

Summary
Early quantitative structure-activity relationship (QSAR) technologies have unsatisfactory versatility and accuracy in fields such as drug discovery because they are based on traditional machine learning and interpretive expert features. The development of Big Data and deep learning technologies significantly improve the processing of unstructured data and unleash the great potential of QSAR. Here we discuss the integration of wet experiments (which provide experimental data and reliable verification), molecular dynamics simulation (which provides mechanistic interpretation at the atomic/molecular levels), and machine learning (including deep learning) techniques to improve QSAR models. We first review the history of traditional QSAR and point out its problems. We then propose a better QSAR model characterized by a new iterative framework to integrate machine learning with disparate data input. Finally, we discuss the application of QSAR and machine learning to many practical research fields, including drug development and clinical trials.
Subject areas: Data analysis in structural biology, Machine learning, Structural biology
Graphical abstract
Data analysis in structural biology; Machine learning; Structural biology
Introduction
Machine learning (ML), with historical breakthroughs being made, has been widely used in Big Data and parallel computation applications such as image recognition, knowledge representation, robotics, autonomous driving, and drug development (Freitag, 2000; Krizhevsky et al., 2017; Kukar et al., 1999; Lecun et al., 2015; Cho et al., 2005). The latter is notoriously inefficient mainly owing to the high development cost (approximately US$2.6 billion for each newly approved drug), low clinical trial success rate (less than 12%), and low return on investment (Wenz, 1982). Computer-aided drug development (design, screening, and testing) may reduce the costs and increase the success rate and investment return (Cheng et al., 2012; Lu et al., 2006; Jain, 2017; Lill and Danielson, 2011; Mcinnes, 2007). Since the 1990s, techniques such as homology modeling, molecular docking, quantitative structure-activity relationship (QSAR) modeling, and molecular dynamics (MD) simulation have been used to research on drug activity mechanisms (Capener et al., 2000; Cavasotto and Phatak, 2009; Edwards et al., 2016; Ewing et al., 2001; Gombar et al., 2004; Hartmman et al., 2013; Kitchen et al., 2004; Krieger et al., 2003; Kwon et al., 2007; Lampi et al., 2010; Morris and Lim-Wilby, 2008; Ohashi and Tanaka, 2010; Rapaport, 2004; Rapaport et al., 2002; Thangapandian et al., 2013; Zheng et al., 2013). Artificial-intelligence (AI)-based big data analyses and high-performance computations have greatly improved the efficiency of drug development, especially in the drug discovery stage. More and more pharmaceutical companies are investing in AI technology. Currently, the value of the medical AI market is approximately US$700 million and is expected to grow at a compound annual rate of 53%, reaching US$8 billion by 2022. More than 35% of this AI market share is taken by drug discovery (Clancey and Shortliffe, 1985; Sondak, 1990). To improve the efficiency of drug discovery and increase the success rate of drug synthesis, many ML companies become specialized in serving pharmaceutical companies; their services include disease target identification, compound screening, de novo drug design, clinical image recognition (Secco et al., 2016), toxicity prediction, and the prediction of absorption, distribution, metabolism, and excretion (ADME) (Hou and Xu, 2002; Kassel, 2004; Li, 2001; Yang et al., 2004). A drug repurposed by the AI company Benevolent (Stephenson et al., 2019), for example, is now in the second phase of clinical trials and is being tested by Johnson & Johnson. However, it is important to be aware of the pros and cons of different AI systems, as they are optimized for specific purposes. By investing in different AI systems, pharmaceutical companies not only can engage multiple areas ranging from drug discovery to clinical trials but also may discover breakthrough treatments for complex diseases.
AI technologies are driven by new ML algorithms, advances in computational power, and ever-increasing experimental data (Bazoon et al., 2002; Pasquier and Hamodrakas, 2009). Advanced biotechnologies such as next-generation sequencing (Dijk et al., 2014; Kim, 2019), cryo-electron microscopy, high-throughput screening (Shin et al., 2016), medical digitization, and internet-of-things infrastructure (Adrian et al., 1984; Atzori et al., 2010; Cai et al., 2019; Dubochet et al., 1988; Feng et al., 2012; Gupta et al., 2009; Li et al., 2013; Song et al., 2019) provide high-quality Big Data for the application of ML-based drug development. ML algorithms can be roughly divided into three kinds: unsupervised, supervised, and reinforced (Ertel, 2017; Figueiredo and Jain, 2002; Kaelbling et al., 1996; Le, 2013; Piotr et al., 2006; Radford et al., 2015; Sahami, 1997; Sasakawa et al., 2010; Turian et al., 2010; Wang et al., 2019; Zhu and Goldberg, 2009). Unsupervised ML can be used to find hidden patterns in medical and biological data and to identify new disease targets (Bailey and Elkan, 1995; Wiskott and Sejnowski, 2002). Supervised ML can be used to predict drug activity, toxicity, and ADME from the existing data of drugs and clinical trials (Carneiro et al., 2007; Conneau et al., 2017; Igual and Seguí, 2017; Møller, 1993). Deep learning (DL), as advanced ML, has unprecedented power to scale up the capabilities of ML. AI is expected to become a major cost-effective, low-risk method in drug virtual screening.
QSAR, first established by Corwin Hansch (Hansch et al., 1962), was a natural extension of physical chemistry into the field of virtual drug screening. After more than 50 years of development with interdisciplinary breakthroughs and community promotion, QSAR has transformed from simple regression analysis (which can only handle similar compounds) to multiple statistics ML technique (which can analyze a very large data set of molecular structures). QSAR models have been widely used to model the biophysical properties of many chemicals (Hopfinger et al., 1997; Jaworska et al., 2005; Sabljić et al., 1995; Wold, 2010) and to assess the potential impacts of medicines, chemicals, and nanomaterials on human health and ecosystems (Alves et al., 2016; In et al., 2012; Karelson et al., 2010; Kim et al., 2015; Shin et al., 2017, 2018; Svetnik et al., 2003). For example, Lee et al. established a QSAR system called MS-HEMs to manage high-energy molecules (Lee et al., 2012). In the field of computer-aided drug design (CADD), QSAR has long been recognized as an effective method of structure- and knowledge-based drug design and optimization (Cramer, 2012). In this article, we review the merits, reliability, and limitations of QSAR to generate new insights into ML-based QSAR models, their integration with experimental or simulation data, and their potential applications (Zhao, 2003).
Structure-based drug design
The key to molecular targeted therapy is the discovery of lead drugs that can inhibit the targeted proteins, which usually entails the screening of a large number of small molecule compounds. Drug screening can be achieved by complex experiments (Larios et al., 2012; Lutz et al., 1996; Pan et al., 2005; Strasser et al., 2003), but their high cost and slow speed prevent high-throughput realization. CADD, through software such as GOLD, SYBYL, DiscoveryStudio, Autodock (Ash et al., 1997; Gao and Huang, 2011; Hashmi, 2007; Trott and Olson, 2010; Verdonk et al., 2010; Wang et al., 2015a; Yang et al., 2011), can rapidly search common libraries (e.g., proprietary libraries, Maybridge commercial library, and Food and Drug Administration drug library) and perform virtual screening (molecular docking and binding free energy evaluation) (Liu, 2016), which are characterized by high speed and high throughput. Compounds with high priority scores are selected for further screening by cellular experiments, animal experiments, or clinical trials, which may finally give rise to lead compounds.
CADD is a structure-based drug design method. Biological/chemical properties, no matter static properties (e.g., chemical composition) or dynamical changes (e.g., the conformational changes of DNA with different ethanol concentrations (Fang et al., 1999)), are fundamentally determined by molecular structure, which underscores the importance of studying structure-property relationship. Molecular mechanics (MM) (Humphrey et al., 1996; Phillips et al., 2010) and quantum mechanics (QM) (Csányi et al., 2004; Habasaki and Okada, 2006; Kwangho Nam and York, 2008; Mochizuki et al., 2007; Noel et al., 2010) are two approaches of MD aiming at the accurate computation of the moving trajectory of every atom in a biomolecule until the movement is stabilized, reaching the stable (low energy) conformation. Owing to the large number of atoms in a biomolecule, an MD simulation is usually computational costly. To expedite drug screening, less accurate but computationally efficient MM force field, such as Merck Molecular Force Fields (MMFF), are generally used (Bret et al., 2000; Cheng et al., 2000; Cieplak et al., 2009; Giese, 2005; Halgren, 2015; Tu and Laaksonen, 2001). Structure-based drug design mainly includes receptor-based methods (molecular docking, de novo design, MD simulation, homology modeling) (Cho et al., 2015; Nam et al., 2003, Nam et al., 2011; Semper et al., 2021) and ligand-based methods (QSAR, pharmacophore, substructure search) (Anderson, 2003; Bohacek et al., 1996; Chang et al., 1992; Greer et al., 1994; Shim et al., 2014; Ma et al., 2012; Verlinde and Hol, 1994). They all involve the interaction between the receptor and ligand, but their focuses are different (Figure 1).
Figure 1.

Classification of drug design methods
Receptor-based methods are mainly based on the three-dimensional structure of the receptor to find a matching ligand. To treat a disease by targeting some human receptor protein, a large number of small-molecule compounds need to be screened by molecular docking or de novo design to find the ones that fit well with the crystal structure of the protein (Butterfoss and Kuhlman, 2006; Degrado, 1997; Lichtenstein et al., 2012). If the crystal structure is unavailable, the structure can usually be estimated by homology modeling, namely, by constructing the human protein structure based on the corresponding structure of another species and the amino acid sequences of both (which only have small differences). Although receptor-based methods are dominant in drug screening, they still have great limitations. First, these methods rarely consider factors such as protein flexibility, the influence of water molecules, solvation effects, and the conformational limitations of the ligand. At present, most molecular docking procedures have certain defects such as the inability to correctly deal with the induced coincidence effect, the solvation effect, and the poor ranking ability of the scoring function. Second, although the efficiency of molecular docking is relatively high, its speed is far from enough for a million-level compound library. Finally, the crystal structure of some proteins has not been solved for any species, making ligand-based methods infeasible. For example, the membrane proteins are difficult to purify and crystallize owing to hydrophobicity.
Ligand-based methods can be used to circumvent the aforementioned problems because they are in principle independent of the receptor protein. They start from a known effective ligand to discover the substructures or structural characteristics that are genuinely responsible for the drug efficacy, which can then be used to guide the selection or design of the analogs of the ligand. In brief, the methods predict new ligands based on the features extracted from a known active ligand. These new ligands are candidate drugs and need to be tested further. Even if the crystal structure of the corresponding receptor is unknown for all the species, the ligands can still be tested by in vitro experiments (e.g., the measurement of binding free energy, pk(a), logP, logD, and other indicators (Bash et al., 2002; Culler et al., 1993; Laitinen et al., 2010; Xing and Glen, 2010)), making the ligand-based methods always useful. Ligand-based methods mainly include pharmacophore-based methods (Kim et al., 2017; Kurogi and Guner, 2001; Lee et al., 2013; Yang, 2010) and QSAR model prediction.
Pharmacophore
Pharmacophore refers to the common characteristics of all the small-molecule compounds actively interacting with the target protein. There must be specific sites in the receptor for small molecules to bind; thus, the ligands binding to the same receptor must have similar chemical substructures. The International Union of Pure and Applied Chemistry (IUPAC) defines pharmacophore as “an ensemble of steric and electronic features that is necessary to ensure the optimal supramolecular interactions with a specific biological target and to trigger (or block) its biological response.” The pharmacophore model uses not only the topology similarity but also the functional similarity of the molecule, allowing for the use of the concept of bioisosterism to make the model more reliable (Wolber and Sippl, 2008). In CADD, the pharmacophore model is mainly applied in three areas:
-
a)
Structure-activity relationship, which is established through the discovery and definition of key pharmacodynamical characteristics of the drug molecules.
-
b)
Scaffold hopping, which is established through the discovery of compounds with novel core structures by modifying the central core structure of a known active compound (Jang et al., 2016).
-
c)
Target fishing, which is established through the prediction of targets of a given compound according to the compound’s pharmacophore characteristics. This also allows for the identification of the potential off-targets (side effects) of the compound so that its candidacy as a lead compound may be eliminated early in time.
To obtain an accurate pharmacophore model, the correct three-dimensional (3D) structure of the compound must first be identified, which necessitates the careful inspection of valence, bond order, protonation state, tautomerism, and stereoisomerism. Although pharmacophore deserves further development, their practical application still faces many difficulties. A pharmacophore is receptor specific and may become useless when the drug target is different from the receptor, even if the difference is small. A pharmacophore is also sensitive to the change of its parental chemical structures. That is, a slightly different chemical may not match the pharmacophore. The matching difficulty is determined by the number of features and the tolerance of the pharmacophore.
Quantitative structure-activity relationship
QSAR is an empirical mathematical model in which regression (Doo Ho Cho and Bum Tae Kim, 2001) and classification (Choi et al., 2010; Choi et al., 2009; Kim et al., 2006a, 2006b; Kim et al., 2008; Lee et al., 2017; You et al., 2015) is performed on many structure-property data to reveal statistically significant correlations between chemical structures and biological properties (Figure 2). A QSAR model can thus predict a new chemical's biological/toxicological properties based solely on the chemical's structure, i.e., without resorting to the time-consuming molecular docking, which makes both the training and application of QSAR highly efficient. This does not necessarily mean that the receptor information cannot be incorporated. Because the structure of a protein is determined by its amino acid (AA) sequence, the structure and activity of the receptor can be taken into account by introducing the AA sequence information into the QSAR model, which may markedly increase the accuracy of model prediction. In summary, QSAR is promising in drug development because it can process a large amount of compounds with high speed and without losing much precision.
Figure 2.

Quantitative structure-activity relationship
(A) The conception of QSAR.
(B) Understanding of QSAR from the perspective of machine learning.
QSAR has received an increasing number of applications in recent years, including drug design, drug toxicity prediction (Wu and Wang, 2018), the study of enzymes' chemical-biological interaction mechanisms, and the prediction of compounds' biological activity. QSAR is generally based on the following three methods: molecular description, chemical similarity search, and ML. They are described in the following.
QSAR: methods based on molecular descriptors
The core of QSAR lies in the acquisition of molecular descriptors (also called chemical descriptors), that is, the extraction of information-rich numerical features from chemical structures (Joung et al., 2012; Kim et al., 2009; Nilakantan et al., 1987; Park et al., 2013; Randić, 1993). The past 50 years has witnessed different types of molecular descriptors, which can be divided into quantitative descriptors (molecular field, molecular shape) and qualitative descriptors (daylight fingerprints, MACCS keys, MDL, Public keys). According to the data type, molecular descriptors can be divided into Boolean (e.g., chiral or not), integer (e.g., the number of rings), real (e.g., molecular weight), vector (e.g., dipole moment), tensor (e.g., electronic susceptibility), scalar field (e.g., electrostatic potential), and vector field (e.g., electrostatic potential gradient). According to the physical meaning, molecular descriptors can be divided into composition descriptors, molecular property descriptors, topological descriptors, and geometric descriptors. According to the structural dimension, molecular descriptors can range from 1D to 6D. Molecular descriptors can further be divided into experimental descriptors (logP, molar refractivity, dipole moment, polarizability, etc.) and theoretical descriptors (e.g., atom number, molecular weight, atom-type count, etc.). These different types of molecular descriptors are widely used in molecular data mining, compound diversity analysis, and compound activity prediction.
2D-QSAR
It is intuitive and concise to distinguish molecular descriptors according to the structural dimension (Azari and Iranmanesh, 2015; Morell et al., 2005; Ivanciuc and Braun, 2006; Karelson et al., 1996; Sandberg et al., 1998; Sheridan et al., 1996). One-dimensional descriptors are scalars that describe aggregated information, such as atom count, bond count, molecular weight, atomic properties, and fragment count (Shimamoto, 1999). Although simple, 1D descriptors are rather degenerative—disparate compounds may have the same value. Therefore, a 1D descriptor is usually used in addition to other high-dimensional descriptors, or combined with other 1D descriptors to form a vector.
Most molecular descriptors in the literature belong to 2D, which defines the connectivity of atoms in a molecule according to the properties of chemical bonds, including topological index, molecular profile, 2D autocorrelation, and chemical fingerprints (Hetényi et al., 2006). Two-dimensional descriptors generally deliver simple and useful structural information, which is rotation and translation invariant and is also invariant when the local structure is optimized. Another important invariance is graphic: the descriptor value does not change with the renumbering of the graphic nodes (vertices), which is very useful for structural differentiation. To facilitate the analysis of the large amount of 2D descriptors, Hong et al. reported the Mold2 system, which can quickly generate 200 types of 2D descriptors for large-scale composite data sets (Hong et al., 2008). Other commonly used commercial software for the generation of 2D descriptors includes the dragon system, which can generate up to 5000 2D molecular descriptors (Mauri et al., 2006). More detailed information about 2D descriptors can be found in the studies by Durant et al., 2002; Duvenaud et al., 2015; Lo et al., 2018; Polanski, 2009; and Rogers and Hahn, 2010.
3D-QSAR
3D-QSAR facilitates the study of QSAR by introducing the 3D structural information of the drug molecule (Cho et al., 2012; Choi et al., 2006). Compared with 2D-QSAR, it has clearer physical meaning and richer information and can better reflect the nonbonding interaction characteristics between the drug and target molecules. Since 1980, 3D-QSAR has gradually replaced 2D-QSAR and become one of the main methods of mechanism-based rational drug design.
The 3D-QSAR chemical descriptors include autocorrelation descriptors, substituent constants, surface and volume descriptors, quantum chemical descriptors, and unique molecular scaffold (Todeschini and Consonni, 2009). These 3D chemical descriptors are useful to derive new drugs from an existing one through scaffold hopping. That is, replacing the existing drug's scaffold with others without significantly affecting the drug's binding activity. The chemical descriptors are obtained by extracting chemical features of the molecules from their conformations (Lipkowitz and Boyd, 2007), which can generally be obtained from crystal structures or from quantum chemistry computation by programs such as CORINA (Gasteiger et al., 1990). The criterion for the determination of molecular conformations is usually the lowest free energy. However, the actual conformation of the molecule corresponds to low energy but not necessarily the lowest one, and a molecule can switch between multiple conformations. Relevant experiments have confirmed that the in vivo active conformation of a drug molecule is generally in a low energy state but not the lowest. As such, it is not guaranteed that the predicted bioactivity can be verified by experiments. These complexities represent the key limitations of generating 3D chemical descriptors.
Commonly used 3D fingerprints include chemical characteristics based on pharmacological patterns, surface properties, molecular volume, and molecular interaction fields, of which the most famous one is based on molecular interaction field (MIF) (Hayakawa et al., 2020), which was realized by Goodford in the GRID program (Goodford, 1985). The MIF fingerprint is obtained by placing the ligand in a rectangular grid with fixed intervals to calculate the fingerprint characteristics of each grid point. In other words, the electrostatic force, 3D coordinates, hydrophilic (hydrophilic) and other dimensions are calculated independently for each grid point. The obtained MIF fingerprint can then be used in a specific 3D-QSAR model to predict the activity relationship between the molecules comprising the complex. More detailed information about 3D-QSAR models can be found in the studies by Baskin and Zhokhova, 2013; Cramer et al., 1988; Datar et al., 2006; Dixon et al., 2006; Gerhard Klebe and Mietzner, 1994; Gohlke and Klebe, 2002; Hopfinger, 1980; Low and Vinter, 2008; Simon et al., 1977; Varela et al., 2012; Veselovsky et al., 2001; and Walters and Hinds, 1994.
Higher dimensional QSAR
Three-dimensional QSAR is computational costly, and its performance is sensitive to the changes in the conformation/orientation of the ligand. To overcome these drawbacks, higher dimensional QSAR models have been developed. Four-dimensional QSAR has solved the conformation/orientation problem by simultaneously considering multiple structural conformations (Andrade et al., 2010). For example, Ash and Fourches calculated a 3D descriptor based on a 20-ns MD simulation trajectory of the atoms of Erk2 (Ash and Fourches, 2017). This 4D (3D space + 1D time) chemical descriptor can effectively distinguish the most active Erk2 inhibitors from those inactive and highly enriched Erk2 inhibitors. Five-dimensional QSAR takes into account factors such as receptor flexibility and inducible fit (Vedani and Dobler, 2002). Six-dimensional QSAR takes into account an additional factor, namely, the effect of solvation on the main receptor-ligand interaction (Vedani et al., 2005).
Search based on compound structure similarity
Chemical similarity search is one of the most popular techniques for drug discovery based on ligands (Willett et al., 1998). It aims to query similar compounds with known active molecules in terms of structure from a database. The basic assumption is that compounds that are similar in function have similar chemical structures. However, this assumption is not always accurate. For example, the "active cliff" effect (Hu and Bajorath, 2020), a situation in which a small modification to a functional group leads to a sudden change in activity, does not meet the aforementioned assumptions and may lead to failure. To evaluate the structural similarity between two molecules, the Tanimoto coefficient (Tc, known as the Jaccard index), a measure of the similarity between two compounds, was proposed (Lipkus, 1999). A higher Tc score indicates that two compounds are more similar, but Tc does not provide details regarding what kind of chemical group they share. More detailed information about 3D chemical similarity can be found in the studies by Bero et al., 2018; Cheeseright et al., 2008; Dossetter et al., 2013; Ferreira and Couto, 2010; Lo et al., 2016; and Rush et al., 2005.
Further considerations
The success of QSAR lies in the fact that the difference in the representation of the ligand structure truly represents the essential difference in the scope of the ligand. For example, the difference in bioactivity of two compounds is solely determined by their structural difference, no matter how complex the subsequent physicochemical and biological interactions are. The ultimate goal of ligand comparison is to maximize the space of positive and negative samples. If the developed QSAR models are expected to be useful for drug synthesis in addition to drug screening, properties need to be considered such as synthesizability, hydrophobicity, drug-likeness, Lipinski rule, and false-positive issues (Alam and Khan, 2017).
Results
QSAR models based on ML
The ML-based QSAR model currently plays a notable role in drug design and screening, property prediction, category prediction, etc. Since the 1990s, ML techniques such as support vector machines (SVMs) and random forests (RFs) have been widely used to discover or design new medicines. For example, its application to a scoring function based on molecular docking has been reported (Lima et al., 2016). Data mining based on chemical graphs can derive a set of two-dimensional or three-dimensional chemical descriptors, which are packaged into chemical fingerprints in various ML models and prediction tasks. A key innovation in this field is the combination of big data and ML, including the mining of gene sequences and protein structure, single-cell sequences, multi-omics interaction data, and the interaction between proteins and genes, which enables the QSAR model to learn and predict a wider range of biological laws.
In the past, QSAR models focused only on the predictive ability of candidate compounds; thus, the commonly used ML models are discriminative prediction models. ML methods are mainly divided into discriminant prediction models and generative models (Jebara and Pentland, 2001). The essence of the discriminative model is to learn the mapping relationship between the characteristics of the characterizing sample and the prediction target from the data, and it is related to pattern recognition, which means that the model can only discriminate and predict. The generative model directly learns the distribution of data from the sample. In addition to being combined with the discriminant model, the generative model can also directly generate new samples based on the learned distribution and is thus used to generate candidate compounds. The methods to learn patterns hidden in data are mainly divided into supervised learning, semisupervised learning, unsupervised learning, and self-supervised learning. Because the QSAR model focuses only on the predictive ability of a model for candidate compounds, we introduce only the discriminant models commonly used under supervised learning. The key to this type of model is the selection of features, the selection of the algorithm itself, and the quality of the training set. Most aspects of feature construction have been included in the chemical descriptor section of the previous chapter. Examples of the extracted features can be found in the literature (e.g., Tomal et al., 2016).
Conformation-related indicators have been taken into account in the QSAR, and conformational calculations for isomers can be calculated using quantum chemistry, semiempirical methods, and molecular dynamics simulations. Because the relative stability of conformational isomerism is related to toxicity detection, the conformation of the compound may change as the solvent environment changes, which affects the three-dimensional correlation characteristics and then impacts the final screening result. Therefore, the methods of calculating the isomer conformations can be used to predict whether the relative stability of the conformation will affect the experimental results. In addition, the ML-based QSAR method can be applied to examine the energy changes between conformational changes, to determine how the conformation is affected by the solvent environment, and to predict the relationship between conformational changes and molecular energy (Lokuge et al., 2010; Singh et al., 2016).
The importance of these features varies in different structure-activity or attributes prediction tasks, but ML can learn from the data which features are more important. Therefore, we need only to exhaust all the features related to the nature of QSAR tasks as much as possible and apply the traditional ML algorithm to learn from the training set.
ML-based QSAR methods are effective under the usual condition that the system under study is so complex that it cannot be physically modeled. Indeed, many physical and chemical properties are difficult to calculate by theoretical methods such as density functional theory (DFT) or MD, but they can usually be computed by cheminformatics models, particularly the water solubility and logP (the logarithm of octanol: water partition coefficient), which are directly related to drug activity. The properties that are indirectly related to drug activity, such as the melting point, solubility, and sublimation energy, can also be calculated by cheminformatics. Of course, the QSAR methods may not be suitable when the underlying mechanisms are sufficiently clear. For example, quantities such as dipole moment, polarizability, and vibration frequency can be directly computed by quantum chemistry.
The characteristics of a compound in the traditional QSAR model are produced in an ideal environment. In reality, however, the solvent environment is not ideal. For example, we know that acidic chemicals are easily absorbed in an acidic environment. Therefore, to achieve the desired effect for a drug in an actual environment, the hydrophilicity and lipophilicity of the drug need to be considered (Ditzinger et al., 2019). The main environmental factors of a solvent can be found in the study by Bruno et al., 2021.
With the aforementioned preparation, the next stage is to build a QSAR model based on ML, which mainly consists of the following steps:
-
(1)
“Molecular coding,” which uses a vector of various characteristics to represent compound molecules, where the chemical characteristics and properties can be learned from the chemical structure or experimental data. The molecular characteristics should be independent as much as possible to avoid excessive correlation. The biological activity data and solvent environmental coefficients should be as accurate and sufficient as possible, and they are best obtained by conventional experiments.
-
(2)
An appropriate number of compounds must be chosen to construct the training/testing sets. Ideally, conformation optimization through MM/QM is carried out first to improve the data quality. Unsupervised learning is used to identify the most relevant attributes and reduce the dimensionality of the feature vector. Supervised ML model is applied to discover an empirical function (explicit or implicit) that can achieve the optimal mapping between the input feature vector and the biological activity.
-
(3)
Internal/external verification of the model is performed to determine its applicability and predictability. After generating exhaustive feature sets, the algorithm determines which feature combinations can be used for new predictions by finding the largest difference between positive and negative samples in the training set; thus, similar data distributions between the training and testing sets should exist. The techniques to detect out of distribution (OOD) can be found in the study by Hendrycks and Gimpel, 2016. The key to constructing a ML model is to constantly add and try more feature combinations, change the divisions of the training set, and adjust the algorithm parameters until the best model is obtained.
ML models trained on specific target sets are not universal, but the latest ML methods are improving versatility. Advances in computing power and software performance have also been used to improve QSAR models. By continuously integrating new algorithms and descriptive features, the model can be continuously optimized. Although the general idea of the ML-based QSAR is the same, the input characteristics may be different for different targets. The latest progress is the ability to learn general representations, such as self-learning, multitask learning, AutoEncoder, Generative Adversarial Networks (GAN), and Bidirectional Encoder Representations from Transformers (BERT) (Caruana, 1997; Devlin et al., 2019; Goodfellow et al., 2014; Liou et al., 2014; Xu et al., 2020). The QSAR learning models still face several problems beyond their construction (Dearden et al., 2009), which are detailed in the study by Artem et al., 2014.
DL invigorates QSAR
With excellent performance in imaging, speech, machine translation, etc (Minar and Naher, 2018), DL has entered into many biological fields, including genes, proteins, metabolites, microbiomes, and population-wide genetic variation, synthetic biology, drug discovery, and diseases (Alkawaa et al., 2018; Golkov et al., 2020; Hill et al., 2018; Zeng et al., 2021). The promising DL methods include capsule networks (Inokuma et al., 2010; Xi et al., 2017), multitask learning (Antropova et al., 2017; Wang et al., 2015b; Zhu et al., 2016), GANs, self-encoding decoders (Marchi et al., 2015; Wang et al., 2018; Xu et al., 2014; Yao et al., 2017; Zhao et al., 2016), Variational AutoEncoders (VAEs) (Panych et al., 2015), Long Short Term Memory Networks (LSTMs) (Baytas et al., 2017; Gers and Schmidhuber, 2001; Graves et al., 2005; Yildirim, 2018), transfer learning (Fernandes et al., 2017; Pan and Yang, 2010; Paul et al., 2016; Zoph et al., 2016), deep neural networks (DNNs) (Yoshioka et al., 2014), and CNNs (Horváth et al., 2017; Luo, 2015; Parashar et al., 2017; Wang, 2013; Xue et al., 2016). Because the biological information is generally rich, multitask learning can be performed to gain a better structural representation. For example, the microbial metabolic network and the loop network formed by the parasitic relationship are very scarce; they can be generated by a GAN. When using DL to predict the protein structure from a gene sequence, VAE can be used to encode compound structures and then be combined with the GAN for learning (Lin, 2009; Widera, 2010; Yu et al., 2015). With switch networks formed by atoms, strategy networks and Monte Carlo trees can be used to predict chemical reactions (Anderson and Long, 2003). CNN can be used to perform learning based on force fields and the protein-ligand complex (Clore and Gronenborn, 2010). A matching network, which is a variant of one-hot learning (Alaya et al., 2019), can be used to evaluate whether a new compound matches the receptor (Deka and Quddus, 2014; Tan et al., 2013). The cheminformatics-based LSTM methods are also frequently reported (Shu et al., 2018). The advantages, notwithstanding, DL entails a large amount of data and computation and is essentially a “black-box”; thus, it cannot replace all traditional ML methods, which are currently irreplaceable owing to better interpretability and lower data requirements.
Biological applications of DL
Since the late 1990s, DL has become a useful tool for drug discovery and can help researchers understand and construct the relationship between a chemical structure and its biological activity (Jing et al., 2018) (Figure 3). In the early days, structure-based methods such as cheminformatics are most successful in drug discovery (Feng et al., 2013; Hwang et al., 2020; Kim et al., 2010; Nam et al., 2014). They were used to conduct chemical structure searches, generate descriptors, construct fingerprints, and analyze chemical similarity. Expert experiences, combined with manually generated explanatory rules, were used to screen compounds. The later DL can capture the nonlinearity and complex relationships in the available data and generate more compact features than those manually generated by experts. Through multilayer networks, DL can directly process raw, unstructured data such as sequences and three-dimensional structures and has better nonlinear fitting capabilities. These multilayer networks are proven to have infinite VC dimensions (Vapnik et al., 1994) and thus can divide any data space. DL has been successfully applied to CADD (Chen, 2014; Quang et al., 2015). Although DL is more suitable for ligand-based drug discovery, there exist several interesting structure-based DL applications. In the AtomNet method (Wallach et al., 2015), for example, the input molecule is discretized into a three-dimensional grid and then sent to a convolutional neural network (CNN) and a fully connected layer to predict the binding affinity. This model needs not to preprocess but uses a learnable representation to identify the pair data of interacting atoms. AtomNet uses the same atom space to describe both the receptor and ligand, which is more natural, requires no preprocessing, and allows for the characterization and regularization of the interaction between the receptor and ligand atoms. To apply AtomNet, the 3D protein-ligand complex needs some conversion and coding (Klebe, 2000). First, the complex is formalized as a cube with 20 angstrom side length, and the cube is placed at the geometric center of the ligand. Then, a 3D grid with 1-angstrom resolution is used to discretize the positions of heavy atoms. This method allows for the representation of an atom as a 4D vector (three coordinates and one feature value) and the whole input as a 4D tensor. As another example, the protein-ligand complexes in the PDBbind database were used to train and test the neural network (Evers et al., 2003). The complexes were protonated and charged using UCSF Chimera with Amber ff14SB for standard residues and AM1-BCC for nonstandard residues and ligands. This study can determine which atom deletion causes the largest decrease in the predicted value. It also found novel interactions between the receptor residues and the ligand: e.g., Tyr693 forms a hydrogen bond with the ligand; Met713 forms a hydrophobic interaction with the ligand.
Figure 3.

Main steps and scenarios of ML-based drug discovery
Another important biological application of DL is the clarification of the molecular mechanisms of how protein structural changes and amino acid mutations cause the change of protein-protein interaction (PPI) networks. In a DL application, instead of using the traditional MM/QM (Vesely, 2001), the protein structure is first divided into fragments and partitions, and then the most effective fragments are predicted. DL can, on the one hand, seek interpretability from the successfully trained model, i.e., the interpretation of the feature contribution of a single sample; the combination of existing field experiences then helps us gain insights into complex relationships. On the other hand, DL can combine raw data to generate specific functional characterization and fragment representation; thus, it can facilitate the study of the mechanism of a protein structure.
Comparing DL with traditional ML
DL and traditional ML have their respective advantages and application suitability (Camacho et al., 2018; Chan et al., 2019; Pei et al., 2019; Yuan et al., 2019). The traditional ML relies more on the domain knowledge (theories and mathematical models), while DL directly extracts patterns, which are not always explainable, from the raw data. For tasks such as image analysis, DL is certainly better; it is particularly useful in de novo molecular design and reaction prediction (Button et al., 2017; Hartenfeller et al., 2012; Kang and Liu, 2021; Schneider and Schneider, 2018). For tasks with structured descriptors as input, DL seems to be at least as effective as traditional ML methods. Although DL can achieve better performance in biological activity prediction through multitasking, ML-based QSAR can also achieve better performance through continuous improvement of the feature engineering, which can be achieved by dividing the protein into fragments and functional regions to remove the influence of the orientation, flexibility, and solvent of a protein molecule. Although DL is better for automatic feature engineering, it entails a large amount of data. In contrast, ML can use a small amount of training data (Altae-Tran et al., 2016).
One common ground of DL and traditional ML is the learning algorithms, including supervised and unsupervised learning. Supervised learning is related to a specific task and thus can obtain task-related feature representations. Its input data must be of a similar structure. If a new task is related to the original one, one can generally use the representations obtained by supervised learning to join the new task to improve the effectiveness of the model. Unsupervised learning can obtain a data-distributed representation of its own existence, which is irrelevant to a specific task. It yields the patterns themselves, that is, the general characteristics of the data set. The use of unsupervised learning is promising to create new synthetic biology samples and then to determine the legibility of the new samples based on the structural knowledge. For example, one can use the general common-sense model learned by unsupervised learning to check whether a synthesized new compound is reasonable. Furthermore, unsupervised training can obtain some general common sense. If this common sense is a prerequisite for certain tasks, then the representations learned can be used to determine whether a sample belongs to this pattern. Multitask learning is a kind of supervised learning with some unsupervised features. It has great advantages when the data are scarce, which is typical in the study of the parasitic relationship between the host and microorganisms in a microbiome.
Combining DL with traditional ML
Although traditional ML and DL have their own advantages and disadvantages, they can be used in combination. For example, the DL-based face recognition is apparently better, but the learned features may be meaningless and the interpretability is thus lacking. This shortcoming can be compensated by the ML-based recognition. Therefore, their combination can achieve a better performance. In the following, such a combined model is outlined (Figure 4).
Figure 4.

ML-DL combined QSAR model
Data-oriented feature extraction
Different models have different requirements of the input data. To implement traditional ML models, the input data should be well prepared to reduce feature dimension and promote feature grouping; the interactions between the features need to be constructed manually, which relies heavily on experience. To implement DL models, raw data are welcome because DL algorithms can automatically extract features. In image recognition, for example, DL can extract hierarchical features from the pixel-level raw data through its convolution kernel. Feature extraction has two types: explicit and implicit. The explicit features, such as chemical descriptors, chemical fingerprints, physical and chemical properties, chargeability, substituent groups, fragments, geometric properties, are usually generated by experts. Being overlap with each other or redundant, these features may need to be processed by applying algorithms such as restricted Boltzmann machines (Pinaya et al., 2016; Zhang et al., 2015) and AutoEncoder to generate new, more compact, distributed features. The implicit feature extraction is very useful in processing sequences such as amino acid sequences, the atomic sequence of a compound, the 3D structure of a protein, and atomic grid structures, where the features can be learned by directly applying sequence processing algorithms including CNN, LSTM, and automatic codecs. For other applications, algorithms such as word2vec (Altszyler et al., 2018; Wang et al., 2016), BERT (Sun et al., 2019), GANs (Yi et al., 2019), and AutoEncoder can be used for unsupervised training.
The dimension of features and the number of data samples must be well balanced. A large feature dimension necessitates the acquisition of more data, which may be costly or even impossible. To solve this problem, the usual method is to filter the features, group them, or apply AutoEncoder or VAE for dimensionality reduction. However, this method usually cannot reduce the amount of data because both unsupervised feature expressions and supervised learning are involved. We therefore propose the following ML-DL combined strategy. By using the traditional ML, various explanatory features related to the training target are first constructed and grouped based on common underlying information; dimensionality reduction is then performed by applying feature selection or AutoEncoder, and the compact and effective explicit features are obtained. By using DL, the other types of data (e.g., sequence and structure data) can be used for unsupervised training to obtain compact and implicitly distributed features. Finally, both the implicit features (which reflect more information about the low-level data) and explicit features (which are more relevant to the task) are sent to the DNN to train the model.
Iterative training to generate more effective features
The explicit features of chemicals, usually in the form of molecular descriptor and fingerprint, are extracted from the structural chemical data and are grouped according to their correlation values. The redundancy of features is reduced by filtering out invalid features by using traditional ML algorithms such as xgboost and gbdt. These steps are iterated several times to determine the features to be input to a module called stacking (Palangi et al., 2014; Zhan et al., 2018; Zhang et al., 2017). The input samples to stacking include both chemical data and the features (sample = data + feature). The samples are divided into N parts, N − 1 of which are used to train a classifier and the remaining one is used to test the prediction made by the trained classifier. In total, N such classifiers are used to obtain N new sets of features, which replace the old sets of features in the next round of iteration.
On the other hand, the implicit features of chemicals are extracted from the nonstructural chemical data such as the chemical sequence, molecular bond, 3D coordinates by using DL methods such as transformer, CNN, and GNN. The samples are then trained by a DNN, whose last layer output is saved and then, together with the above explicit features, is sent to a stacking module consisting of DNN and ML. Through training by the stacking module, a more effective model is obtained.
Iterative integration of different models
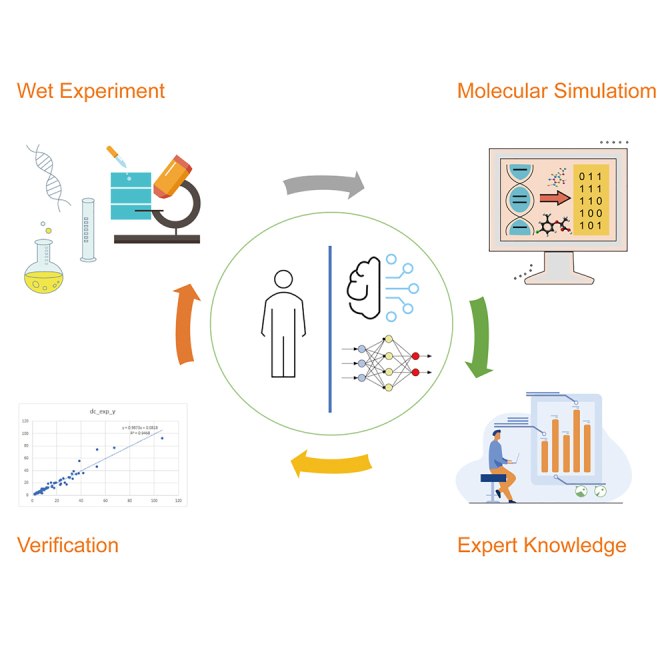
QSAR is based on both experience-driven and data-driven research models, including wet experiments, MD simulation, ML, and DL. Their cooperation makes QSAR increasingly powerful. The experiments usually do not reveal the reaction mechanisms at the molecular and atomic levels, which can be revealed to a certain extent through MD simulation. ML-based QSAR is data driven (good at processing 2D and 3D data) and is oriented by feature extraction. The limitations of traditional ML are that the data are usually low-dimensional and the features must be interpretable and manually constructed. To overcome these limitations, DL is developed to automatically extract features from both unstructured and structured data (which could be high-dimensional). The integration of the four models would make QSAR powerful, for which a framework is provided (Figure 5). In the framework, the four models communicate with each other and adjust themselves iteratively to optimize the synergism of our field experience with ML/DL predictions.
Figure 5.

Iterative integration of wet experiments, MD simulation, traditional ML, and DL
There are two research studies that have implemented part of the framework in Figure 5. Yasushi Okuno's group at Kyoto University proposed an interdisciplinary framework called DEFMap, which combines experimental data, MD simulation, and DL to extract protein dynamic information from cryo-EM density maps (Matsumoto et al., 2021). Shuguang Yuan's group used part of the framework to prove that the frozen structure analyzed in the traditional artificial cell membrane environment is very different from the real cell membrane environment (Zhang et al., 2021), subverting the traditional understanding that the 3D structure of the membrane protein is the same as its physiological state in the artificial cell membrane or precipitant environment.
Conclusions
As a part of Big Data science, ML and DL are based on information theory and data fitting theories, where predictions depend mainly on a large amount of effective and reliable data, high-speed computation (for MM/QM simulations to process experiment data such as those from NMR, X-ray, and cryo-EM), and the number of compounds. These requirements cannot be met in many areas of computational biology; thus, ML/DL does not always work well. This problem can be partly solved in some fields by using powerful public tools such as AlphaFold (Senior et al., 2019). The wide application of ML in many fields depends on its human-like problem-solving capacity, including classification and clustering, explanation and verification, relevance and factor importance. But now the integration of iterative thinking is ever-increasingly important. Only through the cyclical upward process of prediction-verification-update can we form a systematic way of thinking and solve the problem from an overarching perspective.
In this article, we primarily reviewed ML-/DL-based QSAR methods. In addition to traditional descriptors, we emphasized the integration of MD with experimental solvent-related data. We also proposed a better framework that takes advantage of the respective merits of ML and DL to process different forms of data. The framework, through iteratively adjust itself, can potentially reveal the importance of some manual features, the correlation between features, and explain the contribution of different features in a single sample. The obtained embedded features or distributed vectors can help to understand the relationship between the constituent elements and the importance of local structures. The framework would ultimately achieve high efficiency and low cost in the fields of biomedicine and materials, especially in terms of QSAR.
Acknowledgments
This work was partly supported by the National Natural Science Foundation of China (61773196, 32070681), Guangdong Provincial Special Projects on COVID-19 (2020KZDZX1182), Guangdong Provincial Key Laboratory Funds (2019B030301001, 2017B030301018), Shenzhen Research Funds (JCYJ20170817104740861), Shenzhen Peacock Plan (KQTD2016053117035204), and by the Center for Computational Science and Engineering of Southern University of Science and Technology. The kind help of Prof. No's group is acknowledged.
Author contributions
J.M., J.A, K.T.N., X.Z., and G.W. conceived the idea, wrote the article, and prepared the figures. All the authors discussed the results and commented on the manuscript.
Declaration of interests
The authors declare they have no conflict of interest.
Contributor Information
Kyoung Tai No, Email: ktno@yonsei.ac.kr.
Guanyu Wang, Email: wanggy@sustech.edu.cn.
References
- Adrian M., Dubochet J., Lepault J., Mcdowall A.W. Cryo-electron microscopy of viruses. Nature. 1984;308:32–36. doi: 10.1038/308032a0. [DOI] [PubMed] [Google Scholar]
- Alam S., Khan F. 3D-QSAR studies on maslinic acid analogs for anticancer activity against breast cancer cell line MCF-7. Sci. Rep. 2017;7:6019. doi: 10.1038/s41598-017-06131-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alaya M.Z., Bussy S., Gaïffas S., Guilloux A. Binarsity: a penalization for one-hot encoded features. J. Mach. Learn. Res. 2019;20:1–34. [Google Scholar]
- Alkawaa F.M., Chaudhary K., Garmire L.X. Deep learning accurately predicts estrogen receptor status in breast cancer metabolomics data. J. Proteome Res. 2018;17:337–347. doi: 10.1021/acs.jproteome.7b00595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altae-Tran H., Ramsundar B., Pappu A.S., Pande V. Low data drug discovery with one-shot learning. ACS Cent. Sci. 2016;3:283. doi: 10.1021/acscentsci.6b00367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altszyler E., Sigman M., Slezak D.F. Corpus specificity in LSA and Word2vec: the role of out-of-domain documents. RepL4NLP (Association for Computational Linguistics) 2018:1–10. [Google Scholar]
- Alves V.M., Muratov E.N., Capuzzi S.J., Politi R., Low Y., Braga R.C., Zakharov A.V., Sedykh A., Mokshyna E., Farag S. Alarms about structural alerts. Green. Chem. 2016;18:4348–4360. doi: 10.1039/C6GC01492E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson A.C. The process of structure-based drug design. Chem. Biol. 2003;10:787–797. doi: 10.1016/j.chembiol.2003.09.002. [DOI] [PubMed] [Google Scholar]
- Anderson J.B., Long L.N. Direct Monte Carlo simulation of chemical reaction systems: prediction of ultrafast detonations. J. Chem. Phys. 2003;118:3102–3110. [Google Scholar]
- Andrade C.H., Pasqualoto K.F.M., Ferreira E.I., Hopfinger A.J. 4D-QSAR: perspectives in drug design. Molecules. 2010;15:3281–3294. doi: 10.3390/molecules15053281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antropova N., Huynh B., Giger M. Multi-task learning in the computerized diagnosis of breast cancer on DCE-MRIs. arXiv. 2017 arXiv:1701.03882. [Google Scholar]
- Ash J., Fourches D. Characterizing the chemical space of ERK2 Kinase inhibitors using descriptors computed from molecular dynamics trajectories. J. Chem. Inf. Model. 2017;57:1286–1299. doi: 10.1021/acs.jcim.7b00048. [DOI] [PubMed] [Google Scholar]
- Artem C., Muratov E.N., Denis F., Alexandre V., Baskin I.I., Mark C., John D., Paola G., Martin Y.C., Roberto T. QSAR modeling: where have you been? Where are you going to? J. Med. Chem. 2014;57:4977. doi: 10.1021/jm4004285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ash S., Cline M.A., Homer R.W., Tad Hurst A., Smith G.B. SYBYL line notation (SLN): a versatile language for chemical structure representation. J. Chem. Inf. Comput. Sci. 1997;37:71–79. [Google Scholar]
- Atzori L., Iera A., Morabito G. The internet of things: a survey. Comput. Netw. 2010;54:2787–2805. [Google Scholar]
- Azari M., Iranmanesh A. Edge-Wiener descriptors in chemical graph theory: a survey. Curr. Org. Chem. 2015;19:219–239. [Google Scholar]
- Bailey T.L., Elkan C. Unsupervised learning of multiple motifs in biopolymers using expectation maximization. Mach. Learn. 1995;21:51–80. [Google Scholar]
- Bash P.A., Field M.J., Karplus M. Free energy perturbation method for chemical reactions in the condensed phase: a dynamic approach based on a combined quantum and molecular mechanics potential. J. Am. Chem. Soc. 2002;109:8092–8094. [Google Scholar]
- Baskin I.I., Zhokhova Nelly I. The continuous molecular fields approach to building 3D-QSAR models. J. Comput. Aided Mol. Des. 2013;28:427–442. doi: 10.1007/s10822-013-9656-4. [DOI] [PubMed] [Google Scholar]
- Baytas I.M., Xiao C., Zhang X., Wang F., Jain A.K., Zhou J. SIGKDD; 2017. Patient Subtyping via Time-Aware LSTM Networks; pp. 65–74. [Google Scholar]
- Bazoon M., Stacey D.A., Cui C., Harauz G. A hierarchical artificial neural network system for the classification of cervical cells. ICNN. 2002;94:3525–3529. [Google Scholar]
- Bero S., Muda A., Choo Y.-H., Muda N., Pratama S. Weighted Tanimoto coefficient for 3D molecule structure similarity measurement. arXiv. 2018 arXiv:1806.05237. [Google Scholar]
- Bohacek R.S., Mcmartin C., Guida W.C. The art and practice of structure-based drug design: a molecular modeling perspective. Med. Res. Rev. 1996;16:3. doi: 10.1002/(SICI)1098-1128(199601)16:1<3::AID-MED1>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Bret C., Field M.J., Hemmingsen L. A chemical potential equalization model for treating polarization in molecular mechanical force fields. Mol. Phys. 2000;98:751–763. [Google Scholar]
- Bruno C.D., Harmatz J.S., Duan S.X., Zhang Q., Chow C.R., Greenblatt D.J. Effect of lipophilicity on drug distribution and elimination: influence of obesity. Br. J. Clin. Pharmacol. 2021;87:3197–3205. doi: 10.1111/bcp.14735. [DOI] [PubMed] [Google Scholar]
- Butterfoss G.L., Kuhlman B. Computer-based design of novel protein structures. Annu. Rev. Biophys. Biomol. Struct. 2006;35:49. doi: 10.1146/annurev.biophys.35.040405.102046. [DOI] [PubMed] [Google Scholar]
- Button A.L., Hiss J.A., Schneider P., Schneider G. Scoring of de novo designed chemical entities by macromolecular target prediction. Mol. Inf. 2017;36:1600110. doi: 10.1002/minf.201600110. [DOI] [PubMed] [Google Scholar]
- Cai X., Zheng W., Li Z. High-throughput screening strategies for the development of anti-virulence inhibitors against staphylococcus aureus. Curr. Med. Chem. 2019;26:2297–2312. doi: 10.2174/0929867324666171121102829. [DOI] [PubMed] [Google Scholar]
- Camacho D.M., Collins K.M., Powers R.K., Costello J.C., Collins J.J. Next-generation machine learning for biological networks. Cell. 2018;173:1581–1592. doi: 10.1016/j.cell.2018.05.015. [DOI] [PubMed] [Google Scholar]
- Capener C.E., Shrivastava I.H., Ranatunga K.M., Forrest L.R., Smith G.R., Sansom M.S.P. Homology modeling and molecular dynamics simulation studies of an inward rectifier potassium channel. Biophys. J. 2000;78:2929–2942. doi: 10.1016/S0006-3495(00)76833-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carneiro G., Chan A.B., Moreno P.J., Vasconcelos N. Supervised learning of semantic classes for image annotation and retrieval. IEEE Trans. Pattern Anal. Mach. Intell. 2007;29:394–410. doi: 10.1109/TPAMI.2007.61. [DOI] [PubMed] [Google Scholar]
- Caruana R. Multitask Learn. Mach. Learn. 1997;28:41–75. [Google Scholar]
- Cavasotto C.N., Phatak S.S. Homology modeling in drug discovery: current trends and applications. Drug Discov. Today. 2009;14:676–683. doi: 10.1016/j.drudis.2009.04.006. [DOI] [PubMed] [Google Scholar]
- Chan H.C.S., Shan H., Dahoun T., Vogel H., Yuan S. Advancing drug discovery via artificial intelligence. Trends Pharmacol. Sci. 2019;40:592–604. doi: 10.1016/j.tips.2019.06.004. [DOI] [PubMed] [Google Scholar]
- Chang S.-K., Jang M., Han S.Y., Lee J.H., Kang M.H., No K.T. Molecular recognition of butylamines by Calixarens-based ester ligands. Chem. Lett. 1992;21:1937–1940. [Google Scholar]
- Cheeseright T.J., Mackey M.D., Melville J.L., Vinter J.G. FieldScreen: virtual screening using molecular fields. Application to the DUD data set. J. Chem. Inf. Model. 2008;48:2108–2117. doi: 10.1021/ci800110p. [DOI] [PubMed] [Google Scholar]
- Chen Y. UC Irvine; 2014. Machine Learning for Large-Scale Genomics: Algorithms, Models and Applications. [Google Scholar]
- Cheng A., Best S.A., Jr K.M.M., Reynolds C.H. GB/SA water model for the Merck molecular force field (MMFF) J. Mol. Graph. Model. 2000;18:273–282. doi: 10.1016/s1093-3263(00)00038-3. [DOI] [PubMed] [Google Scholar]
- Cheng T., Li Q., Zhou Z., Wang Y., Bryant S.H. Structure-based virtual screening for drug discovery: a problem-centric review. AAPS J. 2012;14:133–141. doi: 10.1208/s12248-012-9322-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho S.G., No K.T., Goh E.M., Kim J.K., Shin J.H., Joo Y.D., Seong S.Y. Optimization of neural networks architecture for impact sensitivity. Bull. Korean Chem. Soc. 2005;26:399–408. [Google Scholar]
- Cho N.C., Cha J.H., Kim H., Kwak J., Kim D., Seo S.H., Shin J.S., Kim T., Park K.D., Lee J. Discovery of 2-aryloxy-4-amino-quinazoline derivatives as novel protease-activated receptor 2 (PAR2) antagonists. Bioorg. Med. Chem. 2015;23:7717–7727. doi: 10.1016/j.bmc.2015.11.016. [DOI] [PubMed] [Google Scholar]
- Cho Y.S., No K.T., Cho K.H. yaInChI: modified InChI string scheme for line notation of chemical structures. SAR QSAR Environ. Res. 2012;23:237–255. doi: 10.1080/1062936X.2012.657677. [DOI] [PubMed] [Google Scholar]
- Choi S.Y., Shin J.H., Ryu C.K., Nam K.Y., No K.T., Park Choo H.Y. The development of 3D-QSAR study and recursive partitioning of heterocyclic quinone derivatives with antifungal activity. Bioorg. Med. Chem. 2006;14:1608–1617. doi: 10.1016/j.bmc.2005.10.010. [DOI] [PubMed] [Google Scholar]
- Choi I., Kim S.Y., Kim H., Kang N.S., Bae M.A., Yoo S.-E., Jung J., No K.T. Classification models for CYP450 3A4 inhibitors and non-inhibitors. Eur. J. Med. Chem. 2009;44:2354–2360. doi: 10.1016/j.ejmech.2008.08.013. [DOI] [PubMed] [Google Scholar]
- Choi I., Kim H., Jung J., Nam K.Y., Yoo S.E., Kang N.S., No K.T. Bayesian model for the classification of GPCR agonists and antagonists. Bull. Korean Chem. Soc. 2010;31:2163–2169. [Google Scholar]
- Cieplak P., Dupradeau F.Y., Duan Y., Wang J. Polarization effects in molecular mechanical force fields. J. Phys. Condens. Matter. 2009;21:333102. doi: 10.1088/0953-8984/21/33/333102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clancey W.J., Shortliffe E.H. Readings in medical artificial intelligence. J. Am. Med. Assoc. 1985;253:3011–3012. [Google Scholar]
- Clore G.M., Gronenborn A.M. Structures of larger proteins, protein-ligand and protein-DNA complexes by multidimensional heteronuclear NMR. Protein Sci. 2010;3:372–390. doi: 10.1002/pro.5560030302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conneau A., Kiela D., Schwenk H., Barrault L., Bordes A. Supervised learning of universal sentence representations from natural language inference data. arXiv. 2017 arXiv:1705.02364. [Google Scholar]
- Cramer R.D. The inevitable QSAR renaissance. J. Comput. Aided Mol. Des. 2012;26:35–38. doi: 10.1007/s10822-011-9495-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer R.D., Patterson D.E., Bunce J.D. Comparative molecular field analysis (CoMFA). 1. Effect of shape on binding of steroids to carrier proteins. J. Am. Chem. Soc. 1988;110:5959–5967. doi: 10.1021/ja00226a005. [DOI] [PubMed] [Google Scholar]
- Csányi G., Albaret T., Payne M.C., De V.A. Learn on the fly": a hybrid classical and quantum-mechanical molecular dynamics simulation. Phys. Rev. Lett. 2004;93:175503. doi: 10.1103/PhysRevLett.93.175503. [DOI] [PubMed] [Google Scholar]
- Culler D., Karp R., Patterson D., Sahay A., Schauser K.E., Santos E., Subramonian R., Von Eicken T. LogP: towards a realistic model of parallel computation. ACM SIGPLAN Not. 1993;28:1–12. [Google Scholar]
- Datar P.A., Khedkar S.A., Malde A.K., Coutinho E.C. Comparative residue interaction analysis (CoRIA): a 3D-QSAR approach to explore the binding contributions of active site residues with ligands. J. Comput. Aided Mol. Des. 2006;20:343–360. doi: 10.1007/s10822-006-9051-5. [DOI] [PubMed] [Google Scholar]
- Dearden J., Cronin M.T.D., Kaiser K. How not to develop a quantitative structure-activity or structure-property relationship (QSAR/QSPR) SAR QSAR Environ. Res. 2009;20:241–266. doi: 10.1080/10629360902949567. [DOI] [PubMed] [Google Scholar]
- Degrado W.F. Proteins from scratch. Science. 1997;278:80–81. doi: 10.1126/science.278.5335.80. [DOI] [PubMed] [Google Scholar]
- Deka L., Quddus M. Network-level accident-mapping: distance based pattern matching using artificial neural network. Accid. Anal. Prev. 2014;65:105–113. doi: 10.1016/j.aap.2013.12.001. [DOI] [PubMed] [Google Scholar]
- Devlin J., Chang M.W., Lee K., Toutanova K. BERT: pre-training of deep bidirectional transformers for language understanding. NAACL (Association for Computational Linguistics) 2019 arXiv:1810.04805. [Google Scholar]
- Dijk E.L.V., Auger H., Yan J., Thermes C. Ten years of next-generation sequencing technology. Trends Genet. 2014;30:418–426. doi: 10.1016/j.tig.2014.07.001. [DOI] [PubMed] [Google Scholar]
- Ditzinger F., Price D.J., Ilie A.R., Köhl N.J., Jankovic S., Tsakiridou G., Aleandri S., Kalantzi L., Holm R., Nair A. Lipophilicity and hydrophobicity considerations in bio-enabling oral formulations approaches – a PEARRL review. J. Pharm. Pharmacol. 2019;71:464–482. doi: 10.1111/jphp.12984. [DOI] [PubMed] [Google Scholar]
- Dixon S.L., Smondyrev A.M., Knoll E.H., Rao S.N., Shaw D.E., Friesner R.A. PHASE: a new engine for pharmacophore perception, 3D QSAR model development, and 3D database screening: 1. Methodology and preliminary results. J. Comput. Aided Mol. Des. 2006;20:647–671. doi: 10.1007/s10822-006-9087-6. [DOI] [PubMed] [Google Scholar]
- Doo Ho Cho S.K.L., Bum Tae Kim K.T.N. Quantitative structure-activity relationship (QSAR) study of new fluorovinyloxyacetamides. Bull. Korean Chem. Soc. 2001;22:388–394. [Google Scholar]
- Dossetter A.G., Griffen E.J., Leach A.G. Matched molecular pair analysis in drug discovery. Drug Discov. Today. 2013;18:724–731. doi: 10.1016/j.drudis.2013.03.003. [DOI] [PubMed] [Google Scholar]
- Dubochet J., Adrian M., Chang J.J., Homo J.C., Lepault J., Mcdowall A.W., Schultz P. Cryo-electron microscopy of vitrified specimens. Q. Rev. Biophys. 1988;21:129–228. doi: 10.1017/s0033583500004297. [DOI] [PubMed] [Google Scholar]
- Durant J.L., Leland B.A., Henry D.R., Nourse J.G. Reoptimization of MDL keys for use in drug discovery. J. Chem. Inf. Comput. Sci. 2002;42:1273–1280. doi: 10.1021/ci010132r. [DOI] [PubMed] [Google Scholar]
- Duvenaud D., Maclaurin D., Aguilera-Iparraguirre J., Gómez-Bombarelli R., Hirzel T., Aspuru-Guzik A., Adams R.P. Convolutional networks on graphs for learning molecular fingerprints. arXiv. 2015 arXiv:1509.09292. [Google Scholar]
- Edwards C.D., Chris L., Peter E., Hessel E.M. Development of a novel quantitative structure-activity relationship model to accurately predict pulmonary absorption and replace routine use of the isolated perfused respiring rat lung model. Pharm. Res. 2016;33:2604–2616. doi: 10.1007/s11095-016-1983-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ertel W. In: Introduction to Artificial Intelligence. Ertel W., editor. Springer; 2017. Reinforcement learning; pp. 289–311. [Google Scholar]
- Evers A., Gohlke H., Klebe G. Ligand-supported homology modelling of protein binding-sites using knowledge-based potentials. J. Mol. Biol. 2003;334:327–345. doi: 10.1016/j.jmb.2003.09.032. [DOI] [PubMed] [Google Scholar]
- Ewing T.J.A., Makino S., Skillman A.G., Kuntz I.D. Dock 4.0: search strategies for automated molecular docking of flexible molecule databases. J. Comput. Aided Mol. Des. 2001;15:411–428. doi: 10.1023/a:1011115820450. [DOI] [PubMed] [Google Scholar]
- Fang Y., Hoh J.H., Spisz T.S. Ethanol-induced structural transitions of DNA on mica. Nucleic Acids Res. 1999;27:1943–1949. doi: 10.1093/nar/27.8.1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng E., Shin W.J., Zhu X., Li J., Ye D., Wang J., Zheng M., Zuo J.P., No K.T., Liu X. Structure-based design and synthesis of C-1- and C-4-modified analogs of zanamivir as neuraminidase inhibitors. J. Med. Chem. 2013;56:671–684. doi: 10.1021/jm3009713. [DOI] [PubMed] [Google Scholar]
- Feng X., Yang L.T., Wang L., Vinel A. Internet of things. Int. J. Commun. Syst. 2012;25:1101–1102. [Google Scholar]
- Fernandes K., Cardoso J.S., Fernandes J. Springer International Publishing); 2017. Transfer Learning with Partial Observability Applied to Cervical Cancer Screening. Pattern Recognition and Image Analysis Lecture Notes in Computer Science; pp. 243–250. [Google Scholar]
- Ferreira J.D., Couto F.M. Semantic similarity for automatic classification of chemical compounds. Plos Comput. Biol. 2010;6:e1000937. doi: 10.1371/journal.pcbi.1000937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueiredo M.A.T., Jain A.K. Unsupervised learning of finite mixture models. IEEE Trans. Pattern Anal. Mach. Intell. 2002;24:381–396. [Google Scholar]
- Freitag D. Machine learning for information extraction in informal domains. Mach. Learn. 2000;39:169–202. [Google Scholar]
- Gao Y.D., Huang J.F. An extension strategy of Discovery Studio 2.0 for non-bonded interaction energy automatic calculation at the residue level. Zool. Res. 2011;32:262–266. doi: 10.3724/SP.J.1141.2011.03262. [DOI] [PubMed] [Google Scholar]
- Gasteiger J., Rudolph C., Sadowski J. Automatic generation of 3D-atomic coordinates for organic molecules. Tetrahedron Comput. Methodol. 1990;3:537–547. [Google Scholar]
- Gerhard Klebe U.A., Mietzner Thomas. Molecular similarity indices in a comparative analysis (CoMSIA) of drug molecules to correlate and predict their biological activity. J. Med. Chem. 1994;37:4130–4146. doi: 10.1021/jm00050a010. [DOI] [PubMed] [Google Scholar]
- Gers F.A., Schmidhuber E. LSTM recurrent networks learn simple context-free and context-sensitive languages. IEEE Trans. Neural Netw. 2001;12:1333–1340. doi: 10.1109/72.963769. [DOI] [PubMed] [Google Scholar]
- Giese T.J. 2005. Development and Validation of New-Generation Molecular Mechanical Force Fields and Semiempirical Hamiltonians. Ph.D. Thesis (University of Minnesota) [Google Scholar]
- Gohlke H., Klebe G. DrugScore Meets CoMFA: adaptation of fields for molecular comparison (AFMoC) or how to tailor knowledge-based pair-potentials to a particular protein. J. Med. Chem. 2002;45:4153–4170. doi: 10.1021/jm020808p. [DOI] [PubMed] [Google Scholar]
- Golkov V., Skwark M.J., Mirchev A., Dikov G., Geanes A.R., Mendenhall J., Meiler J., Cremers D. 3D deep learning for biological function prediction from physical fields. 2020 International Conference on 3D Vision (3DV) 2020 [Google Scholar]
- Gombar V.K., Polli J.W., Humphreys J.E., Wring S.A., Serabjit-Singh C.S. Predicting P-glycoprotein substrates by a quantitative structure-activity relationship model. J. Pharm. Sci. 2004;93:957–968. doi: 10.1002/jps.20035. [DOI] [PubMed] [Google Scholar]
- Goodfellow I.J., Pouget-Abadie J., Mirza M., Xu B., Warde-Farley D., Ozair S., Courville A.C., Bengio Y. Generative adversarial nets. arXiv. 2014 arXiv:1406.2661. [Google Scholar]
- Goodford P.J. A computational procedure for determining energetically favorable binding sites on biologically important macromolecules. J. Med. Chem. 1985;28:849–857. doi: 10.1021/jm00145a002. [DOI] [PubMed] [Google Scholar]
- Graves A., Ndez S., Schmidhuber J. Springer; 2005. Bidirectional LSTM Networks for Improved Phoneme Classification and Recognition. ICANN 2005; pp. 799–804. [Google Scholar]
- Greer J., Erickson J.W., Baldwin J.J., Varney M.D. Application of the three-dimensional structures of protein target molecules in structure-based drug design. J. Med. Chem. 1994;37:1035–1054. doi: 10.1021/jm00034a001. [DOI] [PubMed] [Google Scholar]
- Gupta P.B., Onder T.T., Jiang G., Tao K., Kuperwasser C., Weinberg R.A., Lander E.S. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell. 2009;138:645–659. doi: 10.1016/j.cell.2009.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habasaki J., Okada I. Molecular dynamics simulation of alkali silicates based on the quantum mechanical potential surfaces. Mol. Simul. 2006;9:319–326. [Google Scholar]
- Halgren T.A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 2015;17:490–519. [Google Scholar]
- Hansch C., Maloney P.P., Fujita T., Muir R.M. Correlation of biological activity of phenoxyacetic acids with hammett substituent constants and partition coefficients. Nature. 1962;194:178–180. [Google Scholar]
- Hartenfeller M., Zettl H., Walter M., Rupp M., Reisen F., Proschak E., Weggen S., Stark H., Schneider G. DOGS: reaction-driven de novo design of bioactive compounds. Plos Comput. Biol. 2012;8:e1002380. doi: 10.1371/journal.pcbi.1002380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmman A.P., Jornada D.H., Melo E.B.D. A new, fully validated and interpreted quantitative structure-activity relationship model of p -aminosalicylic acid derivatives as neuraminidase inhibitors. Chem. Pap. 2013;67:556–567. [Google Scholar]
- Hashmi A.S.K. Gold-catalyzed organic reactions. Chem. Rev. 2007;107:3180–3211. doi: 10.1021/cr000436x. [DOI] [PubMed] [Google Scholar]
- Hayakawa D., Sawada N., Watanabe Y., Gouda H. A molecular interaction field describing nonconventional intermolecular interactions and its application to protein–ligand interaction prediction. J. Mol. Graph. Model. 2020;96:107515. doi: 10.1016/j.jmgm.2019.107515. [DOI] [PubMed] [Google Scholar]
- Hendrycks D., Gimpel K. A baseline for detecting misclassified and out-of-distribution examples in neural networks. arXiv. 2016 arXiv:1610.02136. [Google Scholar]
- Hetényi C., Paragi G., Maran U., Timár Z., Karelson M., Penke B. Combination of a modified scoring function with two-dimensional descriptors for calculation of binding affinities of bulky, flexible ligands to proteins. J. Am. Chem. Soc. 2006;128:1233–1239. doi: 10.1021/ja055804z. [DOI] [PubMed] [Google Scholar]
- Hill S.T., Kuintzle R., Teegarden A., Danaee P., Hendrix D.A. A deep recurrent neural network discovers complex biological rules to decipher RNA protein-coding potential. Nucleic Acids Res. 2018;46:8105–8113. doi: 10.1093/nar/gky567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong H., Xie Q., Ge W., Qian F., Fang H., Shi L., Su Z., Perkins R., Tong W. Mold2, molecular descriptors from 2D structures for chemoinformatics and toxicoinformatics. J. Chem. Inf. Model. 2008;48:1337–1344. doi: 10.1021/ci800038f. [DOI] [PubMed] [Google Scholar]
- Hopfinger A.J. A QSAR investigation of dihydrofolate reductase inhibition by Baker Triazines based upon molecular shape analysis. J. Am. Chem. Soc. 1980;102:7196–7206. [Google Scholar]
- Hopfinger A.J., Wang Shen, Tokarski John S., Jin Baiqiang, Albuquerque Magaly, Madhav Prakash J., Duraiswami C. Construction of 3D-QSAR models using the 4D-QSAR analysis formalism. J. Am. Chem. Soc. 1997;119:10509–10524. [Google Scholar]
- Horváth A., Hillmer M., Lou Q., Hu X.S., Niemier M. IEEE; 2017. Cellular Neural Network Friendly Convolutional Neural Networks: CNNs with CNNs. DATE; pp. 145–150. [Google Scholar]
- Hou T., Xu X. ADME evaluation in drug discovery. J. Mol. Model. 2002;8:337–349. doi: 10.1007/s00894-002-0101-1. [DOI] [PubMed] [Google Scholar]
- Hu H., Bajorath J. Activity cliffs produced by single-atom modification of active compounds: systematic identification and rationalization based on X-ray structures. Eur. J. Med. Chem. 2020;207:112846. doi: 10.1016/j.ejmech.2020.112846. [DOI] [PubMed] [Google Scholar]
- Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Hwang S., Shin H.K., Shin S.E., Seo M., Jeon H.N., Yim D.E., Kim D.H., No K.T. PreMetabo: an in silico phase I and II drug metabolism prediction platform. Drug Metab. Pharmacokinet. 2020;35:361–367. doi: 10.1016/j.dmpk.2020.05.007. [DOI] [PubMed] [Google Scholar]
- Igual L., Seguí S. In: Introduction to Data Science. Igual L., Seguí S., editors. Springer; 2017. Supervised learning; pp. 67–96. [Google Scholar]
- In Y.-Y., Lee S.-K., Kim P.-J., No K.-T. Prediction of acute toxicity to fathead minnow by local model based QSAR and global QSAR approaches. Bull. Korean Chem. Soc. 2012;33:613–619. [Google Scholar]
- Inokuma Y., Yoshioka S., Fujita M. A molecular capsule network: guest encapsulation and control of Diels-Alder reactivity. Angew. Chem. 2010;49:8912–8914. doi: 10.1002/anie.201004781. [DOI] [PubMed] [Google Scholar]
- Ivanciuc O., Braun W. Robust quantitative modeling of peptide binding affinities for MHC molecules using physical-chemical descriptors. Protein Pept. Lett. 2006;14:903–916. doi: 10.2174/092986607782110257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain A. Computer aided drug design. J. Phys. Conf. Ser. 2017;884:012072. [Google Scholar]
- Jang J.W., Cho N.C., Min S.J., Cho Y.S., Park K.D., Seo S.H., No K.T., Pae A.N. Novel Scaffold identification of mGlu1 receptor negative allosteric modulators using a hierarchical virtual screening approach. Chem. Biol. Drug Des. 2016;87:239–256. doi: 10.1111/cbdd.12654. [DOI] [PubMed] [Google Scholar]
- Jaworska J., Nikolovajeliazkova N., Aldenberg T. QSAR applicability domain estimation by projection of the training set in descriptor space: a review. Altern. Lab. Anim. 2005;33:445–459. doi: 10.1177/026119290503300508. [DOI] [PubMed] [Google Scholar]
- Jebara T., Pentland A.P. MIT; 2001. Discriminative, Generative and Imitative Learning. Ph.D. thesis. [Google Scholar]
- Jing Y., Bian Y., Hu Z., Wang L., Xie X.Q.S. Deep learning for drug design: an artificial intelligence paradigm for drug discovery in the big data era. AAPS J. 2018;20:58. doi: 10.1208/s12248-018-0210-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joung J.-Y., Kim H.-J., Kim H.-M., Ahn S.-K., Nam K.-Y., No K.-T. Prediction models of P-glycoprotein substrates using simple 2D and 3D descriptors by a recursive partitioning approach. Bull. Korean Chem. Soc. 2012;33:1123–1127. [Google Scholar]
- Kaelbling L.P., Littman M.L., Moore A.P. Reinforcement learning: a survey. J. Artif. Intell. Res. 1996;4:237–285. [Google Scholar]
- Kang P.-L., Liu Z.-P. Reaction prediction via atomistic simulation: from quantum mechanics to machine learning. iScience. 2021;24:102013. doi: 10.1016/j.isci.2020.102013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karelson M., Lobanov V.S., Katritzky A.R. Quantum-chemical descriptors in QSAR/QSPR studies. Chem. Rev. 1996;96:1027–1044. doi: 10.1021/cr950202r. [DOI] [PubMed] [Google Scholar]
- Karelson M., Lobanov V.S., Katritzky A.R. Quantum-chemical descriptors in QSAR/QSPR studies. Cheminform. 2010;96:1027–1044. doi: 10.1021/cr950202r. [DOI] [PubMed] [Google Scholar]
- Kassel D.B. Applications of high-throughput ADME in drug discovery. Curr. Opin. Chem. Biol. 2004;8:339–345. doi: 10.1016/j.cbpa.2004.04.015. [DOI] [PubMed] [Google Scholar]
- Kim J.H. Genome Data Analysis. Springer Singapore; 2019. Next-generation sequencing technology and personal genome data analysis; pp. 17–31. [Google Scholar]
- Kim H.-J., Cho Y.S., Koh H.Y., Kong J.Y., No K.T., Pae A.N. Classification of dopamine antagonists using functional feature hypothesis and topological descriptors. Bioorg. Med. Chem. 2006;14:1454–1461. doi: 10.1016/j.bmc.2005.09.072. [DOI] [PubMed] [Google Scholar]
- Kim H.-J., Choo H., Cho Y.S., Koh H.Y., No K.T., Pae A.N. Classification of dopamine, serotonin, and dual antagonists by decision trees. Bioorg. Med. Chem. 2006;14:2763–2770. doi: 10.1016/j.bmc.2005.11.059. [DOI] [PubMed] [Google Scholar]
- Kim H.J., Park W.K., Yong S.C., Kyoung T.N., Hun Y.K., Choo H., Ae N.P. Classification of piperazinylalkylisoxazole library by RP. Bull. Korean Chem. Soc. 2008;29:111–116. [Google Scholar]
- Kim D.N., Cho K., Oh W.S., Lee C.J., Lee S.K. EaMEAD: activation energy prediction of CYP450 mediated metabolism with effective atomic descriptor. J. Chem. Inf. Model. 2009;49:1643–1654. doi: 10.1021/ci900011g. [DOI] [PubMed] [Google Scholar]
- Kim N.D., Park E.S., Kim Y.H., Moon S.K., Lee S.S., Ahn S.K., Yu D.Y., No K.T., Kim K.H. Structure-based virtual screening of novel tubulin inhibitors and their characterization as anti-mitotic agents. Bioorg. Med. Chem. 2010;18:7092–7100. doi: 10.1016/j.bmc.2010.07.072. [DOI] [PubMed] [Google Scholar]
- Kim K.Y., Shin S.E., No K.T. Assessment of quantitative structure-activity relationship of toxicity prediction models for Korean chemical substance control legislation. Environ. Health Toxicol. 2015;30(Suppl):s2015007. doi: 10.5620/eht.s2015007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.-H., Choi J., Lee K., No K.T. Comparison of three-dimensional ligand-based pharmacophores among 11 phosphodiesterases (PDE 1 to PDE 11) pharmacophores. Bull. Kor. Chem. Soc. 2017;38:1033–1037. [Google Scholar]
- Kitchen D.B., Hélène D., Furr J.R., Jürgen B. Docking and scoring in virtual screening for drug discovery: methods and applications. Nat. Rev. Drug Discov. 2004;3:935–949. doi: 10.1038/nrd1549. [DOI] [PubMed] [Google Scholar]
- Klebe G. Knowledge-based scoring function to predict protein-ligand interactions. J. Mol. Biol. 2000;295:337–356. doi: 10.1006/jmbi.1999.3371. [DOI] [PubMed] [Google Scholar]
- Krieger E., Nabuurs S.B., Vriend G. Homology modeling. Methods Biochem. Anal. 2003;44:509–523. doi: 10.1002/0471721204.ch25. [DOI] [PubMed] [Google Scholar]
- Krizhevsky A., Sutskever I., Hinton G.E. ImageNet classification with deep convolutional neural networks. Commun. ACM. 2017;60:84–90. [Google Scholar]
- Kukar M., Kononenko I., Grošelj C., Kralj K., Fettich J. Analysing and improving the diagnosis of ischaemic heart disease with machine learning. Artif. Intell. Med. 1999;16:25–50. doi: 10.1016/s0933-3657(98)00063-3. [DOI] [PubMed] [Google Scholar]
- Kurogi Y., Guner O.F. Pharmacophore modeling and three-dimensional database searching for drug design using catalyst. Curr. Med. Chem. 2001;8:1035–1055. doi: 10.2174/0929867013372481. [DOI] [PubMed] [Google Scholar]
- Kwangho Nam J.G., York Darrin M. Quantum mechanical/molecular mechanical simulation study of the mechanism of Hairpin ribozyme catalysis. J. Am. Chem. Soc. 2008;130:4680–4691. doi: 10.1021/ja0759141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon Y.E., Park J.Y., No K.T., Shin J.H., Lee S.K., Eun J.S., Yang J.H., Shin T.Y., Kim D.K., Chae B.S. Synthesis, in vitro assay, and molecular modeling of new piperidine derivatives having dual inhibitory potency against acetylcholinesterase and Aβ1–42 aggregation for Alzheimer’s disease therapeutics. Bioorg. Med. Chem. 2007;15:6596–6607. doi: 10.1016/j.bmc.2007.07.003. [DOI] [PubMed] [Google Scholar]
- Laitinen T., Kankare J.A., Peräkylä M. Free energy simulations and MM-PBSA analyses on the affinity and specificity of steroid binding to antiestradiol antibody. Proteins Struct. Funct. Bioinf. 2010;55:34–43. doi: 10.1002/prot.10399. [DOI] [PubMed] [Google Scholar]
- Lampi M.A., Gurska J., Huang X.D., Dixon D.G., Greenberg B.M. A predictive quantitative structure-activity relationship model for the photoinduced toxicity of polycyclic aromatic hydrocarbons to Daphnia magna with the use of factors for photosensitization and photomodification. Environ. Toxicol. Chem. 2010;26:406–415. doi: 10.1897/06-295r.1. [DOI] [PubMed] [Google Scholar]
- Larios, E., Zhang, Y., Yan, K., Di, Z., Ledévédec, S., Groffen, F., and Verbeek, F.J. (2012). Automation in Cytomics: A Modern RDBMS Based Platform for Image Analysis and Management in High-Throughput Screening Experiments.
- Le, Q.V. (2013). Building High-Level Features Using Large Scale Unsupervised Learning.
- Lecun Y., Bengio Y., Hinton G. Deep learning. Nature. 2015;521:436. doi: 10.1038/nature14539. [DOI] [PubMed] [Google Scholar]
- Lee S.-K., Cho S.-G., Park J.-S., Kim K.-Y., No K.-T. MS-HEMs: an on-line management system for high-energy molecules at ADD and BMDRC in Korea. Bull. Kor. Chem. Soc. 2012;33:855–861. [Google Scholar]
- Lee S., Kang Y.M., Park H., Dong M.S., Shin J.M., No K.T. Human nephrotoxicity prediction models for three types of kidney injury based on data sets of pharmacological compounds and their metabolites. Chem. Res. Toxicol. 2013;26:1652–1659. doi: 10.1021/tx400249t. [DOI] [PubMed] [Google Scholar]
- Lee K., You H., Choi J., No K.T. Development of pharmacophore-based classification model for activators of constitutive androstane receptor. Drug Metab. Pharmacokinet. 2017;32:172–178. doi: 10.1016/j.dmpk.2016.11.005. [DOI] [PubMed] [Google Scholar]
- Li A.P. Screening for human ADME/Tox drug properties in drug discovery. Drug Discov. Today. 2001;6:357–366. doi: 10.1016/s1359-6446(01)01712-3. [DOI] [PubMed] [Google Scholar]
- Li K.C., Marcovici P., Phelps A., Potter C., Tillack A., Tomich J., Tridandapani S. Digitization of medicine: how radiology can take advantage of the digital revolution. Acad. Radiol. 2013;20:1479–1494. doi: 10.1016/j.acra.2013.09.008. [DOI] [PubMed] [Google Scholar]
- Lichtenstein B.R., Farid T.A., Kodali G., Solomon L.A., Anderson J.L.R., Sheehan M.M., Ennist N.M., Fry B.A., Chobot S.E., Bialas C. Engineering oxidoreductases: maquette proteins designed from scratch. Biochem. Soc. Trans. 2012;40:561. doi: 10.1042/BST20120067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lill M.A., Danielson M.L. Computer-aided drug design platform using PyMOL. J. Comput. Aided Mol. Des. 2011;25:13–19. doi: 10.1007/s10822-010-9395-8. [DOI] [PubMed] [Google Scholar]
- Lima A.N., Philot E.A., Trossini G.H.G., Scott L.P.B., Maltarollo V.G., Honorio K.M. Use of machine learning approaches for novel drug discovery. Expert Opin. Drug Discov. 2016;11:225–239. doi: 10.1517/17460441.2016.1146250. [DOI] [PubMed] [Google Scholar]
- Lin, M.S. (2009). A physics-based energy function for ab initio protein structure prediction and refinement. Dissertations & Theses - Gradworks.
- Liou C.-Y., Cheng W.-C., Liou J.-W., Liou D.-R. Autoencoder for words. Neurocomputing. 2014;139:84–96. [Google Scholar]
- Lipkowitz, K.B., and Boyd, D.B. (2007). Approaches to three-dimensional quantitative structure-activity relationships.
- Lipkus A.H. A proof of the triangle inequality for the Tanimoto distance. J. Math. Chem. 1999;26:263–265. [Google Scholar]
- Liu A. Jilin University; 2016. The Discovery of Small Molecular Inhibitors of PD-1/pd-L1 Pathway. Doctor. [Google Scholar]
- Lo Y.-C., Senese S., Damoiseaux R., Torres J.Z. 3D chemical similarity networks for structure-based target prediction and Scaffold Hopping. ACS Chem. Biol. 2016;11:2244–2253. doi: 10.1021/acschembio.6b00253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Y.C., Rensi S.E., Wen T., Altman R.B. Machine learning in chemoinformatics and drug discovery. Drug Discov. Today. 2018;102:71. doi: 10.1016/j.drudis.2018.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lokuge S., Hewavitarne H., Wimalaratne P., Ranawana R. Machine learning based Qsar for discovering potential drug candidate from endemic plants of Sri Lanka- case study: Hiv-1 Rt. VCON. 2010;10:12–17. [Google Scholar]
- Low C.M.R., Vinter J.G. Rationalizing the activities of diverse cholecystokinin 2 receptor antagonists using molecular field points. J. Med. Chem. 2008;51:565–573. doi: 10.1021/jm070880t. [DOI] [PubMed] [Google Scholar]
- Lu I.-L., Huang C.-F., Peng Y.-H., Lin Y.-T., Hsieh H.-P., Chen C.-T., Lien T.-W., Lee H.-J., Mahindroo N., Prakash E. Structure-based drug design of a novel family of PPARgamma partial agonists: virtual screening, X-ray crystallography, and in vitro/in vivo biological activities. J. Med. Chem. 2006;49:2703–2712. doi: 10.1021/jm051129s. [DOI] [PubMed] [Google Scholar]
- Luo M. Southern Medical University; 2015. The Research of Brain Tumor Segmentation Based on MRI Multi-Modality Images and 3D-CNNs Features. [Google Scholar]
- Lutz M.W., Menius J.A., Choi T.D., Laskody R.G., Domanico P.L., Goetz A.S., Saussy D.L. Experimental design for high-throughput screening. Drug Discov. Today. 1996;1:277–286. [Google Scholar]
- Ma S.L., Joung J.Y., Lee S., Cho K.H., No K.T. PXR ligand classification model with SFED-weighted WHIM and CoMMA descriptors. SAR QSAR Environ. Res. 2012;23:485–504. doi: 10.1080/1062936X.2012.665385. [DOI] [PubMed] [Google Scholar]
- Marchi E., Vesperini F., Eyben F., Squartini S., Schuller B. A novel approach for automatic acoustic novelty detection using a denoising autoencoder with bidirectional LSTM neural networks. ICASSP. 2015;2015:1996–2000. [Google Scholar]
- Matsumoto S., Ishida S., Araki M., Kato T., Terayama K., Okuno Y. Extraction of protein dynamics information from cryo-EM maps using deep learning. Nat. Mach. Intell. 2021;3:153–160. [Google Scholar]
- Mauri A., Consonni V., Pavan M., Todeschini R., Chemometrics M. DRAGON software: an easy approach to molecular descriptor calculations. MATCH Commun. Math. Comput. Chem. 2006;56:237–248. [Google Scholar]
- Mcinnes C. Virtual screening strategies in drug discovery. Curr. Opin. Chem. Biol. 2007;11:494–502. doi: 10.1016/j.cbpa.2007.08.033. [DOI] [PubMed] [Google Scholar]
- Minar M.R., Naher J. Recent advances in deep learning: an overview. arXiv. 2018 arXiv:1807.08169. [Google Scholar]
- Mochizuki Y., Komeiji Y., Ishikawa T., Nakano T., Yamataka H. A fully quantum mechanical simulation study on the lowest n–π∗ state of hydrated formaldehyde. Chem. Phys. Lett. 2007;437:66–72. [Google Scholar]
- Møller M.F. A scaled conjugate gradient algorithm for fast supervised learning. Neural Netw. 1993;6:525–533. [Google Scholar]
- Morell Christophe, Grand André, Torolabbé A. New dual descriptor for chemical reactivity. J. Phys. Chem. A. 2005;109:205–212. doi: 10.1021/jp046577a. [DOI] [PubMed] [Google Scholar]
- Morris G.M., Lim-Wilby M. Molecular docking. Methods Mol. Biol. 2008;443:365–382. doi: 10.1007/978-1-59745-177-2_19. [DOI] [PubMed] [Google Scholar]
- Nam K.Y., Chang B.H., Han C.K., Ahn S.K., No K.T. Investigation of the protonated state of HIV-1 protease active site. Bull. Korean Chem. Soc. 2003;24:817–823. [Google Scholar]
- Nam K.-Y., Kang J.H., No K.T., Ahn S.K. Identification of Polo-like kinase 1 inhibitors using structure-based molecular design. Bull. Korean Chem. Soc. 2014;35:1929–1930. [Google Scholar]
- Nam Ky-Youb, Oh W.S., Kim C., Song M.Y., Joung J.Y., Kim S.Y., Park J.S., Gang S.M., Cho Y.U., No K.T. Computational drug discovery approach based on nuclear factor-κB pathway dynamics. Bull. Kor. Chem. Soc. 2011;32:1–6. [Google Scholar]
- Nilakantan R., Bauman N., Dixon J.S. Topologial torsion: a new molecular descriptor for sar applications. Comparison with other descriptors. J. Chem. Inf. Model. 1987;27:82–85. [Google Scholar]
- Noel Y., D'Arco P., Demichelis R., Zicovich-Wilson C.M., Dovesi R. On the use of symmetry in the ab initio quantum mechanical simulation of nanotubes and related materials. J. Comput. Chem. 2010;31:855–862. doi: 10.1002/jcc.21370. [DOI] [PubMed] [Google Scholar]
- Ohashi W., Tanaka H. Benefits of pharmacogenomics in drug development—earlier launch of drugs and less adverse events. J. Med. Syst. 2010;34:701–707. doi: 10.1007/s10916-009-9284-7. [DOI] [PubMed] [Google Scholar]
- Palangi H., Deng L., Ward R.K. IEEE; 2014. Recurrent Deep-Stacking Networks for Sequence Classification. ChinaSIP; pp. 510–514. [Google Scholar]
- Pan S.J., Yang Q. A survey on transfer learning. IEEE Trans. Knowl. Data Eng. 2010;22:1345–1359. [Google Scholar]
- Pan S., Zhang H., Rush J., Eng J., Zhang N., Patterson D., Comb M.J., Aebersold R. High throughput proteome screening for biomarker detection. Mol. Cell. Proteomics. 2005;4:182. doi: 10.1074/mcp.M400161-MCP200. [DOI] [PubMed] [Google Scholar]
- Panych L.P., Oesterle C., Zientara G.P., Hennig J. Implementation of a fast gradient-echo SVD encoding technique for dynamic imaging. Magn. Reson. Med. 2015;35:554–562. doi: 10.1002/mrm.1910350415. [DOI] [PubMed] [Google Scholar]
- Parashar A., Rhu M., Mukkara A., Puglielli A., Venkatesan R., Khailany B., Emer J., Keckler S.W., Dally W.J. SCNN: an accelerator for compressed-sparse convolutional neural networks. ACM SIGARCH Computer Architecture News. 2017;45:27–40. arXiv:1708.04485. [Google Scholar]
- Park H., Kim K.K., Kim C., Shin J.-M., No K.T. Descriptor-based profile analysis of kinase inhibitors to predict inhibitory activity and to grasp kinase selectivity. Bull. Korean Chem. Soc. 2013;34:2680–2684. [Google Scholar]
- Pasquier C., Hamodrakas S.J. An hierarchical artificial neural network system for the classification of transmembrane proteins. Protein Eng. 2009;12:631–634. doi: 10.1093/protein/12.8.631. [DOI] [PubMed] [Google Scholar]
- Paul R., Hawkins S.H., Balagurunathan Y., Schabath M.B., Gillies R.J., Hall L.O., Goldgof D.B. Deep feature transfer learning in combination with traditional features predicts survival among patients with lung adenocarcinoma. Tomography. 2016;2:388–395. doi: 10.18383/j.tom.2016.00211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei J., Zheng Z., Merz K.M. Random forest refinement of the KECSA2 knowledge-based scoring function for protein decoy detection. J. Chem. Inf. Model. 2019;59:1919–1929. doi: 10.1021/acs.jcim.8b00734. [DOI] [PubMed] [Google Scholar]
- Phillips J.C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R.D., Kalé L., Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2010;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinaya W.H.L., Gadelha A., Doyle O.M., Noto C., Zugman A., Cordeiro Q., Jackowski A.P., Bressan R.A., Sato J.R. Using deep belief network modelling to characterize differences in brain morphometry in schizophrenia. Sci. Rep. 2016;6:38897. doi: 10.1038/srep38897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piotr Tu, Zhuowen Belongie, Serge Supervised learning of edges and object boundaries. CVPR. 2006;06:1964–1971. [Google Scholar]
- Polanski J. Receptor dependent multidimensional QSAR for modeling drug - receptor interactions. Curr. Med. Chem. 2009;16:3243–3257. doi: 10.2174/092986709788803286. [DOI] [PubMed] [Google Scholar]
- Quang D., Chen Y., Xie X. DANN: a deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics. 2015;31:761–763. doi: 10.1093/bioinformatics/btu703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radford A., Metz L., Chintala S. Unsupervised representation learning with deep convolutional generative adversarial networks. arXiv. 2015 arXiv:1511.06434. [Google Scholar]
- Randić M. Novel molecular descriptor for structure—property studies. Chem. Phys. Lett. 1993;211:478–483. [Google Scholar]
- Rapaport D.C. Cambridge University Press; 2004. The Art of Molecular Dynamics Simulation, 2 Edition. [Google Scholar]
- Rapaport D.C., Blumberg R.L., Mckay S.R., Christian W. The art of molecular dynamics simulation. Comput. Sci. Eng. 2002;1:70–71. [Google Scholar]
- Rogers D., Hahn M. Extended-connectivity fingerprints. J. Chem. Inf. Model. 2010;50:742–754. doi: 10.1021/ci100050t. [DOI] [PubMed] [Google Scholar]
- Rush T.S., Grant J.A., Mosyak L., Nicholls A. A shape-based 3-D Scaffold hopping method and its application to a bacterial protein−protein interaction. J. Med. Chem. 2005;48:1489–1495. doi: 10.1021/jm040163o. [DOI] [PubMed] [Google Scholar]
- Sabljić A., Güsten H., Verhaar H., Hermens J. QSAR modelling of soil sorption. Improvements and systematics of log K OC vs. log K OW correlations. Chemosphere. 1995;31:4489–4514. [Google Scholar]
- Sahami, M. (1997). Supervised and Unsupervised Discretization of Continuous Features.
- Sandberg M., Eriksson L., Jonsson J., Sjöström M., Wold S. New chemical descriptors relevant for the design of biologically active peptides. A multivariate characterization of 87 amino acids. J. Med. Chem. 1998;41:2481–2491. doi: 10.1021/jm9700575. [DOI] [PubMed] [Google Scholar]
- Sasakawa T., Hu J., Hirasawa K. A brainlike learning system with supervised, unsupervised, and reinforcement learning. Electr. Eng. Jpn. 2010;162:32–39. [Google Scholar]
- Schneider P., Schneider G. Polypharmacological drug−target inference for chemogenomics. Mol. Inf. 2018;37:e1800050. doi: 10.1002/minf.201800050. [DOI] [PubMed] [Google Scholar]
- Secco J., Farina M., Demarchi D., Corinto F., Gilli M. IEEE; 2016. Memristor Cellular Automata for Image Pattern Recognition and Clinical Applications. ISCAS 2016; pp. 1378–1381. [Google Scholar]
- Semper C., Watanabe N., Savchenko A. Structural characterization of nonstructural protein 1 from SARS-CoV-2. iScience. 2021;24:101903. doi: 10.1016/j.isci.2020.101903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senior A.W., Evans R., Jumper J., Kirkpatrick J., Sifre L., Green T., Qin C., Žídek A., Nelson A.W.R., Bridgland A. Protein structure prediction using multiple deep neural networks in the 13th critical assessment of protein structure prediction (CASP13) Proteins. 2019;87:1141–1148. doi: 10.1002/prot.25834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheridan R.P., Miller M.D., Underwood D.J., Kearsley S.K. Chemical similarity using geometric atom pair descriptors. J. Chem. Inf. Comput. Sci. 1996;36:128–136. [Google Scholar]
- Shim H.J., Yang H.R., Kim H.I., Kang S.A., No K.T., Jung Y.H., Lee S.T. Discovery of (E)-5-(benzylideneamino)-1H-benzo[d]imidazol-2(3H)-one derivatives as inhibitors PTK-6. Bioorg. Med. Chem. Lett. 2014;24:4659–4663. doi: 10.1016/j.bmcl.2014.08.036. [DOI] [PubMed] [Google Scholar]
- Shimamoto N. One-dimensional diffusion of proteins along DNA. J. Biol. Chem. 1999;274:15293–15296. doi: 10.1074/jbc.274.22.15293. [DOI] [PubMed] [Google Scholar]
- Shin W.J., Nam K.Y., Kim N.D., Kim S.H., No K.T., Seong B.L. Identification of a small benzamide inhibitor of influenza virus using a cell-based screening. Chemotherapy. 2016;61:159–166. doi: 10.1159/000441941. [DOI] [PubMed] [Google Scholar]
- Shin H.K., Kim K.Y., Park J.W., No K.T. Use of metal/metal oxide spherical cluster and hydroxyl metal coordination complex for descriptor calculation in development of nanoparticle cytotoxicity classification model. SAR QSAR Environ. Res. 2017;28:875–888. doi: 10.1080/1062936X.2017.1400998. [DOI] [PubMed] [Google Scholar]
- Shin H.K., Seo M., Shin S.E., Kim K.-Y., Park J.-W., No K.T. Meta-analysis of Daphnia magnananotoxicity experiments in accordance with test guidelines. Environ. Sci. Nano. 2018;5:765–775. [Google Scholar]
- Shu L.O., Ng E.Y.K., Ru S.T., Acharya U.R. Automated diagnosis of arrhythmia using combination of CNN and LSTM techniques with variable length heart beats. Comput. Biol. Med. 2018 doi: 10.1016/j.compbiomed.2018.06.002. S0010482518301446. [DOI] [PubMed] [Google Scholar]
- Simon Z., Badilescu I., Racovitan T. Mapping of dihydrofolate-reductase receptor site by correlation with minimal topological (steric) differences. J. Theor. Biol. 1977;66:485–495. doi: 10.1016/0022-5193(77)90298-3. [DOI] [PubMed] [Google Scholar]
- Singh N., Shah P., Dwivedi H., Mishra S., Tripathi R., Sahasrabuddhe A.A., Siddiqi M.I. Integrated machine learning, molecular docking and 3D-QSAR based approach for identification of potential inhibitors of trypanosomal N-myristoyltransferase. Mol. Biosyst. 2016;12:3711–3723. doi: 10.1039/c6mb00574h. [DOI] [PubMed] [Google Scholar]
- Sondak V.K.S.E. New directions for medical artificial intelligence. Comput. Math. Appl. 1990;20:313–319. [Google Scholar]
- Song S.W., Kim S.D., Oh D.Y., Lee Y., Lee A.C., Jeong Y., Bae H.J., Lee D., Lee S., Kim J. High-throughput screening: one-step generation of a drug-releasing hydrogel microarray-on-a-chip for large-scale sequential drug combination screening. Adv. Sci. 2019;6:1801380. doi: 10.1002/advs.201801380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephenson N., Shane E., Chase J., Rowland J., Ries D., Justice N., Zhang J., Chan L., Cao R. Survey of machine learning techniques in drug discovery. Curr. Drug Metab. 2019;20:185–193. doi: 10.2174/1389200219666180820112457. [DOI] [PubMed] [Google Scholar]
- Strasser P., Fan Q., Martin Devenney A., Weinberg W.H., Nørskov J.K. High throughput experimental and theoretical predictive screening of materials − a comparative study of search strategies for new fuel cell anode catalysts. J. Phys. Chem. B. 2003;107:11013–11021. [Google Scholar]
- Sun F., Liu J., Wu J., Pei C., Lin X., Ou W., Jiang P. BERT4Rec: sequential recommendation with bidirectional encoder representations from transformer. Proceedings of the 28th ACM international conference on information and knowledge management. 2019:1441–1450. [Google Scholar]
- Svetnik V., Liaw A., Tong C., Culberson J.C., Sheridan R.P., Feuston B.P. Random forest: a classification and regression tool for compound classification and QSAR modeling. J. Chem. Inf. Comput. Sci. 2003;43:1947. doi: 10.1021/ci034160g. [DOI] [PubMed] [Google Scholar]
- Tan J.W., Deng S.J., Ye F.W., Zeng D.P. Variability analysis of T network impedance matching. Appl. Mech. Mater. 2013;427-429:620–623. [Google Scholar]
- Thangapandian S., John S., Son M., Arulalapperumal V., Lee K.W. Development of predictive quantitative structure-activity relationship model and its application in the discovery of human leukotriene A4 hydrolase inhibitors. Fut. Med. Chem. 2013;5:27–40. doi: 10.4155/fmc.12.184. [DOI] [PubMed] [Google Scholar]
- Todeschini R., Consonni V. Wiley-VCH; 2009. Molecular Descriptors for Chåemoinformatics. [Google Scholar]
- Tomal J.H., Welch W.J., Zamar R.H. Exploiting multiple descriptor sets in QSAR studies. J. Chem. Inf. Model. 2016;56:501–509. doi: 10.1021/acs.jcim.5b00663. [DOI] [PubMed] [Google Scholar]
- Trott O., Olson A.J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu Y., Laaksonen A. Atomic charges in molecular mechanical force fields: a theoretical insight. Phys. Rev. E: Stat. Nonlinear, Soft Matter Phys. 2001;64:026703. doi: 10.1103/PhysRevE.64.026703. [DOI] [PubMed] [Google Scholar]
- Turian J., Ratinov L., Bengio Y. Proceedings of the 48th Annual Meeting of the Association for Computational Linguistics. 2010. Word representations: a simple and general method for semi-supervised learning; pp. 384–394. [Google Scholar]
- Vapnik V., Levin E., Cun Y. Measuring the VC-dimension of a learning machine. Neural Comput. 1994;6:851–876. [Google Scholar]
- Varela R., Walters W.P., Goldman B.B., Jain A.N. Iterative refinement of a binding pocket model: active computational steering of lead optimization. J. Med. Chem. 2012;55:8926–8942. doi: 10.1021/jm301210j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vedani A., Dobler M. 5D-QSAR: the key for simulating induced fit? J. Med. Chem. 2002;45:2139–2149. doi: 10.1021/jm011005p. [DOI] [PubMed] [Google Scholar]
- Vedani A., Dobler M., Lill M.A. Combining protein modeling and 6D-QSAR. Simulating the binding of structurally diverse ligands to the estrogen receptor. J. Med. Chem. 2005;48:3700–3703. doi: 10.1021/jm050185q. [DOI] [PubMed] [Google Scholar]
- Verdonk M.L., Cole J.C., Hartshorn M.J., Murray C.W., Taylor R.D. Improved protein-ligand docking using GOLD. Proteins Struct. Funct. Bioinf. 2010;52:609–623. doi: 10.1002/prot.10465. [DOI] [PubMed] [Google Scholar]
- Verlinde C.L., Hol W.G. Structure-based drug design: progress, results and challenges. Structure. 1994;2:577–587. doi: 10.1016/s0969-2126(00)00060-5. [DOI] [PubMed] [Google Scholar]
- Veselovsky A.V., Tikhonova O.V., Skvortsov V.S., Medvedev A.E., Ivanov A.S. An approach for visualization of the active site of enzymes with unknown three-dimensional structures. SAR QSAR Environ. Res. 2001;12:345–358. doi: 10.1080/10629360108033243. [DOI] [PubMed] [Google Scholar]
- Vesely F.J. In: Computational Physics: An Introduction. Vesely F.J., editor. Springer US; 2001. Quantum mechanical simulation; pp. 195–214. [Google Scholar]
- Wallach I., Dzamba M., Heifets A. AtomNet: a deep convolutional neural network for bioactivity prediction in structure-based drug discovery. Math. Z. 2015;47:34–46. [Google Scholar]
- Walters D.E., Hinds R.M. Genetically evolved receptor models: a computational approach to construction of receptor models. J. Med. Chem. 1994;37:2527–2536. doi: 10.1021/jm00042a006. [DOI] [PubMed] [Google Scholar]
- Wang S. Qingdao University; 2013. The Study about PET/CT Image Registration Method Based on CNN and Mutual Information. [Google Scholar]
- Wang Q., He J., Wu D., Wang J., Yan J., Li H. Interaction of α-cyperone with human serum albumin: determination of the binding site by using Discovery Studio and via spectroscopic methods. J. Lumin. 2015;164:81–85. [Google Scholar]
- Wang X., Zhang T., Chaim T.M., Zanetti M.V., Davatzikos C. Classification of MRI under the presence of disease heterogeneity using multi-task learning: application to bipolar disorder. Med. Image Comput. Comput. Assist. Interv. 2015;9349:125–132. doi: 10.1007/978-3-319-24553-9_16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Zhang J., An Y., Lin H., Yang Z., Zhang Y., Sun Y. Biomedical event trigger detection by dependency-based word embedding. BMC Med. Genomics. 2016;9:45. doi: 10.1186/s12920-016-0203-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C., Elazab A., Jia F., Wu J., Hu Q. Automated chest screening based on a hybrid model of transfer learning and convolutional sparse denoising autoencoder. Biomed. Eng. Online. 2018;17:63. doi: 10.1186/s12938-018-0496-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Ye Z., Wan P., Zhao J. A survey of dynamic spectrum allocation based on reinforcement learning algorithms in cognitive radio networks. Artif. Intell. Rev. 2019;51:493–506. [Google Scholar]
- Wenz C. Development and drugs: more not less. Nature. 1982;297:173–174. [Google Scholar]
- Widera P. Association for Computing Machinery; 2010. Evolutionary Symbolic Discovery for Bioinformatics, Sysems and Synthetic Biology. GECCO '10; pp. 1991–1998. [Google Scholar]
- Willett P., Barnard J.M., Downs G.M. Chemical similarity searching. J. Chem. Inf. Comput. Sci. 1998;38:983–996. [Google Scholar]
- Wiskott L., Sejnowski T.J. Slow feature analysis: unsupervised learning of invariances. Neural Comput. 2002;14:715. doi: 10.1162/089976602317318938. [DOI] [PubMed] [Google Scholar]
- Wolber Gerhard, Sippl W. In: The Practice of Medicinal Chemistry, 4th Ed. C.G. Wermuth, D. Aldous, P. Raboisson, D. Rognan, editors. Academic Press; 2008. Pharmacophore identification and pseudo-receptor modeling; pp. 489–510. [Google Scholar]
- Wold S. Validation of QSAR's. Mol. Inf. 2010;10:191–193. [Google Scholar]
- Wu Y., Wang G. Machine learning based toxicity prediction: from chemical structural description to transcriptome analysis. Int. J. Mol. Sci. 2018;19:2358. doi: 10.3390/ijms19082358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi E., Bing S., Jin Y. Capsule network performance on complex data. arXiv. 2017 arXiv:1712.03480. [Google Scholar]
- Xing L., Glen R.C. Novel methods for the prediction of logP, pK(a), and logD. J. Chem. Inf. Comput. Sci. 2010;33:231. doi: 10.1021/ci010315d. [DOI] [PubMed] [Google Scholar]
- Xu J., Lei X., Hang R., Wu J. IEEE; 2014. Stacked Sparse Autoencoder (SSAE) based framework for nuclei patch classification on breast cancer histopathology. ISBI 2014; pp. 999–1002. [Google Scholar]
- Xu D., Gopale M., Zhang J., Brown K., Begoli E., Bethard S. Unified medical language system resources improve sieve-based generation and Bidirectional Encoder Representations from Transformers (BERT)–based ranking for concept normalization. J. Am. Med. Inform. Assoc. 2020;27:1510–1519. doi: 10.1093/jamia/ocaa080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue D.X., Zhang R., Feng H., Wang Y.L. CNN-SVM for microvascular morphological type recognition with data augmentation. J. Med. Biol. Eng. 2016;36:755–764. doi: 10.1007/s40846-016-0182-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S.Y. Pharmacophore modeling and applications in drug discovery: challenges and recent advances. Drug Discov. Today. 2010;15:444–450. doi: 10.1016/j.drudis.2010.03.013. [DOI] [PubMed] [Google Scholar]
- Yang L., Yang X., Pu X., Qian Z., Wang K. The absorption, distriution, metabolism, excretion, toxicity (ADME/Tox.) platform construction of novel drugs research. J. Peking Univ. Health Sci. 2004;36:5–8. [Google Scholar]
- Yang Y.F., Wang L., Yan S.J., Zhi L.I., Wang Y.Y., Zhang W.S. Discovery Studio software in the analysis of the blood-brain barrier penetrations of active components of traditional Chinese medicines. Chin. Pharmacol. Bull. 2011;27:739–740. [Google Scholar]
- Yao J., Zhou C., Motani M. Vol. 2017. BIBM; 2017. pp. 886–890. (Spatio-Temporal Autoencoder for Feature Learning in Patient Data with Missing Observations). [Google Scholar]
- Yi X., Walia E., Babyn P. Generative adversarial network in medical imaging: a review. Med. Image Anal. 2019;58:101552. doi: 10.1016/j.media.2019.101552. [DOI] [PubMed] [Google Scholar]
- Yildirim Ö. A novel wavelet sequences based on deep bidirectional LSTM network model for ECG signal classification. Comput. Biol. Med. 2018;96:189. doi: 10.1016/j.compbiomed.2018.03.016. [DOI] [PubMed] [Google Scholar]
- Yoshioka T., Chen X., Gales M.J.F. Impact of single-microphone dereverberation on DNN-based meeting transcription systems. Proc. IEEE Int. Conf. Acoust. Speech Signal Process. 2014:5527–5531. [Google Scholar]
- You H., Lee K., Lee S., Hwang S.B., Kim K.Y., Cho K.H., No K.T. Computational classification models for predicting the interaction of compounds with hepatic organic ion importers. Drug Metab. Pharmacokinet. 2015;30:347–351. doi: 10.1016/j.dmpk.2015.06.004. [DOI] [PubMed] [Google Scholar]
- Yu K., Liu C., Kim B.G., Lee D.Y. Synthetic fusion protein design and applications. Biotechnol. Adv. 2015;33:155–164. doi: 10.1016/j.biotechadv.2014.11.005. [DOI] [PubMed] [Google Scholar]
- Yuan S., Dahoun T., Brugarolas M., Pick H., Filipek S., Vogel H. Computational modeling of the olfactory receptor Olfr73 suggests a molecular basis for low potency of olfactory receptor-activating compounds. Commun. Biol. 2019;2:141. doi: 10.1038/s42003-019-0384-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng M., Li M., Fei Z., Wu F., Li Y., Pan Y., Wang J. A deep learning framework for identifying essential proteins by integrating multiple types of biological information. IEEE ACM Trans. Comput. Bi. 2021;18:296–305. doi: 10.1109/TCBB.2019.2897679. [DOI] [PubMed] [Google Scholar]
- Zhan C., Zhang L., Zhong Z., Didi-Ooi S., Lin Y., Zhang Y., Huang S., Wang C. Deep learning approach in automatic iceberg - ship detection with sar remote sensing data. arXiv. 2018 arXiv:1812.07367. [Google Scholar]
- Zhang C.X., Nan-Nan J.I., Wang G.W. Restricted Boltzmann machines. Chin. J. Eng. Math. 2015;2015:159–173. [Google Scholar]
- Zhang Y., Dijkman P.M., Zou R., Zandl-Lang M., Sanchez R.M., Eckhardt-Strelau L., Köfeler H., Vogel H., Yuan S., Kudryashev M. Asymmetric opening of the homopentameric 5-HT3A serotonin receptor in lipid bilayers. Nat. Commun. 2021;12:1074. doi: 10.1038/s41467-021-21016-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Huang Q., Ma X., Yang Z., Jiang J. 2016 IEEE Trustcom/BigDataSE/ISPA. 2017. Using multi-features and ensemble learning method for imbalanced malware classification; pp. 965–973. [Google Scholar]
- Zhao A. The situation in bioinformatics research and development. China Biotechnol. 2003;23:101–103. [Google Scholar]
- Zhao G., Wang X., Niu Y., Tan L., Zhang S.X. Segmenting brain tissues from Chinese visible human dataset by deep-learned features with stacked autoencoder. Biomed. Res. Int. 2016;2016:1–12. doi: 10.1155/2016/5284586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y., Qiao X., Yang X., Chen G., Li X. Quantitative structure-activity relationship model for bioconcentration factors of halogenated organic compounds. Asian J. Ecotoxicol. 2013;8:772–777. [Google Scholar]
- Zhu X., Goldberg A.B. Introduction to semi-supervised learning. Synth. Lect. Artif. Intell. Mach. Learn. 2009;3:1–130. [Google Scholar]
- Zhu X., Suk H.I., Lee S.W., Shen D. Subspace regularized sparse multi-task learning for multi-class neurodegenerative disease identification. IEEE Trans. Biomed. Eng. 2016;63:607–618. doi: 10.1109/TBME.2015.2466616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoph B., Yuret D., May J., Knight K. Transfer learning for low-resource neural machine translation. arXiv. 2016 arXiv:1604.02201. [Google Scholar]