

Abstract
Clinical and laboratory studies over recent decades have established branched evolution as a feature of cancer. However, while grounded in somatic selection, several lines of evidence suggest a Darwinian model alone is insufficient to fully explain cancer evolution. First, the role of macroevolutionary events in tumour initiation and progression contradicts Darwin's central thesis of gradualism. Whole‐genome doubling, chromosomal chromoplexy and chromothripsis represent examples of single catastrophic events which can drive tumour evolution. Second, neutral evolution can play a role in some tumours, indicating that selection is not always driving evolution. Third, increasing appreciation of the role of the ageing soma has led to recent generalised theories of age‐dependent carcinogenesis. Here, we review these concepts and others, which collectively argue for a model of cancer evolution which extends beyond Darwin. We also highlight clinical opportunities which can be grasped through targeting cancer vulnerabilities arising from non‐Darwinian patterns of evolution.
Keywords: cancer, cancer evolution, cancer therapy, tumour heterogeneity
Subject Categories: Cancer
Evolutionary concepts explaining cancer progression, the selective pressures acting on tumours, and their therapeutic implications are discussed in this comprehensive review.

Introduction
In his revolutionary work (Darwin, 1859), Darwin provided an evolutionary framework which enabled the understanding of somatic selection, diversification and extinction through the application of three key concepts: variation, heredity and selection. More than a 100 years later, the observation of heterogeneity in advanced malignancies led Peter Nowell to hypothesise that tumorigenesis is also an evolutionary process, whereby the same Darwinian principles could be applied to elucidate the mechanisms responsible for cancer formation and development (Nowell, 1976). Owing to Nowell’s seminal work, a Darwinian framework has been historically adopted to develop models of tumour evolution and therapy resistance (Michor et al, 2004; Gatenby & Vincent, 2008; Pepper et al, 2009; Greaves & Maley, 2012) (see Box 1). While gene‐centric Darwinian principles have been shown to explain tumour evolutionary trajectories in multiple instances (Gerlinger & Swanton, 2010; Purushotham & Sullivan, 2010; Gillies et al, 2012), recent studies have suggested additional evolutionary concepts beyond Darwin’s are required to reconcile the full spectrum of evolutionary behaviours in cancer. Specifically, increasing evidence now supports macroevolutionary jumps as a feature of cancer (Stephens et al, 2011; Baca et al, 2013; Sottoriva et al, 2015), which are likely interspaced by phases of microevolutionary gradualism. Furthermore, evidence of discordant inheritance patterns between cells (Decarvalho et al, 2018), and the role of neutral evolution (Ling et al, 2015; Williams et al, 2016; Wu et al, 2016), cell plasticity (Pogrebniak & Curtis, 2018; Mills et al, 2019; Boumahdi & de Sauvage, 2020) and the tumour microenvironment (Coussens & Werb, 2002; Lin & Karin, 2007; Laconi et al, 2020) in cancer demand consideration of a broader set of evolutionary models. Understanding how tumour evolution influences disease progression and how such processes are shaped by environmental factors and treatment remains critical.
Box 1. An historical perspective of tumour evolution and intratumour heterogeneity.
One hundred years have passed since Theodor Boveri set the foundations for much of our understanding of the origins of cancer. He not only hypothesised that cancers arise from normal cells as a consequence of genetic alterations, but also postulated that most tumours and their metastases originate from one cell, a concept that shaped today’s understanding of clonal expansion (Boveri, 2008). The application of evolutionary concepts to understand cancer formation and development can be attributed to Peter Nowell, who pioneered the hypothesis of tumour evolution. Nowell’s model stated that most cancers originate from a single neoplastic cell and evolve through a process of selection for somatic alterations, leading to the proliferation and survival of the most aggressive clones (Nowell, 1976). Despite this conceptual advance, Nowell’s view was largely overlooked, and tumour evolution was traditionally viewed as a linear succession of clonal cell divisions. Under the linear model, alterations accrue in progenitor cells in a stepwise fashion and endow cells with a strong selective advantage, hence enabling previous clones to be outcompeted (Fig 1). As a result, tumours would be composed of clonally identical cells resulting from continuous selective sweeps (Davis et al, 2017). Gloria Heppner challenged this view by demonstrating that tumours are comprised of genetically different subclones exhibiting fundamentally distinct behaviours (Dexter et al, 1978). By applying concepts of population genetics, she described tumours as “societies highly adapted for survival” and recognised that tumours “survive natural and artificial (therapeutic) selection through heterogeneity by producing new variants to "outflank" it.” (Heppner, 1984). These observations supported a model whereby tumours grow in a non‐linear, branched fashion, with multiple subclones derived from a common ancestor eventually diverging and expanding simultaneously with differing fitness (Greaves & Maley, 2012; Swanton, 2012) (Fig 1).
A consequence of branched tumour evolution is intratumour heterogeneity (ITH)—that is the coexistence of molecularly and phenotypically distinct subclones within a tumour. Tumour morphological heterogeneity has been long recognised by pathologists such as Johannes Muller and Rudolf Virchow in their pioneering studies in the 19th century as well as Theodor Boveri (Muller, 1973; Boveri, 2008; Parquet, 2014; Wright, 2014). Those initial discoveries were substantiated by several groups with orthogonal techniques, which provided evidence for intratumour diversity across multiple cancer types (Shapiro et al, 1981; Teixeira et al, 1996; Takahashi et al, 1998; Maley et al, 2006; Cottu et al, 2008). However, it was only with the recent technological innovations (see Box 2), particularly next‐generation sequencing (NGS), that the full breath of ITH could start to be fully resolved (Campbell et al, 2010; Navin et al, 2010; Gerlinger et al, 2012; Nik‐Zainal et al, 2012b; Walter et al, 2012; Patel et al, 2014; Sottoriva et al, 2015; Jamal‐Hanjani et al, 2017; Naxerova et al, 2017; Turajlic et al, 2018; Fittall & Van Loo, 2019; Rambow et al, 2019; Reiter et al, 2019; Gerstung et al, 2020; Machnik & Oleksiewicz, 2020; Marine et al, 2020).
With this review, we discuss our understanding of tumour evolution as a process grounded in Darwinian selection but argue that in light of recent data, we must now incorporate these concepts into a larger conceptual framework inclusive of alternative approaches to fully understand, predict and better respond to cancer evolution and to improve patient outcome.
Darwinian selection as the basis of subclonal diversity
Cancer has been historically viewed from a Darwinian gene‐centric perspective (Greaves & Maley, 2012). Indeed, tumours are frequently typified as a large population of genetically diverse cells giving rise to distinct subpopulations. Subclones will compete with one another for a limited set of nutrients and metabolites and face ever‐shifting selective pressures driven by both endogenous (i.e. microenvironmental pressures and geographical barriers) and exogenous (i.e. therapy) factors (Merlo et al, 2006). The outcome of this competition is the survival of clones adapted to grow under very specific conditions, as Darwinian selection is highly contextual and blind to the future. Many clones that were dominant at one point in time may reach evolutionary dead ends and disappear, while only a minority may be able to persist. Quoting Darwin “One general law, leading to the advancement of all organic beings, namely, multiply, vary, let the strongest live and the weakest die” (Darwin, 1859).
In the one to two decades, direct evidence to support Darwinian evolution in cancer has been reported, principally from studies using next‐generation sequencing (NGS) to perform detailed characterisation of genetic tumour evolution (see Box 2). One of the earliest studies was from Shah et al (2009), where matched primary and metastatic tissue from a lobular breast tumour were sequenced revealing extensive mutational heterogeneity with ∼80% of the non‐synonymous mutations in the metastasis absent from the primary site (Shah et al, 2009). The finding of pervasive heterogeneity in breast cancer has additionally been reported by the extensive work of Kornelia Polyak, which demonstrated that breast tumours were composed of a variety of cell types with distinct morphologies and behaviours, with the source of heterogeneity arising in part from clonal evolution (Campbell & Polyak, 2007). Early evidence of abundant, genetically diverse subpopulations of cells was also revealed through single‐cell sequencing (see Box 2) studies in breast cancer by Nick Navin and others (Navin et al, 2011). Regarding haematological malignancies, Anderson et al. were among the first to show branching evolutionary trajectories in acute lymphoblastic leukaemia (Anderson et al, 2011). Our own work from Gerlinger et al (2012) profiled 30 tumour samples from four renal cell carcinoma patients and revealed that 63 to 69% of all somatic mutations were not detectable across every tumour region (Gerlinger et al, 2012). These observations demonstrated the extent and relevance of branched evolution. Furthermore, evidence of parallel evolution was demonstrated for multiple tumour suppressor genes (SETD2, PTEN, KDM5C), suggesting selective pressures drive inactivation of the same gene multiple times within a single tumour. This report was followed by work from Nik‐Zainal et al (2012b), who studied the life history of 21 breast tumours, identifying extensive genetic variation within individual breast tumours (Nik‐Zainal et al, 2012b). This study also showed further evidence of selection, with each tumour containing a dominant subclonal lineage, representing more than 50% of tumour cells. Extending into further detail on the metastatic process, work from Gundem et al (2015) utilised autopsy sampling in 10 prostate cancer patients to identify metastasis to metastasis seeding as a common event (Gundem et al, 2015). This study emphasised not only the extent of subclonal diversification, but also the complexity of seeding routes to and between metastatic sites. However, these early studies were limited by small sample sizes. Furthermore, the diverse range of cancer types studied meant the nature of specific evolutionary patterns, as generalisable across all tumour types or histology specific, remained undetermined. Despite the limitations, these early NGS studies gave the first direct evidence of extensive genetic subclonal diversification, hence supporting a model of cancer growth as a branched evolutionary process (Fig 1). Furthermore, the demonstration of branched evolution as a feature of solid tumour growth spurred a change in thinking across the community to recognise the importance of Darwinian selection in cancer. Branched evolution has also been shown to be applicable to relatively homogeneous primary tumours and/or metastases, whereby particularly aggressive subclones that may achieve a clonal sweep and present clinically with a homogeneous profile (Reiter et al, 2018) (Fig 1). Clear examples of this are described in pancreatic cancer, where virtually all major driver gene alterations (KRAS, CDKN2A, TP53, SMAD4) are present in the most recent common ancestor and limited evidence of mutational heterogeneity is observed across metastases (Makohon‐Moore et al, 2017). Similar examples are observed in some aggressive renal cell carcinomas, where ∼10–20% of tumours exhibit multiple clonal driver mutations, limited heterogeneity and poor clinical outcome (Turajlic et al, 2018). It is proposed the variation in heterogeneity between tumours may reflect differences in the inherent biology of a given tumour and impact upon the process of metastatic dissemination and clinical outcome (Iacobuzio‐Donahue et al, 2020).
Box 2. Sequencing technologies to assess tumour evolution.
The advent of next‐generation sequencing (NGS) has revolutionised biology. Its high‐throughput, scalability, speed, and cost efficiency enabled researchers to study the genomic and transcriptomic profiles of many human diseases, including cancer, with unprecedented resolution. Studies of cancer evolutionary dynamics and ITH have particularly benefited from these technological advances.
Bulk sequencing
To date, most studies of tumour evolutionary dynamics have relied on the sequencing of bulk tumour samples. While this methodology can only provide an indirect measurement of the subclonal composition of a tumour biopsy, tumour phylogenetic architecture can still be inferred through computational approaches. By sequencing samples at sufficiently high coverage depth, variant allele frequencies (VAFs) can be quantified. VAFs can then be further corrected for tumour purity and local copy number state to derive cancer cell fraction (CCF) values, which intuitively measure what proportion of cancer cells bear a given mutation. CCFs can then be clustered using a Dirichlet process to identify clonal and subclonal populations—based on the assumption that mutations with similar CCFs fit together to form genetically distinct populations. Once the subclonal architecture of a given tumour is defined (i.e. the complete set of detected clones/subclones), phylogenetic techniques can be used to infer the most likely parent to child ordering relationships. This enables phylogenetic tree construction, with each tumour (sub)clone being placed on its relevant branch. Phylogenetic tree construction must adhere to basic principles, such as the pigeonhole principle, which stipulates that if there are m containers (pigeonholes) and n items (pigeons) to go within the containers and if n > m, then there must be a container with more than one item. This principle ensures that the CCF sum of child subclones cannot exceed that of its parental ancestor. Computationally, these methods are implemented within a wide number of tools, and consensus efforts are seeking to drive greater standardisation in methodologies (Schwartz & Schäffer, 2017). It should be noted that key information needed for reconstructing tumour evolution may however be lost in the process, due to confounding effects derived from dealing with mixed cell populations. For instance, alterations only present in low frequency small subclones will likely be confused as noise, posing a significant limitation for tree reconstitution (Cibulskis et al, 2013). Additionally, deconvolution of bulk sequencing data is needed to infer evolutionary dynamics, an approach that may limit the identification of branching events (Malikic et al, 2019).
Single‐cell approaches
The recent development of single‐cell approaches (SCA) has allowed the analysis of the genomes, transcriptomes, epigenomes, proteomes and even metabolomes of individual cancer cells, as well as the TME, with unprecedented resolution (Navin et al, 2011; Hiley et al, 2014; Wang et al, 2014; Navin, 2015). The most widely used SCA has been single‐cell RNA sequencing (scRNA‐seq), which has recently emerged as a valuable tool to study tumour evolutionary dynamics (Nam et al, 2021). Owing to its high resolution, scRNA‐seq permits detection of genes expressed even in small subclones which would be missed by bulk RNA‐seq, allowing, for instance, the identification of minor treatment‐resistant cell populations contributing to therapy failure (Rambow et al, 2018). Additionally, copy number variation and loss of heterozygosity in individual cells can sometimes be inferred through scRNA‐seq, although in most cases the resolution is not sufficient for novel SCNA discovery (Fan et al, 2018). Moreover, by obtaining transcriptomic data from hundreds of individual cells in a range of different evolutionary stages, cells can be ordered in pseudotime and within evolutionary trajectories. This allows the inference of tumour evolutionary history and dynamics, albeit without providing information regarding the direction of such trajectories. To overcome this limitation, RNA velocity analyses can be used. By measuring the relative ratio between intronic and exonic reads, the rate of change in transcript abundance can be inferred, thus providing an estimate of the future transcriptional state of a cell alongside with a better understanding of the cellular transcriptional dynamics (La Manno et al, 2018). Another SCA approach that has emerged in the last decade is single‐cell DNA sequencing (scDNA‐seq), which can provide genomic profiles of individual cells, thereby allowing the inference of genetic phylogeny without any form of deconvolution. Despite its promises, scDNA‐seq still has major limitations. First, the coverage obtained with such approaches tends to be low because of the limited amount of DNA in a single cell, which typically contains a few picograms of genomic DNA, whereas NGS requires nanogram amounts of starting DNA for library preparation. Therefore, a critical step for single‐cell sequencing is whole‐genome amplification to generate sufficient DNA for library construction, a process which is rather error‐prone (Chen et al, 2018). Other frequent issues are an increase rate of false negatives due to allelic dropout and additional noise stemming from accidentally sequencing doublets (Pugh et al, 2008; Lasken, 2013; Navin, 2014; Van Loo & Voet, 2014; Simonsen et al, 2018; Mallory et al, 2020). Interestingly, the weaknesses of scDNA‐seq can be complemented by bulk DNA sequencing and vice versa and the use of both data types for inferring phylogeny of tumours has been shown to provide more accurate results and increased resolution in capturing ITH complexity and evolutionary dynamics (Salehi et al, 2017; Malikic et al, 2019).
Figure 1. Models of tumour evolution.
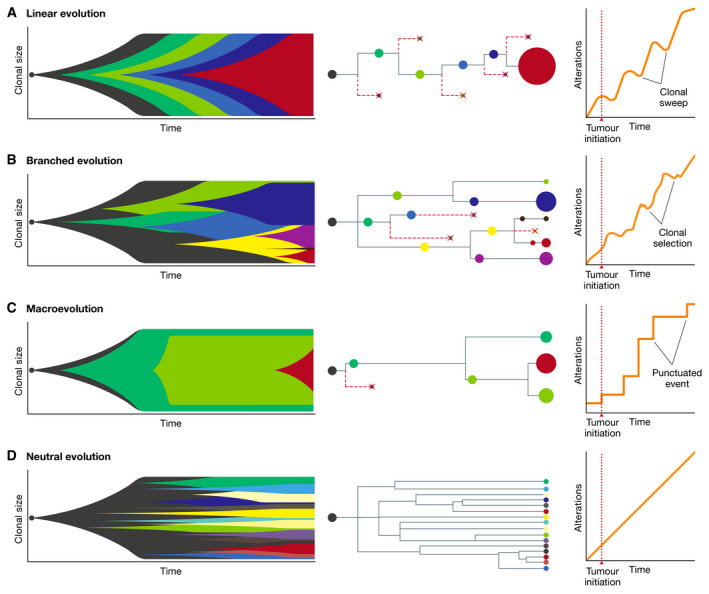
Models of linear evolution (A), branched evolution (B), macroevolution (C) and neutral evolution (D) described by Muller plots representing dynamic changes in clonal size over time (left), clonal lineages and phylogenetic trees (centre) and changes in the number of alterations over time (right). Colours indicate different clones.
However, models of accumulating genetic changes subject to selective pressure are not fully sufficient to explain the full spectrum of cancer evolutionary histories, and increasing evidence points to the existence of non‐Darwinian mechanisms as important features of tumour evolution.
Macroevolution and punctuated events
Neo‐Darwinian models of tumour evolution generally assume that mutations are acquired sequentially in a gradual fashion over time. However, several lines of evidence suggest that in some cases, a large number of genomic aberrations may occur in short bursts of time in cancer cells (Stephens et al, 2011; Baca et al, 2013), as a consequence of chromosomal instability (CIN) (Bakhoum & Landau, 2017), breakage‐fusion‐bridge (BFB) cycles (Gisselsson et al, 2000), chromoplexy (Baca et al, 2013), chromothripsis (Stephens et al, 2011; Notta et al, 2016) and other similar catastrophic events (Fig 2). According to this model, tumour cells alternate long phases of relative mutational equilibrium with short periods of intense evolution, where tumour cells can acquire multiple strong driver events (Cross et al, 2016) (Fig 1). Such examples of saltatory evolution indicate that, at least in cancer, nature can under certain circumstances make jumps, contrary to what Darwin predicted. These observations are reminiscent of the “hopeful monsters” theorised by Richard Goldschmidt, i.e. organisms with a profound mutant genotype compared to their parents that hold the potential to establish a novel evolutionary lineage (Goldschmidt, 1941). Hence, through short and intense bursts of genomic change, cancer cells could potentially obtain greater fitness than would be possible through a gradual accumulation of alterations, owing to the simultaneous acquisition of multiple driver alterations (Korbel & Campbell, 2013). However, the phenotypic impact of such hereditary changes acquired through saltatory evolution, if any at all, will often be deleterious and only in rare instances will it result in an increase in cellular fitness and in the generation of viable “hopeful monsters” (Goldschmidt, 1941; Gerlinger et al, 2014b).
Figure 2. Scales of tumour evolution.
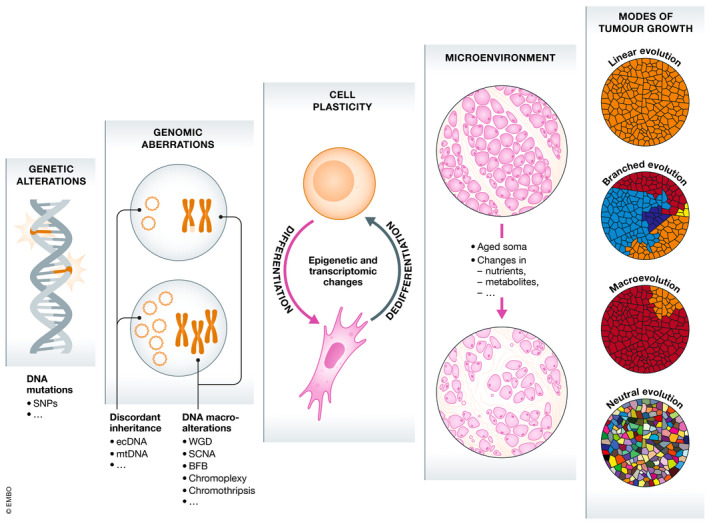
Schematic illustration of the different determinants of tumour evolution, which influence evolutionary trajectories through highly interdependent mechanisms, from a microscopic (left) to a macroscopic (right) scale.
Regarding progression from primary tumour to metastasis and death, increasing evidence implicates macroevolutionary changes as important drivers of progression. For example, in the prospective TRACERx (TRAcking Cancer Evolution through therapy (Rx)) study (Jamal‐Hanjani et al, 2017), elevated copy number heterogeneity was identified as being most strongly associated with recurrence/death risk in non‐small cell lung cancer (NSCLC), whereas single nucleotide variant heterogeneity was non‐significant. Similarly, acquired aneuploidy was frequently detected in recurrent gliomas (Barthel et al, 2019), and genetic diversity alongside chromosomal complexity (characterised by high weighted genome integrity index (Endesfelder et al, 2014)) emerged as significant determinant of poor patient outcome in clear cell renal cell carcinoma (ccRCC) (Turajlic et al, 2018). In ccRCC, losses of chromosomes 9p21.3 (CDKN2A) and 14q31.1 (HIF1A) were specifically associated with reduced survival (Turajlic et al, 2018). The prognostic importance of macroevolutionary changes in the form of somatic copy number alterations (SCNAs), above point mutations, is now becoming increasingly recognised as a pan‐cancer phenomenon (Smith & Sheltzer, 2018). A major outstanding challenge however is minimal mapping of recurrent SCNA cytobands, to find specific causative genes. And even when clear driver genes emerge, as is likely the case for CDKN2A at 9p21 (Smith & Sheltzer, 2018), detailed functional work to delineate the precise mechanisms of disease progression remains to be completed. Additional evidence of punctuated evolution has been reported in prostate cancer, where intense genomic changes occurring over relatively few cataclysmic events, a process termed chromoplexy, were observed (Baca et al, 2013). Similarly, ER/PR/HER2 negative breast cancers were found to undergo short periods of evolution early in tumour development and to remain relatively stable at later stages (Gao et al, 2016). Tumour macroevolution was also found to be driven by chromothripsis, whereby a single catastrophic mutational event is thought to be responsible for the generation of highly complex genomic rearrangements involving dozens of breakpoints (Stephens et al, 2011). This process has been observed in several tumour types, such as bone cancers (Stephens et al, 2011), colon cancers (Kloosterman et al, 2011), neuroblastoma (Molenaar et al, 2012), glioblastoma (Malhotra et al, 2013) and pancreatic cancer (Notta et al, 2016). An extreme case of punctuated evolution, caused by the aforementioned mechanisms, is the “big bang” model, whereby a single or a limited number of catastrophic events and/or genomic crises occurring early in tumourigenesis are responsible for the generation of numerous intermixed subclones that will not substantially evolve over tumour progression due to weak selective pressure (Sottoriva et al, 2015). Such evolutionary dynamics were observed in several cancers, including colon cancers (Sottoriva et al, 2015) and hepatocellular carcinoma (Ling et al, 2015), as well as in pan‐cancer studies (Stephens et al, 2011).
Cancer evolution is conceptually similar to evolution in asexually reproducing organisms, whereby the impact of deleterious alterations in terms of fitness cannot be mitigated through sexual reproduction. A mechanism to alleviate the irreversible detrimental accumulation of alterations (e.g. extensive LOH events) may be whole genome doubling (WGD), a prevalent event in cancer (Storchova & Pellman, 2004; Zack et al, 2013; Dewhurst et al, 2014; Bielski et al, 2018) involving the doubling of the entire genome. The presence of additional, doubled wild‐type alleles as a consequence of WGD could allow the cell to better tolerate LOH events involving essential genes (López et al, 2020). The early occurrence of this event therefore creates a more tolerant and permissive environment which can fuel rapid genomic diversification and CIN, while at the same time may facilitate the sub functionalisation of duplicated genes (Storchova & Pellman, 2004; Huminiecki & Conant, 2012; Dewhurst et al, 2014). Consequently, WGD is often associated with high rates of chromosomal aberrations (Zack et al, 2013; Dewhurst et al, 2014) and with poor prognosis and intrinsic drug resistance (McGranahan et al, 2012; Bielski et al, 2018).
Importantly, certain classes of macroevolutionary events have been shown to be able to trigger other macroevolutionary events. For instance, chromothripsis is prone to arise in genomically unstable cells, such as those harbouring damaged telomeres or with hyperploidy (Mardin et al, 2015). Similarly, BFB cycles have been shown to generate increasing amounts of chromothripsis by providing free DNA ends that can engage in genomic rearrangement and by compromising centromere function (Umbreit et al, 2020). DNA replication stress has been shown to drive genomic instability by promoting both structural and numerical chromosomal aberrations (Burrell et al, 2013) and by triggering single nucleotide‐level mutagenesis mediated via APOBEC3B induction (Kanu et al, 2016), which in turn leads to incomplete replication of genomic DNA (Venkatesan et al, 2021). Relatedly, regional mutational clusters (kataegis) (Stephens et al, 2011) and lesion segregation (Aitken et al, 2020) have also been shown in some instances to be associated with chromothripsis and other rearrangement architectures (Nik‐Zainal et al, 2012a). The combination of such events rapidly accelerates tumour evolution, causing more non‐gradualism than any individual class by itself would.
Discordant inheritance between cells
Recent studies have demonstrated that oncogene amplification within extrachromosomal DNA (ecDNA) is a frequent event in cancer (Verhaak et al, 2019). The existence of chromosomal material in cancer cells outside the autosomal genome has been long recognised, with the first reports of oncogenic ecDNAs going back as far as the 1980s, where sequences resembling MYCN were found in neuroblastoma cell lines (Kohl et al, 1983). However, it was only in the last few years that the frequency and functional relevance of ecDNAs in cancer started to be fully appreciated, thanks to the development of novel techniques such as long‐read whole‐genome sequencing and circular DNA library enrichment (Verhaak et al, 2019). ecDNAs are circular DNA structures located outside of chromosomes of variable size (ranging from 168 kb to 5 Mb, with a median size of 1.26 Mb) (Wu et al, 2019), which can contain one or more oncogenes (Bailey et al, 2020). ecDNAs provide cancer cells with a mechanism to achieve and maintain high copy oncogene amplification and diversity and to drive potent oncogene expression due to highly open chromatin, which allows for increased expression of genes encoded on circular DNA relative to their chromosomal counterparts (Wu et al, 2019; Kim et al, 2020). Importantly, the existence of ecDNAs defies Mendelian genetics. ecDNAs are replicated during S phase, but, owing to the lack of centromeres, they are subject to unequal segregation and are therefore randomly inherited by daughter cells during mitosis. As such, ecDNA‐based oncogene amplification can accelerate tumour evolution through non‐Mendelian mechanisms of inheritance and drive clonal expansion in genomically stable backgrounds (Decarvalho et al, 2018) (Fig 2). Importantly, the random distribution of ecDNAs fosters cell‐to‐cell variability in terms of both copy number and transcriptional levels of oncogenes, enabling tumours to acquire ITH more efficiently than through chromosomal amplifications (Turner et al, 2017; Verhaak et al, 2019). Several pan‐cancer studies revealed a high frequency of ecDNA (albeit at highly variable numbers) across tumour types, particularly in lung, breast and prostate cancer as well as glioblastoma and neuroblastoma (Fan et al, 2011; Turner et al, 2017; Deshpande et al, 2019; Bailey et al, 2020; Koche et al, 2020). Key oncogenes such as MYC, MYCN, EGFR, PDGFRA, MET, HER2, DHFR, CDK4 and MDM2 have been frequently observed on ecDNAs, suggesting that ecDNA‐mediated oncogene amplification is an important driver of tumour development and progression (Decarvalho et al, 2018; Gu et al, 2020). ecDNA amplification in cancer has been shown to increase cell proliferation, invasion and metastatisation and to negatively correlate with overall survival (Bailey et al, 2020). Consequently, ecDNA elimination has been shown to decrease oncogene amplification and to negatively affect cancer cell survival (Shimizu et al, 1998; Nathanson et al, 2014; Clarke et al, 2020; Oobatake & Shimizu, 2020). Furthermore, ecDNAs have been shown to enable tumour adaptation in response to changes in microenvironmental conditions and to selective pressure from therapy (Turner et al, 2017; Decarvalho et al, 2018; Kim et al, 2020), though in some instances ecDNA expression represents a cancer‐specific vulnerability (Nathanson et al, 2014).
Neutral evolution
Neutral evolutionary models of cancer (Ling et al, 2015; Williams et al, 2016) are based on Motoo Kimura's work on population genetics which postulated that the vast majority of molecular alterations are not caused by selection but rather by random fixation of selectively neutral mutations through genetic drift (Kimura, 1983). According to the neutral tumour evolution model, cancer‐driving alterations are selected for and accumulate in a clonal fashion prior to tumour initiation, as a consequence of ageing and carcinogenic insults. Those alterations will be sufficient for tumour formation and development, with little to no contribution of alterations occurring over the course of cancer progression (Fig 1). Therefore, the genetic ITH observed in tumours may often be entirely the result of the random fixation of (nearly) neutral alterations in the population through genetic drift and as such have no functional role in promoting tumour growth and evolution. In one study, multi‐region sequencing of > 300 regions of a patient with hepatocellular carcinoma indicated that there was no particular selection for any clone within the tumour (Ling et al, 2015). In another study, subclonal allele frequencies from the TCGA cohorts were used to conclude that up to one‐third of all tumours do not show indications of subclonal selection but rather evolve in a neutral fashion (Williams et al, 2016). However, several lines of evidence suggest that these results may be an overestimation due to the low resolution of the data used for the study and may suffer from bias in the modelling, since variant abundance distributions may often not provide enough information to exclude selection (Tarabichi et al, 2018; Bozic et al, 2019).
Kimura's neutral theory essentially states that most alterations will be neutral, especially at low population sizes and with weak purifying selection. Most variants will not have any fitness effect, and the rare ones which will have an impact on fitness will predominantly be deleterious, as predicted by mathematical modelling (Cannataro et al, 2017). However, Kimura never excluded the importance of occasional strong positive selection in evolution, especially as a consequence of strong purifying selection (Kimura, 1983). Hence, when applying Kimura's neutral theory to cancer, selective pressures (i.e. microenvironmental changes, metastatisation, therapeutic intervention) must be taken into consideration. Therefore, while a treatment‐naïve tumour could at times evolve in a neutral fashion over the course of its progression, the emergence of strong selective forces, such as therapeutic pressure, may still drive the selection and expansion of previously neutral alterations (Almendro et al, 2014; Williams et al, 2016).
It is also worth noting that the presence of non‐cell‐autonomous drivers in small subpopulations may also give the false impression of neutral evolution (Marusyk et al, 2014). Indeed, Kornelia Polyak’s group demonstrated that tumours can at times be driven by a subclone that does not have higher fitness, but instead stimulates the growth of all tumour cells through non‐cell‐autonomous mechanisms (Marusyk et al, 2014). In such a scenario, it may be misleading to assume that the absence of a predominant clone in a tumour is evidence of neutral evolution, as non‐cell‐autonomous driving of tumour growth can maintain clonal diversity over clinically relevant time frames while simultaneously fuelling tumour growth and evolution.
Non‐genetic determinants of evolution
There is increasing appreciation that tumour evolution is not only driven by mutations and genetic alterations but also from non‐genetic—often non‐heritable—determinants, such as cell plasticity and the tumour microenvironment (TME) (Caiado et al, 2016; Ramón y Cajal et al, 2020) (Fig 2).
Cell plasticity
The notion that cancer cells can dynamically switch from one state to another in response to environmental stresses and therapeutic pressure without genomic alterations is gaining greater recognition (Pogrebniak & Curtis, 2018; Mills et al, 2019; Boumahdi & de Sauvage, 2020) (discussed in other reviews of this series by Milan et al, 2021). This phenomenon, termed cell plasticity, is characterised by a fundamental change in the biological properties of the cell occurring as a consequence of dynamic and reversible epigenetic and transcriptional changes (in sharp contrast to genetic alterations, which have binary and largely irreversible effects) (Calabrese et al, 2020). One of the main advantages of cell plasticity is the ability to swiftly react to dynamic changes in the tumour and in its microenvironment and to engage finely tuned and graded adaptive responses to stressors (i.e. inflammation and therapy) (Rambow et al, 2019). A classic example of cell plasticity is the epithelial–mesenchymal transition (EMT) (Nieto et al, 2016) (extensively covered by Brabletz et al (2021) in another review of this series). Cell plasticity allows for the rapid emergence of different cell states from the same genome, giving rise to a plethora of distinct phenotypes, a process that has shown to be promoted upon therapeutic intervention (Kemper et al, 2014; Gunnarsson et al, 2020; Marine et al, 2020). Cell plasticity has been extensively demonstrated as an adaptive mechanism to escape therapeutic pressure. One of the first evidence of such a mechanism was the identification of drug‐tolerant persisters (DTPs) cells emerging from drug‐sensitive NSCLC cell lines upon exposure to EGFR tyrosine kinase inhibitor (Sharma et al, 2010). This drug‐tolerant phenotype was shown to be transiently acquired and lost by individual cells, thereby demonstrating that cancer cells can dynamically and reversibly develop drug resistance through non‐genetic phenotype switch. Similarly, multiple phenotypically distinct—yet interdependent—drug‐tolerant populations were recently shown to emerge in melanoma PDX models in response to MAPKi treatment through adaptive non‐genetic mechanisms (Rambow et al, 2018). Importantly, although resistant phenotypes are often non‐heritable, they can protect the tumour cell population from eradication thereby increasing overall tumour fitness through non‐Darwinian mechanisms (Kemper et al, 2014; Gunnarsson et al, 2020; Marine et al, 2020). Cell plasticity can therefore enable the emergence of more permanent resistance mechanisms, as was shown in melanoma, whereby therapeutic pressure initially caused the emergence of a transient drug‐tolerant transcriptional state which was later converted into a stably resistant phenotype (Shaffer et al, 2017).
Tumour microenvironment
The observation that healthy tissues often display pervasive somatic mutations, even in cancer driver genes, suggests that genetic mechanisms alone may be not be sufficient to drive malignant transformation (Martincorena et al, 2015, 2018; Teixeira et al, 2019; Yizhak et al, 2019; Yoshida et al, 2020). It has been long noted that the incidence of most cancers uniformly increases after 50 years of age (Siegel et al, 2018b), regardless of the many differences in terms of their cellular origin, of the number of alterations required for their initiation and of the external risk factors (such as smoking, alcohol and UV light) (Rozhok & De Gregori, 2019; Laconi et al, 2020). To explain this discrepancy, James DeGregori’s group demonstrated that cancer incidence rapidly increases with age not only as a consequence of the accumulation of alterations but also (if not predominantly) because of the age‐dependent physiological decline of the soma, combined with a general weakening of the immune system which is less efficient at eliminating altered (precancerous) cells (DeGregori et al, 2020; Laconi et al, 2020). While a young healthy tissue environment may continuously and effectively prevent tumorigenesis, an aged one would provide fertile ground for cancer formation instead. Permissive environments have been shown to result in population expansion in nature, whereby relaxed constraints are known to often spur the establishment, expansion and persistence of particularly versatile species. For instance, a permissive environment was shown to have led to the expansion of two species of parasites in the Arctic as a consequence of climate change (Kutz et al, 2013; Kafle et al, 2020).
Intriguingly, the effect of mutagenic agents was also found to be influenced by the tissue type, as the mutational burden varied greatly based on tissue, suggesting tissue‐specific differences in toxicokinetic, DNA repair activity or of the TME (Riva et al, 2020). Similar evidence comes from the observation that genetic diseases driven by highly penetrant mutations predisposing to cancer invariably lead to very specific cancer types, although the same mutation is shared between all the cells of the organism. This is the case of the NF1 mutation in neurofibromatosis, which often leads to tumours of the nervous system (Gutmann et al, 2017), of the TP53 mutations in the Li‐Fraumeni syndrome, which predispose to breast cancer, adrenocortical carcinomas, central nervous system tumours, osteosarcomas and soft‐tissue sarcomas (Malkin, 2011), and of the BRCA1/2 mutations, which considerably increase the risk of breast cancer (Tung & Garber, 2018). Hence, even highly penetrant cancer‐driving alterations may not be sufficient to drive cancer formation per se. Instead, these examples suggest that oncogenic mutations may often confer a cell type‐specific advantage in terms of fitness rather than a generic selective advantage on all cells and that very specific environmental conditions are needed for tumour initiation. The observation that certain cancer types or cancer subtypes metastasise to specific locations, a fact that cannot be solely be explained by circulation patterns, further reinforces the idea that tumours require particular environments for their growth and survival, even in later stages of cancer evolution (Nguyen et al, 2009). These are only but a few examples of how the TME can affect cancer initiation and evolution, as this topic is amply discussed in other reviews of this series (see Kepp et al, 2021; Parker et al, 2021).
Clinical opportunities: leveraging evolutionary principles
Advances over the last decade have led to a deeper understanding of the molecular pathogenesis of cancer and of its evolution. Small‐molecule inhibitors, such as BRAF and EGFR inhibitors (Bedard et al, 2020), and immune checkpoint inhibitors (CPI), such as anti‐PD‐1/L1 and anti‐CTLA‐4 (Waldman et al, 2020), have revolutionised cancer therapy (Chen et al, 2019). However, despite the significant benefits brought about by these approaches, many advanced tumours—even those with marked initial responses—rapidly develop resistance (Gatenby & Brown, 2020). The dynamic nature of tumour evolution, especially under the strong selective pressure exerted by anti‐cancer therapies, makes longitudinal monitoring necessary (see Box 3) and warrants flexible therapeutic strategies based on the evolutionary changes within a patient's tumour.
Box 3. Sampling techniques to study tumour evolution.
Multiregional (longitudinal) biopsies
Over the last few years, increasing efforts have been put towards sequencing multiple geographically distinct areas of the same tumour and of its metastases and/or samples taken from the same patient at different time points (McGranahan & Swanton, 2015; Chkhaidze et al, 2019). These analyses have proven to be critical for the reconstruction of tumour evolutionary histories, revealing extraordinarily complex and dynamic phylogenetic architectures of cancer subclones and their variegated genetic, epigenetic and transcriptomics profiles. However, multiregional and longitudinal sampling through invasive biopsies is often deemed clinically unfeasible, since the performance status of the patient and the location of the tumour frequently prevent such an approach (Turajlic et al, 2018; Chkhaidze et al, 2019; Litchfield et al, 2020).
Rapid research autopsy
Rapid research autopsy is gaining interest as a powerful technique to assess ITH and tumour evolution through improved tissue sampling, with the potential to overcome the limitations of sampling in living patients in several ways (Duregon et al, 2019). First, they allow the collection of large quantities of tumour tissue, as well as the TME, from multiple body sites from individual patients, supporting a more complete representation of metastatic lesions. Second, enough material can be collected to be used for multiple orthogonal studies (Avigdor et al, 2017; Pisapia et al, 2017; Duregon et al, 2019). Third, these samples can be used for the development of patient‐derived xenografts, organoids and primary cell lines, thus allowing mechanistical validation of the data generated through orthogonal analyses (Grasso et al, 2012; Xie et al, 2015; Krook et al, 2019b). Last, the samples obtained are typically composed of therapy‐resistant cells and can inform about the evolution of drug resistance (Dagogo‐Jack & Shaw, 2018). Such an approach offers an ethical alternative to multiregional biopsy sampling of living patients and can be effectively used to overcome its limitations. Albeit limited in number, the studies performed through rapid research autopsies have substantially increased our understanding of ITH and tumour evolution, and how both processes are affected by therapeutic selective pressure (Campbell et al, 2010; Haffner et al, 2015; Hoadley et al, 2016; Brown et al, 2017; Siegel et al, 2018a; De Mattos‐Arruda et al, 2019). For instance, rapid research autopsies have allowed tracing of the origins of metastases to individual subclones, delineating a precise time course from an initial founder cell to the development of metastases (Yachida et al, 2010; Gerlinger et al, 2012; Aryee et al, 2013; Gerlinger et al, 2014a). It is worth noting that, apart from some notable exceptions (Embuscado et al, 2005; Yachida et al, 2010), the majority of these studies have been performed on therapy‐resistant lesions and might therefore not fully recapitulate the evolutionary processes occurring in drug‐naive cells (Dagogo‐Jack & Shaw, 2018; Duregon et al, 2019; Krook et al, 2019b). Moreover, the execution of such studies requires interdisciplinary collaboration across different medical and scientific disciplines, which can be difficult to be put in place, and necessitate the consent and close involvement of cancer patients and their families.
Blood‐based tests
High frequency monitoring of tumour evolution, especially over the course of therapeutic intervention, is required for the precise understanding of the mechanisms leading to drug resistance. Shedding light on this dynamic process could provide direct evidence of clonal selection and readily warn about the emergence of drug‐resistant clones. Evidently, these types of information could not be obtained through longitudinal multiregional biopsies, both for practical and ethical reasons, nor through rapid research autopsies. In recent years increasing attention has therefore been directed at detecting cancer‐derived components, such as circulating tumour cells (CTCs), as well as tumour‐derived components such as circulating tumour DNA (ctDNA), cell‐free RNAs, extracellular vesicles (EVs), proteins and metabolites (Ramalingam & Jeffrey, 2018; Rossi & Ignatiadis, 2019; Ignatiadis et al, 2021), in patients' bodily fluids. Provided that tumour cells and their components detected in the circulation represent their clonal frequency, blood‐based tests could allow the detection of changes occurring within a patient’s tumour in real time. As such, blood‐based tests have the potential to revolutionise personalised medicine and guide clinical decision‐making through constant assessment of ITH and tumour evolutionary dynamics via non‐invasive and cost‐effective monitoring of tumour growth, minimal residual disease (MRD), metastatisation and response to therapy. Growing evidence supports the feasibility of blood‐based tests to monitor tumour evolution across several tumour types (Misale et al, 2012; Dawson et al, 2013; Aceto et al, 2014; Scher et al, 2015; Siravegna et al, 2015; Abbosh et al, 2017, 2018; Anagnostou et al, 2019; Vidal et al, 2020). Additionally, blood‐based tests were shown to have prognostic value at the time of primary diagnosis and surgery in various solid tumours including breast, bladder, colorectal, head and neck and testicular cancer (Pantel & Alix‐Panabières, 2019; Chen et al, 2020) as well as melanoma (Lee et al, 2019) and NSCLC (Abbosh et al, 2017).
Limiting tumour adaptation
Increasing evidence indicates that both Darwinian and non‐Darwinian adaptive mechanisms can be activated shortly after therapy initiation to cope with the stress caused by the treatment (Hata et al, 2016; Rambow et al, 2018; Russo et al, 2019; Vendramin et al, 2021). The employment of such responses thereby fosters the emergence of a reservoir of drug‐tolerant cellular populations from which therapy‐resistant clones can evolve from. Therefore, therapeutic strategies targeting the mechanisms responsible for drug tolerance may prevent, or at least delay, the evolution of acquired resistance (Fig 3). This strategy was tested in a recent study where the selective pressure exerted by EGFR inhibitors in lung cancer was shown to induce APOBEC3B overexpression, leading to increased cancer cell mutability, fostering tumour adaptation to treatment (preprint: Mayekar et al, 2020). Similar findings were reported in colon cancer, whereby drug‐tolerant cells were shown to temporarily down‐regulate mismatch repair (MMR) and homologous recombination DNA repair genes in favour of error‐prone polymerases as an adaptive response to overcome BRAF and EGFR inhibition (Russo et al, 2019). These findings suggest that, similar to bacteria (Luria & Delbrück, 1943), cancer cells can temporarily enhance their mutability and genetic instability as an adaptive response to overcome therapeutic pressure. Inhibition of these adaptive mechanisms could therefore abolish drug‐driven adaptive mutagenesis and potentially prevent the emergence of drug‐resistant populations.
Figure 3. Evolutionary‐based treatment strategies.
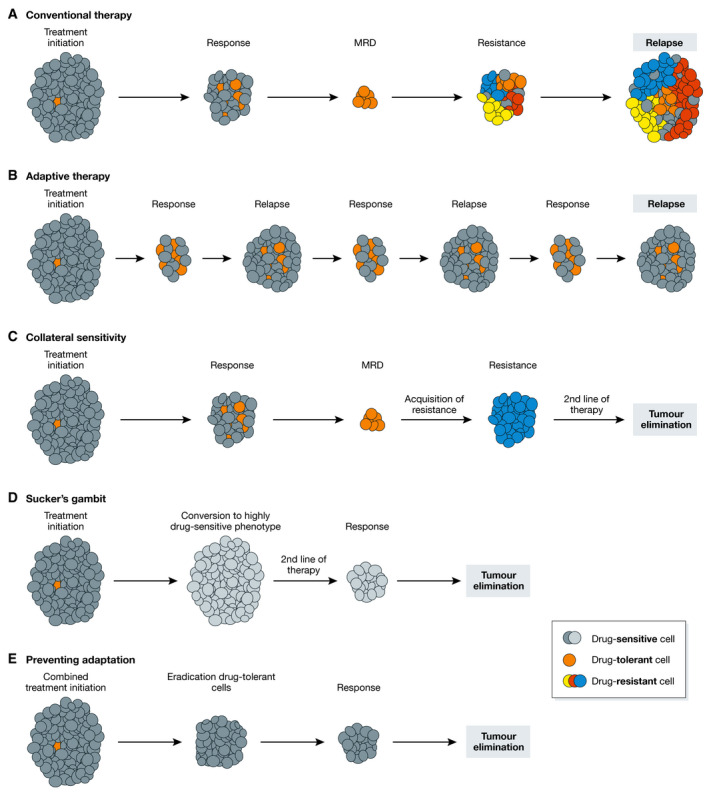
(A) Most conventional therapeutic approaches will lead to marked initial responses but often fail to eliminate—or induce the emergence of—drug‐tolerant cells. This population of minimal residual disease (MRD) represents a reservoir of malignant cells from which drug‐resistant clone(s) may emerge and cause tumour relapse. (B) In adaptive therapy, treatment is discontinued upon reaching a pre‐defined threshold so as not to eliminate all drug‐sensitive cells (dark grey). These cells are then allowed to grow back and are expected to suppress the growth of drug‐tolerant (or therapy‐resistant) clones owing to their higher fitness upon withdrawal. (C) Collateral sensitivities, wherein acquisition of resistance to a first drug induces susceptibility to a second one, can be exploited to target cancer‐specific vulnerabilities and potentially lead to tumour eradication. (D) In the Sucker’s gambit, the first drug forces a phenotypic adaptation that pushes tumour cells into an evolutionary trap by increasing the fitness of a highly drug‐sensitive population (leading to its clonal dominance), which is then eliminated by a second line of treatment. (E) Limiting drug adaptation may prevent the establishment of drug‐tolerant (MRD) and drug‐resistant populations. In this setting, a combination of drugs is used to simultaneously eradicate tumour cells and to prevent the emergence of drug‐induced adaptive responses.
Additionally, there is growing appreciation that non‐Darwinian mechanisms can drive tumour adaptation, especially in the context of therapy resistance, where a clear genetic cause for therapy failure has only been identified in about 60% of tumours. For instance, MAPK inhibition was shown to drive transcriptional reprogramming in melanomas shortly after treatment initiation which resulted in drug tolerance (Smith et al, 2016). Importantly, this response was reversible and non‐mutational in nature. Targeting of a master regulator of this adaptive mechanism was shown to prevent drug tolerance and re‐sensitise melanoma cells to MAPKi (Smith et al, 2016). Similarly, the activation of the integrated stress response (ISR) was observed to foster drug tolerance in melanoma. In this context, ISR activation was found to indirectly enhance mitochondrial translation, making these cells highly vulnerable to mitochondrial translation inhibitors, which were shown to reduce tumour heterogeneity and to prevent acquisition of resistance regardless of the tumour mutational profile or phenotype (Vendramin et al, 2021). The TME was also shown to promote tumour drug tolerance. For instance, paradoxical activation of the fibroblastic stroma in BRAF inhibitor treated melanomas resulted in enhanced matrix remodelling. The remodelled matrix was found to promote BRAF‐independent ERK activation through FAK signalling, thereby enabling drug tolerance and melanoma cell survival. Concomitant inhibition of BRAF and FAK prevented ERK reactivation and led to tumour volume stabilisation (Hirata et al, 2015). Similarly, inhibition of the microenvironment‐produced morphogen EDN3 abrogated transcriptional reprogramming and phenotype switch in melanomas and reduced tumour aggressiveness and growth (Kim et al, 2017).
Exploiting collateral sensitivities
The paradox of treating dynamically evolving cancers is that therapeutic strategies are typically administered in a fixed, sequential schedule in the hope of reducing tumour burden (Gatenby et al, 2009). The integration of evolutionary principles in the design of anti‐cancer therapies may therefore bring about substantial clinical benefits. Knowledge of tumour evolutionary responses to therapy could allow the anticipation of relapse by pre‐emptively targeting drug resistance mechanisms or to exploit collateral sensitivities, i.e. specific weaknesses caused by the development of resistance to a particular treatment (Pluchino et al, 2012; Efferth et al, 2020) (Fig 3). For instance, combining different classes of ABL1 inhibitors with mutually exclusive profiles of resistance mutations in chronic myeloid leukaemia can prevent the emergence of drug‐resistant subclones and lead to durable responses in vivo (Zhao et al, 2016; Wylie et al, 2017). Another example is gefitinib‐resistant NSCLC, which shows collateral sensitivity for TNFα when compared to the non‐resistant counterpart (Ando et al, 2005). A similar strategy is the “sucker's gambit”, in which the initial therapy, rather than killing all cancer cells, aims to push them into an evolutionary cul‐de‐sac by forcing a phenotypic adaptation that is then exploited in second‐line therapy (Merlo et al, 2006; Schweizer et al, 2015) (Fig 3). The first drug will affect most subpopulations and promote the emergence and clonal dominance of a specific clone, which would then be eliminated by the addition of a second drug (West et al, 2019). A limitation of this approach is that it is based on the premise that all cells will develop the same mechanism of resistance, whereas a plethora of in vivo experiments and autopsy studies have demonstrated that most individual tumours can achieve resistance (to chemo‐, targeted and immunotherapy) via multiple distinct routes simultaneously, a process termed polyclonal resistance (Burrell & Swanton, 2014; Shi et al, 2014; Kemper et al, 2015; Faltas et al, 2016; Ascierto et al, 2017; Rambow et al, 2018; Razavi et al, 2018; Krook et al, 2019a; Rosenthal et al, 2019; Sanchez‐Vega et al, 2019).
An alternative strategy, named “benign cell booster”, aims instead to promote the growth of benign, less aggressive tumour subpopulations at the expense of highly malignant populations (Maley et al, 2004). Importantly, such an approach would prevent the emergence of aggressive treatment‐resistant subpopulations and have likely limited adverse effects since it would not be aimed at killing cancer cells but rather select for particularly benign tumour cell populations.
Adaptive therapies
Another intriguing therapeutic approach based on evolutionary principles is adaptive therapy, a therapeutic strategy with the goal of maintaining a stable tumour population below a certain symptomatic threshold, rather than trying to achieve complete tumour elimination, through the use of repeated ON/OFF treatment cycles (Gatenby et al, 2009) (Fig 3). This strategy aims to make use of the understanding of the mechanisms of therapy resistance as well as their “cost” and to exploit the differences in terms of fitness between drug‐sensitive and drug‐resistant subpopulations (Cunningham, 2019). In such a scenario, the optimal dose and treatment duration is not the maximum possible but rather the minimum necessary. In essence, a treatment cycle would last until tumour response (i.e. regression below a certain, pre‐defined threshold) is observed, after which therapy is discontinued. This approach enables minimal tumour cell proliferation to prevent the elimination of all treatment‐sensitive tumour clones, which are expected to have a competitive advantage in terms of fitness over treatment‐resistant populations. Upon relapse, the tumour should mostly be comprised of treatment‐sensitive cells; therefore, a new treatment cycle will be initiated to bring the tumour volume below a symptomatic threshold. Despite its initial promises and successes in in vitro and in vivo models (Enriquez‐Navas et al, 2016; Bacevic et al, 2017), this strategy has not achieve notable results in the clinic, as indefinite tumour control, the goal of such an approach, has not been achieved (Crook et al, 2012; Hussain et al, 2013; Algazi et al, 2020). One limitation of this approach is the lack of tools enabling constant monitoring of tumour subclonal evolution (e.g. via ctDNA/CTCs, see Box 3) over the course of treatment. Another weakness of this strategy is the assumption that drug resistance always comes at a fitness “cost” for the cell, which may not be true in all cases, especially in the context of polyclonal resistance, where different resistance mechanisms may differentially impact cell fitness. Indeed, there is evidence that acquisition of resistance, at least in some instances, may not be associated with a fitness cost at all (Shah et al, 2007; ffrench‐Constant & Bass, 2017; Strobl et al, 2021).
Early detection
Many early‐stage cancers can be cured by surgery alone if removed before metastasis has occurred, by surgery ± adjuvant therapy. Past that point, these approaches are seldomly curative (Siegel et al, 2017; Cohen et al, 2018). Therefore, until more potent and specific therapeutic agents are developed, a promising strategy for reducing cancer mortality is early tumour detection, which has been shown to reduce cancer mortality in several cancer types including lung, breast, bladder, colorectal, head and neck and testicular cancer as well as melanoma (Hawkes, 2019; Lee et al, 2019; Pantel & Alix‐Panabières, 2019; Crosby et al, 2020; de Koning et al, 2020). Early‐stage cancer detection may also allow the identification of tumours in early evolutionary stages, when they may be less resilient to treatment due to their more homogeneous nature (Jamal‐Hanjani et al, 2015). Additionally, detecting pre‐existing therapy‐resistant clones before treatment initiation may allow the employment of individualised therapeutic approaches aimed at eliminating drug‐tolerant/resistant populations. A major evolutionary question, that in the context of early detection remains unaddressed, is timing of tumour initiation and the lead‐time between malignant transformation and clinical presentation. Evolutionary tools such as the molecular clock, originally introduced by Emile Zuckerkandl and Linus Pauling in the 1960s (Morgan, 1998), have recently provided some preliminary estimates using cancer genomics datasets. These approaches take advantage of the somatic mutations acquired by cancer cells with clocklike properties, i.e. a correlation with the chronological age of the person. With this knowledge, mutation counts can be used to infer the timing of biological events, for example 3p loss/5q gain events found in 36% of ccRCC patients were timed back several decades to childhood/adolescence (Mitchell et al, 2018). In another study, a primary colorectal tumour was timed to have likely emerged 5 to 8 years before clinical diagnosis (Lote et al, 2017). In another case, analysis of early NSCLC revealed WGD events and truncal driver events in ex‐smokers dating back more than 20 years prior to tumour detection, indicating a prolonged tumour latency period from the first oncogenic insults to clinical manifestation (De Bruin et al, 2014). Recent large‐scale work from the Pan‐Cancer Analysis of Whole Genomes (PCAWG) consortium in 2,658 tumours also revealed that driver mutations often precede clinical diagnosis by many years, if not decades (Gerstung et al, 2020). While inferred in nature, these studies still provide accumulating evidence that tumour initiation likely dates back years or even decades before clinical presentation, hence suggesting an extended window for cancer early detection, likely through the use of non‐invasive blood‐based tests (see Box 3) (Liu et al, 2020). In addition to genomic data, information on the history of prior mutagenic exposures (i.e. duration of smoking, age of smoking cessation) can be additionally utilised to validate and refine timing inferences (De Bruin et al, 2014). Lastly, as well as absolute timing, the accumulation of mutations can also be timed relative (i.e. before or after) macroevolutionary events such as WGD (López et al, 2020).
Minimal residual disease
Particular attention should also be given to minimal residual disease (MRD) detection and characterisation. MRD represents latent disease, i.e. a small population of undetectable malignant cells that persist or recur following initial surgical or systemic treatment. These cells may represent a reservoir of metastasis competent or drug‐tolerant cells able to drive future tumour relapse (Luskin et al, 2018; Rambow et al, 2018; Chin et al, 2019). Owing to the limited number of cells, it is technically challenging to detect MRD through conventional approaches, such as computed tomography scans. To date, the most promising method to detect MRD is achieved through circulating tumour DNA assessment (see Box 3). This approach has been shown to be successful in several studies in bladder cancer (Dudley et al, 2019), NSCLC (Abbosh et al, 2017; Chaudhuri et al, 2017), colon cancer (Tie et al, 2016), breast cancer (Mounier et al, 2015), oesophageal cancer (Azad et al, 2020) and a range of other solid tumour types (Chin et al, 2019). Importantly, macroevolutionary changes caused by large chromosomal aberrations, WGD or CNAs that appear during metastatic formation have been detected through analysis of circulating tumour cells (CTCs) and circulating tumour DNA (ctDNA) in recent studies performed on colon cancer patients with MRD (Gao et al, 2017; Joosse et al, 2018). Additionally, post‐surgical detection of MRD is a strong predictor of impending disease relapse (Abbosh et al, 2018; Luskin et al, 2018; Azad et al, 2020). Hence, rational targeting of MRD is emerging as a promising strategy to prevent therapy resistance and relapse (Luskin et al, 2018). There are several advantages to treating patients with MRD rather than waiting for clinical relapse to initiate further therapy. First, tracking of MRD allows the identification of alterations emerging in response to treatment. Second, earlier detection of relapse via MRD allows further treatment to be implemented earlier, where a window of more favourable outcome may be achievable when tumour burden and ITH will be lower. Accordingly, the design of personalised ctDNA panels based on patient mutational status could provide a patient‐specific biomarker of tumour response to treatment. For instance, a recent study demonstrated that the use of a personalised ctDNA panel led to detection of breast cancer metastatic recurrence up to two years prior to overt clinical manifestation (Coombes et al, 2019). Last, this approach could allow to adapt therapeutic regimens in the adjuvant setting to eliminate all persisting cells. Regarding predictive MRD tools, the PROSPECT‐C study demonstrated that time to anti‐EGFR treatment failure could be reliably predicted using longitudinal ctDNA sampling in colorectal cancer, hence generating a window of opportunity for future intervention (Khan et al, 2018). Similarly, the IMvigor010 study (Powles et al, 2021) demonstrated that MRD detection through ctDNA detection, using the Signatera assay co‐developed by our laboratory and Natera (Abbosh et al, 2017), could be used to stratify patients who will benefit from atezolizumab (anti‐PD‐L1) postoperatively. Evidence that targeting MRD can prevent relapse comes from the experience of treating patients with completely resected NSCLC (Winton et al, 2005), colon cancer (André et al, 2009) and breast cancer (Albain et al, 2012) with adjuvant (or neoadjuvant) therapy, with promising results coming from in vivo experiments in melanoma (Rambow et al, 2018; Marin‐Bejar et al, 2021; Vendramin et al, 2021). A similar approach is currently being tested in the MERMAID‐1 Phase III clinical trial where the effects of adjuvant treatment with durvalumab (anti‐PD‐L1) plus chemotherapy versus chemotherapy alone on disease‐free survival (DFS) in patients with completely resected stage II‐III NSCLC who show evidence of MRD.
Conclusion and future perspectives
The longstanding assumption that cancer evolution can only be viewed through a Darwinian lens has been shown to be insufficient to explain the complex evolutionary patterns observed in human malignancies. While our understanding of tumour evolution has been built on the foundations of Darwinian selection, increasing evidence indicates that cancer evolutionary dynamics often defy Darwinian principles. The observation that tumours can evolve in a punctuated fashion, rather than in a gradual fashion as Darwin theorised when he stated that “natural selection can act only by taking advantage of slight successive variations; she can never take a leap, but must advance by the shortest and slowest step” (Darwin, 1859), demonstrates that nature does indeed make macroevolutionary jumps. It is also important to consider that the diversity within tumours is not necessarily selectively driven but can also emerge in some tumours as the result of neutral evolutionary dynamics. Evidence of discordant inheritance patterns between cells represents additional proof of the relevance of non‐classical mechanisms in cancer evolution. Furthermore, neo‐Darwinian evolutionary models are traditionally based on a gene‐centric view of evolution, whereas increasing evidence highlights the role of non‐genetic contributors, such as cell plasticity and the tumour microenvironment, as key determinants of cancer evolution. Last, it is becoming increasingly clear that tumorigenesis may in some cases reflect the physiological decline of a tissue, whereby an ageing soma provides a more permissive environment for malignant transformation. It is important to note that individual tumours may not follow a single evolutionary model. Multiple mechanisms may instead be operating either concomitantly or at different stages of progression. Hence, a coherent and unified approach encompassing Darwinian and non‐Darwinian evolutionary theories is needed to fully comprehend the breadth and the depth of cancer evolutionary dynamics.
Research output thus far has revealed that tumour evolution is highly varied in its nature, as well as evidenced how little we still know about cancer evolutionary trajectories. While the studies discussed above have provided critical clues on the driving forces of tumour evolution, they were generally based on individual time points and limited in terms of resolution and cohort size. Larger clinical studies, harnessing the full potential of recent technological innovations, such as single‐cell approaches, making use of multiple longitudinal and post‐mortem approaches, such as blood‐based tests monitoring ctDNA/CTCs and rapid research autopsies, respectively, are thus warranted. The implementation of these approaches will allow us to shed light on the dynamics of cancer evolution and to develop evolutionary‐guided therapies to impact clinical outcomes.
In conclusion, understanding cancer requires an evolutionary perspective. The study of malignant transformation within an evolutionary framework has potential to bring about a substantial improvement in clinical outcomes, by providing novel insights to support personalised cancer treatment, as well anticipating and proactively managing therapeutic resistance.
Conflict of interest
K.L. has a patent on indel burden and CPI response pending and speaker fees from Roche tissue diagnostics, research funding from CRUK TDL/Ono/LifeArc alliance, and a consulting role with Monopteros Therapeutics. C.S. acknowledges grant support from Pfizer, AstraZeneca, Bristol Myers Squibb, Roche‐Ventana, Boehringer‐Ingelheim, Archer Dx Inc (collaboration in minimal residual disease sequencing technologies) and Ono Pharmaceuticals, is an AstraZeneca Advisory Board member and Chief Investigator for the MeRmaiD1 clinical trial. C.S. has consulted for Amgen, AstraZeneca, Bicycle Therapeutics, Bristol Myers Squibb, Celgene, Genentech, GlaxoSmithKline, GRAIL, Illumina, Medixci, Metabomed, MSD, Novartis, Pfizer, Roche‐Ventana and Sarah Cannon Research Institute. C.S. has stock options in Apogen Biotechnologies, Epic Biosciences, GRAIL, and has stock options and is co‐founder of Achilles Therapeutics. C.S. holds patents relating to assay technology to detect tumour recurrence (PCT/GB2017/ 053289); to targeting neoantigens (PCT/EP2016/059401); identifying patent response to immune checkpoint blockade (PCT/EP2016/071471); determining whether HLA LOH is lost in a tumour (PCT/GB2018/052004); predicting survival rates of cancer patients (PCT/GB2020/050221); to treating cancer by targeting insertion/deletion mutations (PCT/GB2018/051893); identifying insertion/deletion mutation targets (PCT/GB2018/051892); methods for lung cancer detection (PCT/US2017/028013); and identifying responders to cancer treatment (PCT/GB2018/051912).
Acknowledgements
R.V. and K.L. are funded by the UK Medical Research Council (MR/V033077/1), the Rosetrees Trust and Cotswold Trust (A2437), the Royal Marsden Cancer Charity (thanks to the Ross Russell family and Macfarlanes donations), Melanoma Research Alliance and Cancer Research UK (C69256/A30194). C.S. is a Royal Society Napier Research Professor (RP150154). This work was supported by the Francis Crick Institute that receives its core funding from Cancer Research UK (FC001169), the UK Medical Research Council (FC001169), and the Wellcome Trust (FC001169). This research was funded in whole, or in part, by the Wellcome Trust (FC001169). For the purpose of Open Access, the author has applied a CC BY public copyright licence to any Author Accepted Manuscript version arising from this submission. C.S. is funded by Cancer Research UK (TRACERx, PEACE and CRUK Cancer Immunotherapy Catalyst Network), Cancer Research UK Lung Cancer Centre of Excellence (C11496/A30025), the Rosetrees Trust, Butterfield and Stoneygate Trusts, NovoNordisk Foundation (ID16584), Royal Society Professorship Enhancement Award (RP/EA/180007), the National Institute for Health Research (NIHR) Biomedical Research Centre at University College London Hospitals, the Cancer Research UK‐University College London Centre, Experimental Cancer Medicine Centre, and the Breast Cancer Research Foundation (BCRF 20‐157). This work was supported by a Stand Up To Cancer‐LUNGevity‐American Lung Association Lung Cancer Interception Dream Team Translational Research Grant (Grant Number: SU2C‐AACR‐DT23‐17 to S.M. Dubinett and A.E. Spira). Stand Up To Cancer is a division of the Entertainment Industry Foundation. Research grants are administered by the American Association for Cancer Research, the Scientific Partner of SU2C. C.S. receives funding from an ERC Advanced Grant (PROTEUS) from the European Research Council under the European Union’s Horizon 2020 research and innovation programme (grant agreement No. 835297).
The EMBO Journal (2021) 40: e108389.
This article is part of the Cancer Reviews 2021 series.
Contributor Information
Kevin Litchfield, Email: k.litchfield@ucl.ac.uk.
Charles Swanton, Email: charles.swanton@crick.ac.uk.
References
- Abbosh C, Birkbak NJ, Wilson GA, Jamal‐Hanjani M, Constantin T, Salari R, Le Quesne J, Moore DA, Veeriah S, Rosenthal Ret al (2017) Phylogenetic ctDNA analysis depicts early‐stage lung cancer evolution. Nature 545: 446–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbosh C, Birkbak NJ, Swanton C (2018) Early stage NSCLC — challenges to implementing ctDNA‐based screening and MRD detection. Nat Rev Clin Oncol 15: 577–586 [DOI] [PubMed] [Google Scholar]
- Aceto N, Bardia A, Miyamoto D, Donaldson M, Wittner B, Spencer J, Yu M, Pely A, Engstrom A, Zhu Het al (2014) Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 158: 1110–1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aitken SJ, Anderson CJ, Connor F, Pich O, Sundaram V, Feig C, Rayner TF, Lukk M, Aitken S, Luft Jet al (2020) Pervasive lesion segregation shapes cancer genome evolution. Nature 583: 265–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albain K, Anderson S, Arriagada R, Barlow W, Bergh J, Bliss J, Buyse M, Cameron D, Carrasco E, Clarke Met al (2012) Comparisons between different polychemotherapy regimens for early breast cancer: meta‐analyses of long‐term outcome among 100 000 women in 123 randomised trials. Lancet 379: 432–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Algazi AP, Othus M, Daud AI, Lo RS, Mehnert JM, Truong T‐G, Conry R, Kendra K, Doolittle GC, Clark JIet al (2020) Continuous versus intermittent BRAF and MEK inhibition in patients with BRAF‐mutated melanoma: a randomized phase 2 trial. Nat Med 26: 1564–1568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almendro V, Cheng Y‐K, Randles A, Itzkovitz S, Marusyk A, Ametller E, Gonzalez‐Farre X, Muñoz M, Russnes H, Helland Ået al (2014) Inference of tumor evolution during chemotherapy by computational modeling and in situ analysis of genetic and phenotypic cellular diversity. Cell Rep 6: 514–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anagnostou V, Forde PM, White JR, Niknafs N, Hruban C, Naidoo J, Marrone K, Sivakumar IKA, Bruhm DC, Rosner Set al (2019) Dynamics of tumor and immune responses during immune checkpoint blockade in non–small cell lung cancer. Cancer Res 79: 1214–1225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson K, Lutz C, van Delft FW, Bateman CM, Guo Y, Colman SM, Kempski H, Moorman AV, Titley I, Swansbury Jet al (2011) Genetic variegation of clonal architecture and propagating cells in leukaemia. Nature 469: 356–361 [DOI] [PubMed] [Google Scholar]
- Ando K, Ohmori T, Inoue F, Kadofuku T, Hosaka T, Ishida H, Shirai T, Okuda K, Hirose T, Horichi Net al (2005) Enhancement of sensitivity to tumor necrosis factor α in non‐small cell lung cancer cells with acquired resistance to gefitinib. Clin Cancer Res 11: 8872–8879 [DOI] [PubMed] [Google Scholar]
- André T, Boni C, Navarro M, Tabernero J, Hickish T, Topham C, Bonetti A, Clingan P, Bridgewater J, Rivera Fet al (2009) Improved overall survival with oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment in stage II or III colon cancer in the MOSAIC trial. J Clin Oncol 27: 3109–3116 [DOI] [PubMed] [Google Scholar]
- Aryee Mj, Liu W, Engelmann Jc, Nuhn P, Gurel M, Haffner Mc, Esopi D, Irizarry Ra, Getzenberg Rh, Nelson Wget al (2013) DNA methylation alterations exhibit intraindividual stability and interindividual heterogeneity in prostate cancer metastases. Sci Transl Med 5: 169ra10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ascierto ML, Makohon‐Moore A, Lipson EJ, Taube JM, McMiller TL, Berger AE, Fan J, Kaunitz GJ, Cottrell TR, Kohutek ZAet al (2017) Transcriptional mechanisms of resistance to anti‐PD‐1 therapy. Clin Cancer Res 23: 3168–3180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avigdor BE, Cimino‐Mathews A, DeMarzo AM, Hicks JL, Shin J, Sukumar S, Fetting J, Argani P, Park BH, Wheelan SJ (2017) Mutational profiles of breast cancer metastases from a rapid autopsy series reveal multiple evolutionary trajectories. JCI Insight 2: e96896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azad TD, Chaudhuri AA, Fang P, Qiao Y, Esfahani MS, Chabon JJ, Hamilton EG, Yang YD, Lovejoy A, Newman AMet al (2020) Circulating tumor DNA analysis for detection of minimal residual disease after chemoradiotherapy for localized esophageal cancer. Gastroenterology 158: 494–505.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baca S, Prandi D, Lawrence M, Mosquera J, Romanel A, Drier Y, Park K, Kitabayashi N, MacDonald T, Ghandi Met al (2013) Punctuated evolution of prostate cancer genomes. Cell 153: 666–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacevic K, Noble R, Soffar A, Wael Ammar O, Boszonyik B, Prieto S, Vincent C, Hochberg ME, Krasinska L, Fisher D (2017) Spatial competition constrains resistance to targeted cancer therapy. Nat Commun 8: 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey C, Shoura MJ, Mischel PS, Swanton C (2020) Extrachromosomal DNA—relieving heredity constraints, accelerating tumour evolution. Ann Oncol 31: 884–893 [DOI] [PubMed] [Google Scholar]
- Bakhoum SF, Landau DA (2017) Chromosomal instability as a driver of tumor heterogeneity and evolution. Cold Spring Harb Perspect Med 7: a029611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barthel FP, Johnson KC, Varn FS, Moskalik AD, Tanner G, Kocakavuk E, Anderson KJ, Abiola O, Aldape K, Alfaro KDet al (2019) Longitudinal molecular trajectories of diffuse glioma in adults. Nature 576: 112–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedard PL, Hyman DM, Davids MS, Siu LL (2020) Small molecules, big impact: 20 years of targeted therapy in oncology. Lancet 395: 1078–1088 [DOI] [PubMed] [Google Scholar]
- Bielski CM, Zehir A, Penson AV, Donoghue MTA, Chatila W, Armenia J, Chang MT, Schram AM, Jonsson P, Bandlamudi Cet al (2018) Genome doubling shapes the evolution and prognosis of advanced cancers. Nat Genet 50: 1189–1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boumahdi S, de Sauvage FJ (2020) The great escape: tumour cell plasticity in resistance to targeted therapy. Nat Rev Drug Discov 19: 39–56 [DOI] [PubMed] [Google Scholar]
- Boveri T (2008) Concerning the origin of malignant tumours by Theodor Boveri. Translated and annotated by Henry Harris. J Cell Sci 121: 1–84 [DOI] [PubMed] [Google Scholar]
- Bozic I, Paterson C, Waclaw B (2019) On measuring selection in cancer from subclonal mutation frequencies. PLoS Comput Biol 15: e1007368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brabletz S, Schuhwerk H, Brabletz T, Stemmler MP (2021) Dynamic EMT: a multi‐tool for tumor progression. EMBO J 10.15252/embj.2021108647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown D, Smeets D, Székely B, Larsimont D, Marcell Szász A, Adnet PY, Rothé F, Rouas G, Nagy ZI, Faragó Zet al (2017) Phylogenetic analysis of metastatic progression in breast cancer using somatic mutations and copy number aberrations. Nat Commun 8: 14944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown TM, Fee E (2014) Rudolf Carl Virchow. Acta Gastroenterol Latinoam 44: 202 [PubMed] [Google Scholar]
- de Bruin EC, McGranahan N, Mitter R, Salm M, Wedge Dc, Yates L, Jamal‐Hanjani M, Shafi S, Murugaesu N, Rowan Ajet al (2014) Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science 346: 251–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrell RA, McClelland SE, Endesfelder D, Groth P, Weller M‐C, Shaikh N, Domingo E, Kanu N, Dewhurst SM, Gronroos Eet al (2013) Replication stress links structural and numerical cancer chromosomal instability. Nature 494: 492–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrell RA, Swanton C (2014) Tumour heterogeneity and the evolution of polyclonal drug resistance. Mol Oncol 8: 1095–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caiado F, Silva‐Santos B, Norell H (2016) Intra‐tumour heterogeneity – going beyond genetics. FEBS J 283: 2245–2258 [DOI] [PubMed] [Google Scholar]
- Calabrese C, Davidson NR, Demircioğlu D, Fonseca NA, He Y, Kahles A, Lehmann K‐V, Liu F, Shiraishi Y, Soulette CMet al (2020) Genomic basis for RNA alterations in cancer. Nature 578: 129–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell LL, Polyak K (2007) Breast tumor heterogeneity: cancer stem cells or clonal evolution? Cell Cycle 6: 2332–2338 [DOI] [PubMed] [Google Scholar]
- Campbell PJ, Yachida S, Mudie LJ, Stephens PJ, Pleasance ED, Stebbings LA, Morsberger LA, Latimer C, McLaren S, Lin M‐Let al (2010) The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature 467: 1109–1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannataro VL, McKinley SA, St. Mary CM (2017) The evolutionary trade‐off between stem cell niche size, aging, and tumorigenesis. Evol Appl 10: 590–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri AA, Chabon JJ, Lovejoy AF, Newman AM, Stehr H, Azad TD, Khodadoust MS, Esfahani MS, Liu CL, Zhou Liet al (2017) Early detection of molecular residual disease in localized lung cancer by circulating tumor DNA profiling. Cancer Discov 7: 1394–1403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen DY, Zhen HF, Qiu Y, Liu P, Zeng P, Xia J, Shi QY, Xie L, Zhu Z, Gao Yet al (2018) Comparison of single cell sequencing data between two whole genome amplification methods on two sequencing platforms. Sci Rep 8: 4963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HZ, Bonneville R, Roychowdhury S (2019) Implementing precision cancer medicine in the genomic era. Semin Cancer Biol 55: 16–27 [DOI] [PubMed] [Google Scholar]
- Chen X, Gole J, Gore A, He Q, Lu M, Min J, Yuan Z, Yang X, Jiang Y, Zhang Tet al (2020) Non‐invasive early detection of cancer four years before conventional diagnosis using a blood test. Nat Commun 11: 3475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin RI, Chen K, Usmani A, Chua C, Harris PK, Binkley MS, Azad TD, Dudley JC, Chaudhuri AA (2019) Detection of solid tumor Molecular Residual Disease (MRD) using Circulating Tumor DNA (ctDNA). Mol Diagnosis Ther 23: 311–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chkhaidze K, Heide T, Werner B, Williams MJ, Huang W, Caravagna G, Graham TA, Sottoriva A (2019) Spatially constrained tumour growth affects the patterns of clonal selection and neutral drift in cancer genomic data. PLoS Comput Biol 15: e1007243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, Getz G (2013) Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 31: 213–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke TL, Tang R, Chakraborty D, Van Rechem C, Ji F, Mishra S, Ma A, Kaniskan HÜ, Jin J, Lawrence MSet al (2020) Histone lysine methylation dynamics control EGFR DNA copy‐number amplification. Cancer Discov 10: 306–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen JD, Li Lu, Wang Y, Thoburn C, Afsari B, Danilova L, Douville C, Javed AA, Wong F, Mattox Aet al (2018) Detection and localization of surgically resectable cancers with a multi‐analyte blood test. Science 359: 926–930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coombes RC, Page K, Salari R, Hastings RK, Armstrong A, Ahmed S, Ali S, Cleator S, Kenny L, Stebbing Jet al (2019) Personalized detection of circulating tumor DNA antedates breast cancer metastatic recurrence. Clin Cancer Res 25: 4255–4263 [DOI] [PubMed] [Google Scholar]
- Cottu PH, Asselah J, Lae M, Pierga JY, Diéras V, Mignot L, Sigal‐Zafrani B, Vincent‐Salomon A (2008) Intratumoral heterogeneity of HER2/neu expression and its consequences for the management of advanced breast cancer [2]. Ann Oncol 19: 596–597 [DOI] [PubMed] [Google Scholar]
- Coussens LM, Werb Z (2002) Inflammation and cancer. Nature 420: 860–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crook JM, O'Callaghan CJ, Duncan G, Dearnaley DP, Higano CS, Horwitz EM, Frymire E, Malone S, Chin J, Nabid Aet al (2012) Intermittent androgen suppression for rising PSA level after radiotherapy. N Engl J Med 367: 895–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosby D, Lyons N, Greenwood E, Harrison S, Hiom S, Moffat J, Quallo T, Samuel E, Walker I (2020) A roadmap for the early detection and diagnosis of cancer. Lancet Oncol 21: 1397–1399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross WCH, Graham TA, Wright NA (2016) New paradigms in clonal evolution: punctuated equilibrium in cancer. J Pathol 240: 126–136 [DOI] [PubMed] [Google Scholar]
- Cunningham JJ (2019) A call for integrated metastatic management. Nat Ecol Evol 3: 996–998 [DOI] [PubMed] [Google Scholar]
- Dagogo‐Jack I, Shaw AT (2018) Tumour heterogeneity and resistance to cancer therapies. Nat Rev Clin Oncol 15: 81–94 [DOI] [PubMed] [Google Scholar]
- Darwin CR (1859) On the origin of species by means of natural selection, or the preservation of favoured races in the struggle for life. London: John Murray. [PMC free article] [PubMed] [Google Scholar]
- Davis A, Gao R, Navin N (2017) Tumor evolution: Linear, branching, neutral or punctuated? Biochim Biophys Acta ‐ Rev Cancer 1867: 151–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson S‐J, Tsui DWY, Murtaza M, Biggs H, Rueda OM, Chin S‐F, Dunning MJ, Gale D, Forshew T, Mahler‐Araujo Bet al (2013) Analysis of circulating tumor DNA to monitor metastatic breast cancer. N Engl J Med 368: 1199–1209 [DOI] [PubMed] [Google Scholar]
- de Koning HJ, van der Aalst CM, de Jong PA, Scholten ET, Nackaerts K, Heuvelmans MA, Lammers J‐W, Weenink C, Yousaf‐Khan U, Horeweg Net al (2020) Reduced lung‐cancer mortality with volume CT screening in a randomized trial. N Engl J Med 382: 503–513 [DOI] [PubMed] [Google Scholar]
- De Mattos‐Arruda L, Sammut S‐J, Ross EM, Bashford‐Rogers R, Greenstein E, Markus H, Morganella S, Teng Y, Maruvka Y, Pereira Bet al (2019) The genomic and immune landscapes of lethal metastatic breast cancer. Cell Rep 27: 2690–2708.e10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- deCarvalho AC, Kim H, Poisson LM, Winn ME, Mueller C, Cherba D, Koeman J, Seth S, Protopopov A, Felicella Met al (2018) Discordant inheritance of chromosomal and extrachromosomal DNA elements contributes to dynamic disease evolution in glioblastoma. Nat Genet 50: 708–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeGregori J, Pharoah P, Sasieni P, Swanton C (2020) Cancer screening, surrogates of survival, and the soma. Cancer Cell 38: 433–437 [DOI] [PubMed] [Google Scholar]
- Deshpande V, Luebeck J, Nguyen NPD, Bakhtiari M, Turner KM, Schwab R, Carter H, Mischel PS, Bafna V (2019) Exploring the landscape of focal amplifications in cancer using AmpliconArchitect. Nat Commun 10: 392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewhurst SM, McGranahan N, Burrell RA, Rowan AJ, Grönroos E, Endesfelder D, Joshi T, Mouradov D, Gibbs P, Ward RLet al (2014) Tolerance of whole‐ genome doubling propagates chromosomal instability and accelerates cancer genome evolution. Cancer Discov 4: 175–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dexter DL, Kowalski HM, Blazar BA, Fligiel Z, Vogel R, Gloria H, Heppner H (1978) Heterogeneity of tumor cells from a single mouse mammary tumor. Cancer Res 38: 3174–3181 [PubMed] [Google Scholar]
- Dudley JC, Schroers‐Martin J, Lazzareschi DV, Shi WY, Chen SB, Esfahani MS, Trivedi D, Chabon JJ, Chaudhuri AA, Stehr Het al (2019) Detection and surveillance of bladder cancer using urine tumor DNA. Cancer Discov 9: 500–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duregon E, Schneider J, DeMarzo AM, Hooper JE (2019) Rapid research autopsy is a stealthy but growing contributor to cancer research. Cancer 125: 2915–2919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efferth T, Saeed MEM, Kadioglu O, Seo EJ, Shirooie S, Mbaveng AT, Nabavi SM, Kuete V (2020) Collateral sensitivity of natural products in drug‐resistant cancer cells. Biotechnol Adv 38: 107342 [DOI] [PubMed] [Google Scholar]
- Embuscado EE, Laheru D, Ricci F, Yun KJ, Witzel SDB, Seigel A, Flickinger K, Hidalgo M, Bova GS, Iacobuzio‐Donahue CA (2005) Immortalizing the complexity of cancer metastasis: Genetic features of lethal metastatic pancreatic cancer obtained from rapid autopsy. Cancer Biol Ther 4: 548–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endesfelder D, Burrell RA, Kanu N, McGranahan N, Howell M, Parker PJ, Downward J, Swanton C, Kschischo M (2014) Chromosomal instability selects gene copy‐number variants encoding core regulators of proliferation in ER+ Breast cancer. Cancer Res 74: 4853–4863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enriquez‐Navas PM, Kam Y, Das T, Hassan S, Silva A, Foroutan P, Ruiz E, Martinez G, Minton S, Gillies RJet al (2016) Exploiting evolutionary principles to prolong tumor control in preclinical models of breast cancer. Sci Transl Med 8 327ra24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faltas BM, Prandi D, Tagawa ST, Molina AM, Nanus DM, Sternberg C, Rosenberg J, Mosquera JM, Robinson B, Elemento Oet al (2016) Clonal evolution of chemotherapy‐resistant urothelial carcinoma. Nat Genet 48: 1490–1499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J, Lee H‐O, Lee S, Ryu D‐E, Lee S, Xue C, Kim SJ, Kim K, Barkas N, Park PJet al (2018) Linking transcriptional and genetic tumor heterogeneity through allele analysis of single‐cell RNA‐seq data. Genome Res 28: 1217–1227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Y, Mao R, Lv H, Xu J, Yan L, Liu Y, Shi M, Ji G, Yu Y, Bai Jet al (2011) Frequency of double minute chromosomes and combined cytogenetic abnormalities and their characteristics. J Appl Genet 52: 53–59 [DOI] [PubMed] [Google Scholar]
- ffrench‐Constant RH, Bass C (2017) Does resistance really carry a fitness cost? Curr Opin Insect Sci 21: 39–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fittall MW, Van Loo P (2019) Translating insights into tumor evolution to clinical practice: promises and challenges. Genome Med 11: 20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao R, Davis A, McDonald TO, Sei E, Shi X, Wang Y, Tsai P‐C, Casasent A, Waters J, Zhang Het al (2016) Punctuated copy number evolution and clonal stasis in triple‐negative breast cancer. Nat Genet 48: 1119–1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Ni X, Guo H, Su Z, Ba Yi, Tong Z, Guo Z, Yao X, Chen X, Yin Jet al (2017) Single‐cell sequencing deciphers a convergent evolution of copy number alterations from primary to circulating tumor cells. Genome Res 27: 1312–1322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatenby RA, Brown JS (2020) Integrating evolutionary dynamics into cancer therapy. Nat Rev Clin Oncol 17: 675–686 [DOI] [PubMed] [Google Scholar]
- Gatenby RA, Silva AS, Gillies RJ, Frieden BR (2009) Adaptive therapy. Cancer Res 69: 4894–4903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatenby R, Vincent T (2008) An evolutionary model for initiation, promotion, and progression in carcinogenesis. Int J Oncol 32: 729–737 [PubMed] [Google Scholar]
- Gerlinger M, Horswell S, Larkin J, Rowan AJ, Salm MP, Varela I, Fisher R, McGranahan N, Matthews N, Santos CRet al (2014a) Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat Genet 46: 225–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlinger M, McGranahan N, Dewhurst SM, Burrell RA, Tomlinson I, Swanton C (2014b) Cancer: evolution within a lifetime. Annu Rev Genet 48: 215–236 [DOI] [PubMed] [Google Scholar]
- Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey Pet al (2012) Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med 366: 883–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlinger M, Swanton C (2010) How Darwinian models inform therapeutic failure initiated by clonal heterogeneity in cancer medicine. Br J Cancer 103: 1139–1143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstung M, Jolly C, Leshchiner I, Dentro SC, Gonzalez S, Rosebrock D, Mitchell TJ, Rubanova Y, Anur P, Yu Ket al (2020) The evolutionary history of 2,658 cancers. Nature 578: 122–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillies RJ, Verduzco D, Gatenby RA (2012) Evolutionary dynamics of carcinogenesis and why targeted therapy does not work. Nat Rev Cancer 12: 487–493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gisselsson D, Pettersson L, Höglund M, Heidenblad M, Gorunova L, Wiegant J, Mertens F, Dal Cin P, Mitelman F, Mandahl N (2000) Chromosomal breakage‐fusion‐bridge events cause genetic intratumor heterogeneity. Proc Natl Acad Sci U S A 97: 5357–5362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldschmidt R (1941) The material basis of evolution. Philos Sci 8: 394 [Google Scholar]
- Grasso CS, Wu Y‐M, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, Quist MJ, Jing X, Lonigro RJ, Brenner JCet al (2012) The mutational landscape of lethal castration‐resistant prostate cancer. Nature 487: 239–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greaves M, Maley CC (2012) Clonal evolution in cancer. Nature 481: 306–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu X, Yu J, Chai P, Ge S, Fan X (2020) Novel insights into extrachromosomal DNA: redefining the onco‐drivers of tumor progression. J Exp Clin Cancer Res 39: 215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundem G, Van Loo P, Kremeyer B, Alexandrov LB, Tubio JMC, Papaemmanuil E, Brewer DS, Kallio HML, Högnäs G, Annala Met al (2015) The evolutionary history of lethal metastatic prostate cancer. Nature 520: 353–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunnarsson EB, De S, Leder K, Foo J (2020) Understanding the role of phenotypic switching in cancer drug resistance. J Theor Biol 490: 110162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutmann DH, Ferner RE, Listernick RH, Korf BR, Wolters PL, Johnson KJ (2017) Neurofibromatosis type 1. Nat Rev Dis Prim 3: 17004 [DOI] [PubMed] [Google Scholar]
- Haffner MC, De Marzo AM, Yegnasubramanian S, Epstein JI, Ballentine Carter H (2015) Diagnostic challenges of clonal heterogeneity in prostate cancer. J Clin Oncol 33: e38–e40 [DOI] [PubMed] [Google Scholar]
- Hata AN, Niederst MJ, Archibald HL, Gomez‐Caraballo M, Siddiqui FM, Mulvey HE, Maruvka YE, Ji F, Bhang H‐E, Krishnamurthy Radhakrishna Vet al (2016) Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat Med 22: 262–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkes N (2019) Cancer survival data emphasise importance of early diagnosis. BMJ 364: l408 [DOI] [PubMed] [Google Scholar]
- Heppner GH (1984) Tumor heterogeneity. Cancer Res 44: 2259–2265 [PubMed] [Google Scholar]
- Hiley C, de Bruin EC, McGranahan N, Swanton C (2014) Deciphering intratumor heterogeneity and temporal acquisition of driver events to refine precision medicine. Genome Biol 15: 453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirata E, Girotti MR, Viros A, Hooper S, Spencer‐Dene B, Matsuda M, Larkin J, Marais R, Sahai E (2015) Intravital imaging reveals how BRAF inhibition generates drug‐tolerant microenvironments with high integrin β1/FAK Signaling. Cancer Cell 27: 574–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoadley KA, Siegel MB, Kanchi KL, Miller CA, Ding Li, Zhao W, He X, Parker JS, Wendl MC, Fulton RSet al (2016) Tumor evolution in two patients with basal‐like breast cancer: a retrospective genomics study of multiple metastases. PLoS Medicine 13: e1002174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huminiecki L, Conant GC (2012) Polyploidy and the evolution of complex traits. Int J Evol Biol 2012: 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain M, Tangen CM, Berry DL, Higano CS, Crawford ED, Liu G, Wilding G, Prescott S, Kanaga Sundaram S, Small EJet al (2013) Intermittent versus continuous androgen deprivation in prostate cancer. N Engl J Med 368: 1314–1325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacobuzio‐Donahue CA, Litchfield K, Swanton C (2020) Intratumor heterogeneity reflects clinical disease course. Nat Cancer 1: 3–6 [DOI] [PubMed] [Google Scholar]
- Ignatiadis M, Sledge GW, Jeffrey SS (2021) Liquid biopsy enters the clinic — implementation issues and future challenges. Nat Rev Clin Oncol 18: 297–312 [DOI] [PubMed] [Google Scholar]
- Jamal‐Hanjani M, Quezada SA, Larkin J, Swanton C (2015) Translational implications of tumor heterogeneity. Clin Cancer Res 21: 1258–1266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamal‐Hanjani M, Wilson GA, McGranahan N, Birkbak NJ, Watkins TBK, Veeriah S, Shafi S, Johnson DH, Mitter R, Rosenthal Ret al (2017) Tracking the evolution of non–small‐cell lung cancer. N Engl J Med 376: 2109–2121 [DOI] [PubMed] [Google Scholar]
- Joosse SA, Souche F‐R, Babayan A, Gasch C, Kerkhoven RM, Ramos J, Fabre J‐M, Riethdorf S, König A, Wikman Het al (2018) Chromosomal aberrations associated with sequential steps of the metastatic cascade in colorectal cancer patients. Clin Chem 64: 1505–1512 [DOI] [PubMed] [Google Scholar]
- Kafle P, Peller P, Massolo A, Hoberg E, Leclerc LM, Tomaselli M, Kutz S (2020) Range expansion of muskox lungworms track rapid arctic warming: implications for geographic colonization under climate forcing. Sci Rep 10: 17323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanu N, Cerone MA, Goh G, Zalmas L‐P, Bartkova J, Dietzen M, McGranahan N, Rogers R, Law EK, Gromova Iet al (2016) DNA replication stress mediates APOBEC3 family mutagenesis in breast cancer. Genome Biol 17: 1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemper K, De Goeje PL, Peeper DS, Van Amerongen R (2014) Phenotype switching: Tumor cell plasticity as a resistance mechanism and target for therapy. Cancer Res 74: 5937–5941 [DOI] [PubMed] [Google Scholar]
- Kemper K, Krijgsman O, Cornelissen‐Steijger P, Shahrabi A, Weeber F, Song J‐Y, Kuilman T, Vis DJ, Wessels LF, Voest EEet al (2015) Intra‐ and inter‐tumor heterogeneity in a vemurafenib‐resistant melanoma patient and derived xenografts. EMBO Mol Med 7: 1104–1118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kepp O, Bezu L, Yamazaki T, Di Virgilio F, Smyth MJ, Kroemer G, Galluzzi L (2021) ATP and cancer immunosurveillance. EMBO J 10.15252/embj.2021108130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan KH, Cunningham D, Werner B, Vlachogiannis G, Spiteri I, Heide T, Mateos JF, Vatsiou A, Lampis A, Damavandi MDet al (2018) Longitudinal liquid biopsy and mathematical modeling of clonal evolution forecast time to treatment failure in the PROSPECT‐C Phase II colorectal cancer clinical trial. Cancer Discov 8: 1270–1285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Nguyen N‐P, Turner K, Wu S, Gujar AD, Luebeck J, Liu J, Deshpande V, Rajkumar U, Namburi Set al (2020) Extrachromosomal DNA is associated with oncogene amplification and poor outcome across multiple cancers. Nat Genet 52: 891–897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim IS, Heilmann S, Kansler ER, Zhang Y, Zimmer M, Ratnakumar K, Bowman RL, Simon‐Vermot T, Fennell M, Garippa Ret al (2017) Microenvironment‐derived factors driving metastatic plasticity in melanoma. Nat Commun 8: 14343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M (1983) The neutral theory of molecular evolution. Cambridge: Cambridge University Press; [Google Scholar]
- Kloosterman WP, Hoogstraat M, Paling O, Tavakoli‐Yaraki M, Renkens I, Vermaat JS, van Roosmalen MJ, van Lieshout S, Nijman IJ, Roessingh Wet al (2011) Chromothripsis is a common mechanism driving genomic rearrangements in primary and metastatic colorectal cancer. Genome Biol 12: R103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koche RP, Rodriguez‐Fos E, Helmsauer K, Burkert M, MacArthur IC, Maag J, Chamorro R, Munoz‐Perez N, Puiggròs M, Dorado Garcia Het al (2020) Extrachromosomal circular DNA drives oncogenic genome remodeling in neuroblastoma. Nat Genet 52: 29–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohl NE, Kanda N, Schreck RR, Bruns G, Latt SA, Gilbert F, Alt FW (1983) Transposition and amplification of oncogene‐related sequences in human neuroblastomas. Cell 35: 359–367 [DOI] [PubMed] [Google Scholar]
- Korbel JO, Campbell PJ (2013) Criteria for inference of chromothripsis in cancer genomes. Cell 152: 1226–1236 [DOI] [PubMed] [Google Scholar]
- Krook MA, Bonneville R, Chen HZ, Reeser JW, Wing MR, Martin DM, Smith AM, Dao T, Samorodnitsky E, Paruchuri Aet al (2019a) Tumor heterogeneity and acquired drug resistance in FGFR2‐fusion‐positive cholangiocarcinoma through rapid research autopsy. Cold Spring Harb Mol Case Stud 5: a004002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krook MA, Chen HZ, Bonneville R, Allenby P, Roychowdhury S (2019b) Rapid research autopsy: piecing the puzzle of tumor heterogeneity. Trends Cancer 5: 1–5 [DOI] [PubMed] [Google Scholar]
- Kutz SJ, Checkley S, Verocai GG, Dumond M, Hoberg EP, Peacock R, Wu JP, Orsel K, Seegers K, Warren ALet al (2013) Invasion, establishment, and range expansion of two parasitic nematodes in the canadian arctic. Glob Chang Biol 19: 3254–3262 [DOI] [PubMed] [Google Scholar]
- La Manno G, Soldatov R, Zeisel A, Braun E, Hochgerner H, Petukhov V, Lidschreiber K, Kastriti ME, Lönnerberg P, Furlan Aet al (2018) RNA velocity of single cells. Nature 560: 494–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laconi E, Marongiu F, DeGregori J (2020) Cancer as a disease of old age: changing mutational and microenvironmental landscapes. Br J Cancer 122: 943–952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasken RS (2013) Single‐cell sequencing in its prime. Nat Biotechnol 31: 211–212 [DOI] [PubMed] [Google Scholar]
- Lee JH, Saw RP, Thompson JF, Lo S, Spillane AJ, Shannon KF, Stretch JR, Howle J, Menzies AM, Carlino MSet al (2019) Pre‐operative ctDNA predicts survival in high‐risk stage III cutaneous melanoma patients. Ann Oncol 30: 815–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin WW, Karin M (2007) A cytokine‐mediated link between innate immunity, inflammation, and cancer. J Clin Invest 117: 1175–1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling S, Hu Z, Yang Z, Yang F, Li Y, Lin P, Chen Ke, Dong L, Cao L, Tao Yet al (2015) Extremely high genetic diversity in a single tumor points to prevalence of non‐Darwinian cell evolution. Proc Natl Acad Sci U S A 112: E6496–E6505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litchfield K, Stanislaw S, Spain L, Gallegos LL, Rowan A, Schnidrig D, Rosenbaum H, Harle A, Au L, Hill SMet al (2020) Representative sequencing: unbiased sampling of solid tumor tissue. Cell Rep 31: 107550 [DOI] [PubMed] [Google Scholar]
- Liu Mc, Oxnard Gr, Klein Ea, Swanton C, Seiden Mv, Liu MC, Oxnard GR, Klein EA, Smith D, Richards Det al (2020) Sensitive and specific multi‐cancer detection and localization using methylation signatures in cell‐free DNA. Ann Oncol 31: 745–759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- López S, Lim EL, Horswell S, Haase K, Huebner A, Dietzen M, Mourikis TP, Watkins TBK, Rowan A, Dewhurst SMet al (2020) Interplay between whole‐genome doubling and the accumulation of deleterious alterations in cancer evolution. Nat Genet 52: 283–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lote H, Spiteri I, Ermini L, Vatsiou A, Roy A, McDonald A, Maka N, Balsitis M, Bose N, Simbolo Met al (2017) Carbon dating cancer: Defining the chronology of metastatic progression in colorectal cancer. Ann Oncol 28: 1243–1249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luria SE, Delbrück M (1943) Mutations of bacteria from virus sensitivity to virus resistance. Genetics 28: 491–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luskin MR, Murakami MA, Manalis SR, Weinstock DM (2018) Targeting minimal residual disease: a path to cure? Nat Rev Cancer 18: 255–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machnik M, Oleksiewicz U (2020) Dynamic signatures of the epigenome: friend or foe? Cells 9: 653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makohon‐Moore AP, Zhang M, Reiter JG, Bozic I, Allen B, Kundu D, Chatterjee K, Wong F, Jiao Y, Kohutek ZAet al (2017) Limited heterogeneity of known driver gene mutations among the metastases of individual patients with pancreatic cancer. Nat Genet 49: 358–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maley CC, Galipeau PC, Finley JC, Wongsurawat VJ, Li X, Sanchez CA, Paulson TG, Blount PL, Risques R‐A, Rabinovitch PSet al (2006) Genetic clonal diversity predicts progression to esophageal adenocarcinoma. Nat Genet 38: 468–473 [DOI] [PubMed] [Google Scholar]
- Maley CC, Reid BJ, Forrest S (2004) Cancer prevention strategies that address the evolutionary dynamics of neoplastic cells: Simulating benign cell boosters and selection for chemosensitivity. Cancer Epidemiol Biomarkers Prev 13: 1375–1384 [PubMed] [Google Scholar]
- Malhotra A, Lindberg M, Faust GG, Leibowitz ML, Clark RA, Layer RM, Quinlan AR, Hall IM (2013) Breakpoint profiling of 64 cancer genomes reveals numerous complex rearrangements spawned by homology‐independent mechanisms. Genome Res 23: 762–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malikic S, Jahn K, Kuipers J, Sahinalp SC, Beerenwinkel N (2019) Integrative inference of subclonal tumour evolution from single‐cell and bulk sequencing data. Nat Commun 10: 2750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malkin D (2011) Li‐Fraumeni syndrome. Genes Cancer 2: 475–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallory XF, Edrisi M, Navin N, Nakhleh L (2020) Methods for copy number aberration detection from single‐cell DNA‐sequencing data. Genome Biol 21: 208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardin BR, Drainas AP, Waszak SM, Weischenfeldt J, Isokane M, Stütz AM, Raeder B, Efthymiopoulos T, Buccitelli C, Segura‐Wang Met al (2015) A cell‐based model system links chromothripsis with hyperploidy. Mol Syst Biol 11: 828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin‐Bejar O, Rogiers A, Dewaele M, Femel J, Karras P, Pozniak J, Bervoets G, Van Raemdonck N, Pedri D, Swings Tet al (2021) Evolutionary predictability of genetic versus nongenetic resistance to anticancer drugs in melanoma. Cancer Cell 10.1016/j.ccell.2021.05.015 [DOI] [PubMed] [Google Scholar]
- Marine JC, Dawson SJ, Dawson MA (2020) Non‐genetic mechanisms of therapeutic resistance in cancer. Nat Rev Cancer 20: 743–756 [DOI] [PubMed] [Google Scholar]
- Martincorena I, Fowler JC, Wabik A, Lawson ARJ, Abascal F, Hall MWJ, Cagan A, Murai K, Mahbubani K, Stratton MRet al (2018) Somatic mutant clones colonize the human esophagus with age. Science 362: 911–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martincorena I, Roshan A, Gerstung M, Ellis P, Van Loo P, McLaren S, Wedge DC, Fullam A, Alexandrov LB, Tubio JMet al (2015) High burden and pervasive positive selection of somatic mutations in normal human skin. Science 348: 880–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marusyk A, Tabassum DP, Altrock PM, Almendro V, Michor F, Polyak K (2014) Non‐cell‐autonomous driving of tumour growth supports sub‐clonal heterogeneity. Nature 514: 54–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayekar MK, Caswell DR, Vokes C‐DNI, Law E‐HEK, Wu W, Hill W, Gronroos E, Rowan A, Al Bakir M, Mccoach CEet al (2020) Targeted cancer therapy induces APOBEC fuelling the evolution of drug resistance. bioRxiv 10.1101/2020.12.18.423280 [PREPRINT] [DOI] [Google Scholar]
- McGranahan N, Burrell RA, Endesfelder D, Novelli MR, Swanton C (2012) Cancer chromosomal instability: therapeutic and diagnostic challenges. EMBO Rep 13: 528–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGranahan N, Swanton C (2015) Biological and therapeutic impact of intratumor heterogeneity in cancer evolution. Cancer Cell 27: 15–26 [DOI] [PubMed] [Google Scholar]
- Merlo LMF, Pepper JW, Reid BJ, Maley CC (2006) Cancer as an evolutionary and ecological process. Nat Rev Cancer 6: 924–935 [DOI] [PubMed] [Google Scholar]
- Michor F, Iwasa Y, Nowak MA (2004) Dynamics of cancer progression. Nat Rev Cancer 4: 197–205 [DOI] [PubMed] [Google Scholar]
- Milan M, Diaferia GR, Natoli G (2021) Tumor cell heterogeneity and its transcriptional bases in pancreatic cancer: a tale of two cell types and their many variants. EMBO J 10.15252/embj.2020107206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills JC, Stanger BZ, Sander M (2019) Nomenclature for cellular plasticity: are the terms as plastic as the cells themselves? EMBO J 38: e103148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misale S, Yaeger R, Hobor S, Scala E, Janakiraman M, Liska D, Valtorta E, Schiavo R, Buscarino M, Siravegna Get al (2012) Emergence of KRAS mutations and acquired resistance to anti‐EGFR therapy in colorectal cancer. Nature 486: 532–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell TJ, Turajlic S, Rowan A, Nicol D, Farmery JHR, O’Brien T, Martincorena I, Tarpey P, Angelopoulos N, Yates LRet al (2018) Timing the landmark events in the evolution of clear cell renal cell cancer: TRACERx renal. Cell 173: 611–623.e17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molenaar JJ, Koster J, Zwijnenburg DA, van Sluis P, Valentijn LJ, van der Ploeg I, Hamdi M, van Nes J, Westerman BA, van Arkel Jet al (2012) Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature 483: 589–593 [DOI] [PubMed] [Google Scholar]
- Morgan GJ (1998) Emile zuckerkandl, linus pauling, and the molecular evolutionary clock, 1959–1965. J Hist Biol 31: 155–178 [DOI] [PubMed] [Google Scholar]
- Mounier A, Georgiev D, Nam KN, Fitz NF, Castranio EL, Wolfe CM, Cronican AA, Schug J, Lefterov I, Koldamova R (2015) Bexarotene‐activated retinoid X receptors regulate neuronal differentiation and dendritic complexity. J Neurosci 35: 11862–11876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller J (1973) On the nature and structural characteristics of cancer: general observations on the minute structure of morbid growths. CA Cancer J Clin 23: 307–312 [Google Scholar]
- Nam AS, Chaligne R, Landau DA (2021) Integrating genetic and non‐genetic determinants of cancer evolution by single‐cell multi‐omics. Nat Rev Genet 22: 3–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathanson DA, Gini B, Mottahedeh J, Visnyei K, Koga T, Gomez G, Eskin A, Hwang K, Wang J, Masui Ket al (2014) Targeted therapy resistance mediated by dynamic regulation of extrachromosomal mutant EGFR DNA. Science 343: 72–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navin NE (2014) Cancer genomics: one cell at a time. Genome Biol 15: 452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navin NE (2015) The first five years of single‐cell cancer genomics and beyond. Genome Res 25: 1499–1507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navin N, Kendall J, Troge J, Andrews P, Rodgers L, McIndoo J, Cook K, Stepansky A, Levy D, Esposito Det al (2011) Tumour evolution inferred by single‐cell sequencing. Nature 472: 90–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navin N, Krasnitz A, Rodgers L, Cook K, Meth J, Kendall J, Riggs M, Eberling Y, Troge J, Grubor Vet al (2010) Inferring tumor progression from genomic heterogeneity. Genome Res 20: 68–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naxerova K, Reiter JG, Brachtel E, Lennerz JK, van de Wetering M, Rowan A, Cai T, Clevers H, Swanton C, Nowak MAet al (2017) Origins of lymphatic and distant metastases in human colorectal cancer. Science 357: 357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen DX, Bos PD, Massagué J (2009) Metastasis: from dissemination to organ‐specific colonization. Nat Rev Cancer 9: 274–284 [DOI] [PubMed] [Google Scholar]
- Nieto MA, Huang RYYJ, Jackson RAA, Thiery JPP (2016) Emt: 2016. Cell 166: 21–45 [DOI] [PubMed] [Google Scholar]
- Nik‐Zainal S, Alexandrov LB, Wedge DC, Van Loo P, Greenman CD, Raine K, Jones D, Hinton J, Marshall J, Stebbings LAet al (2012a) Mutational processes molding the genomes of 21 breast cancers. Cell 149: 979–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nik‐Zainal S, Van Loo P, Wedge DC, Alexandrov LB, Greenman CD, Lau KW, Raine K, Jones D, Marshall J, Ramakrishna Met al (2012b) The life history of 21 breast cancers. Cell 149: 994–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notta F, Chan‐Seng‐Yue M, Lemire M, Li Y, Wilson GW, Connor AA, Denroche RE, Liang S‐B, Brown AMK, Kim JCet al (2016) A renewed model of pancreatic cancer evolution based on genomic rearrangement patterns. Nature 538: 378–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowell PC (1976) The clonal evolution of tumor cell populations. Science 194: 23–28 [DOI] [PubMed] [Google Scholar]
- Oobatake Y, Shimizu N (2020) Double‐strand breakage in the extrachromosomal double minutes triggers their aggregation in the nucleus, micronucleation, and morphological transformation. Genes Chromosom Cancer 59: 133–143 [DOI] [PubMed] [Google Scholar]
- Pantel K, Alix‐Panabières C (2019) Liquid biopsy and minimal residual disease — latest advances and implications for cure. Nat Rev Clin Oncol 16: 409–424 [DOI] [PubMed] [Google Scholar]
- Parker TM, Gupta K, Palma AM, Yekelchyk M, Fisher PB, Grossman SR, Won KJ, Madan E, Moreno E, Gogna R (2021) Cell competition in intratumoral and tumor microenvironment interactions. EMBO J 10.15252/embj.2020107271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT, Martuza RLet al (2014) Single‐cell RNA‐seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344: 1396–1401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepper JW, Findlay CS, Kassen R, Spencer SL, Maley CC (2009) Cancer research meets evolutionary biology. Evol Appl 2: 62–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisapia DJ, Salvatore S, Pauli C, Hissong E, Eng K, Prandi D, Sailer V‐W, Robinson BD, Park K, Cyrta Jet al (2017) Next‐generation rapid autopsies enable tumor evolution tracking and generation of preclinical models. JCO Precis Oncol 2017: 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pluchino KM, Hall MD, Goldsborough AS, Callaghan R, Gottesman MM (2012) Collateral sensitivity as a strategy against cancer multidrug resistance. Drug Resist Updat 15: 98–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pogrebniak KL, Curtis C (2018) Harnessing tumor evolution to circumvent resistance. Trends Genet 34: 639–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powles T, Assaf ZJ, Davarpanah N, Banchereau R, Szabados BE, Yuen KC, Grivas P, Hussain M, Oudard S, Gschwend JEet al (2021) ctDNA guiding adjuvant immunotherapy in urothelial carcinoma. Nature 595: 432–437 [DOI] [PubMed] [Google Scholar]
- Pugh TJ, Delaney AD, Farnoud N, Flibotte S, Griffith M, Li HI, Qian H, Farinha P, Gascoyne RD, Marra MA (2008) Impact of whole genome amplification on analysis of copy number variants. Nucleic Acids Res 36: e80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purushotham AD, Sullivan R (2010) Darwin, medicine and cancer. Ann Oncol 21: 199–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramalingam N, Jeffrey SS (2018) Future of liquid biopsies with growing technological and bioinformatics studies: opportunities and challenges in discovering tumor heterogeneity with single‐cell level analysis. Cancer J 24: 104–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambow F, Marine JC, Goding CR (2019) Melanoma plasticity and phenotypic diversity: therapeutic barriers and opportunities. Genes Dev 33: 1295–1318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambow F, Rogiers A, Marin‐Bejar O, Aibar S, Femel J, Dewaele M, Karras P, Brown D, Chang YH, Debiec‐Rychter Met al (2018) Toward minimal residual disease‐directed therapy in melanoma. Cell 174: 843–855.e19 [DOI] [PubMed] [Google Scholar]
- Ramón y Cajal S, Sesé M, Capdevila C, Aasen T, De Mattos‐Arruda L, Diaz‐Cano SJ, Hernández‐Losa J, Castellví J (2020) Clinical implications of intratumor heterogeneity: challenges and opportunities. J Mol Med 98: 161–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razavi P, Chang MT, Xu G, Bandlamudi C, Ross DS, Vasan N, Cai Y, Bielski CM, Donoghue MTA, Jonsson Pet al (2018) The genomic landscape of endocrine‐resistant advanced breast cancers. Cancer Cell 34: 427–438.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter JG, Baretti M, Gerold JM, Makohon‐Moore AP, Daud A, Iacobuzio‐Donahue CA, Azad NS, Kinzler KW, Nowak MA, Vogelstein B (2019) An analysis of genetic heterogeneity in untreated cancers. Nat Rev Cancer 19: 639–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter JG, Makohon‐Moore AP, Gerold JM, Heyde A, Attiyeh MA, Kohutek ZA, Tokheim CJ, Brown A, DeBlasio RM, Niyazov Jet al (2018) Minimal functional driver gene heterogeneity among untreated metastases. Science 361: 1033–1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riva L, Pandiri AR, Li YR, Droop A, Hewinson J, Quail MA, Iyer V, Shepherd R, Herbert RA, Campbell PJet al (2020) The mutational signature profile of known and suspected human carcinogens in mice. Nat Genet 52: 1189–1197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal R, Cadieux EL, Salgado R, Bakir MA, Moore DA, Hiley CT, Lund T, Tanić M, Reading JL, Joshi Ket al (2019) Neoantigen‐directed immune escape in lung cancer evolution. Nature 567: 479–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi G, Ignatiadis M (2019) Promises and pitfalls of using liquid biopsy for precision medicine. Cancer Res 79: 2798–2804 [DOI] [PubMed] [Google Scholar]
- Rozhok A, De Gregori J (2019) A generalized theory of age‐dependent carcinogenesis. Elife 8: e39950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo M, Crisafulli G, Sogari A, Reilly NM, Arena S, Lamba S, Bartolini A, Amodio V, Magrì A, Novara Let al (2019) Adaptive mutability of colorectal cancers in response to targeted therapies. Science 366: 1473–1480 [DOI] [PubMed] [Google Scholar]
- Salehi S, Steif A, Roth A, Aparicio S, Bouchard‐Côté A, Shah SP (2017) ddClone: joint statistical inference of clonal populations from single cell and bulk tumour sequencing data. Genome Biol 18: 44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez‐Vega F, Hechtman JF, Castel P, Ku GY, Tuvy Y, Won H, Fong CJ, Bouvier N, Nanjangud GJ, Soong Jet al (2019) Egfr and MET amplifications determine response to HER2 inhibition in ERBB2‐amplified esophagogastric cancer. Cancer Discov 9–2: 199–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scher HI, Heller G, Molina A, Attard G, Danila DC, Jia X, Peng W, Sandhu SK, Olmos D, Riisnaes Ret al (2015) Circulating tumor cell biomarker panel as an individual‐level surrogate for survival in metastatic castration‐resistant prostate cancer. J Clin Oncol 33: 1348–1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz R, Schäffer AA (2017) The evolution of tumour phylogenetics: principles and practice. Nat Rev Genet 18: 213–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweizer MT, Antonarakis ES, Wang H, Ajiboye AS, Spitz A, Cao H, Luo J, Haffner MC, Yegnasubramanian S, Carducci MAet al (2015) Effect of bipolar androgen therapy for asymptomatic men with castration‐resistant prostate cancer: results from a pilot clinical study. Sci Transl Med 7: 269ra2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaffer SM, Dunagin MC, Torborg SR, Torre EA, Emert B, Krepler C, Beqiri M, Sproesser K, Brafford PA, Xiao Met al (2017) Rare cell variability and drug‐induced reprogramming as a mode of cancer drug resistance. Nature 546: 431–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah NP, Skaggs BJ, Branford S, Hughes TP, Nicoll JM, Paquette RL, Sawyers CL (2007) Sequential ABL kinase inhibitor therapy selects for compound drug‐resistant BCR‐ABL mutations with altered oncogenic potency. J Clin Invest 117: 2562–2569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah SP, Morin RD, Khattra J, Prentice L, Pugh T, Burleigh A, Delaney A, Gelmon K, Guliany R, Senz Jet al (2009) Mutational evolution in a lobular breast tumour profiled at single nucleotide resolution. Nature 461: 809–813 [DOI] [PubMed] [Google Scholar]
- Shapiro JR, Yung WKA, Shapiro WR, Shapiro JR, Shapiro WR (1981) Isolation, karyotype, and clonal growth of heterogeneous subpopulations of human malignant gliomas. Cancer Res 41: 2349–2359 [PubMed] [Google Scholar]
- Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S, McDermott U, Azizian N, Zou L, Fischbach MAet al (2010) A chromatin‐mediated reversible drug‐tolerant state in cancer cell subpopulations. Cell 141: 69–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H, Hugo W, Kong X, Hong A, Koya RC, Moriceau G, Chodon T, Guo R, Johnson DB, Dahlman KBet al (2014) Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov 4: 80–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu N, Itoh N, Utiyama H, Wahl GM (1998) Selective entrapment of extrachromosomally amplified DNA by nuclear budding and micronucleation during S phase. J Cell Biol 140: 1307–1320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel MB, He X, Hoadley KA, Hoyle A, Pearce JB, Garrett AL, Kumar S, Moylan VJ, Brady CM, Van Swearingen AEDet al (2018a) Integrated RNA and DNA sequencing reveals early drivers of metastatic breast cancer. J Clin Invest 128: 1371–1383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, Jemal A (2017) Cancer statistics, 2017. CA Cancer J Clin 67: 7–30 [DOI] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, Jemal A (2018b) Cancer statistics, 2018. CA Cancer J Clin 68: 7–30 [DOI] [PubMed] [Google Scholar]
- Simonsen AT, Hansen MC, Kjeldsen E, Møller PL, Hindkjær JJ, Hokland P, Aggerholm A (2018) Systematic evaluation of signal‐to‐noise ratio in variant detection from single cell genome multiple displacement amplification and exome sequencing. BMC Genom 19: 681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siravegna G, Mussolin B, Buscarino M, Corti G, Cassingena A, Crisafulli G, Ponzetti A, Cremolini C, Amatu A, Lauricella Cet al (2015) Erratum: clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat Med 21: 827 [DOI] [PubMed] [Google Scholar]
- Smith JC, Sheltzer JM (2018) Systematic identification of mutations and copy number alterations associated with cancer patient prognosis. Elife 7: e39217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MP, Brunton H, Rowling EJ, Ferguson J, Arozarena I, Miskolczi Z, Lee JL, Girotti MR, Marais R, Levesque MPet al (2016) Inhibiting drivers of non‐mutational drug tolerance is a salvage strategy for targeted melanoma therapy. Cancer Cell 29: 270–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sottoriva A, Kang H, Ma Z, Graham TA, Salomon MP, Zhao J, Marjoram P, Siegmund K, Press MF, Shibata Det al (2015) A big bang model of human colorectal tumor growth. Nat Genet 47: 209–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, Pleasance ED, Lau KW, Beare D, Stebbings LAet al (2011) Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 144: 27–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storchova Z, Pellman D (2004) From polyploidy to aneuploidy, genome instability and cancer. Nat Rev Mol Cell Biol 5: 45–54 [DOI] [PubMed] [Google Scholar]
- Strobl MAR, West J, Viossat Y, Damaghi M, Robertson‐Tessi M, Brown JS, Gatenby RA, Maini PK, Anderson ARA (2021) Turnover modulates the need for a cost of resistance in adaptive therapy. Cancer Res 81: 1135–1147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanton C (2012) Intratumor heterogeneity: evolution through space and time. Cancer Res 72: 4875–4882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, Habuchi T, Kakehi Y, Mitsumori K, Akao T, Terachi T, Yoshida O (1998) Clonal and chronological genetic analysis of multifocal cancers of the bladder and upper urinary tract. Cancer Res 58: 5835–5841 [PubMed] [Google Scholar]
- Tarabichi M, Martincorena I, Gerstung M, Leroi AM, Markowetz F, Dentro SC, Leshchiner I, Jolly C, Haase K, Wintersinger Jet al (2018) Neutral tumor evolution? Nat Genet 50: 1630–1633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira MR, Pandis N, Bardi G, Andersen JA, Heim S (1996) Karyotypic comparisons of multiple tumorous and macroscopically normal surrounding tissue samples from patients with breast cancer. Cancer Res 56: 855–859 [PubMed] [Google Scholar]
- Teixeira VH, Pipinikas CP, Pennycuick A, Lee‐Six H, Chandrasekharan D, Beane J, Morris TJ, Karpathakis A, Feber A, Breeze CEet al (2019) Deciphering the genomic, epigenomic, and transcriptomic landscapes of pre‐invasive lung cancer lesions. Nat Med 25: 517–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tie J, Wang Y, Tomasetti C, Li L, Springer S, Kinde I, Silliman N, Tacey M, Wong HL, Christie Met al (2016) Circulating tumor DNA analysis detects minimal residual disease and predicts recurrence in patients with stage II colon cancer. Sci Transl Med 8: 346ra92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tung NM, Garber JE (2018) BRCA1/2 testing: therapeutic implications for breast cancer management. Br J Cancer 119: 141–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turajlic S, Xu H, Litchfield K, Rowan A, Horswell S, Chambers T, O’Brien T, Lopez JI, Watkins TBK, Nicol Det al (2018) Deterministic evolutionary trajectories influence primary tumor growth: TRACERx Renal. Cell 173: 595–610.e11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner KM, Deshpande V, Beyter D, Koga T, Rusert J, Lee C, Li B, Arden K, Ren B, Nathanson DAet al (2017) Extrachromosomal oncogene amplification drives tumour evolution and genetic heterogeneity. Nature 543: 122–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umbreit NT, Zhang C‐Z, Lynch LD, Blaine LJ, Cheng AM, Tourdot R, Sun L, Almubarak HF, Judge K, Mitchell TJet al (2020) Mechanisms generating cancer genome complexity from a single cell division error. Science 368: eaba0712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Loo P, Voet T (2014) Single cell analysis of cancer genomes. Curr Opin Genet Dev 24: 82–91 [DOI] [PubMed] [Google Scholar]
- Vendramin R, Katopodi V, Cinque S, Konnova A, Knezevic Z, Adnane S, Verheyden Y, Karras P, Demesmaeker E, Bosisio FMet al (2021) Activation of the integrated stress response confers vulnerability to mitoribosome‐targeting antibiotics in melanoma. J Exp Med 10.1084/jem.20210571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesan S, Angelova M, Puttick C, Zhai H, Caswell DR, Lu W‐T, Dietzen M, Galanos P, Evangelou K, Bellelli Ret al (2021) Induction of APOBEC3 exacerbates DNA replication stress and chromosomal instability in early breast and lung cancer evolution. Cancer Discov candisc.0725.2020. 10.1158/2159-8290.cd-20-0725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhaak RGW, Bafna V, Mischel PS (2019) Extrachromosomal oncogene amplification in tumour pathogenesis and evolution. Nat Rev Cancer 19: 283–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal J, Taus A, Montagut C (2020) Dynamic treatment stratification using ctDNA. Recent Results Cancer Res 215: 263–273 [DOI] [PubMed] [Google Scholar]
- Waldman AD, Fritz JM, Lenardo MJ (2020) A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol 20: 651–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter MJ, Shen D, Ding Li, Shao J, Koboldt DC, Chen K, Larson DE, McLellan MD, Dooling D, Abbott Ret al (2012) Clonal architecture of secondary acute myeloid leukemia. N Engl J Med 366: 1090–1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Waters J, Leung ML, Unruh A, Roh W, Shi X, Chen K, Scheet P, Vattathil S, Liang Het al (2014) Clonal evolution in breast cancer revealed by single nucleus genome sequencing. Nature 512: 155–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- West JB, Dinh MN, Brown JS, Zhang J, Anderson AR, Gatenby RA (2019) Multidrug cancer therapy in metastatic castrate‐resistant prostate cancer: an evolution‐based strategy. Clin Cancer Res 25: 4413–4421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams MJ, Werner B, Barnes CP, Graham TA, Sottoriva A (2016) Identification of neutral tumor evolution across cancer types. Nat Genet 48: 238–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winton T, Livingston R, Johnson D, Rigas J, Johnston M, Butts C, Cormier Y, Goss G, Inculet R, Vallieres Eet al (2005) Vinorelbine plus cisplatin vs. observation in resected non–small‐cell lung cancer. N Engl J Med 352: 2589–2597 [DOI] [PubMed] [Google Scholar]
- Wright NA (2014) Boveri at 100: cancer evolution, from preneoplasia to malignancy. J Pathol 234: 146–151 [DOI] [PubMed] [Google Scholar]
- Wu CI, Wang HY, Ling S, Lu X (2016) The ecology and evolution of cancer: the ultra‐microevolutionary process. Annu Rev Genet 50: 347–369 [DOI] [PubMed] [Google Scholar]
- Wu S, Turner KM, Nguyen N, Raviram R, Erb M, Santini J, Luebeck J, Rajkumar U, Diao Y, Li Bet al (2019) Circular ecDNA promotes accessible chromatin and high oncogene expression. Nature 575: 699–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wylie AA, Schoepfer J, Jahnke W, Cowan‐Jacob SW, Loo A, Furet P, Marzinzik AL, Pelle X, Donovan J, Zhu Wet al (2017) The allosteric inhibitor ABL001 enables dual targeting of BCR‐ABL1. Nature 543: 733–737 [DOI] [PubMed] [Google Scholar]
- Xie T, Musteanu M, Lopez‐Casas PP, Shields DJ, Olson P, Rejto PA, Hidalgo M (2015) Whole exome sequencing of rapid autopsy tumors and xenograft models reveals possible driver mutations underlying tumor progression. PLoS One 10: e0142631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yachida S, Jones S, Bozic I, Antal T, Leary R, Fu B, Kamiyama M, Hruban RH, Eshleman JR, Nowak MAet al (2010) Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 467: 1114–1117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yizhak K, Aguet F, Kim J, Hess JM, Kübler K, Grimsby J, Frazer R, Zhang H, Haradhvala NJ, Rosebrock Det al (2019) RNA sequence analysis reveals macroscopic somatic clonal expansion across normal tissues. Science 364: eaaw0726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida K, Gowers KHC, Lee‐Six H, Chandrasekharan DP, Coorens T, Maughan EF, Beal K, Menzies A, Millar FR, Anderson Eet al (2020) Tobacco smoking and somatic mutations in human bronchial epithelium. Nature 578: 266–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zack TI, Schumacher SE, Carter SL, Cherniack AD, Saksena G, Tabak B, Lawrence MS, Zhang C‐Z, Wala J, Mermel CHet al (2013) Pan‐cancer patterns of somatic copy number alteration. Nat Genet 45: 1134–1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Sedlak JC, Srinivas R, Creixell P, Pritchard JR, Tidor B, Lauffenburger DA, Hemann MT (2016) Exploiting temporal collateral sensitivity in tumor clonal evolution. Cell 165: 234–246 [DOI] [PMC free article] [PubMed] [Google Scholar]