

Abstract
The Ebola virus VP30 protein interacts with the viral nucleoprotein and with host protein RBBP6 via PPxPxY motifs that adopt non‐canonical orientations, as compared to other proline‐rich motifs. An affinity tag‐purification mass spectrometry approach identified additional PPxPxY‐containing host proteins hnRNP L, hnRNPUL1, and PEG10, as VP30 interactors. hnRNP L and PEG10, like RBBP6, inhibit viral RNA synthesis and EBOV infection, whereas hnRNPUL1 enhances. RBBP6 and hnRNP L modulate VP30 phosphorylation, increase viral transcription, and exert additive effects on viral RNA synthesis. PEG10 has more modest inhibitory effects on EBOV replication. hnRNPUL1 positively affects viral RNA synthesis but in a VP30‐independent manner. Binding studies demonstrate variable capacity of the PPxPxY motifs from these proteins to bind VP30, define PxPPPPxY as an optimal binding motif, and identify the fifth proline and the tyrosine as most critical for interaction. Competition binding and hydrogen‐deuterium exchange mass spectrometry studies demonstrate that each protein binds a similar interface on VP30. VP30 therefore presents a novel proline recognition domain that is targeted by multiple host proteins to modulate viral transcription.
Keywords: Ebola virus, RNA viruses, viral replication, virus–host interactions, VP30
Subject Categories: Microbiology, Virology & Host Pathogen Interaction
Proteomics approaches identify several proline‐rich motif‐containing host cell proteins interacting with the Ebola virus transcription factor VP30, which compete with viral nucleoprotein NP to modulate VP30 phosphorylation and viral RNA synthesis.

Introduction
Zaire ebolavirus (Ebola virus or EBOV), a member of the filovirus family of enveloped, non‐segmented, negative‐sense RNA viruses, is a zoonotic pathogen notable for its propensity to cause outbreaks of severe disease in humans. The public health significance of EBOV is evidenced by past outbreaks where reported case fatality rates ranged up to 90%: by the West Africa EBOV epidemic in 2013–2016, which resulted in more than 28,000 infections and more than 11,000 deaths; by four outbreaks in the Democratic Republic of Congo that have occurred from 2017 to 2021 and the reemergence of EBOV disease in Guinea in 2021 (Bausch et al, 2016; Ilunga Kalenga et al, 2019; Nsio et al, 2019; Adepoju, 2021).
The EBOV genomic RNA is ˜19 kilobases in length and has seven separate transcriptional units (genes) that encode distinct mRNAs. The genome is encapsidated by nucleoprotein (NP) with the resulting ribonucleoprotein serving as the template for RNA synthesis reactions that replicate the viral genomic RNA and transcribe the mRNAs that encode the viral proteins. Viral genome replication requires, in addition to NP, the viral proteins VP35 and L, the viral RNA‐dependent RNA polymerase. Transcription requires these proteins and the EBOV VP30 protein (Muhlberger et al, 1998; Muhlberger et al, 1999).
EBOV VP30 is a zinc‐ and RNA‐binding protein and a component of the viral nucleocapsid complex (Becker et al, 1998; Muhlberger et al, 1999; Modrof et al, 2003; John et al, 2007; Nanbo et al, 2013; Biedenkopf et al, 2016b; Schlereth et al, 2016). VP30 is essential for the virus life cycle (Muhlberger et al, 1999; Enterlein et al, 2006; Halfmann et al, 2008; Biedenkopf et al, 2016a). A critical role for EBOV VP30 is in initiation of viral transcription, a function that is dependent on a stem‐loop structure present at the NP gene start site; disruption of this secondary structure leads to VP30‐independent transcription (Weik, Modrof et al, 2002). EBOV VP30 also facilitates re‐initiation at downstream genes during viral transcription and regulates editing by the viral polymerase during synthesis of nascent mRNAs from the glycoprotein (GP) gene (Martinez et al, 2008; Mehedi et al, 2013).
A substantial body of data implicates VP30 phosphorylation as a regulator of its transcriptional function. Dephosphorylation of VP30 N‐terminal serine and threonine residues or mutation of these residues to alanine increases pro‐transcriptional activity; whereas phosphorylation or mutation to aspartic acid inhibits transcription and promotes viral genome replication (Modrof et al, 2002; Martinez et al, 2011; Biedenkopf et al, 2013; Ilinykh et al, 2014; Biedenkopf et al, 2016a; Lier et al, 2017; Ammosova et al, 2018; Kruse et al, 2018; Tigabu et al, 2018). VP30 interacts with NP, and this influences VP30 phosphorylation levels (Biedenkopf et al, 2013; Kirchdoerfer et al, 2016; Lier et al, 2017; Xu et al, 2017; Kruse et al, 2018). NP recruits the host PP2A‐B56 protein phosphatase through a LxxIxE motif, promoting VP30 dephosphorylation and viral transcription (Kruse et al, 2018). This activity likely explains the importance of VP30–NP interaction for RNA synthesis, as was demonstrated in studies that defined the structure of the VP30–NP interaction interface (Kirchdoerfer et al, 2016; Xu et al, 2017).
Recently, a comprehensive affinity tag‐purification mass spectrometry (AP‐MS) analysis of the EBOV–host protein–protein interactome identified a number of VP30‐interacting host proteins, including retinoblastoma binding protein 6 (RBBP6) (Batra et al, 2018). RBBP6, a multi‐domain protein with E3 ubiquitin ligase activity, has been implicated in cell cycle progression, nucleic acid metabolism, cell proliferation, and differentiation (Ntwasa, 2016). RBBP6 possesses a PPxPxY motif that was demonstrated to interact with VP30 at a site that binds a PPxPxY motif on NP. RBBP6 can compete with NP for binding to VP30, thereby inhibiting viral gene expression. In this study, we characterize VP30 interaction with an additional three host proteins that possess PPxPxY motifs. The data demonstrate that the extended sequence PxPPPPxY mediates optimal binding and that proteins sharing this motif compete with NP for binding to a common site on VP30, alter VP30 phosphorylation, modulate viral mRNA transcription, and influence viral infectivity. These findings reveal a unique virus–host interaction where multiple host factors target the same viral interface to alter replication efficiency.
Results
Multiple host proteins possessing PPxPxY motifs interact with EBOV VP30
A previous study identified RBBP6 as a VP30‐interacting protein and demonstrated that a PPxPxY motif mediates the interaction with VP30; this is reminiscent of a PPxPxY motif present in the EBOV NP protein that binds to VP30 (Fig 1A; Batra et al, 2018). The same study also identified the cellular proteins hnRNP L and hnRNPUL1 in both HEK293T and Huh7 cells and PEG10 in Huh7 cells as VP30 interactors (Batra et al, 2018; Fig 1B). All three of these interactors also contain PPxPxY motifs. To further validate these interactions, FLAG‐tagged host proteins were co‐expressed with HA‐tagged VP30 in HEK293T cells, and pull downs were performed using anti‐FLAG beads (Fig 1C). hnRNP L, hnRNPUL1, and PEG10 each interacted robustly with VP30, similar to RBBP6. The interaction between VP30 and the endogenous host proteins was further addressed by co‐immunoprecipitation. Endogenous RBBP6, hnRNP L, and hnRNPUL1 were each co‐immunoprecipitated in the presence of HA‐VP30 but not in the presence of empty vector (Fig 1D). The HA–VP30 interaction with PEG10 was more equivocal in HEK293T cells. PEG10 is highly expressed in hepatocellular carcinoma cells and was previously identified as an interactor in only Huh7 cells; therefore, we confirmed the interaction of VP30 with endogenous PEG10 in these cells (Fig EV1).
Figure 1. VP30 interacts with multiple host proteins that contain PPxPxY motifs.
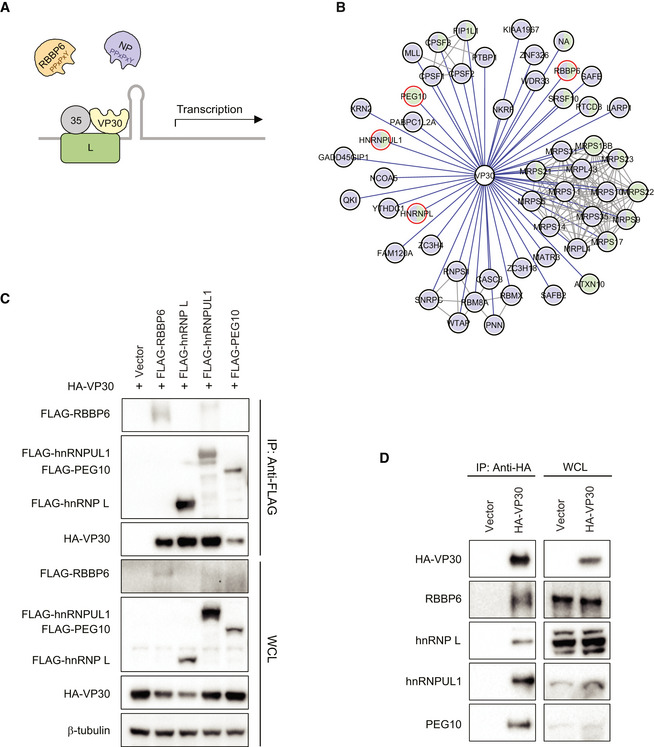
- A model for RBBP6 and NP interaction with VP30 via PPxPxY motifs to regulate viral transcription. Both EBOV NP and human RBBP6 share a common PPxPxY motif for binding to EBOV transcription factor VP30. Interaction between NP and VP30 modulates viral RNA synthesis because RBBP6 outcompetes NP for VP30 binding.
- An EBOV VP30‐human protein–protein interaction network. Purple and green prey nodes indicate that the protein was identified as a VP30 interactor in HEK293T or Huh7 cells, respectively; purple‐green bifurcation indicates that the interaction was identified in both cell types. Gray lines correspond to human–human protein–protein interactions curated in the publicly available CORUM database. Host proteins containing PPxPxY motif(s) are circled in red.
- Co‐immunoprecipitation between VP30 and the indicated PPxPxY‐containing host factors. Empty expression plasmid (Vector) or FLAG‐tagged host protein expression plasmids were co‐transfected with an HA‐VP30 expression plasmid in HEK293T cells. An anti‐FLAG immunoprecipitation (IP: Anti‐FLAG) was performed. Western blots of IP and whole‐cell lysates (WCL) are shown. Anti‐β‐tubulin was used as a loading control for the WCLs.
- Cells were transfected with empty vector or HA‐VP30 expression plasmid, and IPs were performed with anti‐HA antibody (IP: Anti‐HA). Western blotting with anti‐HA tag or antibodies to the indicated host proteins was performed on IP and on whole‐cell lysates (WCL).
Source data are available online for this figure.
Figure EV1. Co‐immunoprecipitation between VP30 and endogenous PEG10.
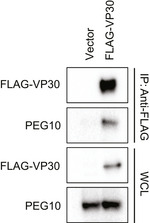
Huh7 cells were transfected with empty vector (Vector) or FLAG‐VP30 expression plasmid. IPs (IP: Anti‐FLAG) and whole‐cell lysates (WCL) were analyzed by Western blotting with anti‐FLAG or anti‐PEG10 antibodies.
Molecular dissection of PPxPxY‐mediated interactions with VP30
There is only one PPxPxY motif within the proline (Pro)‐rich region of RBBP6. However, in hnRNP L, PEG10, and hnRNPUL1, this motif appears two or three times (Fig 2A, left). The amino acid sequences of peptides derived from these host proteins, including 10 amino acids flanking each PPxPxY motif, were aligned (Fig 2A, right). Sequences corresponding to each of these extended peptides (labeled as 1–3) were cloned as N‐terminal GFP fusions. The fusion proteins were tested in co‐immunoprecipitation assays for their ability to bind VP30. In each case, only one of the tested peptides from hnRNP L (hnRNP L_2), hnRNPUL1 (hnRNPUL1_3), and PEG10 (PEG10_2) strongly interacted with VP30, suggesting that the sequence context of the motif plays a significant role in binding (Fig 2B). Also observed was a weak interaction between hnRNPUL1 peptide 1 and VP30.
Figure 2. PPxPxY‐containing peptides derived from host proteins interact with VP30 and modulate EBOV RNA synthesis.
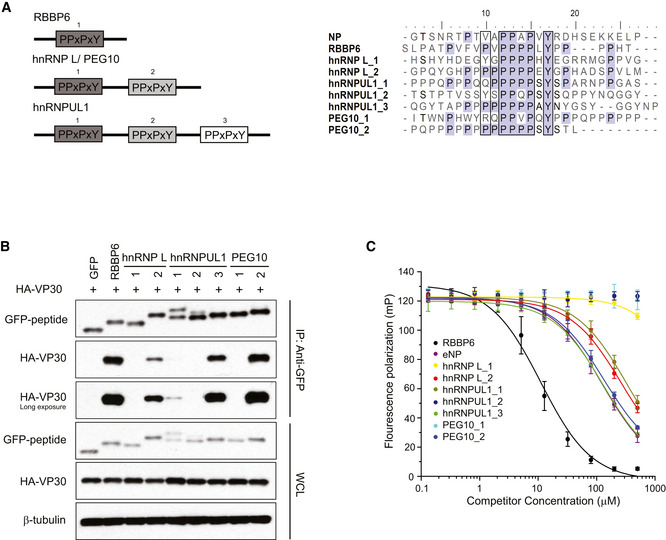
- Schematic representation of the arrangement and number of PPxPxY motifs present in the indicated host proteins (left panel). Multiple sequences aligned using ClustalW, of peptides containing PPxPxY motif derived from cellular proteins and from the EBOV NP protein (right panel).
- Co‐immunoprecipitation between HA‐VP30 and either GFP or GFP‐fused to peptides derived from the host proteins was performed. Western blots of the co‐immunoprecipitation and whole‐cell lysate (WCL) are shown.
- Equilibrium dissociation curves of FITC‐RBBP6 from eVP30130‐272 in the presence of increasing concentrations (0.13–500 μM) of the PPxPxY‐containing peptides. Fluorescence polarization was determined with constant concentrations of FITC‐RBBP6 and eVP30130‐272, at 0.50 μM and 3.8 μM, respectively. Experiments were performed in two independent replicates. Error bars represent standard deviation.
Source data are available online for this figure.
To quantify the strength of individual peptide binding, and to determine whether the peptides can target the VP30 interface bound by RBBP6 and NP, a fluorescence polarization (FP) assay was employed in which a purified C‐terminal domain of EBOV VP30 (eVP30130‐272) and a FITC‐labeled RBBP6 peptide containing the PPxPxY motif were used (Fig 2C). The RBBP6 peptide displayed the highest binding affinity. hnRNPUL1_3 and PEG10_2 demonstrated affinities similar to the NP peptide, whereas hnRNP L_2 and hnRNPUL1_1 exhibited slightly lower affinities. hnRNP L_1, hnRNPUL1_2, and PEG10_1 exhibited minimal to no binding in the FP assays. Overall, these data support the PPxPxY motif as contributing to binding; however, the extended sequence PxPPPPxY and its composition contribute to the strength of interaction.
The EBOV protein VP40 possesses overlapping PTAP and PPEY motifs (PTAPPEY) that function as late domains in viral budding (Licata et al, 2003). To determine whether this sequence mediates an interaction with VP30 and to further address the specificity of Pro‐rich motifs to interact with VP30, EBOV VP40/VP30 co‐immunoprecipitations were performed. VP40 did not co‐precipitate with VP30 (Fig EV2A). Furthermore, fusion of the N‐terminal VP40 PTAPPEY sequence to GFP also did not facilitate co‐precipitation of VP30. Mutation of the late domain motif to contain five proline residues (PPPPPEY) failed to confer interaction. However, mutation of the VP40 peptide to include an additional sixth proline (PPPPPPEY) resulted in interaction (Fig EV2B). These data further support the extended PxPPPPxY motif as requisite for efficient binding to VP30.
Figure EV2. VP30 does not bind to the native VP40 proline‐tyrosine motif but does bind to bat RBBP6.
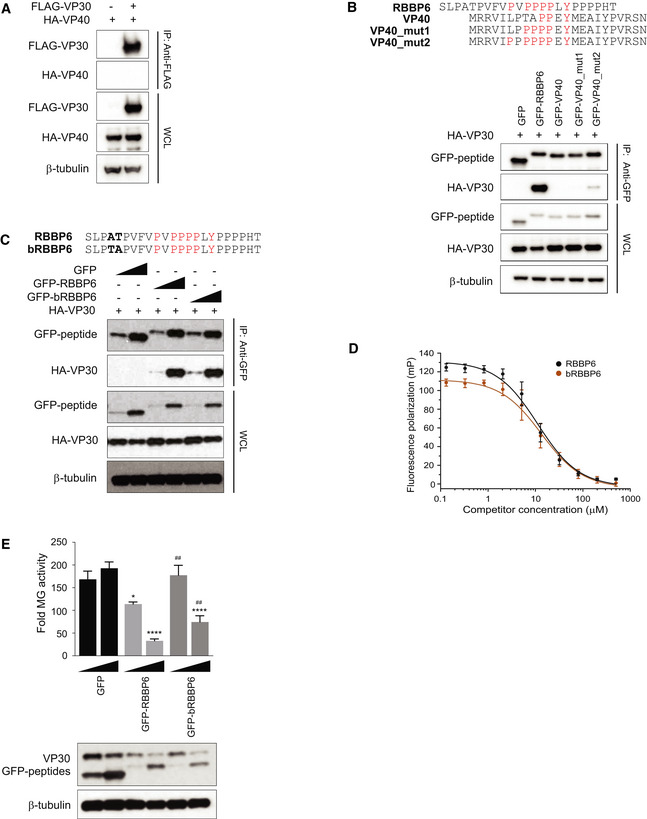
- Co‐immunoprecipitation assay to assess HA–VP40 interaction with FLAG‐VP30. HEK293T cells were transfected with HA‐VP40 plasmid and either empty vector or FLAG‐VP30 plasmid. An anti‐FLAG immunoprecipitation (IP) was performed. IP and WCL were analyzed by Western blotting with anti‐HA and anti‐FLAG antibodies. An anti‐β‐tubulin Western blot provided a loading control.
- Anti‐GFP immunoprecipitation of HA‐VP30 with GFP fusions to wild‐type or mutated VP40‐derived peptides (GFP‐VP40, GFP‐VP40_mut1, or GFP‐VP40_mut2). GFP without a fusion partner (GFP) and GFP fused to RBBP6 peptide (GFP‐RBBP6) served as controls. Immunoblots with anti‐HA, anti‐GFP, and anti‐β‐tubulin are shown.
- GFP fused to peptides derived from human and bat (Rousettus aegyptiacus) RBBP6 were expressed along with HA‐VP30. Co‐immunoprecipitation was performed using anti‐GFP magnetic beads, and representative immunoblots for IP and WCL are shown.
- Equilibrium dissociation curves of FITC‐RBBP6 peptide to eVP30130‐272 as it is outcompeted by increasing concentrations (0.13–500 μM) of human and bat RBBP6 peptides. Fluorescence polarization was determined with constant concentrations of FITC‐RBBP6 and eVP30130‐272, at 0.50 μM and 3.8 μM, respectively. Experiments were performed in two independent replicates. Error bars represent standard deviation.
- MG activity upon titration of GFP fused to human and bat‐derived RBBP6 peptides (12.5 ng and 125 ng). GFP alone was used as a control. Reporter activity was read at 48 h post‐transfection and fold MG activity was calculated relative to a no VP30 control. # denotes statistical significance compared with RBBP6 peptide for each dose. The data represent the mean ± SD from one representative experiment in which each transfection condition was performed in triplicate (n = 3). Each experiment was reproduced in at least two additional, independent experiments (see Appendix Fig S1). Statistical significance was calculated relative to GFP control for each concentration tested using ANOVA with Tukey’s multiple comparisons test. ****P < 0.00005; ## P < 0.005, *P < 0.05
We also tested an RBBP6 peptide derived from a fruit bat (Rousettus aegyptiacus) (bRBBP6 peptide) that serves as MARV reservoir hosts in co‐precipitation assays with VP30. bRBBP6 peptide differs at only two positions from the human RBBP6 peptide and binds robustly to VP30 with a near identical affinity, further substantiating the requirement of the PxPPPPxY motif for optimal binding to VP30 (Fig EV2C and D). The RBBP6 peptide was demonstrated to inhibit EBOV RNA synthesis, as measured by a minigenome (MG) assay (Batra et al, 2018). When titrated in the MG assay, bRBBP6 was modestly less inhibitory to MG activity, as compared to the RBBP6 peptide (Fig EV2E, Appendix Fig S1).
The binding in solution between VP30 and different host peptides, including the interacting RBBP6, hnRNPUL1_1, hnRNPUL1_3, hnRNP L_2, and PEG10_2, and the non‐interacting hnRNPUL1_2, was further characterized with hydrogen‐deuterium exchange (HDX) mass spectrometry. RBBP6, hnRNPUL1_1, hnRNPUL1_3, hnRNP L_2, and PEG10_2 peptides bind nearly identically to VP30 as seen in the HDX kinetic curves, indicating that each peptide binds to a similar region on VP30 (Figs 3A and EV3). Statistical analysis of deuterium uptake differences identified two strong peptide‐binding regions on VP30 from residue L189 to V210 and from residue A224 to D231. The two regions contain seven and four binding residues, respectively, that were identified by X‐ray crystallography (PDB 6E5X; Batra et al, 2018), demonstrating consistency of the two approaches and the ability of HDX to give an accurate picture of the binding interface. The middle region from residue Y211 to E223 presents as a weaker binding region where the differences in HDX are much smaller between bound and unbound, also consistent with the X‐ray structure that show binding via only one residue, R213. Although there is clear binding of residues 224–231, the region 230–237 shows different character in the kinetics of HDX. Its extent for the bound state converges at long times with that of the unbound, indicating a larger off rate and weaker binding at the end of the peptide ligand. We further mapped the HDX differences at 4 h onto the VP30 crystal structure, which illustrates common binding regions among different host factor peptides (Fig 3B).
Figure 3. HDX‐MS analysis of VP30 and host factor peptide interactions.
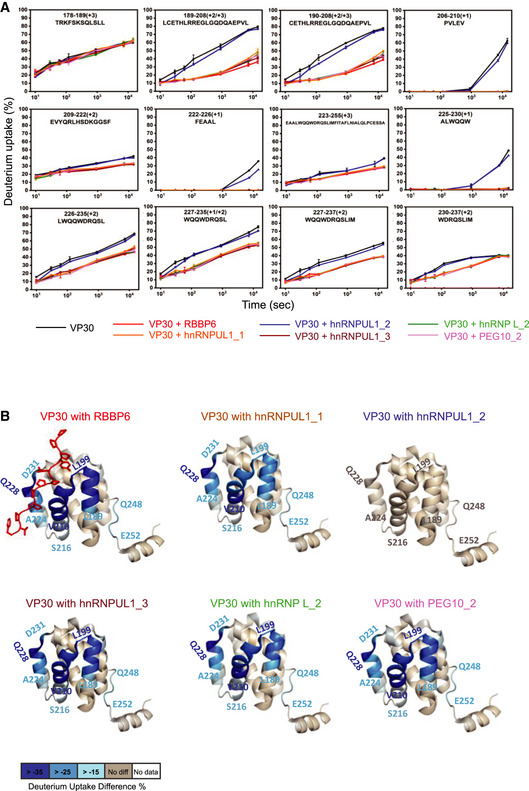
- HDX kinetics data for VP30 alone and for the indicated host peptides incubated with VP30 are shown. VP30 peptide amino acid residue positions and sequences are indicated at the top of each curve. The error bars indicate mean ± SD (n = 3).
- Deuterium uptake differences were mapped onto the VP30 structure (PDB 6E5X), revealing similar binding regions for the peptides. Residue‐level uptake difference was achieved by overlapping peptides.
Figure EV3. HDX kinetic plots for all VP30 peptides.
In addition, the HDX studies revealed another region, residues Q248 to E252, that describes tightening dynamics or remote conformational changes upon peptide binding (Figs 3B and EV4). For hnRNPUL_2, differential HDX kinetic analysis in the absence and presence of the ligand yielded similar HDX kinetics, consistent with the lack of its binding in the pull‐down and fluorescence polarization assays.
Figure EV4. Statistical analysis of deuterium uptake differences of all time points confirms the binding regions.
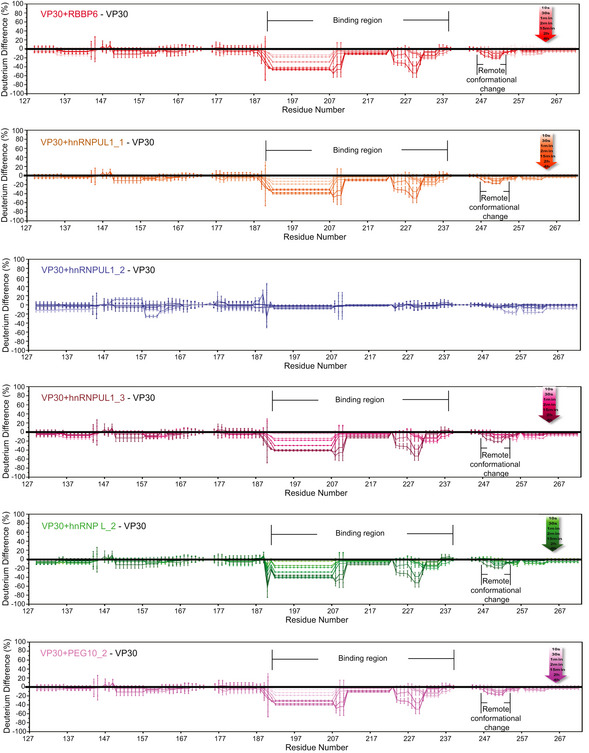
Deuterium uptake differences between different bound VP30 and unbound VP30 were calculated for each time point from 10 s to 4 h, and are depicted by gradient colors. Standard deviation between triplicates was calculated separately for each time point representing the bound and unbound states. Plotted on the graph is a threefold propagation error of each time point as shown by the error bars, giving 99.7% certainty on the observed difference. VP30 bound with RBBP6, hnRNPUL1_1, hnRNPUL1_3, hnRNP L_2, and PEG10_2 showed the same binding regions and similar regions exhibiting remote conformational or dynamics reduction owing to binding. There is no significant difference for VP30 with hnRNPUL1_2; therefore, it served as a negative control.
To determine whether each of the full‐length host proteins target the NP/RBBP6‐binding site on VP30, VP30–host protein interaction was assessed in cell‐based assays in the presence of increasing amounts of NP (Fig 4A). As NP levels increased, VP30 interaction with hnRNP L, hnRNPUL1, and PEG10 was disrupted. As previously reported, increasing amounts of NP could also disrupt the VP30–RBBP6 interaction (Batra et al, 2018). Select point mutations within the NP‐binding cleft on VP30, including E197A, W230A, or combinations of mutations that included these residues, were previously demonstrated to disrupt binding to both NP and RBBP6 (Kirchdoerfer et al, 2016; Xu et al, 2017; Batra et al, 2018). Similarly, E197A, W230A, or combinations of mutations that included these residues exhibited loss of binding to hnRNP L, hnRNPUL1, and PEG10 (Figs 4B and Fig EV5A and B). Interestingly, the VP30 Q229A mutation increased interaction with each of the host proteins. These data further support the existence of a common interaction site on VP30, as well as similar modes of binding.
Figure 4. PPxPxY‐containing host proteins target the NP‐binding cleft on VP30.
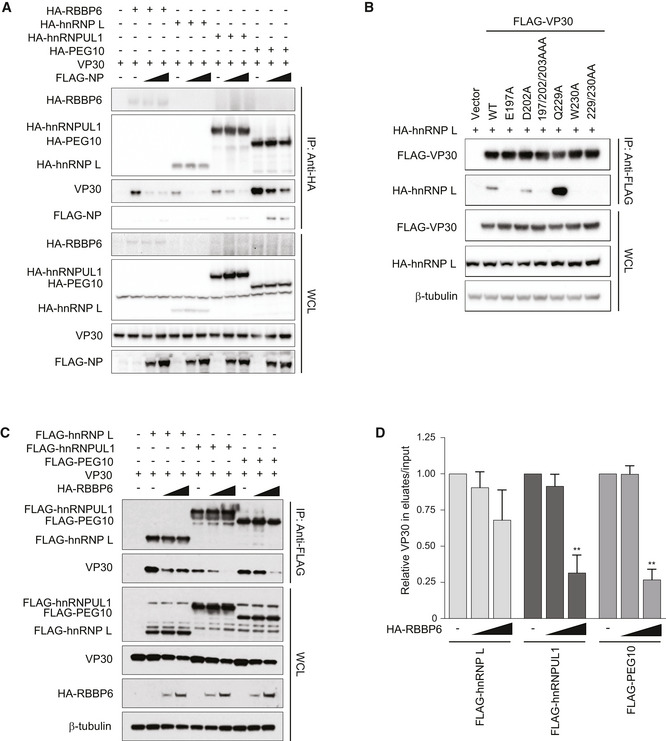
- Co‐immunoprecipitation experiment demonstrating that NP interferes with VP30‐PPxPxY binding. VP30 was co‐expressed with the indicated HA‐tagged PPxPxY proteins and either empty vector (−) or increasing amounts of FLAG‐tagged NP expression plasmid. IPs were performed with anti‐HA antibodies followed by Western blots of IPs or WCL by using anti‐HA, anti‐FLAG, and anti‐VP30 antibodies.
- Representative immunoblots of co‐immunoprecipitations between hnRNP L and the indicated VP30 mutants which were previously described to exhibit varying degrees of NP‐binding activity.
- Co‐immunoprecipitations between VP30 and the indicated host proteins upon titration of RBBP6. VP30 was co‐expressed with FLAG‐tagged PPxPxY‐containing host proteins and either a fivefold or tenfold excess of RBBP6 plasmid. IP was performed using anti‐FLAG beads followed by Western blotting using the indicated antibodies.
- Densitometric analysis of the Western blots shown in panel C. The graph represents the relative amount of VP30 present in the eluates compared with the input. The data represent the mean ± SD from two independent experiments (n = 2). Statistical significance was calculated using ANOVA with Tukey’s multiple comparisons test. **P < 0.005.
Source data are available online for this figure.
Figure EV5. Interaction of VP30 point mutants with hnRNPUL1 and PEG10.
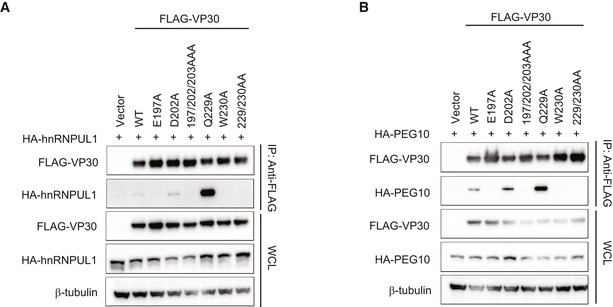
-
A, BRepresentative Western blots of co‐immunoprecipitation experiments to assess interaction between the indicated VP30 mutants and hnRNPUL1 (A) or PEG10 (B).
If there is a shared binding site on VP30, it would be expected that different PxPPPPxY motif‐containing proteins could compete with each other for binding with VP30. To test this possibility, we performed competition co‐immunoprecipitations between VP30 and FLAG‐tagged hnRNP L, hnRNPUL1 or PEG10 in the presence of HA‐tagged RBBP6. Western blotting and densitometric analysis of VP30 levels in eluates versus input suggested that increasing amounts of RBBP6 significantly disrupt the interaction between VP30 and hnRNPUL1 or PEG10 (Figs 4C and D). Competition with hnRNP L was less effective, but as RBBP6 levels increased, hnRNP L binding was modestly reduced. These data further suggest that multiple host proteins may compete with each other in infected cells.
Residues critical for VP30 interaction and MG assay inhibition
To further define the critical residues within the extended PxPPPPxY motif required for binding to VP30, we performed alanine scanning mutagenesis across the proline and tyrosine residues in the RBBP6 peptide and tested the VP30‐binding activity of these mutants in a co‐immunoprecipitation assay. Mutation of the motif (P1xP2P3P4P5xY) at prolines 2 and 5 and at the tyrosine greatly impaired binding (Fig 5A). The critical role of these residues was further supported by FP analysis with wild‐type and mutated RBBP6‐derived peptides (Fig 5B). A peptide with alanine substitution mutations at multiple positions including prolines 1, 3, and 4 fused to GFP fusion peptide resulted in a weaker interaction with VP30 compared with the wild‐type (WT) peptide (Fig 5C). When the mutant‐RBBP6 peptides were analyzed for their ability to inhibit the MG assay as GFP fusions, mutation of the proline residue at position 5 or the tyrosine lost inhibitory activity as compared to the WT RBBP6 peptide (Fig 5D, Appendix Fig S2). Surprisingly, the P2A mutant retained inhibitory activity despite the absence of detectable interaction (Fig 5D, Appendix Fig S2), suggesting a potential off‐target effect.
Figure 5. Defining the contribution of PPxPxY residues to VP30 interaction.
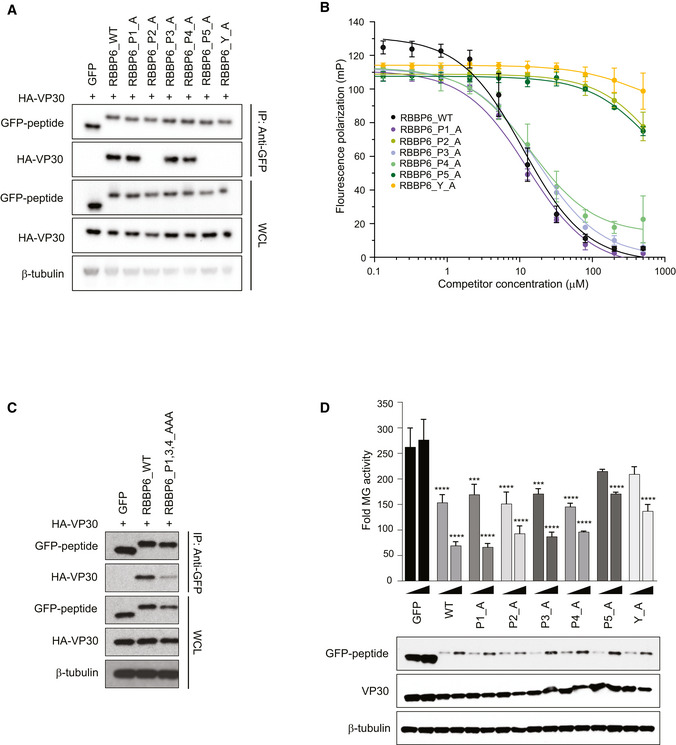
- Co‐immunoprecipitation between HA‐VP30 and either GFP, or GFP‐fused to WT or mutant‐RBBP6 peptides. Each proline or tyrosine residue in the P1xP2P3P4P5xY motif was mutated to alanine, and the indicated mutant peptides were expressed as GFP fusions. Co‐immunoprecipitations of HA‐VP30 with GFP or GFP‐peptide fusions were performed using anti‐GFP magnetic beads (IP: Anti‐GFP). Whole cell lysates (WCL) were blotted with anti‐GFP, anti‐HA, and anti‐β‐tubulin antibodies.
- Equilibrium dissociation curves of FITC‐RBBP6 from eVP30130‐272 as it is outcompeted by increasing concentrations (0.13–500 μM) of RBBP6 alanine mutants. Fluorescence polarization was determined with constant concentrations of FITC‐RBBP6 and eVP30130‐272 at 0.50 µM and 3.8 µM, respectively. Experiments were performed in two independent replicates. Error bars represent the standard deviation.
- Co‐immunoprecipitation between HA‐VP30 and wild‐type (WT) or mutant‐RBBP6 peptides fused to GFP. GFP alone served as a negative control. IP was performed for GFP and blots were probed with anti‐GFP and anti‐HA antibodies. Anti‐β‐tubulin served as a loading control for the whole‐cell lysates.
- Minigenome (MG) activity upon titration of plasmids encoding GFP fused to wild‐type or single point mutant‐RBBP6 peptides. GFP alone or GFP‐RBBP6 peptide plasmids were transfected at 12.5 and 25 ng amounts along with the plasmids of the MG system. The data represent the mean ± SD from one representative experiment in which each transfection condition was performed in triplicate (n = 3). The experiment was reproduced in two additional, independent experiments (see Appendix Fig S2). Statistical significance was calculated using ANOVA with Tukey’s multiple comparisons test. ****P < 0.00005; ***P < 0.0005
Multiple PPxPxY proteins can modulate EBOV RNA synthesis
Based on prior data indicating that RBBP6 inhibits EBOV RNA synthesis, the effect of over‐expression of the additional three host proteins was assessed in the EBOV MG assay. Plasmids expressing hnRNP L, hnRNPUL1, and PEG10 were transfected at two different concentrations along with the plasmids required for the MG assay. Empty vector and RBBP6 were included as controls. MG activity was significantly reduced in hnRNP L‐ and PEG10‐expressing cells (Fig 6A, Appendix Fig S3A and B). In contrast, hnRNPUL1 increased MG activity. Western blotting of lysates from these experiments demonstrated inhibition of viral protein expression at the higher concentration of the hnRNP L, hnRNPUL1, and PEG10 plasmids (Appendix Fig S3A). However, inhibition of viral protein expression was absent at the lower concentrations, where modulation of MG activity was still visible (Fig 6A, Appendix S3A–C).
Figure 6. Functional interaction between VP30 and multiple host proteins that contain PPxPxY motifs.
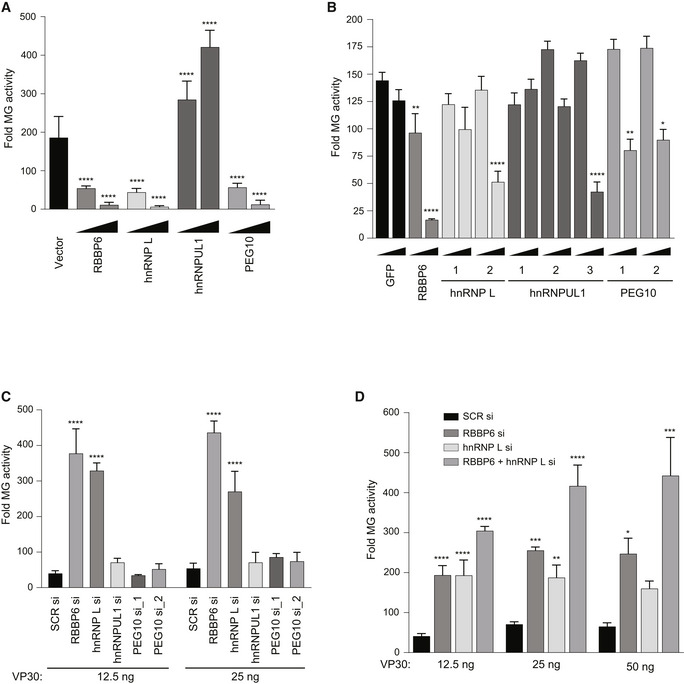
- The effect of over‐expression of the indicated host proteins on EBOV RNA synthesis was assessed using an EBOV MG assay. HEK293T cells were transfected with plasmids expressing components of the MG assay and either 12.5 or 125 ng of expression plasmids for the indicated host factors. Empty expression plasmid (vector) served as a control. Luciferase activity was measured 48 h post‐transfection and fold MG activity was calculated relative to no VP30 control.
- MG activity upon transient expression of GFP or GFP‐fused PPxPxY‐containing peptides. Plasmids expressing GFP‐fused peptides were transfected at 12.5 and 125 ng amounts. Statistical significance was calculated relative to the GFP control for each concentration.
- MG activity upon knockdown of host genes. HEK293T cells were transfected with scrambled siRNA or siRNA targeting RBBP6, hnRNP L, hnRNPUL1, or PEG10. Twenty‐four hours post‐transfection, cells were transfected with MG assay plasmids including VP30 at 12.5 and 25 ng doses.
- MG activity was assessed as in panel C upon single or double knockdown of RBBP6 and hnRNP L.
Data information: For each panel (A–D), the data represent the mean ± SD from one representative experiment in which each transfection condition was performed in triplicate (n = 3). The experiment was reproduced in two additional, independent experiments (see Appendix Figs S3, S4, S8 and S9). Statistical significance was calculated using ANOVA with Tukey’s multiple comparisons test. ****P < 0.00005; ***P < 0.0005, **P < 0.005, *P < 0.05.
We also compared the activity of the single PPxPxY‐motif peptide fusions from hnRNP L, hnRNPUL1, and PEG10 with that of the GFP‐RBBP6 peptide in the EBOV MG assay (Fig 6B, Appendix Fig S4). In this assay, RBBP6 peptide was inhibitory, consistent with prior studies (Batra et al, 2018). Each of the remaining peptides that interacted in the co‐precipitation and FP assays inhibited MG activity (Figs 6B, Appendix Fig S4).
Because VP30 plays roles in viral transcription (mRNA synthesis), the effects of each of the host proteins were compared using the standard MG and with a previously described replication‐deficient minigenome system in which the antigenomic replication promoter was deleted, rendering luciferase expression dependent on the efficiency of viral transcription (Hoenen, Jung et al, 2010). Over‐expression of RBBP6, hnRNP L, and PEG10 significantly reduced MG activity in both systems (Fig EV6A, Appendix Fig S5). In contrast, hnRNPUL1 modestly increased activity in both assays. These data demonstrate that the host proteins can affect EBOV transcription independently of effects on genome replication.
Figure EV6. Effects of host PPxPxY proteins on replication‐defective and VP30‐independent minigenome assays.
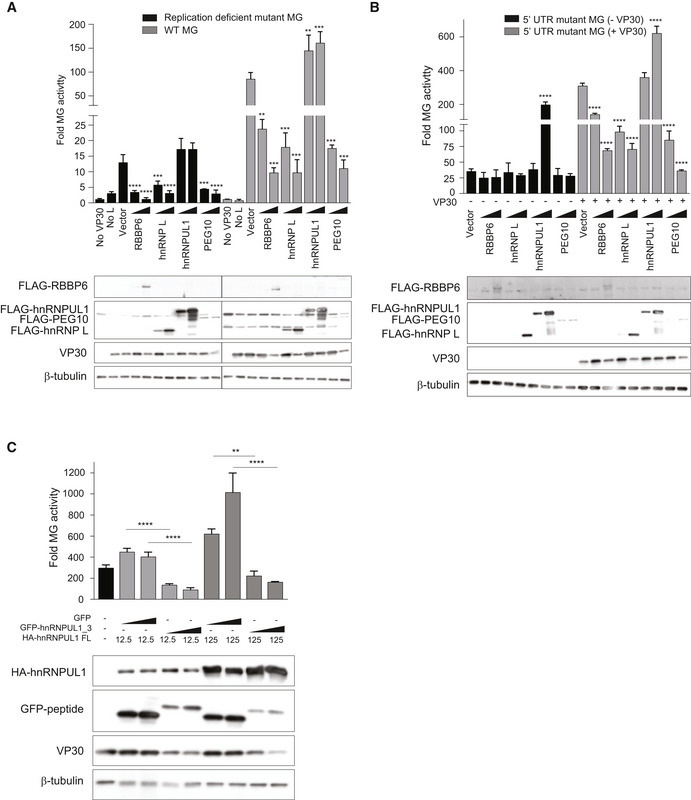
- MG activity upon over‐expression of each of the indicated host proteins using WT or replication‐deficient MG systems. 293T cells were transfected with plasmids encoding FLAG‐tagged host proteins along with the MG plasmids. Fold MG activity was calculated relative to a no VP30 control.
- MG activity upon over‐expression of each of the host protein using the 5’ UTR mutant MG as a template.
- MG activity upon over‐expression of GFP or hnRNPUL1 peptide 3 fused to GFP in combination with full‐length (FL) hnRNPUL1 (doses 12.5 ng and 125 ng). Statistical significance was calculated relative to the GFP control for each concentration tested using ANOVA with Tukey’s multiple comparisons test.
Data information: For each panel (A–C), the data represent the mean ± SD from one representative experiment in which each transfection condition was performed in triplicate (n = 3). Each experiment was reproduced in an additional, independent experiment (see Appendix Figs S5–S7). ****P < 0.00005; ***P < 0.0005, **P < 0.005.
We further used a 5’UTR mutant MG system in which a stem loop at the transcription start site was disrupted, rendering MG activity VP30‐independent (Weik et al, 2002). The signal in the mutant MG assay was not detectably affected by expression of RBBP6, hnRNP L, or PEG10 in the absence of VP30. However, a robust increase in activity was detected upon over‐expression of hnRNPUL1, suggesting that the effects of RBBP6, hnRNP L, and PEG10 are VP30‐dependent but hnRNPUL1 exerts its effects independently of VP30 (Fig EV6B, Appendix Fig S6).
Given the opposite effects of hnRNPUL1 peptide 3 versus full‐length hnRNPUL1, we asked how peptide 3 would affect MG activity in the presence of hnRNPUL1. Compared with the GFP control, the GFP‐peptide 3 fusion exerted a strong inhibitory effect even in the presence of hnRNPUL1, suggesting that peptide 3 exerts a dominant‐negative effect on the activating property of full‐length hnRNPUL1 (Fig EV6C, Appendix Fig S7).
The effect of knocking down endogenous levels of hnRNP L and hnRNPUL1 was also assessed, with RBBP6 siRNA and an irrelevant siRNA serving as controls. Twenty‐four hours post‐siRNA transfection, cells were transfected with the MG assay plasmids. As previously reported, RBBP6 knockdown increased MG assay signal, consistent with RBBP6 acting as an inhibitor of viral RNA synthesis. hnRNP L knockdown also increased the signal (Fig 6C, Appendix Fig S8), indicating it to be an inhibitor. Knockdown of hnRNPUL1 or PEG10 had little to no impact on MG activity (Fig 6C, Appendix Fig S8). To address whether RBBP6 and hnRNP L can act cooperatively, single and double knockdowns were performed in the context of the MG assay. The results demonstrate a roughly additive effect, suggesting that both host proteins can simultaneously modulate viral RNA synthesis (Fig 6D, Appendix Fig S9).
PxPPPPxY motif‐containing host proteins modulate virus infection
To evaluate the impact of the host proteins on EBOV replication, virus infections were performed in the context of siRNA knockdown. HeLa cells were transfected with siRNAs against human hnRNP L, hnRNPUL1, and PEG10, alongside control scrambled siRNA, followed by infection with an EBOV that expresses GFP (EBOV‐EGFP). Twenty‐four hours post‐infection, cells were fixed and quantified for infection by microscopy. Knockdown of hnRNP L strongly enhanced virus infection, whereas hnRNPUL1 siRNA‐treatment was inhibitory, consistent with the MG enhancement seen when hnRNPUL1 was over‐expressed (Fig 7A). Knockdown of PEG10 showed no significant effect on virus replication, consistent with the PEG10 knockdown MG assays. The efficacies of siRNA‐mediated knockdowns were confirmed by Western blotting using specific antibodies (Appendix Fig S10A and B).
Figure 7. PPxPxY‐containing host proteins modulate EBOV infectivity, viral transcription, and VP30 phosphorylation.
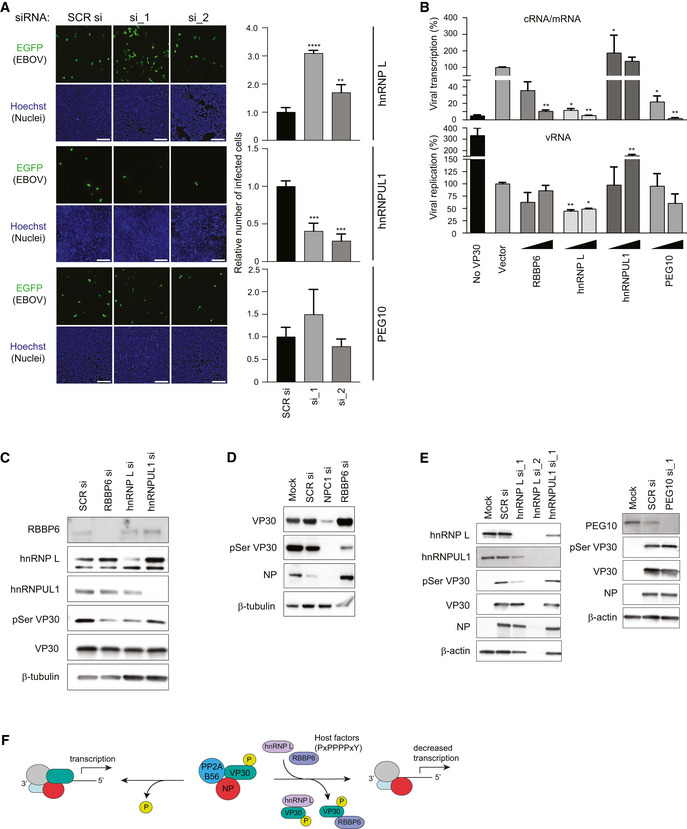
- HeLa cells were transfected with scrambled siRNA (SCR si) or siRNAs‐targeting hnRNP L, hnRNPUL1 or PEG10, 48 h post‐transfection, cells were challenged with EBOV‐EGFP at an MOI of 0.5. 24 h post‐infection, cells were fixed, stained for nuclei with Hoechst dye, and imaged. Scale bar is 200 µm. The graph (right) depicts relative number of infected cells as determined by calculating the ratio of number of infected cells to nuclei and shown relative to SCR siRNA. The data show a representative result of two independent experiments with the mean ± SD calculated from three independently treated wells. Statistical significance was calculated relative to the values obtained in SCR siRNA‐treated cells using ANOVA with Dunnett’s multiple comparisons test. ***P < 0.0001, ***P < 0.001, **P < 0.01
- Levels of transcription and replication in the MG assay upon over‐expression of the indicated host factors. MG assays were performed with 50 and 500 ng of host protein expression plasmid. RNA was isolated and analyzed by qRT–PCR to measure the levels of viral mRNA/cRNA and vRNA. The sample with vector control was set to 100%, and mean ± S.D. values from two independent experiments are shown (each experiment was performed in duplicate, n = 2). **P < 0.005, *P < 0.05. Statistical significance was calculated relative to vector control using ANOVA with Tukey’s multiple comparisons test.
- Immunoblots to assess VP30 and pVP30 levels in lysates from MG assay upon knockdown of RBBP6, hnRNP L, or hnRNPUL1.
- Immunoblots to assess the levels of VP30, pVP30, and NP in EBOV‐infected lysates upon knockdown of NPC1 or RBBP6. HeLa cells were mock transfected or transfected with siRNA targeting NPC1 or RBBP6. 48 h post‐transfection cells were infected with EBOV at MOI = 0.1. At 24 h post‐infection, cells were lysed in TRIzol. Protein was extracted from the TRIzol reagent and analyzed by Western blotting to determine levels of VP30, pSer29 VP30, and NP.
- Western blots detecting pVP30, VP30 and NP levels in EBOV‐infected HeLa cells upon hnRNP L, hnRNPUL1, or PEG10 knockdown.
- Proposed model for inhibition of EBOV replication by host factors containing PxPPPPxY motifs. Host proteins present at varying endogenous levels disrupt VP30–NP interaction thereby preventing VP30 dephosphorylation by NP‐recruited host phosphatase PP2A B56, and therefore inhibit virus transcription.
PxPPPPxY motif‐containing proteins influence VP30 phosphorylation
VP30 undergoes dynamic phosphorylation and dephosphorylation. Phosphorylation occurs via the serine‐arginine protein kinases SRPK1 and SRPK2 which regulate VP30 transcription‐promoting activity (Biedenkopf et al, 2016a; Lier et al, 2017; Takamatsu et al, 2020). NP recruits a host protein phosphatase, protein phosphatase 2A B56 (PP2A B56), resulting in dephosphorylation of NP‐bound VP30, and the resulting dephosphorylated VP30 promotes viral transcription (Kruse et al, 2018). VP30‐interacting host proteins may prevent the dephosphorylation of VP30 by competing with NP and inhibiting transcription. In contrast, elimination of competing host factors might increase VP30 dephosphorylation and enhance viral transcription. To test this, we measured by quantitative RT–PCR the amount of viral mRNA/antigenomic RNA (cRNA) and viral genomic RNA (vRNA) in the context of the MG assay following expression of each host protein. Viral transcription was significantly reduced when RBBP6, hnRNP L, or PEG10 were expressed, whereas the presence of hnRNPUL1 resulted in increased viral transcription and replication (Fig 7B). These findings parallel the effect of these proteins on the luciferase activities in the MG assay (Figs 6A, and EV6A and B). We next used, in the context of siRNA knockdowns, an antibody that specifically recognizes VP30 phosphorylated at Ser29 (Lier et al, 2017; Kruse et al, 2018). Knockdown of RBBP6 and hnRNP L reduced levels of VP30 phosphorylation in MG assays (Fig 7C) as well as in infected cells (Fig 7D and E). hnRNPUL1 and PEG10 knockdown had no significant effect on VP30 phosphorylation compared with control siRNA‐treated cells. These data suggest a model whereby RBBP6 and hnRNP L‐containing PxPPPPxY motifs can disrupt VP30–NP interaction, preventing VP30 dephosphorylation by PP2A B56 and modulating viral transcription (Fig 7F).
Discussion
This study characterized three novel EBOV VP30–host protein–protein interactions (PPIs) involving hnRNP L, hnRNPUL1, and PEG10, all of which have a PPxPxY motif, and demonstrated their functional consequences for EBOV RNA synthesis and infection. All three host proteins were identified in a recent AP‐MS study that defined an EBOV–host PPI network for six EBOV proteins. The network included 194 high‐confidence EBOV–human PPIs; one VP30‐interacting PPxPxY‐containing protein, RBBP6, was functionally characterized (Batra et al, 2018). Sequence analysis of the VP30‐binding peptides derived from RBBP6 and NP identified a common PPxPxY motif. Notably, this motif is conserved among NPs of the Ebolavirus, Marburgvirus, Cuevavirus, and Dianlovirus genera within the filovirus family. Based on the RBBP6 data, the VP30 interaction network was inspected for additional proteins possessing PPxPxY motif(s), identifying hnRNP L and PEG10 as having two such motifs and hnRNPUL1 as having three. Co‐immunoprecipitation studies confirmed the interaction between VP30 and RBBP6, hnRNP L, hnRNPUL1, and PEG10. Several lines of evidence suggest that these proteins target a common interaction interface on VP30. Competition between the PPxPxY proteins and NP for VP30 binding was demonstrated in co‐immunoprecipitation studies. Mutations in VP30 that alter NP binding similarly affect binding to each of the host factors; and competition between the host factors for binding to VP30 could also be demonstrated. Further, a FP assay that measured the capacity of host protein‐derived peptides to disrupt interaction between the C‐terminal domain of VP30 and RBBP6 demonstrated competition of the four host factors and NP for binding to VP30. We also mapped the effect of peptide binding on the conformational dynamics of the VP30 protein by employing HDX‐MS. The distinct patterns of HDX suggested that binding of peptides containing an extended PxPPPPxY motif induce significant changes even at the longer time scales, supporting a conformational change in a nearby region (Q248 to E252). All these findings point to a remarkable circumstance where multiple host proteins can target a critical virus–virus PPI leading to viral RNA synthesis modulation.
Proline‐rich motifs are frequently involved in protein–protein interactions. Domains that bind proline‐rich motifs include Src‐homology 3 (SH3), WW, EVH1, GYF (also known as CD2‐binding domains), UEV domain, and single‐domain profilin proteins (Reviewed in ref. Ball et al, 2005). Structurally, the EBOV VP30 C‐terminal domain is distinct from these previously described proline‐rich motif binding domains in that it is largely α‐helical in nature (Kirchdoerfer et al, 2016; Xu et al, 2017). Further, co‐crystals of the VP30 C‐terminal domain and the NP‐derived PPxPxY peptide or the RBBP6 peptide demonstrated that the PPxPxY motif does not form the type II helix typical of PPxP motifs (Kirchdoerfer et al, 2016; Xu et al, 2017; Batra et al, 2018). Rather, in the VP30‐NP peptide structures, the NP residues are in an extended conformation, making multiple contacts with VP30 residues (Xu et al, 2017). These unique features make understanding the VP30–PPxPxY interaction of particular interest.
The identification of several host proteins that interact with VP30 via PPxPxY motifs allowed us to characterize the basis of this unique Pro‐rich motif interaction. To better understand the specificity of different PPxPxY motifs for VP30, pulldowns were performed using peptide fusions to GFP in which the individual motifs were flanked on each side by ten amino acid residues. These experiments demonstrated differential binding activity. For those proteins with two or three PPxPxY motifs, only a single motif robustly interacted with VP30 (motif 2 for hnRNP L, motif 3 for hnRNPUL1, and motif 2 for PEG10) in each case. FP assay‐based measurements of the equivalent peptides yielded a comparable pattern of interactions and demonstrated a range of binding affinities. The RBBP6 peptide was the tightest binder. hnRNPUL1 motif 3 and PEG10 motif 2 behaved similarly to each other and to the PPxPxY peptide derived from eNP. The hnRNP L motif 2 and hnRNPUL1 motif 1 exhibited comparable binding affinities to each other, consistent with the results of the co‐immunoprecipitation assays, in which hnRNP L motif 2 bound weakly and hnRNPUL1 motif 1 yielded a barely detectable interaction. By aligning the various motifs and ranking them based on affinities, we can see that PxPPPPxY correlated with a stronger interaction. Even among those peptides containing this motif variations were seen, suggesting that diversity in the “x” positions or flanking sequences may also influence binding. Contributions of nearby residues to Pro‐rich motif binding have been demonstrated in other contexts as well (Ball et al, 2005).
Because the RBBP6 peptide consistently showed robust binding to VP30 and the highest inhibitory activity in MG assays, the contribution of individual residues to binding was assessed by mutagenesis. The data support critical roles for the second and fifth proline residues and the tyrosine residue, with each individual mutation abrogating the interaction when assessed either by co‐immunoprecipitation or FP assay. Interestingly, the P2A mutant retained the capacity to inhibit viral RNA synthesis in the MG assay, despite its reduced binding in co‐immunoprecipitation assays. This latter finding could reflect a weaker interaction with VP30 that is nonetheless sufficient to inhibit MG activity. Alternatively, there may be off‐target effects of expressing the peptide fused with GFP that impact viral RNA synthesis. To address specificity of proline‐tyrosine motifs to interact with VP30, a set of experiments was performed with the EBOV matrix protein VP40. EBOV VP40 possess two overlapping late domain motifs 7PTAPPEY13 resembling the PPxPxY motif. The PPxY late domain motif mediates interactions with the WW domain host proteins that facilitate VP40 budding and release from cells (Harty et al, 2000; Timmins et al, 2003; Yasuda et al, 2003; Martin‐Serrano et al, 2005; Han et al, 2016; Han et al, 2017; Liang et al, 2017). Despite the similarity of the VP40 proline‐tyrosine motif to PPxPxY, an interaction between VP30 and VP40 was not detected, either with full‐length VP40 or when a peptide encompassing the late domain motif of VP40 was fused to GFP. This indicates that VP30 does not bind typical WW domain ligands. Mutation of the VP40 peptide to match the PxPPPPxY motif did confer weak binding, further supporting the conclusion that this motif confers binding. However, the relatively weak interaction with this mutated VP40 peptide provides further evidence that the sequence context in which the motif is embedded also influences binding. Cumulatively, these studies further define the VP30 interface as a novel proline‐tyrosine interacting motif and provide a foundation for further elucidation of its structural basis and functional significance.
That the interactions of VP30 with the endogenous host proteins are functionally significant is supported by the demonstrations that modulation of RBBP6, hnRNP L, PEG10, and hnRNPUL1 levels regulate EBOV RNA synthesis, as measured by MG assays and by the demonstration that RBBP6, hnRNP L, and hnRNPUL1 affect EBOV infection. In support of their role as cellular modulators of EBOV replication, over‐expression of full‐length RBBP6, hnRNP L, and PEG10 inhibited EBOV RNA synthesis, whereas transfection of full‐length hnRNPUL1 enhanced MG activity. Similarly, expression of interacting PPxPxY‐containing peptides also inhibited RNA synthesis. That full‐length hnRNPUL1 enhances activity, whereas hnRNP L, PEG10, and the GFP‐peptide fusions, including GFP‐hnRNPUL1 peptide 3 inhibit MG activity, is notable. To clarify the effects of hnRNPUL1 assays, we used a mutant MG assay that functions without a requirement for VP30. In this context, hnRNPUL1 over‐expression still increased MG activity, whereas the effect of the other interactors was lost. hnRNPUL1 over‐expression also enhanced vRNA levels in a MG Assay. This argues for VP30‐indepdendent effects of hnRNPUL1 on replication. Further, the hnRNPUL1 peptide_3 construct retains the capacity to inhibit the standard MG assay when full‐length hnRNPUL1 is over‐expressed. These findings suggest a model where targeting of VP30 by over‐expressing hnRNPUL1 peptide_3 can exert a dominant inhibitory effect over the VP30‐independent activity of hnRNPUL1.
Knockdown of the individual host factors in the context of the MG assay clearly implicates hnRNP L as a modulator of activity, with knockdown increasing signal to a degree that is comparable to that seen upon knockdown of RBBP6. We also asked whether knockdown of both RBBP6 and hnRNP L would result in additive effects. The MG assays performed suggest that this was the case. When knockdown was performed in the context of EBOV infection, hnRNP L was again most clearly implicated as a factor that negatively impacts infection. Notably, hnRNPUL1 knockdown decreased infection, an outcome consistent with the enhancement of MG activity upon over‐expression. Why hnRNPUL1 knockdown affected virus infection but not MG activity is unclear but could reflect levels of expression in the different cell lines used in these studies. Given the VP30‐independent effects hnRNPUL1 in the MG assays, it is likely the effects on virus infection are also VP30‐independent. Even with this “off‐target” effect of hnRNPUL1, our data still suggest that multiple host factors can target the same interface on VP30. How several different host factors together modulate EBOV gene expression will require additional study. However, relative expression levels and localizations in a given cell type will be expected to influence the outcome. Therefore, it remains possible that PEG10 can influence the outcome of infection in tissues where it is highly expressed.
A substantial body of literature implicates VP30 phosphorylation as a regulator of its transcriptional functions (Modrof et al, 2002; Martinez et al, 2011; Biedenkopf et al, 2013; Ilinykh et al, 2014; Biedenkopf et al, 2016a; Lier et al, 2017; Ammosova et al, 2018; Kruse et al, 2018; Tigabu et al, 2018). Further, NP recruits a cellular protein phosphatase to dephosphorylate NP‐bound VP30 (Kruse et al, 2018). Previous studies demonstrated the capacity of RBBP6 to target the NP‐binding site on VP30 and to impair viral RNA synthesis and infectivity (Batra et al, 2018). Data in this study demonstrating modulation of viral RNA synthesis by hnRNP L, hnRNPUL1, and PEG10 suggest a model whereby disruption of the NP–VP30 interaction by a host factor would alter NP‐mediated VP30 dephosphorylation. If levels of phospho‐VP30 rise as a consequence, this would be expected to inhibit virus transcription and gene expression. Fitting this model, expression of RBBP6, hnRNP L, or PEG10 inhibited activity in a replication‐defective MG assay that only reports on transcription. When transcription versus replication was measured by quantitative RT–PCR in the standard MG assay, effects on mRNA synthesis were more dramatic than effects on products of replication. Further, knockdown of endogenous RBBP6 and hnRNP L decreased VP30 phosphorylation levels and increased gene expression in the context of viral infection. These findings support a model where competition for VP30‐NP binding by RBBP6 or hnRNP L inhibits viral transcription by modulating VP30 phosphorylation levels.
It is striking that EBOV maintains a VP30‐NP interface critical for replication and that interface can be directly targeted by multiple host factors. While RBBP6 and hnRNP L appear to act as restriction factors, in that they prevent VP30 dephosphorylation and inhibit viral gene expression, it is also possible that EBOV takes advantage of these interactions as a regulatory mechanism. For example, early in infection, when NP levels are relatively low, the presence of competitors for binding to VP30 could reduce dephosphorylation to dampen viral transcription, perhaps as a means to delay recognition by the immune system. Given that our data suggest cooperation between multiple host factors that inhibit viral transcription, it will be of interest to identify cell types with different levels of the host PPxPxY proteins to determine how this shapes the replication cycle.
Our data do not preclude effects of VP30 on host protein function. RBBP6, hnRNP L, and hnRNPUL1 are each RNA‐binding proteins with a number of biological functions, including the regulation of various RNA processing activities, RNA degradation, pre‐mRNA splicing, and mRNA translation. Beyond this are effects on cellular proliferation, dsDNA break repair and other processes, such as for hnRNP L, on interferon‐β gene expression (Hong et al, 2013; Gurunathan et al, 2015; Peng et al, 2017; Seo et al, 2017; Zhou et al, 2017; Cao et al, 2019). PEG10 is derived from a retrotransposon gag‐pol gene (Xie et al, 2018). Its mRNA contains two open reading frames (ORFs) with the first ORF encoding a gag‐like protein and the second encoding a pol‐like protein. A −1 frameshift occurs during translation leading to production of a gag‐pol fusion protein. PEG10 is highly expressed in the placenta, adrenal gland, ovary, testis, and brain; it plays roles in placenta formation and adipocyte differentiation; and its deletion in mice is embryonic lethal (Hishida et al, 2007; Xie et al, 2018). PEG10 is also upregulated in cancer where it plays a role in promoting cellular proliferation. It remains to be determined whether VP30 interaction modulates the functions of these host proteins and what impact this might have on virus infection.
Materials and Methods
Cell lines
HEK293T (human embryonic kidney SV40 Tag transformed; ATCC, CRL‐3216), Huh7 (a generous gift from the Gordan lab at UCSF), and HeLa cells (ATCC, CCL‐2) were maintained in Dulbecco’s modified Eagle’s medium (Corning Cellgro or Thermo Fisher Scientific) with 10% fetal calf serum (Gibco or Gemini Bio‐Products) at 37°C in a humidified atmosphere with 5% CO2. The cells were monitored regularly for mycoplasma contamination.
Antibodies
The antibodies used in the study include polyclonal rabbit anti‐RBBP6 antibody (Origene Technologies, TA309830, used at 1:250), polyclonal rabbit anti‐hnRNP L (Abcam, ab32680, used at 1:1,000), monoclonal rabbit anti‐hnRNPUL1 (Abcam, ab134954, ab68480, used at 1:1,000), polyclonal mouse anti‐PEG10 (Novus Biologicals, H00023089‐B01P, used at 1:500), monoclonal mouse anti‐FLAG M2 antibody (Sigma‐Aldrich, F1804, used at 1:2,000), polyclonal rabbit anti‐FLAG antibody (Sigma‐Aldrich, F7425, used at 1:2,000), monoclonal mouse anti‐HA antibody (Sigma‐Aldrich, H3663, used at 1:1,000), polyclonal rabbit anti‐HA antibody (Sigma‐Aldrich, H6908, used at 1:1,000), monoclonal mouse anti‐β‐tubulin antibody (Sigma‐Aldrich, T8328, used at 1:5,000), rabbit anti‐β‐actin antibody (CST, 4970S, used at 1:2,000) rabbit anti‐peptide serum raised against EBOV VP30 (2–25aa) (used at 1:5,000) and NP (97–119aa) (used at 1:1,000) and anti‐VP30 pSer VP30 (used at 1:300). The anti‐VP30, ‐NP, and pSer VP30 antibodies have been previously described (Lier et al, 2017; Batra et al, 2018).
Plasmid constructs
The coding sequences of human hnRNP L (NM_001533.2), hnRNPUL1 (NM_007040), and PEG10 (NM_015068.3) were PCR amplified from ORF clones (hnRNP L, GenScript #OHu14072; hnRNPUL1, Origene #RC200576; PEG10, GenScript #OHu101111), and the products were cloned into the mammalian expression plasmid pCAGGS. The amino acid regions containing PPxPxY motifs in hnRNP L (peptide_1: 325–350aa; peptide_2: 360–385aa), hnRNPUL1 (peptide_1: 702–727aa; peptide_2: 742–767aa; peptide_3: 769–794aa), and PEG10 (peptide_1: 672–697aa; peptide_2: 690–708aa) were cloned as fusions to an N‐terminal GFP in pCAGGS.
All expression plasmids for human RBBP6, GFP‐RBBP6 peptide, the EBOV MG assay, and the VP30 mutant constructs (E197A, D202A, E197A/D202A/Q203A, Q229A, W230A, Q229A/W230A) have been previously described (Edwards et al, 2015; Xu et al, 2017). The sequences of the constructs were validated by Sanger DNA sequencing prior to use.
Co‐immunoprecipitation assays
HEK293T cells were transfected with the indicated plasmids using Lipofectamine 2000 (Invitrogen) as per the manufacturer's instructions. 24 h post‐transfection, cells were harvested in NP‐40 lysis buffer (50 mM Tris–Cl pH 7.5, 280 mM NaCl, 0.5% NP‐40, 2 mM EDTA, 10% glycerol) supplemented with cOmplete mini protease inhibitor tablets (Roche). Clarified cell lysates were incubated with anti‐GFP (MBL International Corp.), ‐HA, or ‐FLAG magnetic beads (Sigma‐Aldrich) for 1–2 h at 4°C, followed by washing the magnetic beads with NP‐40 lysis buffer five times. Protein complexes were eluted by direct incubation in 1× SDS loading buffer and heating at 95°C. Eluates and whole‐cell lysates were analyzed by Western blotting using the indicated antibodies.
MG assays
HEK293T cells were transfected with expression plasmids encoding EBOV proteins VP35 (31.25 ng), L (125 ng), NP (62.5 ng), VP30 (12.5 or 25 ng) along with T7 RNA polymerase (50 ng), and a plasmid that produces the negative‐sense minigenome RNA encoding Renilla luciferase (50 ng) flanked by cis‐acting transcription and replication signals from the EBOV genome, by using Lipofectamine 2000 (Thermo Fisher) in 96‐well plates. A plasmid expressing firefly luciferase (1 ng) from an RNA polymerase II promoter was included as an internal control. Forty‐eight hours post‐transfection, reporter activity was measured using the Dual‐luciferase reporter assay system (Promega). Renilla luciferase activity was normalized to firefly luciferase values and plotted as fold MG activity calculated relative to no VP30 control. For some of the MG assays, a 5’ UTR mutant MG (100 ng) (in which a stem loop at transcriptional start site of NP was disrupted) (Weik et al, 2002) or replication‐deficient MG (100 ng) (in which the antigenomic replication promoter was deleted) (Hoenen et al, 2010) were used as a template. The data shown are the mean ± SD from one experiment in which for each condition, three separate transfections (n = 3) were performed in parallel in a representative experiment. Each experiment was performed at least two or three times (n = 2 or 3) with each replicate yielding the equivalent results.
Gene silencing using siRNA
For MG assays, HEK293T cells were transfected with 25 nM of SMARTpool:Accell siRNA targeting RBBP6 (Dharmacon, #E‐006551‐00‐0005), or 10 nM siRNA duplexes targeting hnRNP L (Qiagen, #SI00300475; SI03208527), hnRNPUL1 (Thermo Fisher Scientific, #s21883; s21884) or PEG10 (Qiagen, # SI00682108; SI04369323; SI04217913) (final concentration made up to 25 nM with control siRNA), or 25 nM control ON‐TARGET plus Non‐targeting siRNA (Thermo Fisher Scientific) using Lipofectamine RNAiMAX (Invitrogen). Twenty‐four hours post‐siRNA transfection, cells were transfected with plasmids for the MG assay and luciferase activity was assessed as above. For infection assays, 1.0 × 104 HeLa cells were seeded into 96‐well plates in triplicate. The next day, 40 nM of control siRNA or siRNAs‐targeting hnRNP L or PEG10 were transfected using 1:2 (v/v) Lipofectamine RNAiMAX (Thermo Fisher, 13778150) according to the manufacture’s protocol. 24 h post‐transfection, cells were moved to BSL4 for infection. For hnRNPUL1, double siRNA transfection was conducted to achieve significant inhibition. HeLa cells were seeded into 96‐well plates (1.0 × 104 cells/well). The next day, 10 nM of control siRNA or siRNAs‐targeting hnRNPUL1 were transfected using 1:2 (v/v) Lipofectamine RNAiMAX. 24 h post‐transfection, the cells were transfected again with siRNA using the same protocol. Cells were moved to BSL4 for infection after 24 h after the second transfection.
Western blots
For detection of protein levels in co‐immunoprecipitation eluates and whole‐cell lysates, samples were subjected to SDS–PAGE and Western blotting. All primary and secondary antibodies (HRP‐conjugated, used at 1:5,000) were diluted in 5% non‐fat dry milk in TBST. Imaging was performed using ChemiDoc XRS+ System (Bio‐Rad), KwikQuant Imager (Kindle Biosciences), or X‐ray films.
Fluorescence polarization assay (FPA)
FPA experiments were performed on a Cytation5 plate reader (BioTek) operating on Gen5 software. Excitation and emission wavelengths were set to 485 and 528 nm, respectively, with a band pass of 20 nm. Sample read height was 8.5 nm and G factor was set to 1.26 using the autogain function within the Gen5 software. Experiments were performed using a buffer containing 10 mM HEPES pH 7, 150 mM NaCl, and 2 mM Tris(2‐carboxyethyl)phosphine.
For competition FPA, 0.5 μM of fluorescein isothiocyanate (FITC)‐labeled RBBP6 (FITC‐RBBP6) was incubated with the saturation concentration of 3.8 μM protein and unlabeled PPxPxY‐containing peptide in buffer. Unlabeled PPxPxY‐containing peptides, RBBP6 (Human and Bat), and RBBP6 alanine mutants, were loaded into 96‐well plate, at concentrations ranging from 0.13 μM to 500 μM with 2.5‐fold dilutions. After 20 min of incubation, fluorescence polarization signals were read. Experiments were performed in two independent duplicates, and error bars represent standard deviation. Dissociation constants for competition FPA experiments were fitted with the following equation (Liu et al, 2017):
where F = Fluorescence polarization, F bound = F when FITC‐peptide is completely bound to the receptor protein, F free = F when the FITC‐peptide is free in solution, C tot = the competitor concentration (independent variable), K D = the dissociation constant of the FITC peptide from the protein receptor, K 2 = the dissociation constant of the competitor, and L = the concentration of the FITC peptide. While fitting, L was held constant at 0.5 μM and the K D was held constant at the calculated K D from previous normal FP experiments while the rest of the parameters were varied.
Hydrogen deuterium exchange
VP30130‐272 was incubated individually with host factor peptides RBBP6, hnRNPUL1_1, hnRNPUL1_2, hnRNPUL1_3, hnRNP L_2, and PEG10_2 in the 10 mM HEPES Buffer (150 mM NaCl, pH = 7.0) on ice for 45 min. RBBP6 was at a 4:1 ratio to VP30130‐272, and other host factor peptides were at 20:1 ratio. An aliquot of 2 μl of eVP30 stock solution (40 μM for bound or unbound) was added to 18 μl labeling buffer (10 mM HEPES buffer, 150 mM NaCl in 90% D2O, pH = 7.0) and incubated on ice for 10 s, 30 s, 1 min, 2 min, 15 mins, 2 h, and 4 h. Quenching buffer of 30 μl (50 mM TCEP, 4 M urea in 10 mM HEPES, pH = 2.5) was then added for 30 s on ice to stop the HDX. Samples were then snap‐frozen immediately by immersion in liquid nitrogen and kept at −80°C until mass spectrometric analysis. All measurements were performed in triplicate. Before mass spectrometry, each sample vial was incubated at 25°C for 30 s to thaw, followed by injection into a custom‐built HDX platform. Through a ZORBAX Eclipse XDB C8 column (2.1 mm × 15 mm, Agilent Technologies), protein samples were desalted with 0.1% trifluoroacetic acid in H2O for 3 min. Online digestion was then conducted on an immobilized pepsin column (2 mm × 20 mm) at 200 μl/min flow rate. A Hypersil Gold C18 column (2.1 mm × 50 mm, Thermo Fisher Scientific) with a 9.5‐min‐linear gradient (4–40% acetonitrile with 0.1% formic acid, 200 μl/min flow rate) was used for peptide separation followed by LTQ‐FT mass spectrometry analysis (Thermo Fisher Scientific) with data acquisition at a mass resolving power of 100,000 at m/z 400. Residue‐level HDX data were analyzed and exported from HDExaminer (Sierra Analytics, v2.5.0) by using “heavy” heat map smoothing to show HDX at the peptide and residue levels.
RNA isolation and quantitative RT–PCR
0.5 million HEK293T cells were transfected with expression plasmids encoding EBOV proteins VP35 (125 ng), L (500 ng), NP (250 ng), VP30 (100 ng) along with T7 RNA polymerase (200 ng), and a plasmid that produces the negative‐sense model viral genome RNA encoding Renilla luciferase (200 ng) flanked by cis‐acting transcription and replication signals from the EBOV genome, by using Lipofectamine 2000 (Thermo Fisher) in 12‐well plate. Cells were harvested 48 h post‐transfection and total RNA was isolated from the cells using the RNeasy mini kit (Qiagen) as per the manufacturer’s protocol. Residual plasmid contamination was removed by treatment with ezDNase enzyme (Invitrogen). For cDNA synthesis, ˜1 µg of RNA was used for reverse transcription of either positive strand (cRNA/mRNA) using an oligo(dT) primer or negative‐strand RNA (vRNA) using reverse primer for reporter gene, using SuperScript® IV First‐Strand Synthesis kit (Thermo Fisher Scientific). Subsequently, real‐time PCR was performed using PerfeCTa SYBR Green FastMix (QuantaBio) on a CFX96 Real‐Time PCR detection system (Bio‐Rad) using the primers targeting the reporter gene (5’‐AACGCGGCCTCTTCTTATTT‐3’,5’‐ATTTGCCTGATTTGCCCATA‐3’) and PCR conditions as follows: initial denaturation at 95°C for 30 s and then 40 cycles of 95°C for 5 s, 55°C for 15 s, and 68°C for 10 s. Expression levels were normalized to the β‐actin control (primer sequence: 5’‐ACTGGAACGGTGAAGGTGAC‐3’, 5’‐GTGGACTTGGGAGAGGACTG‐3’). Percent transcription or replication was calculated and set relative to the empty vector control sample.
EBOV infection assays
All studies with infectious EBOV were performed under biosafety level 4 containment at Texas Biomedical Research Institute or at the National Emerging Infectious Diseases Laboratory (NEIDL) at Boston University. A recombinant, fully infectious EBOV virus encoding GFP as an extra gene was used (Ebihara et al, 2007). For siRNA knockdown studies, siRNA‐treated HeLa cells were infected at an MOI of 0.5 for 24 h. Cells were fixed in 10% buffered formalin and stained with the Hoechst 33342 dye. Relative infection rate was calculated as the ratio of EGFP‐positive cells to cell nuclei by analyzing ≥ 7,000 cells. siRNA‐mediated knockdown was assessed by Western blotting the cell lysates collected at the same time as fixation. Three independent experiments were performed for each siRNA tested.
Isolation of proteins from EBOV‐infected lysates
HeLa cells were infected with EBOV‐GFP at an MOI of 0.1. Twenty‐four hours post‐infection, cells were lysed in TRIzol reagent and proteins were isolated as described (Batra et al, 2018).
Statistical analysis
All statistical analysis was performed using GraphPad Prism 7, using appropriate parameters as indicated in the figure legends and text. Data values were considered significantly different if the P‐value was < 0.05. Unless stated otherwise, ANOVA with Tukey’s multiple comparisons test was used.
Conflict of interest
The authors declare that they have no conflict of interest. The Krogan Laboratory has received research support from Vir Biotechnology and F. Hoffmann‐La Roche. Nevan Krogan has consulting agreements with the Icahn School of Medicine at Mount Sinai, New York, Maze Therapeutics and Interline Therapeutics, is a shareholder of Tenaya Therapeutics and has received stocks from Maze Therapeutics and Interline Therapeutics.
Supporting information
Review Process File
Appendix
Expanded View Figures PDF
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 4
Source Data for Expanded View Figures
Acknowledgments
This study was supported by NIH grants R01AI143292 to GKA, CFB, and NJK and P01AI120943 to GKA, CFB, DWL, MLG, and RAD. It was also supported by Department of the Defense, Defense Threat Reduction Agency grant HDTRA1‐16‐1‐0033 to C.F.B. and G.K.A. Operations support of the National Emerging Infectious Diseases Laboratories (NEIDL) was provided by NIAID/NIH grant UC7AI095321. The mass spectrometry was supported by NIGMS of the NIH (grants P41GM103422 and R24GM136766). We thank Megan R. Edwards for critical reading of the manuscript.
Author contributions
Experiments: JB, HM, GIS, MA, OS, NM, MZ, DL, and CGW; Data analysis: JB, HM, MA, OS, GIS, NM, MZ, DL, MLG, RAD, DWL, GKA, NJK, and CFB; Experiment design: JB, HM, OS, MA, MLG, DWL, RAD, GKA, NJK, and CFB; Critical reagents: NB and SB; Manuscript writing: JB, GKA, and CFB.
The EMBO Journal (2021) 40: e105658.
Contributor Information
Robert A Davey, Email: radavey@bu.edu.
Gaya K Amarasinghe, Email: gamarasinghe@wustl.edu.
Nevan J Krogan, Email: nevan.krogan@ucsf.edu.
Christopher F Basler, Email: cbasler@gsu.edu.
Data availability
This study includes no data deposited in external repositories.
References
- Adepoju P (2021) Ebola returns to Guinea and DR Congo. Lancet 397: 781 [DOI] [PubMed] [Google Scholar]
- Ammosova T, Pietzsch CA, Saygideger Y, Ilatovsky A, Lin X, Ivanov A, Kumari N, Jerebtsova M, Kulkarni A, Petukhov Met al (2018) Protein phosphatase 1‐targeting small‐molecule C31 inhibits Ebola virus replication. J Infect Dis 218: S627–S635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball LJ, Kuhne R, Schneider‐Mergener J, Oschkinat H (2005) Recognition of proline‐rich motifs by protein‐protein‐interaction domains. Angew Chem Int Ed Engl 44: 2852–2869 [DOI] [PubMed] [Google Scholar]
- Batra J, Hultquist JF, Liu D, Shtanko O, Von Dollen J, Satkamp L, Jang GM, Luthra P, Schwarz TM, Small GIet al (2018) Protein interaction mapping identifies RBBP6 as a negative regulator of Ebola Virus replication. Cell 175: 1917–1930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bausch DG, Rojek A, Scheld WM, Hughes JM, Whitley RJ (2016) West Africa 2013: re‐examining Ebola. Microbiol Spectr 4: 4.3.39. 10.1128/microbiolspec.EI10-0022-2016 [DOI] [PubMed] [Google Scholar]
- Becker S, Rinne C, Hofsass U, Klenk HD, Muhlberger E (1998) Interactions of Marburg virus nucleocapsid proteins. Virology 249: 406–417 [DOI] [PubMed] [Google Scholar]
- Biedenkopf N, Hartlieb B, Hoenen T, Becker S (2013) Phosphorylation of Ebola virus VP30 influences the composition of the viral nucleocapsid complex: impact on viral transcription and replication. J Biol Chem 288: 11165–11174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biedenkopf N, Lier C, Becker S (2016a) Dynamic phosphorylation of VP30 is essential for Ebola virus life cycle. J Virol 90: 4914–4925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biedenkopf N, Schlereth J, Grunweller A, Becker S, Hartmann RK (2016b) RNA binding of Ebola virus VP30 is essential for activating viral transcription. J Virol 90: 7481–7496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L, Liu S, Li Y, Yang G, Luo Y, Li S, Du H, Zhao Y, Wang D, Chen Jet al (2019) The nuclear matrix protein SAFA surveils viral RNA and facilitates immunity by activating antiviral enhancers and super‐enhancers. Cell Host Microbe 26: 369–384. [DOI] [PubMed] [Google Scholar]
- Ebihara H, Theriault S, Neumann G, Alimonti J, Geisbert J, Hensley L, Groseth A, Jones S, Geisbert T, Kawaoka Yet al (2007) In vitro and in vivo characterization of recombinant Ebola viruses expressing enhanced green fluorescent protein. J Infect Dis 196(Suppl 2): S313–S322 [DOI] [PubMed] [Google Scholar]
- Edwards MR, Pietzsch C, Vausselin T, Shaw ML, Bukreyev A, Basler CF (2015) High‐throughput minigenome system for identifying small‐molecule inhibitors of Ebola virus replication. ACS Infect Dis 1: 380–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enterlein S, Volchkov V, Weik M, Kolesnikova L, Volchkova V, Klenk HD, Muhlberger E (2006) Rescue of recombinant Marburg virus from cDNA is dependent on nucleocapsid protein VP30. J Virol 80: 1038–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurunathan G, Yu Z, Coulombe Y, Masson JY, Richard S (2015) Arginine methylation of hnRNPUL1 regulates interaction with NBS1 and recruitment to sites of DNA damage. Sci Rep 5: 10475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halfmann P, Kim JH, Ebihara H, Noda T, Neumann G, Feldmann H, Kawaoka Y (2008) Generation of biologically contained Ebola viruses. Proc Natl Acad Sci USA 105: 1129–1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Z, Sagum CA, Bedford MT, Sidhu SS, Sudol M, Harty RN (2016) ITCH E3 ubiquitin ligase interacts with Ebola virus VP40 to regulate budding. J Virol 90: 9163–9171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Z, Sagum CA, Takizawa F, Ruthel G, Berry CT, Kong J, Sunyer JO, Freedman BD, Bedford MT, Sidhu SSet al (2017) Ubiquitin ligase WWP1 interacts with Ebola virus VP40 to regulate egress. J Virol 91: e00812‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harty RN, Brown ME, Wang G, Huibregtse J, Hayes FP (2000) A PPxY motif within the VP40 protein of Ebola virus interacts physically and functionally with a ubiquitin ligase: implications for filovirus budding. Proc Natl Acad Sci USA 97: 13871–13876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hishida T, Naito K, Osada S, Nishizuka M, Imagawa M (2007) peg10, an imprinted gene, plays a crucial role in adipocyte differentiation. FEBS Lett 581: 4272–4278 [DOI] [PubMed] [Google Scholar]
- Hoenen T, Jung S, Herwig A, Groseth A, Becker S (2010) Both matrix proteins of Ebola virus contribute to the regulation of viral genome replication and transcription. Virology 403: 56–66 [DOI] [PubMed] [Google Scholar]
- Hong Z, Jiang J, Ma J, Dai S, Xu T, Li H, Yasui A (2013) The role of hnRPUL1 involved in DNA damage response is related to PARP1. PLoS One 8: e60208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilinykh PA, Tigabu B, Ivanov A, Ammosova T, Obukhov Y, Garron T, Kumari N, Kovalskyy D, Platonov MO, Naumchik VSet al (2014) Role of protein phosphatase 1 in dephosphorylation of Ebola virus VP30 protein and its targeting for the inhibition of viral transcription. J Biol Chem 289: 22723–22738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilunga Kalenga O, Moeti M, Sparrow A, Nguyen VK, Lucey D, Ghebreyesus TA (2019) The ongoing Ebola epidemic in the democratic republic of Congo, 2018–2019. N Engl J Med 381: 373–383 [DOI] [PubMed] [Google Scholar]
- John SP, Wang T, Steffen S, Longhi S, Schmaljohn CS, Jonsson CB (2007) Ebola virus VP30 is an RNA binding protein. J Virol 81: 8967–8976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchdoerfer RN, Moyer CL, Abelson DM, Saphire EO (2016) The Ebola virus VP30‐NP interaction is a regulator of viral RNA synthesis. PLoS Pathog 12: e1005937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruse T, Biedenkopf N, Hertz EPT, Dietzel E, Stalmann G, Lopez‐Mendez B, Davey NE, Nilsson J, Becker S (2018) The Ebola virus nucleoprotein recruits the host PP2A‐B56 phosphatase to activate transcriptional support activity of VP30. Mol Cell 69: 136–145 [DOI] [PubMed] [Google Scholar]
- Liang J, Sagum CA, Bedford MT, Sidhu SS, Sudol M, Han Z, Harty RN (2017) Chaperone‐mediated autophagy protein BAG3 negatively regulates Ebola and Marburg VP40‐mediated egress. PLoS Pathog 13: e1006132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licata JM, Simpson‐Holley M, Wright NT, Han Z, Paragas J, Harty RN (2003) Overlapping motifs (PTAP and PPEY) within the Ebola virus VP40 protein function independently as late budding domains: involvement of host proteins TSG101 and VPS‐4. J Virol 77: 1812–1819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lier C, Becker S, Biedenkopf N (2017) Dynamic phosphorylation of Ebola virus VP30 in NP‐induced inclusion bodies. Virology 512: 39–47 [DOI] [PubMed] [Google Scholar]
- Liu G, Nash PJ, Johnson B, Pietzsch C, Ilagan MXG, Bukreyev A, Basler CF, Bowlin TL, Moir DT, Leung DWet al (2017) A Sensitive in vitro high‐throughput screen to identify pan‐filoviral replication inhibitors targeting the VP35‐NP interface. ACS Infect Dis 3: 190–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez MJ, Biedenkopf N, Volchkova V, Hartlieb B, Alazard‐Dany N, Reynard O, Becker S, Volchkov V (2008) Role of Ebola virus VP30 in transcription reinitiation. J Virol 82: 12569–12573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez MJ, Volchkova VA, Raoul H, Alazard‐Dany N, Reynard O, Volchkov VE (2011) Role of VP30 phosphorylation in the Ebola virus replication cycle. J Infect Dis 204(Suppl 3): S934–S940 [DOI] [PubMed] [Google Scholar]
- Martin‐Serrano J, Eastman SW, Chung W, Bieniasz PD (2005) HECT ubiquitin ligases link viral and cellular PPXY motifs to the vacuolar protein‐sorting pathway. J Cell Biol 168: 89–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehedi M, Hoenen T, Robertson S, Ricklefs S, Dolan MA, Taylor T, Falzarano D, Ebihara H, Porcella SF, Feldmann H (2013) Ebola virus RNA editing depends on the primary editing site sequence and an upstream secondary structure. PLoS Pathog 9: e1003677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modrof J, Becker S, Muhlberger E (2003) Ebola virus transcription activator VP30 is a zinc‐binding protein. J Virol 77: 3334–3338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modrof J, Muhlberger E, Klenk HD, Becker S (2002) Phosphorylation of VP30 impairs ebola virus transcription. J Biol Chem 277: 33099–33104 [DOI] [PubMed] [Google Scholar]
- Muhlberger E, Lotfering B, Klenk HD, Becker S (1998) Three of the four nucleocapsid proteins of Marburg virus, NP, VP35, and L, are sufficient to mediate replication and transcription of Marburg virus‐specific monocistronic minigenomes. J Virol 72: 8756–8764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhlberger E, Weik M, Volchkov VE, Klenk HD, Becker S (1999) Comparison of the transcription and replication strategies of marburg virus and Ebola virus by using artificial replication systems. J Virol 73: 2333–2342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanbo A, Watanabe S, Halfmann P, Kawaoka Y (2013) The spatio‐temporal distribution dynamics of Ebola virus proteins and RNA in infected cells. Sci Rep 3: 1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nsio J, Kapetshi J, Makiala S, Raymond F, Tshapenda G, Boucher N, Corbeil J, Okitandjate A, Mbuyi G, Kiyele Met al (2019) 2017 Outbreak of Ebola virus disease in northern democratic republic of Congo. J Infect Dis 221: 701–706. [DOI] [PubMed] [Google Scholar]
- Ntwasa M (2016) Retinoblastoma binding protein 6, another p53 monitor. Trends Cancer 2: 635–637 [DOI] [PubMed] [Google Scholar]
- Peng YP, Zhu Y, Yin LD, Zhang JJ, Wei JS, Liu X, Liu XC, Gao WT, Jiang KR, Miao Y (2017) PEG10 overexpression induced by E2F–1 promotes cell proliferation, migration, and invasion in pancreatic cancer. J Exp Clin Cancer Res 36: 30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlereth J, Grunweller A, Biedenkopf N, Becker S, Hartmann RK (2016) RNA binding specificity of Ebola virus transcription factor VP30. RNA Biol 13: 783–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo JY, Kim DY, Kim SH, Kim HJ, Ryu HG, Lee J, Lee KH, Kim KT (2017) Heterogeneous nuclear ribonucleoprotein (hnRNP) L promotes DNA damage‐induced cell apoptosis by enhancing the translation of p53. Oncotarget 8: 51108–51122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamatsu Y, Krahling V, Kolesnikova L, Halwe S, Lier C, Baumeister S, Noda T, Biedenkopf N, Becker S (2020) Serine‐arginine protein kinase 1 regulates Ebola virus transcription. MBio 11: e02565‐19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tigabu B, Ramanathan P, Ivanov A, Lin X, Ilinykh PA, Parry CS, Freiberg AN, Nekhai S, Bukreyev A (2018) Phosphorylated VP30 of marburg virus is a repressor of transcription. J Virol 92: e00426‐18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmins J, Schoehn G, Ricard‐Blum S, Scianimanico S, Vernet T, Ruigrok RW, Weissenhorn W (2003) Ebola virus matrix protein VP40 interaction with human cellular factors Tsg101 and Nedd4. J Mol Biol 326: 493–502 [DOI] [PubMed] [Google Scholar]
- Weik M, Modrof J, Klenk HD, Becker S, Muhlberger E (2002) Ebola virus VP30‐mediated transcription is regulated by RNA secondary structure formation. J Virol 76: 8532–8539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie T, Pan S, Zheng H, Luo Z, Tembo KM, Jamal M, Yu Z, Yu Y, Xia J, Yin Qet al (2018) PEG10 as an oncogene: expression regulatory mechanisms and role in tumor progression. Cancer Cell Int 18: 112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Luthra P, Wu C, Batra J, Leung DW, Basler CF, Amarasinghe GK (2017) Ebola virus VP30 and nucleoprotein interactions modulate viral RNA synthesis. Nat Commun 8: 15576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda J, Nakao M, Kawaoka Y, Shida H (2003) Nedd4 regulates egress of Ebola virus‐like particles from host cells. J Virol 77: 9987–9992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Li Qi, He J, Zhong L, Shu F, Xing R, Lv D, Lei B, Wan Bo, Yang Yuet al (2017) HnRNP‐L promotes prostate cancer progression by enhancing cell cycling and inhibiting apoptosis. Oncotarget 8: 19342–19353 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Review Process File
Appendix
Expanded View Figures PDF
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 4
Source Data for Expanded View Figures
Data Availability Statement
This study includes no data deposited in external repositories.