

Abstract
Background
Platelets play a key role in hemostasis through plug formation and secretion of their granule contents at sites of endothelial injury. Defects in von Willebrand factor (VWF), a platelet α‐granule protein, are implicated in von Willebrand disease (VWD), and may lead to defective platelet adhesion and/or aggregation. Studying VWF quantity and subcellular localization may help us better understand the pathophysiology of VWD.
Objective
Quantitative analysis of the platelet α‐granule compartment and VWF storage in healthy individuals and VWD patients.
Patients/Methods
Structured illumination microscopy (SIM) was used to study VWF content and organization in platelets of healthy individuals and patients with VWD in combination with established techniques.
Results
SIM capably quantified clear morphological and granular changes in platelets stimulated with proteinase‐activated receptor 1 (PAR‐1) activating peptide and revealed a large intra‐ and interdonor variability in VWF‐positive object numbers within healthy resting platelets, similar to variation in secreted protein acidic and rich in cysteine (SPARC). We subsequently characterized VWD platelets to identify changes in the α‐granule compartment of patients with different VWF defects, and were able to stratify two patients with type 3 VWD rising from different pathological mechanisms. We further analyzed VWF storage in α‐granules of a patient with homozygous p.C1190R using electron microscopy and found discrepant VWF levels and different degrees of multimerization in platelets of patients with heterozygous p.C1190 in comparison to VWF in plasma.
Conclusions
Our findings highlight the utility of quantitative imaging approaches in assessing platelet granule content, which may help to better understand VWF storage in α‐granules and to gain new insights in the etiology of VWD.
Keywords: blood platelets, optical imaging, type 3, von Willebrand disease, von Willebrand factor
Essentials.
Characterizing platelet granule content is essential to better understand platelet disorders.
Structured illumination microscopy (SIM) allows quantitative study of platelet cargo like von Willebrand factor (VWF).
SIM identifies different VWF alterations in platelets of patients with type 3 von Willebrand disease .
Electron microscopy and multimer analysis implicates defective VWF storage in patients with p.C1190.
1. INTRODUCTION
Platelets are circulating, anucleate cells that play a key role in the hemostatic system. Platelets bud off from megakaryocytes into the circulation and contain several different types of granules.1 The most abundant organelle in platelets is the α‐granule, which contains many important hemostatic and inflammatory proteins such as von Willebrand factor (VWF), fibrinogen, and platelet factor 4.1 Other proteins such as secreted protein acidic and rich in cysteine (SPARC; or osteonectin) have less well‐defined roles in hemostasis but are nevertheless abundant in platelet α‐granules.2, 3, 4, 5
Upon tissue injury, platelets adhere to damaged endothelium through extracellular matrix collagen and VWF via various glycoprotein receptors on the platelet membrane, followed by secretion of their granular contents into the thrombus environment.6 This further enhances the hemostatic response through release of adhesion proteins (like VWF and fibrinogen) and potent signaling molecules, that is, thrombin, ADP, and thromboxane A2.1, 7 Regulation of platelet granule biogenesis and release is essential for normal hemostasis, as defects in these pathways can lead to bleeding problems.6, 8 Mechanisms that control these biological processes have been gradually elucidated, yet routine techniques often fail to identify the underlying defects in patients.
With the advent of super‐resolution light microscopy techniques, we are able to image platelets with sufficient resolution to discern individual granule cargo while obtaining high‐throughput quantitative light microscopy data.9 Structured illumination microscopy (SIM) is a relatively quick and easy‐to‐use super‐resolution method, which makes it suitable to image platelets of a large number of healthy individuals and patients. A previous study has successfully used SIM‐based quantification of dense‐granule content to identify Hermansky‐Pudlak syndrome patients.10 However, quantitative studies of platelet α‐granule content and subcellular localization are lacking and will be essential to better understand bleeding disorders where platelet α‐granules or their content are affected.10, 11
Von Willebrand disease (VWD) is the most common inherited bleeding disorder, hallmarked by defects of VWF that may consequently lead to defective platelet adhesion and/or aggregation.12, 13 VWD is classified in subtypes based on either a quantitative (type 1 or 3) or qualitative (type 2) defect of VWF, determined through diagnostic assays that measure circulating levels and function.14 However, mutations can also cause intracellular defects that lead to inadequate synthesis, secretion, or multimerization.12 Patients with the same type of VWD and similar circulating VWF levels can have a very different bleeding phenotype,13 suggesting VWF in cellular compartments like platelet α‐granules may play a role in the pathogenesis of VWD.
To study VWF in α‐granules, we have developed an analysis tool based on SIM imaging of platelets of healthy individuals and people with VWD. Large data sets of immunofluorescence‐based data were processed simultaneously through an analysis workflow that facilitates quantitative study of platelet α‐granule content. We quantified morphological shape change and α‐granule content release in proteinase‐activated receptor 1 (PAR‐1) stimulated platelets and assessed the variability in α‐granule constituents across healthy individuals. We then focused on patients with VWD with severe quantitative defects (type 3) and qualitative multimerization defects (type 2A) in VWF and assessed changes in their platelet compartment. We generated quantitative platelet α‐granule cargo data in conjunction with electron microscopy (EM) and VWF multimer analysis, showing the viability of a SIM‐based approach in the characterization of the platelet α‐granule compartment and VWF storage in α‐granules in VWD.
2. METHODS
A full description of flow cytometry, VWF multimer analysis, and EM are available in the Supporting Information (Methods).
2.1. Patients and healthy donors
Peripheral blood was obtained from consenting healthy donors and patients with VWD. Patients were included at the Department of Hematology in the Erasmus University Medical Center, Rotterdam, via the Prospective Von Willebrand Disease in the Netherlands Study (WiN‐Pro). This is an ongoing nationwide prospective‐ observational cohort study (NCT03521583), performed according to the Declaration of Helsinki, and approved by the institutional ethics board. Clinical data shown here was collected as part of the WiN‐Pro study protocol or obtained retrospectively from patient files (Table 1).
TABLE 1.
Characterization of patients with VWD
| Patient | Family | Age | Sex | Blood group |
VWD type |
VWF:Aga [0.60‐1.40] |
VWF:Aba [0.60‐1.40] |
VWF:CBa [0.60‐1.40] |
FVIII:Ca [0.60‐1.40] |
VWFpp |
Platelet countb [150‐400] |
BS | Mutation | DNA |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | … | 33 | M | O | 3 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 302 | 34 | Δexon 4‐5 +p.L2306R fs*4 compound het | Del exon4‐5 +c.6917delT, exon 40 |
| 2 | 2 + 3 + 4 | 14 | F | B | 3 | 0.05 | 0.04 | … | 0.04 | 0.00 | 328 | 20 | p.C1190R hom | c.3568T>C, exon 27 |
| 3 | 2 + 3 + 4 | 18 | M | A | 2A | 0.29 | 0.12 | … | 0.52 | 0.39 | 294 | 4 | p.C1190R het | c.3568T>C, exon 27 |
| 4 | 2 + 3 + 4 | 43 | F | O | 2A | 0.24 | 0.05 | 0.10 | 0.42 | 0.45 | 261 | 3 | p.C1190R het | c.3568T>C, exon 27 |
| 5 | … | 57 | F | O | 2A | 0.84 | 0.25 | 0.23 | 0.57 | 0.89 | 316 | 10 | p.C1190Y het | c.3569G>A, exon 27 |
Clinical and laboratory data is shown for all five patients with VWD included in this study.
Reference values are shown in brackets.
Abbreviations: BS, bleeding score as determined by the ISTH Bleeding Assessment Tool53; FVIII:C, factor VIII activity (IU/mL); het, heterozygous, hom, homozygous; VWF:Ab, von Willebrand factor activity (IU/mL); VWF:Ag, von Willebrand factor antigen (IU/mL); VWF:CB, von Willebrand factor collagen binding (IU/mL); VWFpp, von Willebrand factor propeptide levels (IU/mL).
Historically lowest levels.
Average over last four most‐recent measurements (*109/L).
2.2. Platelet preparation
Whole blood was spun for 20 minutes at 120 g without brake to generate platelet‐rich plasma (PRP). Any subsequent centrifugation steps to wash platelet suspensions were performed at 2000 g for 8 minutes as detailed below. PRP was immediately fixed in 1% formaldehyde for 5 minutes, then washed three times in washing buffer (36 mM citric acid, 103 mM NaCl, 5 mM KCl, 5 mM EDTA, 5.6 mM glucose, pH 6.5). For activation experiments, PRP was first incubated with either phosphate buffered saline (PBS) or 10 µM PAR‐1 AP (Peptides International, Louisville, KY, USA; cat# PAR‐3676‐PI) for 30 minutes at 37℃, without stirring or agitation.
2.3. Immunofluorescent staining
All incubations were done at room temperature unless otherwise stated. 9 mm2 1.5H high‐precision coverslips (Paul Marienfeld, Lauda‐Königshofen, Germany; cat#OTMS20B) were coated with poly‐l‐lysine (Sigma‐Aldrich, St. Louis, MO, USA; cat# P0899; 100 µg/mL in PBS) and seeded with 3 million platelets per sample. Platelets were permeabilized in Perm/Quench (50 mM NH4Cl, 0.1% saponin), washed three times in PGAS (PBS with 0.2% gelatin, 0.02% azide, and 0.02% saponin), and finally stored in PGAS at 4℃ until immunostaining.15 Samples were stained and imaged within 1 week. Coverslips containing platelets were probed with antibodies (Table S1) and washed three times with PGAS in between steps. Initial staining setups were controlled using primary or secondary antibodies only and did not yield any immunopositivity. Finally, slides were dipped in PBS, mounted in mowiol (Sigma‐Aldrich; cat# 214590), and sealed with nail polish before imaging.
2.4. Structured illumination and confocal microscopy
SIM images were acquired on an Elyra PS1 (Zeiss, Jena, Germany) microscope through a 63× plan‐apochromat DIC 1.4 NA lens, using five phases and five rotations of the illumination pattern. Diode lasers (100 mW) with a wavelength of 488, 561, and 633 nm were used with an appropriate emission filter (BP 420–480 +LP750, BP 495–575 +LP 750, LP 655, respectively) in the light path. Laser power and gain were optimized per sample to ensure reconstruction quality. At least three representative fields of view with full Z‐stacks were collected per donor, using an interval of 110 nm between slices (≈50 slices per Z‐stack, or 5.5 µm). After image acquisition, samples were reconstructed with state‐of‐the‐art SIM reconstruction using Zen Software (Zeiss). From the same sections, confocal images were taken at standardized laser settings. Multiple Z‐stacks were taken per sample on a SP5 confocal microscope (Leica, Wetzlar, Germany).
2.5. Image analysis
For analysis of reconstructed SIM data, we applied a semi‐automated analysis method using Fiji software16 in combination with published plugins as well as in‐house written macro code (Figure S1). Quality of reconstruction was assessed with the SIM‐check plugin17 while lateral resolution was calculated to be ≈120 nm using Fourier ring correlation18 (Figure S2).
In the analysis, single platelets were first segmented on the basis of α‐tubulin staining in a two‐dimensional average intensity projection and thresholded on the basis of the Li algorithm,19 excluding platelets that were not completely in the field of view. Baseline correction was performed by subtracting a fixed value (determined by reconstruction software) from the whole image. Background correction for granular staining was then performed for all images individually by normalizing the whole image to the highest background level in the stack (determined by finding an area of background in the slice with the lowest average signal, usually in the middle of a stack).
Next, we segmented unique immunopositive structures, termed objects, per granular channel in full three‐dimensional (3D) voxel space using a 3D maxima finder and 3D watershed20 and applied Moments thresholding.21 Noise parameters were determined by selecting the best fit (highest derivative point of a plot of threshold parameter vs number of objects) to filter out any quantification of residual noise. Three‐dimensional morphometrics were calculated from the segmented immunopositive objects with relative volume sizes represented in voxels.20
We also quantified unique immunopositive objects in 2D space on an average‐intensity projection of each granular channel to assess spatial distribution. Similarly, we used a 2D maxima finder and determined noise parameters through best fit (highest derivative point of a plot of threshold parameter vs number of objects). For each unique 2D object we then calculated minimal distances to the nearest object using the in‐house Nearest Neighbour plugin, or minimal distances to the α‐tubulin mask (Nearest Edge) (code available at https://github.com/ErasmusOIC/NearestNeighbour). Finally, we assessed colocalization of separate granular stains using an object‐based method by calculating minimal distances of unique objects in channel 1 (eg, VWF) to the nearest object in channel 2 (eg, SPARC).22, 23, 24, 25
Analysis and quantification of unique objects in confocal images was largely similar but adapted to a 2D approach using maximum intensity projections for all channels. In brief, segmentation of single platelets using α‐tubulin was performed using the Otsu thresholding algorithm.26 Spatial distributions were calculated as described for reconstructed SIM images. Confocal mean fluorescence intensity (MFI) measurements were performed on an average of three sequential slices.
A detailed description of all parameters measured per platelet and per sample is presented in Table S2.
2.6. Statistical analysis
Continuous variables are presented as mean ± standard deviation, whereas categorical variables are presented as number and proportion. To compare two or more groups, we used nested t test or analysis of variance using a mixed‐effects model (Prism version 8; GraphPad Software, La Jolla, CA, USA). Correlation was performed using Spearman’s ranked correlation. Poisson regression was used in regression analysis. A P value <.05 was considered significant.
3. RESULTS
3.1. Quantitative imaging of alpha‐granule content by super‐resolution microscopy
To quantitatively study platelets and their α‐granule cargo, we used SIM imaging and compared it to standard confocal imaging (Figure 1A). For the analysis, we developed a semi‐automated workflow in Fiji software to annotate and quantify unique intracellular structures (termed objects) in full 3D voxel space from reconstructed SIM images (Table S2). In this 3D‐SIM workflow, of which one field of view is shown (Figure 1B), platelets were stained for α‐tubulin to depict their unique marginal band (Figure 1C), while VWF and SPARC were immunolabeled to study the α‐granule compartment (Figure 1D,E). We found that SPARC stained more diffusely, while VWF staining was concentrated into discrete volumes (Figure 1D). Additionally, we detected VWF‐positive volumes predominantly in close proximity to SPARC‐positive staining or in an outline of SPARC‐positive objects through the Z‐axis (Figure 1E). This was also evident from a 3D perspective (Video S1). Unique objects were quantified per granular staining and per platelet (Figure 1C,D), and we found that the number of VWF‐ and SPARC‐positive structures per platelet was slightly different but correlated closely (Figure 1D,F). Our quantification of unique objects is two‐ to fourfold lower than α‐granule numbers estimated from historical EM stereology approaches.27, 28, 29, 30 The total cargo volume, which is independent of optimal segmentation of unique structures, showed a slightly larger discrepancy between VWF and SPARC (Figure 1F). These staining and volume differences are likely due to the reported eccentric localization of VWF within the α‐granule, as opposed to most other α‐granule proteins like SPARC.29, 30, 31 As SPARC occupies a larger area inside α‐granules, this explains the larger volume measurements in comparison with VWF. Some studies have previously argued that VWF and other proteins may be stored in subpopulations of α‐granules,32, 33 potentially representing subpopulations with distinct functions. However, recent EM work of whole platelets has shown that VWF is present in every α‐granule, but due to its eccentric localization this becomes only apparent in 3D‐EM reconstructions29 (Figure 1E). Morphological SIM analysis of VWF and SPARC staining with fibrinogen or P‐selectin yields a similar pattern (Figure S3).
FIGURE 1.
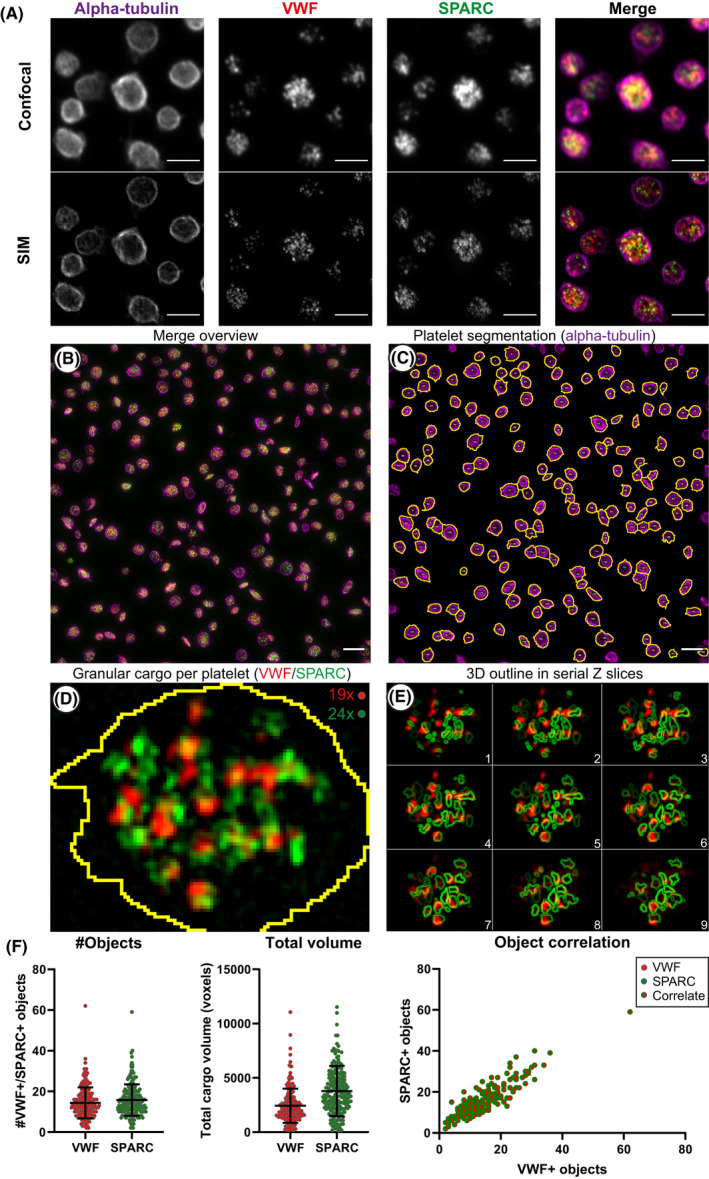
Differences between confocal and structured illumination microscopy (SIM) imaging, and analysis workflow of platelet and granule morphometrics. Resolution differences between confocal and SIM are illustrated in resting platelets. (A). Shown are the four main steps (B‐D) for ImageJ‐based processing of super‐resolution data. One field of view is shown (B). First, platelets are segmented through automated thresholding on alpha‐tubulin staining, shown in magenta, segmentation line in yellow (C). Each individual platelet (n = 205) is then assessed for granular staining using von Willebrand factor (VWF) (D; shown in red) and secreted protein acidic and rich in cysteine (SPARC) (D; shown in green). Overlap between VWF (object) and SPARC (outline) is shown in serial slices in (E). VWF‐ or SPARC‐positive volumes of fluorescence are then separated in 3D through 3D‐based plugins, counted and processed for morphometry (F). Scale bar is 3 µm in panel (A), 5 µm in panel (B‐C); both show an average intensity projection. One slice is shown for panel (D). Brightness and contrast were adjusted to enhance visibility of the design. Quantification of unique VWF‐positive (red) and SPARC‐positive (green) objects their total volume, and correlation per platelet is illustrated in (F) for one image. Data are represented as mean ± standard deviation
In a quantitative confocal microscopy‐based analysis of the same data set, we detected less VWF‐positive objects (SIM, 14.4; confocal, 8.1) and less SPARC‐positive objects (SIM, 15.8; confocal, 8.4) (Figure 4A). In similar fashion, we observed an overestimation of the total volume by 2.5× in confocal images (Figure 4B) indicating that SIM is more capable to segregate individual objects as well as to better estimate the volume of granule constituents.
FIGURE 4.
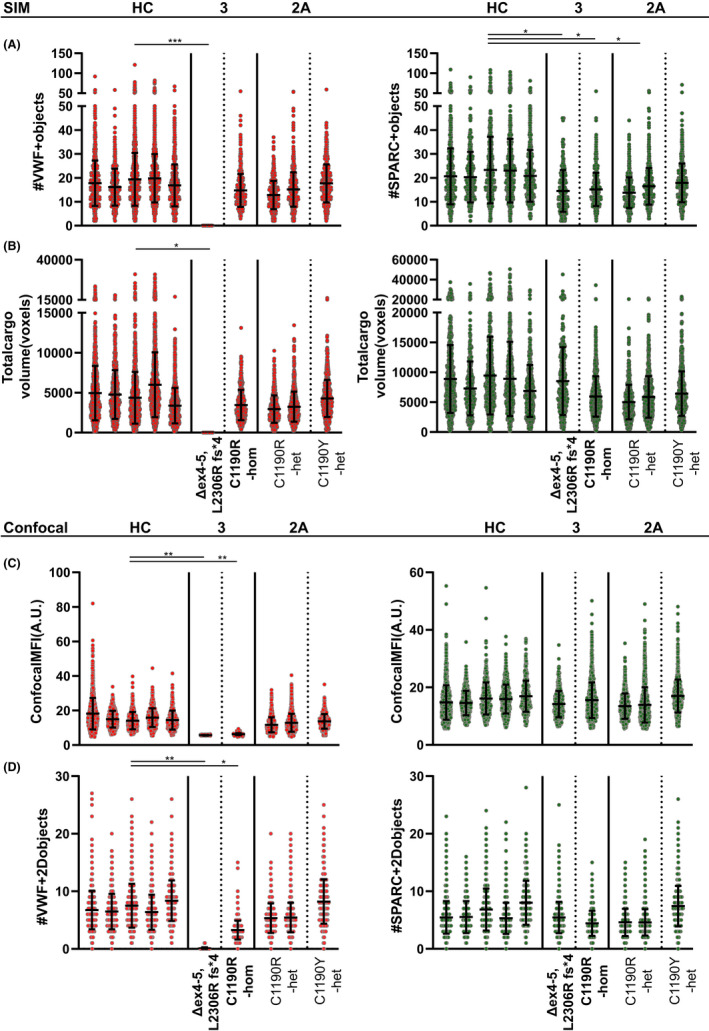
(Previous page) Quantification of α‐granule content of von Willebrand disease (VWD) platelets using super‐resolution and confocal microscopy. Von Willebrand factor (red) and secreted protein acidic and rich in cysteine (SPARC; green) α‐granule cargo was quantified for all patients through 3D structured illumination microscopy (A‐B) and 2D confocal (C‐D) workflows and compared to healthy control platelets (n = 5310 platelets over five donors). Patients with type 3 VWD are marked in bold text (Δex4‐5, L2306R fs*4; n = 290; homozygous C1190R‐hom, n = 578 platelets), while patients with type 2A are nonbold (C1190R‐het (n = 535/921) and C1190Y‐het (n = 749)). Object numbers (A/D) were assessed through both techniques while total volume (B) and mean fluorescence intensity (C) were exclusively measured through SIM and confocal microscopy, respectively. Data are represented as mean ± standard deviation. *P < .05, **P < .01, ***P < .001
3.2. Platelet stimulation by PAR‐1 activating peptide triggers VWF+granule release, marginal band compression, and α‐granule content reorganization
Next, we tested whether our analysis was able to identify clear changes in platelet morphology and α‐granule content after platelet activation in vitro. Activation of platelets with PAR‐1 activating peptide (PAR‐1‐AP), a strong agonist of the PAR‐1 receptor, led to a reduction of VWF‐positive objects (rest 14.04 vs PAR‐1‐AP 7.43, P = .03) that signified α‐granule release (Figure 2A,B). Degranulation was confirmed through assessment of P‐selectin exposure by flow cytometry, which also showed no signs of platelet aggregation (Figure S5). Confocal 2D analysis found only a trend toward lower levels (Figure S6A,B), which is most likely due to the lower dynamic range in which we can detect granular structures in confocal microscopy. Additionally, we were able to quantify compression of the marginal band area in activated platelets (rest 7.21 µm2 vs PAR‐1‐AP 5.69 µm2, P = .03) (Figure 2C), which is thought to play a role in α‐granule exocytosis.34 Finally, we detected a decrease in total VWF‐positive volume in stimulated platelets, albeit not significant due to a high variation across experiments (Figure 2D).
FIGURE 2.
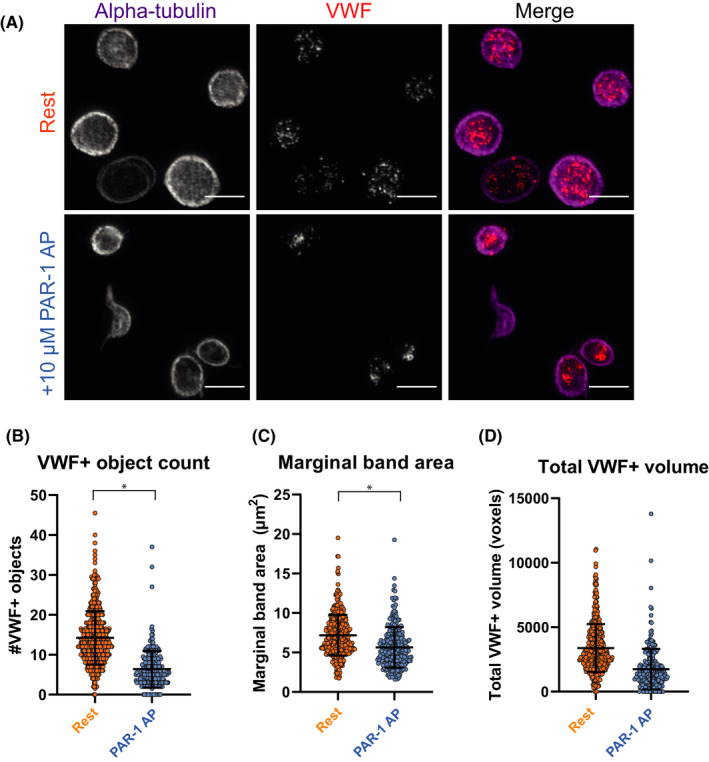
Quantification of granular and morphological changes in activated platelets using structured illumination microscopy. Platelets were incubated with 10 µM proteinase‐activated receptor 1 activating peptide (PAR‐1‐AP; blue) or phosphate buffered saline (PBS; orange, “rest”) for 10 min at 37℃, imaged and processed through our analysis workflow. One representative image of several platelets is shown per condition (A). VWF‐positive object numbers (B) and marginal band area were quantified and compared between resting (n = 663) and stimulated platelets (n = 429) (C). Total VWF‐positive volume was assessed in (D). Data is represented as mean ± standard deviation. Scale bar is 3 µm in panel (A); brightness and contrast were equally amplified for both panels to enhance visibility. *P < .05
Poisson regression analysis showed that the relation between object numbers and marginal band area was significantly different between both conditions (Figure S6C). We were also able to detect changes in the spatial distribution of unique objects in relation to each other or to the marginal band in PAR‐1–stimulated platelets (Figure S6D). Combined, these findings are in accordance with an α‐granule release phenotype and may even suggest some degree of granule reorganization in response to PAR‐1‐AP triggering. Potentially, granule reorganization signifies some degree of compound exocytosis as described previously,35 but this cannot be concluded from our data alone.
3.3. Intra‐ and interindividual variability in α‐granule cargo of healthy donor platelets
We subsequently applied SIM analysis on the alpha‐granule compartment in platelets of healthy donors, generating data on >5000 platelets and 100 000 granular objects (per staining) over five individuals (Figure 3; Figure S7, healthy control [HC]). We observed a large variation in the number of VWF‐ and SPARC‐positive objects in healthy, resting platelets (VWF, 18.0 ± 8.9; SPARC, 21.6 ± 12.1) (Figure 4A). VWF‐ and SPARC‐positive total volume similarly varied across individuals (Figure 4B). Furthermore, object numbers and volumes were correlated to marginal band area in all donors (Figure S8A‐D), in line with earlier studies that showed that the number of α‐granules is related to platelet size.29 Together, our data provide quantitative evidence for a highly heterogeneous storage capacity of individual healthy platelets, which is proportional to platelet size.
FIGURE 3.
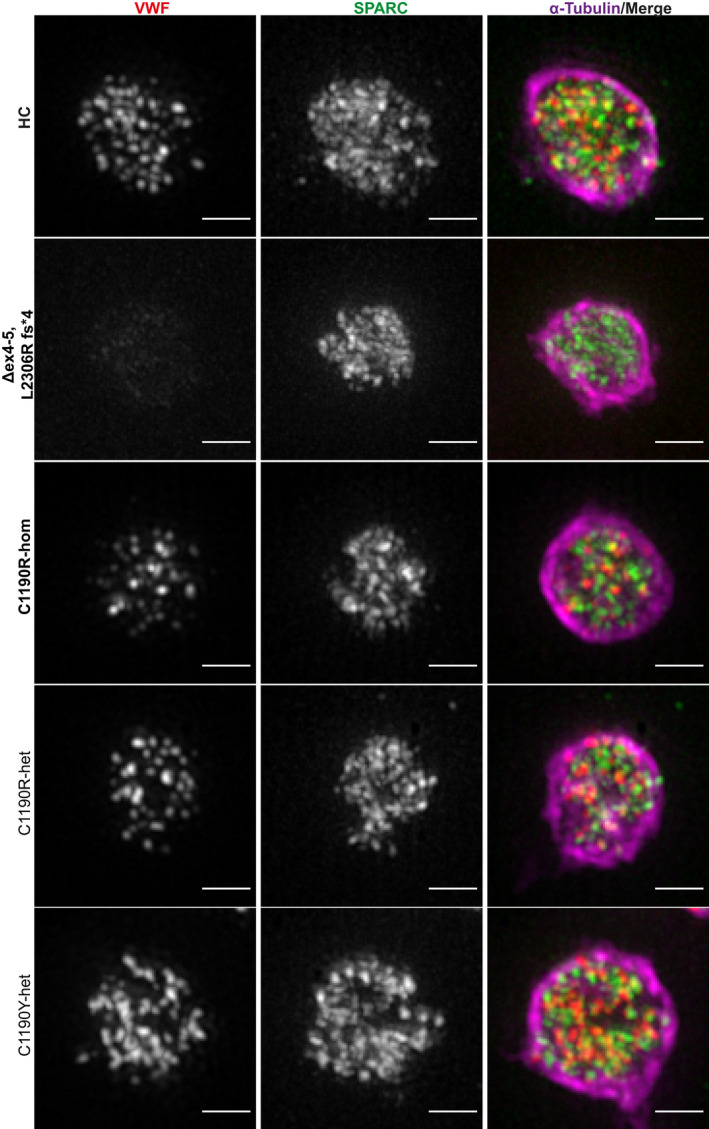
Detailed super‐resolution images of representative platelets from patients with von Willebrand disease (VWD). Platelets from healthy controls (HC), patients with type 3 VWD (Δex4‐5, L2306R fs*4; C1190R‐hom; in bold) and type 2A VWD (C1190R‐het, C1190Y‐het) were stained for VWF (red), SPARC (green), and α‐tubulin (magenta) and were imaged through structured illumination microscopy. Scale bar is 1 µm
3.4. Quantitative differences in platelet VWF storage in type 3 and 2A VWD platelets
To address whether quantitative or qualitative defects in VWF lead to abnormalities in platelet α‐granule cargo we applied our analysis to patients with VWD (Table 1, Figures 3 and 4), analyzing more than 500 platelets and 10 000 granular objects per individual patient (in all but one with >250 platelets). We first studied a compound heterozygous patient with type 3 VWD with complete absence of plasma VWF due to a deletion of exons 4 and 5 and a frameshift mutation (c.6917delT p.L2306Rfs*4) in the other allele, both leading to early termination signals. In endothelial cells, ablation of VWF expression leads to absence of their VWF‐containing secretory organelles, the Weibel‐Palade bodies.36, 37, 38 As expected, we did not detect any VWF‐positive objects (Figure 4A; Δex4‐5, L2306R fs*4), which is also evident from the overview images (Figure 3, Δex4‐5, L2306R fs*4). However, SPARC‐positive objects were still present (Δex4‐5, L2306R fs*4 14.5), and the total SPARC‐positive volume was similar to healthy individuals (HC 8285 voxels vs Δex4‐5, L2306R fs*4 8524 voxels) (Figure 4A,B), indicating that despite the absence of VWF, other α‐granule proteins are organized normally in this patient.
We also analyzed another patient with type 3 VWD with a homozygous missense mutation at p.C1190 in the D3 domain (c.3568T>C; p.C1190R), which is thought to affect VWF multimerization (Figure 3).39, 40 Interestingly, in this patient we found a relatively normal number of VWF‐ and SPARC‐positive objects (HC 18.0 vs C1190R‐hom 14.7) (Figure 4A). This was not apparent from confocal MFI measurements, in which patients with type 3 were very close to baseline intensities (Δex4‐5, L2306R fs*4 5.66 vs C1190R‐hom 6.27) (Figure 4C, Figure S9). Quantification of unique VWF‐positive objects through confocal microscopy seemed to correlate directly to staining intensity rather than accurately reflecting the number of objects/granular structures (Figure 4D). This suggests that p.C1190R homozygous platelets probably contain some residual VWF, and it highlights that our workflow is capable of discriminating these patients with type 3 VWD stemming from distinct molecular defects.
Additionally, we investigated three patients with type 2A VWD with a heterozygous mutation at the same position p.C1190 into R or Y (Table 1), two of whom are part of the same family as the homozygous p.C1190R patient. We found that two patients with the heterozygous p.C1190R mutation had relatively normal VWF‐ and SPARC‐positive object numbers and total volumes compared to healthy individuals (Figures 3 and 4A,B). Although the number of SPARC‐positive objects was significantly lower in the patients (HC 21.6; C1190R‐het 15.1, P = .03), the biological implications of such minor differences are unclear. Interestingly, object numbers were closer to those of healthy individuals (and not significantly different) in the patient with a heterozygous p.C1190Y mutation (VWF, 17.7; SPARC, 17.91). Parallel confocal analysis to obtain information on average protein levels (Figure 4C,D) showed that the decrease in MFI (HC 15.52; C1190R‐hom 6.27; C1190R‐het 12.30; C1190Y‐het 13.71) and VWF‐positive object numbers (HC 7.56; C1190R‐hom 2.75; C1190R‐het 5.32; C1190Y‐het 8.2) followed the same pattern as the plasma VWF levels in these patients (Table 1). In contrast to VWF, SPARC levels were not significantly different across any of the patients, which is in line with the minimal differences observed by SIM (Figure 4C,D). We found no evidence of altered VWF‐SPARC colocalization across controls and patients (Figure S10).
3.5. Normal α‐granules but reduced VWF content in p.C1190R homozygous platelets
To assess whether the residual VWF observed in p.C1190R homozygous platelets is in fact localized in α‐granules, we studied platelet VWF and α‐granules by immuno‐EM (Figure 5A). VWD platelets and α‐granules looked morphologically normal as assessed through thin sections: In line with our SIM analysis, we found similar numbers of morphologically identifiable α‐granules between HC and VWD platelets (HC 10.6 vs C1190R‐hom 9.3) (Figure 5B). In healthy platelets, VWF immuno‐gold labeling was mostly confined to α‐granules, and, as reported earlier,31 on one side of the α‐granule (Figure 5A *). VWF in homozygous p.C1190R platelets was also found in α‐granules but in much lower quantities (Figure 5A). Some labeling was also observed in areas outside α‐granules, but whether this is mislocalization of VWF or low‐level aspecific background labeling (from the antibody or secondary protein A gold‐labeled reagent) remains unclear. Semiquantitative analysis of VWF labeling in morphologically identifiable α‐granules identified a strong reduction of VWF present in p.C1190R homozygous platelets compared to HCc, both in total and per α‐granule (Figure 5C). Occasionally, we observed VWF labeling in structures that resemble the open canalicular system (Figure 5A #), although this did not appear to differ between patient and control platelets.
FIGURE 5.
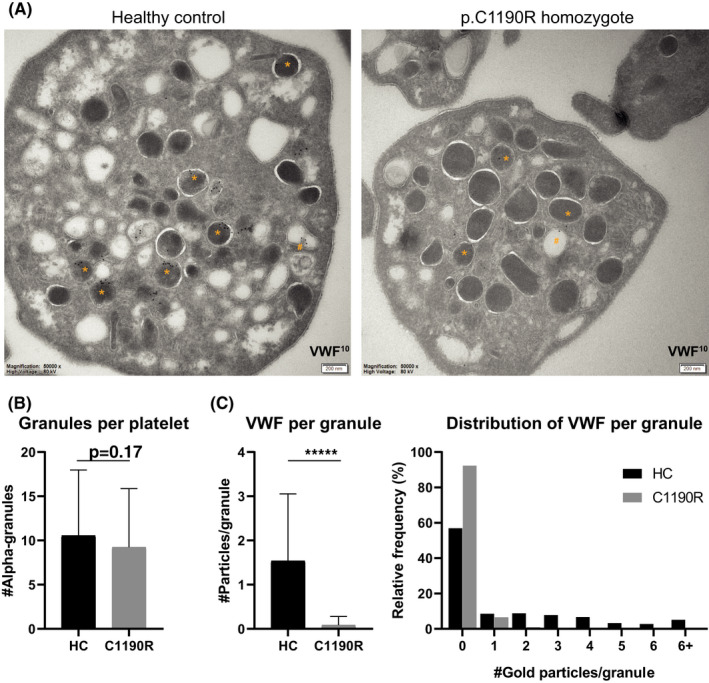
Morphological evaluation of α‐granules and von Willebrand factor (VWF) localization in the p.C1190R homozygous patient. Platelets from a healthy control (HC; n = 73) and homozygous p.C1190R patient (C1190R, n = 75) were assessed through immuno‐electron microscopy for α‐granule numbers and subcellular localization of VWF (A). Immuno‐gold labeling for VWF is shown in α‐granules (*) and open canalicular system (#). Morphologically identifiable α‐granules were quantified (B) and scored for VWF staining inside these granules (C). Data are represented as mean ± standard deviation. ****P < 0.0001
3.6. Intracellular and extracellular defects in VWF multimers caused by mutations at p.C1190
Finally, we analyzed VWF multimers in plasma and platelets (Figure 6). Consistent with the strong reduction of VWF content that we observed via platelet imaging, we were able to detect very little VWF in platelets of the patient with homozygous p.C1190R (Figure 6A, lane 2). A similar pattern was observed in plasma (Figure 6A, lane 7). Any residual VWF in platelets and plasma of this patient consisted of low‐molecular‐weight multimers, which points to reduced multimerization of VWF that most probably already occurs intracellularly (Figure 6B,C). Platelet VWF from all three patients with type 2A showed a normal multimer pattern similar to HCs (Figure 6B; lanes 1, 3–5), while plasma VWF was lacking high‐molecular‐weight (HMW) multimers in patients with p.C1190R and p.C1190Y heterozygotes (Figure 6A,C; lanes 6, 8‐10). Intruigingly, platelet and plasma VWF migrated differently through the gel, which may point at significant molecular differences such as differential glycosylation or processing between megakaryocytes and endothelial cells.41, 42
FIGURE 6.
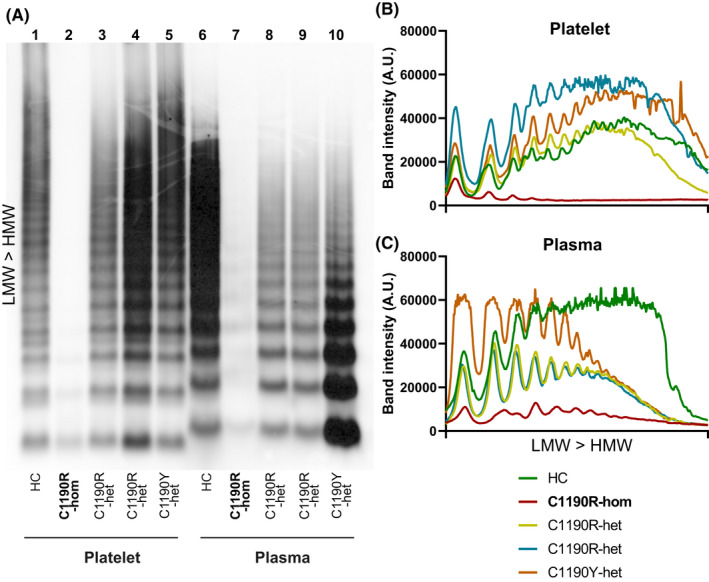
Von Willebrand factor (VWF) multimer analysis of patients with p.C1190. Platelets (lanes 1‐5) and plasma (lanes 6‐10) from a healthy control (HC) all patients with p.C1190 were analyzed for VWF multimer patterns (A). Patient with type 3 VWD (C1190R‐hom) is marked as bold text to distinguish from heterozygous C1190R (C1190R‐het) and C1190Y (C1190Y‐het). Line intensity plots were generated and plotted separately for platelet lysates (B) and plasma (C), with each individual color coded. Patterns were illustrated from low‐molecular‐weight (LMW) to high‐molecular‐weight (HMW) VWF multimers (B‐C), which is bottom to top on the blot (A)
4. DISCUSSION
We report the application of a quantitative methodology to assess platelet α‐granule content in platelets from healthy individuals and patients with VWD based on super‐resolution microscopy. Using a semiautomated analysis workflow, we generated quantitative data sets of platelet α‐granule proteins of hundreds of platelets per donor. We validated our approach in vitro measuring PAR‐1‐AP–induced degranulation of activated platelets, and assessed variation of α‐granule cargo in and across healthy individuals. Finally, we quantitatively characterized the α‐granule compartment of a subset of patients with VWD. SIM data revealed distinct alterations between and within different VWD subtypes that we ultimately characterized through EM and VWF multimer analysis. Our findings illustrate the utility of quantitative imaging approaches to study and pinpoint platelet α‐granule alterations in patients with VWD.
We found a large inter‐ and intraindividual variability in α‐granule cargo across healthy individuals, which demonstrates the heterogeneity of platelets in circulation, as platelets gradually release granule content and undergo morphological changes during aging.43, 44, 45 Our findings also highlight the importance of generating large data sets when interpreting platelet morphology or granule cargo data and the caveat of drawing conclusions based on limited sample sizes. In contrast, detailed analysis of thin sections generated through EM‐based methods may yield data of even higher resolution than our current approach but is labor intensive, which limits the cell volume and numbers that can be analyzed simultaneously. As such, SIM‐based quantitative granule cargo studies will play an important role in parallel to established methods to further unravel α‐granule biology and processes that affect platelet α‐granule content.
While SIM provides powerful quantitative immunofluorescence‐based data of platelet granule cargo, the separation of unique structures is not optimal due to its resolution limitations, particularly in the Z‐axis.9 For example, the average object numbers we quantified in our studies are two‐ to fourfold lower than estimated by EM.29 α‐Granules can be found in 30 nm proximity of one another,29 which is below the resolution level of SIM (~100 nm in XY). Therefore, differences between the EM and SIM approaches may be explained by the incomplete separation of unique cargo‐containing α‐granules in SIM, thereby leading to lower quantifiable object numbers. Nevertheless, we have presented data in our work that strongly correlates with previous SIM and EM findings,29, 46 showing that our estimation of unique granular structures defined by unique volumes of fluorescence can accurately distinguish changes in the α‐granule compartment of platelets. We were able to identify more unique objects by SIM than by confocal microscopy, implying that our approach is an improvement of previous light microscopy–based methods to discern individual granular structures. While confocal microscopy provides more linear intensity data that is suitable for direct comparison of labeling density, SIM is more capable of defining unique structures independently of signal intensity. Thus, we think using both imaging modalities in tandem is a powerful tool to assess α‐granule content. For those applications in which demands for increased resolution outweigh quantitative advantages or speed of SIM, super‐resolution microscopy approaches with even higher resolving power such as stimulated emission depletion and stochastic optical reconstruction microscopy may be more applicable.47, 48
We used a SIM‐based technique to assess the α‐granule compartment in patients with VWD, as the role that platelet VWF has in this disease is still elusive.49, 50, 51, 52, 53, 54, 55, 56, 57 Our quantitative analysis showed that platelets of both patients with type 3 VWD contained normal numbers and regular distribution of SPARC‐positive objects, which indicates that in these patients α‐granule formation and storage of other α‐granule proteins was not disturbed. This is in support of an earlier EM study that showed that α‐granules in patients with type 3 VWD appear morphologically normal.56 Taken together, this shows that unlike endothelial Weibel‐Palade bodies, whose biogenesis is driven by VWF biosynthesis,36, 38, 58 defective or severely reduced VWF synthesis does not impact the formation of its storage organelles in platelets.
SIM‐based quantitative analysis of platelet VWF content, in contrast to SPARC, highlighted profound differences between the two patients with type 3 VWD. These patients have severe quantitative VWF deficiencies that are underpinned by different pathogenic mechanisms. In one patient, compound heterozygous for two separate null alleles (Δex4‐5, L2306Rfs*4), we were unable to detect any VWF in platelets, consistent with an absence of protein synthesis because of early termination signals in both alleles. Conversely, VWF‐positive objects were found in platelets of the other patient with type 3 VWD with a homozygous missense mutation at p.C1190R in the D3 domain. Heterozygous mutations at this position40, 59, 60 and at other cysteine residues within this domain12 have been previously linked to type 2A VWD, a subtype that is characterized by low platelet binding activity of VWF due to loss of HMW multimers (also Figure 6). Lower multimers in type 2A VWD arise from either intracellular trafficking and/or multimerization defects during VWF biosynthesis (group I) or an increased sensitivity to extracellular proteolysis by ADAMTS‐13 (group II).61 The cysteines in the D3 domain form a complex network of intra‐ and interchain disulfide bonds that support the alignment and N‐terminal oligomerization of pro‐VWF dimers.62 The contribution of intrachain disulfide bonds, including the one formed by C1190, are not clearly defined, except that heterologous expression of various type 2A missense mutants at D3 cysteines in this region invariably leads to low‐molecular‐weight multimers.40, 60, 63, 64 This is reflected by the absence of medium‐molecular‐weight and HMW VWF multimers in the platelets of the patient with homozygous p.C1190R, which also points to a defect occurring during VWF biosynthesis. Despite the strongly reduced synthesis of VWF and its apparent loss of multimerization, our combined SIM and EM data (Figures 4A,C and 5A) points toward targeting of homozygous p.C1190R VWF to a normal number of α‐granules, albeit in reduced quantities. These data are in line with a previous study that showed that platelets of patients with type 3 VWD with the P2808Lfs*24 mutation, which presumably impairs VWF dimerization, still contained VWF in granular structures that could be mobilized by platelet activation.57 From this study it remained unclear whether VWF was incorporated in α‐granules and whether the lack of VWF multimers in plasma also manifested in platelets. Regardless, both this previous and our current study suggest that multimerization per se is not a prerequisite for VWF to enter platelet α‐granules.
Despite the reduction of HMW VWF multimers in their plasma, patients heterozygous for p.C1190R or p.C1190Y had platelet VWF multimers that were indistinguishable from healthy controls. De Jong et al59 recently tested a single nucleotide polymorphism–based approach to specifically silence expression of an autosomal dominant VWD allele in blood outgrowth endothelial cells of the same patient with type 2A with p.C1190Y included in our study. VWF multimers from heterozygous p.C1190Y endothelial cells showed an overabundance of dimeric VWF and pronounced retention of VWF in the endoplasmic reticulum (ER), both of which were alleviated by specific silencing of the p.C1190Y allele. Accumulation of dimeric VWF, which possibly consists of ER‐retained mutant wild‐type heterodimers,65 was not observed in platelets in our current work. This discrepancy may be explained by retention of this pool in megakaryocytes during proplatelet formation. However, we cannot exclude the possibility that this is the result of differences in disulfide exchange or posttranslational modifications of VWF between megakaryocytes and endothelial cells. Similarly, we found that platelet‐ and plasma‐derived VWF migrated differently through a multimer gel, suggestive of altered processing between both cell types. Judging by the presence of HMW multimers in platelets as well as endothelial cells of this patient, reduction of VWF multimer size in plasma cannot be fully explained by biosynthetic defects alone but potentially also by a reduced half‐life of circulating HMW multimers. Finally, we also observed a discrepancy between VWF levels in platelets of the patients with p.C1190Y and p.C1190R heterozygotes, which was consistent with their plasma VWF levels (Table 1). This is in line with previous observations suggesting that missense mutations at p.C1190 can have differential effects based on the substitution.40
The potential diagnostic utility of our approach was demonstrated by the quantification of granular abnormalities in patients with VWD with different etiologies. Currently, we are collecting and analyzing platelet data from additional patients with VWD to answer follow‐up questions. For example, studying the distribution of VWF in platelets may provide an easily accessible way of studying VWF synthesis defects. Additionally, tools to explain variability in bleeding phenotypes of patients with VWD are lacking, for which studies in larger groups need to be done to address the link between platelet VWF and bleeding severity. The possibility to rapidly image and analyze large numbers of platelets in 3D in a large field of view with SIM will be an important advantage versus other super‐resolution methods when analyzing platelet content and morphology in large cohort studies48 and may also be useful to study inherited platelet storage pool disorders stemming from variants such as GFI1b, NBEAL2, or GATA1. We envision SIM in parallel to confocal imaging to be considered as a diagnostic tool for complex cases, such as unexplained storage pool disease, similar to current EM‐based diagnostics.
One promising example to better characterize the bleeding phenotype in patients stems from our current study in relation to the Bowman study.57 Patients with the p.P2808Lfs*24 mutation had very low VWF plasma levels, but their bleeding scores were relatively low (median, 8.5) compared to other patients with type 3 VWD, or the patient with Δex4‐5 described in our study (bleeding score = 34, Table 1). Similarly, the p.C1190R homozygote in our work had but a moderate bleeding phenotype (score = 20, Table 1). It remains to be determined whether (release of) residual platelet VWF contributes to the milder phenotype of p.P2808Lfs*24 and p.C1190R homozygous patients in comparison to other patients with type 3 VWD, or whether it potentially is a surrogate marker of VWD severity. However, our combined findings suggest that studying VWF content in platelets of patients with VWD could have predictive value in telling apart patients with severe VWD with severe and mild bleeding phenotypes.
In summary, we developed a quantitative methodology to evaluate alpha‐granule constituents through super‐resolution imaging which can be used for the characterization of the platelet α‐granule compartment and VWF storage in healthy individuals and patients with VWD.
RELATIONSHIP DISCLOSURE
The authors declare no conflicts of interest related to the subject of this study.
AUTHOR CONTRIBUTIONS
MS performed in vitro experiments and imaging and designed the Fiji‐based analysis. FA included patients for the study and obtained clinical and laboratory data. PEB performed VWF multimer analysis. JAS and ABH helped design the imaging setup and analysis. CdH and JK performed immuno‐EM imaging and analysis. FWGL, JV, AJG J, and RB supervised experimental design, analysis design, and selection of patients. All authors provided input and made suggestions for improvement of the manuscript.
Supporting information
Fig S1
Fig S2
Fig S3
Fig S4
Fig S5
Fig S6
Fig S7
Fig S8
Fig S9
Fig S10
Table S1‐S2
Video S1
ACKNOWLEDGMENTS
We thank I. van Moort for on‐site blood collection.
Swinkels M, Atiq F, Bürgisser PE, et al. Quantitative 3D microscopy highlights altered von Willebrand factor α‐granule storage in patients with von Willebrand disease with distinct pathogenic mechanisms. Res Pract Thromb Haemost. 2021;5:e12595. 10.1002/rth2.12595
Arend Jan Gerard Jansen and Ruben Bierings: Shared last authorship.
Handling Editor: Yotis Senis
Funding information
This work was supported by grants from the Landsteiner Stichting voor Bloedtransfusie Research (LSBR‐1707) (R.B.) and an EHA Clinical Research Fellowship (AJGJ). The WiN‐Pro study was supported (in part) by research funding from the Dutch Hemophilia Foundation (Stichting Haemophilia) (FA) and CSL Behring (FWGL, unrestricted grant)
Contributor Information
Maurice Swinkels, @MauriceSwinkels.
Johan A. Slotman, @JohanSlotman.
Judith Klumperman, @JKlumperman.
Frank W. G. Leebeek, @FLeebeek.
Jan Voorberg, @VoorbergJ.
Arend Jan Gerard Jansen, @ajgjansen.
Ruben Bierings, Email: r.bierings@erasmusmc.nl, @rbierings.
REFERENCES
- 1.Yadav S, Storrie B. The cellular basis of platelet secretion: emerging structure/function relationships. Platelets. 2017;28:108‐118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kelm RJ Jr, Mann KG. Human platelet osteonectin: release, surface expression, and partial characterization. Blood. 1990;75:1105‐1113. [PubMed] [Google Scholar]
- 3.Breton‐Gorius J, Clezardin P, Guichard J, et al. Localization of platelet osteonectin at the internal face of the alpha‐granule membranes in platelets and megakaryocytes. Blood. 1992;79:936‐941. [PubMed] [Google Scholar]
- 4.Chen CI, Federici AB, Cramer EM, et al. Studies of multimerin in patients with von Willebrand disease and platelet von Willebrand factor deficiency. Br J Haematol. 1998;103:20‐28. [DOI] [PubMed] [Google Scholar]
- 5.Hayward CP, Cramer EM, Kane WH, et al. Studies of a second family with the Quebec platelet disorder: evidence that the degradation of the alpha‐granule membrane and its soluble contents are not secondary to a defect in targeting proteins to alpha‐granules. Blood. 1997;89:1243‐1253. [PubMed] [Google Scholar]
- 6.Karampini E, Bierings R, Voorberg J. Orchestration of primary hemostasis by platelet and endothelial lysosome‐related organelles. Arterioscler Thromb Vasc Biol. 2020;40:1441‐1453. [DOI] [PubMed] [Google Scholar]
- 7.Heemskerk JW, Mattheij NJ, Cosemans JM. Platelet‐based coagulation: different populations, different functions. J Thromb Haemost. 2013;11:2‐16. [DOI] [PubMed] [Google Scholar]
- 8.Ambrosio AL, Di Pietro SM. Storage pool diseases illuminate platelet dense granule biogenesis. Platelets. 2017;28:138‐146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schermelleh L, Ferrand A, Huser T, et al. Super‐resolution microscopy demystified. Nat Cell Biol. 2019;21:72‐84. [DOI] [PubMed] [Google Scholar]
- 10.Westmoreland D, Shaw M, Grimes W, et al. Super‐resolution microscopy as a potential approach to diagnosis of platelet granule disorders. J Thromb Haemost. 2016;14:839‐849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pluthero FG, Kahr WHA. Imaging platelets and megakaryocytes by high‐resolution laser fluorescence microscopy. Methods Mol Biol. 2018;1812:13‐31. [DOI] [PubMed] [Google Scholar]
- 12.de Jong A, Eikenboom J. Von Willebrand disease mutation spectrum and associated mutation mechanisms. Thromb Res. 2017;159:65‐75. [DOI] [PubMed] [Google Scholar]
- 13.Leebeek FW, Eikenboom JC. Von Willebrand’s disease. N Engl J Med. 2016;375:2067‐2080. [DOI] [PubMed] [Google Scholar]
- 14.Leebeek FWG, Atiq F. How I manage severe von Willebrand disease. Br J Haematol. 2019;187:418‐430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schillemans M, Karampini E, van den Eshof BL, et al. Weibel‐Palade body localized syntaxin‐3 modulates Von Willebrand factor secretion from endothelial cells. Arterioscler Thromb Vasc Biol. 2018;38:1549‐1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schindelin J, Arganda‐Carreras I, Frise E, et al. Fiji: an open‐source platform for biological‐image analysis. Nat Methods. 2012;9:676‐682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ball G, Demmerle J, Kaufmann R, Davis I, Dobbie IM, Schermelleh L. SIMcheck: a toolbox for successful super‐resolution structured illumination microscopy. Sci Rep. 2015;5:15915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Banterle N, Bui KH, Lemke EA, Beck M. Fourier ring correlation as a resolution criterion for super‐resolution microscopy. J Struct Biol. 2013;183:363‐367. [DOI] [PubMed] [Google Scholar]
- 19.Sezgin M, Sankur B. Survey over image thresholding techniques and quantitative performance evaluation. J Electron Imaging. 2004;13:146‐168. [Google Scholar]
- 20.Ollion J, Cochennec J, Loll F, Escude C, Boudier T. TANGO: a generic tool for high‐throughput 3D image analysis for studying nuclear organization. Bioinformatics. 2013;29:1840‐1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tsai WH. Moment‐preserving thresholding ‐ a new approach. Comput Vision Graph. 1985;29:377‐393. [Google Scholar]
- 22.Aaron JS, Taylor AB, Chew TL. Image co‐localization ‐ co‐occurrence versus correlation. J Cell Sci. 2018;131(3):jcs211847. [DOI] [PubMed] [Google Scholar]
- 23.Coltharp C, Yang X, Xiao J. Quantitative analysis of single‐molecule superresolution images. Curr Opin Struct Biol. 2014;28:112‐121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nicovich PR, Owen DM, Gaus K. Turning single‐molecule localization microscopy into a quantitative bioanalytical tool. Nat Protoc. 2017;12:453‐460. [DOI] [PubMed] [Google Scholar]
- 25.Lagache T, Sauvonnet N, Danglot L, Olivo‐Marin JC. Statistical analysis of molecule colocalization in bioimaging. Cytometry A. 2015;87:568‐579. [DOI] [PubMed] [Google Scholar]
- 26.Otsu N. Threshold selection method from gray‐level histograms. IEEE T Syst Man Cyb. 1979;9:62‐66. [Google Scholar]
- 27.Harrison P, Savidge GF, Cramer EM. The origin and physiological relevance of alpha‐granule adhesive proteins. Br J Haematol. 1990;74:125‐130. [DOI] [PubMed] [Google Scholar]
- 28.King SM, Reed GL. Development of platelet secretory granules. Semin Cell Dev Biol. 2002;13:293‐302. [DOI] [PubMed] [Google Scholar]
- 29.Pokrovskaya ID, Yadav S, Rao A, et al. 3D ultrastructural analysis of alpha‐granule, dense granule, mitochondria, and canalicular system arrangement in resting human platelets. Res Pract Thromb Haemost. 2020;4:72‐85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van Nispen tot Pannerden H, de Haas F, Geerts W, Posthuma G, van Dijk S, Heijnen HF. The platelet interior revisited: electron tomography reveals tubular alpha‐granule subtypes. Blood. 2010;116:1147‐1156. [DOI] [PubMed] [Google Scholar]
- 31.Cramer EM, Meyer D, le Menn R, Breton‐Gorius J. Eccentric localization of von Willebrand factor in an internal structure of platelet alpha‐granule resembling that of Weibel‐Palade bodies. Blood. 1985;66:710‐713. [PubMed] [Google Scholar]
- 32.Battinelli EM, Thon JN, Okazaki R, et al. Megakaryocytes package contents into separate alpha‐granules that are differentially distributed in platelets. Blood Adv. 2019;3:3092‐3098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Italiano JE Jr, Richardson JL, Patel‐Hett S, et al. Angiogenesis is regulated by a novel mechanism: pro‐ and antiangiogenic proteins are organized into separate platelet alpha granules and differentially released. Blood. 2008;111:1227‐1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Diagouraga B, Grichine A, Fertin A, Wang J, Khochbin S, Sadoul K. Motor‐driven marginal band coiling promotes cell shape change during platelet activation. J Cell Biol. 2014;204:177‐185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eckly A, Rinckel JY, Proamer F, et al. Respective contributions of single and compound granule fusion to secretion by activated platelets. Blood. 2016;128:2538‐2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Groeneveld DJ, van Bekkum T, Dirven RJ, et al. Angiogenic characteristics of blood outgrowth endothelial cells from patients with von Willebrand disease. J Thromb Haemost. 2015;13:1854‐1866. [DOI] [PubMed] [Google Scholar]
- 37.Schillemans M, Karampini E, Kat M, Bierings R. Exocytosis of Weibel‐Palade bodies: how to unpack a vascular emergency kit. J Thromb Haemost. 2019;17:6‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schillemans M, Kat M, Westeneng J, et al. Alternative trafficking of Weibel‐Palade body proteins in CRISPR/Cas9‐engineered von Willebrand factor‐deficient blood outgrowth endothelial cells. Res Pract Thromb Haemost. 2019;3:718‐732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Springer TA. von Willebrand factor, Jedi knight of the bloodstream. Blood. 2014;124:1412‐1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schneppenheim R, Michiels JJ, Obser T, et al. A cluster of mutations in the D3 domain of von Willebrand factor correlates with a distinct subgroup of von Willebrand disease: type 2A/IIE. Blood. 2010;115:4894‐4901. [DOI] [PubMed] [Google Scholar]
- 41.McGrath RT, van den Biggelaar M, Byrne B, et al. Altered glycosylation of platelet‐derived von Willebrand factor confers resistance to ADAMTS13 proteolysis. Blood. 2013;122:4107‐4110. [DOI] [PubMed] [Google Scholar]
- 42.McGrath RT, McRae E, Smith OP, O’Donnell JS. Platelet von Willebrand factor–structure, function and biological importance. Br J Haematol. 2010;148:834‐843. [DOI] [PubMed] [Google Scholar]
- 43.Rijkers M, van den Eshof BL, van der Meer PF, et al. Monitoring storage induced changes in the platelet proteome employing label free quantitative mass spectrometry. Sci Rep. 2017;7:11045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rijkers M, van der Meer PF, Bontekoe IJ, et al. Evaluation of the role of the GPIb‐IX‐V receptor complex in development of the platelet storage lesion. Vox Sang. 2016;111:247‐256. [DOI] [PubMed] [Google Scholar]
- 45.Shrivastava M. The platelet storage lesion. Transfus Apher Sci. 2009;41:105‐113. [DOI] [PubMed] [Google Scholar]
- 46.Sehgal S, Storrie B. Evidence that differential packaging of the major platelet granule proteins von Willebrand factor and fibrinogen can support their differential release. J Thromb Haemost. 2007;5:2009‐2016. [DOI] [PubMed] [Google Scholar]
- 47.Ronnlund D, Yang Y, Blom H, Auer G, Widengren J. Fluorescence nanoscopy of platelets resolves platelet‐state specific storage, release and uptake of proteins, opening up future diagnostic applications. Adv Healthc Mater. 2012;1:707‐713. [DOI] [PubMed] [Google Scholar]
- 48.Montague SJ, Lim YJ, Lee WM, Gardiner EE. Imaging platelet processes and function‐current and emerging approaches for imaging in vitro and in vivo. Front Immunol. 2020;11:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Casonato A, Cattini MG, Daidone V, Pontara E, Bertomoro A, Prandoni P. Diagnostic value of measuring platelet von Willebrand factor in von Willebrand disease. PLoS One. 2016;11:e0161310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gralnick HR, Rick ME, McKeown LP, et al. Platelet von Willebrand factor: an important determinant of the bleeding time in type I von Willebrand’s disease. Blood. 1986;68:58‐61. [PubMed] [Google Scholar]
- 51.Rodeghiero F, Castaman G, Ruggeri M, Tosetto A. The bleeding time in normal subjects is mainly determined by platelet von Willebrand factor and is independent from blood group. Thromb Res. 1992;65:605‐615. [DOI] [PubMed] [Google Scholar]
- 52.Verhenne S, Denorme F, Libbrecht S, et al. Platelet‐derived VWF is not essential for normal thrombosis and hemostasis but fosters ischemic stroke injury in mice. Blood. 2015;126:1715‐1722. [DOI] [PubMed] [Google Scholar]
- 53.Nurden P, Chretien F, Poujol C, Winckler J, Borel‐Derlon A, Nurden A. Platelet ultrastructural abnormalities in three patients with type 2B von Willebrand disease. Br J Haematol. 2000;110:704‐714. [DOI] [PubMed] [Google Scholar]
- 54.Nurden P, Debili N, Vainchenker W, et al. Impaired megakaryocytopoiesis in type 2B von Willebrand disease with severe thrombocytopenia. Blood. 2006;108:2587‐2595. [DOI] [PubMed] [Google Scholar]
- 55.Nurden P, Gobbi G, Nurden A, et al. Abnormal VWF modifies megakaryocytopoiesis: studies of platelets and megakaryocyte cultures from patients with von Willebrand disease type 2B. Blood. 2010;115:2649‐2656. [DOI] [PubMed] [Google Scholar]
- 56.Nurden P, Nurden AT, La Marca S, Punzo M, Baronciani L, Federici AB. Platelet morphological changes in 2 patients with von Willebrand disease type 3 caused by large homozygous deletions of the von Willebrand factor gene. Haematologica. 2009;94:1627‐1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bowman ML, Pluthero FG, Tuttle A, et al. Discrepant platelet and plasma von Willebrand factor in von Willebrand disease patients with p.Pro2808Leufs*24. J Thromb Haemost. 2017;15:1403‐1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Denis CV, Andre P, Saffaripour S, Wagner DD. Defect in regulated secretion of P‐selectin affects leukocyte recruitment in von Willebrand factor‐deficient mice. Proc Natl Acad Sci U S A. 2001;98:4072‐4077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.de Jong A, Dirven RJ, Boender J, et al. Ex vivo improvement of a von Willebrand disease type 2A phenotype using an allele‐specific small‐interfering RNA. Thromb Haemost. 2020;120:1569‐1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jacobi PM, Gill JC, Flood VH, Jakab DA, Friedman KD, Haberichter SL. Intersection of mechanisms of type 2A VWD through defects in VWF multimerization, secretion, ADAMTS‐13 susceptibility, and regulated storage. Blood. 2012;119:4543‐4553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lyons SE, Bruck ME, Bowie EJ, Ginsburg D. Impaired intracellular transport produced by a subset of type IIA von Willebrand disease mutations. J Biol Chem. 1992;267:4424‐4430. [PubMed] [Google Scholar]
- 62.Dong X, Leksa NC, Chhabra ES, et al. The von Willebrand factor D'D3 assembly and structural principles for factor VIII binding and concatemer biogenesis. Blood. 2019;133:1523‐1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Eikenboom JC, Matsushita T, Reitsma PH, et al. Dominant type 1 von Willebrand disease caused by mutated cysteine residues in the D3 domain of von Willebrand factor. Blood. 1996;88:2433‐2441. [PubMed] [Google Scholar]
- 64.Tjernberg P, Vos HL, Castaman G, Bertina RM, Eikenboom JC. Dimerization and multimerization defects of von Willebrand factor due to mutated cysteine residues. J Thromb Haemost. 2004;2:257‐265. [DOI] [PubMed] [Google Scholar]
- 65.Bodo I, Katsumi A, Tuley EA, Eikenboom JC, Dong Z, Sadler JE. Type 1 von Willebrand disease mutation Cys1149Arg causes intracellular retention and degradation of heterodimers: a possible general mechanism for dominant mutations of oligomeric proteins. Blood. 2001;98:2973‐2979. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Fig S3
Fig S4
Fig S5
Fig S6
Fig S7
Fig S8
Fig S9
Fig S10
Table S1‐S2
Video S1