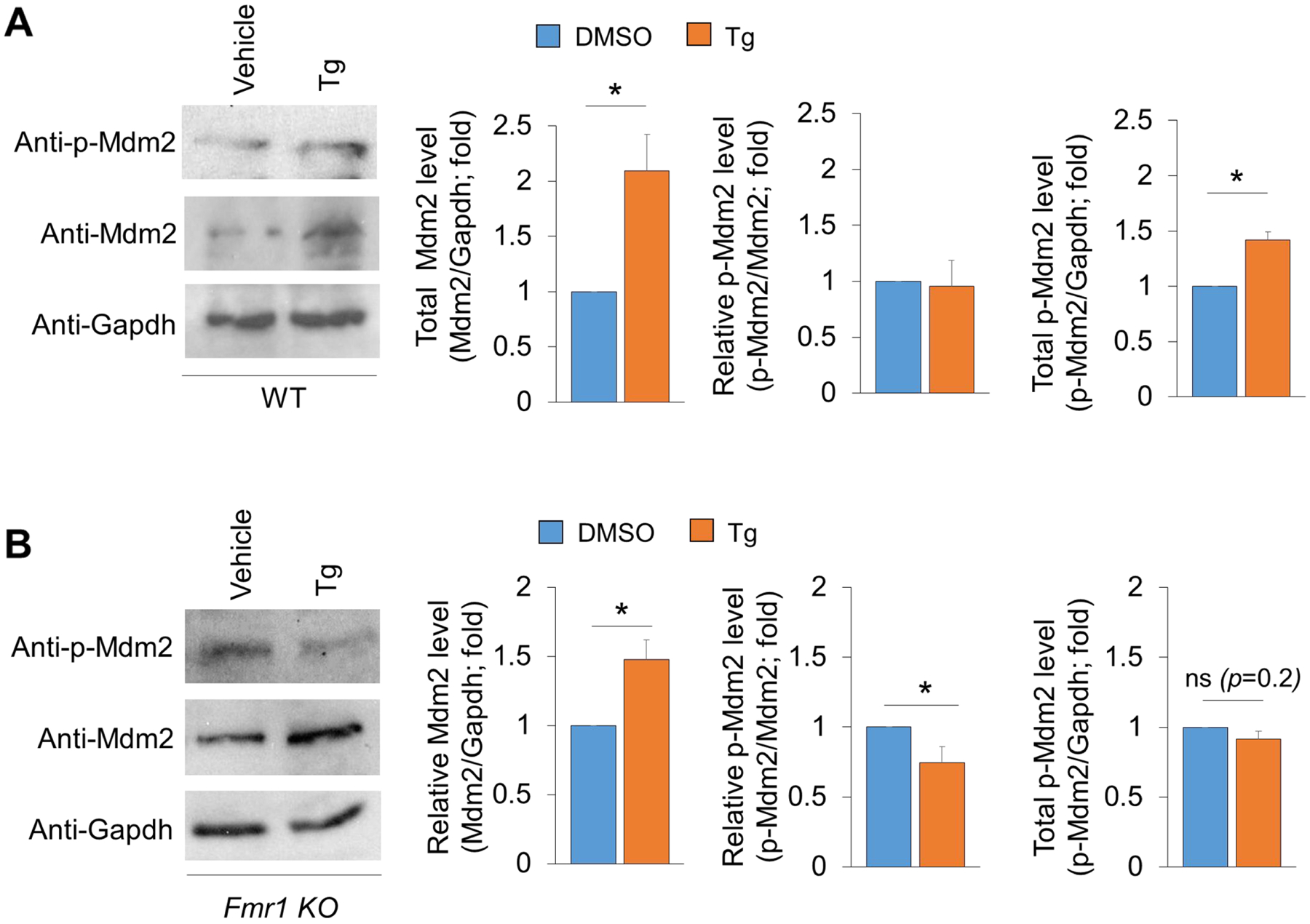
(A, B) Representative western blots and quantification of Mdm2, p-Mdm2 and Gapdh from wild-type (A) and Fmr1 KO (B) cortical neuron cultures treated with vehicle (DMSO) or Thapsigargin (Tg, 1 μM) for 1 h at DIV14. (n = 3–6). Student’s t-test was used. Data are represented as mean ± SEM with *P<0.05, ns: non-significant.
To test whether impaired Mdm2 signaling is crucial to acute ER stress–induced maintenance of neural network synchronization that we observed in Fig. 3, we obtained a conditional Mdm2 knockdown mouse model by crossing Mdm2 floxed mice (Mdm2f/f) with Emx1-Cre mice to obtain Mdm2f/+-Emx-Cre+ (Mdm2-cKD) and Mdm2f/+-Emx-Cre− (Mdm2-WT) mice. Emx1-Cre can confer Mdm2 reduction in the cortex and hippocampus, primarily in excitatory neurons, beginning as early as embryonic day 10.5 (E10.5) (Gorski et al., 2002; Young et al., 2007). We used heterozygous mice (Mdm2f/+) to avoid potential apoptosis caused by complete Mdm2 knockout (Jones et al., 1995; Montes de Oca Luna et al., 1995). The knockdown efficiency of Mdm2 in Mdm2-cKD in cortical neuron cultures at DIV 14 is approximately 45% in comparison to the Mdm2-WT cultures, when cultures are prepared on postnatal day 0, as we have shown previously (Liu et al., 2017). We then measured the cross-electrode synchronization in Mdm2-WT and Mdm2-cKD cultures at DIV 13–14 treated with vehicle (DMSO) or Tg (1 μM) for 1 hour. As shown (Fig. 5), whereas Mdm2-WT cultures exhibited maintained synchronization following the Tg treatment, similar to what we have observed in wild-type cultures (Fig. 3A), Mdm2-cKD cultures exhibited a significant elevation of synchronization following the Tg treatment that is similar to what we have observed in Fmr1 KO cultures (Fig. 3B). These results indicate that impaired Mdm2 signaling likely contributes to abnormally elevated synchronization in Fmr1 KO cultures following the induction of acute ER stress.
Figure 5. The maintenance of neural network synchronization following acute ER stress induction requires Mdm2.
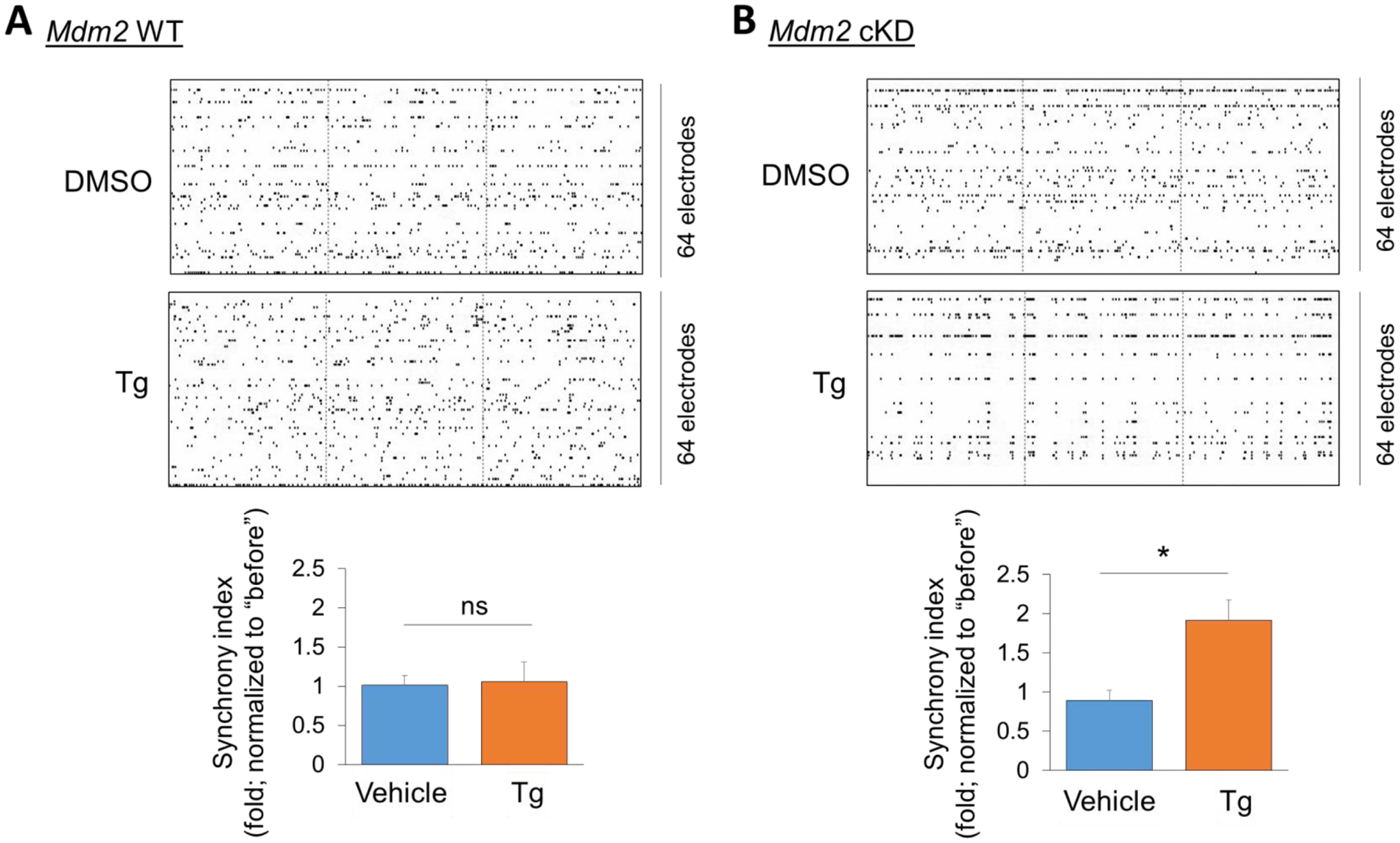
(A, B) Top: Representative raster plots of spontaneous spikes from Mdm2-WT (A) and Mdm2-cKD (B) cortical neuron cultures treated with vehicle (DMSO) or Thapsigargin (Tg, 1 μM) for 1 h at DIV14. Bottom: Quantification of synchrony index by comparing ‘after treatment’ to ‘before treatment’ of the same cultures (n = 6 independent cultures). Student’s t-test was used. Data are represented as mean ± SEM with *P<0.05, ns: non-significant.
Acute ER stress induces Fmr1-dependent p53 ubiquitination and transient protein synthesis
Activated, phosphorylated, Mdm2 is known to trigger ubiquitination and inactivation of tumor suppressor p53. Our previous work has demonstrated that acute ER stress–induced p53 ubiquitination is necessary for a transient protein synthesis. To this end, we aim to test whether this phenomenon is impaired in Fmr1 KO neurons. As shown (Fig. 6), Tg treatment successfully induced ubiquitination of p53 (Fig 6A), downregulation of p53 (Fig. 6B), as well as transient protein synthesis, measured by puromycin labeling (Fig. 6C), in wild-type cultures, as we have previously observed (Liu et al., 2019). However, in Fmr1 KO cultures, Tg treatment abnormally reduced ubiquitination of p53 (Fig 6D). While the total level of p53 was not significantly affected (Fig. 6E), the transient protein synthesis was abnormally reduced in Fmr1 KO cultures following Tg treatment (Fig. 6F). These data further confirm our observation of an impaired response to acute ER stress in Fmr1 KO cultures.
Figure 6. Acute ER stress-induced ubiquitination of p53, down-regulation of p53 as well as transient protein synthesis are impaired in Fmr1 KO neurons.
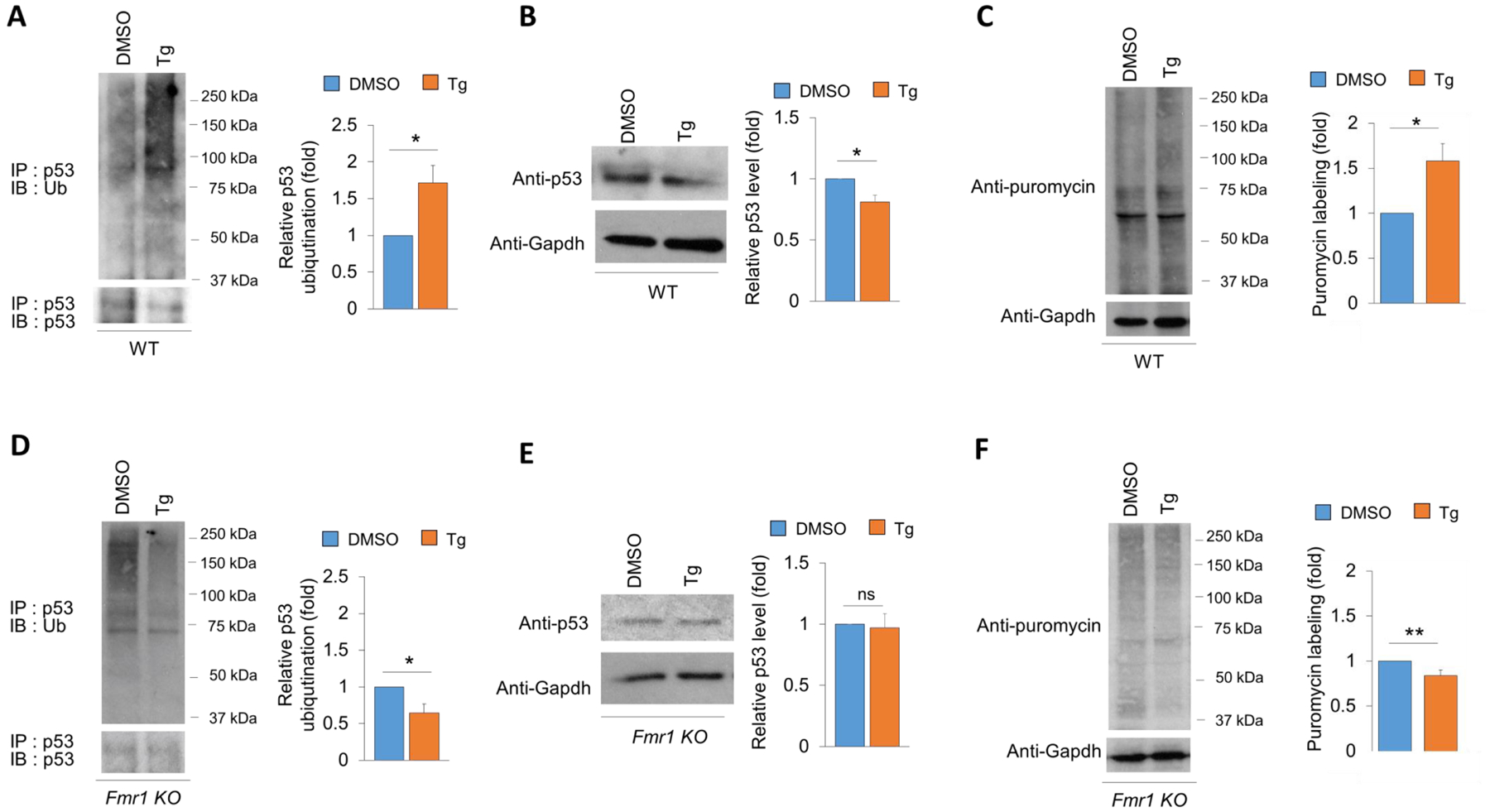
(A, D) Representative western blots of Ubiquitin and p53 after immunoprecipitation with anti-p53 antibody using lysates from wild-type (A) and Fmr1 KO (D) cortical neuron cultures treated with vehicle (DMSO) or Tg for 1 hour. Quantification is performed by first normalizing ubiquitinated p53 (IP:p53, IB:Ub) to immunoprecipitated p53 (IP:p53, IB:p53), followed by normalizing Tg-treated group to DMSO-treated group within the same genotype (n = 5 for both wild-type and Fmr1 KO). (B, E) Representative western blots of p53 and Gapdh from wild-type (B) and Fmr1 KO (E) cortical neuron cultures treated with vehicle (DMSO) or Tg for 1 hour. Quantification is shown on the right (n = 4 and 5 for wild-type and Fmr1 KO, respectively). (C, F) Representative western blots of puromycin and Gapdh, and quantification after 1-hour labeling of puromycin in wild-type (C) and Fmr1 KO (F) cortical neuron cultures treated with vehicle (DMSO) or Tg for 1 hour (n = 6 and 9 for wild-type and Fmr1 KO, respectively). Student’s t-test was used. Data are represented as mean± SEM with *P<0.05, **P<0.01, ns: non-significant.
Inhibition of p53 restores acute ER stress–induced maintenance of neural network synchronicity and reduction of seizure activity
To this point, our results suggest that Fmr1 functions to prevent an elevation of neural network synchronization and to reduce seizure susceptibility. To confirm that these defects are caused by a failure in activation of Mdm2-p53 signaling, we employed a widely used p53 transcriptional inhibitor, Pifithrin-α (1 μM), to mimic inactivation of p53 when p53 is ubiquitinated and downregulated in wild-type cultures (Fig. 6). We first applied Pifithrin-α in Mdm2-cKD cultures and performed MEA recordings as we had done for Fig. 3. As shown (Fig. 7A), Pifithrin-α was able to blunt the abnormally elevated neural network synchronization induced by the Tg treatment. Most importantly, when we performed the same experiment in Fmr1 KO cultures, we were also able to correct the abnormally elevated neural network synchronization induced by the Tg treatment. These results indicate the possibility that Pifithrin-α may be able to reduce abnormally elevated seizure activity in Fmr1 KO mice following preconditioning to acute ER stress that we have observed (Fig. 1). To test this possibility, we intraperitoneally injected Fmr1 KO mice with saline, Tg (2 mg/kg), Pifithrin-α (2 mg/kg), or Tg + Pifithrin-α. Three hours after the injection, the mice were injected again with kainic acid (30 mg/kg). Immediately following the second injection, seizure behavior was closely monitored and scored. As shown (Fig. 8), the Fmr1 KO mice that received Tg exhibited a significantly enhanced latency to stage 4 seizures and a significantly delayed onset of stage 5 seizures, as we had observed in WT mice (Fig. 1). Pifithrin-α alone slightly delayed the seizure activity, as we observed previously (Jewett et al., 2016). Altogether, our data demonstrated that Fmr1 functions to prevent an increase in neural network synchronization and reduce the seizure susceptibility through Mdm2-p53 signaling. These findings also suggest an impaired response to cellular stress in Fmr1 KO and a novel deficit potentially contributing to the imbalanced neuronal excitability in FXS.
Figure 7. Pharmacological inhibition of p53 restores the maintenance of neural network synchronization following acute ER stress induction in Mdm2-cKD and Fmr1 KO cultures.
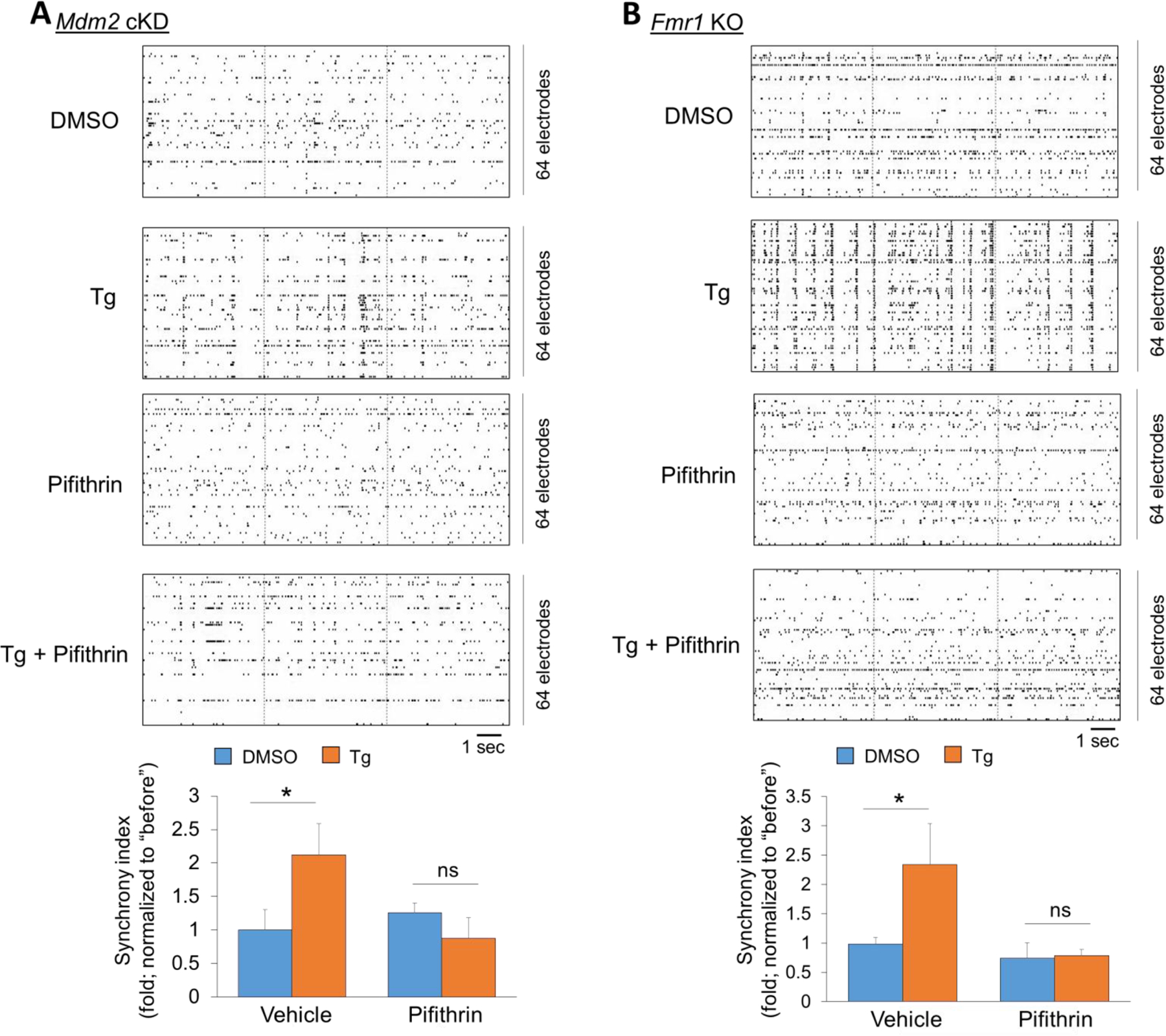
(A, B) Top: Representative raster plots of spontaneous spikes from Mdm2-cKD (A) and Fmr1 KO (B) cortical neuron cultures treated with vehicle (DMSO), Thapsigargin (Tg, 1 μM), Pifithrin-α (Pifithrin, 1 μM) or Pifithrin+Tg, for 1 h at DIV14. Bottom: Quantification of synchrony index by comparing ‘after treatment’ to ‘before treatment’ of the same cultures (n = 9–10 independent cultures). A two-way ANOVA with Tukey test were used. Data are represented as mean ± SEM with *P<0.05, ns: non-significant.
Figure 8. Pharmacological inhibition of p53 restores the sensitivity of seizure severity to acute ER stress in Fmr1 KO mice.
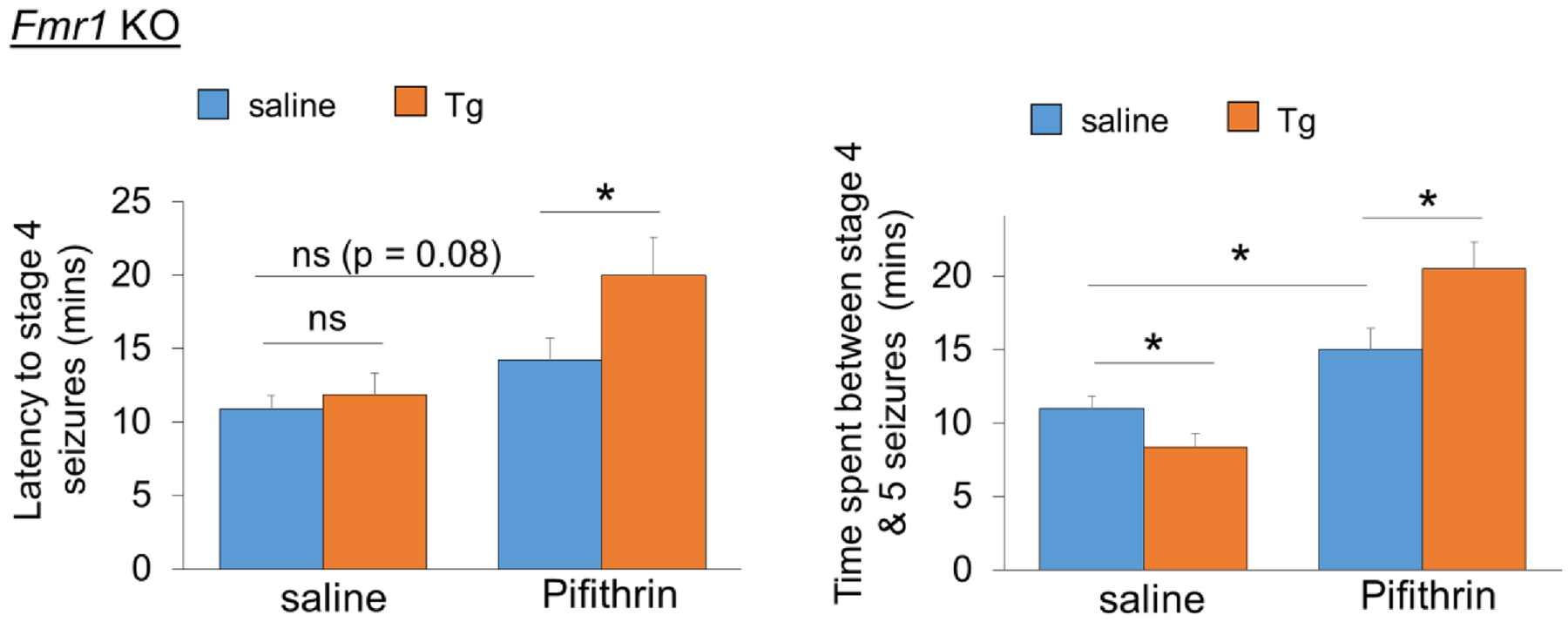
Quantification of latency to stage 4 seizures and the time spent between stage 4 to 5 seizures from 3-weeks old Fmr1 KO mice intraperitoneally injected with saline, Thapsigargin (Tg; 2 mg/kg), Pifithrin-α (Pifithrin; 2 mg/kg) or Tg+Pifithrin for 3 hours followed by injections with KA (30 mg/kg). n = 7–8 mice per group. A two-way ANOVA with Tukey test were used. Data are represented as mean ± SEM with *P<0.05, ns: non-significant.
DISCUSSION
Our study reveals that the animal model of FXS, the Fmr1 KO, exhibits impaired response to acute ER stress that ultimately contributes to abnormally elevated neural network synchronization and seizure susceptibility. Because our previous work has shown that seizure itself can induce ER stress and that the induced Mdm2-p53 signaling following a seizure functions to homeostatically reduce neuronal excitability (Liu et al., 2019), our current data indicate a possibility that Fmr1 KO may have difficulty in homeostatically reducing excitability following a seizure attack. This is consistent with the literature showing epileptic discharges in patients and animal models of FXS (Chuang et al., 2005; Heard et al., 2014). Because Fmr1 is an autism spectrum disorder (ASD) gene that functionally interacts with many other ASD-linked genes, and because imbalanced neuronal excitability and elevated seizure activity are also seen in other ASDs, we aim to understand, as a future direction, whether altered ER stress response and excitability regulation are common and similarly impaired in different ASDs. One of the other ASDs that could be investigated is tuberous sclerosis complex (TSC). TSC results from a mutation of either TSC1 or TSC2, which leads to a dysregulated mammalian target of rapamycin (mTOR) signaling (Tsai et al., 2012b). Patients with TSC exhibit hyperexcitability (Zhao and Yoshii, 2019) and TSC1/2 functionally interacts with p53 to regulate cell differentiation and survival (Armstrong et al., 2017). In addition, mTOR signaling is highly associated with ER stress response (Novotna et al., 2016; Yang et al., 2018). These connections indicate that ER stress response and Mdm2-p53 signaling may be common neuronal excitability regulators that are similarly dysregulated in different psychiatric and neurological disorders. We expect continuous work in the future to address this question.
Imbalanced neuronal excitability has also been linked to cognitive deficits and memory impairment in FXS and other ASDs (Brennan et al., 2006). This is particularly pertinent to hippocampal synaptic plasticity mediated by multiple activity-dependent proteins, such as metabotropic glutamate receptors (mGluRs) and myocyte enhancer factor 2 (MEF2), with which Mdm2 is known to functionally interact. For example, Mdm2 can be downregulated upon activation of group 1 mGluRs and such downregulation contributes to mGluR-dependent de novo protein synthesis through reduced association of Mdm2 in the ribosomes (Liu et al., 2017). On the other hand, Mdm2 can be dephosphorylated upon activation of MEF2, and that leads to activity-dependent synapse elimination through ubiquitination of the post-synaptic density protein 95 (PSD-95) (Tsai et al., 2017; Tsai et al., 2012a). Importantly, both mechanisms mentioned above are impaired in Fmr1 KO mice, indicating a general deficit of activity-dependent regulation in FXS through Mdm2. However, given the multifunctional, substrate-dependent, role of Mdm2 in the brain and the phosphorylation state of Mdm2 that is differentially regulated during different plasticity mechanisms, Mdm2 is unlikely an ideal therapeutic target for FXS. Nevertheless, despite this limitation, it does not change the fact that Mdm2 is a massive regulator in FMRP-dependent neuronal plasticity. Because FMRP interconnects with many other ASD-linked genes to regulate neurodevelopment, continuous study of Mdm2 in the nervous system is crucial and could help us better understand other ASDs.
In the field of cancer biology, p53 is a well-characterized transcription factor (Devine and Dai, 2013). The ubiquitination of p53 by Mdm2 leads to inactivation of p53-dependent transcriptional regulation and ultimately alters the cell cycle (Goldberg et al., 2002; Vazquez et al., 2008; Zhang and Zhang, 2008). Unbiased screening of direct p53 target mRNAs has revealed many brain-enriched genes (Li et al., 2015; Wei et al., 2006). However, the function and regulation of Mdm2-p53 signaling was largely unclear in terminally differentiated neuronal cells before our previous work to demonstrate the roles of p53 in neuronal excitability regulation (Jewett et al., 2018; Lee et al., 2018; Liu et al., 2020). Specifically, we have shown that chronic activity stimulation triggers ubiquitination of p53 and a reduction of that contributes to an impairment in homeostatic downscaling in Fmr1 KO neurons (Jewett et al., 2018; Lee et al., 2018). Together with our current study, our research suggests that an altered transcriptional program involving p53 is crucial to imbalanced neuronal plasticity in FXS. However, because elevated p53 ubiquitination and reduced expression of p53 have been observed in adult mouse neural stem cells (NSCs) of Fmr1 KO mice (Li et al., 2016), inhibiting p53 activity through the use of Pifithrin-α, as we have done in the current study, may dampen the production of new neurons in FXS if the treatment is prolonged. This also reflects on the complexity of cell type–specific alterations in FXS and many other neurodevelopmental disorders. Identifying the differentially regulated target genes of p53 in different cell population in the nervous system of FXS will improve our understanding about p53 and potential therapeutic development for FXS.
ACKNOWLEDGEMENT:
The authors are grateful to the technical support by Bailey Metcalf (University of Illinois at Urbana-Champaign).
FUNDING:
This work is supported by the startup fund provided by the School of Molecular and Cellular Biology, University of Illinois at Urbana-Champaign, and by National Institute of Health (R01NS105615, R01MH124827 and R21MH122840 to N-P.T.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF COMPETING INTEREST:
The authors declare that they have no conflict of interest.
REFERENCES
- Amaral DG, et al. , 2008. Neuroanatomy of autism. Trends Neurosci. 31, 137–45. [DOI] [PubMed] [Google Scholar]
- Armstrong LC, et al. , 2017. Heterozygous loss of TSC2 alters p53 signaling and human stem cell reprogramming. Hum Mol Genet. 26, 4629–4641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashley CT Jr., et al. , 1993. FMR1 protein: conserved RNP family domains and selective RNA binding. Science. 262, 563–6. [DOI] [PubMed] [Google Scholar]
- Beique JC, et al. , 2011. Arc-dependent synapse-specific homeostatic plasticity. Proc Natl Acad Sci U S A. 108, 816–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry-Kravis E, et al. , 2010. Seizures in fragile X syndrome: characteristics and comorbid diagnoses. Am J Intellect Dev Disabil. 115, 461–72. [DOI] [PubMed] [Google Scholar]
- Brennan FX, et al. , 2006. Fmr1 knockout mice are impaired in a leverpress escape/avoidance task. Genes Brain Behav. 5, 467–71. [DOI] [PubMed] [Google Scholar]
- Bui A, et al. , 2015. Microcircuits in Epilepsy: Heterogeneity and Hub Cells in Network Synchronization. Cold Spring Harb Perspect Med. 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang SC, et al. , 2005. Prolonged epileptiform discharges induced by altered group I metabotropic glutamate receptor-mediated synaptic responses in hippocampal slices of a fragile X mouse model. J Neurosci. 25, 8048–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyne AN, et al. , 2015. Fragile X protein mitigates TDP-43 toxicity by remodeling RNA granules and restoring translation. Febs j. 24, 6886–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devine T, Dai MS, 2013. Targeting the ubiquitin-mediated proteasome degradation of p53 for cancer therapy. Curr Pharm Des. 19, 3248–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Cristo G, et al. , 2018. KCC2, epileptiform synchronization, and epileptic disorders. Prog Neurobiol. 162, 1–16. [DOI] [PubMed] [Google Scholar]
- Di Marco B, et al. , 2021. Fragile X mental retardation protein (FMRP) and metabotropic glutamate receptor subtype 5 (mGlu5) control stress granule formation in astrocytes. Mol Autism. 154, 105338. [DOI] [PubMed] [Google Scholar]
- Eagleman DE, et al. , 2020. Unbiased proteomic screening identifies a novel role for the E3 ubiquitin ligase Nedd4-2 in translational suppression during ER stress. J Neurochem. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggermont JJ, 2006. Properties of correlated neural activity clusters in cat auditory cortex resemble those of neural assemblies. J Neurophysiol. 96, 746–64. [DOI] [PubMed] [Google Scholar]
- Ferraro TN, et al. , 2001. Quantitative genetic study of maximal electroshock seizure threshold in mice: evidence for a major seizure susceptibility locus on distal chromosome 1. Genomics. 75, 35–42. [DOI] [PubMed] [Google Scholar]
- Gao M, et al. , 2010. A specific requirement of Arc/Arg3.1 for visual experience-induced homeostatic synaptic plasticity in mouse primary visual cortex. J Neurosci. 30, 7168–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson JR, et al. , 2008. Imbalance of neocortical excitation and inhibition and altered UP states reflect network hyperexcitability in the mouse model of fragile X syndrome. J Neurophysiol. 100, 2615–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg Z, et al. , 2002. Tyrosine phosphorylation of Mdm2 by c-Abl: implications for p53 regulation. EMBO J. 21, 3715–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonçalves JT, et al. , 2013. Circuit level defects in the developing neocortex of Fragile X mice. Nat Neurosci. 16, 903–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorski JA, et al. , 2002. Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emx1-expressing lineage. J Neurosci. 22, 6309–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross C, et al. , 2011. Fragile X mental retardation protein regulates protein expression and mRNA translation of the potassium channel Kv4.2. J Neurosci. 31, 5693–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, et al. , 2016. Selective Disruption of Metabotropic Glutamate Receptor 5-Homer Interactions Mimics Phenotypes of Fragile X Syndrome in Mice. J Neurosci. 36, 2131–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heard TT, et al. , 2014. EEG abnormalities and seizures in genetically diagnosed Fragile X syndrome. Int J Dev Neurosci. 38, 155–60. [DOI] [PubMed] [Google Scholar]
- Hu JH, et al. , 2010. Homeostatic scaling requires group I mGluR activation mediated by Homer1a. Neuron. 68, 1128–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacoangeli A, Tiedge H, 2013. Translational control at the synapse: role of RNA regulators. Trends Biochem Sci. 38, 47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jewett KA, et al. , 2016. Feedback modulation of neural network synchrony and seizure susceptibility by Mdm2-p53-Nedd4-2 signaling. Mol Brain. 9, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jewett KA, et al. , 2018. Dysregulation and restoration of homeostatic network plasticity in fragile X syndrome mice. Neuropharmacology. 138, 182–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SN, et al. , 1995. Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature. 378, 206–8. [DOI] [PubMed] [Google Scholar]
- Keith DJ, et al. , 2012. Palmitoylation of A-kinase anchoring protein 79/150 regulates dendritic endosomal targeting and synaptic plasticity mechanisms. J Neurosci. 32, 7119–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korb E, et al. , 2013. Arc in the nucleus regulates PML-dependent GluA1 transcription and homeostatic plasticity. Nat Neurosci. 16, 874–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KY, Chung HJ, 2014. NMDA receptors and L-type voltage-gated Ca(2)(+) channels mediate the expression of bidirectional homeostatic intrinsic plasticity in cultured hippocampal neurons. Neuroscience. 277, 610–23. [DOI] [PubMed] [Google Scholar]
- Lee KY, et al. , 2018. Loss of fragile X protein FMRP impairs homeostatic synaptic downscaling through tumor suppressor p53 and ubiquitin E3 ligase Nedd4-2. Hum Mol Genet. 27, 2805–2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KY, et al. , 2015. N-methyl-D-aspartate receptors mediate activity-dependent down-regulation of potassium channel genes during the expression of homeostatic intrinsic plasticity. Mol Brain. 8, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitt P, et al. , 2004. Regulation of neocortical interneuron development and the implications for neurodevelopmental disorders. Trends Neurosci. 27, 400–6. [DOI] [PubMed] [Google Scholar]
- Li Y, et al. , 2015. p53 Enables metabolic fitness and self-renewal of nephron progenitor cells. Development. 142, 1228–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, et al. , 2016. MDM2 inhibition rescues neurogenic and cognitive deficits in a mouse model of fragile X syndrome. Sci Transl Med. 8, 336ra61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu DC, et al. , 2019. Novel roles of ER stress in repressing neural activity and seizures through Mdm2- and p53-dependent protein translation. PLoS Genet. 15, e1008364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu DC, et al. , 2017. Mdm2 mediates FMRP- and Gp1 mGluR-dependent protein translation and neural network activity. Hum Mol Genet. 26, 3895–3908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu DC, et al. , 2020. Chronic Activation of Gp1 mGluRs Leads to Distinct Refinement of Neural Network Activity through Non-Canonical p53 and Akt Signaling. J Neurochem. 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovelace JW, et al. , 2016. Matrix metalloproteinase-9 deletion rescues auditory evoked potential habituation deficit in a mouse model of Fragile X Syndrome. Neurobiol Dis. 89, 126–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin BS, et al. , 2014. Deficient tonic GABAergic conductance and synaptic balance in the fragile X syndrome amygdala. J Neurophysiol. 112, 890–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalon A, et al. , 2012. Chronic pharmacological mGlu5 inhibition corrects fragile X in adult mice. Neuron. 74, 49–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montes de Oca Luna R, et al. , 1995. Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature. 378, 203–6. [DOI] [PubMed] [Google Scholar]
- Novotna B, et al. , 2016. Activation of the ER stress and calcium signaling in angiomyolipoma. Neoplasma. 63, 687–95. [DOI] [PubMed] [Google Scholar]
- Pfeiffer BE, et al. , 2010. Fragile X mental retardation protein is required for synapse elimination by the activity-dependent transcription factor MEF2. Neuron. 66, 191–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racine RJ, 1972. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol. 32, 281–94. [DOI] [PubMed] [Google Scholar]
- Repicky S, Broadie K, 2009. Metabotropic glutamate receptor-mediated use-dependent down-regulation of synaptic excitability involves the fragile X mental retardation protein. J Neurophysiol. 101, 672–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronesi JA, et al. , 2012. Disrupted Homer scaffolds mediate abnormal mGluR5 function in a mouse model of fragile X syndrome. Nat Neurosci. 15, 431–40, s1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronesi JA, Huber KM, 2008. Metabotropic glutamate receptors and fragile x mental retardation protein: partners in translational regulation at the synapse. Sci Signal. 1, pe6. [DOI] [PubMed] [Google Scholar]
- Rotschafer SE, Razak KA, 2014. Auditory processing in fragile x syndrome. Front Cell Neurosci. 8, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen M, et al. , 2019. Reduced mitochondrial fusion and Huntingtin levels contribute to impaired dendritic maturation and behavioral deficits in Fmr1-mutant mice. Nat Neurosci. 22, 386–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd JD, et al. , 2006. Arc/Arg3.1 mediates homeostatic synaptic scaling of AMPA receptors. Neuron. 52, 475–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soden ME, Chen L, 2010. Fragile X protein FMRP is required for homeostatic plasticity and regulation of synaptic strength by retinoic acid. J Neurosci. 30, 16910–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taha MS, et al. , 2021. Novel FMRP interaction networks linked to cellular stress. 288, 837–860. [DOI] [PubMed] [Google Scholar]
- Tian X, et al. , 2010. MEF-2 regulates activity-dependent spine loss in striatopallidal medium spiny neurons. Mol Cell Neurosci. 44, 94–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai NP, et al. , 2017. FMRP-dependent Mdm2 dephosphorylation is required for MEF2-induced synapse elimination. Hum Mol Genet. 26, 293–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai NP, et al. , 2012a. Multiple autism-linked genes mediate synapse elimination via proteasomal degradation of a synaptic scaffold PSD-95. Cell. 151, 1581–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai PT, et al. , 2012b. Autistic-like behaviour and cerebellar dysfunction in Purkinje cell Tsc1 mutant mice. Nature. 488, 647–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utami KH, et al. , 2020. Elevated de novo protein synthesis in FMRP-deficient human neurons and its correction by metformin treatment. 11, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez A, et al. , 2008. The genetics of the p53 pathway, apoptosis and cancer therapy. Nat Rev Drug Discov. 7, 979–87. [DOI] [PubMed] [Google Scholar]
- Vislay RL, et al. , 2013. Homeostatic responses fail to correct defective amygdala inhibitory circuit maturation in fragile X syndrome. J Neurosci. 33, 7548–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei CL, et al. , 2006. A global map of p53 transcription-factor binding sites in the human genome. Cell. 124, 207–19. [DOI] [PubMed] [Google Scholar]
- Yang F, et al. , 2018. Uncoupling of ER/Mitochondrial Oxidative Stress in mTORC1 Hyperactivation-Associated Skin Hypopigmentation. J Invest Dermatol. 138, 669–678. [DOI] [PubMed] [Google Scholar]
- Young KM, et al. , 2007. Subventricular zone stem cells are heterogeneous with respect to their embryonic origins and neurogenic fates in the adult olfactory bulb. J Neurosci. 27, 8286–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, et al. , 2014. Dendritic channelopathies contribute to neocortical and sensory hyperexcitability in Fmr1(−/y) mice. Nat Neurosci. 17, 1701–9. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Zhang R, 2008. Proteasome activator PA28 gamma regulates p53 by enhancing its MDM2-mediated degradation. EMBO J. 27, 852–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao JP, Yoshii A, 2019. Hyperexcitability of the local cortical circuit in mouse models of tuberous sclerosis complex. Mol Brain. 12, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong X, et al. , 2012. MeCP2 phosphorylation is required for modulating synaptic scaling through mGluR5. J Neurosci. 32, 12841–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, et al. , 2017. Epilepsy-associated gene Nedd4-2 mediates neuronal activity and seizure susceptibility through AMPA receptors. PLoS Genet. 13, e1006634. [DOI] [PMC free article] [PubMed] [Google Scholar]