

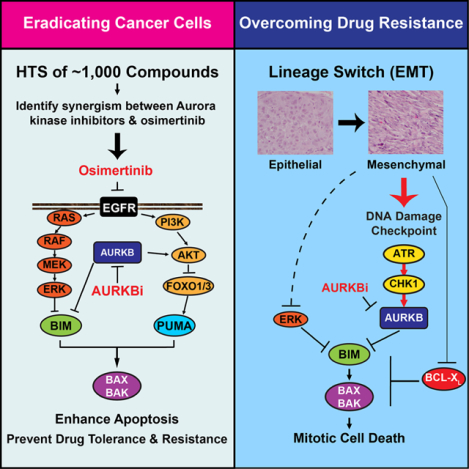
SUMMARY
The clinical success of EGFR inhibitors in EGFR-mutant lung cancer is limited by the eventual development of acquired resistance. We hypothesize that enhancing apoptosis through combination therapies can eradicate cancer cells and reduce the emergence of drug tolerant persisters. Through high-throughput screening of a custom library of ~1,000 compounds, we discover Aurora B kinase inhibitors as potent enhancers of osimertinib-induced apoptosis. Mechanistically, Aurora B inhibition stabilizes BIM through reduced Ser87 phosphorylation, and transactivates PUMA through FOXO1/3. Importantly, osimertinib-resistance caused by epithelial–mesenchymal transition (EMT) activates the ATR-CHK1-Aurora B signaling cascade and thereby engenders hypersensitivity to respective kinase inhibitors by activating BIM-mediated mitotic catastrophe. Combined inhibition of EGFR and Aurora B not only efficiently eliminates cancer cells but also overcomes resistance beyond EMT.
Graphical Abstract
Blurb:
Tanaka et al. identify Aurora kinase inhibitors as potent enhancers of osimertinib-induced apoptosis by HTS. Concurrent inhibition of EGFR and AURKB maximizes BIM- and PUMA-mediated apoptosis to eradicate cancer cells. Osimertinib-resistance caused by EMT activates ATR-CHK1-AURKB and engenders hypersensitivity to these kinase inhibitors by activating BIM-mediated mitotic catastrophe.
INTRODUCTION
The discovery of targetable molecular alterations in genes, such as epidermal growth factor receptor (EGFR), has driven the evolution of targeted therapies for non-small cell lung cancer (NSCLC) (Lynch et al., 2004; Paez et al., 2004). However, the clinical success of EGFR tyrosine kinase inhibitors (TKI) in EGFR-mutant NSCLC is limited by the eventual development of acquired resistance (Camidge et al., 2014; Garraway and Jänne, 2012; Rotow and Bivona, 2017; Yu et al., 2013). Among the resistance mechanisms, a “second-site mutation” in EGFR, T790M, accounts for ~50–60% of acquired resistance after the first- and second-generation EGFR TKI treatment. To overcome this major resistance mechanism, the third-generation EGFR inhibitors, including rociletinib (Walter et al., 2013) and osimertinib (Osi) (Cross et al., 2014), have been developed. Osi is not only effective in T790M-acquired resistance but also superior to earlier generation EGFR TKIs as first-line therapy (Ramalingam et al., 2020; Soria et al., 2018). Unfortunately, acquired resistance to osi inevitably occurs and the resistance mechanisms are heterogeneous, among which osi resistance secondary mutations in EGFR (C797X, L718X, G724X, etc.) are identified only in a subset of patients (10–26%) (Leonetti et al., 2019; Thress et al., 2015; Tumbrink et al., 2021). Overall, it has become evident that targeting EGFR mutations alone is unlikely to cure EGFR-mutant NSCLC patients.
Induction of cancer cell apoptosis is integral to the success of targeted cancer therapy (Hata et al., 2015). The BCL-2 family proteins are central regulators of TKI-induced apoptosis (Czabotar et al., 2014; Hata et al., 2015; Jeng et al., 2018). BAX and BAK are the essential effectors of mitochondrial outer membrane permeabilization (MOMP) whereas BCL-2/BCL-XL/MCL-1 prevents MOMP. BH3s relay upstream apoptotic signals to initiate apoptosis by either activating BAX/BAK directly or inactivating BCL-2/BCL-XL/MCL-1 (Czabotar et al., 2014; Jeng et al., 2018). In response to apoptotic signals, the activator BH3s (BID, BIM, PUMA, and NOXA) directly activate BAX/BAK to induce BAX/BAK homo-oligomerization, leading to MOMP (Chen et al., 2015; Kim et al., 2006; Ren et al., 2010). We and others have identified BIM and PUMA as key apoptotic effectors for TKI-induced killing of EGFR-mutant NSCLC both in vitro and in vivo (Bean et al., 2013; Costa et al., 2007; Cragg et al., 2007; Gong et al., 2007). Specifically, inhibition of the MEK-ERK signaling cascade induces BIM whereas antagonizing the PI3K-AKT signaling axis triggers nuclear translocation of FOXO1/3 that transactivates PUMA (Bean et al., 2013).
Acquired resistance to EGFR TKIs occurs through the selection of pre-existing resistant clones as well as the evolution of drug-tolerant persisters (DTPs) that survive treatment through adaptive mechanisms (Hata et al., 2016; Sharma et al., 2010). Over time, DTPs can acquire resistance through mutational or non-mutational mechanisms (Hata et al., 2016; Rotow and Bivona, 2017). We hypothesize that enhancing apoptosis through the early administration of combination therapies could effectively eradicate cancer cells, and thereby prevent the ensuing emergence of drug-tolerant and resistant clones during treatment. In this study, an integrated high-throughput drug screening and mechanistic elucidation approach was employed to identify cell death mechanism-based combination therapies that enhance the proapoptotic effect of osi to eradicate cancer cells.
RESULTS
HTS identifies Aurora kinase inhibitors as potent enhancers of osimertinib-induced apoptosis
To identify combination strategies that enhance the proapoptotic effect of osi in EGFR-mutant NSCLC, we performed high-throughput screening (HTS) using a custom library of ~1,000 compounds encompassing inhibitors of 200 targets across more than 20 signaling pathways, small molecule modulators of epigenetics, FDA-approved drugs, and natural products (Inoue-Yamauchi et al., 2017). H1975 (EGFR L858R/T790M) cells were treated with each compound from this library ± osi, and the fold inhibition of growth by the combination therapy compared to each monotherapy was determined (Figure 1A). Among the top 25 hit compounds that enhanced osi-induced growth inhibition, four distinct inhibitor classes were identified, targeting Aurora kinases (AK), IGF1 receptor (IGF1R), SRC family kinases (SFK), or the PI3K/AKT/mTOR pathway (Figure 1B). We further assessed the efficacy of inhibitors targeting previously reported resistance mechanisms to EGFR TKIs, which revealed that inhibitors of AK, IGF1R, mTOR, PI3K, NFκB, and FGFR significantly enhanced osi-induced growth inhibition (Figure 1C). HTS was also performed in HCC827 that harbors EGFR exon 19 deletion. Eight overlapping compounds were identified among the top fifty hits in H1975 and HCC827, including three AK inhibitors (AKi), one IGF1R inhibitor, two PI3K inhibitors, and two mTOR inhibitors (Figure 1D). Because our HTS assessed growth inhibition rather than direct cell death-inducing activity, annexin-V staining was employed to quantify apoptosis (Figure 1E). Indeed, the combination of osi and AK or IGF1R inhibitors induced robust apoptosis in H1975 (Figure 1E). SFK inhibitors failed to enhance osi-induced apoptosis, indicating that this combination mainly inhibited cell proliferation. Inhibition of PI3K or AKT only slightly increased osi-induced apoptosis whereas mTOR inhibition had no effect. Furthermore, the MEK inhibitor trametinib had a minimal impact on osi-induced apoptosis (Figure S1). These findings were further assessed using one patient-derived EGFR-mutant NSCLC cell line, ECLC26 (L858R), and two EGFR exon 19 deletion lines, HCC827 and PC9 (Figure 1E). In summary, our initial HTS and subsequent validation studies identified AKi as potent enhancers of osi-induced apoptosis across different EGFR-mutant NSCLC cell lines.
Figure 1. HTS identifies Aurora kinase inhibitors as potent enhancers of osimertinib-induced apoptosis in EGFR-mutant lung cancer.
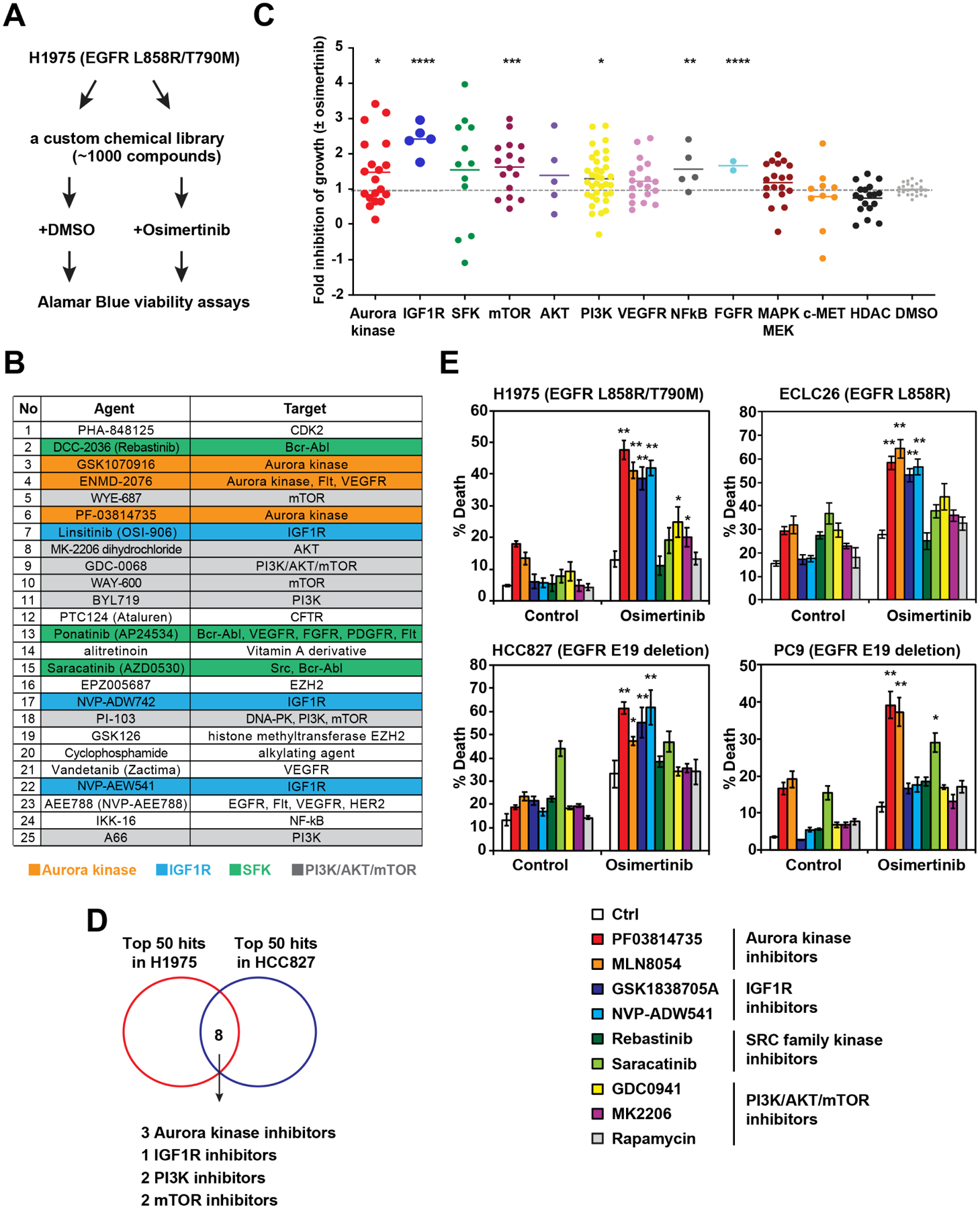
(A) A schematic of HTS to identify agents that enhance osimertinib (osi)-induced growth inhibition. H1975 was treated with each compound from the library (2 μM) ± osi (2 μM) in duplicate. Cell viability was assessed by alamarBlue assays at 72 h.
(B) Top 25 agents that enhance osi-induced growth inhibition of H1975. Green cluster, SRC family kinase inhibitors; orange, Aurora kinase inhibitors (AKi); gray, PIK3/AKT/mTOR inhibitors; and blue, IGF1R inhibitors.
(C) An overview of growth inhibition of H1975 by various pathway inhibitors ± osi. Fold inhibition of growth by the combination of each compound with osi compared to each compound alone was normalized against that by osi. *, P<0.05; **, P<0.01; ***, P<0.001; ****, P<0.0001 (Mann-Whitney U test).
(D) A Venn diagram of top 50 agents that enhance osi-induced growth inhibition of H1975 and HCC827.
(E) Cells were treated with the indicated compounds ± osi for 48 h. Cell death was quantified by annexin-V (AV) staining (mean ± s.d., n=3). *, P<0.05; **, P<0.01 (Student’s t-test).
See also Figure S1.
AURKB inhibition enhances osimertinib-induced apoptosis synergistically through BIM and PUMA induction
To evaluate whether osi and AKi synergize, we assessed the EC50 of osi ± PF03814735 (PF), an AKi, in H1975. PF reduced the EC50 of osi by 50-fold in H1975 (Figure 2A), of which the combination index (CI) was 0.46. Of note, CI < 0.8 is considered synergistic (Chou, 2010). Although PF had a minimal effect on the clonogenic growth of H1975, it completely eliminated the emergence of osi-tolerant persisters (Figure 2B). Our prior study demonstrated critical crosstalk between the EGFR signal transduction pathway and the BCL-2 family-regulated apoptotic program (Figure 2C) (Bean et al., 2013). Accordingly, we investigated whether and how PF modulates these pathways to enhance osi-induced apoptosis (Figure 2D). As expected, osi reduced ERK and AKT phosphorylation and thereby induced BIMEL (BIM) and PUMA in H1975 (Figure 2D). BIMEL is the predominant isoform of BIM in most cell lines and tissues (Czabotar et al., 2014). PF increased BIM and PUMA proteins and slightly reduced AKT phosphorylation. Importantly, PF further enhanced osi-induced upregulation of BIM and PUMA proteins as well as suppression of AKT and FOXO1/3 phosphorylation while having a minimal effect on ERK phosphorylation (Figure 2D). PF-mediated induction of PUMA but not BIM occurred primarily at the level of transcription (Figure 2E). PF and/or osi had a minimal impact on the expression of other BCL-2 family proteins (Figure 2D). Functionally, knockdown (KD) of BIM or PUMA greatly reduced apoptosis triggered by PF and/or osi (Figure 2F). Consistent with our reported FOXO1/3-mediated upregulation of PUMA (Bean et al., 2013), KD of FOXO1/3 abrogated PF and/or osi-mediated induction of PUMA (Figure 2G). Together, these data indicate that PF and osi cooperate to inhibit AKT and thereby reduce FOXO1/3 phosphorylation, leading to FOXO1/3-mediated induction of PUMA.
Figure 2. AURKB inhibition enhances osimertinib-induced apoptosis through BIM and PUMA induction.
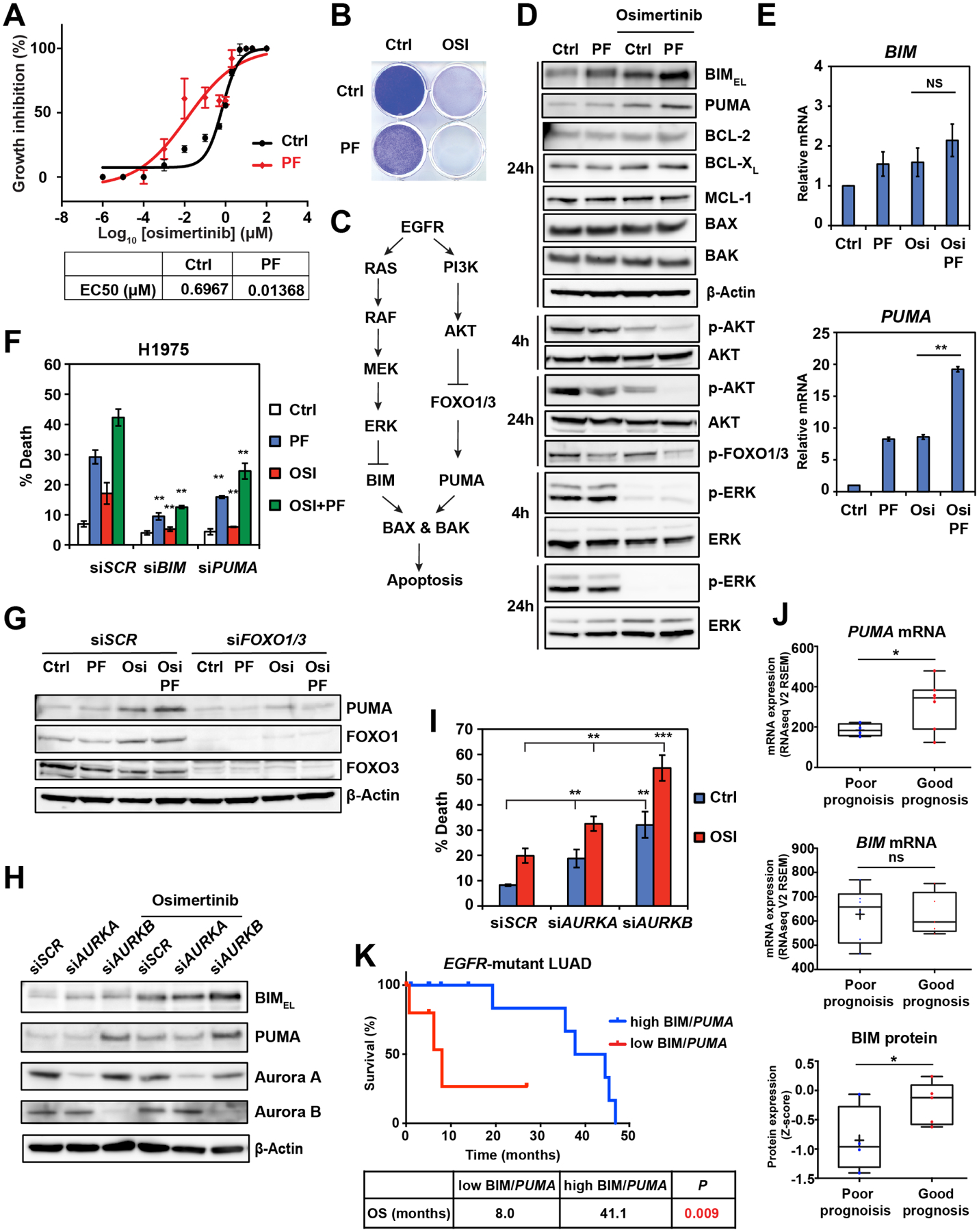
(A) H1975 was treated with osi at the indicated concentrations ± PF03814735 (PF, 2 μM). EC50 was assessed by CellTiter-Glo assays at 72 h (mean ± s.d., n=3).
(B) H1975 was treated with osi (1 μM) and/or PF (2 μM). Colonies were stained with crystal violet after 14 days.
(C) A schematic demonstrating the crosstalk between the EGFR signal transduction pathway and BCL-2 family-regulated apoptosis.
(D) H1975 treated with the indicated agents was assessed by immunoblots.
(E) qRT-PCR for BIM and PUMA mRNA in H1975 treated with the indicated agents (2 μM) for 24 h. Data were normalized against β-Actin (mean ± s.d., n=3). **, P<0.01; ns, not significant (Student’s t-test).
(F) H1975, transfected with the indicated siRNAs, was treated with the indicated agents (2 μM) for 48 h. Cell death was quantified by AV staining (mean ± s.d., n=3). **, P<0.01 (Student’s t-test).
(G) H1975, transfected with the indicated siRNAs, was treated with the indicated agents (2 μM) for 24 h and assessed by immunoblots.
(H-I) H1975, transfected with the indicated siRNAs, was treated with osi (2 μM) and assessed by immunoblots at 24 h. Cell death was quantified by AV staining at 48 h (mean ± s.d., n=3). **, P<0.01; ***, P<0.001 (Student’s t-test).
(J) Comparison of PUMA mRNA, BIM mRNA, and BIM protein expression in tumors from EGFR-mutant lung adenocarcinoma (LUAD) patients with a good or poor prognosis in TCGA (n=22). *, P<0.05 (Student’s t-test).
(K) Kaplan-Meier analysis of overall survival in EGFR-mutant LUAD patients from TCGA based on the expression of BIM protein and PUMA mRNA. Blu, high BIM protein or PUMA mRNA (n = 13); Red, low BIM protein or PUMA mRNA (n = 6). P = 0.009 (Log-rank test).
See also Figure S2.
Aurora A (AURKA) and Aurora B (AURKB) kinases play important roles in mitosis (Carmena et al., 2009). AURKA associates with the spindle poles to regulate mitotic entry, centrosome maturation, and spindle assembly; AURKB is a member of the Chromosomal Passenger Complex that regulates chromosome segregation and cytokinesis. Because most AKi including PF display significant cross-reactivity against both AURKA and AURKB, we sought to determine which of these factors is primarily responsible for the synergistic effect of PF with osi using a genetic approach. KD of AURKB but not AURKA recapitulated PF in enhancing osi-medicated induction of BIM and PUMA (Figure 2H). Consequently, KD of AURKB greatly enhanced osi-induced apoptosis whereas KD of AURKA exhibited a minor impact (Figure 2I). Similar results were obtained in ECLC26 using two independent siRNA oligos against AURKA or AURKB (Figure S2). Collectively, our data indicate that combined inhibition of AURKB and EGFR synergistically induces apoptosis in EGFR-mutant NSCLC through BIM and PUMA induction.
To further probe the correlation between BIM/PUMA and clinical outcome, we assessed data from EGFR-mutant lung adenocarcinomas (LUAD) in TCGA. Significantly higher PUMA mRNA was detected in tumors from patients with a good prognosis compared to those from patients with a poor prognosis whereas no significant difference in BIM mRNA was noted (Figure 2J). Notably, significantly higher BIM protein levels were detected in tumors from patients with a good prognosis compared to those from patients with a poor prognosis (Figure 2J), supporting the critical regulation of BIM protein stability by the EGFR signaling pathway. In line with these findings, patients with low expression of either BIM protein or PUMA mRNA had significantly shorter overall survival compared to those with high expression (8.0 months vs. 41.1 months, P=0.009, Figure 2K).
AURKB inhibition reduces BIM S87 phosphorylation and stabilizes BIM protein
Because PF and osi-mediated induction of BIM is mainly at the protein level (Figures 2D and 2E), we next investigated whether PF ± osi affected BIM protein stability. The half-life of BIM protein was determined using the protein synthesis inhibitor emetine. BIM protein degradation was delayed upon either PF or osi treatment, and the combination further extended the half-life of BIM protein (Figures 3A and 3B). It has been reported that phosphorylation of BIM by ERK on S69 and RSK on S93/S94/S98 targets BIM for βTrCP-mediated ubiquitination and degradation (Dehan et al., 2009; Luciano et al., 2003). Notably, PF had a minimal impact on ERK signaling (Figure 2D). Because BIM degradation is often mediated by phosphorylation-dependent ubiquitination, we explored whether AURKB directly phosphorylates BIM, leading to BIM degradation. Prediction of potential kinase phosphorylation motifs in BIM protein was performed using the SCANCITE 4.0 software, which identified AURKA, ERK1, and AURKB as the top three kinases that phosphorylate BIM on S87, S69, and S87, respectively (Figure 3C). Consistent with this prediction, BIM phosphorylation on S69 and S87 was reduced by osi and PF, respectively (Figure 3D). Inhibition of BIM phosphorylation by osi on S69 conferred more BIM stabilization than that on S87 by PF whereas combined inhibition had a greater effect (Figure 3D), which mirrored their effects on the half-life of BIM protein (Figure 3B). Specifically, KD of AURKB but not AURKA reduced BIM S87 phosphorylation (Figure 3E), implying that PF-mediated regulation of BIM is mediated through AURKB inhibition. Because AURKB is most active during mitosis, we examined whether arresting cells in mitosis would activate AURKB and thereby increase BIM S87 phosphorylation. Indeed, nocodazole induced mitotic arrest of H1975 (Figure S3A) and resulted in increased AURKB autophosphorylation and BIM S87 phosphorylation (Figure 3F). BIM S69 phosphorylation was also increased but to a lesser extent. In stark contrast, the CDK4/6 inhibitor (CDK4/6i) palbociclib arrested H1975 in G1 (Figure S3A), abrogated both AURKB and AURKA autophosphorylation, and greatly reduced BIM S87 phosphorylation (Figure 3F). BIM S69 phosphorylation was also reduced. Consequently, BIM protein was downregulated in cells arrested in mitosis and upregulated in cells arrested in G1 (Figure 3F).
Figure 3. AURKB inhibition reduces BIM S87 phosphorylation and stabilizes BIM protein.
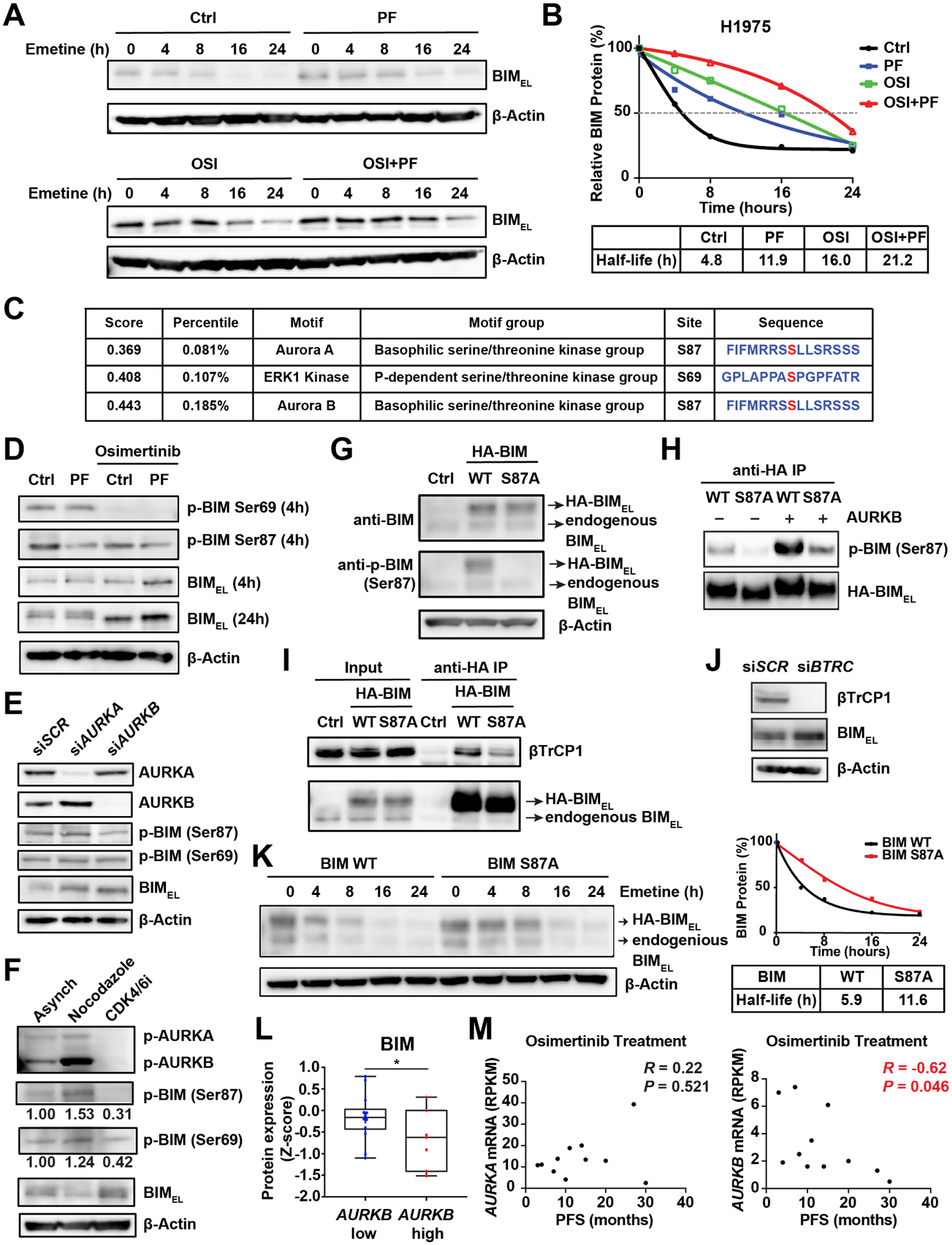
(A) H1975 was treated with osi (2 μM) and/or PF (2 μM) for 4 h, followed by the addition of emetine (20 μg/ml) to inhibit translation. Immunoblot analyses were performed at the indicated times.
(B) The half-life of BIM protein upon treatment with the indicated agents as in (A). The anti-BIM immunoblots shown in (A) were quantified by the ImageJ software and plotted with respect to time. Data shown are the mean of two independent experiments.
(C) Prediction of potential kinase phosphorylation motifs in BIM using the SCANCITE 4.0 software.
(D) H1975 treated with the indicated agents as in (A) for 4 or 24 h was assessed by immunoblots.
(E) H1975, transfected with the indicated siRNAs, was assessed by immunoblots.
(F) H1975 treated with nocodazole (50 ng/ml) for 16 h or the CDK4/6 inhibitor palbociclib (1 μM) for 24 h was assessed by immunoblots.
(G) BAX−/−BAK−/− H1975 was transduced with retrovirus expressing HA-tagged WT BIM or BIM S87A mutant and assessed by immunoblots.
(H) H1975 as in (G) was treated with palbociclib (1 μM) for 24 h and subjected to anti-HA immunoprecipitation. The immunoprecipitates were incubated with recombinant AURKB in the presence of ATP and assessed by immunoblots.
(I) H1975 as in (G) was subjected to anti-HA immunoprecipitation. The input (5%) and immunoprecipitates were assessed by immunoblots.
(J) H1975, transfected with the indicated siRNAs, was assessed by immunoblots.
(K) H1975 as in (G) was treated with emetine (20 μg/ml) and assessed by immunoblots. The BIM protein levels were quantified by the ImageJ software and plotted with respect to time. Data shown are the mean of two independent experiments.
(L) The expression of BIM protein in EGFR-mutant LUAD with high or low expression of AURKB mRNA from TCGA (n=22). *, P<0.05 (Student’s t-test).
(M) Scatter plots showing the correlation between progression-free survival (PFS) following osi treatment and pretreatment mRNA levels of AURKA or AURKB in EGFR-mutant LUAD patients (n=11).
See also Figure S3.
To gain insight into BIM degradation regulated by S87 phosphorylation, HA-tagged wild-type (WT) or S87A mutant BIM were stably expressed in BAX−/−BAK−/− H1975 because BIM overexpression killed H1975 through BAX/BAK (Figure 3G). The anti-phospho-BIM S87 antibody specifically detected WT but not S87A BIM (Figure 3G). Importantly, in vitro kinase assays demonstrated direct phosphorylation of BIM by recombinant AURKB, which was greatly diminished by the S87A mutation (Figure 3H). Although recombinant AURKA could also phosphorylate BIM on S87 in vitro, its efficacy was much lower than that of AURKB (Figure S3B), which is consistent with the notion that KD of AURKB but not AURKA reduced BIM S87 phosphorylation (Figure 3E). Mechanistically, the S87A mutation in BIM greatly reduced its interaction with βTrCP1 (Figure 3I) and KD of BTRC (βTrCP1) stabilized BIM protein (Figure 3J). Consequently, the S87A BIM protein has a longer half-life than WT BIM (Figure 3K). Consistent with the negative regulation of BIM stability by AURKB, a negative correlation between BIM protein levels and AURKB mRNA levels was observed in the TCGA EGFR-mutant LUAD (Figure 3L). Furthermore, the pretreatment mRNA levels of AURKB but not AURKA negatively correlated with progression-free survival (PFS) in a cohort of EGFR-mutant NSCLC patients treated with osi (Roper et al., 2020) (Figure 3M). Collectively, these results support a model in which BIM S87 phosphorylation by AURKB mediates its binding to βTrCP1 and degradation via SCFβTrCP. Accordingly, genetic and chemical inhibition of AURKB reduces BIM S87 phosphorylation, leading to BIM stabilization.
Ability of Aurora kinase inhibitors to enhance osimertinib-induced apoptosis correlates with their expeditious inhibition of AURKB
Our mechanistic studies revealed that targeting AURKB has greater proapoptotic effect than targeting AURKA. Because our HTS showed that some AKi were more potent than others in enhancing osi-induced growth inhibition (Figure 1C), we asked whether this is due to their differential inhibition of AURKB. In line with our HTS, PF, MLN8054, GSK1070916, and hesperidin enhanced osi-induced apoptosis in both H1975 and ECLC26 (Figures 4A and S4A). In contrast, MLN8237, AZD1152, CCT137690, and MK5108 failed to do so. Based on these results, we divided these AKi into the enhancer and non-enhancer groups of osi-induced apoptosis. The enhancer group was superior to the non-enhancer group in reducing the emergence of DTPs (Figure 4B).
Figure 4. Aurora kinase inhibitors are divided into enhancer and non-enhancer groups of osimertinib-induced apoptosis.
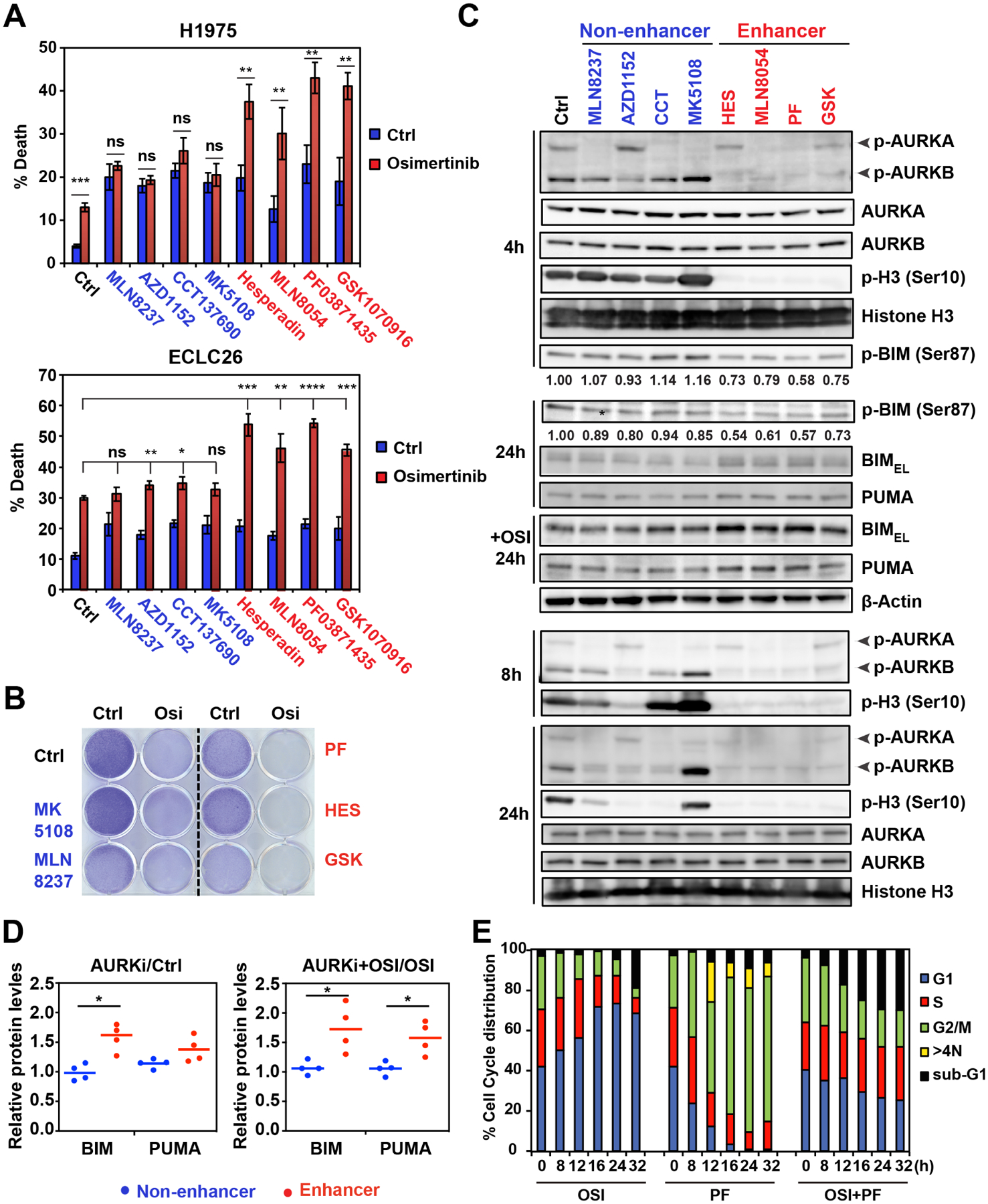
(A) H1975 and ECLC26 were treated with the indicated AKi (1 μM) ± osi (1 μM) for 48 h. Cell death was quantified by AV staining (mean ± s.d., n=3). *, P<0.05; **, P<0.01; ***, P<0.001; ****, P<0.0001 (Student’s t-test).
(B) H1975 was treated with the indicated agents (1 μM) and colonies were stained with crystal violet after 14 days.
(C) H1975 treated as in (A) was assessed by immunoblots at the indicated times.
(D) The anti-BIM and anti-PUMA immunoblots shown in C were quantified by the ImageJ software. Data presented are the ratio of BIM and PUMA protein levels upon the treatment of AKi compared to control and the ratio upon the treatment of both AKi and osi compared to osi alone. *, P<0.05 (Student’s t-test).
(E) H1975 was treated with osi (1 μM) and/or PF (1 μM) and subjected to cell-cycle analysis using propidium iodide (PI) staining at the indicated times.
See also Figure S4.
We next assessed the intracellular activity of these inhibitors against AURKA versus AURKB. Consistent with reported findings (Mortlock et al., 2007; Shimomura et al., 2010), MK5108 and AZD1152 selectively inhibited AURKA and AURKB autophosphorylation, respectively; while the remaining agents displayed variable cross-reactivity against both AURKA and AURKB (Figure 4C). There was no association between AURKA inhibition and the efficacy of enhancing osi-induced apoptosis. Strikingly, the enhancer group abrogated AURKB autophosphorylation and histone H3S10 phosphorylation (a substrate of AURKB) within 4 h (Figure 4C), indicative of effective inhibition of AURKB. Consequently, reduced BIM S87 phosphorylation was noted within 4 h, which in turn increased BIM protein. It is noteworthy that osi-induced inhibition of BIM S69 phosphorylation occurred within 4 h (Figure 3D), suggesting that concurrent inhibition of BIM phosphorylation on both S69 and S87 within 4 h is required to fully stabilize BIM for inducing apoptosis. Quantification of BIM and PUMA protein levels at 24 h showed that the enhancer group significantly increased BIM and further cooperated with osi to induce both BIM and PUMA (Figure 4D), which is in accordance with their proapoptotic cooperation with osi (Figure 4A). Although AZD1152 selectively inhibited AURKB (Figure 4C), strong inhibition of both AURKB autophosphorylation and H3S10 phosphorylation was not observed until 8 h post-treatment of AZD1152, and AZD1152 neither induced BIM/PUMA nor cooperated with osi to induce apoptosis (Figures 4A and 4C). These data further support the importance of concurrent inhibition of EGFR and AURKB within 4 h to maximize apoptotic induction.
Because osi and PF are known to block cell cycle at different phases, we next determined whether combined osi and PF exerted a specific effect on cell cycle progression. As expected, osi induced G1 arrest whereas PF resulted in mitotic arrest (Figures 4E and S4B). Remarkably, combined inhibition of EGFR and AK counteracted the respective cell cycle blockade, leading to normal cell cycle progression (Figures 4E and S4B). Given that osi-induced G1 arrest is expected to lower AK activity, it may be more advantageous to inhibit AK before cells enter G1 arrest. It was reported that AURKA is activated in DTPs, which drives the evolution of resistance to third-generation TKIs in EGFR-mutant NSCLC (Shah et al., 2019). This prompted us to investigate whether AURKB is activated in DTPs and whether DTPs are dependent on AURKB for survival. Consistent with the quiescent or G1 arrest status of DTPs as reported (Sharma et al., 2010), both AURKA and AURKB autophosphorylation was greatly reduced in DTPs of H1975, PC9, and ECLC26 (Figure S4C), which in turn resulted in reduced BIM S87 phosphorylation (Figure S4D). Consequently, DTPs were not sensitized to AKi (Figure S4E). Overall, these findings suggest that concurrent inhibition of EGFR and AURKB is required to augment apoptotic induction, eradicating cancer cells upfront to reduce the emergence of drug tolerance and resistance.
Osimertinib-resistant cells exhibit EMT and become vulnerable to AURKB inhibition
To this end, we demonstrated that combined inhibition of EGFR and AURKB efficiently eliminate TKI-naïve cancer cells. We next asked whether this combination is also effective against osi-resistant cells. Osi-resistant H1975 (H1975R) and ECLC26 (ECLC26R) cell lines were generated through stepwise dose escalation (Figure 5A). These cells displayed cross-resistance to rociletinib (Figure S5A). MSK-IMPACT assays detected additional mutations with low allele frequencies in ECLC26R but not in H1975R (Table S1). Additional copy number alterations were also detected in both models. However, none of these alterations have obvious roles in osi resistance (Figures S5B and S5C, and data not shown). To further characterize the resistance mechanisms, RNA-seq was performed. Gene set enrichment analysis (GSEA) revealed upregulation of the epithelial-to-mesenchymal transition (EMT) signature in osi-resistant cells (Figure S5D and Table S2). EMT is a known resistance mechanism to EGFR TKIs (Sequist et al., 2011; Shibue and Weinberg, 2017). Both H1975R and ECLC26R lost E-cadherin and increased vimentin expression (Figure 5B). Interestingly, H1975R and ECLC26R became highly sensitive to AKi PF and MLN8054 (Figures 5C and S5E). In contrast, inhibitors of IGF1R, SFK, and PI3K/mTOR/AKT induced minimal apoptosis ± osi (Figure 5C). Mechanistically, PF induced BIM/PUMA and slightly suppressed AKT phosphorylation in H1975R and ECLC26R (Figure 5D). In accordance with the inability of osi to induce apoptosis in H1975R and ECLC26R, osi neither reduced ERK/AKT phosphorylation nor induced BIM/PUMA (Figure 5D).
Figure 5. Osimertinib-resistant cells exhibit EMT and become hypersensitive to AURKB inhibition.
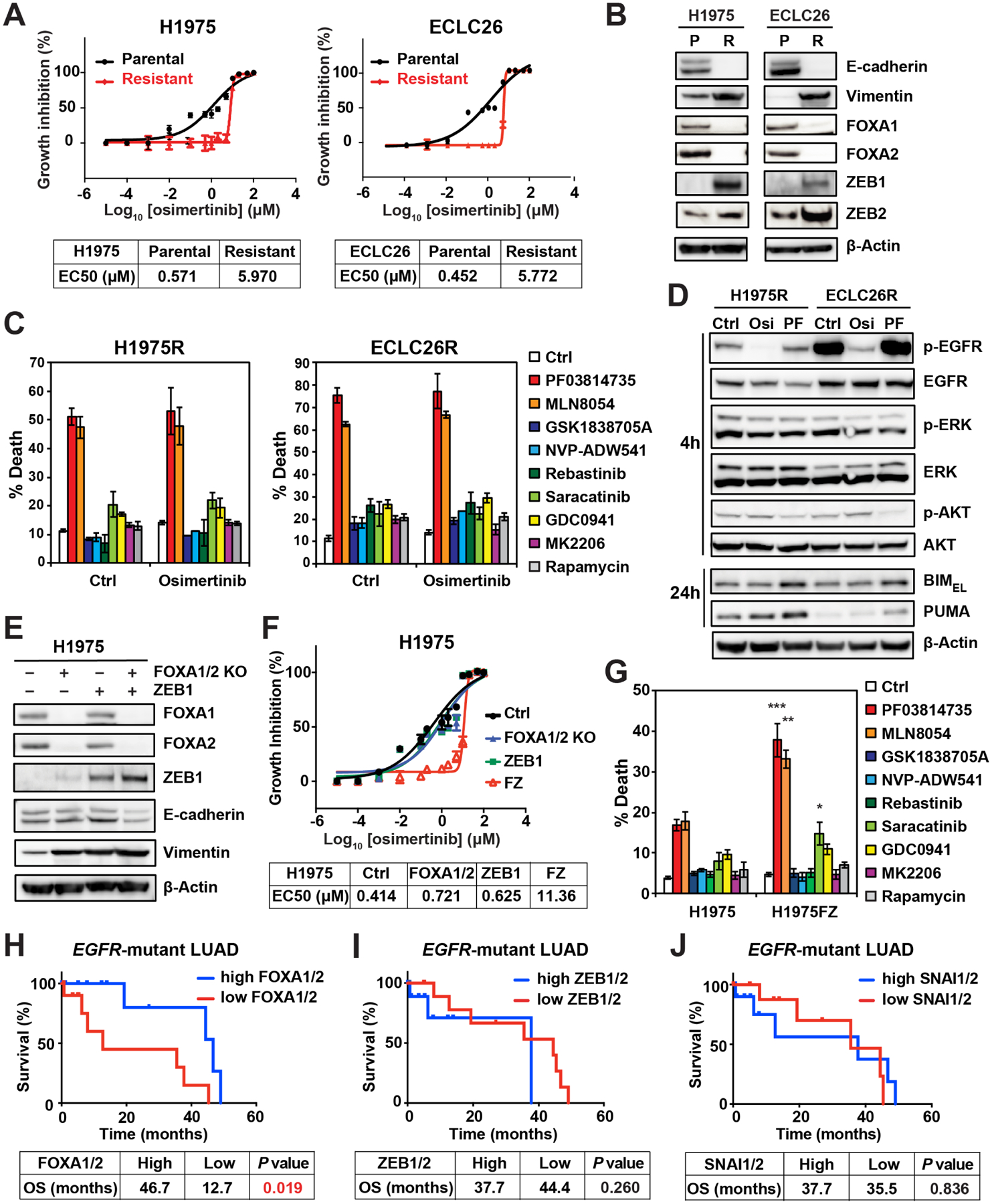
(A) EC50 of osi in the indicated cell lines was assessed by CellTiter-Glo assays at 72 h (mean ± s.d., n=3).
(B) The parental (P) and osi-resistant (R) H1975 and ECLC26 were assessed by immunoblots.
(C) H1975R and ECLC26R were treated with the indicated agents (2 μM) for 48 h. Cell death was quantified by AV staining (mean ± s.d., n=3).
(D) H1975R and ECLC26R treated with the indicated agents (1 μM) for 4 or 24 h were assessed by immunoblots.
(E-F) H1975 cells with CRISPR/Cas9-mediated KO of both FOXA1 and FOXA2, retroviral transduction of ZEB1, and both (H1975FZ) were assessed by immunoblots in (E). EC50 was assessed by CellTiter-Glo assays at 72 h in (F) (mean ± s.d., n=3).
(G) H1975 and H1975FZ were treated with the indicated agents (2 μM) for 48 h. Cell death was quantified by AV staining (mean ± s.d., n=3). *, P<0.05; **, P<0.01; ***, P<0.001 (Student’s t-test, comparing H1975 to H1975FZ).
(H-J) Kaplan-Meier analysis of overall survival in EGFR-mutant LUAD patients from TCGA (n=22) based on the expression of FOXA1/2 (H), ZEB1/2 (I), and SNAI1/2 (J) mRNA. Blue, the top 50% highly expressed; Red, the bottom 50%. P values were calculated by the Log-rank test.
To establish a causal relationship between EMT and osi resistance as well as sensitivity to AKi, we sought to induce EMT in H1975 by perturbing the EMT master regulators. Analysis of RNA-seq revealed upregulation of transcriptional repressors ZEB1 and ZEB2 that promote EMT (Shibue and Weinberg, 2017), and downregulation of pioneer transcription factors FOXA1 and FOXA2 that are required for respiratory epithelial differentiation (Wan et al., 2005) in both H1975R and ECLC26R. These findings were further validated by immunoblots (Figure 5B). Because there were no concordant changes of SNAIL, SLUG, and TWIST expression in both H1975R and ECLC26R, we focused on FOXA1/2 and ZEB1/2. Retroviral transduction of ZEB1 and/or CRISPR/Cas9-mediated knockout (KO) of FOXA1/2 were performed in H1975. Either FOXA1/2 KO or ZEB1 overexpression increased vimentin without altering E-cadherin expression (Figure 5E). Notably, combined FOXA1/2 KO and ZEB1 overexpression (H1975FZ) not only reduced E-cadherin but further increased vimentin (Figure 5E). H1975FZ became resistant to osi whereas either FOXA1/2 KO or ZEB1 overexpression alone was insufficient to confer osi resistance (Figure 5F). Similar to H1975R, H1975FZ was also sensitized to AKi, but not to other pathway inhibitors (Figure 5G). These data support that EMT is responsible for the sensitization of osi-resistant cells to AKi.
We next investigated whether FOXA1/2 and ZEB1/2 mRNA levels have any prognostic value for EGFR-mutant LUAD patients in TCGA. Indeed, patients with low FOXA1/2 expression had significantly shorter overall survival compared to those with high expression (Figure 5H), while ZEB1/2 expression had no significant impact on the survival (Figure 5I). In addition, the expression of SNAI1 (SNAIL) and SNAI2 (SLUG) had no prognostic value (Figure 5J). Our findings highlight the important cooperation between FOXA1/2 and ZEB1/2 in driving EMT and the development of therapeutic resistance.
Inhibition of ATR-CHK1-AURKB induces BIM-mediated mitotic cell death in osimertinib-resistant EMT cells
To interrogate whether AURKA or AURKB is crucial to the survival of osi-resistant EMT cells, we assessed the proapoptotic activity of various AKi in H1975R and ECLC26R. Strikingly, both H1975R and ECLC26R showed increased apoptosis compared to parental cells upon treatment with all the AKi with the exception of MK5108 (Fig. 6A), a highly selective AURKA inhibitor (Figure 4C). In ECLC26R, MK5108 was the only inhibitor that failed to inhibit AURKB autophosphorylation and H3S10 phosphorylation (Figure 6B), indicating that osi-resistant EMT cells become highly vulnerable to AURKB rather than AURKA inhibition. Notably, similar inhibitory profiles of AURKA versus AURKB by AKi were observed in both ECLC26R and H1975 (Figures 4C and 6B). We further demonstrated that KD of AURKB induced robust apoptosis in H1975R whereas KD of AURKA had a lesser impact (Figure 6C).
Figure 6. Inhibition of ATR-CHK1-AURKB induces BIM-mediated mitotic cell death in osimertinib-resistant EMT cells.
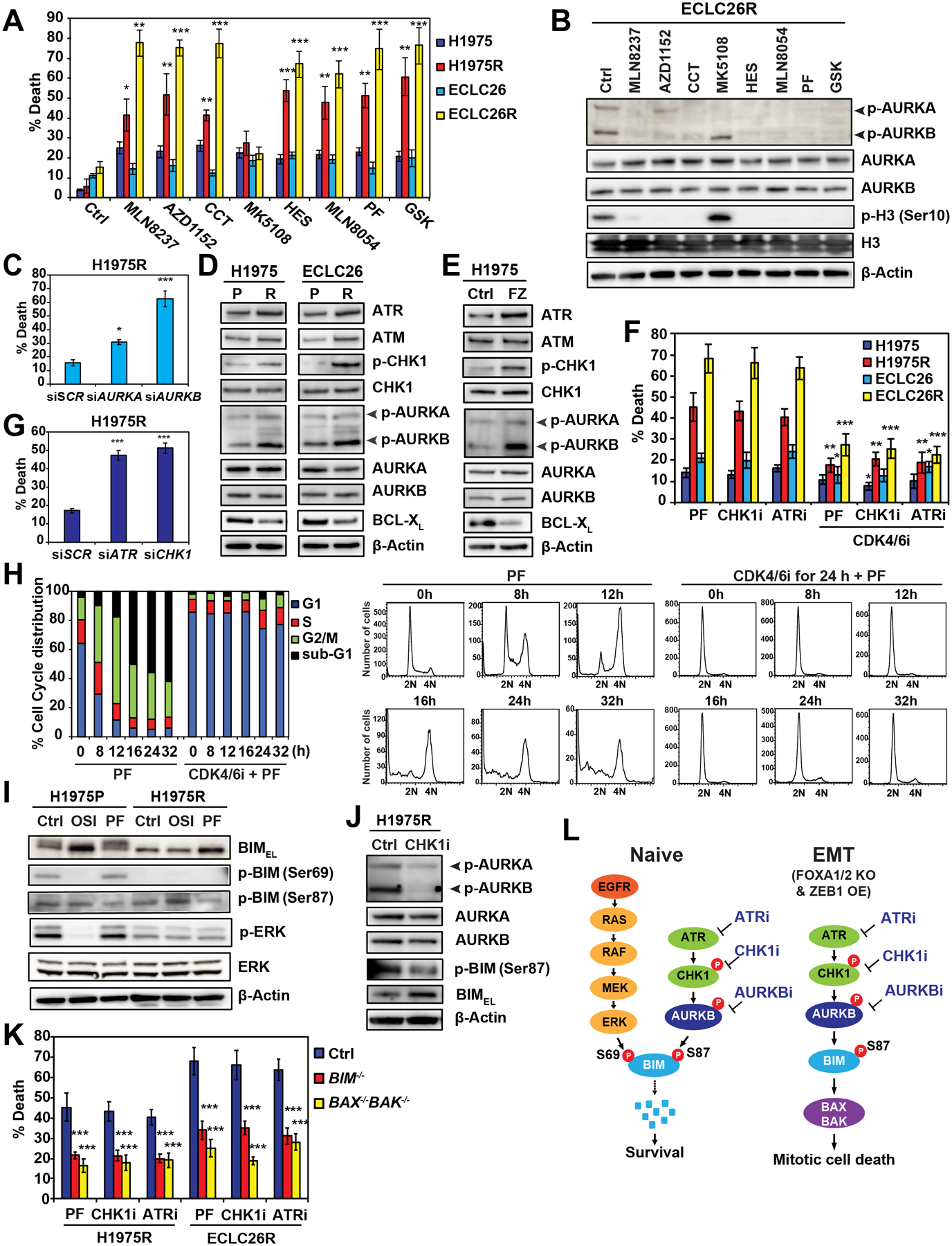
(A) The parental and osi-resistant H1975 and ECLC26 were treated with the indicated AKi (1 μM) for 48 h. Cell death was quantified by AV staining (mean ± s.d., n=3). *, P<0.05; **, P<0.01; ***, P<0.001 (Student’s t-test).
(B) ECLC26R treated with the indicated AKi (1 μM) for 24 h was assessed by immunoblots.
(C) H1975R was transfected with the indicated siRNAs and cell death was quantified by AV staining at 48 h (mean ± s.d., n=3). *, P<0.05; ***, P<0.001 (Student’s t-test).
(D) H1975, H1975R, ECLC26, and ECLC26R were assessed by immunoblots.
(E) H1975FZ was assessed by immunoblots.
(F) H1975, H1975R, ECLC26, and ECLC26R were treated with PF (1 μM), the CHK1 inhibitor LY2603618 (1 μM), or the ATR inhibitor VX-970 (2 μM) for 48 h. Alternatively, cells were pretreated with palbociclib (1 μM) for 24 h and then treated with PF, LY2603618, or VX-970 for 48 h. Cell death was quantified by AV staining (mean ± s.d., n=3). *, P<0.05; **, P<0.01; ***, P<0.001 (Student’s t-test, comparing without to with palbociclib).
(G) H1975R was transfected with the indicated siRNAs and cell death was quantified by AV staining at 72 h (mean ± s.d., n=3). ***, P<0.001 (Student’s t-test).
(H) ECLC26R ± palbociclib (1 μM) pretreatment for 24 h was treated with PF (1 μM) and subjected to cell-cycle analysis using PI staining at the indicated times.
(I) H1975P and H1975R treated with the indicated agents (1 μM) for 4 h were assessed by immunoblots.
(J) H1975R ± LY2603618 (1 μM) for 4 or 24 h was assessed by immunoblots.
(K) H1975R and ECLC26R with retroviral transduction of sgRNA against LacZ, BIM, or BAX/BAK were treated as in (F) for 48 h. Cell death was quantified by AV staining (mean ± s.d., n=3). ***, P<0.001 (Student’s t-test).
(L) A schematic depicts the regulation of BIM phosphorylation by the EGFR-RAS-RAF-MEK-ERK and the ATR-CHK1-AURKB signaling pathways in naïve versus EMT cells to induce BAX/BAK-dependent mitotic cell death.
To understand the molecular basis of hypersensitivity of osi-resistant EMT cells to AURKB inhibition, we analyzed RNA-seq and identified ATM and ATR upregulation in both H1975R and ECLC26R compared to parental cells (Figure S6A), which was further validated by immunoblots (Figure 6D). It is known that ATR, via its downstream effector CHK1, regulates AURKB to control cell division (Mackay and Ullman, 2015; Petsalaki et al., 2011). Accordingly, increased phosphorylation of CHK1 on S345, a known ATR-mediated phosphorylation, was observed in both H1975R and ECLC26R (Figure 6D). Furthermore, robust induction of AURKB autophosphorylation was seen in both H1975R and ECLC26R (Figure 6D), indicating that AURKB was activated in osi-resistant EMT cells. AURKA autophosphorylation was not obviously altered. These results revealed activation of the ATR-CHK1-AURKB signaling cascade in osi-resistant EMT cells. Notably, osi-resistant EMT cells exhibited abnormal spindle assembly, increased chromosomal segregation errors, and increased γH2AX (Figures S6B, S6C, and S6D). We then investigated whether the demonstrated vulnerability of H1975FZ to AKi (Figure 5G) is due to activation of ATR-CHK1-AURKB. Indeed, combined FOXA1/2 KO and ZEB1 overexpression increased ATR expression, CHK1 phosphorylation, and AURKB autophosphorylation in H1975 (Figure 6E). Altogether, activation of ATR-CHK1-AURKB correlated well with the sensitization to AURKB inhibition. Analysis of TCGA dataset revealed that ZEB2 expression positively correlated with ATR expression in EGFR-mutant LUAD (Table S3). In addition, ZEB2 expression positively correlated with both ATR and ATM expression, ZEB1 expression positively correlated with ATM expression, and FOXA2 expression negatively correlated with ATR expression in the entire TCGA-LUAD cohort (Table S4).
Given that ATR and CHK1 are activated in osi-resistant EMT cells and inhibitors of ATR/CHK1 activate mitotic catastrophe or mitotic cell death (Ma et al., 2011; Pilié et al., 2019), we sought to determine whether H1975R and ECLC26R are also sensitive to ATR/CHK1 inhibitors and whether inhibition of ATR-CHK1-AURKB induces mitotic catastrophe. Indeed, the ATR inhibitor VX-970 and the CHK1 inhibitor LY2603618 induced cell death only in H1975R and ECLC26R but not in parental cells (Figure 6F). Furthermore, KD of ATR or CHK1 was sufficient to induce death of H1975R (Figure 6G). To determine whether inhibition of ATR-CHK1-AURKB induces mitotic cell death in H1975R and ECLC26R, we explored whether preventing the entry of PF-treated cells into mitosis using CDK4/6i could mitigate PF-induced cell death. Cell cycle profiling confirmed that PF induced mitotic arrest of ECLC26R within 12 h followed by a time-dependent accumulation of dying sub-G1 population up to 32 h (Figure 6H). Importantly, co-treatment of cells with CDK4/6i and PF arrested ECLC26R in G1 (Figure 6H) and abrogated PF-induced cell death (Figure 6F). Likewise, co-treatment of CDK4/6i greatly reduced cell death triggered by ATR/CHK1 inhibitors (Figure 6F). Collectively, these findings support that inhibition of ATR-CHK1-AURKB induces mitotic cell death in osi-resistant EMT cells.
We next determined whether the hypersensitivity of osi-resistant EMT cells to ATR-CHK1-AURKB inhibitors is due to altered expressions of BCL-2 family that lower the apoptotic threshold. RNA-seq analysis discovered downregulation of antiapoptotic BCL-XL in both H1975R and ECLC26R (Figure S6A), which was further validated by immunoblots in these cells as well as in H1975FZ (Figures 6D and 6E). No concordant changes of other BCL-2 family members were identified in both models. As BIM protein is normally downregulated during mitosis (Figure 3F), it is conceivable that AURKB inhibition-mediated stabilization of BIM would be sufficient to trigger mitotic cell death in EMT cells where the apoptotic threshold is lowered due to BCL-XL downregulation (Chen et al., 2015). We next examined whether osi-resistant cells have altered BIM S69 phosphorylation in addition to their activation of ATR-CHK1-AURKB. Interestingly, BIM phosphorylation on S69 was diminished in H1975R compared to H1975 parental (H1975P) cells, consistent with the observed reduced ERK activation (Figure 6I). As a result, PF-mediated inhibition of BIM S87 phosphorylation increased more BIM protein in H1975R than H1975P where BIM could still be degraded through S69 phosphorylation (Figure 6I). In line with these findings, the CHK1 inhibitor LY2603618 reduced AURKB autophosphorylation and thereby BIM S87 phosphorylation, leading to BIM stabilization (Figure 6J). To further establish the role of BIM and its downstream effectors BAX/BAK in regulating mitotic cell death upon inhibition of ATR-CHK1-AURKB, CRISPR/Cas9-mediated KO of BIM or BAX/BAK was performed in H1975R and ECLC26R. Indeed, KO of BIM as well as double KO of BAX/BAK protected H1975R and ECLC26R from mitotic cell death triggered by ATR-CHK1-AURKB inhibitors (Figures 6K, S6E, and S6F). Overall, our studies uncovered previously unrecognized regulatory phosphorylation of BIM in the control of mitotic catastrophe (Figure 6L).
Aurora kinase inhibition improves the therapeutic efficacy of osimertinib in xenograft models
Next, we determined whether combined osi and PF displays better in vivo therapeutic efficacy than osi alone in mouse xenograft models. Mice bearing parental H1975 xenografts were treated with vehicle, osi, PF, or the combination for 28 days. Both monotherapy and combination therapy significantly suppressed tumor growth without overt toxicity and weight loss (Figures 7A and S7A). Importantly, waterfall plot analyses of tumor size changes showed that all tumors treated with the combination regressed more than 50% whereas approximately half of the tumors treated with either monotherapy failed to regress after 28 days (Figure 7B). We then explored whether differential tumor regrowth occurs after discontinuation of treatment. Consistent with the ability of combined osi and PF to reduce the emergence of DTPs in vitro (Figures 2B and 4B), the combination therapy significantly reduced tumor regrowth compared to osi or PF alone (Figure 7A). The therapeutic efficacy was further demonstrated in a patient-derived xenograft (PDX) model ECLC26. Again, combined osi and PF had better therapeutic efficacy than osi or PF alone (Figures 7C and S7C).
Figure 7. Aurora kinase inhibition improves the therapeutic efficacy of osimertinib in xenograft models.
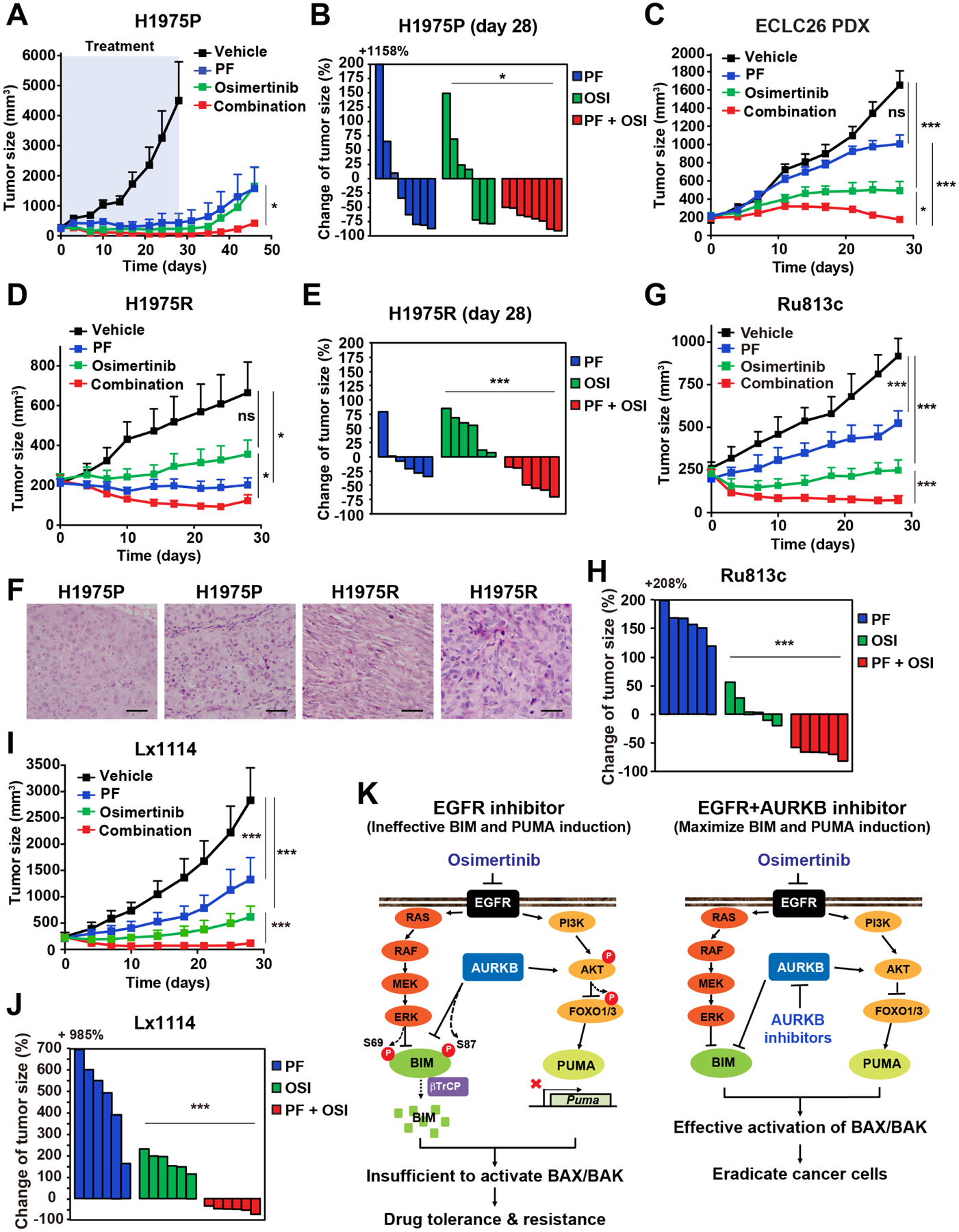
(A) Athymic nude mice bearing H1975 xenografts were treated with vehicle, osi (5 mg/kg), PF (20 mg/kg), or the combination for 28 days. Tumor volumes were measured twice weekly by caliper (mean ± SEM, n = 6–8 for each group). *, P < 0.05 (two-way ANOVA).
(B) Waterfall plot of changes in tumor volume after 28 days of treatment in (A). *, P < 0.05 (Student’s t-test).
(C) NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice bearing patient-derived ECLC26 xenografts were treated as in (A) (mean ± SEM, n = 8–12 for each group). *, P < 0.05; ***, P < 0.001 (two-way ANOVA).
(D) Athymic nude mice bearing H1975R xenografts were treated as in (A) (mean ± SEM, n = 6–8 for each group). *, P < 0.05 (two-way ANOVA).
(E) Waterfall plot of changes in tumor volume in (D). ***, P < 0.001 (Student’s t-test).
(F) Representative H&E images of xenografts established using either H1975 or H1975R cells. Scale bars, 200 μm.
(G) NSG mice bearing patient-derived Ru813c xenografts were treated as in (A) (mean ± SD, n = 6 for each group). ***, P < 0.001 (two-way ANOVA).
(H) Waterfall plot of changes in tumor volume in (G). ***, P < 0.001 (Student’s t-test).
(I) NSG mice bearing patient-derived Lx1114 xenografts were treated as in (A) (mean ± SD, n = 6 for each group). ***, P < 0.001 (two-way ANOVA).
(J) Waterfall plot of changes in tumor volume in (I). ***, P < 0.001 (Student’s t-test).
(K) A schematic summarizing how EGFR and AURKB regulate the proapoptotic activity of BIM and PUMA.
To determine whether the combination is still effective in cancer that has acquired resistance to osi through EMT, we established xenografts in mice using H1975R cells (Figures 7D, 7E, and S7B). Histological examination of H1975R xenografts showed spindle-shaped malignant cells with mesenchymal morphology to a variable extent, which is in stark contrast to the typical LUAD observed in H1975P xenografts (Figure 7F and data not shown). Either PF alone or the combination significantly suppressed tumor growth whereas osi alone had limited efficacy (Figures 7D and 7E). As EMT-mediated resistance has been reported to be reversible upon withdrawal of EGFR inhibitors (Shah et al., 2019), it is conceivable that the resistance phenotype of H1975R may not be fully maintained during the establishment of PDXs such that the combination therapy is required to fully inhibit tumor growth.
Finally, we assessed the therapeutic efficacy of osi and/or PF in PDX models derived from two EGFR-mutant (exon 19 deletion) NSCLC patients experiencing disease progression on osi (Figures 7G–J). Ru813c was derived from a patient whose primary lung tumor harbored concurrent mutations in EGFR (exon 19 deletion) and NF2 (Q125*). No new genetic alternations were identified by MSK-IMPACT in Ru813c after disease progression (Table S5). On the other hand, new mutations in KEAP1 (p.G509R) and RTEL1 (p.V271M) were identified in Lx1114 after disease progression (Table S5). Although the contributions of these mutations to osi resistance remained to be further characterized, the combination of osi and PF potently inhibited the growth of both PDX models (Figures 7G–J). Overall, our data indicate that the combination of osi and PF is an effective therapeutic strategy for both treatment-naïve and osi-resistant EGFR-mutant NSCLC regardless of EMT.
DISCUSSION
The discoveries of individual oncogenes and paired development of targeted therapies specific to each driver mutation have revolutionized cancer therapy, laying the foundation of precision cancer medicine (Weinstein, 2002). However, these therapies are rarely, if ever, curative. In most EGFR-mutant NSCLC patients, resistance arises after a dramatic initial response to EGFR TKIs followed by stable minimal residual disease and subsequent development of drug-resistant tumors. One explanation for such observations is the inability of EGFR TKIs to eradicate all tumor cells. It is conceivable that enhancing apoptosis through upfront combination therapies will eradicate cancer cells and thereby reduce the emergence of drug resistance. Here, we have performed unbiased HTS to identify therapeutic agents that synergize with EGFR TKIs to eradicate EGFR-mutant NSCLC. This approach led to the discovery of AKi as a rational combination strategy with osi to maximize the induction of BIM and PUMA, two proapoptotic sentinels that interconnect the EGFR signal transduction pathway and BAX/BAK-dependent apoptotic program (Figure 7K). Both genetic perturbation and chemical inhibition of AURKA versus AURKB using a panel of AKi indicate that AURKB inhibition is primarily responsible for the synergistic proapoptotic effect with osi. Importantly, we found that osi-resistant EMT cells were highly sensitive to AURKB inhibition due to the intrinsic activation of ATR-CHK1-AURKB signaling cascade. Hence, combined inhibition of EGFR and AURKB can serve as a reinforcement to eliminate any drug-resistant clones that may have emerged from DTPs. Moreover, this combination strategy is effective in two osi-resistant PDXs in which the resistance mechanisms are unrelated to EMT.
In addition to the known regulation of BIM degradation through ERK-mediated phosphorylation of S69 (Dehan et al., 2009; Luciano et al., 2003), we showed that AURKB phosphorylated BIM on S87, leading to its interaction with βTrCP1 and degradation via SCFβTrCP. Interestingly, only the AKi that are able to inhibit AURKB and BIM S87 phosphorylation within 4 h, a time point when osi-mediated inhibition of BIM S69 phosphorylation occurs, could enhance osi-induced apoptosis (Figure 4). These findings indicate that concurrent inhibition of BIM phosphorylation on both S69 and S87 within 4 h is required to fully stabilize BIM, leading to robust activation of BAX/BAK. Of note, prolonged osi treatment would induce G1 arrest (Figure 4E), which would inhibit AURKB as we demonstrated for CDK4/6i (Figure 3F). Hence, AKi that exhibit delayed inhibition of AURKB and BIM phosphorylation are unlikely to synergize with osi. We have previously demonstrated that inhibition of PI3K-AKT triggers nuclear translocation of FOXO1/3 to transactivate PUMA (Bean et al., 2013). Here, we showed that AURKB inhibition reduced the phosphorylation of AKT and FOXO1/3 and induced PUMA transcription. How AURKB regulates AKT signaling remains to be determined. Of note, it has been reported that AURKA can activate AKT signaling (Yao et al., 2009). Although previous studies have demonstrated that BIM mRNA can predict both clinical response and survival benefits for EGFR-mutant NSCLC patients (Costa et al., 2014; Faber et al., 2011), BIM protein levels may be a better prognostic marker given the important regulation of BIM protein stability.
Recently, lineage plasticity or transdifferentiation has emerged as a mode of targeted therapy evasion in various cancers (Boumahdi and de Sauvage, 2019; Quintanal-Villalonga et al., 2020; Shibue and Weinberg, 2017). In EGFR-mutant NSCLC, two well-known examples are EMT and transformation to small cell lung cancer (SCLC) (Niederst et al., 2015; Sequist et al., 2011). The observation that EMT almost universally occurs in various EGFR-mutant NSCLC cell lines following osi exposure strongly supports that EMT is a principal adaptive response to EGFR inhibition (Kurppa et al., 2020; Shah et al., 2019; Zhang et al., 2021). In contrast to prior studies of EMT that have mainly focused on the transcriptional repressors ZEB1/2, SNAIL, SLUG, and TWIST (Shibue and Weinberg, 2017; Stemmler et al., 2019), we found that concurrent ZEB1 overexpression and loss of the epithelial lineage-specifying pioneer factors FOXA1/2 are required to fully activate EMT and confer osi resistance (Figures 5E and 5F). Furthermore, the mRNA levels of FOXA1/2 but not ZEB1/2 or SNAIL/SLUG are prognostic for EGFR-mutant LUAD patients in TCGA (Figures 5H, 5I, and 5J). Importantly, combined FOXA1/2 KO and ZEB1 overexpression activate the ATR-CHK1-AURKB DNA damage checkpoint response (DDCR) and sensitize cancer cells to mitotic catastrophe upon inhibition of ATR-CHK1-AURKB. Given that EGFR inhibition would reduce BIM S69 phosphorylation and increase BIM, activation of ATR-CHK1-AURKB may be an adaptation mechanism to evade apoptosis by downregulating BIM through S87 phosphorylation. Interestingly, it has been reported that RB1-deficient tumors express high levels of CHK1 and are vulnerable to CHK1 and AURKB inhibition (Oser et al., 2019; Witkiewicz et al., 2018). Furthermore, deletion of RB1, which is pathognomonic of SCLC transformation (Niederst et al., 2015), sensitized PC9 and H1975 to AURKB inhibition (Oser et al., 2019). Altogether, these findings suggest that activation of ATR-CHK1-AURKB may constitute a common cellular adaptation to lineage switch.
Although activation of mitotic catastrophe by ATR/CHK1 inhibitors has gained attention as an important anticancer strategy, the underlying molecular mechanisms are not well understood (Ma et al., 2011; Pilié et al., 2019). Our studies serendipitously identified that AURKB-mediated phosphorylation of BIM functions downstream of ATR-CHK1 to govern the activation of BAX/BAK-dependent mitotic cell death. BIM appears to sit at the crossroads between the EGFR-RAS-RAF-MEK-ERK signaling pathway and the ATR-CHK1-AURKB DDCR to integrate environmental cues for cell death decision (Figure 6L). In osi-sensitive EGFR-mutant NSCLC, AURKB inhibition is insufficient to stabilize BIM due to continuous degradation of BIM through S69 phosphorylation (Figure 6L). In contrast, osi-resistant EMT cells become vulnerable to mitotic catastrophe upon inhibition of ATR-CHK1-AURKB due to the intrinsic activation of DDCR, reduced ERK signaling, and downregulation of pro-survival BCL-XL. Although our studies clearly demonstrated a predominant role of AURKB in BIM S87 phosphorylation, AURKA can also phosphorylate BIM on S87 but with lower efficiency. AURKA appears to play a less important and possibly redundant role. Activation of AURKA was recently reported to drive the evolution of resistance to third-generation EGFR inhibitors in NSCLC (Shah et al., 2019); however, AURKB activation is more obvious than AURKA activation in our models. Activation of AURKB has been reported as a non-genetic resistance mechanism to both first- and third-generation TKIs and TKI-resistant cells become sensitized to AURKB inhibitors (Bertran-Alamillo et al., 2019).
In conclusion, our HTS and cell death mechanism-based studies identified AURKB as a crucial target to prevent the critical initial adaptation upon osi treatment by maximizing BIM- and PUMA-initiated apoptosis, as well as to overcome lineage plasticity-mediated resistance that activates the ATR-CHK1-AURKB DDCR pathway. Our studies uncovered a previously unrecognized role of upstream BIM and downstream BAX/BAK in mitotic cell death triggered by ATR-CHK1-AURKB inhibitors. Our data suggest that combined inhibition of EGFR and AURKB, two distinct pathways, is more effective in eliminating tumor cells than improving the inhibition of the same signaling axis, such as combined inhibition of EGFR and MEK or combined inhibition of EGFR and PI3K/AKT/mTOR (Figures 1E and S1). We identified PF03814735 as a potent AURKB inhibitor in combination with osi. Notably, this combination strategy is also effective for osi-resistant PDX models regardless of EMT (Figures 7G–J). A phase I trial of PF03814735 reported 19 solid tumor cases achieving stable disease with a clinically manageable adverse effect profile, with toxicities non-overlapping with those of EGFR-TKIs (Schoffski et al., 2011). Clinical evaluation will be needed to determine the efficacy and tolerability of this cell death mechanism-based therapeutic strategy for EGFR-mutant lung cancer.
STAR METHODS
RESOURCE AVAILABILITY
LEAD CONTACT
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Emily H. Cheng (chenge1@mskcc.org).
MATERIALS AVAILABILITY
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
DATA AND CODE AVAILABILITY
Raw data for RNA-seq have been deposited at https://www.ncbi.nlm.nih.gov/sra/?term=PRJNA736433.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animal Models
All animal experiments were performed in accordance with the Institutional Animal Care and Use Committee at Memorial Sloan Kettering Cancer Center. Animals were allowed to acclimate for at least 5 days before initiation of the study. Female 7–8 week-old athymic nude mice (NU(NCr)-Foxn1nu, Charles River) were injected subcutaneously with 5×106 H1975 or H1975R cells in 0.2 mL 50% Matrigel (BD Biosciences). The PDX models (ECLC26, Ru813c, and Lx1114) were derived from EGFR-mutant lung adenocarcinoma patients in accordance with the MSKCC Institutional Review Board approved tissue collection protocol with informed consent from the patient and propagated in female 7–8 week-old NSG mice (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ, Jackson Laboratories). The PDX models were characterized by MSK-IMPACT assays (Zehir et al., 2017). Tumor growth was monitored twice weekly by calipers (tumor volume = length × width2 / 2). When tumors reached an average of 250.7 ± 18.8 mm3 for H1975, 217.8 ± 15.4 mm3 for H1975R, 197.0 ± 10.4 mm3 for ECLC26, 224.7 ± 6.5 mm3 for Ru813c, and 230.7 ± 12.5 mm3 for Lx1114, tumor-bearing mice were randomly assigned to receive either vehicle, osimertinib (5 mg/kg), PF03814735 (20 mg/kg), or combined osimertinib and PF03814735 (n = 6 – 12 per group). The vehicle for osimertinib is 1% polysorbate 80 and that for PF03814735 is 12.5% ethanol, 12.5% cremophor, and 75% Saline (0.9%). All agents were administered by oral gavage once daily at a weekly schedule of 5 days on and 2 days off or 4 days on and 3 days off.
Cell Line Authentication
H1975 (human lung adenocarcinoma, female origin), HCC827 (human lung adenocarcinoma, female origin), and NIH3T3 were obtained from the American Type Culture Collection (ATCC) and cultured according to the recommendations of ATCC. PC9 (human lung adenocarcinoma, male origin) was obtained from Dr. David Scheinberg at Memorial Sloan Kettering Cancer Center and cultured as described (Bean et al., 2013). ECLC26 cell line was derived from an EGFR-mutant (L858R) female lung adenocarcinoma patient in accordance with the MSKCC Institutional Review Board approved tissue collection protocol with informed consent from the patient. ECLC26 cells were cultured in advanced DMEM/F12 supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin, 1% non-essential amino acids, and 1% L-glutamine (Thermo Fisher Scientific). To generate osimertinib-resistance cells, individual cell lines were treated with increasing concentrations of osimertinib starting at 300 nM, followed by a stepwise dose escalation every 2–3 days up to 4–10 μM. The osimertinib-resistant H1975 and ECLC26 as well as their parental counterparts were characterized by MSK-IMPACT assays (Zehir et al., 2017).
METHOD DETAILS
High-Throughput Screening
HTS was performed using an automated 384-well platform as previously described (Inoue-Yamauchi et al., 2017). H1975 and HCC827 were screened against a custom library (Selleck Chemicals), encompassing inhibitors of 200 targets across more than 20 signaling pathways, small molecule modulators of epigenetics, FDA-approved drugs, and natural products, in combination with osimertinib. One thousand cells were plated per each well and treated with each library compound at 2 μM in the absence (DMSO) or presence of 2 μM osimertinib. Compounds were screened in duplicate and the growth inhibition was assessed by alamarBlue assays at 3 days post drug treatment for H1975 or at 2 days post drug treatment for HCC827. For H1975, the assay reproducibility was R2 = 0.929 and R2 = 0.912 among DMSO controls and osimertinib, respectively. For HCC827, the assay reproducibility was R2 = 0.597 and R2 = 0.894 among DMSO controls and osimertinib, respectively. The chemicals used for HTS are summarized in Table S6.
Plasmid Construction, CRISPR/Cas9-Mediated Genome Editing, and RNA Interference
Human BIM or ZEB1 was tagged with Flag-HA at the N-terminus and cloned into MSCV-Hygro (Takara Bio). The S87A mutant of BIM were generated by PCR-based site-directed mutagenesis. Human wild-type EGFR, EGFR L858R, EGFR P919T, or EGFR L858R/P919T were cloned into pBABE-Puro (Addgene). For CRISPR/Cas9-mediaed knockout, sgRNAs were designed using Optimized CRISPR Design (https://zlab.bio/guide-design-resources) and cloned into lentiCRISPRv2 (Sanjana et al., 2014). All constructs were confirmed by DNA sequencing. Lentivirus was produced by co-transfection of 293T cells with pCMVDR8.2 and pHCMV.VSVG using Lipofectamine 2000 (Thermo Fisher Scientific) as described (Wang et al., 2017). siRNA oligos (Thermo Fisher Scientific) were reversely transfected with Lipofectamine RNAiMAX (Thermo Fisher Scientific) to a final concentration of 10 nM. The sequences of sgRNAs and siRNAs were summarized in the key resources table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-BIM (22–40) | EMD Biosciences | Cat# 202000; RRID: AB_565367 |
| Rabbit polyclonal anti-BIM (22–40) | Covance | COVQ21008 |
| Rabbit monoclonal anti-PUMA (D30C10) | Cell Signaling Technology | Cat# 12450; RRID: AB_2797920 |
| Hamster monoclonal anti-BCL-2 (6C8) | BD Biosciences | Cat# 551051; RRID: AB_394018 |
| Rabbit polyclonal anti-BCL-XL | Cell Signaling Technology | Cat# 2762; RRID: AB_10694844 |
| Rabbit polyclonal anti-MCL-1 (S-19) | Santa Cruz Biotechnology | Cat# sc-819; RRID: AB_2144105 |
| Rabbit polyclonal anti-BAX | Cell Signaling Technology | Cat# 2772; RRID: AB_10695870 |
| Rabbit monoclonal anti-BAK (D4E4) | Cell Signaling Technology | Cat# 12105; RRID: AB_2716685 |
| Rabbit monoclonal anti-phospho AKT (Ser473) | Cell Signaling Technology | Cat# 4058; RRID: AB_331168 |
| Rabbit polyclonal anti-AKT | Cell Signaling Technology | Cat# 9272; RRID: AB_329827 |
| Rabbit polyclonal anti-phospho ERK1/2 | Cell Signaling Technology | Cat# 9101; RRID: AB_331646 |
| Mouse monoclonal anti-ERK1/2 (3A7) | Cell Signaling Technology | Cat# 9107; RRID: AB_10695739 |
| Rabbit polyclonal anti-phospho BIM (Ser69) | Cell Signaling Technology | Cat# 4581; RRID: AB_2065179 |
| Rabbit polyclonal anti-phospho BIM (Ser87) | Bioss Inc | Cat# bs-3012R |
| Rabbit polyclonal anti-phospho FOXO1/3 (Thr24/Thr32) | Cell Signaling Technology | Cat# 9464; RRID: AB_329842 |
| Rabbit monoclonal anti-FOXO1 (C29H4) | Cell Signaling Technology | Cat# 2880; RRID: AB_2106495 |
| Rabbit polyclonal anti-FOXO3 | Millipore | Cat# 07–702; RRID: AB_441949 |
| Mouse monoclonal anti-ZEB1 (H-3) | Santa Cruz Biotechnology | Cat# sc-515797 |
| Mouse monoclonal anti-ZEB2 (E-11) | Santa Cruz Biotechnology | Cat# sc-271984; RRID: AB_10708399 |
| Mouse monoclonal anti-E-cadherin | BD Biosciences | Cat# 610182; RRID: AB_397581 |
| Rabbit monoclonal anti-Vimentin (D21H3) | Cell Signaling Technology | Cat# 5741; RRID: AB_10695459 |
| Rabbit monoclonal anti-AURKA (D3E4Q) | Cell Signaling Technology | Cat# 14475; RRID: AB_2665504 |
| Rabbit polyclonal anti-AURKB | Cell Signaling Technology | Cat# 3094; RRID: AB_10695307 |
| Rabbit monoclonal anti-phospho AURKA/AURKB/AURKC (Thr288/Thr232/Thr398) | Cell Signaling Technology | Cat# 2914; RRID: AB_2061631 |
| Mouse monoclonal anti-Histone H3 (1B1B2) | Cell Signaling Technology | Cat# 14269; RRID: AB_2756816 |
| Rabbit polyclonal anti-phospho Histone H3 (Ser10) | Millipore | Cat# 06–570; RRID: AB_310177 |
| Rabbit polyclonal anti-ATM | Bethyl Laboratories | Cat# A300-136A; RRID: AB_155872 |
| Rabbit monoclonal anti-ATR (E1S3S) | Cell Signaling Technology | Cat# 13934; RRID: AB_2798347 |
| Mouse monoclonal anti-CHK1 | Cell Signaling Technology | Cat# 2360; RRID: AB_2080320 |
| Rabbit monoclonal anti- phospho CHK1 (Ser345) | Cell Signaling Technology | Cat# 2348; RRID: AB_331212 |
| Rabbit monoclonal anti-βTRCP (D13F10) | Cell Signaling Technology | Cat# 4394; RRID: AB_10545763 |
| Mouse monoclonal anti-β-Actin | Sigma | Cat# A1978; RRID: AB_476692 |
| Rabbit monoclonal anti-phospho EGFR (Y1068) | Cell Signaling Technology | Cat# 3777; RRID: AB_2798512 |
| Rabbit monoclonal anti-EGFR(L858R mutant specific) (43B2) | Cell Signaling Technology | Cat# 3197; RRID: AB_1903955 |
| Rabbit monoclonal anti-phospho H2A.X | Cell Signaling Technology | Cat# 9718; RRID: AB_2118009 |
| Mouse monoclonal anti-alpha-tubulin (DM1A) | Sigma | Cat# T9026; RRID: AB_477593 |
| Mouse monoclonal anti-HA (12CA5) | Kim et al., 2006 | N/A |
| Biological Samples | ||
| ECLC26 patient-derived xenograft model | This paper | N/A |
| Ru813c patient-derived xenograft model | Dr. Charles Rudin (MSKCC) | N/A |
| Lx1114 patient-derived xenograft model | Dr. Charles Rudin (MSKCC) | N/A |
| Chemicals, Peptides, and Recombinant proteins | ||
| HTS chemical library | Selleck Chemicals | Table S6 |
| Osimertinib | Selleck Chemicals | Cat# S7297 |
| PF03814735 | Selleck Chemicals | Cat# S2725 |
| Rociletinib | Selleck Chemicals | Cat# S7284 |
| MLN8054 | Selleck Chemicals | Cat# S1100 |
| MLN8237 | Selleck Chemicals | Cat# S1133 |
| AZD1152 | Selleck Chemicals | Cat# S1147 |
| CCT137690 | Selleck Chemicals | Cat# S2744 |
| MK5108 | Selleck Chemicals | Cat# S2770 |
| Hesperadin | Selleck Chemicals | Cat# S1529 |
| GSK1070916 | Selleck Chemicals | Cat# S2740 |
| GSK1838705A | Selleck Chemicals | Cat# S2703 |
| NVP-AEW541 | Selleck Chemicals | Cat# S1034 |
| Rebastinib | Selleck Chemicals | Cat# S2634 |
| Saracatinib | Selleck Chemicals | Cat# S1006 |
| GDC0941 | Selleck Chemicals | Cat# S1065 |
| MK2206 | Selleck Chemicals | Cat# S1078 |
| Rapamycin | Selleck Chemicals | Cat# S1039 |
| CDK4/6 inhibitor (Palbociclib) | Selleck Chemicals | Cat# S1116 |
| CHK1 inhibitor (LY2603618) | Selleck Chemicals | Cat# S2626 |
| ATR inhibitor (VX-970) | Selleck Chemicals | Cat# S7102 |
| Nocodazole | Selleck Chemicals | Cat# S2775 |
| Trametinib | Selleck Chemicals | Cat# S2673 |
| Emetine | EMD Millipore | Cat# 324693 |
| Recombinant human AURKB protein | Thermo Fisher Scientific | Cat# PV6130 |
| Recombinant human AURKA protein | Thermo Fisher Scientific | Cat# PV3612 |
| Critical commercial assays | ||
| CellTiter-Glo Luminescent Cell Viability Assay | Promega | Cat# G7572 |
| alamarBlue Viability Assay | Thermo Fisher Scientific | DAL1100 |
| Annexin V-FITC | BioVision | Cat# 1001–1000 |
| Annexin V-Cy3 | BioVision | Cat# 1002–1000 |
| Deposited Data | ||
| RNA-seq | This paper | https://www.ncbi.nlm.nih.gov/sra/?term=PRJNA736433 |
| Experimental Models: Cell lines | ||
| H1975: human EGFR-mutant NSCLC | ATCC | ATCC Cat# CRL-5908; RRID: CVCL_1511 |
| HCC827: human EGFR-mutant NSCLC | ATCC | ATCC Cat# CRL-2868; RRID: CVCL_2063 |
| PC9: human EGFR-mutant NSCLC | Dr. David Scheinberg (MSKCC) | RRID: CVCL_B260 |
| ECLC26: human EGFR-mutant NSCLC | This paper | N/A |
| 293T | ATCC | ATCC Cat# CRL-11268; RRID: CVCL_1926 |
| NIH3T3 | ATCC | ATCC Cat# CRL-1658; RRID: CVCL_0594 |
| Experimental Models: Organisms/Strains | ||
| Mouse: Athymic nude: NU(NCr)-Foxn1nu | Charles River Laboratories | CR: 490 |
| Mouse: NSG: NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ | The Jackson Laboratory | JAX: 005557 |
| Oligonucleotides | ||
| qRT-PCR: BIM | 5′-CCAGCACCCATGAGTTGTGACAA-3′ | |
| qRT-PCR: BIM | 5′-GCGTTAAACTCGTCTCCAATACGCC-3′ | |
| qRT-PCR: PUMA | 5′-ACGACCTCAACGCACAGTACG-3′ | |
| qRT-PCR: PUMA | 5′-GTAAGGGCAGGAGTCCCATGATG-3′ | |
| qRT-PCR: ACTB | 5′-TCCCTGGAGAAGAGCTACGAG-3′ | |
| qRT-PCR: ACTB | 5′-AGGAAGGAAGGCTGGAAGAGTG-3′ | |
| siRNA: AURKA | 5’-GAGUCUACCUAAUUCUGGAtt-3’ | Ambion Silencer Select Cat# s196 |
| siRNA: AURKA | 5’-ACAUACCAAGAGACCUACAtt-3’ | Ambion Silencer Select custom synthesis |
| siRNA: AURKB | 5’-CCUGCGUCUCUACAACUAUtt-3’ | Ambion Silencer Select Cat# s17611 |
| siRNA: AURKB | 5’-UCGUCAAGGUGGACCUAAAtt-3’ | Ambion Silencer Select Cat# s17612 |
| siRNA: BIM | 5’-CAACCACUAUCUCAGUGCAtt-3′ | Ambion Silencer Select Cat# s223065 |
| siRNA: PUMA | 5’-GCCUGUAAGAUACUGUAUAtt-3’ | Ambion Silencer Select Cat# s25840 |
| sgRNA: BAX | CAAGCGCATCGGGGACGAAC | |
| sgRNA: BAK | ACGGCAGCTCGCCATCATCG | |
| sgRNA: FOXA1 | CGCAGTAGCCGGCATGCCGG | |
| sgRNA: FOXA2 | ATGAACATGTCGTCGTACGT | |
| sgRNA: BIM | GCCCAAGAGTTGCGGCGTAT | |
| sgRNA: LacZ | TGCGAATACGCCCACGCGAT | |
| Recombinant DNA | ||
| MSCV-Hygro | Takara Bio | Cat# 634401 |
| lentiCRISPR_v2 vector | Addgene | Cat# 49535 |
| pBABE-Puro | Addgene | Cat# 1764 |
| MSCV-Hygro-N-Flag-HA-BIM | This paper | N/A |
| MSCV-Hygro-N-Flag-HA-BIM_S87A | This paper | N/A |
| MSCV-Hygro-N-Flag-HA-ZEB1 | This paper | N/A |
| pBABE-Puro-EGFR | This paper | N/A |
| pBABE-Puro-EGFR_L858R | This paper | N/A |
| pBABE-Puro-EGFR_P919T | This paper | N/A |
| pBABE-Puro-EGFR_L858R/P919T | This paper | N/A |
| Software and Algorithms | ||
| Graphpad Prism 8 | GraphPad | http://www.graphpad.com/scientific-software/prism/ |
| FlowJo | FlowJo LLC | www.flowjo.com |
| ImageJ software | ImageJ open source | http://imagej.net/Welcome |
| Trimmomatic | (Bolger et al., 2014) | http://www.usadellab.org/cms/?page=trimmomatic |
| STAR | (Dobin et al., 2013) | https://github.com/alexdobin/STAR |
| HTSeq | (Anders et al., 2015) | https://htseq.readthedocs.io/en/master/ |
| DESeq2 | (Love et al., 2014) | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| GSEA | (Subramanian et al., 2005) | http://software.broadinstitute.org/gsea/index.jsp |
| Optimized CRISPR Design | CRISPR Design Tool MIT | https://zlab.bio/guide-design-resources |
Reverse Transcription and Quantitative Real-Time PCR
Total RNA was extracted from cells or tissues using TRIzol (Thermo Fisher Scientific) according to the manufacturer’s instructions. Reverse transcription was performed with oligo-dT plus random decamer primers (Thermo Fisher Scientific) using Superscript II (Thermo Fisher Scientific). Quantitative PCR was performed with SYBR green master mix (Thermo Fisher Scientific) in duplicate using the indicated gene specific primers on a ViiA 7 Real-Time PCR System (Thermo Fisher Scientific). Data were analyzed as described previously by normalization against β-Actin. Primers for qRT-PCR are listed in the key resources table.
Cell Viability Assays
H1975, HCC827, PC9, ECLC26 cells were plated in 12-well plates at 8 × 104 or 105 cells / well and treated with the indicated agents next day. At the indicated time points, cell death was quantified by Annexin V (Bio Vision) staining, followed by flow cytometric analyses using a LSRFortessa (BD Biosciences). Data were analyzed using FACSDiva (BD biosciences). For EC50 determination, cell viability was assessed by the CellTiter-Glo luminescence assays (Promega) using 96-well plates and a luminescent plate reader (SpectraMax M2e, Molecular Devices). EC50 value was calculated using Prism software (GraphPad).
Clonogenic Assays
H1975 cells were plated in 12-well plates at 104 cells / well and treated with the indicated agents next day. Medium with fresh drugs was changed every 3–5 days. After cells were exposed to drugs for 14 days, cells in plates were fixed with methanol and then stained with crystal violet. Pictures of stained cells were taken using an EPSON Perfection V600 scanner.
Cell-Cycle Analysis
Cells were seeded in 6- or 12-well plates with 30–50% confluency and treated with the indicated agents next day. After the indicated times post-drug treatment, cells were harvested and stained with propidium iodide (25 μg/ml) for 15 minutes at room temperature and analyzed by flow cytometry using a LSRFortessa (BD Biosciences). Cell cycle subpopulations were calculated using the cell cycle module in FlowJo software.
Immunoblot Analysis
Cells were lysed in RIPA buffer and protein concentration was determined by BCA kit (Pierce). 25–40 μg of proteins were resolved by 10% or 4–12% NuPAGE gels (Thermo Fisher Scientific) and transferred onto PVDF membranes (Immobilon-P, Millipore). Antibody detection was accomplished using enhanced chemiluminescence method (Western Lightning, PerkinElmer) and LAS-3000 Imaging system (FUJIFILM). To quantify protein expression ratios, immunoblots were assessed by ImageJ software.
Co-Immunoprecipitation
The BAX−/−BAK−/− H1975 cells stably expressing N-terminal HA-tagged wild-type or S87A mutant BIM were lysed in 0.2% NP-40 isotonic buffer (0.2% NP-40, 142mM KCl, 5mM MgCl2, 1mM EGTA, 20mM HEPES, pH 7.5) supplemented with complete protease inhibitor cocktail (Roche) and subjected to anti-HA (12CA5) immunoprecipitation as described (Kim et al., 2006). Immunoprecipitates were resolved by 10% NuPAGE gels (Thermo Fisher Scientific) and analyzed by immunoblots.
In vitro Kinase Assays
The BAX−/−BAK−/− H1975 cells stably expressing HA-tagged wild-type or S87A mutant BIM were treated with 1 μM palbociclib for 24 h. Cells were harvested and then lysed in 0.2% NP-40 buffer (0.2% NP-40, 142mM KCl, 5mM MgCl2, 1mM EGTA, 20mM HEPES, pH 7.5) supplemented with complete protease inhibitor cocktail (Roche), phosphatase inhibitor cocktail 1 (Sigma), and phosphatase inhibitor cocktail 2 (Sigma). Cell lysates containing ~300 μg protein were immunoprecipitated with anti-HA antibody (12CA5) and captured by protein A agarose matrix (Piece). After washing for 3 times with 0.8 mL 0.2% NP-40 buffer and one more wash with 1 mL kinase reaction buffer (5 mM β-glycerophosphate, 2 mM DTT, 0.1 mM Na3VO4, 10 mM MgCl2, 25 mM Tris-HCl, pH 7.5), the beads were incubated with 20 μL kinase reaction buffer supplemented with 300 ng recombinant AURKB or AURKA protein (Thermo Fisher Scientific) and 200 μM ATP at 30°C for 30 minutes as described (Xie et al., 2013). Control experiments were carried out in a similar way without AURKB or AURKA protein. The reaction product was then resolved by 10% NuPAGE gels (Thermo Fisher Scientific) and analyzed by immunoblots.
RNA-seq and GSEA
Total RNA was extracted using TRIzol (Thermo Fisher Scientific) and cleaned up using RNeasy Mini Kit (Qiagen). Library preparation and sequencing were performed by the Integrated Genomics Operation Core Facility at MSKCC. After RiboGreen quantification and quality control by Agilent BioAnalyzer, 498–500ng of total RNA underwent polyA selection and cDNA synthesis according to instructions provided by Kapa Biosystems (KAPA Stranded mRNA-Seq). Library preparation and barcoding was performed using the KAPA Hyper Prep Kit (Kapa Biosystems) according to the manufacturer’s protocol with 8 cycles of PCR. Samples were sequenced on a HiSeq 2500 in High Output mode in a PE50 run, using the TruSeq SBS Kit v4 (Illumina). An average of 46 million paired reads was generated per sample. Following trimming and quality filtration with Trimmomatic (Bolger et al., 2014), reads were aligned against the hg19 version of the human genome with STAR (Dobin et al., 2013). HTSeq (Anders et al., 2015) was used to count reads overlapping with exons for each RefSeq annotated gene. Differential expression analysis was performed with DESeq2 (Love et al., 2014) and differentially expressed genes (FDR < 0.05) were subjected to Gene Set Enrichment Analysis (GSEA) (Subramanian et al., 2005).
QUANTIFICATION AND STATISTICAL ANALYSIS
When comparing two groups for cell viability, mRNA and protein expression, and tumor volume changes, statistical significance was calculated using unpaired two-tailed Student’s t-tests. Mann-Whitney U test was performed when the data was non-parametric (Figure 1C, analysis of HTS). Two-way ANOVA was used when comparing three or more groups for xenograft tumor growth (Figures 7A, 7C, 7D, 7G, and 7I). When comparing the survival data in Kaplan-Meier analysis, Log-rank test was used. The statistical tests used are indicated in the figure legends. All statistical analyses were generated using GraphPad Prism 8.4.1 software. Data were presented as mean ± s.d. with P < 0.05 considered statistically significant. Statistical significance was denoted as *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Supplementary Material
Table S1. Related toFigure 5. Summary of mutations and copy number alterations (CNA) comparing parental H1975 or ECLC26 cells with osimertinib-resistant H1975 (H1975R) or ECLC26 (ECLC26R) cells using MSK-IMPACT assays.
Table S6. Related to STAR MethodsKey Resources Table. Summary of chemicals used for HTS.
Highlights: (each bullet point is up to 85 characters – including spaces).
Aurora kinase inhibitors combined with osimertinib prevent and overcome resistance
Concurrent inhibition of EGFR and AURKB maximizes BIM- and PUMA-mediated apoptosis
EMT activates ATR-CHK1-AURKB and sensitizes cancer cells to these kinase inhibitors
Mitotic catastrophe triggered by ATR-CHK1-AURKB inhibitors involves BIM and BAX/BAK
ACKNOWLEDGEMENTS
This work was supported by the NIH grants (R01 CA125562 and R01 CA252658) and Lung Cancer Discovery Award from American Lung Association to E. Cheng. This work was also supported by the NCI Cancer Center Support Grant (P30CA008748). K. Tanaka was supported by the Japan Society for the Promotion of Science.
DECLARATION OF INTERESTS
H.A.Y. has consulted for AstraZeneca, Daiichi, Janssen, and Blueprint Medicine; she has received research funding from AstraZeneca, Daiichi, Janssen, Pfizer, Novartis, Cullinan, and Lilly. C.M.R. has consulted for AbbVie, Amgen, AstraZeneca, Epizyme, Genentech/Roche, Ipsen, Jazz, Lilly, and Syros; he serves on the scientific advisory boards of Bridge Medicines, Earli, and Harpoon Therapeutics. M.G.K. has consulted for AstraZeneca, Daiichi-Sankyo, Janssen, Novartis, Pfizer, Regeneron, and Sanofi/Genzyme. U.G. has a clinical trial agreement with AstraZeneca and received research funding from AstraZeneca, Esanex, and Aurigene; he is a current employee of Bristol Myers Squibb. J.J.H. has consulted for Eisai and BostonGene; he has received clinical trial funding from Bristol Myers Squibb, Merck, AstraZeneca, Exelixis, Calithera, and SillaJen; he has received research funding from Merck, BostonGene, and TScan.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Anders S, Pyl PT, and Huber W (2015). HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bean GR, Ganesan YT, Dong Y, Takeda S, Liu H, Chan PM, Huang Y, Chodosh LA, Zambetti GP, Hsieh JJ, and Cheng EH (2013). PUMA and BIM are required for oncogene inactivation-induced apoptosis. Sci Signal 6, ra20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertran-Alamillo J, Cattan V, Schoumacher M, Codony-Servat J, Gimenez-Capitan A, Cantero F, Burbridge M, Rodriguez S, Teixido C, Roman R, et al. (2019). AURKB as a target in non-small cell lung cancer with acquired resistance to anti-EGFR therapy. Nat Commun 10, 1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boumahdi S, and de Sauvage FJ (2019). The great escape: tumour cell plasticity in resistance to targeted therapy. Nature Reviews Drug Discovery, 1–18. [DOI] [PubMed] [Google Scholar]
- Camidge DR, Pao W, and Sequist LV (2014). Acquired resistance to TKIs in solid tumours: learning from lung cancer. Nature reviews Clinical oncology 11, 473. [DOI] [PubMed] [Google Scholar]
- Carmena M, Ruchaud S, and Earnshaw WC (2009). Making the Auroras glow: regulation of Aurora A and B kinase function by interacting proteins. Current opinion in cell biology 21, 796–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HC, Kanai M, Inoue-Yamauchi A, Tu HC, Huang Y, Ren D, Kim H, Takeda S, Reyna DE, Chan PM, et al. (2015). An interconnected hierarchical model of cell death regulation by the BCL-2 family. Nature cell biology 17, 1270–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou TC (2010). Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res 70, 440–446. [DOI] [PubMed] [Google Scholar]
- Costa C, Molina MA, Drozdowskyj A, Gimenez-Capitan A, Bertran-Alamillo J, Karachaliou N, Gervais R, Massuti B, Wei J, Moran T, et al. (2014). The impact of EGFR T790M mutations and BIM mRNA expression on outcome in patients with EGFR-mutant NSCLC treated with erlotinib or chemotherapy in the randomized phase III EURTAC trial. Clinical cancer research : an official journal of the American Association for Cancer Research 20, 2001–2010. [DOI] [PubMed] [Google Scholar]
- Costa DB, Halmos B, Kumar A, Schumer ST, Huberman MS, Boggon TJ, Tenen DG, and Kobayashi S (2007). BIM mediates EGFR tyrosine kinase inhibitor-induced apoptosis in lung cancers with oncogenic EGFR mutations. PLoS Med 4, 1669–1679; discussion 1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cragg MS, Kuroda J, Puthalakath H, Huang DC, and Strasser A (2007). Gefitinib-induced killing of NSCLC cell lines expressing mutant EGFR requires BIM and can be enhanced by BH3 mimetics. PLoS Med 4, 1681–1689; discussion 1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross DA, Ashton SE, Ghiorghiu S, Eberlein C, Nebhan CA, Spitzler PJ, Orme JP, Finlay MR, Ward RA, Mellor MJ, et al. (2014). AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov 4, 1046–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czabotar PE, Lessene G, Strasser A, and Adams JM (2014). Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nature reviews Molecular cell biology 15, 49–63. [DOI] [PubMed] [Google Scholar]
- Dehan E, Bassermann F, Guardavaccaro D, Vasiliver-Shamis G, Cohen M, Lowes KN, Dustin M, Huang DC, Taunton J, and Pagano M (2009). betaTrCP- and Rsk1/2-mediated degradation of BimEL inhibits apoptosis. Mol Cell 33, 109–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber AC, Corcoran RB, Ebi H, Sequist LV, Waltman BA, Chung E, Incio J, Digumarthy SR, Pollack SF, Song Y, et al. (2011). BIM expression in treatment-naive cancers predicts responsiveness to kinase inhibitors. Cancer Discov 1, 352–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garraway LA, and Jänne PA (2012). Circumventing cancer drug resistance in the era of personalized medicine. Cancer discovery 2, 214–226. [DOI] [PubMed] [Google Scholar]
- Gong Y, Somwar R, Politi K, Balak M, Chmielecki J, Jiang X, and Pao W (2007). Induction of BIM is essential for apoptosis triggered by EGFR kinase inhibitors in mutant EGFR-dependent lung adenocarcinomas. PLoS Med 4, e294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hata AN, Engelman JA, and Faber AC (2015). The BCL2 Family: Key Mediators of the Apoptotic Response to Targeted Anticancer Therapeutics. Cancer Discov 5, 475–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hata AN, Niederst MJ, Archibald HL, Gomez-Caraballo M, Siddiqui FM, Mulvey HE, Maruvka YE, Ji F, Bhang HE, Krishnamurthy Radhakrishna V, et al. (2016). Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat Med 22, 262–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue-Yamauchi A, Jeng PS, Kim K, Chen H-C, Han S, Ganesan YT, Ishizawa K, Jebiwott S, Dong Y, and Pietanza MC (2017). Targeting the differential addiction to antiapoptotic BCL-2 family for cancer therapy. Nature Communications 8, ncomms16078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeng PS, Inoue-Yamauchi A, Hsieh JJ, and Cheng EH (2018). BH3-Dependent and Independent Activation of BAX and BAK in Mitochondrial Apoptosis. Current Opinion in Physiology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Rafiuddin-Shah M, Tu HC, Jeffers JR, Zambetti GP, Hsieh JJ, and Cheng EH (2006). Hierarchical regulation of mitochondrion-dependent apoptosis by BCL-2 subfamilies. Nature cell biology 8, 1348–1358. [DOI] [PubMed] [Google Scholar]
- Kurppa KJ, Liu Y, To C, Zhang T, Fan M, Vajdi A, Knelson EH, Xie Y, Lim K, and Cejas P (2020). Treatment-induced tumor dormancy through YAP-mediated transcriptional reprogramming of the apoptotic pathway. Cancer Cell 37, 104–122. e112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonetti A, Sharma S, Minari R, Perego P, Giovannetti E, and Tiseo M (2019). Resistance mechanisms to osimertinib in EGFR-mutated non-small cell lung cancer. Br J Cancer 121, 725–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luciano F, Jacquel A, Colosetti P, Herrant M, Cagnol S, Pages G, and Auberger P (2003). Phosphorylation of Bim-EL by Erk1/2 on serine 69 promotes its degradation via the proteasome pathway and regulates its proapoptotic function. Oncogene 22, 6785–6793. [DOI] [PubMed] [Google Scholar]
- Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, et al. (2004). Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. The New England journal of medicine 350, 2129–2139. [DOI] [PubMed] [Google Scholar]
- Ma CX, Janetka JW, and Piwnica-Worms H (2011). Death by releasing the breaks: CHK1 inhibitors as cancer therapeutics. Trends in molecular medicine 17, 88–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay DR, and Ullman KS (2015). ATR and a Chk1-Aurora B pathway coordinate postmitotic genome surveillance with cytokinetic abscission. Molecular biology of the cell 26, 2217–2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortlock AA, Foote KM, Heron NM, Jung FH, Pasquet G, Lohmann J-JM, Warin N, Renaud F, De Savi C, and Roberts NJ (2007). Discovery, synthesis, and in vivo activity of a new class of pyrazoloquinazolines as selective inhibitors of aurora B kinase. Journal of medicinal chemistry 50, 2213–2224. [DOI] [PubMed] [Google Scholar]
- Niederst MJ, Sequist LV, Poirier JT, Mermel CH, Lockerman EL, Garcia AR, Katayama R, Costa C, Ross KN, and Moran T (2015). RB loss in resistant EGFR mutant lung adenocarcinomas that transform to small-cell lung cancer. Nature communications 6, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oser MG, Fonseca R, Chakraborty AA, Brough R, Spektor A, Jennings RB, Flaifel A, Novak JS, Gulati A, Buss E, et al. (2019). Cells Lacking the RB1 Tumor Suppressor Gene Are Hyperdependent on Aurora B Kinase for Survival. Cancer Discov 9, 230–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, et al. (2004). EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 304, 1497–1500. [DOI] [PubMed] [Google Scholar]
- Petsalaki E, Akoumianaki T, Black EJ, Gillespie DA, and Zachos G (2011). Phosphorylation at serine 331 is required for Aurora B activation. Journal of Cell Biology 195, 449–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilié PG, Tang C, Mills GB, and Yap TA (2019). State-of-the-art strategies for targeting the DNA damage response in cancer. Nature Reviews Clinical Oncology 16, 81–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintanal-Villalonga Á, Chan JM, Yu HA, Pe’er D, Sawyers CL, Sen T, and Rudin CM (2020). Lineage plasticity in cancer: a shared pathway of therapeutic resistance. Nature reviews Clinical oncology 17, 360–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramalingam SS, Vansteenkiste J, Planchard D, Cho BC, Gray JE, Ohe Y, Zhou C, Reungwetwattana T, Cheng Y, Chewaskulyong B, et al. (2020). Overall Survival with Osimertinib in Untreated, EGFR-Mutated Advanced NSCLC. The New England journal of medicine 382, 41–50. [DOI] [PubMed] [Google Scholar]
- Ren D, Tu HC, Kim H, Wang GX, Bean GR, Takeuchi O, Jeffers JR, Zambetti GP, Hsieh JJ, and Cheng EH (2010). BID, BIM, and PUMA are essential for activation of the BAX- and BAK-dependent cell death program. Science 330, 1390–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roper N, Brown A-L, Wei JS, Pack S, Trindade C, Kim C, Restifo O, Gao S, Sindiri S, and Mehrabadi F (2020). Clonal evolution and heterogeneity of osimertinib acquired resistance mechanisms in EGFR mutant lung cancer. Cell reports medicine 1, 100007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotow J, and Bivona TG (2017). Understanding and targeting resistance mechanisms in NSCLC. Nature Reviews Cancer 17, 637. [DOI] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nature methods 11, 783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoffski P, Jones SF, Dumez H, Infante JR, Van Mieghem E, Fowst C, Gerletti P, Xu H, Jakubczak JL, English PA, et al. (2011). Phase I, open-label, multicentre, dose-escalation, pharmacokinetic and pharmacodynamic trial of the oral aurora kinase inhibitor PF-03814735 in advanced solid tumours. Eur J Cancer 47, 2256–2264. [DOI] [PubMed] [Google Scholar]
- Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P, Bergethon K, Shaw AT, Gettinger S, and Cosper AK (2011). Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Science translational medicine 3, 75ra26–75ra26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah KN, Bhatt R, Rotow J, Rohrberg J, Olivas V, Wang VE, Hemmati G, Martins MM, Maynard A, Kuhn J, et al. (2019). Aurora kinase A drives the evolution of resistance to third-generation EGFR inhibitors in lung cancer. Nat Med 25, 111–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S, McDermott U, Azizian N, Zou L, Fischbach MA, et al. (2010). A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 141, 69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibue T, and Weinberg RA (2017). EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nature reviews Clinical oncology 14, 611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimomura T, Hasako S, Nakatsuru Y, Mita T, Ichikawa K, Kodera T, Sakai T, Nambu T, Miyamoto M, and Takahashi I (2010). MK-5108, a highly selective Aurora-A kinase inhibitor, shows antitumor activity alone and in combination with docetaxel. Molecular cancer therapeutics 9, 157–166. [DOI] [PubMed] [Google Scholar]
- Soria JC, Ohe Y, Vansteenkiste J, Reungwetwattana T, Chewaskulyong B, Lee KH, Dechaphunkul A, Imamura F, Nogami N, Kurata T, et al. (2018). Osimertinib in Untreated EGFR-Mutated Advanced Non-Small-Cell Lung Cancer. The New England journal of medicine 378, 113–125. [DOI] [PubMed] [Google Scholar]
- Stemmler MP, Eccles RL, Brabletz S, and Brabletz T (2019). Non-redundant functions of EMT transcription factors. Nature cell biology 21, 102–112. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thress KS, Paweletz CP, Felip E, Cho BC, Stetson D, Dougherty B, Lai Z, Markovets A, Vivancos A, Kuang Y, et al. (2015). Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat Med 21, 560–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumbrink HL, Heimsoeth A, and Sos ML (2021). The next tier of EGFR resistance mutations in lung cancer. Oncogene 40, 1–11. [DOI] [PubMed] [Google Scholar]
- Walter AO, Sjin RT, Haringsma HJ, Ohashi K, Sun J, Lee K, Dubrovskiy A, Labenski M, Zhu Z, Wang Z, et al. (2013). Discovery of a mutant-selective covalent inhibitor of EGFR that overcomes T790M-mediated resistance in NSCLC. Cancer Discov 3, 1404–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan H, Dingle S, Xu Y, Besnard V, Kaestner KH, Ang S-L, Wert S, Stahlman MT, and Whitsett JA (2005). Compensatory roles of Foxa1 and Foxa2 during lung morphogenesis. Journal of Biological Chemistry 280, 13809–13816. [DOI] [PubMed] [Google Scholar]
- Wang GX, Tu H-C, Dong Y, Skanderup AJ, Wang Y, Takeda S, Ganesan YT, Han S, Liu H, and Hsieh JJ (2017). ΔNp63 inhibits oxidative stress-induced cell death, including ferroptosis, and cooperates with the BCL-2 family to promote clonogenic survival. Cell reports 21, 2926–2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein IB (2002). Addiction to oncogenes--the Achilles heal of cancer. Science 297, 63–64. [DOI] [PubMed] [Google Scholar]
- Witkiewicz AK, Chung S, Brough R, Vail P, Franco J, Lord CJ, and Knudsen ES (2018). Targeting the vulnerability of RB tumor suppressor loss in triple-negative breast cancer. Cell reports 22, 1185–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie H, Lee M-H, Zhu F, Reddy K, Peng C, Li Y, Kim DJ, Li X, Kang S, and Li H (2013). Identification of an Aurora kinase inhibitor specific for the Aurora B isoform. Cancer research 73, 716–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao JE, Yan M, Guan Z, Pan CB, Xia LP, Li CX, Wang LH, Long ZJ, Zhao Y, Li MW, et al. (2009). Aurora-A down-regulates IkappaBalpha via Akt activation and interacts with insulin-like growth factor-1 induced phosphatidylinositol 3-kinase pathway for cancer cell survival. Mol Cancer 8, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu HA, Arcila ME, Rekhtman N, Sima CS, Zakowski MF, Pao W, Kris MG, Miller VA, Ladanyi M, and Riely GJ (2013). Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clinical cancer research : an official journal of the American Association for Cancer Research 19, 2240–2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, Srinivasan P, Gao J, Chakravarty D, Devlin SM, et al. (2017). Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med 23, 703–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Maity TK, Ross KE, Qi Y, Cultraro CM, Bahta M, Pitts S, Keswani M, Gao S, Nguyen KDP, et al. (2021). Alterations in the Global Proteome and Phosphoproteome in Third Generation EGFR TKI Resistance Reveal Drug Targets to Circumvent Resistance. Cancer Res 81, 3051–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Related toFigure 5. Summary of mutations and copy number alterations (CNA) comparing parental H1975 or ECLC26 cells with osimertinib-resistant H1975 (H1975R) or ECLC26 (ECLC26R) cells using MSK-IMPACT assays.
Table S6. Related to STAR MethodsKey Resources Table. Summary of chemicals used for HTS.
Data Availability Statement
Raw data for RNA-seq have been deposited at https://www.ncbi.nlm.nih.gov/sra/?term=PRJNA736433.