

Abstract
Dysregulation of the astrocytic glutamate transporter excitatory amino acid transporter 2 (EAAT2) is associated with several neurological disorders, including Parkinson’s disease, Alzheimer’s disease, and manganism, the latter induced by chronic exposure to high levels of manganese (Mn). Mechanisms of Mn-induced neurotoxicity include impairment in EAAT2 function secondary to the activation of the transcription factor Yin Yang 1 (YY1) by nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB). However, the upstream mechanisms by which Mn-induced NF-κB activates YY1 remain has yet to be elucidated. In the present study, we used the H4 human astrocyte cell line to test if Mn activates YY1 through the canonical NF-κB signaling pathway, leading to EAAT2 repression. The results demonstrate that Mn exposure induced phosphorylation of the upstream kinase IκB kinase IKK-β, leading to NF-κB p65 translocation, increased YY1 promoter activity, mRNA/protein levels, and consequently repressed EAAT2. Results also demonstrated that Mn-induced oxidative stress and subsequent TNF-α production was upstream of IKK-β activation, as antioxidants attenuated Mn-induced TNF-α production and IKK-β activation. Moreover, the TNF-α inhibition attenuated Mn-induced activation of IKK-β and YY1. Taken together, Mn-induced oxidative stress and TNF-α mediates activation of NF-κB signaling and YY1 upregulation, leading to the repression of EAAT2. Thus, targeting reactive oxygen species (ROS), TNF-α and IKK-β may attenuate Mn-induced YY1 activation and consequent EAAT2 repression.
Keywords: Manganese, oxidative stress, TNF-α, EAAT2, NF-κB, YY1
1. Introduction
Glutamate, the primary excitatory neurotransmitter of the central nervous system (CNS), has been implicated in several neurological disorders, including Parkinson’s disease (PD), Alzheimer’s disease (AD), schizophrenia, and epilepsy [1–3]. Thus, glutamate transporters serve as critical regulators of CNS homeostasis, where they maintain optimal extracellular glutamate concentration via glutamate-glutamine cycling [4, 5]. In particular, astrocytic glutamate transporters known as excitatory amino acid transporters (EAATs) rapidly reuptake synaptic glutamate after signal termination, preventing excitotoxic neuronal injury [6, 7]. Among the five identified EAAT subtypes in humans, EAAT2 is the primary astrocytic glutamate transporter in the CNS, accounting for over 90% of synaptic glutamate reuptake [8]. While the mechanisms of neurodegenerative diseases are not well understood, the dysregulation of EAAT2 and consequent neuronal excitotoxicity may play a significant role in neuropathogenesis [9–11]. Therefore, examining the mechanisms involved in EAAT2 impairment is critical to identifying new therapeutic targets in the treatment of neurological disorders associated with astrocytic glutamate transporter dysregulation.
Chronic exposure to the heavy metal manganese (Mn) leads to a neurological disorder known as manganism, which shares pathological features with sporadic PD [12, 13]. In addition to extrapyramidal motor dysregulation, Mn targets astrocytes and induces toxicity via oxidative stress, inflammation, apoptosis, and impairment of glutamate transport [14–16]. Our previous studies have shown that Mn dysregulates glutamate transporters, decreasing EAAT2 mRNA and protein levels and impairing astrocytic glutamate uptake [17]. Yet the mechanism by which Mn induces transcriptional repression of EAAT2 has yet to be elucidated. Notably, Mn exposure upregulates the transcription factors nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and Yin Yang 1 (YY1), which mediate the expression and function of glutamate transporters both in vitro and in vivo [17–19].
The EAAT2 promoter contains a consensus site for YY1, a multifunctional transcription factor involved in embryogenesis, neuronal differentiation, and oncogenesis [17, 20]. Mn exposure increases YY1 promoter activity and mRNA/protein levels, as well as enhances its binding to the EAAT2 promoter, repressing EAAT2 gene expression and leading to decreased glutamate uptake and neuronal excitotoxicity due to excess synaptic glutamate [21]. In agreement with these findings, studies demonstrate that mutation of the YY1 binding site restores EAAT2 expression and function, suggesting that YY1 may serve as a critical mediator of Mn-induced neurotoxicity [17].
Previous studies have reported that the YY1 promoter contains two NF-κB binding sites, and mutation of the functional NF-κB binding site decreases YY1 promoter activity [17, 22]. Additionally, pharmacological induction of NF-κB resulted in the upregulation of YY1 in C2C12 myoblasts, and shRNA knockdown of p65 attenuated increases in YY1 expression [22]. In vivo, p65−/− mice showed decreased YY1 mRNA and protein levels, suggesting that NF-κB is a critical regulator of YY1 expression [22].
NF-κB signaling may be activated via canonical or non-canonical pathways [23–25]. The canonical NF-κB pathway is induced in response to toxicant exposure, cellular stressors, or proinflammatory stimuli, including reactive oxygen species (ROS) and tumor necrosis factor-alpha (TNF-α), and this pathway involves phosphorylation of the beta subunit of the IκB kinase (IKK) complex (IKK-β) [26–28]. Notably, NF-κB can also negatively regulate EAAT2 via TNF-α [29, 30]. As Mn toxicity potentiates the rapid release of ROS and TNF-α, Mn-induced YY1 upregulation may be induced by the IKK-β-mediated activation of canonical NF-κB signaling in response to oxidative stress and TNF-α [31, 32].
In the present study, we investigated the role of the canonical NF-κB pathway in Mn-induced YY1 activation. Our results demonstrate that Mn exposure induced oxidative stress and TNF-α production, leading to canonical phosphorylation of the upstream kinase IKK-β, NF-κB p65 nuclear translocation, increased YY1 promoter activity and mRNA/protein levels, and consequent EAAT2 repression. Pharmacological inhibition along this pathway attenuated Mn-induced YY1 activation and restored EAAT2. Taken together, our findings demonstrate that Mn-induced YY1 activation is critically mediated by canonical NF-κB signaling induced by oxidative stress and TNF-α, leading to the repression of EAAT2.
2. Materials and Methods
2.1. Materials
Cell culture media and reagents were purchased from Gibco (Carlsbad, CA). Manganese (II) chloride (MnCl2), N-Acetyl-L-cysteine (NAC), α-tocopherol (AT), protease inhibitor cocktail, and dimethyl sulfoxide (DMSO) were purchased from MilliporeSigma (St. Louis, MO). The TNF-α inhibitor (E)-4-2-4-chloro-3-nitrophenyl (C-87) and the IKK-β inhibitor IKK16 were purchased from Tocris (Littleton, CO). The chloromethyl derivative of 2′,7′-dichlorodihydrofluorescein diacetate (CM-H2DCFDA) was purchased from Life Technologies, Inc. (Carlsbad, CA). Antibodies for EAAT2 (ab41621), IKK-β (ab32135) and phospho-IKK-β (ab59195) were obtained from Abcam (Cambridge, MA); antibodies for NF-κB p65 (sc-8008), YY1 (sc-7341), β-actin (sc-47778), and histone H4 (sc-25260) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Bright-Glo luciferase reporter assay kits were purchased from Promega (Madison, WI), and an RNA isolation kit was obtained from Qiagen (Valencia, CA). The TNF-α standard tetramethylbenzidine (TMB) enzyme-linked immunosorbent assay (ELISA) development kit for human samples (900-T25) was purchased from PeproTech (Rocky Hill, NJ). All chemicals were prepared in phosphate-buffered saline (PBS), double-distilled H2O or DMSO, according to the manufacturer’s instructions, and diluted to working concentration in Opti-MEM prior to use.
2.2. Cell Culture
H4 human astrocytes were obtained from obtained from American Type Culture Collection (ATCC, Manassas, VA) and grown in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin. After dissociation with 0.025% Trypsin-0.1g/l EDTA, cells were plated in 24-well plates for luciferase assays, 6-well plates for mRNA and protein analysis, and 100×20 mm dishes for cellular fractionation and immunoprecipitation assays. Cultures were maintained at 37°C in a 95% air, 5% CO2 incubator.
2.3. Luciferase assay
H4 astrocytes were cultured in 500 μL of growth medium in 24-well plates, then transfected with 0.5 μg of NF-κB, YY1, or EAAT2 luciferase vectors. The NF-κB reporter vector was purchased from Clontech, the YY1 wild-type promoter vector was provided by Dr. Guttridge (Ohio State University), and the EAAT2 promoter vector was a gift from Dr. Baldwin (University of North Carolina at Chapel Hill). Transfections were performed with Lipofectamine 3000 reagent (Invitrogen) in MEM supplemented with 5% FBS. Following transfection, cells were treated with the designated compounds in Opti-MEM and incubated for the indicated time points (see figures and legends). Following treatment, luciferase activity was measured via the Bright-Glo luciferase kit (Promega) in accordance with the manufacturer’s protocol.
2.4. Cell lysate preparation and western blot analysis
Cells were treated with the designated compounds in Opti-MEM (see figures and legends), then washed twice with ice-cold PBS. Following treatment, cells were lysed with radioimmunoprecipitation assay (RIPA) buffer containing a protease inhibitor cocktail. Protein concentration of the cell lysate was determined via bicinchoninic acid (BCA) assay (ThermoFisher) according to manufacturer’s instructions and 30 μg of protein were mixed with Laemmli sample buffer containing 5% β-mercaptoethanol. After heating at 95°C for 5 minutes, the samples were run on a 10% SDS-PAGE gel under reducing conditions and electrophoretically transferred to a nitrocellulose membrane. Western blot analysis was performed using primary antibodies at a 1:1000 dilution and horseradish peroxidase (HRP) - conjugated secondary antibodies were used at a 1:5000 dilution. The proteins were then detected using the SuperSignal™ West Pico PLUS chemiluminescent detection kit (ThermoFisher Scientific, Rockford, IL) and a Molecular Imager ChemiDoc XRS+ System (BioRad, Hercules, CA).
2.5. Cellular fractionation
Cells were washed twice with ice-cold PBS and lysed in a hypotonic buffer (10 mM HEPES-KOH pH 7.9, 10 mM KCl, 1.5 mM MgCl2) containing 0.5% NP-40 (Millipore Sigma). After 15 minutes on ice, the cell lysate was centrifuged for five (5) minutes at 2,500 rpm at 4°C. The supernatant was retained as the cytoplasmic fraction, and the nuclear pellet was washed with hypotonic buffer without detergent. The pelleted nuclei were then incubated in a hypertonic buffer (20 mM HEPES-KOH pH 7.9, 0.4 M NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, and 25% glycerol) on ice for 30 min with periodic vortexing. After centrifugation at 20,000 × g for 10 min at 4°C, the supernatant was collected as the nuclear fraction. Protein concentration was determined by BCA assay.
2.6. Quantitative real-time PCR (qPCR)
Following treatment with the indicated compounds, cells were washed twice with ice-cold PBS and total RNA was extracted using the RNeasy Mini RNA Isolation kit (Qiagen). The purified RNA (2 μg) was transcribed to cDNA using a high-capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA). Quantitative real-time PCR (qPCR) was performed for YY1, EAAT2 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) using the CFX96 real-time PCR detection system (BioRad). The PCR reaction mixture contained 1 μg of cDNA template, 0.4 μM of the designated primers and iQ SYBR Green Supermix (BioRad), for a final volume of 10 μL. The primer pairs were as follows: YY1, 5’-GCG ACG ACG ACT ACA TTG AAC-3’ (forward) and 5’-TTC TTG CCG CTC TTC TTG CC-3’; EAAT2, 5’-CAA CAG AGC CCT CTC TGA ATA C-3’ (forward) and 5’- GTA GGG TGG ATG GGA TAC AAT G -3’ (reverse); GAPDH, 5’-CTC TGC TCC TCC TGT TCG AC -3’ (forward) and 5’-GCG CCC AAT ACG ACC AAA TC-3’ (reverse). The qPCR parameters utilized were one cycle at 95°C for 10 minutes, 40 cycles at 95°C for 15 seconds, and 60°C for 1 minute. GAPDH was utilized as an internal control. Following PCR, mRNA levels were analyzed with Bio-Rad CFX Manager Version 3.1.
2.7. ELISA
Mn-induced TNF-α release from H4 cells was measured using a human TNF-α ELISA kit (PeproTech). H4 cells were grown in 6-well plates and treated with the indicated compounds, and cell-free media (1 mL/well) were collected. The ELISA was performed according to the manufacturer’s instructions, and the concentration of secreted TNF-α was determined using a multimode microplate reader (Molecular Devices) set to 450 nm, with wavelength correction at 620 nm.
2.8. Measurement of ROS
The generation of ROS after Mn exposure was utilized as an indicator of oxidative stress and measured via the CM-H2DCFDA ROS molecular probe (Life Technologies). Astrocytes exposed to Mn (250 μM) for 3 hours. Following treatment, cells were washed with PBS and 2.5 μM of CM-H2DCFDA was added for 10 minutes. Endpoint fluorescence was determined using the Spectramax i3x Multi-mode microplate reader (Molecular Devices, San Jose, CA) at an excitation/emission wavelength of 485/527 nm.
2.9. Statistical analysis
All data are expressed as mean ± standard deviation (SD). Statistical analyses were carried out via Student t-test or one-way analysis of variance (ANOVA), followed by a Tukey’s post hoc test using GraphPad Prism Software version 6.0 (San Diego, CA). A p-value of less than 0.05 (p <0.05) was considered statistically significant. The data are representative of three independent experiments.
3. Results
3.1. Mn induces ROS production and phosphorylation of IKK-β.
Mn accumulates in mitochondria and induces the rapid release of ROS in various neural cell types including neurons [32] and astrocytes [33]. In the present study, we tested if Mn induces ROS and phosphorylates IKK-β, a kinase of IκB in the canonical NF-κB pathway, using a pathologically relevant concentration of Mn (250 μM) [18, 34–36]. As shown in Fig. 1A, Mn induced ROS production within 3 hours of incubation. Additionally, Mn induced phosphorylation of IKK-β within the same period (Fig. 1B), indicating that Mn induces ROS and activates IKK-β concomitantly and warranting further investigation of the role of ROS and IKK-β in Mn-induced YY1 upregulation via NF-κB activation.
Fig. 1. Mn induces ROS production and IKK-β phosphorylation.
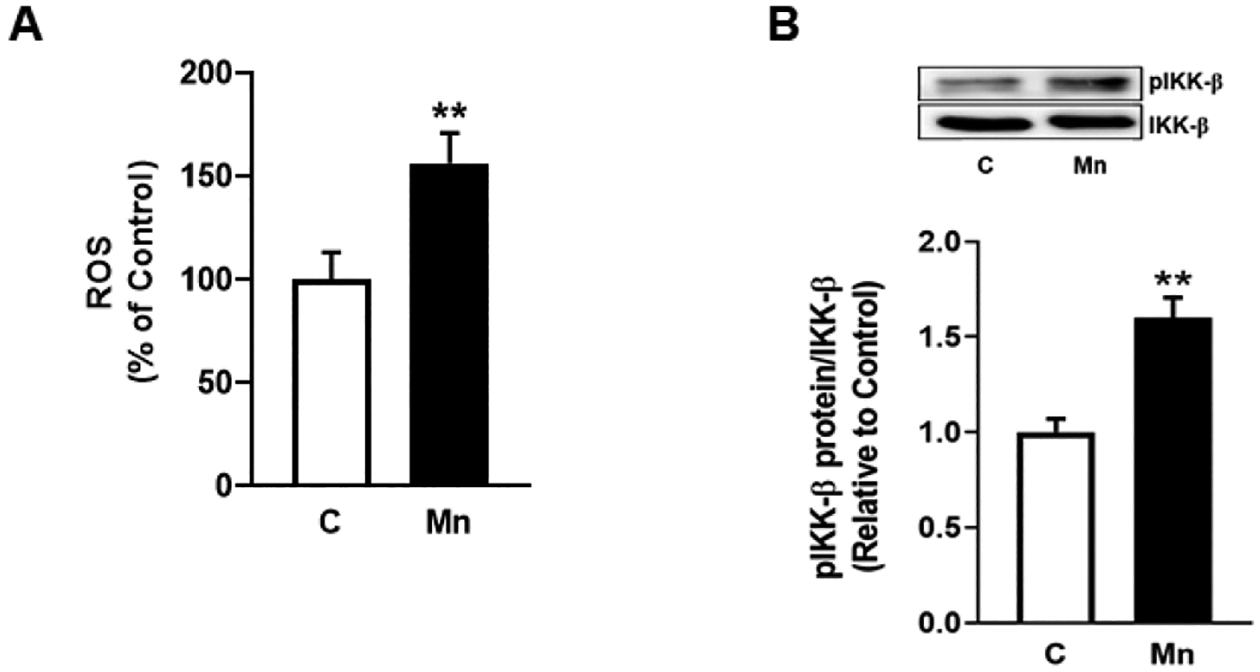
(A) Human H4 astrocytes were exposed to Mn (250 μM) for 3h, and cell lysates were analyzed for ROS via the DCF-DA assay. (B) Astrocytes were exposed to Mn (250 μM) for 3h and cell lysates were analyzed for phosphorylated IKK-β protein expression by western blotting. (**p<0.01, compared to the control; Unpaired student’s t-test; N=3).
3.2. Mn induces NF-κB activation via phosphorylation of IKK-β.
Our previous results established that NF-κB is a critical mediator of Mn-induced YY1 upregulation by directly binding to the YY1 promoter [18, 37]. To further investigate the mechanism by which Mn activates the NF-κB pathway to upregulate YY1, resulting in impairment of EAAT2, we assessed the effects of IKK16, a selective inhibitor of IKK-β, on Mn-induced activation of NF-κB. As shown in Fig. 2A, treatment of astrocytes with IKK16 significantly attenuated Mn-induced increases in IKK-β phosphorylation, but did not completely inhibit Mn-induced increases in NF-κB reporter activity (Fig. 2B) or NF-κB p65 nuclear translocation (Fig. 2C). These results indicate that Mn-induced IKK-β phosphorylation occurs upstream of canonical NF-κB activation.
Fig. 2. Mn induces canonical NF-κB activation via phosphorylation of IKK-β.

(A) Human H4 astrocytes were pre-treated with the selective IKK-β inhibitor IKK16 (200 nM, 1h), followed by Mn exposure (250 μM, 3h), and cell lysates were analyzed for phosphorylated IKK-β protein expression by western blotting. (B) After overnight transfection with an NF-κB reporter vector, astrocytes were treated with IKK16 and Mn for the indicated time periods, followed by the luciferase assay. (C) Astrocytes were treated with IKK16 and Mn as previously described, then cell lysates were fractionated and analyzed for NF-κB p65 protein expression in the nuclear fraction by western blotting. Histone H4 was used as a loading control. (*p<0.05, **p<0.01, ***p<0.001, compared to the control; #significantly different # p<0.05, ## p<0.01, ### p<0.01; ANOVA followed by Tukey’s post hoc test; N=3).
3.3. IKK-β inhibition attenuates Mn-induced YY1 activation.
Studies have also demonstrated that Mn-induced activation of YY1 leads to repression of EAAT2, but the mechanisms have yet to be characterized [38]. Notably, the YY1 promoter contains an NF-κB binding site, and mutation of this site decreases YY1 promoter activity [22]. Based on these previous findings, we tested if Mn induces YY1 upregulation via phosphorylation of IKK-β. The results showed that inhibition of IKK-β with IKK16 partially, but significantly attenuated Mn-induced increases in YY1 promoter activity (Fig. 3A), mRNA (Fig. 3B) and protein (Fig. 3B) levels, indicating that IKK-β phosphorylation is critically involved in Mn-induced YY1 activation.
Fig. 3. IKK-β inhibition attenuates Mn-induced YY1 activation.
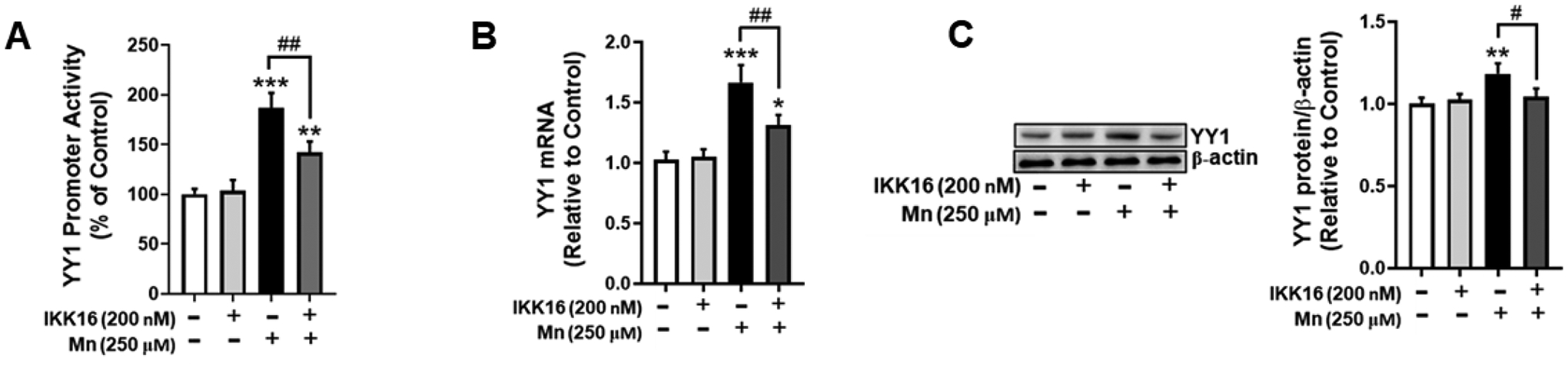
(A) Astrocytes were transfected overnight with a YY1 promoter vector, followed by pre-treatment with IKK16 (200 nM, 1h) and Mn exposure (250 μM, 3h). YY1 promoter activity was measured via the luciferase assay. Astrocytes were pre-treated with IKK16 (200 nM, 1h) and Mn (250 μM, 6h), and cell lysates were analyzed for YY1 (B) mRNA and (C) protein expression via qPCR and western blotting, respectively. (*p<0.05, **p<0.01, ***p<0.001, compared to the control; #significantly different # p<0.05, ## p<0.01; ANOVA followed by Tukey’s post hoc test; N=3).
3.4. Antioxidants attenuate Mn-induced YY1 activation via NF-κB
Mn induces ROS production, leading to oxidative stress and proinflammatory signaling [39]. Studies have demonstrated that oxidative stress can also induce phosphorylation of IKK-β and consequent activation of the NF-κB pathway [40, 41]. Accordingly, we tested the efficacy of antioxidant NAC in attenuating Mn-induced IKK-β phosphorylation. As shown in Fig. 4, NAC completely inhibited Mn-induced IKK-β phosphorylation (Fig. 4A), and partially attenuated NF-κB reporter activity (Fig. 4B) and NF-κB p65 nuclear translocation (Fig. 4C).
Fig. 4. Treatment with N-acetylcysteine (NAC) attenuates Mn-induced YY1 activation via NF-κB.
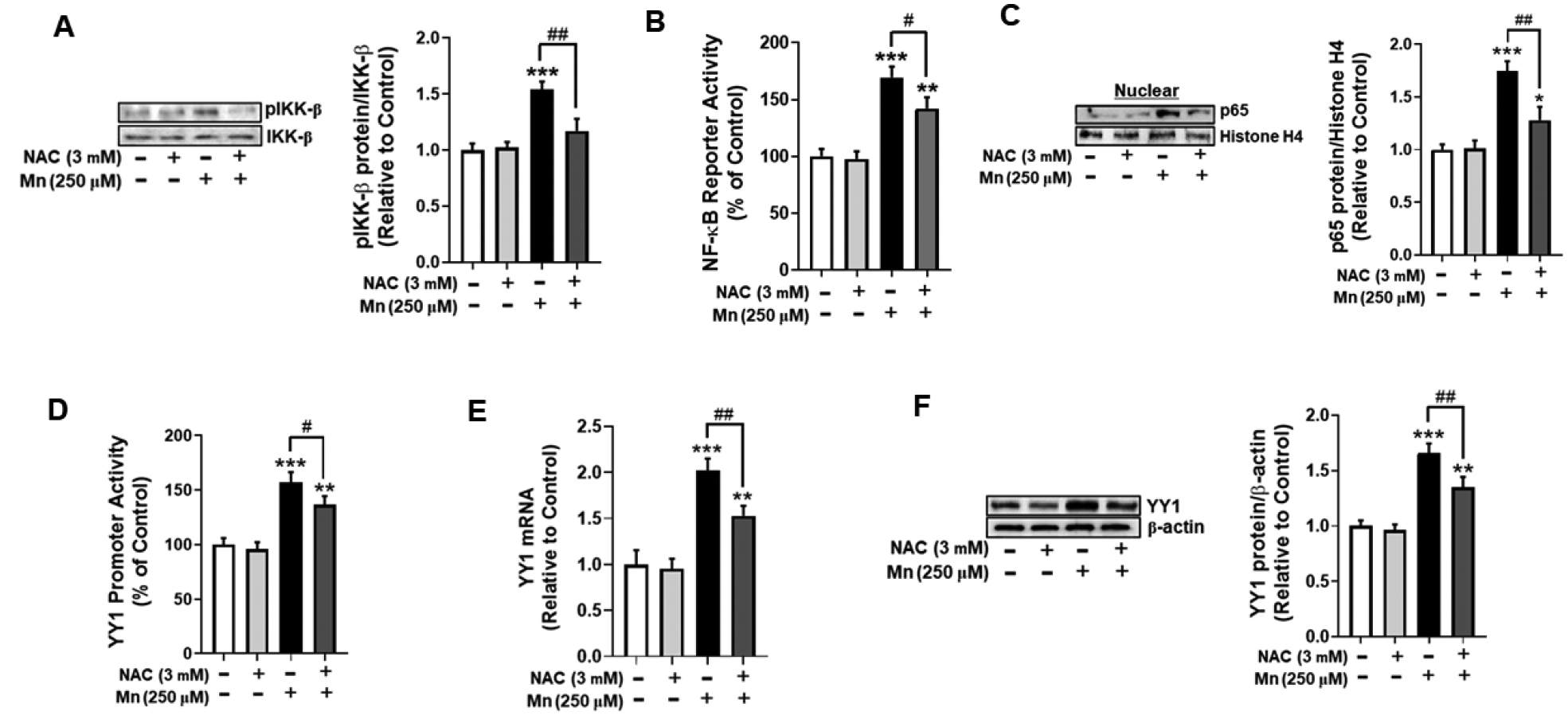
(A) Astrocytes were pre-treated with NAC (3 mM, 30 min) and Mn (250 μM, 3h), and cell lysates were analyzed for phosphorylated IKK-β protein expression by western blotting. (B) After overnight transfection with an NF-κB reporter vector, astrocytes were pre-treated with NAC (3 mM, 30 min) and Mn (250 μM, 3h), followed by the luciferase assay. (C) Astrocytes were treated with NAC and Mn as previously described, and cell lysates were fractionated and analyzed for NF-κB p65 protein expression in the nuclear fraction by western blotting. Histone H4 was used as a loading control. (D) After overnight transfection with a YY1 promoter vector, astrocytes were pre-treated with NAC (3 mM, 30 min) and Mn (250 μM, 3h) followed by the luciferase assay. (E) YY1 mRNA levels were measured following NAC (3 mM, 30 min) and Mn (250 μM, 6h) treatment by real-time qPCR (normalized to GAPDH). (F) Astrocytes were treated with NAC and Mn as previously indicated, and cell lysates were analyzed for YY1 protein expression by western blotting. (*p<0.05, **p<0.01, ***p<0.001, compared to the control; #significantly different # p<0.05, ## p<0.01; ANOVA followed by Tukey’s post hoc test; N=3).
Given that the YY1 promoter contains an NF-κB binding site which is critical for YY1 activation [22], we tested if NAC blocked Mn-induced YY1 activation. Treatment with NAC partially, but significantly attenuated Mn-induced YY1 promoter activity (Fig. 4D) and increases in YY1 mRNA (Fig. 4E) and protein levels (Fig. 4F). These findings indicate that Mn-induced oxidative stress, at least in part, mediates YY1 activation via the canonical NF-κB pathway.
We also tested if another antioxidant, AT, attenuated the Mn-induced IKK-β/NF-κB/YY1 cascade to further confirm the role of ROS in this pathway. Results showed that AT completely blocked Mn-induced IKK-β phosphorylation (Fig. 5A), and partially attenuated NF-κB reporter activity (Fig. 5B) and NF-κB p65 nuclear translocation (Fig. 5C). Additionally, treatment with AT also partially, but significantly attenuated Mn-induced increases in YY1 promoter activity (Fig. 5D), mRNA (Fig. 5E) and protein levels (Fig. 5F), indicating that Mn-induced oxidative stress critically mediates YY1 activation, at least in part by canonical NF-κB activation.
Fig. 5. Treatment with α-tocopherol (AT) attenuates Mn-induced YY1 activation via NF-κB.
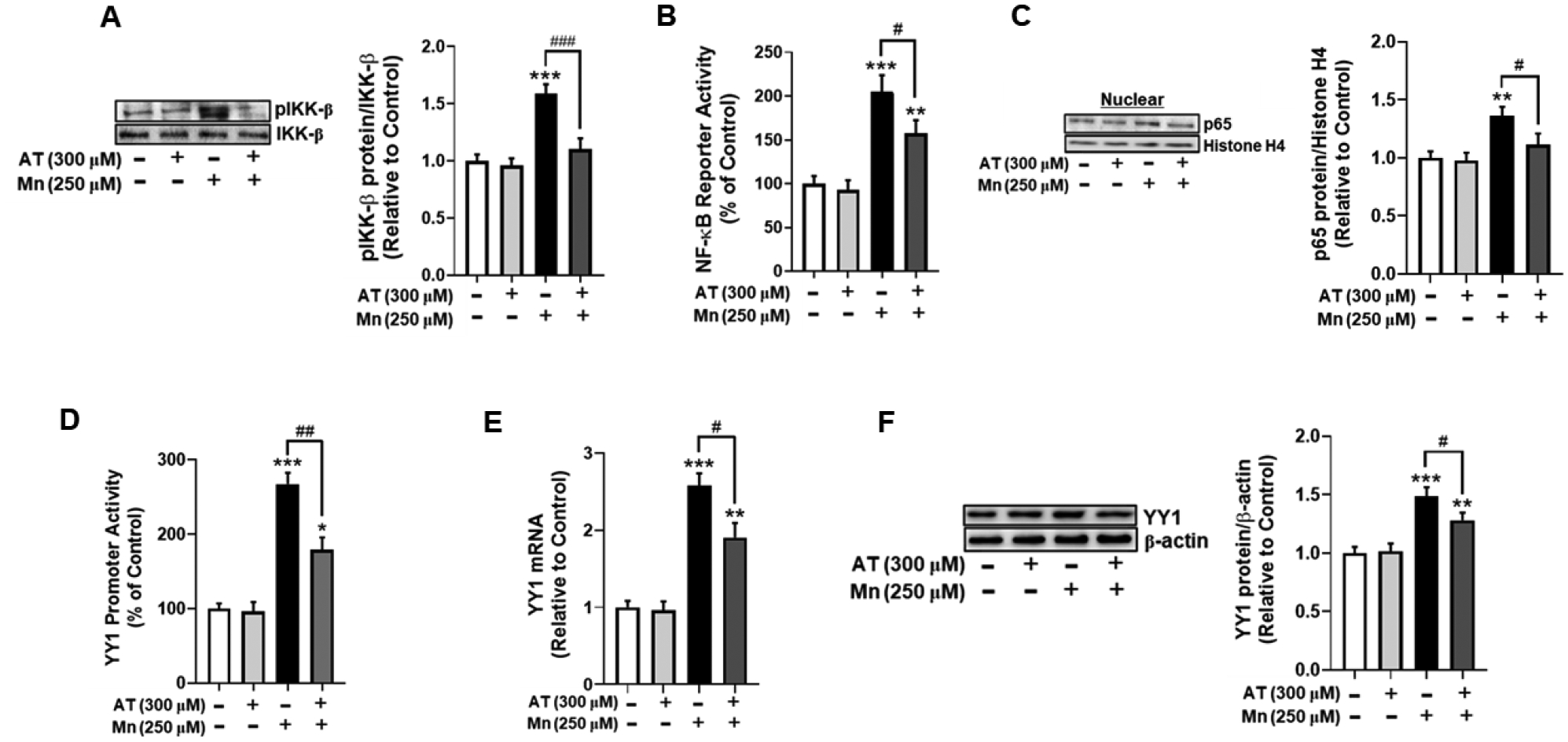
(A) Astrocytes were pre-treated with AT (300 μM, 30 min) and Mn (250 μM, 3h), and cell lysates were analyzed for phosphorylated IKK-β protein expression by western blotting. (B) After overnight transfection with an NF-κB reporter vector, astrocytes were pre-treated with AT (300 μM, 30 min) and exposed to Mn (250 μM, 3h), followed by the luciferase assay. (C) Astrocytes were treated with AT and Mn as previously described, and cell lysates were fractionated and analyzed for NF-κB p65 protein expression in the nuclear fraction by western blotting. Histone H4 was used as a loading control. (D) After overnight transfection with a YY1 promoter vector, astrocytes were pre-treated with AT (300 μM, 30 min) and Mn (250 μM, 3h), and a luciferase assay was performed. (E) After treatment with AT (300 μM, 30 min) and Mn (250 μM, 6h), YY1 mRNA level were measured by real-time qPCR (normalized to GAPDH). (F) Astrocytes were treated with AT and Mn as previously indicated, and cell lysates were analyzed for YY1 protein expression by western blotting. (*p<0.05, **p<0.01, ***p<0.001, compared to the control; #significantly different # p<0.05, ## p<0.01, ### p<0.001; ANOVA followed by Tukey’s post hoc test; N=3).
3.5. TNF-α mediates Mn-induced YY1 activation via canonical NF-κB signaling.
Previous results have shown that Mn induces the production of TNF-α and ROS, leading to a proinflammatory feedback loop of neuroinflammation and oxidative stress [39]. Thus, we tested if Mn-induced production of proinflammatory cytokine TNF-α initiates activation of the IKK-β-NF-κB pathway [39] using the TNF-α inhibitor C-87. The results showed that pre-treatment of astrocytes with C-87 partially, but significantly attenuated Mn-induced IKK-β phosphorylation (Fig. 6A), NF-κB reporter activity (Fig. 6B), and NF-κB p65 nuclear translocation (Fig. 6C), indicating that Mn activates the IKK-β-NF-κB pathway at least in part via TNF-α.
Fig. 6. TNF-α inhibition attenuates Mn-induced YY1 activation via canonical NF-κB signaling.
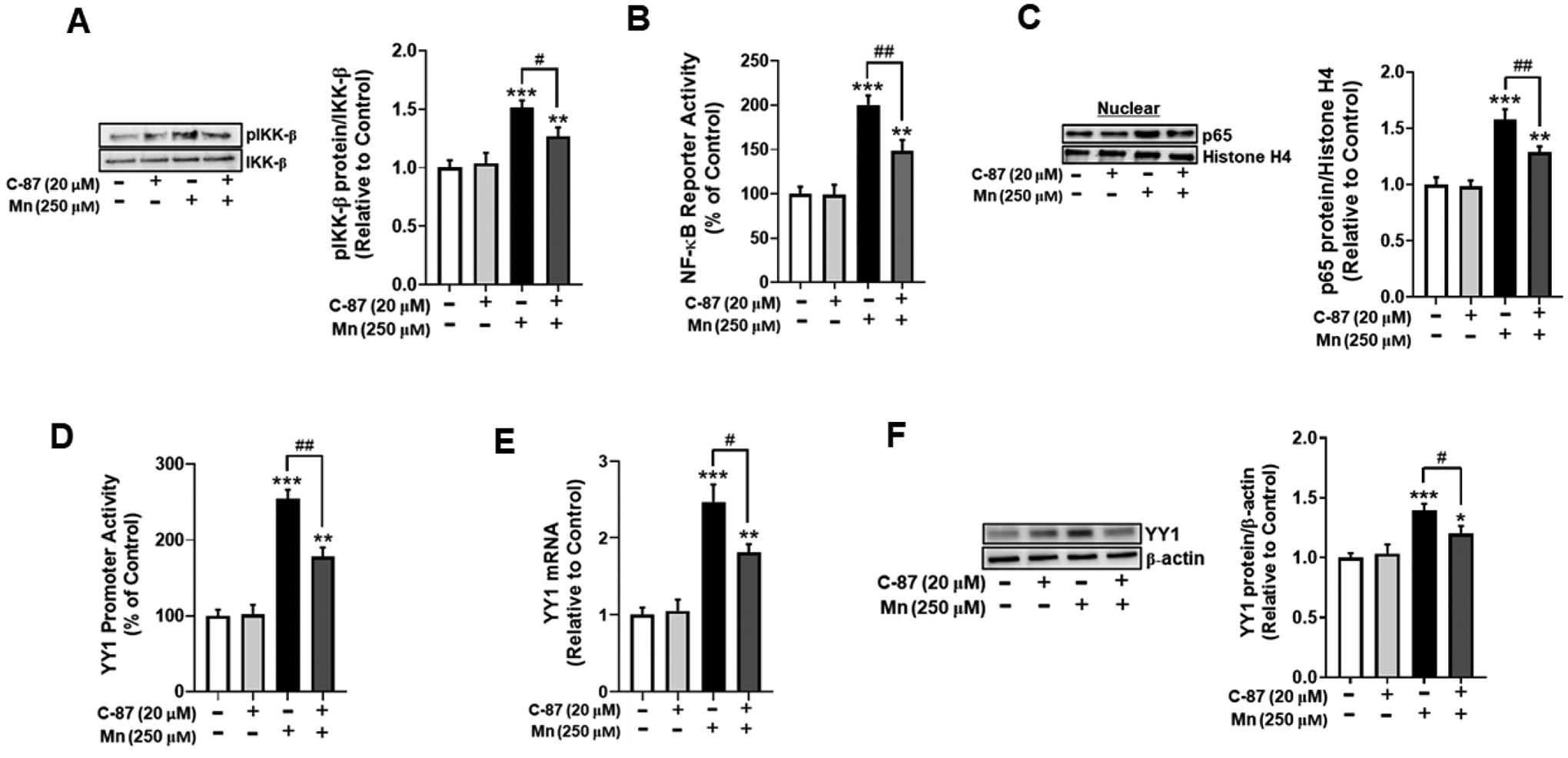
(A) Human H4 astrocytes were pre-treated with the TNF-α inhibitor C-87 (20 μM) for 30 min, followed by Mn exposure (250 μM) for 3 hours, and cell lysates were analyzed for phosphorylated IKK-β protein expression by western blotting. (B) After overnight transfection with an NF-κB reporter vector, astrocytes were pre-treated with C-87 (20 μM, 30 min) and Mn (250 μM, 3h), followed by the luciferase assay. (C) Astrocytes were treated with C-87 and Mn as previously described, and cell lysates were fractionated and analyzed for NF-κB p65 protein expression in the nuclear fraction by western blotting. Histone H4 was used as a loading control. (D) After overnight transfection with a YY1 promoter vector, astrocytes were treated with C-87 (20 μM, 30 min) and Mn (250 μM, 3h), and a luciferase assay was performed. (E) After treatment with C-87(20 μM, 30 min) and Mn (250 μM, 6h), YY1 mRNA levels were measured by real-time qPCR (normalized to GAPDH). (F) Astrocytes were treated with C-87 and Mn as previously indicated, and cell lysates were analyzed for YY1 protein expression by western blotting. (*p<0.05, **p<0.01, ***p<0.001, compared to the control; #significantly different # p<0.05, ## p<0.01; ANOVA followed by Tukey’s post hoc test; N=3).
We also tested if C-87 inhibited Mn-induced YY1 activation. The results showed that C-87 partially, but significantly attenuated increases in Mn-induced YY1 promoter activity (Fig. 6D), mRNA (Fig. 6E) and protein levels (Fig. 6F), indicating that Mn-induced TNF-α is involved in YY1 activation via NF-κB.
3.6. ROS is upstream of Mn-induced TNF-α production.
To further delineate the sequential events of Mn-induced ROS and TNF-α in IKK-β phosphorylation, we determined whether the antioxidants NAC and AT attenuated Mn-induced TNF-α production. As shown in Fig. 7, both NAC and AT attenuated Mn-induced TNF-α production, indicating that oxidative stress is upstream of TNF- α release in Mn neurotoxicity.
Fig. 7. ROS is upstream of Mn-induced TNF-α production.
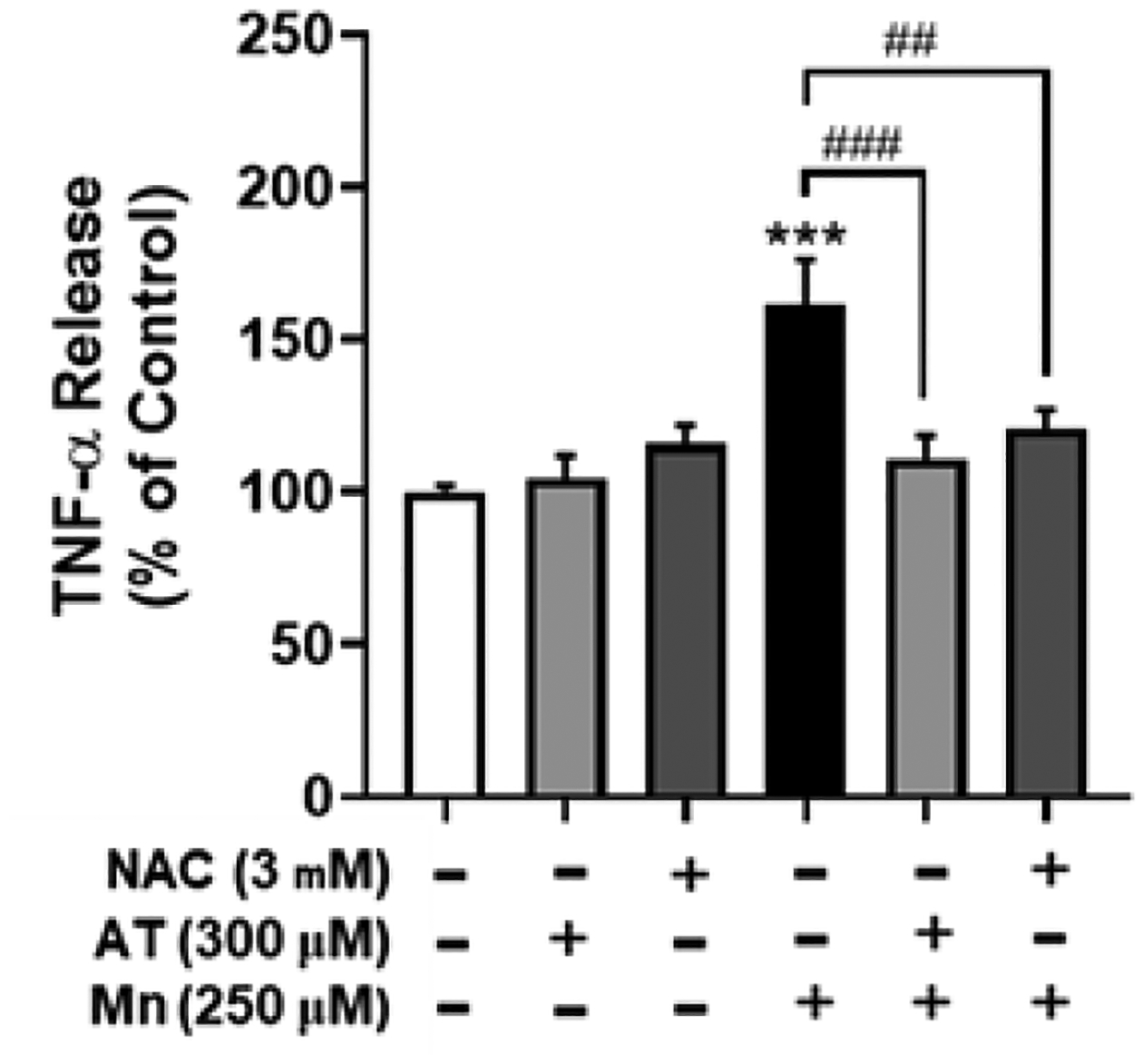
H4 astrocytes were pre-treated with NAC (3 mM, 30 min) or AT (300 μM, 30 min) prior to Mn exposure (250 μM, 3h), followed by assessment of TNF-α levels by the ELISA assay. (***p<0.001, compared to the control; #significantly different ## p<0.01, ### p<0.001; ANOVA followed by Tukey’s post hoc test; N=3).
3.7. IKK-β phosphorylation mediates Mn-induced repression of EAAT2.
Although NF-κB is a positive regulator of EAAT2 expression in astrocytes by pharmacological agents such as ceftriaxone with extended period of treatment [37], NF-κB is also involved in Mn-induced rapid activation of YY1, leading to decreases in EAAT2 [30]. Thus, we tested if Mn-induced IKK-β phosphorylation mediated the Mn-induced decrease in EAAT2 expression. The results showed that inhibition of IKK-β partially, but significantly attenuated Mn-induced decreases in EAAT2 promoter activity (Fig. 8A), mRNA (Fig. 8B) and protein (Fig. 8C) levels, indicating that the IKK-β-NF-κB pathway is, at least in part, involved in Mn-induced EAAT2 repression via YY1.
Fig. 8. IKK-β phosphorylation mediates Mn-induced repression of EAAT2.

(A) Astrocytes were transfected overnight with an EAAT2 promoter vector, then incubated with IKK16 (200 nM, 1h) and Mn (250 μM, 6h), followed by the luciferase assay. Astrocytes were treated with IKK16 and Mn for the indicated time periods, and EAAT2 mRNA and protein levels were measured via real-time (B) qPCR and (C) western blotting, respectively. (# p<0.05, ## p<0.01, ### p<0.001, compared to the control; @significantly different @ p<0.05; ANOVA followed by Tukey’s post hoc test; N=3).
3.8. Oxidative stress mediates Mn-induced repression of EAAT2.
We also tested if the antioxidants NAC and AT attenuate Mn-induced decreases in EAAT2. Treatment of astrocytes with NAC partially, but significantly attenuated Mn-induced decreases in EAAT2 promoter activity (Fig. 9A), mRNA and protein levels (Figs. 9B and C). Likewise, treatment with AT also partially attenuated Mn-induced decreases in EAAT2 promoter activity (Fig. 9D), mRNA and protein levels (Figs. 9E and F). These findings indicate that Mn-induced oxidative stress promotes EAAT2 repression, at least in part by activation of NF-κB signaling and consequent YY1 upregulation.
Fig. 9. Oxidative stress mediates Mn-induced repression of EAAT2.

(A) After overnight transfection with a EAAT2 promoter vector, astrocytes were treated with NAC (3 mM, 30 min) and Mn (250 μM, 6h) followed by the luciferase assay. (B) EAAT2 mRNA levels were measured following NAC (3 mM, 30 min) and Mn (250 μM, 12h) treatment by real-time qPCR (normalized to GAPDH). (C) Astrocytes were treated with NAC and Mn as previously indicated, and cell lysates were analyzed for EAAT2 protein expression by western blotting. (D) After overnight transfection with an EAAT2 promoter vector, astrocytes were treated with AT (300 μM, 30 min) and Mn (250 μM, 6h), and a luciferase assay was performed. (E) After treatment with AT (300 μM, 30 min) and Mn (250 μM, 12h), EAAT2 mRNA levels were measured by real-time qPCR (normalized to GAPDH). (F) Astrocytes were treated with AT and Mn as previously indicated, and cell lysates were analyzed for EAAT2 protein expression by western blotting. (#p <0.05, ## p<0.01, ### p<0.001, compared to the control; @significantly different @ p<0.05, @@ p<0.01, @@@ p<0.001; ANOVA followed by Tukey’s post hoc test; N=3).
3.9. TNF-α mediates Mn-induced repression of EAAT2.
We tested if TNF-α mediates Mn-induced decreases in EAAT2 expression. The results showed that inhibition of TNF-α with C-87 partially, but significantly attenuated Mn-induced decreases in EAAT2 promoter activity (Fig. 10A), mRNA (Fig. 10B), and protein (Fig. 10C) levels, indicating that TNF-α mediates Mn-induced EAAT2 repression, at least in part through NF-κB mediated YY1 activation.
Fig. 10. TNF-α mediates Mn-induced repression of EAAT2.
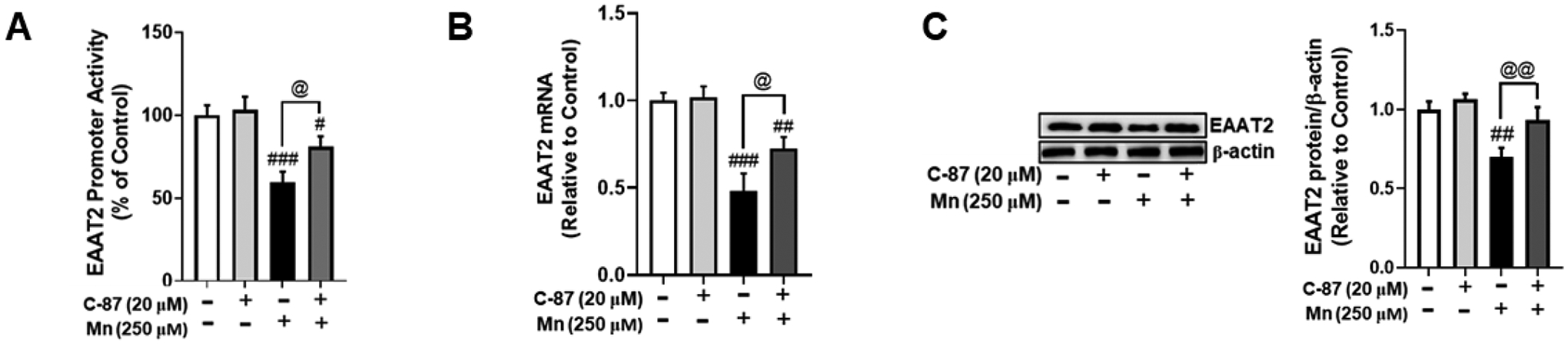
(A) After overnight transfection with an EAAT2 promoter vector, astrocytes were treated with the TNF-α inhibitor C-87 (20 μM, 30 min) and Mn (250 μM, 6h) followed by the luciferase assay. (B) EAAT2 mRNA levels were measured following C-87 (20 μM, 30 min) and Mn (250 μM, 12h) treatment by real-time qPCR (normalized to GAPDH). (C) Astrocytes were treated with C-87 and Mn as previously indicated, and cell lysates were analyzed for EAAT2 protein expression by western blotting. (# p<0.05, ## p<0.01, ### p<0.001, compared to the control; @significantly different @ p<0.05, @@ p<0.01; ANOVA followed by Tukey’s post hoc test; N=3).
4. Discussion
Our novel findings establish for the first time that Mn-induced YY1 activation is mediated by canonical NF-κB signaling in human astrocytes. Further, Mn-induced ROS was an initiating cellular event which increased TNF-α production, leading to canonical NF-κB pathway activation and consequent YY1 upregulation. Accordingly, inhibition of Mn-induced oxidative stress, TNF-α production and IKK-β activation attenuated Mn-induced YY1 activation, potentially leading to repression of EAAT2.
Aberrant astrocytic glutamate transporter EAAT2 and consequent neuronal excitotoxicity have been implicated in several neurological disorders, including PD, AD, and manganism [42]. As EAAT2 reuptakes nearly 90% of synaptic glutamate, it is crucial to delineate the mechanisms which impair EAAT2 expression and function [43]. While several studies have reported Mn-induced EAAT2 dysregulation, the molecular and cellular mechanisms of Mn-induced EAAT2 repression have yet to be completely understood [21, 42, 44]. Our findings reveal that Mn activates YY1 via the canonical NF-κB pathway to potentially repress EAAT2 provide novel information regarding the mechanisms of Mn neurotoxicity and identify new therapeutic targets in the treatment of neurological disorders associated with glutamate transporter impairment.
Our previous studies showed that Mn represses EAAT2 via activation of YY1 in murine primary astrocytes, and NF-κB plays a critical role in this activation by binding to the YY1 promoter via TNF-α [17]. Our current study demonstrates that Mn induces the early activation of canonical NF-κB signaling via phosphorylation of the upstream kinase IKK-β, leading to YY1 upregulation in astrocytes. Further, Mn-induced ROS production and consequent TNF-α release is the initiating cellular event leading to activation of NF-κB signaling.
Studies have demonstrated that canonical NF-κB signaling is rapid and transient, requiring the phosphorylation of IKK-β [45]. Our results indicate that Mn exposure induces IKK-β phosphorylation within 3 hours, leading to upregulation of NF-κB and activation of YY1. The selective IKK-β inhibitor IKK16 partially, but significantly attenuated these effects, further supporting that Mn induces initiation of the canonical NF-κB pathway. Moreover, since canonical NF-κB signaling is initiated by ROS and TNF-α [46, 47], our results that treatment with antioxidants blocked IKK-β phosphorylation and partially attenuated increases in NF-κB signaling provide further evidence that canonical NF-κB signaling is involved in Mn-induced NF-κB activation. Furthermore, our findings that TNF-α inhibition only partially attenuated IKK-β phosphorylation and consequent NF-κB/YY1 activation suggest that Mn-induced TNF-α is important, but not the sole mediator of Mn-induced canonical NF-κB signaling and YY1 upregulation.
Oxidative stress and inflammation are associated with neurodegeneration in postmortem studies and animal models, and previous research has established both oxidative stress and inflammation as mechanisms of Mn-induced neurotoxicity [15, 48–50]. Studies have shown that antioxidants such as NAC and AT protect against oxidative stress and inflammation in vivo, and attenuate oxidative damage and neurodegeneration in aging rat brains and in clinical studies [51–54]. Accordingly, our results that the antioxidants NAC and AT inhibited Mn-induced phosphorylation of IKK-β and partially attenuated Mn-induced NF-κB signaling and YY1 upregulation suggest that Mn-induced ROS mediates YY1 activation, at least in part via the canonical NF-κB pathway. Moreover, these antioxidants partially, but significantly attenuated Mn-induced downregulation of EAAT2 expression, providing further evidence of the role of oxidative stress in Mn-induced activation of the canonical NF-κB/YY1 cascade and subsequent EAAT2 repression.
The results also demonstrate that Mn-induced ROS production is upstream of TNF-α since antioxidants attenuated Mn-induced TNF-α production, suggesting that Mn-induced oxidative stress may be the initial event leading to YY1 upregulation and EAAT2 repression. Given that NF-κB can directly repress EAAT2 via TNF-α, it is possible that the Mn-induced repression of EAAT2 occurs independently of YY1 [29, 30]. Accordingly, the present findings do not provide direct evidence for the role of YY1 in Mn-induced decreases in EAAT2 expression via oxidative stress or TNF-α, but the current results support that activation of the canonical NF-κB pathway via ROS/TNF-α is critical to Mn-induced YY1 upregulation and consequent impairment of EAAT2.
Notably, our findings indicate that the inflammatory IKK-β/NF-κB pathway may not be the only means by which Mn induces YY1 activation and subsequent EAAT2 repression. As the selective inhibition of IKK-β via IKK16 only partially blocked increases in NF-κB promoter activity and nuclear translocation, it is possible that Mn may also activate NF-κB and YY1 through non-canonical signaling, a pathway mediated by the kinase IKK-α, or as of yet other unknown pathways. Moreover, the results that antioxidants and the TNF-α inhibitor C-87 only partially attenuated Mn-induced NF-κB/YY1 activation and decreases in EAAT2 indicate that there are other mechanisms, in addition to oxidative stress and proinflammatory cytokines, which mediate this effect. Further investigation is warranted to understand if the mechanisms by which Mn-induced YY1 upregulation and subsequent EAAT2 downregulation are independent of canonical NF-κB signaling, or if other factors work in tandem with NF-κB.
Taken together, our novel findings demonstrate that Mn activates YY1 at least in part via canonical NF-κB signaling, leading to decreases in EAAT2 mRNA and protein levels as shown in the mechanistic model for Mn-induced YY1 activation (Fig. 11). The results from the present study provide novel insight into the mechanisms of Mn-induced YY1 activation and provide new molecular targets in the development of therapies to treat neurological disorders associated with EAAT2 dysregulation.
Fig. 11. Proposed mechanism for Mn-induced YY1 activation via NF-κB.

Mn induces the release of ROS and TNF-α, leading to activation of the NF-κB pathway after phosphorylation of the upstream kinase IKK-β. After IKK-β phosphorylation, NF-κB p65 translocates to the nucleus, where it interacts with YY1 and enhances its promoter activity and mRNA/protein levels, leading to downstream repression of EAAT2 expression and function. ROS, reactive oxygen species; IKK-β: IκB kinase-β; NAC, N-acetyl cysteine; AT, α-tocopherol.
Highlights.
Mn activates transcription factor YY1 via the canonical NF-κB pathway
Mn phosphorylates IKK-β via ROS and TNF-α production to activate NF-κB
Mn-induced oxidative stress is an initiating event which increases TNF-α levels
Inhibition of NF-κB signaling attenuates Mn’s YY1 activation and EAAT2 impairment
Acknowledgements
The study was supported in part by National Institutes of Health Research Grants, NIEHS R01 ES024756, R01 ES031282, R01 ES010563, R01 ES020852, NIMHD U54 MD007582, and NCI SC1 CA200519.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- 1.Buchanan RJ, et al. , Glutamate and GABA concentration changes in the globus pallidus internus of Parkinson’s patients during performance of implicit and declarative memory tasks: a report of two subjects. Neurosci Lett, 2015. 589: p. 73–8. [DOI] [PubMed] [Google Scholar]
- 2.Dedeurwaerdere S, et al. , In the grey zone between epilepsy and schizophrenia: alterations in group II metabotropic glutamate receptors. Acta Neurol Belg, 2015. 115(3): p. 221–32. [DOI] [PubMed] [Google Scholar]
- 3.Jahng GH, et al. , Glutamine and Glutamate Complex, as Measured by Functional Magnetic Resonance Spectroscopy, Alters During Face-Name Association Task in Patients with Mild Cognitive Impairment and Alzheimer’s Disease. J Alzheimers Dis, 2016. 53(2): p. 745. [DOI] [PubMed] [Google Scholar]
- 4.Walls AB, et al. , The glutamine-glutamate/GABA cycle: function, regional differences in glutamate and GABA production and effects of interference with GABA metabolism. Neurochem Res, 2015. 40(2): p. 402–9. [DOI] [PubMed] [Google Scholar]
- 5.Todd AC and Hardingham GE, The Regulation of Astrocytic Glutamate Transporters in Health and Neurodegenerative Diseases. Int J Mol Sci, 2020. 21(24). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shigeri Y, Seal RP, and Shimamoto K, Molecular pharmacology of glutamate transporters, EAATs and VGLUTs. Brain Res Brain Res Rev, 2004. 45(3): p. 250–65. [DOI] [PubMed] [Google Scholar]
- 7.Magi S, et al. , Excitatory Amino Acid Transporters (EAATs): Glutamate Transport and Beyond. Int J Mol Sci, 2019. 20(22). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rao P, et al. , Designing Novel Nanoformulations Targeting Glutamate Transporter Excitatory Amino Acid Transporter 2: Implications in Treating Drug Addiction. J Pers Nanomed, 2015. 1(1): p. 3–9. [PMC free article] [PubMed] [Google Scholar]
- 9.Pajarillo E, et al. , The role of astrocytic glutamate transporters GLT-1 and GLAST in neurological disorders: Potential targets for neurotherapeutics. Neuropharmacology, 2019. 161: p. 107559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lauriat TL and McInnes LA, EAAT2 regulation and splicing: relevance to psychiatric and neurological disorders. Mol Psychiatry, 2007. 12(12): p. 1065–78. [DOI] [PubMed] [Google Scholar]
- 11.Takahashi K, Foster JB, and Lin CL, Glutamate transporter EAAT2: regulation, function, and potential as a therapeutic target for neurological and psychiatric disease. Cell Mol Life Sci, 2015. 72(18): p. 3489–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bowman AB, et al. , Role of manganese in neurodegenerative diseases. J Trace Elem Med Biol, 2011. 25(4): p. 191–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martins AC Jr., et al. , Manganese-induced neurodegenerative diseases and possible therapeutic approaches. Expert Rev Neurother, 2020. 20(11): p. 1109–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sidoryk-Wegrzynowicz M, et al. , Manganese disrupts astrocyte glutamine transporter expression and function. J Neurochem, 2009. 110(3): p. 822–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Milatovic D, et al. , Oxidative damage and neurodegeneration in manganese-induced neurotoxicity. Toxicol Appl Pharmacol, 2009. 240(2): p. 219–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pajarillo E, et al. , 17beta-estradiol and tamoxifen protect mice from manganese-induced dopaminergic neurotoxicity. Neurotoxicology, 2018. 65: p. 280–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Karki P, et al. , Yin Yang 1 is a repressor of glutamate transporter EAAT2, and it mediates manganese-induced decrease of EAAT2 expression in astrocytes. Mol Cell Biol, 2014. 34(7): p. 1280–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Karki P, et al. , Transcriptional Regulation of the Astrocytic Excitatory Amino Acid Transporter 1 (EAAT1) via NF-kappaB and Yin Yang 1 (YY1). J Biol Chem, 2015. 290(39): p. 23725–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ramesh GT, Ghosh D, and Gunasekar PG, Activation of early signaling transcription factor, NF-kappaB following low-level manganese exposure. Toxicol Lett, 2002. 136(2): p. 151–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shi J, et al. , The role of YY1 in oncogenesis and its potential as a drug target in cancer therapies. Curr Cancer Drug Targets, 2015. 15(2): p. 145–57. [DOI] [PubMed] [Google Scholar]
- 21.Karki P, et al. , Role of transcription factor yin yang 1 in manganese-induced reduction of astrocytic glutamate transporters: Putative mechanism for manganese-induced neurotoxicity. Neurochem Int, 2015. 88: p. 53–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang H, et al. , NF-kappaB regulation of YY1 inhibits skeletal myogenesis through transcriptional silencing of myofibrillar genes. Mol Cell Biol, 2007. 27(12): p. 4374–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dewert N, et al. , The IKKalpha-dependent non-canonical pathway of NF-kappaB activation is constitutively active and modulates progression-related functions in a subset of human melanomas. Arch Dermatol Res, 2016. 308(10): p. 733–742. [DOI] [PubMed] [Google Scholar]
- 24.Kendellen MF, et al. , Canonical and non-canonical NF-kappaB signaling promotes breast cancer tumor-initiating cells. Oncogene, 2014. 33(10): p. 1297–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ziegler LS, et al. , Attenuation of canonical NF-kappaB signaling maintains function and stability of human Treg. FEBS J, 2021. 288(2): p. 640–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li ZQ, et al. , Activation of the canonical nuclear factor-kappaB pathway is involved in isoflurane-induced hippocampal interleukin-1beta elevation and the resultant cognitive deficits in aged rats. Biochem Biophys Res Commun, 2013. 438(4): p. 628–34. [DOI] [PubMed] [Google Scholar]
- 27.Bloom MJ, et al. , The effects of IKK-beta inhibition on early NF-kappa-B activation and transcription of downstream genes. Cell Signal, 2019. 55: p. 17–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Israel A, The IKK complex, a central regulator of NF-kappaB activation. Cold Spring Harb Perspect Biol, 2010. 2(3): p. a000158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gupta RK and Prasad S, Differential regulation of GLT-1/EAAT2 gene expression by NF-kappaB and N-myc in male mouse brain during postnatal development. Neurochem Res, 2014. 39(1): p. 150–60. [DOI] [PubMed] [Google Scholar]
- 30.Sitcheran R, et al. , Positive and negative regulation of EAAT2 by NF-kappaB: a role for N-myc in TNFalpha-controlled repression. EMBO J, 2005. 24(3): p. 510–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Crittenden PL and Filipov NM, Manganese-induced potentiation of in vitro proinflammatory cytokine production by activated microglial cells is associated with persistent activation of p38 MAPK. Toxicol In Vitro, 2008. 22(1): p. 18–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moyano P, et al. , Manganese induced ROS and AChE variants alteration leads to SN56 basal forebrain cholinergic neuronal loss after acute and long-term treatment. Food Chem Toxicol, 2019. 125: p. 583–594. [DOI] [PubMed] [Google Scholar]
- 33.Vicente-Gutierrez C, et al. , Abrogating mitochondrial ROS in neurons or astrocytes reveals cell-specific impact on mouse behaviour. Redox Biol, 2021. 41: p. 101917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kumar KK, et al. , Cellular manganese content is developmentally regulated in human dopaminergic neurons. Sci Rep, 2014. 4: p. 6801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Song D, et al. , FOXO3 promoted mitophagy via nuclear retention induced by manganese chloride in SH-SY5Y cells. Metallomics, 2017. 9(9): p. 1251–1259. [DOI] [PubMed] [Google Scholar]
- 36.Yin Z, et al. , Ferroportin is a manganese-responsive protein that decreases manganese cytotoxicity and accumulation. J Neurochem, 2010. 112(5): p. 1190–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Karki P, et al. , cAMP response element-binding protein (CREB) and nuclear factor kappaB mediate the tamoxifen-induced up-regulation of glutamate transporter 1 (GLT-1) in rat astrocytes. J Biol Chem, 2013. 288(40): p. 28975–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hou X, et al. , Adenosine Receptor A1-A2a Heteromers Regulate EAAT2 Expression and Glutamate Uptake via YY1-Induced Repression of PPARgamma Transcription. PPAR Res, 2020. 2020: p. 2410264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu B, et al. , IKKbeta regulates the expression of coagulation and fibrinolysis factors through the NF-kappaB canonical pathway in LPS-stimulated alveolar epithelial cells type II. Exp Ther Med, 2019. 18(4): p. 2859–2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang KS, et al. , Imperatorin efficiently blocks TNF-alpha-mediated activation of ROS/PI3K/Akt/NF-kappaB pathway. Oncol Rep, 2017. 37(6): p. 3397–3404. [DOI] [PubMed] [Google Scholar]
- 41.Yang J, et al. , Feed-forward signaling of TNF-alpha and NF-kappaB via IKK-beta pathway contributes to insulin resistance and coronary arteriolar dysfunction in type 2 diabetic mice. Am J Physiol Heart Circ Physiol, 2009. 296(6): p. H1850–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lin CL, et al. , Glutamate transporter EAAT2: a new target for the treatment of neurodegenerative diseases. Future Med Chem, 2012. 4(13): p. 1689–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim K, et al. , Role of excitatory amino acid transporter-2 (EAAT2) and glutamate in neurodegeneration: opportunities for developing novel therapeutics. J Cell Physiol, 2011. 226(10): p. 2484–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tinkov AA, et al. , Molecular Targets of Manganese-Induced Neurotoxicity: A Five-Year Update. Int J Mol Sci, 2021. 22(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zarnegar BJ, et al. , Noncanonical NF-kappaB activation requires coordinated assembly of a regulatory complex of the adaptors cIAP1, cIAP2, TRAF2 and TRAF3 and the kinase NIK. Nat Immunol, 2008. 9(12): p. 1371–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fischer R and Maier O, Interrelation of oxidative stress and inflammation in neurodegenerative disease: role of TNF. Oxid Med Cell Longev, 2015. 2015: p. 610813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Morgan MJ and Liu ZG, Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res, 2011. 21(1): p. 103–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bosco DA, et al. , Elevated levels of oxidized cholesterol metabolites in Lewy body disease brains accelerate alpha-synuclein fibrilization. Nat Chem Biol, 2006. 2(5): p. 249–53. [DOI] [PubMed] [Google Scholar]
- 49.Keeney PM, et al. , Parkinson’s disease brain mitochondrial complex I has oxidatively damaged subunits and is functionally impaired and misassembled. J Neurosci, 2006. 26(19): p. 5256–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schwalm MT, et al. , Acute brain inflammation and oxidative damage are related to long-term cognitive deficits and markers of neurodegeneration in sepsis-survivor rats. Mol Neurobiol, 2014. 49(1): p. 380–5. [DOI] [PubMed] [Google Scholar]
- 51.Abdel-Wahab WM and Moussa FI, Neuroprotective effect of N-acetylcysteine against cisplatin-induced toxicity in rat brain by modulation of oxidative stress and inflammation. Drug Des Devel Ther, 2019. 13: p. 1155–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Garg G, et al. , N-acetyl-l-cysteine attenuates oxidative damage and neurodegeneration in rat brain during aging. Can J Physiol Pharmacol, 2018. 96(12): p. 1189–1196. [DOI] [PubMed] [Google Scholar]
- 53.Monti DA, et al. , N-Acetyl Cysteine Is Associated With Dopaminergic Improvement in Parkinson’s Disease. Clin Pharmacol Ther, 2019. 106(4): p. 884–890. [DOI] [PubMed] [Google Scholar]
- 54.Ulatowski LM and Manor D, Vitamin E and neurodegeneration. Neurobiol Dis, 2015. 84: p. 78–83. [DOI] [PubMed] [Google Scholar]