

Abstract
The process of epithelial–mesenchymal transition (EMT) is fundamental for embryonic morphogenesis. Cells undergoing it lose epithelial characteristics and integrity, acquire mesenchymal features, and become motile. In cancer, this program is hijacked to confer essential changes in morphology and motility that fuel invasion. In addition, EMT is increasingly understood to orchestrate a large variety of complementary cancer features, such as tumor cell stemness, tumorigenicity, resistance to therapy and adaptation to changes in the microenvironment. In this review, we summarize recent findings related to these various classical and non‐classical functions, and introduce EMT as a true tumorigenic multi‐tool, involved in many aspects of cancer. We suggest that therapeutic targeting of the EMT process will—if acknowledging these complexities—be a possibility to concurrently interfere with tumor progression on many levels.
Keywords: EMT, MET, ZEB1, ZEB2, SNAIL, SLUG, TWIST, cancer, invasion, metastasis, signaling pathways, cell plasticity, tumor stemness, hybrid EMT, partial EMT
Subject Categories: Cancer; Cell Adhesion, Polarity & Cytoskeleton; Chromatin, Epigenetics, Genomics & Functional Genomics
This review contrasts classical and non‐classical roles of epithelial‐mesenchymal transition in diverse aspects of cancer cell biology.

Introduction
Epithelial‐to‐mesenchymal transition (EMT) describes the transdifferentiation of stationary epithelial cells to a mesenchymal, motile phenotype and was initially observed in early development (Hay, 1995). Here, EMT contributes to embryonal processes like gastrulation, neural crest formation, or heart development (Thiery et al, 2009; Nieto et al, 2016). The program is also crucial for physiological processes like wound healing (Arnoux et al, 2008) and tissue homeostasis (Ahmed et al, 2006). Importantly, pathological reactivation of the EMT process plays a fundamental role in diseases like organ fibrosis or cancer progression to metastasis (Fig 1A), which is the focus of this review.
Figure 1. Classical EMT functions and cancer.
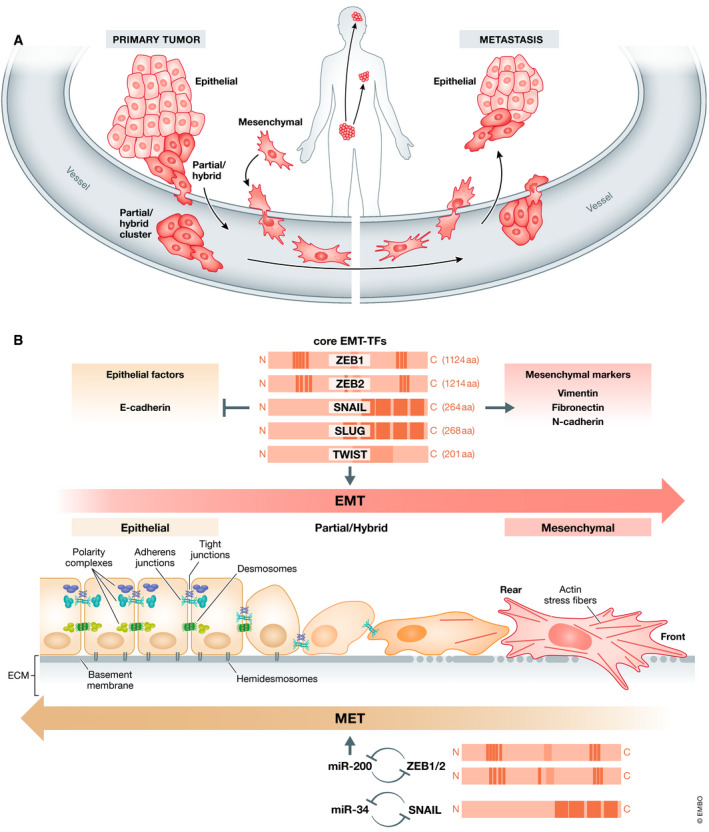
(A) EMT frequently occurs at the invasive front of epithelial tumors, destroys the well‐defined epithelial structures, and allows the cancer cells to migrate, invade the tissue, and intravasate in blood or lymphatic vessels. Tumor cells on their way through the body can travel as mesenchymal single cells, as cell clusters exhibiting partial EMT or as more epithelial cell clusters headed by a mesenchymal leader cell. At the secondary site, the cells extravasate and colonize the distant organ, where MET allows the outgrowth to macrometastases. (B) EMT is induced mainly by a set of transcription factors (EMT‐TFs) like ZEB1, ZEB2, SNAIL, SLUG and TWIST that differ in protein structure, size, and individual functions. All of them are repressors of epithelial factors like E‐cadherin and activate mesenchymal markers like Vimentin, Fibronectin or N‐cadherin. Epithelial cells displaying apical–basal polarity are held together by tight junctions, adherens junctions, and desmosomes and are anchored to the underlying basement membrane by hemidesmosomes. They express three different polarity complexes that together with the junctional molecules maintain epithelial cell polarity. In the classical EMT, expression of EMT‐TFs leads to inhibition of major components of these epithelial structures and concomitantly activates the expression of genes associated with the mesenchymal state. Cells gain front–rear polarity, display actin stress fibers, become motile and acquire invasive capacities. Notably, tumor cells very rarely switch to a completely mesenchymal phenotype, but fluently convert between various intermediate states displaying certain mesenchymal features but keeping partial sets of epithelial characteristics. Further, EMT is a reversible process. Mesenchymal cells can revert to the epithelial state undergoing MET. An important role in the execution of MET is played by microRNAs of the miR‐200 and mir‐34 families that are regulated in double‐negative feedback loops with the EMT‐TFs ZEB1/2 and SNAIL, respectively, that serve to reinforce either the epithelial or the mesenchymal state.
Cancer is an extremely complex and diverse disease not only varying between entities, but also within the same entity, between different subtypes, and even within subtypes. Notably, tumors display not only spatial but also temporal heterogeneity within the same individual that can be elicited, e.g., via the occurrence of consecutive mutations and clonal evolution (McGranahan & Swanton, 2017). However, cancer cell plasticity, allowing continuous and reversible adaption to ever‐changing conditions, is mediated by the EMT process that is not genetically fixed, depending on accumulating mutations, but is epigenetically orchestrated by signals from the microenvironment, rendering the whole program reversible (by activating mesenchymal–epithelial transition; MET) and highly dynamic (see overview in Fig 2).
Figure 2. Dynamic EMT as a multi‐tool for tumor progression.

Overview summarizing the multiple oncogenic EMT functions in the course of tumor progression. The classical EMT functions allow cancer cell to migrate, invade, intra‐ and extravasate blood and lymphatic vessels. At the distant sites, MET enables the outgrowth of macrometastases. The non‐classical EMT traits support tumor initiation as well as metastatic colonization. Throughout the whole process of tumor progression, they help the cells to cope with changing conditions by metabolic reprogramming, enhanced survival via altered DNA repair and prevention of cell death, immune evasion and improved resistance to chemo‐ and radiotherapy. Importantly, EMT is not only supporting the cancer cells to handle changing environmental conditions, but is also induced by extracellular signals from, e.g., CAFs or immune cells in the microenvironment or by therapeutic approaches.
The EMT program is mainly executed by a core set of EMT‐activating transcription factors (EMT‐TFs) including SNAIL (also SNAI1) and SLUG (also SNAI2), the basic helix–loop–helix factors TWIST1 (TWIST) and TWIST2 and the zinc finger E‐Box binding homeobox factors ZEB1 and ZEB2. All these factors share the ability to repress epithelial genes like the E‐cadherin encoding gene CDH1 via binding to E‐Box motifs in their cognate promoter regions (Nieto et al, 2016) as shown for SNAIL (Batlle et al, 2000; Cano et al, 2000), TWIST (Yang et al, 2004), ZEB1 (Eger et al, 2005), and ZEB2 (Comijn et al, 2001). In parallel, the EMT‐TFs directly or indirectly activate genes associated with a mesenchymal phenotype, including VIM (Vimentin), FN1 (Fibronectin), and CDH2 (N‐cadherin) (Nieto et al, 2016; Dongre & Weinberg, 2019) (Fig 1B). However, many functions are not shared, but executed by distinct EMT‐TFs, as they differ in aspects like expression patterns or extremely different protein size and structure (Stemmler et al, 2019).
Beyond the “classical” EMT traits, the gain of motility, and invasive capacities, the widespread importance of EMT‐TFs in cancer biology is indicated by additional pleiotropic functions (Brabletz et al, 2018). EMT‐TFs have been shown to maintain stemness properties and to increase tumorigenicity, linking them to the concept of cancer stem cells (CSCs). Additionally, EMT‐TFs are involved in DNA repair, the escape from senescence and apoptosis, therapy resistance, and immune evasion resulting in a pro‐survival phenotype, providing an advantage under various types of stress conditions.
Altogether, the classical EMT functions together with the very diverse non‐redundant context‐dependent non‐classical functions of EMT‐TFs that are dynamically regulated also by the tumor microenvironment (TME) allow the cancer cells to permanently adapt to changing conditions (Puisieux et al, 2014). Consequently, therapeutic intervention with EMT/plasticity will provide the opportunity to fight many aspects of tumor progression at a single blow.
In this review, we summarize all these different EMT functions in cancer biology and highlight the resulting clinical implications.
Classical/core EMT functions
Migration and invasion
In a normal epithelial tissue, the cells form continuous protective sheets that are crucial for their structural integrity. The connection between epithelial cells is mediated by different cell junction complexes including adherens junction, desmosomes, and tight junctions that seal the cells, are located apical to the adherens junctions to support epithelial polarity and constitute a barrier for solutes and water. This apical‐basal polarity is fundamental for tissue function, and has been defined as “asymmetry” within cells and epithelial tissues. Polarity complexes, including the Par, the Crumbs, and the Scribble complexes, ensure the proper organization of apical versus basolateral domains in the epithelial cell (Huang et al, 2012). Of note, some of the genes that control epithelial cell polarity also regulate spindle orientation and division mode in stem cells (Martin‐Belmonte & Perez‐Moreno, 2011) (Fig 1B).
Elicited by the TME, the activation of EMT is fundamental for the transition toward malignancy, accompanied by substantial cellular changes on multiple levels. Cell–cell contacts become deconstructed through the repression of CDH1, encoding epithelial cadherin (E‐cadherin), the main constituent of adherens junctions, and of genes coding for other epithelial junction molecules.
As a consequence of the disintegration of all epithelial junction complexes and of direct transcriptional repression of several members of the Crumbs and the Scribble complexes, apical–basal polarity is lost (Aigner et al, 2007; Moreno‐Bueno et al, 2008; Spaderna et al, 2008; Lamouille et al, 2014) (Fig 1B). This coincides with profound cytoskeletal reorganization like apical constriction, the formation of actin stress fibers, and the conversion of cell morphology from cuboidal or columnar shapes to more spindle‐like elongated forms with front–rear polarity, resulting in a gain of motility for the tumor cells (Moreno‐Bueno et al, 2008). Newly formed actin‐rich membrane protrusions like lamellipodia and filopodia support cell movement. To invade the surrounding tissues, e.g., TWIST or ZEB1 also induce the formation of invadopodia, specialized filopodia with proteolytic function (Yilmaz & Christofori, 2009; Eckert et al, 2011; Ridley, 2011; Sundararajan et al, 2015). This is supported by induction of matrix metalloproteases by EMT‐TFs, essential for degradation of the basement membrane and the extracellular matrix of adjacent tissues (Miyoshi et al, 2004; Miyoshi et al, 2005; Huang et al, 2009) (Fig 1B). Additionally, EMT inducers prevent basement membrane synthesis by directly repressing the transcription of its components (Spaderna et al, 2006). All these classical EMT events cause the loss of epithelial integrity of the tissue, allow invasion and dissemination of cancer cells, and thus execute the first step of the metastatic cascade (Fig 1A).
MET: metastatic colonization and outgrowth
In 1990, Fearon and Vogelstein proposed a meanwhile classical genetic model for colorectal tumorigenesis. They described tumor development to be the continuous deterioration of initially normal epithelial cells toward greater malignancy driven by the stepwise accumulation of mutations and hypothesized that this process perpetuates during the last step of tumor progression from an established malignant carcinoma to distant metastases, implying that distant metastases are the most degenerated tissues of the malignancy (Fearon & Vogelstein, 1990). However, all efforts to identify specific metastasis‐associated mutations remained unsuccessful.
Rather, already twenty years ago, it was observed that metastases, compared with the de‐differentiated nature of the invasion front of the primary tumor, exhibit a re‐differentiated epithelial morphology, similar to the center of the primary tumor (Brabletz et al, 2001). These findings led to the hypothesis that de‐differentiation and EMT during invasion is a transient condition and that an opposing process of epithelial re‐differentiation or MET needs to be initiated and is advantageous for the formation of distant macrometastases (Figs 1A and 2). But why do metastases re‐differentiate? Invasive, de‐differentiated cancer cells were shown to be growth arrested, whereas proliferation was detected in re‐differentiated metastasis, suggesting that EMT must be reversed in order to allow colonization and growth (Brabletz et al, 2001). This is supported by the fact that EMT‐TFs can directly inhibit proliferation (Thiery et al, 2009) (Fig 3). There are also several publications confirming the relevance of MET for metastatic outgrowth (Chaffer et al, 2006; Korpal et al, 2011; Ocana et al, 2012; Tsai et al, 2012). Importantly, the occurrence of MET is in perfect accordance with the failure to identify EMT‐causing mutations, but is attributed to the transcriptional and epigenetic regulation of the process.
Figure 3. Cellular plasticity of tumor cells is governed by EMT and provided in a window of partial EMT.
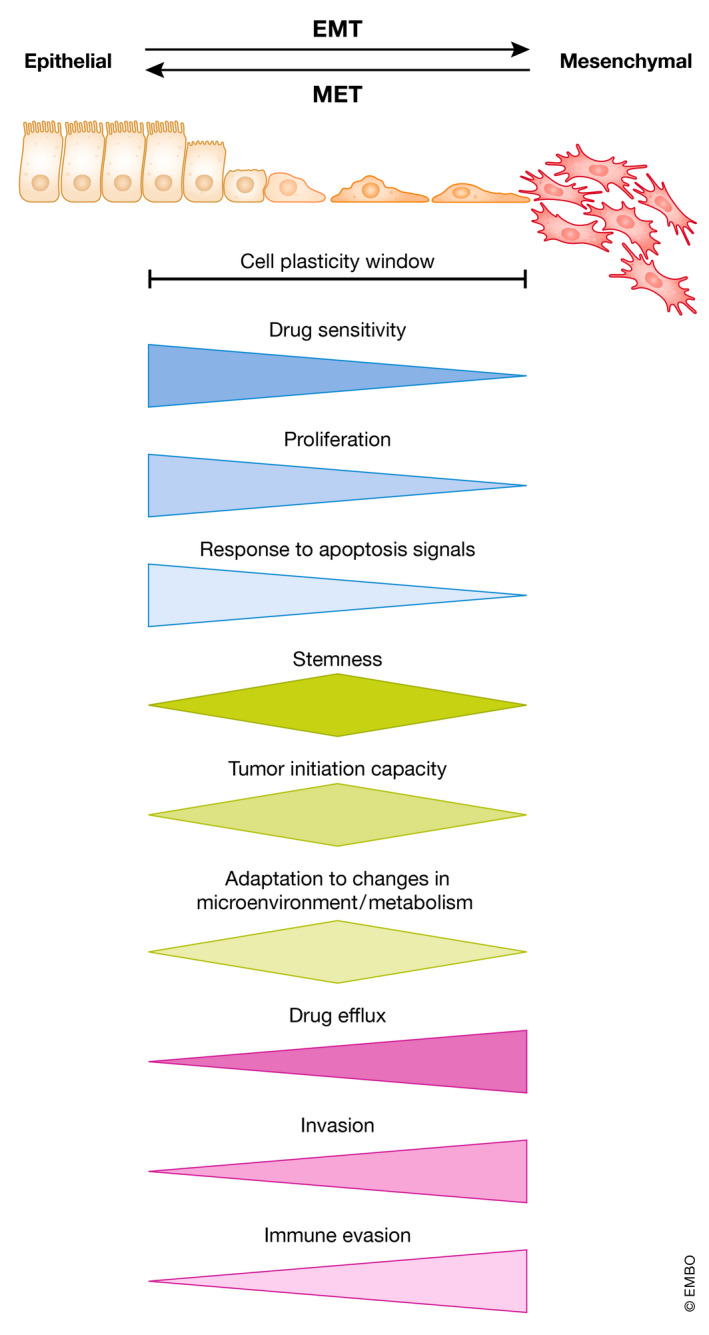
Dynamic induction of EMT and MET changes cellular phenotypes of carcinoma cells. Drug sensitivity, proliferation, and response to apoptosis signals are highest in more epithelial states, whereas drug efflux, invasion, and immune evasion are highest in more mesenchymal states. A hybrid EMT state provides maximal stemness, tumor initiation capacity, and ability to adapt to environmental changes. Note that in extreme epithelial and mesenchymal states, features like stemness, tumor initiation, and colonization are lost.
Whereas EMT induction has been investigated in great detail (see below), less is known about the trigger of the reverse MET process. Is it just lack of EMT‐inducing stimuli coupled to reduced expression of EMT‐TFs? Several studies could show that knockdown of the EMT‐TF ZEB1 is sufficient to elicit MET in vitro in carcinoma cell lines of different entities (Eger et al, 2005; Spaderna et al, 2006; Spaderna et al, 2008) and that depletion of Zeb1 in a genetic mouse model of pancreatic cancer fixes the tumor cells in an epithelial MET state (Krebs et al, 2017). Once expression of ZEB‐family members (ZEB1 and ZEB2) declines, their reduction is reinforced by a double‐negative feedback loop with the miR‐200 family of microRNAs. During EMT, ZEB factors transcriptionally repress the genes of all miR‐200 family members. Vice versa, miR‐200 family members inhibit expression of ZEB1/2 at the post‐transcriptional level (Fig 1B). Thus, the bidirectional transitions between EMT and MET states are potentiated by the ZEB/miR‐200 circuit (Bracken et al, 2008; Burk et al, 2008; Wellner et al, 2009). This branch of MET was shown to be inducible via the tumor‐suppressor p53, that directly activates miR‐200s transcription (Kim et al, 2011). Upregulation of miR‐200 family members has different consequences. It exerts both an invasion‐ and migration‐inhibiting, thus tumor‐suppressing function (Peter, 2009), and promotes metastatic colonization (Korpal et al, 2011). Importantly, these findings are not controversial, but highlight the dynamic nature of the EMT‐MET process adapting the tumor cells to the respective conditions. Similar to the ZEB/miR‐200 negative feedback loop, SNAIL and the miR‐34 family constitute additional epithelial plasticity regulatory loops (Siemens et al, 2011; Diaz‐Lopez et al, 2014) (Fig 1B). Of note, the described microRNAs target not only EMT‐TFs themselves, but also genes like BMI1, CD44, CD133, JAG1, or MYC that are involved in tumor‐relevant “non‐classical” EMT functions like stemness and survival as discussed below (Brabletz & Brabletz, 2010; Brabletz et al, 2011).
Since the metastatic cascade is an extremely complex and slow multistep process that can take many years and requires very different, sometimes apparently opposing traits from the tumor cells in a spatiotemporal manner, valid investigation remains challenging and must rely on mouse tumor models. Recently, several studies were focusing on the spatiotemporal regulation of EMT/MET using elegant mouse models. In 2012, Tsai et al (2012) demonstrated that EMT induction, in this case via TWIST, supports tumor cell dissemination in a mouse model of skin cancer, but subsequent Twist1 downregulation and MET was necessary for colonization and formation of macrometastases (Tsai et al, 2012) (Fig 3). Another study described the necessity of an EMT in the MMTV‐PyMT mouse model of breast cancer to disseminate to the lung. Here, the EMT‐state cancer cells at the metastatic niche activate local fibroblasts that in turn induce epithelial re‐differentiation (MET) for outgrowth of macrometastases (del Pozo Martin et al, 2015). Further, Esposito and colleagues found that E‐selectin mediated cell adhesion of bone vascular niche cells to cancer cells elicits MET and promotes bone metastasis (Esposito et al, 2019).
In summary, many reports support the occurrence and significance of MET in metastatic colonization and outgrowth (Fig 3). However, its regulation and detailed functions still need further investigation.
Partial EMT
EMT and MET have long been viewed as a binary switch between separate epithelial or mesenchymal cell populations. In the past, this narrow perspective challenged the concept of EMT as a major means in metastasis. One example for this is the analysis of Fischer et al using Fsp1‐Cre driven lineage tracing in a genetic mouse model of breast cancer. Based on the finding that lung metastases mainly consist of tumor cells that had never switched to a full mesenchymal Fsp1+ phenotype, the authors concluded that EMT is not important for this process (Fischer et al, 2015).
However, nowadays it is accepted that, although reactivated in many cancer types, EMT is rarely fully executed in tumor cells, and end‐stage markers such as Vimentin are often not expressed. The process is rather gradual and often remains incomplete, termed partial or hybrid EMT (Nieto et al, 2016; Pastushenko & Blanpain, 2019; Yang et al, 2020) (Fig 1B). Over the last years, more and more studies report in vivo detection of cancer cells carrying a combination of both epithelial and mesenchymal markers. Already in the 1990s, early analyses reported observations of cancer cells exhibiting only partial loss of E‐cadherin during invasion (Mareel et al, 1992; Birchmeier & Behrens, 1994). Later, additional publications described circulating tumor cells (CTCs) with simultaneous expression of epithelial and mesenchymal markers (Yu et al, 2013). Similarly, partial EMT of cells within a tumor was identified by co‐expression of epithelial (EpCAM+) and mesenchymal (Vim+) marker genes in an autochthonous murine prostate cancer model (Ruscetti et al, 2015). Partial EMT was also linked to metastasis via single‐cell transcriptomics in head and neck cancer (Puram et al, 2017). Another group identified different tumor transition states occurring during EMT in cancer progression and introduced the term “hybrid” EMT (Pastushenko et al, 2018; Pastushenko & Blanpain, 2019; Pastushenko et al, 2021) (Fig 1B). Moreover, Bornes and colleagues demonstrated that Fsp1‐Cre mediated lineage tracing as used by Fischer et al (2015) is incapable in detecting the majority of disseminating cells that are in a partial/hybrid EMT state (Fischer et al, 2015; Bornes et al, 2019). Importantly, this lack of full EMT does not mean that the process is not important. There is direct in vivo evidence that, e.g., the lack of Zeb1 expression in a genetic mouse model of pancreatic cancer that traps the cancer cells in an epithelial phenotype profoundly suppresses invasion and metastasis (Krebs et al, 2017). In mouse mammary carcinoma cells, SNAIL expression is crucial for Zeb1 induction and metastatic dissemination (Ye et al, 2015). Similarly, in an oncogene‐induced mammary tumor model, Xu et al (2017) found TWIST to be required for the expression of other EMT‐TFs in a small subset of tumor cells to induce partial EMT and metastasis (Xu et al, 2017).
Interestingly, different modes of cancer cell invasion that involve EMT at various levels have been observed. Cancer cells can invade in groups forming rather epithelial clusters. Indeed, this type of “collective migration” might be even more common than single‐cell dissemination, as supported by several studies using lineage tracing approaches and detection of clustered tumor cells in circulation (Friedl et al, 2012; Aceto et al, 2014; Cheung et al, 2016). Nevertheless, despite the epithelial appearance, either characteristic of a partial EMT is also detectable in these migrating cell clusters (Aiello et al, 2018), or the epithelial clusters follow mesenchymal “leader” cells that pave the way for their more epithelial “follower” cells (Matise et al, 2012; Chen et al, 2020) (Fig 1A).
Non‐classical EMT features
Besides the classical features to drive changes in cellular phenotypes, EMT is also involved in regulating various additional aspects of tumorigenesis (Fig 2).
Regulation of stemness
Normal tissue homeostasis is dependent on the function of stem cells that serve as a source to replenish dying committed and terminally differentiated cells in a tissue. The observation that tumor heterogeneity is maintained after single tumor cell transplantations into mice, prompted to adapt normal stem cell biology also to tumors (Reya et al, 2001). Simplified, cancer cell stemness is measured by the capability of cell fractions to form tumors in mice. Strikingly, tumor initiation capacity is increasing with EMT activation by stem cell features provided by EMT‐TFs within the cell plasticity window (Puisieux et al, 2014; Nieto et al, 2016; Shibue & Weinberg, 2017; Wilson et al, 2020) (Fig 3). Overexpression of SNAI1, TWIST1, and ZEB1 in breast cancer is sufficient to increase the CD44+/CD24lo stem cell pool, resulting in increased sphere forming capacity in vitro, as well as elevated tumorigenicity and metastasis in vivo (Mani et al, 2008; Morel et al, 2008). Moreover, SLUG determines stem cell states in the healthy mammary gland and converts luminal cells to stem cells in both healthy and tumor cell settings (Guo et al, 2012). Similar results were obtained for pancreatic cancer, where ZEB1 expression is a key determinant of cancer cell stemness and the reciprocal ZEB1/miR‐200 feedback loop is controlling the expression of genes of stemness factors including BMI1 and SOX2 (Shimono et al, 2009; Wellner et al, 2009; Krebs et al, 2017). Similarly, in squamous cell carcinoma (SCC) TWIST is regulating BMI1 to cooperatively repress CDKN2A (p16INK4A) and promote tumor initiation capacities (Yang et al, 2010). Recently, the protocadherin FAT1 was identified as one additional player in regulating EMT and stemness. FAT1 is frequently inactivated resulting in increased tumor stemness and metastasis in a mouse model of SCC involving CAMK2, CD44, and SRC activities that induce nuclear translocation of YAP1 and ZEB1 (Pastushenko et al, 2021). Interestingly, another important EMT promoting transcription factor, PRRX1, promotes EMT during development and in breast cancer cells, but its loss is required for cancer cell metastasis, thereby uncoupling EMT/migration from stemness features (Ocana et al, 2012). However, PRRX1 isoform switching is the driving force of invasion and metastasis in pancreatic cancer (Takano et al, 2016). Mechanistically, defined cellular states within the EMT cell plasticity realm correlate with gradually increasing efficiencies in metastatic seeding and invasion (Pastushenko et al, 2018), in line with the idea of partial/hybrid EMT states.
In addition to the regulation of tumor stemness and colonization (Shibue & Weinberg, 2009; Shibue et al, 2013; Nieto et al, 2016; Stemmler et al, 2019; Wilson et al, 2020), EMT is involved in driving tumor initiation and malignant transformation. TWIST1 and ZEB1 upregulation in RAS transformed mammary and bronchial epithelial cells is sufficient to unleash malignancy favoring formation of aggressive undifferentiated tumors (Morel et al, 2012; Liu et al, 2014b; Larsen et al, 2016). Moreover, EMT can induce reduction in KRAS dependency of tumor cells (KRAS addiction) and different ZEB1 thresholds drive KRAS‐dependent tumor initiation and metastasis capacities (Singh et al, 2009; Liu et al, 2014b). The oncogenic effects of EMT‐TF activities are even more evident by ectopic activation of Zeb2 in the intestinal epithelium of transgenic mice. Elevation of ZEB2 generates invasive colorectal cancer in absence of cooperating genetic defects (Slowicka et al, 2020).
Therapy resistance
Loss of durable therapeutic efficacy and tumor relapse after initial successful treatment are major obstacles in the battle against cancer. Conventional therapy is favorably eliminating differentiated non‐stem cell‐like cells, but fails to deplete cancer cells with stem cell properties. EMT activation in these cells is controlling therapy resistance on multiple levels (Singh & Settleman, 2010; Shibue & Weinberg, 2017; Santamaria et al, 2019; Dudas et al, 2020). EMT gene signatures and acquisition of therapy resistance are strongly correlated, e.g., for standard and for targeted therapy with EGFR or PI3K inhibitors (Creighton et al, 2009; Farmer et al, 2009; Byers et al, 2013). For example, gemcitabine‐resistant Panc1 pancreatic cancer cells exhibit EMT characteristics and become sensitized upon ZEB1 knockdown (Wellner et al, 2009). Mechanistically, common routes toward drug resistance include increasing drug efflux or evading apoptosis and anoikis (Fig 3). The former is mediated by activating the expression of members of the ATP‐binding cassette (ABC) transporter family, which are transcriptionally regulated by the EMT‐TFs TWIST, SNAIL, and FOXC1 (Aller et al, 2009; Singh & Settleman, 2010; Saxena et al, 2011). SLUG and SNAIL contribute to evading therapy‐induced apoptosis by interfering with p53 function, repression of the tumor‐suppressor PTEN, or upregulation of the pro‐survival protein BCL‐XL (Vega et al, 2004; Escriva et al, 2008; Kurrey et al, 2009; Wu et al, 2015; Cao et al, 2016).
Experimentally transformed HMLER breast cancer cells with increased EMT features display about 10‐fold increase in IC50 doses of many chemotherapeutics (Gupta et al, 2009; Singh et al, 2009; Singh & Settleman, 2010; Tulchinsky et al, 2019). In lineage tracing experiments, GFP‐labeled mesenchymal tumor cells in PyMT tumors display high resistance to cyclophosphamide treatment (Fischer et al, 2015). In non‐small‐cell lung cancer (NSCLC), activation of the AXL receptor tyrosine kinase in the course of EMT provides resistance to EGFR and PI3K inhibition, dependent on sustained KRAS activity (Singh et al, 2009; Sequist et al, 2011; Zhang et al, 2012; Byers et al, 2013; Tulchinsky et al, 2019). Furthermore, ZEB1 downregulation through HDAC class I inhibition or DNA demethylation resensitizes pancreatic cancer and osteosarcoma cells to chemotherapy (Meidhof et al, 2015; Ruh et al, 2021). Docetaxel‐resistance of lung adenocarcinoma cells and paclitaxel‐resistance of ovarian cancer cells can be reverted by blocking ZEB1 expression, providing increased survival of lung metastasis‐bearing mice upon treatment (Ren et al, 2013; Sakata et al, 2017). In BRAF‐mutant melanoma, ZEB1 is sufficient and only partly compensated by TWIST to promote an undifferentiated p75high neural crest stem cell‐like state that is characterized by resistance to MAPK and BRAF inhibitors (Richard et al, 2016; Shaffer et al, 2017; Rambow et al, 2018). In contrast, TWIST and SNAIL dampen the response to gemcitabine in an autochthonous mouse model of pancreatic cancer as Snai1 and Twist1 knockout results in prolonged survival in gemcitabine‐treated tumor mice (Zheng et al, 2015). Similarly, in NSCLC TWIST inhibition leads to sensitization to tyrosine kinase inhibitors of mutant EGFR (Yochum et al, 2019). In glioblastoma, the EMT‐regulated shift toward a more stem‐cell‐like phenotype correlates with increased radioresistance. This is mediated by elevated STAT3 which is directly binding to the SNAI2 promoter for transcriptional activation (Lin et al, 2018). Collectively, EMT induction, increase in cancer stem cell features, and acquisition of therapy resistance are strongly connected and EMT‐TFs are central players in orchestrating these processes.
EMT induced by therapy
As outlined before, one feature of EMT is to provide resistance to several treatments, including chemo‐, radio‐, and immunotherapy. Of note, also the therapy itself can induce an EMT phenotype with more aggressive features that involves activation of EMT promoting pathways via TGFβ, NF‐κB, WNT, FGF, and EGF/HER2 (see below) (Terry et al, 2017; Redfern et al, 2018). Therapy‐induced selection of cells with EMT characteristics, eventually displaying gain of ZEB1 and VIM, but repressed CDH1, MIR200C and MIR205 expression, was observed in vitro after long‐term exposures to gemcitabine in BxPC3 pancreatic cancer cells and to Docetaxel in PC3 and DU‐145 prostate cancer cells (Wellner et al, 2009; Puhr et al, 2012). Similarly, withdrawal of BRAF and MEK inhibition results in an enhanced motility and invasion through reactivation of ERK1/2 and AKT/mTOR in vitro (Norz et al, 2015). Treatment of mice suffering from CRC liver metastasis, with a vascular disruptive agent leads to a transient accumulation of nuclear β‐catenin and upregulation of ZEB1, which reverts after treatment. This indicates that EMT induction might display adoption to cope with therapy‐induced cellular stress (Fifis et al, 2013; Terry et al, 2017; Redfern et al, 2018). Supporting this hypothesis, inhibition of factors involved in therapy‐induced DNA damage response (DDR), like CHK1 activation by ATR inhibitors, results in EMT and ZEB1 upregulation in CRC cells, reduced phosphorylation of CHK1 and decreased sensitivity to ATR inhibition (Song et al, 2018; Zhang et al, 2018). Observations in patients support a clinical relevance of EMT‐associated pro‐survival stress adaptation, exemplified by analysis of post‐therapy specimens, e.g., in esophageal and breast cancer after chemotherapy and in CRC after radiotherapy, showing decreased E‐cadherin and elevated SNAIL and Vimentin levels (Creighton et al, 2009; Kawamoto et al, 2012; Hara et al, 2014).
Metabolic reprogramming
Metabolic rewiring is one important feature of cancer cells to cope with the excessive needs for energy and nutrients, enabling unlimited proliferation in conditions of rather restricted oxygen supply. In support of the metastasis cascade, reprogramming of glucose, lipid, and amino acid metabolism is tethered to activation of the EMT program and orchestrated by core EMT‐TFs (Kang et al, 2019; Georgakopoulos‐Soares et al, 2020; Sun & Yang, 2020; Bergers & Fendt, 2021). Cancer cell‐mediated changes in metabolism are well described in glycolysis and triglyceride pathways, favoring the “Warburg effect”, i.e., energy‐inefficient glycolysis even under normoxic conditions (Sun & Yang, 2020). This is another example how cancer cells adopt a developmental program of enhanced aerobic glycolysis that favors EMT and migration in the embryo (Youssef & Nieto, 2020). Upregulation of glucose transporters GLUT1 and GLUT3 is further promoting glycolysis. It was demonstrated in NSCLC that the increase in GLUT3 correlates with poor prognosis and EMT activation, mediated by activation of ZEB1 (Masin et al, 2014). On the contrary, metabolic abnormalities, e.g., in the tricarboxylic acid (TCA) cycle, can facilitate EMT. For example, loss of fumarate hydratase (FH) leads to aberrant methylation of a miR‐200 family promoter region, resulting in enhanced expression of ZEB factors and EMT induction. Fumarate accumulation by repression of FH by the chromatin modifier LSH subsequently increases α‐ketoglutarate (α‐KG) levels. This affects IKKα‐dependent EMT by promoting IKKα binding to and activation of EMT‐associated gene promoters (He et al, 2016; Sciacovelli et al, 2016; Sciacovelli & Frezza, 2017; Georgakopoulos‐Soares et al, 2020). Interestingly, ZEB1 is essential to allow metabolic plasticity. ZEB1 deficient pancreatic cancer cell lines are incapable of compensatory increasing glycolysis when oxidative phosphorylation is blocked, highlighting poor glycolytic reserve (Krebs et al, 2017). A systematic approach to categorize metabolic rewiring during EMT focused on gene expression alterations in high grade malignancies and identified a mesenchymal metabolic signature of 44 genes. Upregulation of this group of genes is correlated with a mesenchymal phenotype and is controlled by TWIST1 in mammary epithelial cells (Shaul et al, 2014).
In addition to carbohydrate pathways, EMT‐associated changes in fatty acid, lipid and glycosphingolipid metabolisms are detected (Kang et al, 2019; Georgakopoulos‐Soares et al, 2020; Hua et al, 2020; Bergers & Fendt, 2021). The morphological changes of cancer cells during EMT are accompanied by alterations in membrane fluidity and lipid composition. This requires the elevation of available fatty acids and is achieved for example by increased expression of the fatty acid translocase CD36. The presence of CD36 promotes EMT in HCC and is crucial for metastatic colonization of oral cancer (Nath et al, 2015; Pascual et al, 2017; Bergers & Fendt, 2021). Elevated free fatty acids (FFAs) lead to increased SRC and MMP9 activity that accelerates metastasis (Gubelmann et al, 2014; Jiang et al, 2020). In addition, the synthesis, storage and use of long‐chain polyunsaturated fatty acids (PUFA) is elevated in the therapy‐resistant EMT cell state. Consequently, the cells become dependent on elimination of resultant reactive lipid peroxides by Glutathione peroxidase 4 (GPX4) to evade the iron‐dependent cell death pathway ferroptosis (Viswanathan et al, 2017). In summary, these analyses are a few examples demonstrating the reciprocal connection of EMT, cell plasticity, and metabolic reprogramming and how they synergize toward malignant progression (Fig 3).
EMT and immune evasion
During tumor progression, cancer cells develop a variety of strategies to withstand immunosurveillance and adaptive anti‐tumor immunity that is induced by tumor neo‐antigens. Most commonly pursued strategies include hiding the neo‐antigen spectrum and formatting of an immunosuppressive environment, e.g., by hijacking negative immunological feedbacks referred to as “immune checkpoints” (Terry et al, 2017; Kudo‐Saito et al, 2021). Interestingly, these unwanted processes often occur in strong correlation with EMT, and EMT‐TFs have repeatedly been shown to co‐regulate them directly or indirectly (Terry et al, 2017; Dongre & Weinberg, 2019; Jiang & Zhan, 2020; Kudo‐Saito et al, 2021). For instance, defects in the immunoproteasome, that generates peptides for antigen presentation, were associated with a more mesenchymal phenotype in NSCLC (Tripathi et al, 2016). A study using the PyMT mouse model of breast cancer demonstrated a remarkable anti‐correlation of mesenchymal expression profiles and MHC I expression (Dongre et al, 2017). Thus, human leukocyte antigen (HLA) class I complexes and major histocompatibility complex I (MHC I), required for antigen presentation, can apparently be deregulated by EMT‐related mechanisms. Furthermore, utilizing breast cancer models in vitro and in vivo demonstrated that more mesenchymal tumor cells promote the generation and recruitment of regulatory T cells (Tregs) (Joffroy et al, 2010; Akalay et al, 2013; Dongre et al, 2017), which are an integral part of immune checkpoints able to inhibit cytotoxic T cells (CTL). In this regard, one of such intrinsic “brake” regulatory mechanisms of CTLs is activated by binding of their programmed cell death receptor (PD1) to its ligand PD‐L1 provided by Tregs and myeloid antigen presenting cells. Tumor cells can hijack these immune checkpoints mainly by upregulation of CD274 (PD‐L1), which is strongly promoted by the EMT program (Zou et al, 2016; Terry et al, 2017; Jiang & Zhan, 2020; Kudo‐Saito et al, 2021). In NSCLC, PD‐L1 is activated by deregulation of the ZEB1/miR‐200 axis in more mesenchymal cells (Chen et al, 2014; Jung et al, 2020). This regulation is likely independent of a general EMT program, since ZEB1 loss cannot be compensated by SNAIL, SLUG, or TWIST (Noman et al, 2017). Kudo‐Saito showed that SNAI1 expression in melanoma promotes induction of Tregs via Thrombospondin1 activation that results in resistance to immunotherapy (Kudo‐Saito et al, 2009). Based on these findings, assessing the EMT signatures of, e.g., bladder cancer and melanoma may help to select patients for immune checkpoint blockers (Hugo et al, 2016; Kardos et al, 2016). In summary, EMT cells evade T‐cell anti‐tumor immunity either via downregulation of antigen presentation or activation of immune checkpoints.
EMT and DNA integrity
Genomic instability is a common feature in cancer with evidence of contribution of EMT (Nieto et al, 2016; Yang et al, 2020) and has frequently been observed in pre‐malignant mesenchymal cells in vitro as well as in tumors (Massague, 2012; Comaills et al, 2016; Caramel et al, 2018; Wang et al, 2019a). Although EMT stimuli are normally anti‐proliferative, some cancer cells maintain cell doubling, perhaps at the expense of genome stability (Omabe et al, 2021). The involved mechanisms overriding the anti‐proliferative signals and the generation of genomic aberrations are poorly understood. Usually, DNA repair, cell cycle checkpoints, senescence and cell death in response to endogenous (e.g., during DNA replication) and exogenous DNA damages (e.g., during chemotherapy) are orchestrated by the DDR, which is a conserved, multifactorial and context‐dependent signaling network (Halazonetis et al, 2008; Omabe et al, 2021). Recent evidence suggests that EMT‐TFs differentially affect the DDR. TWIST promotes chromosomal instability (CIN) by repressing mitotic checkpoint factors in colorectal cancer (CRC), whereas SLUG facilitates homologous recombination‐directed DNA repair (HDR) during replication stress in mammary epithelial cells (Gross et al, 2019; Khot et al, 2020). Hence, regulation of the DDR by EMT‐TFs may influence cancer treatment. In breast cancer cells, ZEB1 promotes radioprotection by driving the expression of the apical DDR kinase ATM via complex formation with p300 and PCAF at the ATM promoter as well as by stabilizing the mediator kinase CHK1 via sequestration of its deubiquitinase USP7 (Zhang et al, 2014; Zhang et al, 2018). In triple‐negative breast cancer, ZEB1 was shown to directly repress POLQ (Polθ) expression to prevent usage of the highly mutagenic alternative end joining (a‐EJ or microhomology‐mediated end joining) for the repair of endogenously occurring DSBs, thereby supporting the maintenance of genomic stability (Prodhomme et al, 2021). Intriguingly, during breast cancer development, the accumulation of CIN is apparently prevented, if oncogene activation had taken place in mammary stem cells, engaging a ZEB1‐driven preemptive program with upregulation of the anti‐oxidant MSRB3. This mitigates oncogene‐induced replication stress to overcome oncogene‐induced senescence (OIS) in TP53 wild‐type cells (Halazonetis et al, 2008) (Morel et al, 2017). Along that line, SNAIL and TWIST regulate p53 and RB‐dependent pathways to modulate OIS (Ansieau et al, 2008; Puisieux et al, 2014). In addition, SNAIL and SLUG alter the downstream DDR‐driven cell death in MCF7 breast cancer cells by repressing pro‐apoptotic effectors (Kajita et al, 2004). In summary, tumor cells utilize specific DDR‐related functions of EMT‐TFs to evade oncosuppressive mechanisms.
Box 1 Signaling pathways activating EMT in cancer .
TGFβ
TGFβ is the major extrinsic signal of EMT in tumors, secreted by many cell types in the TME, but the role of TGFβ signaling in tumor initiation and progression is very complex as it shows a biphasic function with opposing effects. During tumor initiation, TGFβ promotes differentiation, cell cycle arrest, senescence and apoptosis in pre‐malignant epithelial cells whereas during later stages TGFβ acts as inducer of EMT and promotes metastasis (Massague, 2012; Seoane & Gomis, 2017; Ramachandran et al, 2018; Derynck et al, 2021; Liu et al, 2021) (Fig 4A). Moreover, TGFβ acts in a very context‐dependent manner in crosstalk with other pathways, including Hippo, WNT, Notch, Hedgehog, ERK, p38 MAPK, PI3K/AKT and RHO‐like GTPases to enhance their function (Bakin et al, 2000; Zavadil & Bottinger, 2005; Hao et al, 2019; Derynck et al, 2021; Liu et al, 2021) (Fig 4A). In transformed mammary epithelial cells, execution of TGFβ‐mediated EMT depends on autocrine WNT activity and inhibition of these autocrine cues reduces tumorigenicity and metastasis in vivo (Scheel et al, 2011). TGFβ induces mesenchymal genes like VIM and FN1 as well as EMT‐TFs SNAI1, SNAI2, TWIST1 and ZEB1 which form a feedback loop by activating TGFβ ligand expression to self‐enforce and maintain TGFβ signaling (Dongre & Weinberg, 2019). Initial TGFβ receptor activation in breast or skin cancer results in transcriptional activation of SNAI1, combined with sumoylation of SNAIL at K234 fostering SMAD/SNAIL complex formation that promotes EMT (Hoot et al, 2008; Vincent et al, 2009; Gudey et al, 2017) (Fig 4B). Similarly, ZEB proteins have SMAD‐interacting domains and TGFβ, in crosstalk with MAPK signaling, utilizes SMAD3/SMAD4/ZEB1/2 complexes to regulate EMT‐related gene expression (Postigo, 2003; Shirakihara et al, 2007). Very strikingly, in pancreatic cancer, TGFβ‐mediated EMT depends almost exclusively on ZEB1, since Zeb1 knockout in KPC cells blocks more than 90% of TGFβ‐induced alterations in gene expression, despite of Snai1 upregulation during TGFβ treatment (Krebs et al, 2017).
Notch
Like TGFβ, the Notch pathway is important for differentiation processes during development that in part involve EMT by SNAI1 and SNAI2 (Timmerman et al, 2004; Niessen et al, 2008). In cancer, a Notch‐dependent EMT program is in part executed by cooperating with TGFβ signaling (Yuan et al, 2014; Bray, 2016). Activation of the Notch pathway by Delta/Jagged ligands results in processing and release of the Notch ICD transducer that in turn interacts with SMADs, in addition to canonical RBP/J cofactor binding (Fig 4A and B). This crosstalk is essential for mesenchymal gene activation and regulates bone metastasis in NSCLC (Blokzijl et al, 2003; Xie et al, 2012; Liu et al, 2014a; Yuan et al, 2014). While in NSCLC Notch3 is crucial for invasion and EMT by directly regulating ZEB1 transcription, in stratified squamous epithelia Notch3 promotes terminal differentiation. Oncogene‐induced NOTCH1 in SCC is activating ZEB1 expression, which in turn downregulates NOTCH3 to promote stem cell features and block differentiation (Liu et al, 2014a; Natsuizaka et al, 2017). Moreover, the ZEB1/miR‐200 feedback loop further modulates Notch signaling activity as downstream Notch components including MAML2/3 are miR‐200 targets, resulting in enhanced Notch activation upon ZEB1 induction (Brabletz et al, 2011) (Fig 4B). Pancreatic cancer metastasis is promoted by a Notch‐dependent inflammatory feedback loop that involves recruitment and activation of M2‐like tumor‐promoting macrophages and EMT via IL6/STAT3 signaling (Geng et al, 2021).
Hippo/YAP/TAZ
Deregulation of the Hippo pathway was shown to promote tumorigenesis as well (Moroishi et al, 2015; Ansari et al, 2019) (Fig 4A). YAP/TAZ overexpression and activation is crucial to mediate EMT and cell plasticity in skin cancer and is sufficient to induce EMT in MCF10A non‐cancerous mammary epithelial cells. YAP was shown to cooperate with KRAS for progression of pancreatic cancer and TAZ is a promoter of proliferation, EMT and stemness in breast and oral cancer (Zhang et al, 2009; Shao et al, 2014; Li et al, 2015; Debaugnies et al, 2018). The function of YAP/TAZ is dependent on the TEAD‐binding domain and in breast cancer, TEAD2 is key to orchestrate EMT by regulating nuclear localization of YAP and TAZ (Lamar et al, 2012; Diepenbruck et al, 2014). In the very aggressive subgroup of Claudin‐low breast cancer, YAP and ZEB1 interact to drive expression of a common target gene subset to promote EMT and metastasis (Lehmann et al, 2016). Moreover, YAP forms a TEAD/YAP/AP1 ternary complex to regulate activity of distal enhancers, whereas ZEB1 and YAP are part of a multimeric complex with AP1 factors independent of direct ZEB1 DNA binding to drive gene expression on a genome‐wide level (Zanconato et al, 2015; Feldker et al, 2020) (Fig 4B). Wang and colleagues demonstrated that in breast cancer, also TWIST is involved in YAP/TAZ‐mediated EMT orchestrated by TWIST‐dependent activation of the GPCR member PAR1 (Wang et al, 2020).
WNT
The role of WNT/β‐catenin signaling in tumorigenesis and metastasis is highlighted by mutations of APC that preclude the control of the signaling competent cytoplasmic pool of β‐catenin (Fig 4A). Moreover, the WNT target G‐coupled receptor LGR5 labels stem cells in several organs as well as cancer stem cells in tumors (Clevers, 2006). Interestingly, LGR5 expression and WNT activity are crucial for disease progression, induction of EMT, stemness and metastasis, which is not provided by LGR5‐negative tumor cells and depletion of Lgr5+ cells in mice blocks metastasis (de Sousa e Melo et al, 2017), though a Lgr5‐ state is detected in circulating CSCs (Fumagalli et al, 2020). In breast cancer, reduction in WNT signaling by addition of secreted negative regulators of the pathways, like DKK1 or SFRP1 can promote reduction in migration and invasion, whereas inhibition of SFRP1, and thus elevated WNT signaling, can support an EMT phenotype and sensitize to TGFβ‐induced EMT (Gauger et al, 2011; Scheel et al, 2011). Intracellularly, the WNT‐induced block of GSK3β is not only promoting β‐catenin cytoplasmic accumulation, but also stabilizes SNAIL (Zhou et al, 2004). ZEB1 is transcriptionally activated via β‐catenin/TCF4 binding to the ZEB1 promoter (Sanchez‐Tillo et al, 2011). Furthermore, increased ZEB1 in turn represses MIR200A expression which leads to de‐repression of CTNNB1 (β‐catenin) thereby fueling this feedback loop (Su et al, 2012; Liu et al, 2013) (Fig 4B).
RTK signaling (induced by, e.g., EGF, FGF, IGF, HGF and PDGF)
Receptor tyrosine signaling has pleiotropic functions in proliferation and differentiation during development and homeostasis as well as in cancer initiation and progression. Growth factors like EGF, FGF, HGF and PDGF act via their cognate receptors and promote EMT either in support of TGFβ or alone via MAPK/ERK, PI3K/AKT and mTORC signaling (Lamouille & Derynck, 2007; Lamouille et al, 2012; Dongre & Weinberg, 2019) (Fig 4A). The importance of RTK signaling is best described by the key function of RAS (KRAS, HRAS, NRAS) and BRAF oncogenes. One of them is mutated in most cancer types, leading to continuous pathway activation. SNAI1 and SNAI2 are direct targets of mutant KRAS and BRAF activities and ZEB1/2 is regulated downstream by ERK2, but not ERK1 (Shin et al, 2010; Makrodouli et al, 2011). EGF induces EMT by upregulation of SNAI1, TWIST1 and ZEB1 in different cell lines through MAPK/ERK as well as JNK/STAT3 activation (Lu et al, 2003). In particular, EGF is cooperating with other signaling pathways, e.g., IL6R/STAT3 and TGFβ, resulting in CDH1 downregulation and enhanced cell motility and invasion (Lo et al, 2007; Tian et al, 2007; Uttamsingh et al, 2008; Colomiere et al, 2009). Phosphorylation of SMAD2/3 and nuclear co‐localization with SNAIL was induced by activation of EGFR in MCF7 and MDA‐MB‐231 breast cancer cells (Kim et al, 2016). EGF‐induced EMT seems to require the function of the RNA‐binding protein Musashi2 (MSI2) by interaction with ZEB1 to facilitate ZEB1‐ERK/MAPK signaling in pancreatic cancer (Sheng et al, 2020). Similar examples exist for activation of FGFR, IGFR, PDGFR and c‐Met (HGF receptor) (Grotegut et al, 2006; Yang et al, 2006; Graham et al, 2008; Devarajan et al, 2012; Zhu et al, 2016; Maehara et al, 2017; Zheng et al, 2019).
Cytokines and hypoxia
Several inflammatory cytokines, produced by cells in the TME, are contributing to cellular plasticity and an invasive phenotype (Fig 4A). Most prominently TNFα and IL6, but also IL1β have critical functions and promote stemness, invasion and regulation of genes involved in EMT mediated by SNAIL, TWIST and ZEB1 as demonstrated in colon, breast and renal cell carcinoma (Bates & Mercurio, 2003; Sullivan et al, 2009; Ho et al, 2012; Li et al, 2012; Miao et al, 2014; Ieda et al, 2019). IL6 signals through STAT3 that directly activates SNAI1 and ZEB1 expression, whereas TNFα facilitates TGFβ and PI3K/AKT signaling that inhibits GSK3β (Bates & Mercurio, 2003; Sullivan et al, 2009; Ho et al, 2012; Miao et al, 2014) (Fig 4A). Interestingly, an autocrine feedback loop is generated in tumor cells to enhance an inflammatory phenotype, since SNAIL and ZEB1/2 are able to activate IL6 expression in head and neck squamous carcinoma and breast cancer (Lyons et al, 2008; Katsura et al, 2017).
In addition to growth factors, chemokines and cytokines, hypoxia and HIF1α are well known to induce EMT as well (Hapke & Haake, 2020). In this scenario, EMT is induced by inefficient neo‐angiogenesis and deprivation of pericytes in the tumor (Hapke & Haake, 2020). Activated HIF1α or HIF2α directly or indirectly regulate SNAI1, TWIST1 and ZEB1 expression which fosters stemness and metastasis formation in bladder, ovarian, gastric and breast cancer (Yang et al, 2008; Cooke et al, 2012; Liu et al, 2017; Hapke & Haake, 2020; Zhang et al, 2020).
In summary, data that have been collected over decades provide a comprehensive picture in which EMT in tumorigenesis and metastasis is induced by integration of many pathways and growth factors. Moreover, synergism and the crosstalk of several signaling networks highlight that cellular plasticity in epithelial tumor cells renders the promotion of a cancer stem cell‐like, immune evading, multi‐resistant, invasive phenotype, able to adapt to changing conditions in the tumor and the metastatic niches. However, detailed mechanistic insight is still lacking to understand the complex crosstalks of signaling pathways that drive EMT and metastasis.
The tumor microenvironment: Stroma‐tumor crosstalk to foster EMT
The TME is the source of many different signaling cues that have been implicated in EMT induction, including TGFβ, WNT, RTK, Hippo, Notch and cytokine signaling (Box 1, Fig 4). The TME consists of extracellular matrix and non‐transformed cells that are recruited to the tumor and adopt a variety of abnormal phenotypes instructed by tumor/stroma crosstalk. Cell types include fibroblasts, cells of the immune system, endothelial cells, adipocytes and neuronal cells (Fig 5). The complex crosstalk of the entity of these cell types and the ECM is providing pro‐ and anti‐tumorigenic functions. Stroma composition has a strong impact on tumor aggressiveness and carries prognostic value (Hanahan & Weinberg, 2011; Jin & Jin, 2020). Moreover, individual stromal cell types are contributing to EMT and metastasis.
Figure 4. Signaling pathways and their crosstalk during EMT induction.
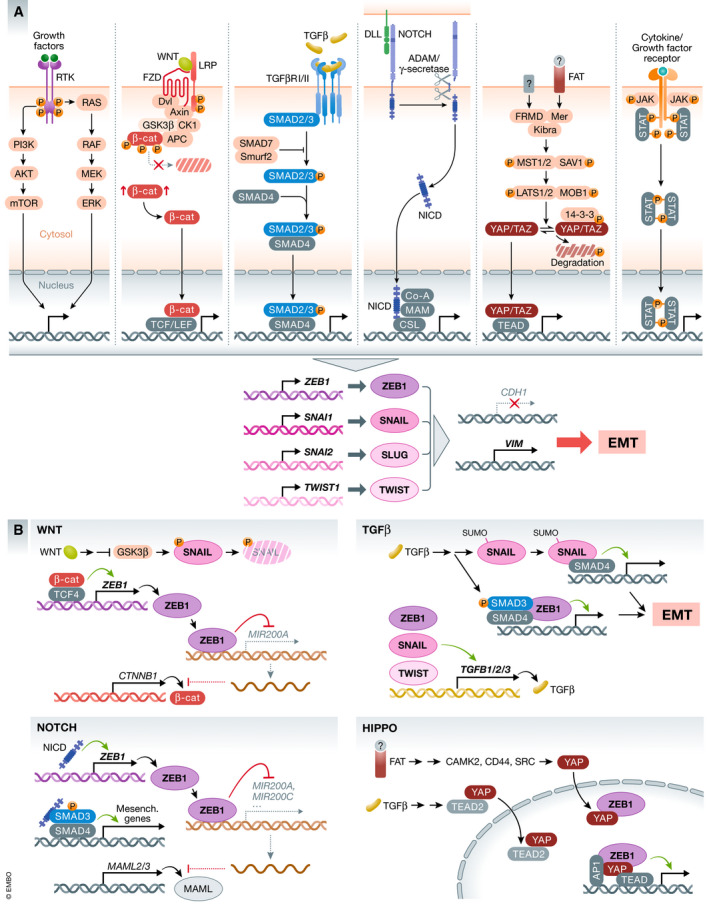
(A) Canonical receptor tyrosine kinase pathways activated by EGF/EGFR, PDGF/PDGFR, HGF/c‐Met and others, Wnt, TGFβ, Notch, Hippo, and Cytokine signaling (e.g., IL6, TNFα) and their nuclear effectors are shown, which all promote expression of core EMT‐TFs. Activation of EMT‐TFs results in execution of the EMT program, exemplified by regulated CDH1 and VIM expression. EMT is promoted by crosstalk of signaling pathways on multiple levels already in the cytoplasm (not shown). (B) Examples of EMT gene regulation of individual pathways and input from other signal transduction pathways. Effects on direct gene activation and repression are shown by green arrows and red block connections, respectively. For details, see text.
Figure 5. Cell types in the tumor microenvironment and contribution to EMT.
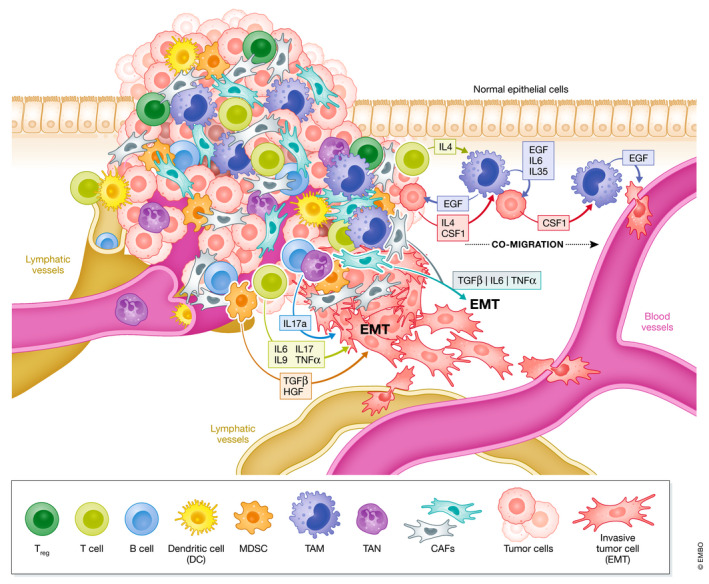
Cells of the hematopoietic lineage, fibroblasts and others contribute to the TME. Exemplified modes of action and cytokines secreted from individual cell types are depicted which promote EMT, migration, invasion, and intravasation of tumor cells into blood vessels. EMT induction and co‐migration can be induced by TAMs and specific lymphocyte/tumor cell crosstalk.
CAFs
Cancer‐associated fibroblasts (CAFs) display a very heterogenous population generated by reprogramming and expansion of either local tissue‐resident or recruited fibroblasts from the periphery, including transdifferentiation of adipocytes, pericytes, monocytes, and mesenchymal stem cells (LeBleu & Kalluri, 2018; Sahai et al, 2020). CAFs can be categorized into different subpopulations with specific functions and spatial organization (Lambrechts et al, 2018; Elyada et al, 2019; Sahai et al, 2020). They serve as a primary source of EMT‐inducing signaling molecules, like IL6, TNFα and TGFβ, which promote a mesenchymal phenotype of tumor cells (Fig 5). Consequently, as an example they increase chemoresistance in NSCLC, regulate metastasis in ovarian cancer and promote oncogenic potential of adjacent epithelia in gastric squamous cell carcinoma (Bhowmick et al, 2004; Yu et al, 2014; Calon et al, 2015; Shintani et al, 2016; Zhao et al, 2017; Goulet et al, 2019). As demonstrated for prostate cancer, the entry into TGFβ‐mediated EMT initiated by CAFs is dependent on changes in DNA methylation by DNMTA3A, silencing epithelial genes like CDH1 and GRHL2 (Pistore et al, 2017). TGFβ secretion by CAFs and EMT signatures in CRC are correlated with poor prognosis and promote metastasis (Calon et al, 2015; Tauriello et al, 2018). Interestingly, WNT signaling is involved in orchestrating CAF diversity and crosstalk to CRC tumor cells. In a subcutaneous organoid model, SFRP1 secretion induces a switch from contractile myCAFs to inflammatory iCAFs, accompanied by increasing EMT signatures in tumor cells (Mosa et al, 2020). Moreover, CAFs can impinge on anti‐tumor immunity and on success of anti‐CSFR1 immunotherapy (Kumar et al, 2017; Tauriello et al, 2018), supported by the finding that collagen 1 (COL1) deposition by myCAFs is crucial to augment an immunosuppressive TME (Chen et al, 2021).
TAMs
Macrophages in the TME are contributing to EMT on multiple levels. In a simplified model, macrophage‐tumor crosstalk generates a variety of tumor‐associated macrophage (TAM) phenotypes, with an M1‐like state promoting elimination of immunogenic tumor cells and a contrasting pro‐tumorigenic M2‐like state (Gonzalez et al, 2018). In crosstalk with other cell types and in the presence of TGFβ, metastasis is induced by CD4+ T‐cell derived IL4 and macrophage‐derived WNT7B/EGF as well as CSF1 from tumor cells. These ligands induce co‐migration of tumor cells and macrophages toward blood vessels. TAMs near blood vessels create “the tumor microenvironment of metastasis” enabling tumor cell intravasation (Joyce & Pollard, 2009; Noy & Pollard, 2014; Gonzalez et al, 2018) (Fig 5). TAMs can also directly express and secrete TGFβ and cytokines to induce EMT. In F9 teratocarcinoma, NMuMG cells and hepatocellular carcinoma (HCC), intratumoral or cocultured macrophages are secreting TGFβ to promote EMT (Bonde et al, 2012; Fan et al, 2014). In CRC, EMT is further accelerated by TNFα‐secreting macrophages, inducing p38 MAPK signaling in tumor organoids (Bates & Mercurio, 2003). Moreover, in renal clear cell carcinoma, NSCLC, colon and breast cancer IL6 and IL35 are produced by macrophages and promote EMT by prostaglandin/β‐catenin and JAK2‐STAT6‐GATA3 signaling, respectively (Zhang et al, 2019b; Sun et al, 2020) (Che et al, 2017; Lee et al, 2018).
MDSCs
Myeloid‐derived suppressor cells (MDSCs) are a heterogenous cell population originating from neutrophils and monocytes. They are expanding in cancer patients and are involved in immune cell suppression, specifically of T cells. Accordingly, they promote tumor cell immune escape and block cytotoxic T‐cell activity (Gabrilovich & Nagaraj, 2009; Gabrilovich, 2017). Moreover, they directly induce EMT via release of different growth factors and cytokines, including TGFβ and HGF, as well as the high mobility group protein B1 (HMGB1) which acts through toll‐like receptors (TLRs) and leads to induction of SNAI1, MMP7 and NFKB1 (NF‐κB) (Trovato et al, 2020) (Fig 5). At the primary tumor, specific monocytic CXCR2‐expressing MDSCs (mMDSCs) promote EMT and they are recruited by breast cancer cells via secretion of CXCL1/2, whereas MET at pulmonary metastatic sites is induced by granulocytic MDSCs (gMDSCs) (Ouzounova et al, 2017; Zhu et al, 2017; Taki et al, 2018; Trovato et al, 2020). Of note, MDSCs become activated and expand upon operational stress during primary breast cancer resection and start to secrete TGFβ, VEGF and IL10 to promote lung metastasis in a 4T1 orthotopic breast cancer model (Ma et al, 2019).
TANs
Neutrophils are promoting metastasis at the pre‐metastatic niche in lungs and liver by suppressing T cells (Gonzalez et al, 2018). In addition, they are involved in promoting EMT at the primary site. Akin to TAMs, tumor‐associated neutrophils (TANs) can polarize between extreme states “N1” TANs with anti‐tumor and pro‐immune functions, induced by IFN‐β, and “N2” TANs with pro‐tumorigenic and pro‐metastatic functions. “N2” TANs are induced by TGFβ (Gonzalez et al, 2018; Masucci et al, 2019; Wu & Zhang, 2020). Neutrophils in the stroma of gastric cancer (GC) produce IL17a and CXCL5 both promoting EMT, indicated by upregulation of VIM and ZEB1, whereas CDH1 is repressed (Fig 5). Anti‐IL17a treatment in vitro blocks EMT in neutrophil‐GC co‐cultures (Li et al, 2019; Mao et al, 2020; Wu & Zhang, 2020). In MCF7 breast cancer cells, EMT is induced by TIMP‐1 expression secreted by neutrophils. TAN‐MCF7 interaction creates an enforcing feedback‐loop as MCF7 cells with a more mesenchymal phenotype activate TIMP‐1 expression in neutrophils by CD90 during direct cell contact. Blocking CD90 reduces the number of metastasis in a 4T1 orthotopic injection model in mice (Wang et al, 2019b; Wu & Zhang, 2020).
T cells
CD4+ and CD8+ T cells are the most prominent effectors of anti‐tumor immune response. High levels of T‐cell infiltration usually correlate with better prognosis; however, their activity is dependent on immune regulatory and suppressive mechanisms including Treg activities (Joyce & Pollard, 2009; Gonzalez et al, 2018). Moreover, some examples demonstrate direct contribution of T cells to EMT. In the presence of isolated effector CD4+ T cells, pancreatic cancer cells show upregulation of VIM, L1CAM and ZEB1, combined with a more migratory phenotype, which was reverted by blocking IL6 and TNFα (Goebel et al, 2015). In breast cancer, also CD8+ cytotoxic T cells induce EMT, specifically during relapse and loss of a HER2/neu antigen‐mediated response, resulting in a more stem cell‐like phenotype with increased therapy resistance (Kmieciak et al, 2007; Santisteban et al, 2009). In lung cancer, specific T‐lymphocyte subsets induce EMT in co‐culture experiments via IL9 and IL17 secretion and block of IL9/IL17 reduced EMT and metastasis (Salazar et al, 2020) (Fig 5).
Therapeutic options to target EMT
As outlined in the previous chapters, various aspects of EMT are contributing to poor outcome, including resistance to treatment. Since therapeutic intervention with standard care, targeted therapy, irradiation, and surgery is often promoting EMT, major benefit is expected from therapeutic approaches that prevent or reverse the fatal effects of EMT. Standard treatment for targeting differentiated highly proliferative cancer cells combined with specific therapy to either block/revert an EMT/stemness/therapy‐resistant state or to target specific, yet unidentified, vulnerabilities of such exquisitely plastic cells seems advantageous. Several molecules to target EMT have been or are currently in clinical trials whereas others are already in use. These include blocking of upstream pathways which promote tumorigenesis also beyond EMT by ligand‐neutralizing antibodies, decoy receptors or inhibitors to block TGFβ, NF‐κB, EGFR, c‐MET, WNT and Notch signaling (comprehensively reviewed in (Redfern et al, 2018; Dudas et al, 2020; Jonckheere et al, 2021).
Due to difficulties in targeting transcription factors, i.e., EMT‐TFs, directly, another promising approach is to apply modified synthetic miRNAs that interfere with EMT‐TFs on a post‐transcriptional level. Preclinical data demonstrate that miR‐34 and miR‐200s administration will attenuate SNAIL and ZEB1 protein levels, respectively, and inhibit stemness and metastasis (Pecot et al, 2013; Cortez et al, 2014). Liposomal miR‐34 (MRX34) is already used in clinical trials with effects on tumor growth and metastasis (Zhang et al, 2019a).
Another interesting approach to block cell plasticity and to resensitize tumor cells to standard therapy is to induce re‐ or transdifferentiation. Interfering with chromatin remodeling by using HDAC inhibition or DNA‐demethylating agents has been proven valid methods to induce differentiation. In pancreatic cancer, the application of the HDAC class I inhibitor mocetinostat results in de‐repression of ZEB1‐inhibiting miRNAs, ZEB1 downmodulation, MET, and resensitization to gemcitabine in mice (Meidhof et al, 2015). Reactivating expression of silenced genes has a similar effect in osteosarcoma. Demethylation of the imprinted DLK‐DIO3 locus by 5‐Azacitidine reactivates expression of ZEB1‐silencing miRNAs, induces osteogenic and adipogenic differentiation, and sensitizes to doxorubicin treatment (Ruh et al, 2021). The idea of transdifferentiation was nicely demonstrated by Ishay‐Ronen et al, showing that EMT‐derived breast cancer cells can be differentiated into post‐mitotic functional adipocytes by rosiglitazone in vivo, resulting in reduced metastasis (Ishay‐Ronen et al, 2019). Another group of molecules that induce re‐differentiation and drug sensitivity includes compounds generated for different purposes, like metformin, used for type2 diabetes, or herbal components, like curcumin (Kothari et al, 2014; Dudas et al, 2020; Jonckheere et al, 2021). Metformin leads to reduction in SNAIL, TWIST, and ZEB1 levels, and curcumin inhibits EMT by downregulation of NF‐κB/SNAIL in breast cancer and reactivation of epithelial miRNAs (Kothari et al, 2014; Dudas et al, 2020).
With all these promising approaches to block and reverse EMT, it is important to consider potential adverse effects. Although EMT is harmful during tumorigenesis and EMT‐TFs contribute to poor prognosis, they play very important physiological roles. EMT is involved in wound closure and repair after injury and EMT‐TFs have important functions in stem cell homeostasis and differentiation, e.g., in melanocytes, muscle, bone and the nervous system, as well as in innate and adaptive immune responses. Accordingly, global interference with the context‐dependent favorable actions of EMT‐TF by special therapies may well affect normal tissue homeostasis. Likewise, miRNA‐based applications will simultaneously regulate other targets with unpredicted outcome. On the contrary, therapeutic intervention that favors MET, might promote colonization and metastasis of circulating tumor cells, leading to opposing effects. Hence, a very careful evaluation and examination of adverse effects is needed to promote such approaches in the clinic. Moreover, we are only beginning to understand the complex nature of the multi‐tool EMT in cancer and integration of aspects of multiple pathways and of TME diversity are the main challenges toward a more comprehensive understanding of metastasis for identifying novel treatment approaches.
Conflict of interest
The authors declare that they have no conflict of interest.
Acknowledgments
We apologize to our colleagues whose work could not be cited owing to space limitations. This work was supported by the German Research Foundation (TRR305 TP A03, A04, B01, and B07, FOR2438 P04 and BR1399/9‐1, BR1399/10‐1, BR4145/1‐1, and BR4145/2‐1), the Wilhelm Sander Stiftung (2020.039.1), and the European Union’s Horizon 2020 research and innovation program under the Marie Skłodowska‐Curie grant agreement (No 861196; PRECODE). Open Access funding enabled and organized by Projekt DEAL.
The EMBO Journal (2021) 40: e108647.
This article is part of the Cancer Reviews 2021 series.
Contributor Information
Simone Brabletz, Email: simone.brabletz@fau.de.
Marc P. Stemmler, Email: marc.stemmler@fau.de.
References
- Aceto N, Bardia A, Miyamoto D, Donaldson M, Wittner B, Spencer J, Yu M, Pely A, Engstrom A, Zhu Het al (2014) Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 158: 1110–1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed N, Maines‐Bandiera S, Quinn MA, Unger WG, Dedhar S, Auersperg N (2006) Molecular pathways regulating EGF‐induced epithelio‐mesenchymal transition in human ovarian surface epithelium. Am J Physiol Cell Physiol 290: C1532–1542 [DOI] [PubMed] [Google Scholar]
- Aiello NM, Maddipati R, Norgard RJ, Balli D, Li J, Yuan S, Yamazoe T, Black T, Sahmoud A, Furth EEet al (2018) EMT subtype influences epithelial plasticity and mode of cell migration. Dev Cell 45: 681–695.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aigner K, Dampier B, Descovich L, Mikula M, Sultan A, Schreiber M, Mikulits W, Brabletz T, Strand D, Obrist Pet al (2007) The transcription factor ZEB1 (deltaEF1) promotes tumour cell dedifferentiation by repressing master regulators of epithelial polarity. Oncogene 26: 6979–6988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akalay I, Janji B, Hasmim M, Noman MZ, André F, De Cremoux P, Bertheau P, Badoual C, Vielh P, Larsen AKet al (2013) Epithelial‐to‐mesenchymal transition and autophagy induction in breast carcinoma promote escape from T‐cell‐mediated lysis. Cancer Res 73: 2418–2427 [DOI] [PubMed] [Google Scholar]
- Aller SG, Yu J, Ward A, Weng Y, Chittaboina S, Zhuo R, Harrell PM, Trinh YT, Zhang Q, Urbatsch ILet al (2009) Structure of P‐glycoprotein reveals a molecular basis for poly‐specific drug binding. Science 323: 1718–1722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansari D, Ohlsson H, Althini C, Bauden M, Zhou Q, Hu D, Andersson R (2019) The hippo signaling pathway in pancreatic cancer. Anticancer Res 39: 3317–3321 [DOI] [PubMed] [Google Scholar]
- Ansieau S, Bastid J, Doreau A, Morel A‐P, Bouchet BP, Thomas C, Fauvet F, Puisieux I, Doglioni C, Piccinin Set al (2008) Induction of EMT by twist proteins as a collateral effect of tumor‐promoting inactivation of premature senescence. Cancer Cell 14: 79–89 [DOI] [PubMed] [Google Scholar]
- Arnoux V, Nassour M, L'Helgoualc'h A, Hipskind RA, Savagner P (2008) Erk5 controls Slug expression and keratinocyte activation during wound healing. Mol Biol Cell 19: 4738–4749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakin AV, Tomlinson AK, Bhowmick NA, Moses HL, Arteaga CL (2000) Phosphatidylinositol 3‐kinase function is required for transforming growth factor beta‐mediated epithelial to mesenchymal transition and cell migration. J Biol Chem 275: 36803–36810 [DOI] [PubMed] [Google Scholar]
- Bates RC, Mercurio AM (2003) Tumor necrosis factor‐alpha stimulates the epithelial‐to‐mesenchymal transition of human colonic organoids. Mol Biol Cell 14: 1790–1800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batlle E, Sancho E, Franci C, Dominguez D, Monfar M, Baulida J, Garcia De Herreros A (2000) The transcription factor snail is a repressor of E‐cadherin gene expression in epithelial tumour cells. Nat Cell Biol 2: 84–89 [DOI] [PubMed] [Google Scholar]
- Bergers G, Fendt SM (2021) The metabolism of cancer cells during metastasis. Nat Rev Cancer 21: 162–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhowmick NA, Chytil A, Plieth D, Gorska AE, Dumont N, Shappell S, Washington MK, Neilson EG, Moses HL (2004) TGF‐beta signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science 303: 848–851 [DOI] [PubMed] [Google Scholar]
- Birchmeier W, Behrens J (1994) Cadherin expression in carcinomas: role in the formation of cell junctions and the prevention of invasiveness. Biochim Biophys Acta 1198: 11–26 [DOI] [PubMed] [Google Scholar]
- Blokzijl A, Dahlqvist C, Reissmann E, Falk A, Moliner A, Lendahl U, Ibanez CF (2003) Cross‐talk between the Notch and TGF‐beta signaling pathways mediated by interaction of the Notch intracellular domain with Smad3. J Cell Biol 163: 723–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonde AK, Tischler V, Kumar S, Soltermann A, Schwendener RA (2012) Intratumoral macrophages contribute to epithelial‐mesenchymal transition in solid tumors. BMC Cancer 12: 35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornes L, van Scheppingen RH, Beerling E, Schelfhorst T, Ellenbroek SIJ, Seinstra D, van Rheenen J (2019) Fsp1‐mediated lineage tracing fails to detect the majority of disseminating cells undergoing EMT. Cell Rep 29: 2565–2569.e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brabletz S, Bajdak K, Meidhof S, Burk U, Niedermann G, Firat E, Wellner U, Dimmler A, Faller G, Schubert Jet al (2011) The ZEB1/miR‐200 feedback loop controls Notch signalling in cancer cells. EMBO J 30: 770–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brabletz S, Brabletz T (2010) The ZEB/miR‐200 feedback loop–a motor of cellular plasticity in development and cancer? EMBO Rep 11: 670–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brabletz T, Jung A, Reu S, Porzner M, Hlubek F, Kunz‐Schughart LA, Knuechel R, Kirchner T (2001) Variable beta‐catenin expression in colorectal cancers indicates tumor progression driven by the tumor environment. Proc Natl Acad Sci U S A 98: 10356–10361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brabletz T, Kalluri R, Nieto MA, Weinberg RA (2018) EMT in cancer. Nat Rev Cancer 18: 128–134 [DOI] [PubMed] [Google Scholar]
- Bracken CP, Gregory PA, Kolesnikoff N, Bert AG, Wang J, Shannon MF, Goodall GJ (2008) A double‐negative feedback loop between ZEB1‐SIP1 and the microRNA‐200 family regulates epithelial‐mesenchymal transition. Cancer Res 68: 7846–7854 [DOI] [PubMed] [Google Scholar]
- Bray SJ (2016) Notch signalling in context. Nat Rev Mol Cell Biol 17: 722–735 [DOI] [PubMed] [Google Scholar]
- Burk U, Schubert J, Wellner U, Schmalhofer O, Vincan E, Spaderna S, Brabletz T (2008) A reciprocal repression between ZEB1 and members of the miR‐200 family promotes EMT and invasion in cancer cells. EMBO Rep 9: 582–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byers LA, Diao L, Wang J, Saintigny P, Girard L, Peyton M, Shen Li, Fan Y, Giri U, Tumula PKet al (2013) An epithelial‐mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin Cancer Res 19: 279–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calon A, Lonardo E, Berenguer‐Llergo A, Espinet E, Hernando‐Momblona X, Iglesias M, Sevillano M, Palomo‐Ponce S, Tauriello DVF, Byrom Det al (2015) Stromal gene expression defines poor‐prognosis subtypes in colorectal cancer. Nat Genet 47: 320–329 [DOI] [PubMed] [Google Scholar]
- Cano A, Perez‐Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, Portillo F, Nieto MA (2000) The transcription factor snail controls epithelial‐mesenchymal transitions by repressing E‐cadherin expression. Nat Cell Biol 2: 76–83 [DOI] [PubMed] [Google Scholar]
- Cao Z, Livas T, Kyprianou N (2016) Anoikis and EMT: lethal "Liaisons" during cancer progression. Crit Rev Oncog 21: 155–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caramel J, Ligier M, Puisieux A (2018) Pleiotropic roles for ZEB1 in cancer. Cancer Res 78: 30–35 [DOI] [PubMed] [Google Scholar]
- Chaffer CL, Brennan JP, Slavin JL, Blick T, Thompson EW, Williams ED (2006) Mesenchymal‐to‐epithelial transition facilitates bladder cancer metastasis: role of fibroblast growth factor receptor‐2. Cancer Res 66: 11271–11278 [DOI] [PubMed] [Google Scholar]
- Che D, Zhang S, Jing Z, Shang L, Jin S, Liu F, Shen J, Li Y, Hu J, Meng Qet al (2017) Macrophages induce EMT to promote invasion of lung cancer cells through the IL‐6‐mediated COX‐2/PGE2/beta‐catenin signalling pathway. Mol Immunol 90: 197–210 [DOI] [PubMed] [Google Scholar]
- Chen BJ, Wu JS, Tang YJ, Tang YL, Liang XH (2020) What makes leader cells arise: Intrinsic properties and support from neighboring cells. J Cell Physiol 235: 8983–8995 [DOI] [PubMed] [Google Scholar]
- Chen L, Gibbons DL, Goswami S, Cortez MA, Ahn Y‐H, Byers LA, Zhang X, Yi X, Dwyer D, Lin Wet al (2014) Metastasis is regulated via microRNA‐200/ZEB1 axis control of tumour cell PD‐L1 expression and intratumoral immunosuppression. Nat Commun 5: 5241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Kim J, Yang S, Wang H, Wu CJ, Sugimoto H, LeBleu VS, Kalluri R (2021) Type I collagen deletion in alphaSMA(+) myofibroblasts augments immune suppression and accelerates progression of pancreatic cancer. Cancer Cell 39: 548–565.e546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung KJ, Padmanaban V, Silvestri V, Schipper K, Cohen JD, Fairchild AN, Gorin MA, Verdone JE, Pienta KJ, Bader JSet al (2016) Polyclonal breast cancer metastases arise from collective dissemination of keratin 14‐expressing tumor cell clusters. Proc Natl Acad Sci U S A 113: E854–E863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clevers H (2006) Wnt/beta‐catenin signaling in development and disease. Cell 127: 469–480 [DOI] [PubMed] [Google Scholar]
- Colomiere M, Ward AC, Riley C, Trenerry MK, Cameron‐Smith D, Findlay J, Ackland L, Ahmed N (2009) Cross talk of signals between EGFR and IL‐6R through JAK2/STAT3 mediate epithelial‐mesenchymal transition in ovarian carcinomas. Br J Cancer 100: 134–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comaills V, Kabeche L, Morris R, Buisson R, Yu M, Madden MW, LiCausi JA, Boukhali M, Tajima K, Pan Set al (2016) Genomic instability is induced by persistent proliferation of cells undergoing epithelial‐to‐mesenchymal transition. Cell Rep 17: 2632–2647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comijn J, Berx G, Vermassen P, Verschueren K, van Grunsven L, Bruyneel E, Mareel M, Huylebroeck D, van Roy F (2001) The two‐handed E box binding zinc finger protein SIP1 downregulates E‐cadherin and induces invasion. Mol Cell 7: 1267–1278 [DOI] [PubMed] [Google Scholar]
- Cooke V, LeBleu V, Keskin D, Khan Z, O'Connell J, Teng Y, Duncan M, Xie L, Maeda G, Vong Set al (2012) Pericyte depletion results in hypoxia‐associated epithelial‐to‐mesenchymal transition and metastasis mediated by met signaling pathway. Cancer Cell 21: 66–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez MA, Valdecanas D, Zhang X, Zhan Y, Bhardwaj V, Calin GA, Komaki R, Giri DK, Quini CC, Wolfe Tet al (2014) Therapeutic delivery of miR‐200c enhances radiosensitivity in lung cancer. Mol Ther 22: 1494–1503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creighton CJ, Li X, Landis M, Dixon JM, Neumeister VM, Sjolund A, Rimm DL, Wong H, Rodriguez A, Herschkowitz JIet al (2009) Residual breast cancers after conventional therapy display mesenchymal as well as tumor‐initiating features. Proc Natl Acad Sci U S A 106: 13820–13825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Sousa e Melo F, Kurtova AV, Harnoss JM, Kljavin N, Hoeck JD, Hung J, Anderson JE, Storm EE, Modrusan Z, Koeppen Het al (2017) A distinct role for Lgr5(+) stem cells in primary and metastatic colon cancer. Nature 543: 676–680 [DOI] [PubMed] [Google Scholar]
- Debaugnies M, Sanchez‐Danes A, Rorive S, Raphael M, Liagre M, Parent MA, Brisebarre A, Salmon I, Blanpain C (2018) YAP and TAZ are essential for basal and squamous cell carcinoma initiation. EMBO Rep 19: e45809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Pozo Martin Y, Park D, Ramachandran A, Ombrato L, Calvo F, Chakravarty P, Spencer‐Dene B, Derzsi S, Hill C, Sahai Eet al (2015) Mesenchymal cancer cell‐stroma crosstalk promotes niche activation, epithelial reversion, and metastatic colonization. Cell Rep 13: 2456–2469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derynck R, Turley SJ, Akhurst RJ (2021) TGFbeta biology in cancer progression and immunotherapy. Nat Rev Clin Oncol 18: 9–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devarajan E, Song YH, Krishnappa S, Alt E (2012) Epithelial‐mesenchymal transition in breast cancer lines is mediated through PDGF‐D released by tissue‐resident stem cells. Int J Cancer 131: 1023–1031 [DOI] [PubMed] [Google Scholar]
- Diaz‐Lopez A, Moreno‐Bueno G, Cano A (2014) Role of microRNA in epithelial to mesenchymal transition and metastasis and clinical perspectives. Cancer Manag Res 6: 205–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diepenbruck M, Waldmeier L, Ivanek R, Berninger P, Arnold P, van Nimwegen E, Christofori G (2014) Tead2 expression levels control the subcellular distribution of Yap and Taz, zyxin expression and epithelial‐mesenchymal transition. J Cell Sci 127: 1523–1536 [DOI] [PubMed] [Google Scholar]
- Dongre A, Rashidian M, Reinhardt F, Bagnato A, Keckesova Z, Ploegh HL, Weinberg RA (2017) Epithelial‐to‐mesenchymal transition contributes to immunosuppression in breast carcinomas. Cancer Res 77: 3982–3989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dongre A, Weinberg RA (2019) New insights into the mechanisms of epithelial‐mesenchymal transition and implications for cancer. Nat Rev Mol Cell Biol 20: 69–84 [DOI] [PubMed] [Google Scholar]
- Dudas J, Ladanyi A, Ingruber J, Steinbichler TB, Riechelmann H (2020) Epithelial to mesenchymal transition: a mechanism that fuels cancer radio/chemoresistance. Cells 9: 428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckert MA, Lwin TM, Chang AT, Kim J, Danis E, Ohno‐Machado L, Yang J (2011) Twist1‐induced invadopodia formation promotes tumor metastasis. Cancer Cell 19: 372–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eger A, Aigner K, Sonderegger S, Dampier B, Oehler S, Schreiber M, Berx G, Cano A, Beug H, Foisner R (2005) DeltaEF1 is a transcriptional repressor of E‐cadherin and regulates epithelial plasticity in breast cancer cells. Oncogene 24: 2375–2385 [DOI] [PubMed] [Google Scholar]
- Elyada E, Bolisetty M, Laise P, Flynn WF, Courtois ET, Burkhart RA, Teinor JA, Belleau P, Biffi G, Lucito MSet al (2019) Cross‐species single‐cell analysis of pancreatic ductal adenocarcinoma reveals antigen‐presenting cancer‐associated fibroblasts. Cancer Discov 9: 1102–1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escrivà M, Peiró S, Herranz N, Villagrasa P, Dave N, Montserrat‐Sentís B, Murray SA, Francí C, Gridley T, Virtanen Iet al (2008) Repression of PTEN phosphatase by Snail1 transcriptional factor during gamma radiation‐induced apoptosis. Mol Cell Biol 28: 1528–1540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito M, Mondal N, Greco TM, Wei Y, Spadazzi C, Lin S‐C, Zheng H, Cheung C, Magnani JL, Lin S‐Het al (2019) Bone vascular niche E‐selectin induces mesenchymal‐epithelial transition and Wnt activation in cancer cells to promote bone metastasis. Nat Cell Biol 21: 627–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Q‐M, Jing Y‐Y, Yu G‐F, Kou X‐R, Ye F, Gao Lu, Li R, Zhao Q‐D, Yang Y, Lu Z‐Het al (2014) Tumor‐associated macrophages promote cancer stem cell‐like properties via transforming growth factor‐beta1‐induced epithelial‐mesenchymal transition in hepatocellular carcinoma. Cancer Lett 352: 160–168 [DOI] [PubMed] [Google Scholar]
- Farmer P, Bonnefoi H, Anderle P, Cameron D, Wirapati P, Becette V, André S, Piccart M, Campone M, Brain Eet al (2009) A stroma‐related gene signature predicts resistance to neoadjuvant chemotherapy in breast cancer. Nat Med 15: 68–74 [DOI] [PubMed] [Google Scholar]
- Fearon ER, Vogelstein B (1990) A genetic model for colorectal tumorigenesis. Cell 61: 759–767 [DOI] [PubMed] [Google Scholar]
- Feldker N, Ferrazzi F, Schuhwerk H, Widholz SA, Guenther K, Frisch I, Jakob K, Kleemann J, Riegel D, Bonisch Uet al (2020) Genome‐wide cooperation of EMT transcription factor ZEB1 with YAP and AP‐1 in breast cancer. EMBO J 39: e103209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fifis T, Nguyen L, Malcontenti‐Wilson C, Chan LS, Nunes Costa PL, Daruwalla J, Nikfarjam M, Muralidharan V, Waltham M, Thompson EWet al (2013) Treatment with the vascular disruptive agent OXi4503 induces an immediate and widespread epithelial to mesenchymal transition in the surviving tumor. Cancer Med 2: 595–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer KR, Durrans A, Lee S, Sheng J, Li F, Wong STC, Choi H, El Rayes T, Ryu S, Troeger Jet al (2015) Epithelial‐to‐mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 527: 472–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedl P, Locker J, Sahai E, Segall JE (2012) Classifying collective cancer cell invasion. Nat Cell Biol 14: 777–783 [DOI] [PubMed] [Google Scholar]
- Fumagalli A, Oost KC, Kester L, Morgner J, Bornes L, Bruens L, Spaargaren L, Azkanaz M, Schelfhorst T, Beerling Eet al (2020) Plasticity of Lgr5‐negative cancer cells drives metastasis in colorectal cancer. Cell Stem Cell 26: 569–578.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabrilovich DI (2017) Myeloid‐derived suppressor cells. Cancer Immunol Res 5: 3–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabrilovich DI, Nagaraj S (2009) Myeloid‐derived suppressor cells as regulators of the immune system. Nat Rev Immunol 9: 162–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauger KJ, Chenausky KL, Murray ME, Schneider SS (2011) SFRP1 reduction results in an increased sensitivity to TGF‐beta signaling. BMC Cancer 11: 59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng Y, Fan J, Chen L, Zhang C, Qu C, Qian L, Chen K, Meng Z, Chen Z, Wang P (2021) A notch‐dependent inflammatory feedback circuit between macrophages and cancer cells regulates pancreatic cancer metastasis. Cancer Res 81: 64–76 [DOI] [PubMed] [Google Scholar]
- Georgakopoulos‐Soares I, Chartoumpekis DV, Kyriazopoulou V, Zaravinos A (2020) EMT factors and metabolic pathways in cancer. Front Oncol 10: 499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goebel L, Grage‐Griebenow E, Gorys A, Helm O, Genrich G, Lenk L, Wesch D, Ungefroren H, Freitag‐Wolf S, Sipos Bet al (2015) CD4(+) T cells potently induce epithelial‐mesenchymal‐transition in premalignant and malignant pancreatic ductal epithelial cells‐novel implications of CD4(+) T cells in pancreatic cancer development. Oncoimmunology 4: e1000083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez H, Hagerling C, Werb Z (2018) Roles of the immune system in cancer: from tumor initiation to metastatic progression. Genes Dev 32: 1267–1284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goulet CR, Champagne A, Bernard G, Vandal D, Chabaud S, Pouliot F, Bolduc S (2019) Cancer‐associated fibroblasts induce epithelial‐mesenchymal transition of bladder cancer cells through paracrine IL‐6 signalling. BMC Cancer 19: 137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham TR, Zhau HE, Odero‐Marah VA, Osunkoya AO, Kimbro KS, Tighiouart M, Liu T, Simons JW, O'Regan RM (2008) Insulin‐like growth factor‐I‐dependent up‐regulation of ZEB1 drives epithelial‐to‐mesenchymal transition in human prostate cancer cells. Cancer Res 68: 2479–2488 [DOI] [PubMed] [Google Scholar]
- Gross KM, Zhou W, Breindel JL, Ouyang J, Jin DX, Sokol ES, Gupta PB, Huber K, Zou L, Kuperwasser C (2019) Loss of slug compromises DNA damage repair and accelerates stem cell aging in mammary epithelium. Cell Rep 28: 394–407.e6 [DOI] [PubMed] [Google Scholar]
- Grotegut S, von Schweinitz D, Christofori G, Lehembre F (2006) Hepatocyte growth factor induces cell scattering through MAPK/Egr‐1‐mediated upregulation of Snail. EMBO J 25: 3534–3545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubelmann C, Schwalie PC, Raghav SK, Röder E, Delessa T, Kiehlmann E, Waszak SM, Corsinotti A, Udin G, Holcombe Wet al (2014) Identification of the transcription factor ZEB1 as a central component of the adipogenic gene regulatory network. Elife 3: e03346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudey SK, Sundar R, Heldin CH, Bergh A, Landstrom M (2017) Pro‐invasive properties of Snail1 are regulated by sumoylation in response to TGFbeta stimulation in cancer. Oncotarget 8: 97703–97726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Keckesova Z, Donaher J, Shibue T, Tischler V, Reinhardt F, Itzkovitz S, Noske A, Zürrer‐Härdi U, Bell Get al (2012) Slug and Sox9 cooperatively determine the mammary stem cell state. Cell 148: 1015–1028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg RA, Lander ES (2009) Identification of selective inhibitors of cancer stem cells by high‐throughput screening. Cell 138: 645–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halazonetis TD, Gorgoulis VG, Bartek J (2008) An oncogene‐induced DNA damage model for cancer development. Science 319: 1352–1355 [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144: 646–674 [DOI] [PubMed] [Google Scholar]
- Hao Y, Baker D, Ten Dijke P (2019) TGF‐beta‐mediated epithelial‐mesenchymal transition and cancer metastasis. Int J Mol Sci 20: 2767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hapke RY, Haake SM (2020) Hypoxia‐induced epithelial to mesenchymal transition in cancer. Cancer Lett 487: 10–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara J, Miyata H, Yamasaki M, Sugimura K, Takahashi T, Kurokawa Y, Nakajima K, Takiguchi S, Mori M, Doki Y (2014) Mesenchymal phenotype after chemotherapy is associated with chemoresistance and poor clinical outcome in esophageal cancer. Oncol Rep 31: 589–596 [DOI] [PubMed] [Google Scholar]
- Hay ED (1995) An overview of epithelio‐mesenchymal transformation. Acta Anat (Basel) 154: 8–20 [DOI] [PubMed] [Google Scholar]
- He X, Yan B, Liu S, Jia J, Lai W, Xin X, Tang C‐e, Luo D, Tan T, Jiang Yet al (2016) Chromatin remodeling factor LSH drives cancer progression by suppressing the activity of fumarate hydratase. Cancer Res 76: 5743–5755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho MY, Tang SJ, Chuang MJ, Cha TL, Li JY, Sun GH, Sun KH (2012) TNF‐alpha induces epithelial‐mesenchymal transition of renal cell carcinoma cells via a GSK3beta‐dependent mechanism. Mol Cancer Res 10: 1109–1119 [DOI] [PubMed] [Google Scholar]
- Hoot KE, Lighthall J, Han G, Lu SL, Li A, Ju W, Kulesz‐Martin M, Bottinger E, Wang XJ (2008) Keratinocyte‐specific Smad2 ablation results in increased epithelial‐mesenchymal transition during skin cancer formation and progression. J Clin Invest 118: 2722–2732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua W, Ten Dijke P, Kostidis S, Giera M, Hornsveld M (2020) TGFbeta‐induced metabolic reprogramming during epithelial‐to‐mesenchymal transition in cancer. Cell Mol Life Sci 77: 2103–2123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CH, Yang WH, Chang SY, Tai SK, Tzeng CH, Kao JY, Wu KJ, Yang MH (2009) Regulation of membrane‐type 4 matrix metalloproteinase by SLUG contributes to hypoxia‐mediated metastasis. Neoplasia 11: 1371–1382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang RY, Guilford P, Thiery JP (2012) Early events in cell adhesion and polarity during epithelial‐mesenchymal transition. J Cell Sci 125: 4417–4422 [DOI] [PubMed] [Google Scholar]
- Hugo W, Zaretsky JM, Sun Lu, Song C, Moreno BH, Hu‐Lieskovan S, Berent‐Maoz B, Pang J, Chmielowski B, Cherry Get al (2016) Genomic and transcriptomic features of response to anti‐PD‐1 therapy in metastatic melanoma. Cell 165: 35–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ieda T, Tazawa H, Okabayashi H, Yano S, Shigeyasu K, Kuroda S, Ohara T, Noma K, Kishimoto H, Nishizaki Met al (2019) Visualization of epithelial‐mesenchymal transition in an inflammatory microenvironment‐colorectal cancer network. Sci Rep 9: 16378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishay‐Ronen D, Diepenbruck M, Kalathur RKR, Sugiyama N, Tiede S, Ivanek R, Bantug G, Morini MF, Wang J, Hess Cet al (2019) Gain fat‐lose metastasis: converting invasive breast cancer cells into adipocytes inhibits cancer metastasis. Cancer Cell 35: 17–32.e6 [DOI] [PubMed] [Google Scholar]
- Jiang YP, Tang YL, Wang SS, Wu JS, Zhang M, Pang X, Wu JB, Chen Y, Tang YJ, Liang XH (2020) PRRX1‐induced epithelial‐to‐mesenchymal transition in salivary adenoid cystic carcinoma activates the metabolic reprogramming of free fatty acids to promote invasion and metastasis. Cell Prolif 53: e12705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Zhan H (2020) Communication between EMT and PD‐L1 signaling: new insights into tumor immune evasion. Cancer Lett 468: 72–81 [DOI] [PubMed] [Google Scholar]
- Jin MZ, Jin WL (2020) The updated landscape of tumor microenvironment and drug repurposing. Signal Transduct Target Ther 5: 166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joffroy CM, Buck MB, Stope MB, Popp SL, Pfizenmaier K, Knabbe C (2010) Antiestrogens induce transforming growth factor beta‐mediated immunosuppression in breast cancer. Cancer Res 70: 1314–1322 [DOI] [PubMed] [Google Scholar]
- Jonckheere S, Adams J, De Groote D, Campbell K, Berx G, Goossens S (2021) Epithelial‐Mesenchymal Transition (EMT) as a therapeutic target. Cells Tissues Organs 1–26. 10.1159/000512218 [DOI] [PubMed] [Google Scholar]
- Joyce JA, Pollard JW (2009) Microenvironmental regulation of metastasis. Nat Rev Cancer 9: 239–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung AR, Jung CH, Noh JK, Lee YC, Eun YG (2020) Epithelial‐mesenchymal transition gene signature is associated with prognosis and tumor microenvironment in head and neck squamous cell carcinoma. Sci Rep 10: 3652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajita M, McClinic KN, Wade PA (2004) Aberrant expression of the transcription factors snail and slug alters the response to genotoxic stress. Mol Cell Biol 24: 7559–7566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang H, Kim H, Lee S, Youn H, Youn B (2019) Role of metabolic reprogramming in Epithelial(‐)Mesenchymal Transition (EMT). Int J Mol Sci 20: 2042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kardos J, Chai S, Mose LE, Selitsky SR, Krishnan B, Saito R, Iglesia MD, Milowsky MI, Parker JS, Kim WYet al (2016) Claudin‐low bladder tumors are immune infiltrated and actively immune suppressed. JCI Insight 1: e85902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsura A, Tamura Y, Hokari S, Harada M, Morikawa M, Sakurai T, Takahashi K, Mizutani A, Nishida J, Yokoyama Yet al (2017) ZEB1‐regulated inflammatory phenotype in breast cancer cells. Mol Oncol 11: 1241–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamoto A, Yokoe T, Tanaka K, Saigusa S, Toiyama Y, Yasuda H, Inoue Y, Miki C, Kusunoki M (2012) Radiation induces epithelial‐mesenchymal transition in colorectal cancer cells. Oncol Rep 27: 51–57 [DOI] [PubMed] [Google Scholar]
- Khot M, Sreekumar D, Jahagirdar S, Kulkarni A, Hari K, Faseela EE, Sabarinathan R, Jolly MK, Sengupta K (2020) Twist1 induces chromosomal instability (CIN) in colorectal cancer cells. Hum Mol Genet 29: 1673–1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kong J, Chang H, Kim H, Kim A (2016) EGF induces epithelial‐mesenchymal transition through phospho‐Smad2/3‐Snail signaling pathway in breast cancer cells. Oncotarget 7: 85021–85032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T, Veronese A, Pichiorri F, Lee TJ, Jeon Y‐J, Volinia S, Pineau P, Marchio A, Palatini J, Suh S‐Set al (2011) p53 regulates epithelial‐mesenchymal transition through microRNAs targeting ZEB1 and ZEB2. J Exp Med 208: 875–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kmieciak M, Knutson KL, Dumur CI, Manjili MH (2007) HER‐2/neu antigen loss and relapse of mammary carcinoma are actively induced by T cell‐mediated anti‐tumor immune responses. Eur J Immunol 37: 675–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korpal M, Ell BJ, Buffa FM, Ibrahim T, Blanco MA, Celià‐Terrassa T, Mercatali L, Khan Z, Goodarzi H, Hua Yet al (2011) Direct targeting of Sec23a by miR‐200s influences cancer cell secretome and promotes metastatic colonization. Nat Med 17: 1101–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kothari AN, Mi Z, Zapf M, Kuo PC (2014) Novel clinical therapeutics targeting the epithelial to mesenchymal transition. Clin Transl Med 3: 35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs AM, Mitschke J, Lasierra Losada M, Schmalhofer O, Boerries M, Busch H, Boettcher M, Mougiakakos D, Reichardt W, Bronsert Pet al (2017) The EMT‐activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat Cell Biol 19: 518–529 [DOI] [PubMed] [Google Scholar]
- Kudo‐Saito C, Ozaki Y, Imazeki H, Hayashi H, Masuda J, Ozawa H, Ogiwara Y (2021) Targeting oncoimmune drivers of cancer metastasis. Cancers (Basel) 13: 554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudo‐Saito C, Shirako H, Takeuchi T, Kawakami Y (2009) Cancer metastasis is accelerated through immunosuppression during Snail‐induced EMT of cancer cells. Cancer Cell 15: 195–206 [DOI] [PubMed] [Google Scholar]
- Kumar V, Donthireddy L, Marvel D, Condamine T, Wang F, Lavilla‐Alonso S, Hashimoto A, Vonteddu P, Behera R, Goins MAet al (2017) Cancer‐associated fibroblasts neutralize the anti‐tumor effect of CSF1 receptor blockade by inducing PMN‐MDSC infiltration of tumors. Cancer Cell 32: 654–668.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurrey NK, Jalgaonkar SP, Joglekar AV, Ghanate AD, Chaskar PD, Doiphode RY, Bapat SA (2009) Snail and slug mediate radioresistance and chemoresistance by antagonizing p53‐mediated apoptosis and acquiring a stem‐like phenotype in ovarian cancer cells. Stem Cells 27: 2059–2068 [DOI] [PubMed] [Google Scholar]
- Lamar JM, Stern P, Liu H, Schindler JW, Jiang ZG, Hynes RO (2012) The Hippo pathway target, YAP, promotes metastasis through its TEAD‐interaction domain. Proc Natl Acad Sci U S A 109: E2441–E2450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambrechts D, Wauters E, Boeckx B, Aibar S, Nittner D, Burton O, Bassez A, Decaluwé H, Pircher A, Van den Eynde Ket al (2018) Phenotype molding of stromal cells in the lung tumor microenvironment. Nat Med 24: 1277–1289 [DOI] [PubMed] [Google Scholar]
- Lamouille S, Connolly E, Smyth JW, Akhurst RJ, Derynck R (2012) TGF‐beta‐induced activation of mTOR complex 2 drives epithelial‐mesenchymal transition and cell invasion. J Cell Sci 125: 1259–1273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamouille S, Derynck R (2007) Cell size and invasion in TGF‐beta‐induced epithelial to mesenchymal transition is regulated by activation of the mTOR pathway. J Cell Biol 178: 437–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamouille S, Xu J, Derynck R (2014) Molecular mechanisms of epithelial‐mesenchymal transition. Nat Rev Mol Cell Biol 15: 178–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen JE, Nathan V, Osborne JK, Farrow RK, Deb D, Sullivan JP, Dospoy PD, Augustyn A, Hight SK, Sato Met al (2016) ZEB1 drives epithelial‐to‐mesenchymal transition in lung cancer. J Clin Invest 126: 3219–3235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBleu VS, Kalluri R (2018) A peek into cancer‐associated fibroblasts: origins, functions and translational impact. Dis Model Mech 11: dmm029447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CC, Lin JC, Hwang WL, Kuo YJ, Chen HK, Tai SK, Lin CC, Yang MH (2018) Macrophage‐secreted interleukin‐35 regulates cancer cell plasticity to facilitate metastatic colonization. Nat Commun 9: 3763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann W, Mossmann D, Kleemann J, Mock K, Meisinger C, Brummer T, Herr R, Brabletz S, Stemmler MP, Brabletz T (2016) ZEB1 turns into a transcriptional activator by interacting with YAP1 in aggressive cancer types. Nat Commun 7: 10498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Cong X, Gao H, Lan X, Li Z, Wang W, Song S, Wang Y, Li C, Zhang Het al (2019) Tumor‐associated neutrophils induce EMT by IL‐17a to promote migration and invasion in gastric cancer cells. J Exp Clin Cancer Res 38: 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Wang L, Pappan L, Galliher‐Beckley A, Shi J (2012) IL‐1beta promotes stemness and invasiveness of colon cancer cells through Zeb1 activation. Mol Cancer 11: 87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Wang Y, Zhu Y, Yuan C, Wang D, Zhang W, Qi B, Qiu J, Song X, Ye Jet al (2015) The Hippo transducer TAZ promotes epithelial to mesenchymal transition and cancer stem cell maintenance in oral cancer. Mol Oncol 9: 1091–1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JC, Tsai JT, Chao TY, Ma HI, Liu WH (2018) The STAT3/slug axis enhances radiation‐induced tumor invasion and cancer stem‐like properties in radioresistant glioblastoma. Cancers 10: 512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Ruan B, You N, Huang Q, Liu W, Dang Z, Xu W, Zhou T, Ji R, Cao Yet al (2013) Downregulation of miR‐200a induces EMT phenotypes and CSC‐like signatures through targeting the beta‐catenin pathway in hepatic oval cells. PLoS One 8: e79409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Chen X, Wang Y, Qu Z, Lu Q, Zhao J, Yan X, Zhang H, Zhou Y (2014a) Notch3 is important for TGF‐beta‐induced epithelial‐mesenchymal transition in non‐small cell lung cancer bone metastasis by regulating ZEB‐1. Cancer Gene Ther 21: 364–372 [DOI] [PubMed] [Google Scholar]
- Liu S, Ren J, Ten Dijke P (2021) Targeting TGFbeta signal transduction for cancer therapy. Signal Transduct Target Ther 6: 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Lu X, Huang Li, Wang W, Jiang G, Dean KC, Clem B, Telang S, Jenson AB, Cuatrecasas Met al (2014b) Different thresholds of ZEB1 are required for Ras‐mediated tumour initiation and metastasis. Nat Commun 5: 5660 [DOI] [PubMed] [Google Scholar]
- Liu Z, Tu K, Wang Y, Yao B, Li Q, Wang L, Dou C, Liu Q, Zheng X (2017) Hypoxia accelerates aggressiveness of hepatocellular carcinoma cells involving oxidative stress, epithelial‐mesenchymal transition and non‐canonical hedgehog signaling. Cell Physiol Biochem 44: 1856–1868 [DOI] [PubMed] [Google Scholar]
- Lo HW, Hsu SC, Xia W, Cao X, Shih JY, Wei Y, Abbruzzese JL, Hortobagyi GN, Hung MC (2007) Epidermal growth factor receptor cooperates with signal transducer and activator of transcription 3 to induce epithelial‐mesenchymal transition in cancer cells via up‐regulation of TWIST gene expression. Cancer Res 67: 9066–9076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z, Ghosh S, Wang Z, Hunter T (2003) Downregulation of caveolin‐1 function by EGF leads to the loss of E‐cadherin, increased transcriptional activity of beta‐catenin, and enhanced tumor cell invasion. Cancer Cell 4: 499–515 [DOI] [PubMed] [Google Scholar]
- Lyons JG, Patel V, Roue NC, Fok SY, Soon LL, Halliday GM, Gutkind JS (2008) Snail up‐regulates proinflammatory mediators and inhibits differentiation in oral keratinocytes. Cancer Res 68: 4525–4530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Wang M, Yin T, Zhao Y, Wei X (2019) Myeloid‐derived suppressor cells promote metastasis in breast cancer after the stress of operative removal of the primary cancer. Front Oncol 9: 855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maehara O, Suda G, Natsuizaka M, Ohnishi S, Komatsu Y, Sato F, Nakai M, Sho T, Morikawa K, Ogawa Ket al (2017) Fibroblast growth factor‐2‐mediated FGFR/Erk signaling supports maintenance of cancer stem‐like cells in esophageal squamous cell carcinoma. Carcinogenesis 38: 1073–1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makrodouli E, Oikonomou E, Koc M, Andera L, Sasazuki T, Shirasawa S, Pintzas A (2011) BRAF and RAS oncogenes regulate Rho GTPase pathways to mediate migration and invasion properties in human colon cancer cells: a comparative study. Mol Cancer 10: 118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani SA, Guo W, Liao M‐J, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin Met al (2008) The epithelial‐mesenchymal transition generates cells with properties of stem cells. Cell 133: 704–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Z, Zhang J, Shi Y, Li W, Shi H, Ji R, Mao F, Qian H, Xu W, Zhang X (2020) CXCL5 promotes gastric cancer metastasis by inducing epithelial‐mesenchymal transition and activating neutrophils. Oncogenesis 9: 63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mareel M, Vleminckx K, Vermeulen S, Bracke M, Van Roy F (1992) E‐cadherin expression: a counterbalance for cancer cell invasion. Bull Cancer 79: 347–355 [PubMed] [Google Scholar]
- Martin‐Belmonte F, Perez‐Moreno M (2011) Epithelial cell polarity, stem cells and cancer. Nat Rev Cancer 12: 23–38 [DOI] [PubMed] [Google Scholar]
- Masin M, Vazquez J, Rossi S, Groeneveld S, Samson N, Schwalie PC, Deplancke B, Frawley LE, Gouttenoire J, Moradpour Det al (2014) GLUT3 is induced during epithelial‐mesenchymal transition and promotes tumor cell proliferation in non‐small cell lung cancer. Cancer Metab 2: 11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massague J (2012) TGFbeta signalling in context. Nat Rev Mol Cell Biol 13: 616–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masucci MT, Minopoli M, Carriero MV (2019) Tumor associated neutrophils. Their role in tumorigenesis, metastasis, prognosis and therapy. Front Oncol 9: 1146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matise LA, Palmer TD, Ashby WJ, Nashabi A, Chytil A, Aakre M, Pickup MW, Gorska AE, Zijlstra A, Moses HL (2012) Lack of transforming growth factor‐beta signaling promotes collective cancer cell invasion through tumor‐stromal crosstalk. Breast Cancer Res 14: R98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGranahan N, Swanton C (2017) Clonal heterogeneity and tumor evolution: past, present, and the future. Cell 168: 613–628 [DOI] [PubMed] [Google Scholar]
- Meidhof S, Brabletz S, Lehmann W, Preca BT, Mock K, Ruh M, Schuler J, Berthold M, Weber A, Burk Uet al (2015) ZEB1‐associated drug resistance in cancer cells is reversed by the class I HDAC inhibitor mocetinostat. EMBO Mol Med 7: 831–847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao JW, Liu LJ, Huang J (2014) Interleukin‐6‐induced epithelial‐mesenchymal transition through signal transducer and activator of transcription 3 in human cervical carcinoma. Int J Oncol 45: 165–176 [DOI] [PubMed] [Google Scholar]
- Miyoshi A, Kitajima Y, Sumi K, Sato K, Hagiwara A, Koga Y, Miyazaki K (2004) Snail and SIP1 increase cancer invasion by upregulating MMP family in hepatocellular carcinoma cells. Br J Cancer 90: 1265–1273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi A, Kitajima Y, Kido S, Shimonishi T, Matsuyama S, Kitahara K, Miyazaki K (2005) Snail accelerates cancer invasion by upregulating MMP expression and is associated with poor prognosis of hepatocellular carcinoma. Br J Cancer 92: 252–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morel A‐P, Ginestier C, Pommier RM, Cabaud O, Ruiz E, Wicinski J, Devouassoux‐Shisheboran M, Combaret V, Finetti P, Chassot Cet al (2017) A stemness‐related ZEB1‐MSRB3 axis governs cellular pliancy and breast cancer genome stability. Nat Med 23: 568–578 [DOI] [PubMed] [Google Scholar]
- Morel A‐P, Hinkal GW, Thomas C, Fauvet F, Courtois‐Cox S, Wierinckx A, Devouassoux‐Shisheboran M, Treilleux I, Tissier A, Gras Bet al (2012) EMT inducers catalyze malignant transformation of mammary epithelial cells and drive tumorigenesis towards claudin‐low tumors in transgenic mice. PLoS Genet 8: e1002723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morel AP, Lievre M, Thomas C, Hinkal G, Ansieau S, Puisieux A (2008) Generation of breast cancer stem cells through epithelial‐mesenchymal transition. PLoS One 3: e2888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno‐Bueno G, Portillo F, Cano A (2008) Transcriptional regulation of cell polarity in EMT and cancer. Oncogene 27: 6958–6969 [DOI] [PubMed] [Google Scholar]
- Moroishi T, Hansen CG, Guan KL (2015) The emerging roles of YAP and TAZ in cancer. Nat Rev Cancer 15: 73–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosa MH, Michels BE, Menche C, Nicolas AM, Darvishi T, Greten FR, Farin HF (2020) A Wnt‐induced phenotypic switch in cancer‐associated fibroblasts inhibits EMT in colorectal cancer. Cancer Res 80: 5569–5582 [DOI] [PubMed] [Google Scholar]
- Nath A, Li I, Roberts LR, Chan C (2015) Elevated free fatty acid uptake via CD36 promotes epithelial‐mesenchymal transition in hepatocellular carcinoma. Sci Rep 5: 14752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natsuizaka M, Whelan KA, Kagawa S, Tanaka K, Giroux V, Chandramouleeswaran PM, Long A, Sahu V, Darling DS, Que Jet al (2017) Interplay between Notch1 and Notch3 promotes EMT and tumor initiation in squamous cell carcinoma. Nat Commun 8: 1758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niessen K, Fu Y, Chang L, Hoodless PA, McFadden D, Karsan A (2008) Slug is a direct Notch target required for initiation of cardiac cushion cellularization. J Cell Biol 182: 315–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieto MA, Huang RY, Jackson RA, Thiery JP (2016) Emt: 2016. Cell 166: 21–45 [DOI] [PubMed] [Google Scholar]
- Noman MZ, Janji B, Abdou A, Hasmim M, Terry S, Tan TZ, Mami‐Chouaib F, Thiery JP, Chouaib S (2017) The immune checkpoint ligand PD‐L1 is upregulated in EMT‐activated human breast cancer cells by a mechanism involving ZEB‐1 and miR‐200. Oncoimmunology 6: e1263412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norz D, Grottke A, Bach J, Herzberger C, Hofmann BT, Nashan B, Jucker M, Ewald F (2015) Discontinuing MEK inhibitors in tumor cells with an acquired resistance increases migration and invasion. Cell Signal 27: 2191–2200 [DOI] [PubMed] [Google Scholar]
- Noy R, Pollard JW (2014) Tumor‐associated macrophages: from mechanisms to therapy. Immunity 41: 49–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ocana OH, Corcoles R, Fabra A, Moreno‐Bueno G, Acloque H, Vega S, Barrallo‐Gimeno A, Cano A, Nieto MA (2012) Metastatic colonization requires the repression of the epithelial‐mesenchymal transition inducer Prrx1. Cancer Cell 22: 709–724 [DOI] [PubMed] [Google Scholar]
- Omabe K, Uduituma S, Igwe D, Omabe M (2021) Deeper insight in metastatic cancer progression;Epithelial‐to‐mesenchymal transition and genomic instability: implications on treatment resistance. Curr Mol Med 21. 10.2174/1566524021666210202114844 [DOI] [PubMed] [Google Scholar]
- Ouzounova M, Lee E, Piranlioglu R, El Andaloussi A, Kolhe R, Demirci MF, Marasco D, Asm I, Chadli A, Hassan KAet al (2017) Monocytic and granulocytic myeloid derived suppressor cells differentially regulate spatiotemporal tumour plasticity during metastatic cascade. Nat Commun 8: 14979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual G, Avgustinova A, Mejetta S, Martín M, Castellanos A, Attolini C‐O, Berenguer A, Prats N, Toll A, Hueto JAet al (2017) Targeting metastasis‐initiating cells through the fatty acid receptor CD36. Nature 541: 41–45 [DOI] [PubMed] [Google Scholar]
- Pastushenko I, Blanpain C (2019) EMT transition states during tumor progression and metastasis. Trends Cell Biol 29: 212–226 [DOI] [PubMed] [Google Scholar]
- Pastushenko I, Brisebarre A, Sifrim A, Fioramonti M, Revenco T, Boumahdi S, Van Keymeulen A, Brown D, Moers V, Lemaire Set al (2018) Identification of the tumour transition states occurring during EMT. Nature 556: 463–468 [DOI] [PubMed] [Google Scholar]
- Pastushenko I, Mauri F, Song Y, de Cock F, Meeusen B, Swedlund B, Impens F, Van Haver D, Opitz M, Thery Met al (2021) Fat1 deletion promotes hybrid EMT state, tumour stemness and metastasis. Nature 589: 448–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pecot CV, Rupaimoole R, Yang Da, Akbani R, Ivan C, Lu C, Wu S, Han H‐D, Shah MY, Rodriguez‐Aguayo Cet al (2013) Tumour angiogenesis regulation by the miR‐200 family. Nat Commun 4: 2427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter ME (2009) Let‐7 and miR‐200 microRNAs: guardians against pluripotency and cancer progression. Cell Cycle 8: 843–852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pistore C, Giannoni E, Colangelo T, Rizzo F, Magnani E, Muccillo L, Giurato G, Mancini M, Rizzo S, Riccardi Met al (2017) DNA methylation variations are required for epithelial‐to‐mesenchymal transition induced by cancer‐associated fibroblasts in prostate cancer cells. Oncogene 36: 5551–5566 [DOI] [PubMed] [Google Scholar]
- Postigo AA (2003) Opposing functions of ZEB proteins in the regulation of the TGFbeta/BMP signaling pathway. EMBO J 22: 2443–2452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prodhomme MK, Pommier RM, Franchet C, Fauvet F, Bergoglio V, Brousset P, Morel AP, Brunac AC, Devouassoux‐Shisheboran M, Petrilli Vet al (2021) EMT transcription factor ZEB1 represses the mutagenic POLtheta‐mediated end‐joining pathway in breast cancers. Cancer Res 81: 1595–1606 [DOI] [PubMed] [Google Scholar]
- Puhr M, Hoefer J, Schafer G, Erb HH, Oh SJ, Klocker H, Heidegger I, Neuwirt H, Culig Z (2012) Epithelial‐to‐mesenchymal transition leads to docetaxel resistance in prostate cancer and is mediated by reduced expression of miR‐200c and miR‐205. Am J Pathol 181: 2188–2201 [DOI] [PubMed] [Google Scholar]
- Puisieux A, Brabletz T, Caramel J (2014) Oncogenic roles of EMT‐inducing transcription factors. Nat Cell Biol 16: 488–494 [DOI] [PubMed] [Google Scholar]
- Puram SV, Tirosh I, Parikh AS, Patel AP, Yizhak K, Gillespie S, Rodman C, Luo CL, Mroz EA, Emerick KSet al (2017) Single‐cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer. Cell 171: 1611–1624.e24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran A, Vizan P, Das D, Chakravarty P, Vogt J, Rogers KW, Muller P, Hinck AP, Sapkota GP, Hill CS (2018) TGF‐beta uses a novel mode of receptor activation to phosphorylate SMAD1/5 and induce epithelial‐to‐mesenchymal transition. Elife 7: e31756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambow F, Rogiers A, Marin‐Bejar O, Aibar S, Femel J, Dewaele M, Karras P, Brown D, Chang YH, Debiec‐Rychter Met al (2018) Toward minimal residual disease‐directed therapy in melanoma. Cell 174: 843–855.e19 [DOI] [PubMed] [Google Scholar]
- Redfern AD, Spalding LJ, Thompson EW (2018) The Kraken Wakes: induced EMT as a driver of tumour aggression and poor outcome. Clin Exp Metastasis 35: 285–308 [DOI] [PubMed] [Google Scholar]
- Ren J, Chen Y, Song H, Chen L, Wang R (2013) Inhibition of ZEB1 reverses EMT and chemoresistance in docetaxel‐resistant human lung adenocarcinoma cell line. J Cell Biochem 114: 1395–1403 [DOI] [PubMed] [Google Scholar]
- Reya T, Morrison SJ, Clarke MF, Weissman IL (2001) Stem cells, cancer, and cancer stem cells. Nature 414: 105–111 [DOI] [PubMed] [Google Scholar]
- Richard G, Dalle S, Monet MA, Ligier M, Boespflug A, Pommier RM, de la Fouchardiere A, Perier‐Muzet M, Depaepe L, Barnault Ret al (2016) ZEB1‐mediated melanoma cell plasticity enhances resistance to MAPK inhibitors. EMBO Mol Med 8: 1143–1161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley AJ (2011) Life at the leading edge. Cell 145: 1012–1022 [DOI] [PubMed] [Google Scholar]
- Ruh M, Stemmler MP, Frisch I, Fuchs K, van Roey R, Kleemann J, Roas M, Schuhwerk H, Eccles RL, Agaimy Aet al (2021) The EMT transcription factor ZEB1 blocks osteoblastic differentiation in bone development and osteosarcoma. J Pathol 254: 199–211 [DOI] [PubMed] [Google Scholar]
- Ruscetti M, Quach B, Dadashian EL, Mulholland DJ, Wu H (2015) Tracking and functional characterization of epithelial‐mesenchymal transition and mesenchymal tumor cells during prostate cancer metastasis. Cancer Res 75: 2749–2759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM, Fearon D, Greten FR, Hingorani SR, Hunter Tet al (2020) A framework for advancing our understanding of cancer‐associated fibroblasts. Nat Rev Cancer 20: 174–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakata J, Utsumi F, Suzuki S, Niimi K, Yamamoto E, Shibata K, Senga T, Kikkawa F, Kajiyama H (2017) Inhibition of ZEB1 leads to inversion of metastatic characteristics and restoration of paclitaxel sensitivity of chronic chemoresistant ovarian carcinoma cells. Oncotarget 8: 99482–99494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar Y, Zheng X, Brunn D, Raifer H, Picard F, Zhang Y, Winter H, Guenther S, Weigert A, Weigmann Bet al (2020) Microenvironmental Th9 and Th17 lymphocytes induce metastatic spreading in lung cancer. J Clin Invest 130: 3560–3575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez‐Tillo E, de Barrios O, Siles L, Cuatrecasas M, Castells A, Postigo A (2011) beta‐catenin/TCF4 complex induces the epithelial‐to‐mesenchymal transition (EMT)‐activator ZEB1 to regulate tumor invasiveness. Proc Natl Acad Sci U S A 108: 19204–19209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santamaria PG, Moreno‐Bueno G, Cano A (2019) Contribution of epithelial plasticity to therapy resistance. J Clin Med 8: 676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santisteban M, Reiman JM, Asiedu MK, Behrens MD, Nassar A, Kalli KR, Haluska P, Ingle JN, Hartmann LC, Manjili MHet al (2009) Immune‐induced epithelial to mesenchymal transition in vivo generates breast cancer stem cells. Cancer Res 69: 2887–2895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena M, Stephens MA, Pathak H, Rangarajan A (2011) Transcription factors that mediate epithelial‐mesenchymal transition lead to multidrug resistance by upregulating ABC transporters. Cell Death Dis 2: e179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheel C, Eaton E, Li S‐J, Chaffer C, Reinhardt F, Kah K‐J, Bell G, Guo W, Rubin J, Richardson Aet al (2011) Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell 145: 926–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciacovelli M, Frezza C (2017) Fumarate drives EMT in renal cancer. Cell Death Differ 24: 1–2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciacovelli M, Gonçalves E, Johnson TI, Zecchini VR, da Costa ASH, Gaude E, Drubbel AV, Theobald SJ, Abbo SR, Tran MGBet al (2016) Fumarate is an epigenetic modifier that elicits epithelial‐to‐mesenchymal transition. Nature 537: 544–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seoane J, Gomis RR (2017) TGF‐beta family signaling in tumor suppression and cancer progression. Cold Spring Harb Perspect Biol 9: a022277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sequist LV, Waltman BA, Dias‐Santagata D, Digumarthy S, Turke AB, Fidias P, Bergethon K, Shaw AT, Gettinger S, Cosper AKet al (2011) Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med 3: 75ra26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaffer SM, Dunagin MC, Torborg SR, Torre EA, Emert B, Krepler C, Beqiri M, Sproesser K, Brafford PA, Xiao Met al (2017) Rare cell variability and drug‐induced reprogramming as a mode of cancer drug resistance. Nature 546: 431–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao D, Xue W, Krall E, Bhutkar A, Piccioni F, Wang X, Schinzel A, Sood S, Rosenbluh J, Kim Jet al (2014) KRAS and YAP1 converge to regulate EMT and tumor survival. Cell 158: 171–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaul Y, Freinkman E, Comb W, Cantor J, Tam W, Thiru P, Kim D, Kanarek N, Pacold M, Chen Wet al (2014) Dihydropyrimidine accumulation is required for the epithelial‐mesenchymal transition. Cell 158: 1094–1109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng W, Shi X, Lin Y, Tang J, Jia C, Cao R, Sun J, Wang G, Zhou L, Dong M (2020) Musashi2 promotes EGF‐induced EMT in pancreatic cancer via ZEB1‐ERK/MAPK signaling. J Exp Clin Cancer Res 39: 16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibue T, Brooks MW, Weinberg RA (2013) An integrin‐linked machinery of cytoskeletal regulation that enables experimental tumor initiation and metastatic colonization. Cancer Cell 24: 481–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibue T, Weinberg RA (2009) Integrin beta1‐focal adhesion kinase signaling directs the proliferation of metastatic cancer cells disseminated in the lungs. Proc Natl Acad Sci U S A 106: 10290–10295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibue T, Weinberg RA (2017) EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol 14: 611–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimono Y, Zabala M, Cho RW, Lobo N, Dalerba P, Qian D, Diehn M, Liu H, Panula SP, Chiao Eet al (2009) Downregulation of miRNA‐200c links breast cancer stem cells with normal stem cells. Cell 138: 592–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin S, Dimitri CA, Yoon SO, Dowdle W, Blenis J (2010) ERK2 but not ERK1 induces epithelial‐to‐mesenchymal transformation via DEF motif‐dependent signaling events. Mol Cell 38: 114–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shintani Y, Fujiwara A, Kimura T, Kawamura T, Funaki S, Minami M, Okumura M (2016) IL‐6 secreted from cancer‐associated fibroblasts mediates chemoresistance in NSCLC by increasing epithelial‐mesenchymal transition signaling. J Thorac Oncol 11: 1482–1492 [DOI] [PubMed] [Google Scholar]
- Shirakihara T, Saitoh M, Miyazono K (2007) Differential regulation of epithelial and mesenchymal markers by deltaEF1 proteins in epithelial mesenchymal transition induced by TGF‐beta. Mol Biol Cell 18: 3533–3544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siemens H, Jackstadt R, Hunten S, Kaller M, Menssen A, Gotz U, Hermeking H (2011) miR‐34 and SNAIL form a double‐negative feedback loop to regulate epithelial‐mesenchymal transitions. Cell Cycle 10: 4256–4271 [DOI] [PubMed] [Google Scholar]
- Singh A, Greninger P, Rhodes D, Koopman L, Violette S, Bardeesy N, Settleman J (2009) A gene expression signature associated with "K‐Ras addiction" reveals regulators of EMT and tumor cell survival. Cancer Cell 15: 489–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A, Settleman J (2010) EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene 29: 4741–4751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slowicka K, Petta I, Blancke G, Hoste E, Dumas E, Sze M, Vikkula H, Radaelli E, Haigh JJ, Jonckheere Set al (2020) Zeb2 drives invasive and microbiota‐dependent colon carcinoma. Nature Cancer 1: 620–634 [DOI] [PubMed] [Google Scholar]
- Song NA, Jing W, Li CE, Bai M, Cheng YU, Li H, Hou K, Li Y, Wang K, Li Zet al (2018) ZEB1 inhibition sensitizes cells to the ATR inhibitor VE‐821 by abrogating epithelial‐mesenchymal transition and enhancing DNA damage. Cell Cycle 17: 595–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaderna S, Schmalhofer O, Hlubek F, Berx G, Eger A, Merkel S, Jung A, Kirchner T, Brabletz T (2006) A transient, EMT‐linked loss of basement membranes indicates metastasis and poor survival in colorectal cancer. Gastroenterology 131: 830–840 [DOI] [PubMed] [Google Scholar]
- Spaderna S, Schmalhofer O, Wahlbuhl M, Dimmler A, Bauer K, Sultan A, Hlubek F, Jung A, Strand D, Eger Aet al (2008) The transcriptional repressor ZEB1 promotes metastasis and loss of cell polarity in cancer. Cancer Res 68: 537–544 [DOI] [PubMed] [Google Scholar]
- Stemmler MP, Eccles RL, Brabletz S, Brabletz T (2019) Non‐redundant functions of EMT transcription factors. Nat Cell Biol 21: 102–112 [DOI] [PubMed] [Google Scholar]
- Su J, Zhang A, Shi Z, Ma F, Pu P, Wang T, Zhang J, Kang C, Zhang Q (2012) MicroRNA‐200a suppresses the Wnt/beta‐catenin signaling pathway by interacting with beta‐catenin. Int J Oncol 40: 1162–1170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan NJ, Sasser AK, Axel AE, Vesuna F, Raman V, Ramirez N, Oberyszyn TM, Hall BM (2009) Interleukin‐6 induces an epithelial‐mesenchymal transition phenotype in human breast cancer cells. Oncogene 28: 2940–2947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun D, Luo T, Dong P, Zhang N, Chen J, Zhang S, Dong L, Janssen HLA, Zhang S (2020) M2‐polarized tumor‐associated macrophages promote epithelial‐mesenchymal transition via activation of the AKT3/PRAS40 signaling pathway in intrahepatic cholangiocarcinoma. J Cell Biochem 121: 2828–2838 [DOI] [PubMed] [Google Scholar]
- Sun NY, Yang MH (2020) Metabolic reprogramming and epithelial‐mesenchymal plasticity: opportunities and challenges for cancer therapy. Front Oncol 10: 792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundararajan V, Gengenbacher N, Stemmler MP, Kleemann JA, Brabletz T, Brabletz S (2015) The ZEB1/miR‐200c feedback loop regulates invasion via actin interacting proteins MYLK and TKS5. Oncotarget 6: 27083–27096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano S, Reichert M, Bakir B, Das KK, Nishida T, Miyazaki M, Heeg S, Collins MA, Marchand B, Hicks PDet al (2016) Prrx1 isoform switching regulates pancreatic cancer invasion and metastatic colonization. Genes Dev 30: 233–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taki M, Abiko K, Baba T, Hamanishi J, Yamaguchi K, Murakami R, Yamanoi K, Horikawa N, Hosoe Y, Nakamura Eet al (2018) Snail promotes ovarian cancer progression by recruiting myeloid‐derived suppressor cells via CXCR2 ligand upregulation. Nat Commun 9: 1685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauriello DVF, Palomo‐Ponce S, Stork D, Berenguer‐Llergo A, Badia‐Ramentol J, Iglesias M, Sevillano M, Ibiza S, Canellas A, Hernando‐Momblona Xet al (2018) TGFbeta drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 554: 538–543 [DOI] [PubMed] [Google Scholar]
- Terry S, Savagner P, Ortiz‐Cuaran S, Mahjoubi L, Saintigny P, Thiery JP, Chouaib S (2017) New insights into the role of EMT in tumor immune escape. Mol Oncol 11: 824–846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiery JP, Acloque H, Huang RY, Nieto MA (2009) Epithelial‐mesenchymal transitions in development and disease. Cell 139: 871–890 [DOI] [PubMed] [Google Scholar]
- Tian YC, Chen YC, Chang CT, Hung CC, Wu MS, Phillips A, Yang CW (2007) Epidermal growth factor and transforming growth factor‐beta1 enhance HK‐2 cell migration through a synergistic increase of matrix metalloproteinase and sustained activation of ERK signaling pathway. Exp Cell Res 313: 2367–2377 [DOI] [PubMed] [Google Scholar]
- Timmerman LA, Grego‐Bessa J, Raya A, Bertran E, Perez‐Pomares JM, Diez J, Aranda S, Palomo S, McCormick F, Izpisua‐Belmonte JCet al (2004) Notch promotes epithelial‐mesenchymal transition during cardiac development and oncogenic transformation. Genes Dev 18: 99–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi SC, Peters HL, Taguchi A, Katayama H, Wang H, Momin A, Jolly MK, Celiktas M, Rodriguez‐Canales J, Liu Het al (2016) Immunoproteasome deficiency is a feature of non‐small cell lung cancer with a mesenchymal phenotype and is associated with a poor outcome. Proc Natl Acad Sci U S A 113: E1555–E1564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trovato R, Cane S, Petrova V, Sartoris S, Ugel S, De Sanctis F (2020) The engagement between MDSCs and metastases: partners in crime. Front Oncol 10: 165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai JH, Donaher JL, Murphy DA, Chau S, Yang J (2012) Spatiotemporal regulation of epithelial‐mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell 22: 725–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tulchinsky E, Demidov O, Kriajevska M, Barlev NA, Imyanitov E (2019) EMT: a mechanism for escape from EGFR‐targeted therapy in lung cancer. Biochim Biophys Acta Rev Cancer 1871: 29–39 [DOI] [PubMed] [Google Scholar]
- Uttamsingh S, Bao X, Nguyen KT, Bhanot M, Gong J, Chan JL, Liu F, Chu TT, Wang LH (2008) Synergistic effect between EGF and TGF‐beta1 in inducing oncogenic properties of intestinal epithelial cells. Oncogene 27: 2626–2634 [DOI] [PubMed] [Google Scholar]
- Vega S, Morales AV, Ocana OH, Valdes F, Fabregat I, Nieto MA (2004) Snail blocks the cell cycle and confers resistance to cell death. Genes Dev 18: 1131–1143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent T, Neve EP, Johnson JR, Kukalev A, Rojo F, Albanell J, Pietras K, Virtanen I, Philipson L, Leopold PLet al (2009) A SNAIL1‐SMAD3/4 transcriptional repressor complex promotes TGF‐beta mediated epithelial‐mesenchymal transition. Nat Cell Biol 11: 943–950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore‐Ludlow B, Kaffenberger SD, Eaton JK, Shimada K, Aguirre AJet al (2017) Dependency of a therapy‐resistant state of cancer cells on a lipid peroxidase pathway. Nature 547: 453–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Xiang D, Liu B, He A, Randle HJ, Zhang KX, Dongre A, Sachs N, Clark AP, Tao Let al (2019a) Inadequate DNA damage repair promotes mammary transdifferentiation, leading to BRCA1 breast cancer. Cell 178: 135–151.e19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Chen J, Yang L, Li J, Wu W, Huang M, Lin L, Su S (2019b) Tumor‐contacted neutrophils promote metastasis by a CD90‐TIMP‐1 JUXTACRINE‐paracrine loop. Clin Cancer Res 25: 1957–1969 [DOI] [PubMed] [Google Scholar]
- Wang Y, Liao R, Chen X, Ying X, Chen G, Li M, Dong C (2020) Twist‐mediated PAR1 induction is required for breast cancer progression and metastasis by inhibiting Hippo pathway. Cell Death Dis 11: 520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellner U, Schubert J, Burk UC, Schmalhofer O, Zhu F, Sonntag A, Waldvogel B, Vannier C, Darling D, Hausen AZet al (2009) The EMT‐activator ZEB1 promotes tumorigenicity by repressing stemness‐inhibiting microRNAs. Nat Cell Biol 11: 1487–1495 [DOI] [PubMed] [Google Scholar]
- Wilson MM, Weinberg RA, Lees JA, Guen VJ (2020) Emerging mechanisms by which EMT programs control stemness. Trends Cancer 6: 775–780 [DOI] [PubMed] [Google Scholar]
- Wu DW, Lee MC, Hsu NY, Wu TC, Wu JY, Wang YC, Cheng YW, Chen CY, Lee H (2015) FHIT loss confers cisplatin resistance in lung cancer via the AKT/NF‐kappaB/Slug‐mediated PUMA reduction. Oncogene 34: 2505–2515 [DOI] [PubMed] [Google Scholar]
- Wu L, Zhang XH (2020) Tumor‐associated neutrophils and macrophages‐heterogenous but not chaotic. Front Immunol 11: 553967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie M, Zhang L, He CS, Xu F, Liu JL, Hu ZH, Zhao LP, Tian Y (2012) Activation of Notch‐1 enhances epithelial‐mesenchymal transition in gefitinib‐acquired resistant lung cancer cells. J Cell Biochem 113: 1501–1513 [DOI] [PubMed] [Google Scholar]
- Xu Y, Lee DK, Feng Z, Xu Y, Bu W, Li Y, Liao L, Xu J (2017) Breast tumor cell‐specific knockout of Twist1 inhibits cancer cell plasticity, dissemination, and lung metastasis in mice. Proc Natl Acad Sci U S A 114: 11494–11499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Antin P, Berx G, Blanpain C, Brabletz T, Bronner M, Campbell K, Cano A, Casanova J, Christofori Get al (2020) Guidelines and definitions for research on epithelial‐mesenchymal transition. Nat Rev Mol Cell Biol 21: 341–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A, Weinberg RA (2004) Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 117: 927–939 [DOI] [PubMed] [Google Scholar]
- Yang L, Lin C, Liu ZR (2006) P68 RNA helicase mediates PDGF‐induced epithelial mesenchymal transition by displacing Axin from beta‐catenin. Cell 127: 139–155 [DOI] [PubMed] [Google Scholar]
- Yang M‐H, Hsu D‐S, Wang H‐W, Wang H‐J, Lan H‐Y, Yang W‐H, Huang C‐H, Kao S‐Y, Tzeng C‐H, Tai S‐Ket al (2010) Bmi1 is essential in Twist1‐induced epithelial‐mesenchymal transition. Nat Cell Biol 12: 982–992 [DOI] [PubMed] [Google Scholar]
- Yang MH, Wu MZ, Chiou SH, Chen PM, Chang SY, Liu CJ, Teng SC, Wu KJ (2008) Direct regulation of TWIST by HIF‐1alpha promotes metastasis. Nat Cell Biol 10: 295–305 [DOI] [PubMed] [Google Scholar]
- Ye X, Tam WL, Shibue T, Kaygusuz Y, Reinhardt F, Ng Eaton E, Weinberg RA (2015) Distinct EMT programs control normal mammary stem cells and tumour‐initiating cells. Nature 525: 256–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz M, Christofori G (2009) EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev 28: 15–33 [DOI] [PubMed] [Google Scholar]
- Yochum ZA, Cades J, Wang H, Chatterjee S, Simons BW, O’Brien JP, Khetarpal SK, Lemtiri‐Chlieh G, Myers KV, Huang E‐Bet al (2019) Targeting the EMT transcription factor TWIST1 overcomes resistance to EGFR inhibitors in EGFR‐mutant non‐small‐cell lung cancer. Oncogene 38: 656–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youssef KK, Nieto MA (2020) Glucose metabolism takes center stage in epithelial‐mesenchymal plasticity. Dev Cell 53: 133–135 [DOI] [PubMed] [Google Scholar]
- Yu M, Bardia A, Wittner BS, Stott SL, Smas ME, Ting DT, Isakoff SJ, Ciciliano JC, Wells MN, Shah AMet al (2013) Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 339: 580–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Xiao CH, Tan LD, Wang QS, Li XQ, Feng YM (2014) Cancer‐associated fibroblasts induce epithelial‐mesenchymal transition of breast cancer cells through paracrine TGF‐beta signalling. Br J Cancer 110: 724–732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan X, Wu H, Han N, Xu H, Chu Q, Yu S, Chen Y, Wu K (2014) Notch signaling and EMT in non‐small cell lung cancer: biological significance and therapeutic application. J Hematol Oncol 7: 87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanconato F, Forcato M, Battilana G, Azzolin L, Quaranta E, Bodega B, Rosato A, Bicciato S, Cordenonsi M, Piccolo S (2015) Genome‐wide association between YAP/TAZ/TEAD and AP‐1 at enhancers drives oncogenic growth. Nat Cell Biol 17: 1218–1227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zavadil J, Bottinger EP (2005) TGF‐beta and epithelial‐to‐mesenchymal transitions. Oncogene 24: 5764–5774 [DOI] [PubMed] [Google Scholar]
- Zhang D, Yang L, Liu X, Gao J, Liu T, Yan Q, Yang X (2020) Hypoxia modulates stem cell properties and induces EMT through N‐glycosylation of EpCAM in breast cancer cells. J Cell Physiol 235: 3626–3633 [DOI] [PubMed] [Google Scholar]
- Zhang H, Liu CY, Zha ZY, Zhao B, Yao J, Zhao S, Xiong Y, Lei QY, Guan KL (2009) TEAD transcription factors mediate the function of TAZ in cell growth and epithelial‐mesenchymal transition. J Biol Chem 284: 13355–13362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Liao Y, Tang L (2019a) MicroRNA‐34 family: a potential tumor suppressor and therapeutic candidate in cancer. J Exp Clin Cancer Res 38: 53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Wei Y, Wang Li, Debeb BG, Yuan Y, Zhang J, Yuan J, Wang M, Chen D, Sun Yet al (2014) ATM‐mediated stabilization of ZEB1 promotes DNA damage response and radioresistance through CHK1. Nat Cell Biol 16: 864–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Liu L, Lai W, Zeng Y, Xu H, Lan Q, Su P, Chu Z (2019b) Interaction with tumorassociated macrophages promotes PRL3induced invasion of colorectal cancer cells via MAPK pathwayinduced EMT and NFkappaB signalinginduced angiogenesis. Oncol Rep 41: 2790–2802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Zhang Z, Zhang Q, Zhang Q, Sun P, Xiang R, Ren G, Yang S (2018) ZEB1 confers chemotherapeutic resistance to breast cancer by activating ATM. Cell Death Dis 9: 57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Lee JC, Lin L, Olivas V, Au V, LaFramboise T, Abdel‐Rahman M, Wang X, Levine AD, Rho JKet al (2012) Activation of the AXL kinase causes resistance to EGFR‐targeted therapy in lung cancer. Nat Genet 44: 852–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Ji G, Le X, Luo Z, Wang C, Feng M, Xu L, Zhang Y, Lau Wb, Lau Bet al (2017) An integrated analysis identifies STAT4 as a key regulator of ovarian cancer metastasis. Oncogene 36: 3384–3396 [DOI] [PubMed] [Google Scholar]
- Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, Sugimoto H, Wu CC, LeBleu VS, Kalluri R (2015) Epithelial‐to‐mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 527: 525–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Lu G, Yao Y, Gu W (2019) An autocrine IL‐6/IGF‐1R loop mediates EMT and promotes tumor growth in non‐small cell lung cancer. Int J Biol Sci 15: 1882–1891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou BP, Deng J, Xia W, Xu J, Li YM, Gunduz M, Hung MC (2004) Dual regulation of Snail by GSK‐3beta‐mediated phosphorylation in control of epithelial‐mesenchymal transition. Nat Cell Biol 6: 931–940 [DOI] [PubMed] [Google Scholar]
- Zhu G, Li X, Guo B, Ke Q, Dong M, Li F (2016) PAK5‐mediated E47 phosphorylation promotes epithelial‐mesenchymal transition and metastasis of colon cancer. Oncogene 35: 1943–1954 [DOI] [PubMed] [Google Scholar]
- Zhu H, Gu Y, Xue Y, Yuan M, Cao X, Liu Q (2017) CXCR2(+) MDSCs promote breast cancer progression by inducing EMT and activated T cell exhaustion. Oncotarget 8: 114554–114567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou W, Wolchok JD, Chen L (2016) PD‐L1 (B7–H1) and PD‐1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci Transl Med 8: 328rv324 [DOI] [PMC free article] [PubMed] [Google Scholar]