

Abstract
The spike protein of severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) plays a crucial role in mediating viral entry into host cells. However, whether it contributes to pulmonary hyperinflammation in patients with coronavirus disease 2019 is not well known. In this study, we developed a spike protein–pseudotyped (Spp) lentivirus with the proper tropism of the SARS‐CoV‐2 spike protein on the surface and determined the distribution of the Spp lentivirus in wild‐type C57BL/6J male mice that received an intravenous injection of the virus. Lentiviruses with vesicular stomatitis virus glycoprotein (VSV‐G) or with a deletion of the receptor‐binding domain (RBD) in the spike protein [Spp (∆RBD)] were used as controls. Two hours postinfection (hpi), there were 27‐75 times more viral burden from Spp lentivirus in the lungs than in other organs; there were also about 3‐5 times more viral burden from Spp lentivirus than from VSV‐G lentivirus in the lungs, liver, kidney, and spleen. Deletion of RBD diminished viral loads in the lungs but not in the heart. Acute pneumonia was observed in animals 24 hpi. Spp lentivirus was mainly found in SPC+ and LDLR+ pneumocytes and macrophages in the lungs. IL6, IL10, CD80, and PPAR‐γ were quickly upregulated in response to infection in the lungs as well as in macrophage‐like RAW264.7 cells. Furthermore, forced expression of the spike protein in RAW264.7 cells significantly increased the mRNA levels of the same panel of inflammatory factors. Our results demonstrated that the spike protein of SARS‐CoV‐2 confers the main point of viral entry into the lungs and can induce cellular pathology. Our data also indicate that an alternative ACE2‐independent viral entry pathway may be recruited in the heart and aorta.
Keywords: COVID‐19, inflammation, lung, SARS‐CoV‐2, spike
Abbreviations
- ACE2
angiotensin‐converting enzyme 2
- CD
cluster of differentiation
- COVID‐19
coronavirus disease 2019
- CVD
cardiovascular disorders
- DM
diabetes mellitus
- EGFP
enhanced green fluorescent protein
- HIV
human immunodeficiency virus
- hpi
hours postinfection
- IL
interleukin
- LDLR
low‐density lipoprotein receptor
- MRC1
mannose receptor C‐type 1
- NRP
neuropilin
- PPAR‐γ
peroxisome proliferator‐activated receptor gamma
- RBD
receptor‐binding domain
- RIPA
radioimmunoprecipitation assay
- SARS‐CoV‐2
severe acute respiratory syndrome coronavirus 2
- SPC
surfactant protein C
- Spp (∆RBD)
Spp lentivirus with deletion of RBD in spike protein
- Spp
spike protein–pseudotyped
- TGF‐β
transforming growth factor beta
- TMPRSS2
host transmembrane serine protease 2
- TNF‐α
tumor necrosis factor α
- VSV‐G
vesicular stomatitis virus glycoprotein
1. INTRODUCTION
Coronavirus disease 19 (COVID‐19) is a significant threat to global health. So far, more than 64 million infections and 1.5 million victims have been reported worldwide in 191 countries and regions. The US alone had registered more than 14 million cases and 271 000 deaths as of December 4, 2020.1 Severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) is the causative organism for COVID‐19.2 SARS‐CoV‐2, a positive‐sense single‐stranded RNA virus, is the newest and seventh known coronavirus that is capable of infecting humans.2, 3 The SARS‐CoV2 spike (S) protein can be cleaved by furin to produce subunits S1 and S2 to form a trimer that can mediate viral entry into host cells via surface angiotensin‐converting enzyme 2 (ACE2).4 Host transmembrane serine protease 2 (TMPRSS2) is also capable of promoting SARS‐CoV‐2 entry into target cells by cleavage of the S2’ site in the S2 subunit.5, 6 ACE2 and TMPRSS2 have been found to be coexpressed in lung type II pneumocytes, ileal absorptive enterocytes, and nasal goblet secretory cells,7 which are thought to be host determinants for viral infection in the initial stage. Recently, neuropilin (NRP) 1 has been identified as a second host factor facilitating SARS‐CoV‐2 entry into target cells; this appears to be ACE2 independent, because a different binding motif is used.8, 9, 10
COVID‐19 patients may be asymptomatic or symptomatic. The mortality rate of COVID‐19 varies in different geographic locations and patient populations.1 Patients with metabolic‐associated preconditions such as hypertension, cardiovascular disorders (CVD), obesity, and diabetes mellitus (DM) are prone to developing more severe symptoms.11 COVID‐19 patients also show decreases in serum lipid levels.12, 13 SARS‐CoV‐2–induced hyperinflammation of the lungs is considered to cause disease progression. The molecular mechanisms of SARS‐CoV‐2–induced pathologies in patients have just begun to be elucidated. In this study, we investigated whether the S protein of SARS‐CoV‐2 interacts with macrophages and induces acute lung inflammation in vivo using a spike protein–pseudotyped (Spp) lentivirus.
2. MATERIALS AND METHODS
2.1. Materials
Phoenix cells and Dulbecco's Modified Eagle medium (DMEM) were purchased from ATCC (Manassas, Virginia). Lentiviral vector pLV‐mCherry and vesicular stomatitis virus glycoprotein (VSV‐G) expression vector pMD2.G were obtained from Addgene (Watertown, Massachusetts). The coding sequence for the fusion of the SARS‐CoV‐2 S gene (GenBank: QHU36824.1) with a C‐terminal His‐tag (Genscript, Piscataway, New Jersey) was synthesized in vitro after codon optimization for expression in human cells. The sequence was cloned into a pcDNA3.1 vector to obtain a pcDNA‐spike plasmid. Anti‐spike S1 subunit, anti‐low‐density lipoprotein receptor (LDLR), anti‐mannose receptor C‐type 1 (MRC1), anti‐surfactant protein C (SPC), anti‐cluster of differentiation (CD) 68, and anti‐human immunodeficiency virus (HIV)‐1 p24 antibodies were obtained from Novus Biologicals (Littleton, Colorado). Anti‐His‐tag antibodies were obtained from Proteintech (Rosemont, Illinois) and Thermo Fisher Scientific (Waltham, Massachusetts). Primers were synthesized by Integrated DNA Technologies (Coralville, Iowa), and the primer sequences are listed in Table S1. The RNA extraction kit was obtained from Zymo Research (Irvine, California). The reverse transcription (RT) kit was obtained from Takara Bio USA (Mountain View, California). The SYBR green master mix was obtained from Bio‐Rad (Hercules, California). Wild‐type C57BL/6J mice were purchased from the Jackson Laboratory (Bar Harbor, Maine).
2.2. Generation of pseudotyped lentivirus
A SARS‐CoV‐2 S gene fusion with a C‐terminal 12x His‐tag was synthesized and cloned into a pcDNA3.1 vector to generate a pcDNA‐spike plasmid. An S gene with deletion of receptor‐binding domain (RBD) (residues Arg319‐Phe541) was subsequently introduced into the pcDNA‐spike plasmid to generate a pcDNA‐spike RBD‐deleted (∆RBD) plasmid. Phoenix cells were grown in DMEM containing 10% fetal bovine serum (FBS) and cotransfected with pLV‐mCherry, pcDNA‐spike, and pcDNA‐spike (∆RBD) or pMD2.G vector using a calcium phosphate kit (Thermo Fisher Scientific, Waltham, Massachusetts). Supernatant with the viruses produced—Spp, Spp (∆RBD), or VSV‐G lentiviruses—was harvested 72 hours post‐transfection, clarified by centrifuging at 5000 g for 15 minutes, and then filtered through a 0.45‐µm filter disk. The viruses were collected by ultracentrifugation at 24 000 rpm for 2 hours using a Beckman SW41 rotor. The viral pellets were resuspended in cold phosphate‐buffered saline (PBS) buffer and stored at −80℃ before use. The viral particle number was determined using a real‐time reverse‐transcriptase polymerase chain reaction (RT‐PCR) assay to quantify the RNA copies of mCherry.
2.3. Surface protein biotinylation of Spp and immunoprecipitation
S protein biotinylation of the Spp lentivirus was performed using EZ‐Link Sulfo‐NHS‐LC‐Biotinylation kit (Thermo Fisher Scientific, Waltham, Massachusetts). Briefly, intact Spp lentivirus was incubated with Sulfo‐NHS‐LC‐Biotin in a PBS buffer for 30 minutes at room temperature. A protein desalting spin column (Thermo Fisher, Waltham, Massachusetts) was used to remove the unlabeled Sulfo‐NHS‐LC‐Biotin. The biotinylated surface proteins of the Spp lentivirus were extracted using a radioimmunoprecipitation assay (RIPA) buffer. For immunoprecipitation, about 10 µg of total lysate protein from the Spp lentivirus was incubated with 0.5 µg of anti‐S2 or anti‐His antibody for 2 hours at 4℃. The immunoprecipitated protein complex was pulled down using protein G agarose beads (Thermo Fisher Scientific, Waltham, Massachusetts), washed thoroughly with a RIPA buffer, dissolved in a protein loading dye, and analyzed using a western blot assay.
2.4. Intravenous viral administration in vivo
The animal protocol was approved by the University of South Carolina institutional animal care and use committee (IACUC). Male wild‐type C57BL/6J male mice (5‐6 weeks old) were intravenously administered 100 µL of Spp or VSV‐G lentivirus (8 × 108 viral particles) via tail venous or retro‐orbital injection. In order to achieve 80% power to detect a standardized effect size (mean/standard deviation) of 0.80 at an alpha = 0.05 level, we assigned 6‐7 animals per group. The animals were sacrificed at 2 or 24 hours postinfection (hpi) and perfused with 50 mL PBS per mouse. Tissues were collected, including the lungs, heart, liver, kidney, aorta, and spleen. RNA samples from the aorta were unavailable for some animals due to the challenges of the collecting procedure for tissue of such a small size. In general, one part of each tissue was used for RNA extraction, which was followed by a real‐time RT‐PCR assay to determine the number of viral particles in the tissue. The other part of each tissue was fixed, embedded, and used for histology and immunohistochemistry. We used male mice in this study in order to minimize potential hormonal effects on the inflammatory response induced by the Spp lentivirus.
2.5. RAW264.7 cell culture, viral uptake, and electroporation
Macrophage‐like RAW264.7 (RAW) cells (TIB‐71, ATCC, Manassas, Virginia) were cultured in DMEM (10% FBS) medium. The cells were changed into 2% FBS DMEM medium and left overnight prior to the viral uptake assay. Spp or VSV‐G lentivirus was added into the RAW cells (4.8 × 107 particles per well) in a 12‐well plate with 90% confluence and incubated for 2 or 16 hours. Following treatment, the cells were washed with PBS three times, and RNA was extracted using an RNA extraction kit (Zymo, Irvine, California). In a parallel experiment, RAW cells (5 × 106) were electroporated (BTX/Harvard Bioscience, Inc, Holliston, Massachusetts) with a pcDNA3.1 empty vector (Thermo Fisher Scientific, Waltham, Massachusetts), enhanced green fluorescent protein (EGFP)‐N2 control plasmid (Takara Bio, Mountain View, CA, USA), or pcDNA‐spike plasmid (10 µg) using the following parameters: 2‐mm gap cuvette, 250‐µL sample volume, 120 V. The cells were harvested 48 hours after electroporation for analysis.
2.6. Real‐time RT‐PCR and immunohistochemistry assay
To generate cDNA, 1.0‐5 µg of RNA was reverse transcribed in a 20‐µL reaction containing 1× RT buffer (Clontech, Mountain View, California), 0.5 mM deoxynucleotide triphosphates (dNTPs), 0.5 µg of oligo‐dT 15‐mer and random primer mixture, 20 units of RNasin, and 5 units of SMART Moloney murine leukemia virus reverse transcriptase (Takara Bio, Mountain View, California). The RT reaction was carried out at 42℃ for 2 hours. Seven housekeeping genes were screened for the normalization controls: GAPDH, Rps18, Ppia, Nono, Rpp30, Alas2, and β‐actin. We found that Rps18 and Nono showed the most stable expression levels in tissues crossing various samples (data not shown); both Rps18 and Nono were then used as controls to normalize the amplification data. Expression levels of a panel of 23 inflammatory genes (Table S1) were determined using real‐time RT‐PCR. The reaction for the multiplex real‐time PCR assays contained 1 × SYBR Green qPCR Master Mix (Bio‐Rad Laboratories, Hercules, California), 10 ng of each template, and 10 pmol of each specific primer in a total volume of 25 µL in a 96‐well format. Each reaction was performed in duplicate under identical conditions. The PCR conditions were one cycle at 95℃ for 2 minutes followed by 40 cycles of 15 s at 95℃ and 60 s at 60℃. Relative quantification of the real‐time PCR was based on the amplification efficiency of the target and reference genes and the cycle number at which fluorescence crossed a prescribed background level, the cycle threshold (Ct).
For the immunoblot assays, cell lysates were extracted from RAW cells using RIPA lysis buffer (Santa Cruz Biotechnology, Inc, Dallas, Texas). Proteins were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS‐PAGE) and transferred onto polyvinylidene fluoride (PVDF) membranes. Anti‐spike protein subunit S1, anti‐HIV‐1 p24, or anti‐His antibodies were used to detect the expression of viral proteins; following this, HRP‐labeled secondary antibodies were used. Images were acquired using a Bio‐Rad gel imaging system (Hercules, California).
Lung tissues from mice were fixed in 10% buffered formalin (Fisher Scientific, Pittsburgh, Pennsylvania) and embedded in OCT media for frozen sections. Sections approximately 6‐µm thick were cut and collected. The sections were blocked with 5% donkey serum and then incubated in a humidified chamber overnight at 4℃ with primary antibodies. Then the sections were rinsed and incubated with fluorescent conjugated secondary antibodies for 2 hours at room temperature. Images were acquired using an ImageXpress Pico cell imaging system (Molecular Devices, San Jose, CA, USA) or a confocal microscopy system (Carl Zeiss AG, Oberkochen, Germany).
2.7. Statistical analyses
All statistical analyses were performed in Origin 2019. Paired t test or one‐way ANOVA was used for two groups or multiple comparisons, respectively. Data are presented as mean ± SD, and P < .05 was considered as significant.
3. RESULTS
3.1. Tissue distributions of Spp in infected mice
We generated Spp lentivirus from Phoenix cells. Spike protein was shown to be cleaved into S1 and S2 subunits assembled on the Spp lentivirus that were produced in Phoenix cells by immunoblot analysis (Figure 1A). No evidence of full‐length, noncleaved spike protein was detected in the Spp lentivirus (Figure 1A). Using a helper vector expressing VSV‐G, a lentivirus was obtained as a control; this is referred to as VSV‐G lentivirus. Gag‐p24 is the capsid core shell protein in both the Spp and VSV‐G lentiviruses (Figure 1A). In order to confirm the surface assembly of S protein on the Spp lentivirus, we performed a surface protein biotinylation assay. We used anti‐S2 or anti‐His antibodies to immunoprecipitate the biotinylated S protein from the Spp lentivirus and then carried out a western blot test. We found that both anti‐His and anti‐S2 antibodies detected an S2 subunit with a higher molecular weight in the immunoprecipitated complexes than the unbiotinylated control S2 protein, demonstrating successful biotinylation of surface S2 protein from Spp lentivirus (Figure 1B).
FIGURE 1.
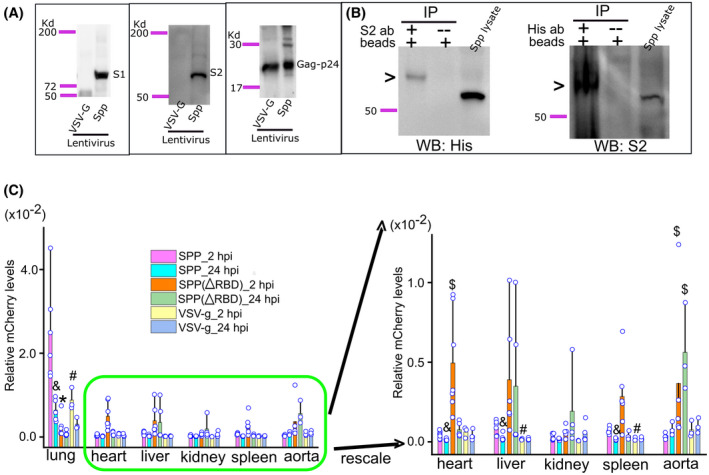
Systemic dissemination of Spp, Spp (∆RBD), and VSV‐G lentiviruses in mice. A, Detection of S1 and S2 subunits in Spp lentivirus by western blot using a specific antibody against the S1 subunit and an anti‐His antibody recognizing the S2 subunit. Both Spp and VSV‐G lentiviruses have a gag‐p24 protein. B, A demonstration of surface S protein on Spp lentivirus. Biotinylation of surface proteins of Spp lentivirus was followed by immunoprecipitation and western blot using an anti‐His or anti‐S2 antibody. A biotinylated S2 protein, indicating by a high molecular weight band by arrows, was detected from immunoprecipitated complexes. C, Spp group shows a predominant distribution in the lungs after intravenous administration. &, P < .05, Spp lentivirus viral burden as compared with VSV‐g lentivirus in the same tissue at 2 hpi; #, P < .05, Spp lentivirus viral burden at 24 hpi as compared with 2 hpi in the same tissue; $, P < .05, Spp (∆RBD) lentivirus viral burden as compared with the Spp group in the heart or aorta at 2 or 24 hpi
Spp or VSV‐G lentivirus (8 × 108 particles) was intravenously injected into male C57BL/6J mice. The animals were sacrificed at 2 or 24 hpi, and various tissues were collected for determination of the viral load. We did not find any systemic discrepancy for viral distribution in animals that received a tail vein injection compared to those that received retro‐orbital administration (data not shown). Therefore, we combined the data from both administration methods for the final analysis. At 2 hpi, the highest viral loads for both the Spp and VSV‐G lentiviruses were in the lungs. Spp lentivirus had 27, 33, 55, 71, and 74 times the viral load in the lungs than it had in the liver, spleen, heart, aorta, and kidney, respectively (P < .05, n = 3‐7 mice); Spp lentivirus also had 2.8, 4.1, 4.5, and 5.7 times the viral load than VSV‐G lentivirus had in the lungs, spleen, kidney, and liver, respectively (P < .05, n = 3‐7 mice) (Figure 1C). At 24 hpi, the viral loads for Spp lentivirus decreased significantly in the lungs, heart, liver, kidney, and spleen (P < .05, n = 3‐7 mice) (Figure 1C). Deletion of RBD in the Spp lentivirus diminished the viral load in the lungs (P < .05, n = 5‐6 mice) at both 2 and 24 hpi (Figure 1C). However, Spp (∆RBD) lentivirus showed a significant increase in viral load in the heart at 2 hpi (P < .05, n = 7 mice) and in the aorta at both 2 and 24 hpi (P < .05, n = 3‐5 mice) as compared with Spp lentivirus (Figure 1C), indicating an alternative ACE2‐independent viral entry pathway might be recruited in these organs.
3.2. Pathological features of pneumonia in Spp‐infected mice
We then investigated whether the male mice treated with Spp virus acquired pneumonia. There were no evident histological changes in the lungs at 2 hpi in either the Spp or VSV‐G groups. At 24 hpi in the Spp group, many pathological changes were evident in the lungs, including multifocal lesions, inflammatory cell infiltrations, thickened alveolar walls, perivascular and peribronchial infiltrations, and fibroplasia with exudation of fibrin and proteins (Figure 2). We observed only mild inflammation, such as mildly thickened alveolar walls in the lungs, in the VSV‐G group at 24 hpi (Figure 2). This indicates that Spp but not VSV‐G lentivirus is capable of inducing acute and diffuse pneumonia in mouse lungs with pathological manifestations very similar to those observed in severe COVID‐19 patients. These data also suggest that the S protein of SARS‐CoV‐2 plays an important role in the development of acute pneumonia.
FIGURE 2.
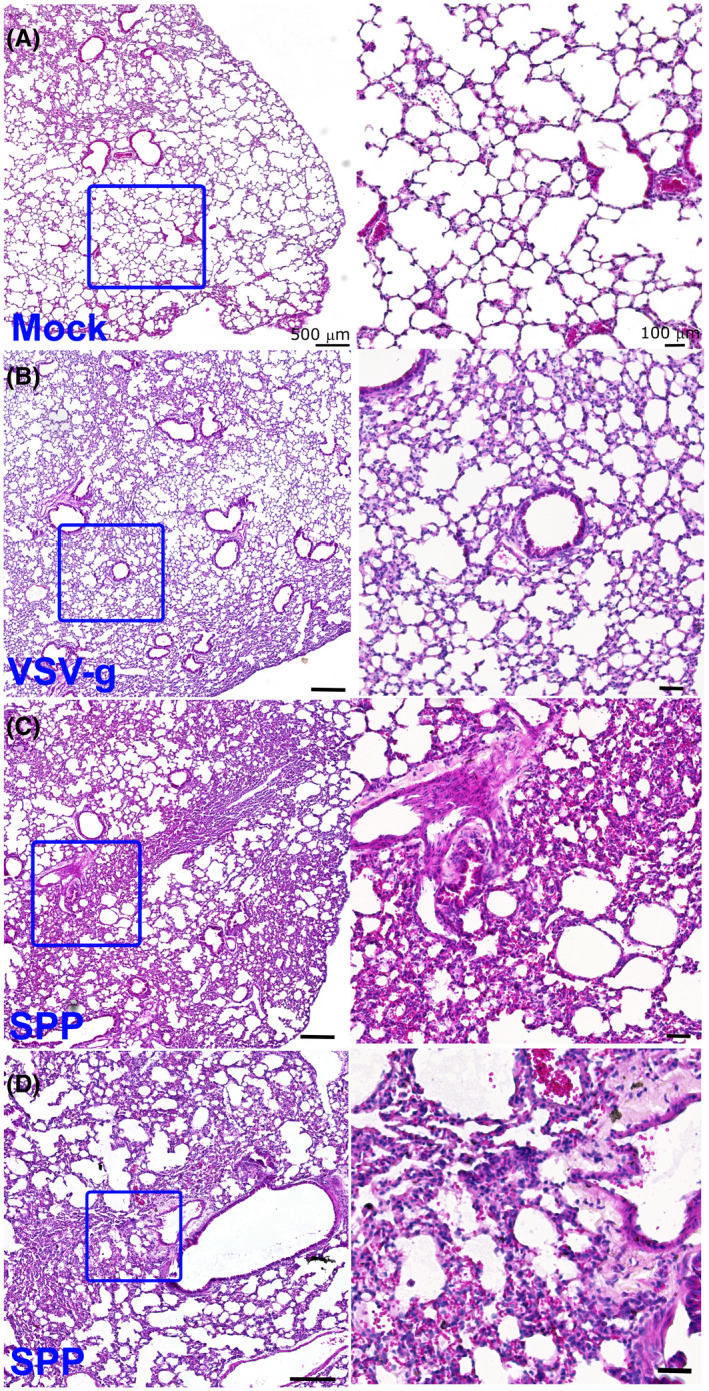
Mice acquired acute pneumonia at 24 hpi with Spp lentivirus. Histological analysis of lungs in control mice (A) or mice receiving VSV‐G lentivirus (B) or Spp lentivirus (C and D) shows acute and diffuse inflammatory responses in the lungs at 24 hpi with Spp lentivirus. Multifocal lesions, inflammatory cell infiltrations, thickened alveolar walls, perivascular and peribronchial infiltrations, and fibroplasia with exudation of fibrin and proteins were observed in the lungs of mice infected with Spp lentivirus. Right panel is the magnification of the boxed area in the left panel. Scale bar: 500 µm (left panel) and 100 µm (right panel)
3.3. Cellular colocalization of Spp lentivirus in the lungs
Next, we examined the cellular distribution of Spp lentivirus in the lungs. Spp viral antigen was detected by an anti‐His antibody. LDLR is expressed in type II alveolar epithelial cells and macrophages in the lungs.14, 15 The majority of cells that demonstrated Spp‐lentivirus uptake (His+) (82.3% ± 11.4%) were LDLR+ cells, while 84.4% ± 14.9% of LDLR+ cells showed uptake of Spp lentivirus (Figure 3). In addition, most His+ (81.9% ± 8.4%) cells were SPC+ cells; 86.4% ± 11.2% of SPC+ cells were His+ cells (Figure 3). We then examined the types of macrophages with uptake of Spp lentivirus in the lungs using the macrophage markers CD68 and MRC1. We found that 10% ± 4.4% of cells with evidence of Spp lentivirus uptake were CD68+ macrophages, while 38.3% ± 19.3% of CD68+ macrophages showed uptake of Spp lentivirus (Figure 3). However, we found little evidence of MRC1+ macrophages with uptake of Spp lentivirus (Figure 3).
FIGURE 3.
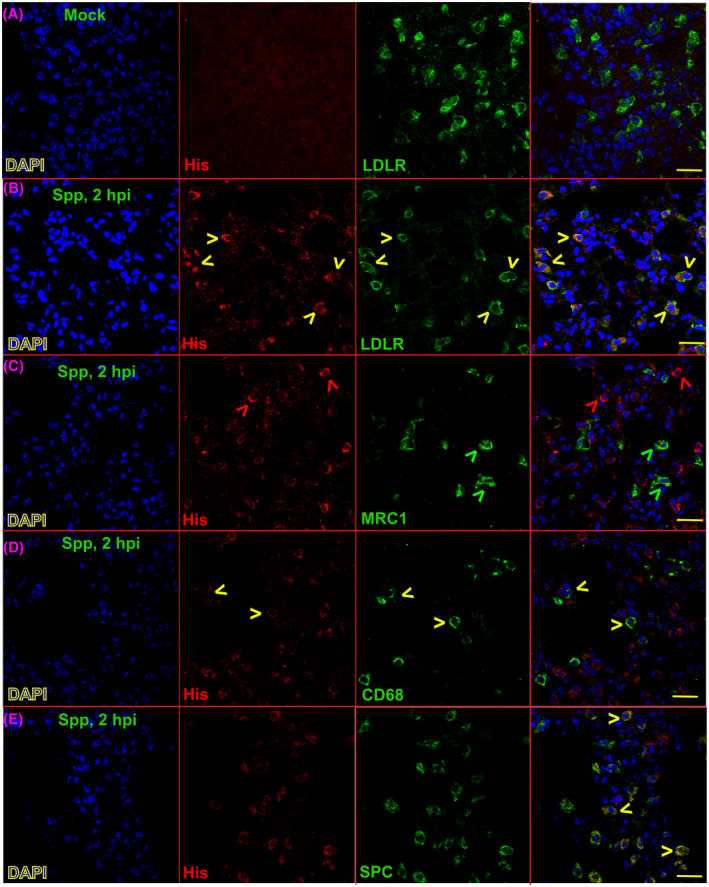
Cellular colocalizations of Spp lentivirus and various lung markers in the lungs 2 hpi. An anti‐His‐tag antibody was used to recognize S‐fusion protein in the Spp lentivirus. LDLR, MRC1, CD68, and SPC markers were used to stain type II alveolar cells and subtypes of macrophages in the lungs. Yellow arrows indicate cells positive for both His‐tag and corresponding lung markers, including LDLR, MRC1, CD68, and SPC. Red or green arrows indicate cells positive for His‐tag or corresponding lung markers only, respectively. Blue: DAPI staining for nuclei. Scale bar: 20 µm
3.4. Dysregulation of inflammatory cytokines in the lungs in Spp‐infected mice
Next, we attempted to assess which inflammatory factors were induced by Spp lentivirus. We examined expression levels of a panel of 23 genes that are representative inflammatory markers in macrophages (Table S1). Two hours after viral administration, the mRNA levels of interleukin (IL)6, IL10, CD80, and peroxisome proliferator‐activated receptor gamma (PPAR‐γ) showed a significant and rapid increase in the lungs in Spp lentivirus‐infected mice but not in VSV‐g lentivirus‐infected mice as compared with untreated control mice (Figure 4). Levels of tumor necrosis factor α (TNF‐α) were elevated in the mice at both 2 and 24 hpi with Spp and VSV‐g lentiviruses as compared with untreated control mice (Figure 4). Transforming growth factor beta (TGF‐β) increased significantly in the lungs of Spp lentivirus‐infected mice only at 24 hpi as compared with untreated control mice and mice at 2 hpi (Figure 4). We did not find significant changes in expression levels of other inflammatory markers that we examined (data not shown). These data suggest that Spp lentivirus infection induced a rapid increase in inflammatory factors IL6, IL10, CD80, and PPAR‐γ.
FIGURE 4.
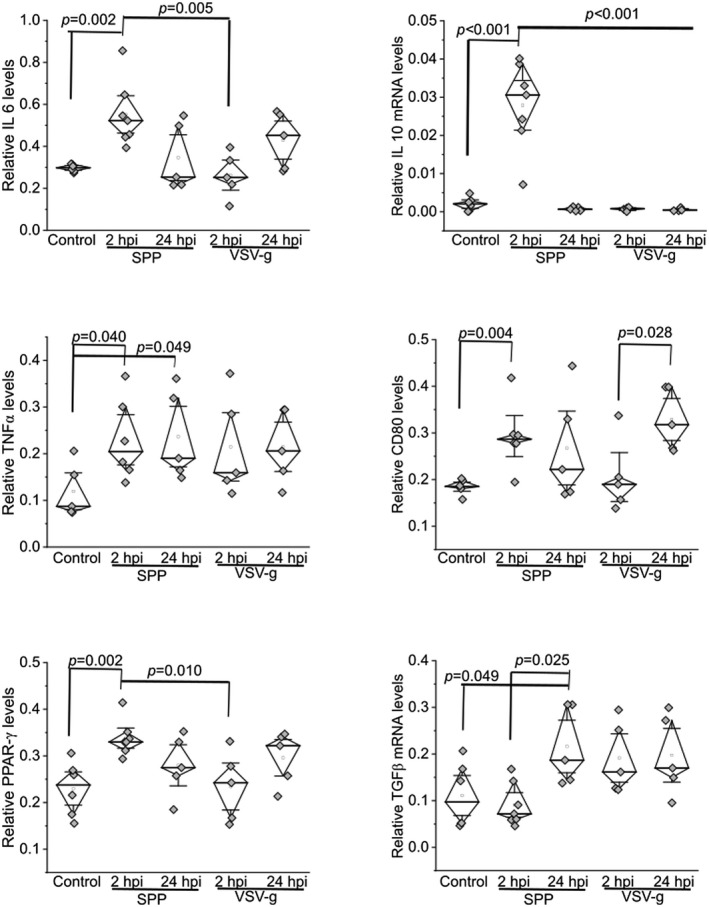
Upregulation of inflammatory factors in the lungs of mice after administration of Spp or VSV‐G lentivirus at 2 or 24 hpi. The relative mRNA levels of IL6, IL10, TNFα, CD80, PPAR‐γ, and TGFβ were determined using a real‐time RT‐PCR assay. The mRNA level of each target gene was normalized to Rps18 levels
3.5. Dysregulation of inflammatory cytokines in RAW cells caused by spike protein
In order to investigate potential immunomodulatory function induced by S protein, we infected RAW cells with Spp or VSV‐g lentivirus. The mRNA levels of IL6, IL10, CD80, and PPAR‐γ were significantly higher in the Spp lentivirus‐infected group at 2 hpi than in the untreated controls and the VSV‐G group at 2 hpi (Figure 5). The mRNA levels of these markers were higher in the Spp group at 16 hpi than at 2 hpi (Figure 5).
FIGURE 5.
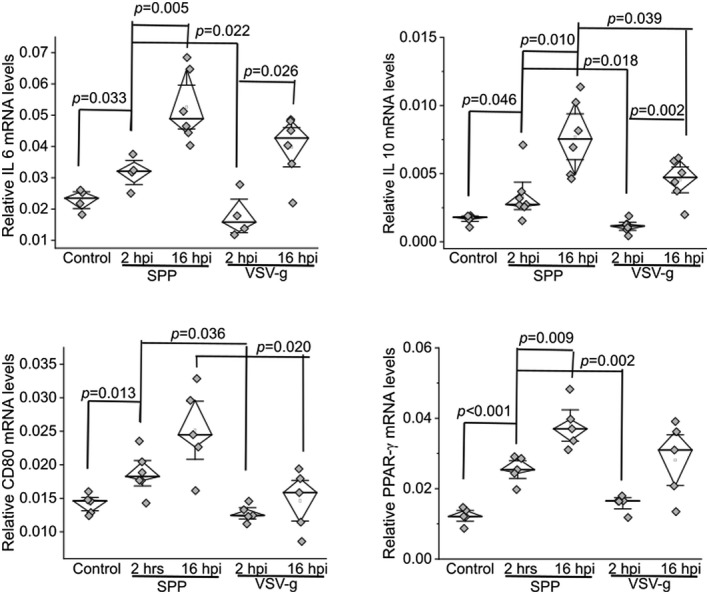
Upregulation of inflammatory factors in RAW cells after being infected with Spp or VSV‐G lentivirus at 2 or 16 hpi. Relative mRNA levels of IL6, IL10, CD80, and PPAR‐γ were determined using a real‐time RT‐PCR assay. The mRNA level of each target gene was normalized to Rps18 levels
Next, we electroporated RAW cells with pcDNA‐spike expression plasmid or two control plasmids, pcDNA and EGFP‐N2. The expression of EGFP 2 days postelectroporation showed 30%‐40% transfection efficiency in RAW cells (Figure 6). The mRNA levels of IL6, IL10, CD80, and PPAR‐γ were significantly higher in the S‐protein‐expression group than in the pcDNA or EGFP‐N2 groups (Figure 6).
FIGURE 6.
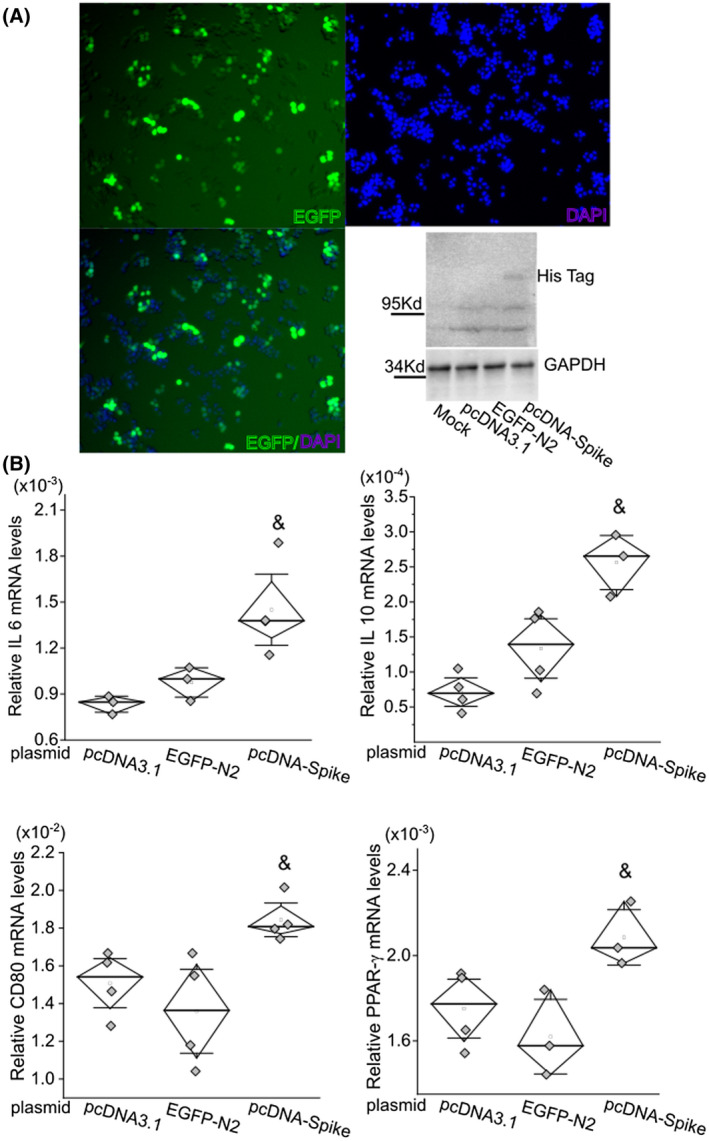
S protein of SARS‐CoV‐2 only induces upregulation of inflammatory factors in RAW cells. A, Expression of EGFP‐N2 after electroporation of targeted plasmid into RAW cells to estimate transfection efficiency. The expression of S protein in RAW cells was detected using an anti‐His antibody by western blot test. Electroporation of pcDNA3.1 and EGFP‐N2 plasmids was used as negative controls. B, mRNA levels of IL6, IL10, CD80, and PPAR‐γ were significantly upregulated after forced expression of S protein. The mRNA level of each target gene was normalized to Rps18 levels. &, P < .05 as compared with control groups with electroporation of pcDNA3.1 blank vector or EGFP‐N2 plasmid
4. DISCUSSION
There is an urgent priority for the global healthcare and research communities to mitigate the current COVID‐19 pandemic. Animal models that can replicate viral transmission and subsequent pathological development are critical for this effort to understand the pathogenic mechanisms of this disorder and develop effective antiviral countermeasures. However, the handling of specimens infected with SARS‐CoV‐2 requires high‐security biosafety level 3 (BSL3) facilities and BSL3 work practices. We have developed an Spp lentivirus that confers the spike protein on the viral surface in order to investigate host tropism in a BSL2 setting. More importantly, using this virus, we have demonstrated the pathogenicity of the S protein in isolated cells and in an animal model. Therefore, our data prove that the Spp lentivirus, although it cannot completely replicate the infectious pathway and pathological process in humans, is a very useful system for specifically investigating S‐protein‐mediated cell type susceptibility, host tropism for infection, and pathogenicity.
The SARS‐CoV‐2 RNA virus can be detected in swab samples from the upper respiratory tract for more than 80% of affected patients.16, 17, 18 Viral RNAs are rarely detectable in the blood of asymptomatic patients or nonhospitalized patients16 but have been found more frequently in 10.5%‐67% hospitalized patients with severe symptoms.17, 18, 19 Using a supersensitive RT‐PCR RUO assay (low limit of detection at 625 copies of SARS‐CoV‐2 RNA/mL), SARS‐CoV‐2 has been detected in the blood of 53% of mild‐to‐moderate patients and 88% of critically ill patients, with the level of virus being associated with the disease severity.20 The viral shedding time (from positive to negative) of blood is shorter than that of a nasal swab.18 An animal study has also shown that intranasal inoculation of SARS‐CoV‐2 in a transgenic mouse model expressing human ACE2 resulted in high levels of viral infection in the lungs, with spread to other organs.21 These reports demonstrate that SARS‐CoV‐2 can be disseminated from the respiratory system into the circulatory system and to many other vital organs during the late stages of the disease; this is one crucial determinant for whether a patient will develop severe symptoms. Therefore, it is necessary to recapitulate the disease progression under this viremic condition in an animal model. In this study, we intravenously administrated Spp lentivirus into male mice and attempted to develop an animal model to mimic part of these critical conditions in patients. Our data show a high preference for the Spp lentivirus to reside in lung tissues and induction of acute pneumonia, which is thought to be mediated or contributed by the S protein. Therefore, although it may not fully simulate the nature of the entry pathway of SARS‐CoV‐2 through the upper airway into the lungs in the initial stage of the disease, this study has provided valuable information relevant to the pathological progression of patients with viremia.
The S protein is thought to be the crucial glycoprotein on the surface of SARS‐CoV‐2 for mediating viral entry into host cells. It can bind to ACE2 in host cells; this is followed by an aid of TMPRSS2 cleavage, leading to endocytosis of the viral/receptor complex in the host cells. ACE2 is present in type II alveolar cells, macrophages, and endothelial cells in the lungs, which causes them to be targeted by SARS‐CoV‐2. Indeed, SARS‐CoV‐2 has been found in pneumocytes and endothelial cells in autopsy studies of COVID‐19 patients.22 In this study, we showed that Spp lentivirus was mainly present in LDLR+ type II alveolar cells and macrophages in the lungs. Further evidence demonstrated that type II alveolar cells (SPC+) are the predominant cell type responsible for uptake of Spp lentivirus. These cells also express ACE2.23, 24 Although rodent ACE2 receptors have shown a much lower affinity to the SARS‐CoV‐2 spike protein as compared with human ACE2 receptors,21, 25, 26 our data suggest that the ACE2 receptor confers the main point of viral entry into the lungs of mice, because deletion of the RBD domain of the S protein diminished viral uptake. Unexpectedly, deletion of the RBD domain increased viral load in the heart and aorta, indicating that an alternative ACE2‐independent pathway may be recruited in these organs. We speculate that other receptors or cofactors may facilitate S‐protein‐mediated viral entry into host cells in the cardiovascular system; for example, the NRP1‐mediated entry pathway.8, 9, 10 These detailed mechanisms need to be elucidated in the future. Nevertheless, these data provide insight into the possibility that multiple factors/pathways are involved in SARS‐CoV‐2 entry into various target cells. The Spp lentivirus will allow us to investigate the mechanisms of viral entry into target cells in greater detail in future studies.
Because the Spp lentivirus is a replication‐deficient virus, it is unable to generate a full spectrum of pathological changes in host cells. We did not observe any inflammatory responses in the lungs of the mice at 2 hpi for the Spp or VSV‐g groups. At 24 hpi with Spp but not VSV‐g lentivirus, the mice developed acute, diffuse, and evident lung inflammation. Pneumonia was transient and resolved 7 days after Spp lentivirus administration (data not shown). This study demonstrated that the Spp lentivirus is able to replicate a portion of the acute lung pathology caused by SARS‐CoV‐2 in humans. SARS‐CoV‐2–induced hyperinflammation in the lungs is considered to cause disease progression. For example, the occurrence of acute and diffuse lung injuries is supported by evidence of upregulation of a series of serum cancer biomarkers in patients.27 Our data show that a portion of M1 but not M2 macrophages exhibited rapid uptake of Spp lentivirus in the lungs (by 2 hpi), providing direct evidence to support this notion. The severity of the acute lung inflammation induced in our mouse model was greater in the Spp group than in the VSV‐g group. Furthermore, a panel of inflammatory factors was upregulated in the lungs following Spp lentivirus infection; the same panel exhibited different patterns in the lungs with VSV‐g infection. Considering that the S protein is the only difference between the Spp and VSV‐g lentiviruses, these data lead us to speculate that the S protein may play a unique role in the development of this lung pathology. Indeed, overexpression of the S protein in RAW cells can induce upregulation of the same panel of inflammatory factors (IL6, IL10, CD80, and PPAR‐γ), demonstrating that the S protein has a role in inducing intracellular pathological alterations. The detailed mechanisms underlying the way in which the S protein contributes to inflammatory reactions in macrophages are unknown and will be studied in the future.
COVID‐19 patients with metabolic‐associated preconditions have a high risk of developing more severe symptoms. Our recent studies have shown that decreased levels of low‐ and high‐density lipoprotein cholesterol levels are associated with severity and mortality of COVID‐1912, 13; this has been confirmed by many other reports.28, 29, 30, 31 The etiology of lipid abnormality in COVID‐19 patients is likely multifactorial, including factors such as liver dysfunction, cytokine storm‐induced alterations of lipid metabolism, virus‐induced aberrant modulations in cholesterol synthesis, and increased free radical signaling to facilitate the modification and degradation of LDL‐c.32, 33 The S protein has a binding pocket for fatty acids, steroids, and cholesterol to modulate entrance into host cells.34, 35 In this study, our data showed that LDLR+ cells had the highest level of Spp lentivirus uptake in the lungs. This correlation, together with previous reports, indicates that lipid metabolism is tightly associated with SARS‐CoV‐2–induced acute lung pathology. These data also suggest the possibility that LDLR may be directly involved in viral uptake; this will be investigated in the future with the Spp lentivirus.
The limitations of this study include the following. First, the Mus musculus strain as an animal model for SARS‐CoV‐2 viral entry studies is limited because of the low affinity of the endogenous ACE2 receptor to the RBD domain of the spike protein. However, this has a minimal effect on the study of various pathological functions of the S protein apart from mediating viral entry which were explored in the current study. Second, veinous viral administration is not the natural way of acquiring SARS‐CoV‐2 infection. However, this approach can directly help recapitulate some phenotypes of COVID‐19 patients that develop viremia.
In conclusion, our data show that the Spp lentivirus induced an acute and transient inflammatory response in the lungs of male mice. The Spp lentivirus preferentially targets those macrophages and pneumocytes with expression of LDLR in the lungs and leads to upregulation of IL6, IL10, CD80, and PPAR‐γ. In addition, forced expression of the S protein can cause elevation of IL6, IL10, CD80, and PPAR‐γ in RAW cells. Our results demonstrated that the S protein of SARS‐CoV‐2 confers the main point of viral entry into the lungs and can activate macrophages and contribute to the induction of acute inflammation in the lungs. Our data also indicate that an alternative ACE2‐independent viral entry pathway may be recruited in the heart and aorta.
DISCLAIMER
Any views expressed here represent personal opinion and do not necessarily reflect those of the US Department of Health and Human Services or the United States federal government.
PRIOR PUBLICATION
None of the material in this manuscript has been published or is under consideration for publication elsewhere, including the Internet.
DISCLOSURES
The authors have stated explicitly that there is no conflict of interest in connection with this article.
AUTHOR CONTRIBUTIONS
Cao and Tian performed in vivo studies; Nguyen performed in vitro RAW cell studies; Zhang generated Spp and VSV‐G lentiviruses; Gao and Yin participated in cell culture experiments and real‐time RT‐PCR and immunoblot assays; Carver, Fan, Albrecht, and Cui analyzed the data and revised the manuscript; Tan and Cui provided funding; and Tan designed and supervised the research.
Supporting information
Table S1
ACKNOWLEDGMENTS
We greatly appreciate Dr Mitzi Nagarkatti and Dr Juhua Zhou from the Department of Pathology, Microbiology, and Immunology at the University of South Carolina School of Medicine for their kind and generous help with real‐time PCR assays. We are also thankful for the support and assistance from the Instrumentation Resource Facility at the University of South Carolina School of Medicine. We are grateful for the support of Dr Igor Roninson and the COBRE Center for Targeted Therapeutics at the University of South Carolina.
Cao X, Tian Y, Nguyen V, et al. Spike protein of SARS‐CoV‐2 activates macrophages and contributes to induction of acute lung inflammation in male mice. The FASEB J. 2021;35:e21801. 10.1096/fj.202002742RR
Xiaoling Cao, Yan Tian, and Vi Nguyen contributed equally to this work.
Funding information
This work was supported by the National Institutes of Health (NIH) Grant No AR073172 to WT, the NIH Centers of Biomedical Research Excellence (COBRE) Grant No P20GM109091 pilot award to WT, NIH Grant No. HL131667 to T. C., and the Department of Defense Congressionally Directed Medical Research Programs (CDMRP) Grant No. W81XWH1810096 to WT.
REFERENCES
- 1.CoronavirusResourceCenter . COVID‐19 Dashboard by the Center for Systems Science and Engineering (CSSE) at Johns Hopkins University (JHU); 2020. [Google Scholar]
- 2.Coronaviridae Study Group of the International Committee on Taxonomy of Viruses . The species Severe acute respiratory syndrome‐related coronavirus: classifying 2019‐nCoV and naming it SARS‐CoV‐2. Nat Microbiol. 2020;5:536‐544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhou P, Yang XL, Wang XG, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;588(7836):E6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, function, and antigenicity of the SARS‐CoV‐2 spike glycoprotein. Cell. 2020;181(2):281‐292.e286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hoffmann M, Kleine‐Weber H, Pohlmann S. A multibasic cleavage site in the spike protein of SARS‐CoV‐2 is essential for infection of human lung cells. Mol Cell. 2020;78:779‐784.e775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bestle D, Heindl MR, Limburg H, et al. TMPRSS2 and furin are both essential for proteolytic activation of SARS‐CoV‐2 in human airway cells. Life Sci Alliance 2020;3(9):e202000786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ziegler C, Allon SJ, Nyquist SK, et al. SARS‐CoV‐2 receptor ACE2 is an interferon‐stimulated gene in human airway epithelial cells and is enriched in specific cell subsets across tissues. Cell. 2020;181(5):1016‐1035.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Daly JL, Simonetti B, Klein K, et al. Neuropilin‐1 is a host factor for SARS‐CoV‐2 infection. Science. 2020;370(6518):861‐865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cantuti‐Castelvetri L, Ojha R, Pedro LD, et al. Neuropilin‐1 facilitates SARS‐CoV‐2 cell entry and infectivity. Science. 2020;370(6518):856‐860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moutal A, Martin LF, Boinon L, et al. SARS‐CoV‐2 spike protein co‐opts VEGF‐A/Neuropilin‐1 receptor signaling to induce analgesia. Pain. 2020;162(1):243‐252.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Richardson S, Hirsch JS, Narasimhan M, et al. Presenting characteristics, comorbidities, and outcomes among 5700 patients hospitalized with COVID‐19 in the New York City area. JAMA. 2020;323(20):2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fan J, Wang H, Ye G, et al. Low‐density lipoprotein is a potential predictor of poor prognosis in patients with coronavirus disease 2019. Metabolism. 2020;107:154243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wei X, Zeng W, Su J, et al. Hypolipidemia is associated with the severity of COVID‐19. J Clin Lipidol. 2020;14(3):297‐304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Voyno‐Yasenetskaya TA, Dobbs LG, Erickson SK, Hamilton RL. Low density lipoprotein‐ and high density lipoprotein‐mediated signal transduction and exocytosis in alveolar type II cells. Proc Natl Acad Sci U S A. 1993;90:4256‐4260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gowdy KM, Fessler MB. Emerging roles for cholesterol and lipoproteins in lung disease. Pulm Pharmacol Ther. 2013;26:430‐437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Corman VM, Rabenau HF, Adams O, et al. SARS‐CoV‐2 asymptomatic and symptomatic patients and risk for transfusion transmission. Transfusion. 2020;60(6):1119‐1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen W, Lan Y, Yuan X, et al. Detectable 2019‐nCoV viral RNA in blood is a strong indicator for the further clinical severity. Emerg Microbes Infect. 2020;9(1):469‐473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fang Z, Zhang Y, Hang C, Ai J, Li S, Zhang W. Comparisons of viral shedding time of SARS‐CoV‐2 of different samples in ICU and non‐ICU patients. J Infect. 2020;81(1):147‐178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Buetti N, Patrier J, Le Hingrat Q, et al. Risk factors for SARS‐CoV‐2 detection in blood of critically ill patients. Clin Infect Dis. 2020;72(10):e690‐e691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Veyer D, Kerneis S, Poulet G, et al. Highly sensitive quantification of plasma SARS‐CoV‐2 RNA shelds light on its potential clinical value. Clin Infect Dis. 2020. Aug 17;ciaa1196. 10.1093/cid/ciaa1196. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Winkler ES, Bailey AL, Kafai NM, et al. SARS‐CoV‐2 infection of human ACE2‐transgenic mice causes severe lung inflammation and impaired function. Nat Immunol. 2020;21:1327‐1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fox SE, Akmatbekov A, Harbert JL, Li G, Quincy Brown J, Vander Heide RS. Pulmonary and cardiac pathology in African American patients with COVID‐19: an autopsy series from New Orleans. Lancet Respir Med. 2020;8:681‐686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wiener RS, Cao YX, Hinds A, Ramirez MI, Williams MC. Angiotensin converting enzyme 2 is primarily epithelial and is developmentally regulated in the mouse lung. J Cell Biochem. 2007;101:1278‐1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Keidar S, Gamliel‐Lazarovich A, Kaplan M, et al. Mineralocorticoid receptor blocker increases angiotensin‐converting enzyme 2 activity in congestive heart failure patients. Circ Res. 2005;97:946‐953. [DOI] [PubMed] [Google Scholar]
- 25.Li W, Greenough TC, Moore MJ, et al. Efficient replication of severe acute respiratory syndrome coronavirus in mouse cells is limited by murine angiotensin‐converting enzyme 2. J Virol. 2004;78:11429‐11433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Letko M, Marzi A, Munster V. Functional assessment of cell entry and receptor usage for SARS‐CoV‐2 and other lineage B betacoronaviruses. Nat Microbiol. 2020;5:562‐569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wei X, Su J, Yang K, et al. Elevations of serum cancer biomarkers correlate with severity of COVID‐19. J Med Virol. 2020;92(10):2036‐2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu X, Chen D, Wu L, He G, Ye W. Declined serum high density lipoprotein cholesterol is associated with the severity of COVID‐19 infection. Clin Chim Acta. 2020;510:105‐110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sorokin AV, Karathanasis SK, Yang ZH, Freeman L, Kotani K, Remaley AT. COVID‐19—Associated dyslipidemia: implications for mechanism of impaired resolution and novel therapeutic approaches. FASEB J. 2020;34(8):9843‐9853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang G, Zhang Q, Zhao X, et al. Low high‐density lipoprotein level is correlated with the severity of COVID‐19 patients: an observational study. Lipids Health Dis. 2020;19:204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tanaka S, De Tymowski C, Assadi M, et al. Lipoprotein concentrations over time in the intensive care unit COVID‐19 patients: results from the ApoCOVID study. PLoS ONE. 2020;15:e0239573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bojkova D, Klann K, Koch B, et al. Proteomics of SARS‐CoV‐2‐infected host cells reveals therapy targets. Nature. 2020;583(7816):469‐472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cao X, Yin R, Albrecht H, Fan D, Tan W. Cholesterol: a new game player accelerating vasculopathy caused by SARS‐CoV‐2? Am J Physiol Endocrinol Metab. 2020;319:E197‐E202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Toelzer C, Gupta K, Yadav SKN, et al. Free fatty acid binding pocket in the locked structure of SARS‐CoV‐2 spike protein. Science. 2020;370:725‐730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shoemark D, Colenso C, Toelzer C, et al. Molecular simulations suggest vitamins, retinoids and steroids as ligands binding the free fatty acid pocket of SARS‐CoV‐2 SPIKE PROTEIN. Angew Chem Int Ed. 2021;60:7098‐7110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1