

Abstract
A multi‐pronged approach with help in all forms possible is essential to completely overcome the Covid‐19 pandemic. There is a requirement to research as many new and different types of approaches as possible to cater to the entire world population, complementing the vaccines with promising results. The need is also because SARS‐CoV‐2 has several unknown or variable facets which get revealed from time to time. In this work, in silico scientific findings are presented, which are indicative of the potential for the use of the LL‐37 human anti‐microbial peptide as a therapeutic against SARS‐CoV‐2. This indication is based on the high structural similarity of LL‐37 to the N‐terminal helix, with which the virus interacts, of the receptor for SARS‐CoV‐2, Angiotensin Converting Enzyme 2. Moreover, there is positive prediction of binding of LL‐37 to the receptor‐binding domain of SARS‐CoV‐2; this is the first study to have described this interaction. In silico data on the safety of LL‐37 are also reported. As Vitamin D is known to upregulate the expression of LL‐37, the vitamin is a candidate preventive molecule. This work provides the possible basis for an inverse correlation between Vitamin D levels in the body and the severity of or susceptibility to Covid‐19, as widely reported in literature. With the scientific link put forth herein, Vitamin D could be used at an effective, medically prescribed, safe dose as a preventive. The information in this report would be valuable in bolstering the worldwide efforts to eliminate the pandemic as early as possible.
Keywords: Covid‐19, in silico structural analysis, LL‐37, molecular docking, therapeutic, vitamin D
1. INTRODUCTION
The world is plagued with the peculiar and daunting problem that a vast population of humans does not mount an effective innate immune response against SARS‐CoV‐2. 1 World‐wide efforts have come/are coming to fruition for the development of vaccines and drugs. Yet, there could be challenges to surmount, confounded by the nature and behavior of SARS‐CoV‐2. 2 Even with the promising results of vaccines developed in the world, it is well recognized that the need for additional modalities is essential, due to the sheer enormity of the problem. It could take a long time before Covid‐19 is wiped from the face of Earth in totality. Also, by now, it is known that the virus is notoriously dynamic and also that there are several unknowns. SARS‐CoV‐2 has hypervariable genomic hotspots, evolves rapidly by mutations, and there is variability in epidemiologic and clinical attributes. The picture is not fully known about the human immune response to the virus and neither are its clinical manifestations completely understood. 3 Thus, to minimize the period of suffering and bring the ultimate goal closer, there should be research in the direction of as many different types of approaches that can be thought of, including non‐typical approaches. In this work, a different possibility is presented, in the form of the human anti‐microbial peptide LL‐37 as a therapeutic, the reasoning for which is collated and elucidated below.
SARS‐CoV‐2 attaches to the cell surface receptor Angiotensin Converting Enzyme 2 (ACE2) by the binding of the Receptor Binding Domain (RBD) of its Spike protein to ACE2. The N‐terminal helix (NTH) of the peptidase domain of ACE2 is mainly responsible for the interaction with the RBD. 4 The Receptor Binding Motif (RBM) within the RBD, forms a concavity into which the NTH fits in. Most of the residues of ACE2 that form interactions with the RBD are present in the N‐terminal helix. A stretch of ACE2 from Ser19 to Asn53 comprises the NTH. 5
LL‐37, a 37 amino acid peptide, produced from the cleavage of an 18 kDa polypeptide, hCAP18, is an anti‐microbial peptide (AMP) present in humans. 6 It is produced not only by multiple immune cells like NK cells, B cells, mast cells, and so forth, but also by epithelial cells present in the skin and the respiratory tract. 7 LL‐37 is majorly present extracellularly. 8 Within cells, it is present in its proform in granules. LL‐37 can get internalized into human macrophages where it can act in clearance of intracellular pathogens. 9 LL‐37 is internalized by human mast cells which it induces to release nucleic acids. 10 Besides the action of LL‐37 in destabilizing membranes of pathogens, LL‐37 has also been reported to be a cell‐penetrating peptide (CPP). 11 The physiological concentration of LL‐37 is known to be in the range of 2–5 μg/ml. However, in case of infection its concentration could rise up to 20 μg/ml. 12 LL‐37 also exhibits antiviral activity against several viruses by interacting with and destabilizing the viral envelopes. 13 LL‐37 has also been found to interfere with the replication of several single‐stranded enveloped RNA viruses similar to SARS‐CoV‐2, such as Respiratory Syncytial Virus, Influenza A hepatitis C virus, Dengue virus, HIV‐1 and Vaccinia Virus. 14 Furthermore, LL‐37 has been found to inhibit viral binding to host cells. LL‐37 inhibits Dengue virus type 2 from entering cells by binding to the E protein of the virus. 15 Binding of Respiratory Syncytial Virus to epithelial cells is diminished by LL‐37. 16 LL‐37 also had an inhibitory effect on the binding of non‐enveloped Enterovirus 71 to U251 cells. 17 A metabolite of Vitamin D3, 1α,25‐dihydroxy vitamin D3 (1,25D3), is known for its role in transcriptional regulation of many genes of the immune system. 18 Genes regulated by 1,25D3 also code for anti‐microbial peptides. Human antimicrobial peptide hCAP‐18/LL‐37 gene is one such gene which is upregulated by Vitamin D. 19
Moving on to the logic of this study, a large collection of evidence has mounted that there is a correlation between Covid‐19 and Vitamin D. In PubMed, there are numerous articles reporting or discussing such correlation. To give an example, in an Israeli population‐based analysis involving 7807 people, 10% individuals who were Covid‐19 positive had lower 25(OH) Vitamin D levels in the plasma than the individuals who were tested negative and the finding was statistically significant. 20 In another work, which is most relevant to our study, the binding of LL‐37 to enveloped viruses and their consequent disablement has been described and it has been postulated that Vitamin D deficiency manifests as aggravated Covid‐19 through consequent low expression of cathelicidin. 14 Besides severity of disease, a recent study 21 implicates an increased risk of contracting Covid‐19 when there is deficiency of Vitamin D. Also, several studies have found Vitamin D deficiency to be linked with greater risk of viral respiratory tract infections. 22 In our study, the question was asked—Could this association be something that could be tapped for developing a potential therapy? LL‐37 is known to have a helical structure 23 as depicted in Figure 1 (Panel I). The actual part of ACE2 that the RBD binds to is the N‐terminal helix (NTH) (Figure 1, Panel I) of ACE2.
FIGURE 1.
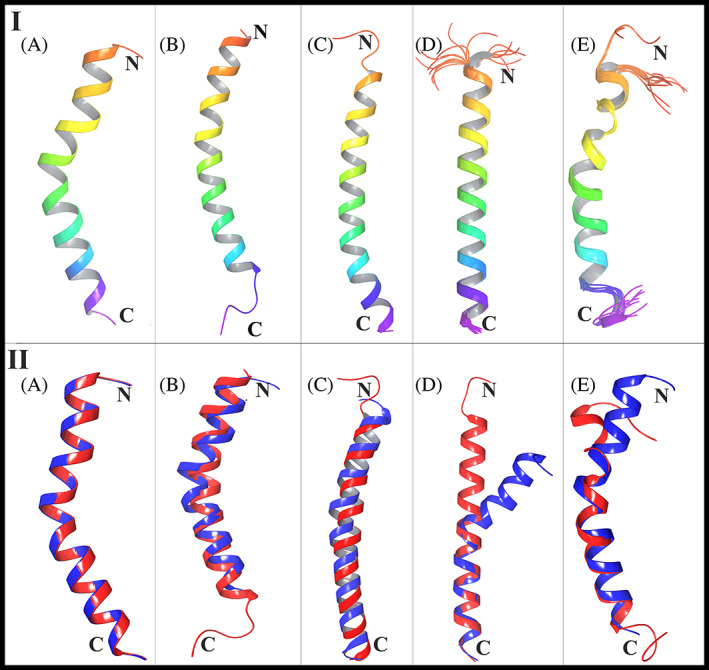
Panel I: Secondary structures of NTH and peptides used in the study (A) The secondary structure of NTH was obtained by extracting residues 19–53 of hACE2 using Maestro from PDB ID 6LZG from https://www.rcsb.org/. (B) The structure of the LL‐37 peptide (2K6O) from https://www.rcsb.org/. (C) The structure of the PACAP‐38 peptide (2D2P) from https://www.rcsb.org/. (D) The structure of the Amylin endocrine hormone (2KB8) from https://www.rcsb.org/. (E) The structure of the GLP‐2 peptide (2L63) from https://www.rcsb.org/. Panel II: Structural alignment of hACE2 NTH with (A) hACE2 NTH, (B) LL‐37, (C) PACAP‐38, (D) Amylin and (E) GLP‐2. The structure of hACE2 NTH is represented in blue color and aligned peptides are shown in red color
Thus, a hypothesis was made that LL‐37, could be binding to RBD and if true, this could be one of the most effective way in which LL‐37 could interfere with SARS‐CoV‐2 by binding to the receptor‐binding domain (RBD) of the virus. Therefore, in this work, in silico studies were undertaken, mainly to test the hypothesis put forth and to unearth the possibility, if any, of binding of LL‐37 to RBD. A great value of this approach to develop a treatment would be that a molecule that is already present in the body's circulation could be used as a therapeutic, as the safety criteria for the therapeutic would be more likely to be met than extraneous peptides or those not already present in the circulation.
The Spike protein RBD is amply glycosylated 24 and the glycan moieties are considered to be steric hindrances for neutralizing antibodies to bind—a mechanism of immune evasion by the virus. The RBD gets exposed when the virus is about to bind to ACE2; this is referred to as the “UP” state of the RBD, as against the unavailable “DOWN” state, these states being alternated by the control of glycans. 25 The development of neutralizing antibodies as a therapeutic depends on the “Open” or available state of the RBD as against its “closed” state. 26 So, in the UP or Open state of the RBD, LL‐37 could also bind to it.
2. METHODS
2.1. Retrieval of 3‐D structures and their preparation
The secondary structure of the N‐terminal helix (NTH) of ACE2 was obtained by extracting residues 19–53 of hACE2 from PDB ID 6LZG from the Protein Data Bank (PDB) of the Research Collaboratory for Structural Bioinformatics (https://www.rcsb.org/), using the Maestro software (Schrödinger Release 2020‐2: Maestro, Schrödinger, LLC, New York, NY, 2020). The NMR structure of human LL‐37 and two X‐ray crystal structures of SARS‐CoV‐2 Receptor‐Binding Domain (RBD) in complex with its receptor, that is, human ACE2 (hACE2) were retrieved from the PDB database (https://www.rcsb.org/) with PDB ID: 2K6O, 27 6LZG, 28 and 6M0J, 4 respectively. Another three peptides that are structurally similar to LL‐37 have been used as “controls.” The basis for selection of these three peptides is explained in the Structural Alignment Results section. The three‐dimensional structures of these three peptides viz. Pituitary adenylate cyclase‐activating peptide‐38 (PACAP‐38) (PDB ID: 2D2P), 29 Amylin endocrine hormone (PDB ID: 2KB8) 30 and Glucagon‐like peptide‐2 (GLP‐2) (PDB ID: 2L63) 31 were retrieved from the PDB database. The sequences of NTH and the peptides are given in Table 1(A) and their structures are shown in Figure 1 (Panel I). The Schrödinger's Protein Preparation Wizard tool 32 (Schrödinger Release 2020‐2: Protein Preparation Wizard, Schrödinger, LLC, New York, NY, 2020) was used to prepare the downloaded PDB structures for structural correctness with respect to optimization of H‐bonds and energy minimization of heavy atoms by using OPLS‐2005 force field, resulting into high‐confidence structures for further molecular docking studies.
TABLE 1.
(A) The amino acid sequences of NTH and the peptides used in this study. (B) Quantitative assessment, using TM‐Align server, of hACE2 NTH Structural Similarity with peptides from RCSB (https://www.rcsb.org/) selected on the basis as described in the Results. The reference protein was NTH of ACE2. The descriptions are from (https://www.rcsb.org/)
| Panel A | |
|---|---|
| Structure | Amino acid sequence |
| NTH | STIEEQAKTFLDKFNHEAEDLFYQSSLASWNYNTN |
| 2K6O (LL‐37) | LLGD FFRKSKEKIGKEFKRIVQRIKDFLRNLVP RTES |
| 2D2P | HSDGIFTDSYSRYRKQMAVKKYLAAVLGKRYKQRVKNKX |
| 2KB8 | KCNTATCATQRLANFLVHSSNNFGAILSSTNVGSNTY |
| 2L63 | HADGSFSDEMNTILDNLAARDFINWLIQTKITD |
| Panel B | |||||
|---|---|---|---|---|---|
| Structure | Peptide length | TM‐score | RMSD (Å) | Aligned length | Description |
| N‐terminal helix | 35 | 1 | 0 | 35 | N‐terminal helix of ACE2 (from 6LZG) |
| Strict search | |||||
| 2K6O (LL‐37) | 37 | 0.68037 | 1.5 | 32 | Anti‐microbial peptide |
| 2D2P | 38 | 0.56736 | 1.62 | 34 | Pituitary adenylate cyclase activating polypeptide‐38 |
| 1GOE | 42 | 0.51713 | 2.61 | 33 | Corticotropin releasing hormone |
| 1G09 | 42 | 0.52615 | 2.61 | 33 | Corticotropin releasing hormone |
| 2RMY | 34 | 0.55258 | 1.52 | 27 | N‐BAR domain |
| 2KB8 | 37 | 0.54169 | 1.87 | 31 | Amylin endocrine hormone |
| 2N7I | 37 | 0.59781 | 2.33 | 32 | Prolactin receptor transmembrane domain |
| 2LAT | 37 | 0.69158 | 0.94 | 31 | Human minimembrane protein Ost4 |
| Relaxed search | |||||
| 2L63 | 33 | 0.44354 | 1.8 | 23 | Glucagon‐like peptide‐2 |
| 6ITH | 35 | 0.55154 | 1.64 | 32 | Trans‐membrane domain of syndecan‐2 |
| 1R02 | 33 | 0.32949 | 2.35 | 22 | Orexin‐A: Regulator of Appetite and Wakefulness |
| 2L77 | 39 | 0.49377 | 2.2 | 25 | PAP248‐286 |
| 2RND | 34 | 0.51577 | 1.16 | 25 | N‐terminal BAR peptide |
| 5NAO | 35 | 0.54347 | 1.41 | 28 | TLR4 transmembrane domain (624–657) |
| 6AHZ | 35 | 0.59246 | 1.58 | 29 | Polysialyltransferase Domain (PSTD) in Polysialyltransferase ST8siaIV |
| 6F46 | 32 | 0.39864 | 1.43 | 22 | Transmembrane helix of BclxL |
Abbreviation: RMSD, root mean square deviation of the alignments.
2.2. Quantitative assessment of NTH structure similarity with LL‐37, PACAP‐38, amylin, and GLP‐2
Firstly, the four peptide sequences were compared with the NTH sequence using BLASTP (National Center for Biotechnology Information, U.S. National Library of Medicine). To assess the structural similarity between N‐terminal Helix (NTH) of hACE2 with LL‐37, and the three control peptides, the quantitative assessment of protein structure similarity was carried out using TM‐Align online server 33 (https://zhanglab.ccmb.med.umich.edu/TM-Align/). The TM‐Align program produces the optimized residual alignment between two structures depending on their structural similarity followed by structural superimposition and gives the TM‐score which is a measure of the structural similarity. TM‐score gives the values in between 0 and 1, where 0 indicates the given structures are structurally different while 1 indicates perfect alignment and implies that both the structures are identical to each other. If the TM‐score is higher than 0.5, then it signifies that similar structural folds are present in both the structures and a score < 0.3 indicates random structural similarity. Further, a greater value for TM‐score signifies stronger structural similarity. 34 CATH (Database based on Class, Architecture, Topology, and Homologous superfamily) and SCOP (Structural classification of Proteins database) are commonly used standards for comparison and classification of protein structures. 35 For CATH, a TM‐score of 0.5 means that it is 37% probable that the two structures fall into to the same topology family; when the TM‐score is 0.6, this probability elevates to 80%. When TM‐score is <0.4, it means that are nearly zero protein pairs in the same SCOP Fold family. When the TM‐score is >0.6, the probability of the two proteins being in the same SCOP Fold is raised to >65%. TM‐Align also classifies the alignment of amino acid pairs according to a distance cut‐off “d,” distinguishing the pairs with d < 5.0 Å from those with d > 5.0 Å, the former being the better aligned pairs than the latter.
2.3. Macromolecular or protein–protein docking
For the macromolecular docking of given structures, we used the HDOCK online server for blind docking (http://hdock.phys.hust.edu.cn/) to predict their binding complexes with binding affinity. HDOCK predicts the intermolecular interactions at the interface of two proteins in binding complexes through a hybrid algorithm. 36 The HDOCK algorithm executes rigid docking by considering both the receptor and ligand as rigid molecules. We performed macromolecular docking of LL‐37 (PDB ID: 2K6O) with RBD of SARS‐CoV‐2 (from PDB ID: 6LZG and 6MOJ). For this, we separated the RBD‐ACE2 complex structures using Maestro software and used only RBD structural coordinates for docking. Then, these docking results were compared with positive and control dockings. Positive control docking, that is, docking of SARS‐CoV‐2 RBD with its receptor hACE2 was done by taking the 6LZG PDB complex, separating the two protein structures and then docking them. For control dockings, the three control peptides were docked with SARS‐CoV‐2 RBD (from 6LZG). Also, as a different type of control, LL‐37 was docked with the Spike protein region from residue 96 to residue 318, which is adjacent (immediately upstream) to RBD by extracting the structure of this region from the full spike protein structure of SARS‐CoV‐2 (PDB ID: 6Z97), 37 using Maestro software. All docked complexes were downloaded from the HDOCK server and their intermolecular interaction patterns were analyzed using Maestro.
2.4. Explicit MD simulation and MM/GBSA
To analyze the binding strength of LL‐37, NTH and control peptides with SARS‐CoV‐2 RBD, 100 ns molecular dynamic (MD) simulations were implemented with the help of Desmond software. 38 These docked complexes were prepared for MD simulation using the system builder of a panel of Desmond software. All the complex systems were immersed in a solvated cubic box of size 10 Å using explicit solvent, that is, the TIP3P water model. The suitable counter ions (Na+/Cl−) were added to the solvated system to neutralize the absolute charge of the complex system. The steepest descent energy minimization method was utilized during 100 ns MD simulation along with the OPLS‐2005 force field. 39 Also, all the systems were simulated using NPT (N/number of atoms, P/pressure, T/temperature) ensemble at 300 K temperature and 1 bar pressure. The Martyna‐Tobias‐Klein barostat method and Noose‐Hoover chain thermostat method were used to control the pressure and the temperature during the 100 ns MD simulation. The short‐range interactions, that is, columbic interactions were maintained at 9.0 Å cut‐off radius, and to integrate the equations of motion leap‐frog algorithm was used with 2 fs time step.
During the time of 100 ns MD, structural changes in RBD were monitored by comparing with docked complex conformation. Also, the binding strengths of NTH and peptides were analyzed through MM/GBSA (Molecular Mechanics, The Generalized Born Model, and Solvent Accessibility) using the Prime 40 module of Schrodinger software. Prime MM/GBSA works with the following equation by calculating the number of energies including polar solvation energies, non‐polar solvation energies, and potential energies of the complex system.
where G complex represents the protein‐ligand complex energy, G protein represents the unbound (free) protein energy and G ligand symbolizes the free ligand energy.
3. RESULTS
3.1. Structural alignment of LL‐37, PACAP‐38, amylin, and GLP‐2, with NTH
BLASTP gave the result “No significant sequence similarity” (to NTH) for all peptides. The validation of the TM‐Align server was done by giving the same hACE2 NTH structure as structure one and structure two inputs to the server. The resulting TM‐score of this alignment was 1, with root mean square deviation (RMSD) 0 Å and this confirmed the accuracy of the TM‐Align server. Structural similarity searches were performed in the RCSB PDB to make the study more stringent by finding structures similar to LL‐37 under (a) strict and (b) relaxed criteria. From the searches, structures that were isolated, that is, not in complex or as dimer/tetramer, and so forth, as that could change their native structure; in the range 37 plus/minus five residues so that they are closer to LL‐37; and those that were Solution NMR structures (as solution NMR gives the structure in native state) were selected. Structural alignment of each was done with NTH of ACE2. The results are tabulated in Table 1(B). The score for LL‐37 was 0.68037, so it falls under the high similarity category according to CATH and SCOP. It shows that LL‐37 has a significantly higher score than all (except 2LAT), even when compared with structures that were already similar to itself.
The structural alignments generated from the TM‐Align server are shown in Figure 1 (Panel II). It can be seen that LL‐37 even shares a similar bend in the helix as NTH, and this could help in fitting of LL‐37 into the concavity of RBD. Also, although the peptide Amylin has aligned with NTH with a score a little greater than 0.5, it can be seen from the figure that there is a clear bifurcation of the two structures toward the N‐terminus. This study suggests that the reported human anti‐microbial peptide LL‐37 is structurally very close to hACE2 NTH and this feature could facilitate its binding to RBD. For each peptide, the amino acids that aligned with those of NTH are shown in Figure 2 (Panel I). It can be seen that LL‐37 and PACAP‐38 have a greater number of aligned residues with d < 5.0 Å, and hence, better alignment than Amylin and GLP‐2. Now, those structures with alignment RMSD > 2.0 Å and/or those that are not found as free peptides in the circulation (which includes 2LAT), were eliminated. Thus, 2D2P, 2KB8, and 2L63 were selected as control peptides for docking studies.
FIGURE 2.
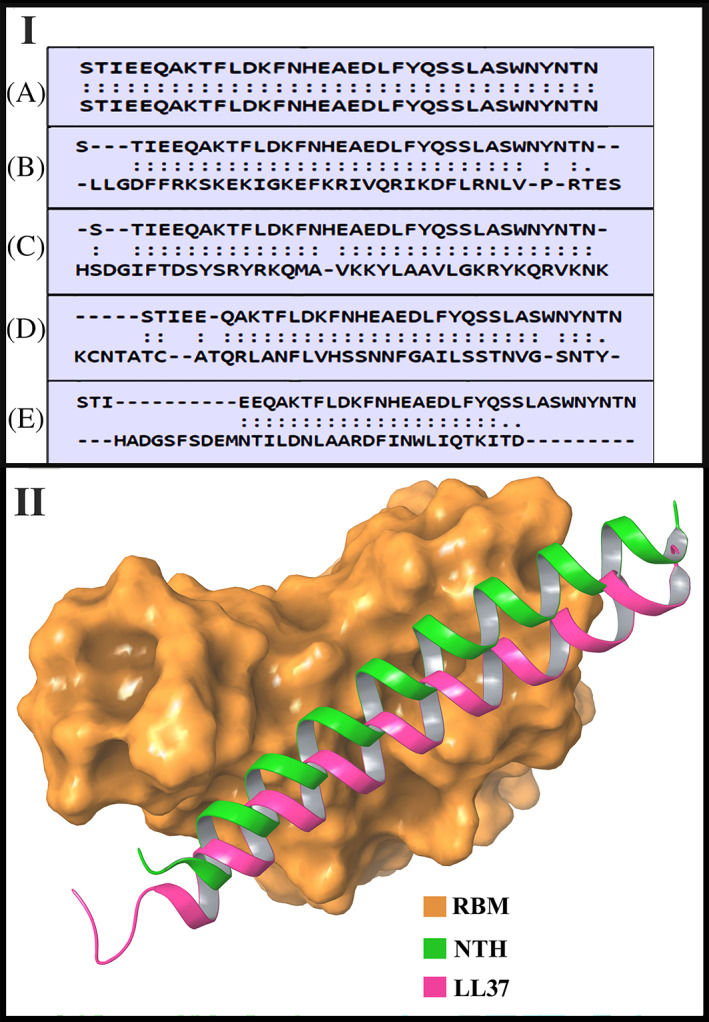
Panel I: The amino acids participating in the structural alignment of the different peptides with NTH. In each case, the top sequence is of NTH. The bottom sequence is of the following peptides (A) NTH (B) LL‐37 (C) PACAP‐38 (D) Amylin (E) GLP‐2. Two dots between a pair of aligned residues indicates d < 5.0 Å as explained in the Methods. A single dot shows other aligned residue pairs and dashes mean no structural alignment. Panel II: Depiction of the binding positions of NTH (extracted from ACE2 of PDB ID 6LZG complex) and docked LL‐37 (PDB ID 2K6O) across the surface of the receptor binding motif (RBM) of the RBD from PDB ID 6LZG
3.2. Intermolecular interactions between hACE2 and RBD (positive control)
The already determined crystal structure of SARS‐CoV‐2 RBD complexed with hACE2 (PDB ID: 6LZG) was used for docking analysis. The complexed structure was split into separate SARS‐CoV‐2 RBD and separate hACE2 structure and subjected to macromolecular docking calculations. The docking results gave a structurally very similar complex as compared to the crystal structure (PDB ID: 6LZG) with a docking score of −334.45. Intermolecular interactions between these docked complexes are summarized in Table 2(A). The N‐Terminal Helix of hACE2 is exposed toward the Receptor Binging Motif (RBM) of SARS‐CoV‐2 RBD and this interface makes a very stable complex by forming 15 different interactions comprising of nine hydrogen bond interactions, four aromatic hydrogen bonds, and two salt bridges. From the table, it can be seen that nine of the interactions predicted by our docking were common with the RBD‐ACE2 interactions in the experimentally determined RBD‐ACE2 complex structures. 4 , 41 , 42 This indicates the validity of the docking method used in this work.
TABLE 2.
(A) Detailed intermolecular interaction analysis of the hACE2‐SARS‐CoV‐2 RBD complex. Interactions which were common with the RBD‐ACE2 crystal structure determined by Shang et al, 41 are in Green. The interaction common with the RBD‐ACE2 crystal structure reported by Lan et al 4 is in Red, and the one common with RBD‐ACE2 cryo‐electron microscopy structure described by Yan et al 42 is in Blue. Interactions not found in any of these three reports are in Black. (B) Detailed intermolecular interaction analysis of the complex of NTH alone with RBD of SARS‐CoV‐2
| Panel A | |||
|---|---|---|---|
| Interacting residues (−334.45) a | |||
| hACE2 (PDB ID: 6LZG) | SARS‐CoV‐2 RBD (PDB ID: 6LZG) | Bond type | Bond distance (Å) |
| Ser19 | Ala475 | HB | 2.70 |
| Tyr83 | Asn487 | HB | 2.22 |
| Tyr83 | Asn487 | Ar‐HB | 3.20 |
| Phe28 | Tyr489 | Ar‐HB | 3.51 |
| Lys31 | Glu484 | Salt‐Bridge | 3.79 |
| Thr27 | Phe456 | Ar‐HB | 2.94 |
| Asp30 | Phe456 | Ar‐HB | 3.03 |
| Asp30 | Lys417 | Salt‐Bridge | 2.30 |
| Asp30 | Lys417 | HB | 1.36 |
| Glu35 | Gln493 | HB | 1.93 |
| Gln42 | Gly446 | HB | 1.66 |
| Lys353 | Gly496 | HB | 1.69 |
| Gln42 | Gln498 | HB | 2.17 |
| Lys353 | Gln498 | HB | 1.80 |
| Lys353 | Gly502 | HB | 2.10 |
| Panel B | |||
|---|---|---|---|
| Interacting residues (−224.14) a | |||
| NTH (PDB ID: 6LZG) | RBD (PDB ID: 6LZG) | Type of bond | Bond distance (Å) |
| Ile21 | Gln498 | HB | 2.74 |
| Phe28 | Tyr449 | Ar‐HB | 2.67 |
| Phe32 | Gln493 | Ar‐HB | 2.54 |
| Phe32 | Arg403 | Pi‐cation | 5.58 |
| Asn33 | Tyr505 | HB | 1.82 |
| Ser47 | Tyr421 | HB | 1.88 |
| Leu39 | Phe456 | Ar‐HB | 2.33 |
| Asn51 | Ser459 | HB | 1.70 |
Abbreviations: Ar‐HB, aromatic hydrogen bond; HB, hydrogen bond.
Docking score.
3.3. Intermolecular interactions between LL‐37 and RBD (Test)
The docked complexes of LL‐37‐RBD6LZG and LL‐37‐RBD6M0J gave docking scores of −253.75 and −238.18, respectively (a more negative value indicates stronger binding and stable complex). The intermolecular interactions between LL‐37 and SARS‐CoV‐2 RBD from 6LZG and 6M0J PDB are illustrated in Figure 3 (Panel I). In the LL‐37‐RBD (RBD from PDB ID: 6LZG) complex, there are eight interactions including three hydrogen bonds, three aromatic hydrogen bonds, and two salt bridges and the detailed interaction analysis is summarized in Table 3(A). In the LL‐37‐RBD (RBD from PDB ID: 6M0J) complex also, eight interactions were observed at the interface including two hydrogen bonds, four aromatic hydrogen bond interactions and two salt bridges. In Figure 2 (Panel II), the positions of NTH (extracted from ACE2 of 6LZG) and LL‐37 docked with the RBD from 6LZG, across the surface of the receptor binding motif (RBM) of the RBD from 6LZG are shown. From this figure, it is apparent that LL‐37 could hinder the binding of RBD to ACE2 by occupying the region where NTH interacts with RBD.
FIGURE 3.
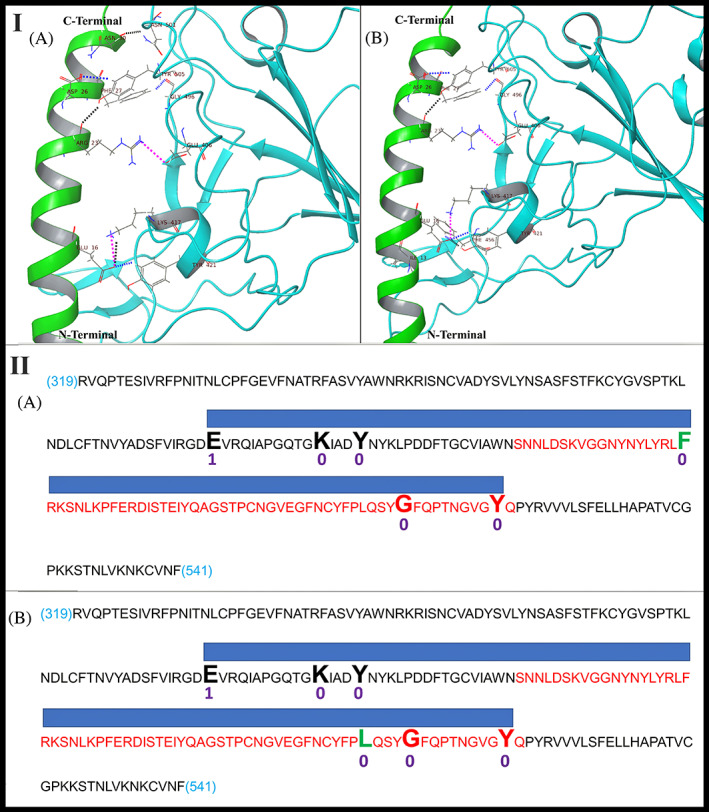
Panel I: Intermolecular interactions and binding mode of LL‐37 (green color) with (A) SARS‐CoV‐2 RBD from 6LZG PDB (cyan color); (B) SARS‐CoV‐2 RBD from 6M0J PDB (cyan color). Hydrogen bonds, aromatic hydrogen bonds and salt bridges are represented by dashed black, blue and pink colored lines respectively. Panel II: Diagrammatic representation of molecular docking of LL‐37 to RBD. Sequence: RBD sequence (319 to 541 of UniProt ID PODTC2 common to 6MOJ and 6LZG); Red sequence: RBM sequence; Large font bold residues: Contacted by LL‐37; Blue bar: Contact region covered by LL‐37. (A) LL‐37 bound to RBD of 6MOJ (Test1); Black/red residues: Common with LL‐37_RBD of 6LZG docking; Green residue: Not common with LL‐37_RBD of 6LZG docking; (B) LL‐37 bound to RBD OF 6LZG (Test2); Black/red residues: Common with LL‐37_RBD of 6MOJ docking; Green residue: Not common with LL‐37_RBD of 6MOJ docking. Numbers below residues: Number of mutations in 3564 SARS‐CoV‐2 genomes
TABLE 3.
(A) Detailed intermolecular interaction analysis of the LL‐37‐SARS‐CoV‐2 RBD complex. (B) Detailed intermolecular interaction analysis of the complexes of PACAP‐38, Amylin, GLP‐2 with SARS‐CoV‐2 RBD, and LL‐37 with the region adjacent (immediately upstream) to RBD
| Panel A | |||
|---|---|---|---|
| Interacting residues | Bond type | Bond distance (Å) | |
| LL‐37 (PDB ID: 2K6O) | SARS‐CoV‐2 RBD (PDB ID: 6LZG) (−253.75) a | ||
| Asn30 | Asn501 | HB | 1.67 |
| Phe27 | Gly496 | Ar‐HB | 2.75 |
| Asp26 | Tyr505 | Ar‐HB | 2.73 |
| Arg23 | Tyr505 | HB | 2.48 |
| Arg23 | Glu406 | Salt Bridge | 4.12 |
| Glu16 | Lys417 | Salt Bridge | 3.18 |
| Glu16 | Lys417 | HB | 2.53 |
| Glu16 | Tyr421 | Ar‐HB | 1.75 |
| LL‐37 (PDB ID: 2K6O) | SARS‐CoV‐2 RBD (PDB ID: 6M0J) (−238.18) a | ||
|---|---|---|---|
| Asp26 | Tyr505 | Ar‐HB | 2.96 |
| Phe27 | Gly496 | Ar‐HB | 2.82 |
| Arg23 | Tyr505 | HB | 2.74 |
| Arg23 | Glu406 | Salt Bridge | 3.25 |
| Glu16 | Phe456 | Ar‐HB | 3.38 |
| Glu16 | Lys417 | Salt Bridge | 3.09 |
| Glu16 | Tyr421 | Ar‐HB | 2.25 |
| Glu16 | Tyr421 | HB | 1.94 |
| Panel B | |||
|---|---|---|---|
| Interacting residues | Bond type | Bond distance (Å) | |
| PACAP‐38 (PDB ID: 2D2P) | SARS‐CoV‐2 RBD (PDB ID: 6LZG) (−230.30) a | ||
| Lys15 | Leu455 | HB | 1.56 |
| Arg12 | Phe456 | Ar‐HB | 2.63 |
| Tyr13 | Tyr489 | Pi‐Pi | 5.46 |
| Arg30 | Gln498 | 2 HB | 1.71, 2.08 |
| Amylin (PDB ID: 2KB8) | SARS‐CoV‐2 RBD (PDB ID: 6LZG) (−229.10) a | ||
|---|---|---|---|
| Phe23 | Gly446 | Ar‐HB | 2.63 |
| Phe23 | Tyr449 | Ar‐HB | 3.55 |
| Ser19 | Tyr449 | HB | 1.90 |
| Asn14 | Tyr505 | Ar‐HB | 2.37 |
| Asn14 | Arg403 | HB | 2.09 |
| Lys1 | Asp420 | Salt Bridge | 4.27 |
| Lys1 | Asn460 | HB | 1.15 |
| GLP‐2 (PDB ID: 2L63) | SARS‐CoV‐2 RBD (PDB ID: 6LZG) (−202.59) a | ||
|---|---|---|---|
| Phe22 | Asn450 | Ar‐HB | 2.85 |
| His1 | Asn343 | HB | 2.23 |
| His1 | Glu340 | Ar‐HB | 3.28 |
| LL‐37 (PDB ID: 2K6O) (PDB ID: 2K6O) | SARS‐CoV‐2 region adjacent to RBD (PDB ID: 6LZG) (−209.74) a | ||
|---|---|---|---|
| Phe17 | Phe238 | Pi‐Pi | 4.80 |
| Arg7 | Asp138 | Salt Bridge | 3.13 |
Abbreviations: Ar‐HB, aromatic hydrogen bond; HB, hydrogen bond; Pi‐Pi, non‐covalent interaction between aromatic rings.
Docking score.
From this docking study, it is clear that the residues from LL‐37, viz., Glu16, Arg23, Asp26 and Phe27, also residues from SARS‐CoV‐2 RBD, viz., Glu406, Lys417, Tyr421, Gly 496, and Tyr505 are very important residues for binding as these interactions were observed in both the Test complexes. The regions of RBD spanned by LL‐37 in each case are shown diagrammatically in Figure 3 (Panel II). Thus, five residues of RBD contacted by LL‐37 were found reproducible between the two test dockings. This finding also demonstrates that LL‐37 binding to RBD appears to be specific. The presence of two salt bridges in both the Test complexes predicts thermostability of the binding of LL‐37 to RBD. Comparing the docking of LL‐37 with RBD and hACE2 with RBD, hACE2 forms a very stable complex with RBD (−334.45 docking score), as not only the N‐terminal helix but other secondary structure elements of hACE2 are exposed to the surface binding site of SARS‐CoV‐2 RBD. Despite the weaker binding energy of the LL‐37‐RBD complex, the stability of this complex is very similar to the stability of the hACE2‐RBD complex because the scoring function of any docking program not only considers the number of interactions but also takes into consideration the Van der Waals' and electrostatic interactions, the decrease in entropy which occurs when the ligand binds, the hydrogen bonds and solvation factor. As seen in Table 2A, not only the NTH of hACE2 contributed in the binding but other residues are also involved in the binding with SARS‐CoV‐2 RBD resulting in a lower binding energy as compared to LL‐37‐RBD complex. LL‐37 consists of only a helix for docking with RBD, therefore the docking score for this complex was −253.75. To validate this point, only the NTH was extracted from the ACE2 of 6LZG and was docked with the RBD of 6LZG. The docked model which showed the binding position of NTH similar to the NTH position when the entire ACE2 binds to RBD as in determined structures (Figure 2, Panel II) was analyzed, and its docking score was −224.14. The detailed interactions are given in Table 2(B), from which it is seen that the interactions are mostly not common with the experimentally determined interactions. This would be due to the NTH alone docking in a similar but not perfectly the same position as when in the entire ACE2, due to the surrounding influences of ACE2.
3.4. Intermolecular interactions between other peptides and RBD (Control)
The docked complex of 2D2P‐RBD6LZG gives a docking score of −230.30. Five interactions including three hydrogen bonds, one aromatic hydrogen bond, and Pi‐Pi interaction were detected at the interface of the 2D2P‐RBD complex, and the detailed interaction analysis is summarized in Table 3(B). The docked complex of 2KB8‐RBD6LZG gives −229.10 as docking score. Seven interactions were observed at the interface including three hydrogen bonds, three aromatic hydrogen bond interactions, and one salt bridge. 2L63 forms only three interactions (two aromatic hydrogen bonds and one hydrogen bond) with RBD of SARS‐CoV‐2 and gives a docking score of −202.59. No salt bridges are formed. The intermolecular interactions between 2D2P, 2KB8, 2L63 and SARS‐CoV‐2 RBD from 6LZG PDB are illustrated in Figure 4 (Panel I). There was no commonality between RBD residues interacted with by NTH and by any of the Test or Control peptides. Figure 4 (Panel II) diagrammatically shows the alignments of the regions of RBD spanned by the control peptides. We also explored the binding stability of LL‐37 peptide toward the region which is immediately adjacent and upstream to SARS‐CoV‐2 RBD. This docking result gives a docking score of −209.74 by forming two interactions at the interface of this complex (Table 3(B), Figure 4 (Panel I)). Thus, LL‐37 showed poor binding to a region adjacent to RBD and this also is indicative of the specificity of LL‐37 binding to RBD.
FIGURE 4.
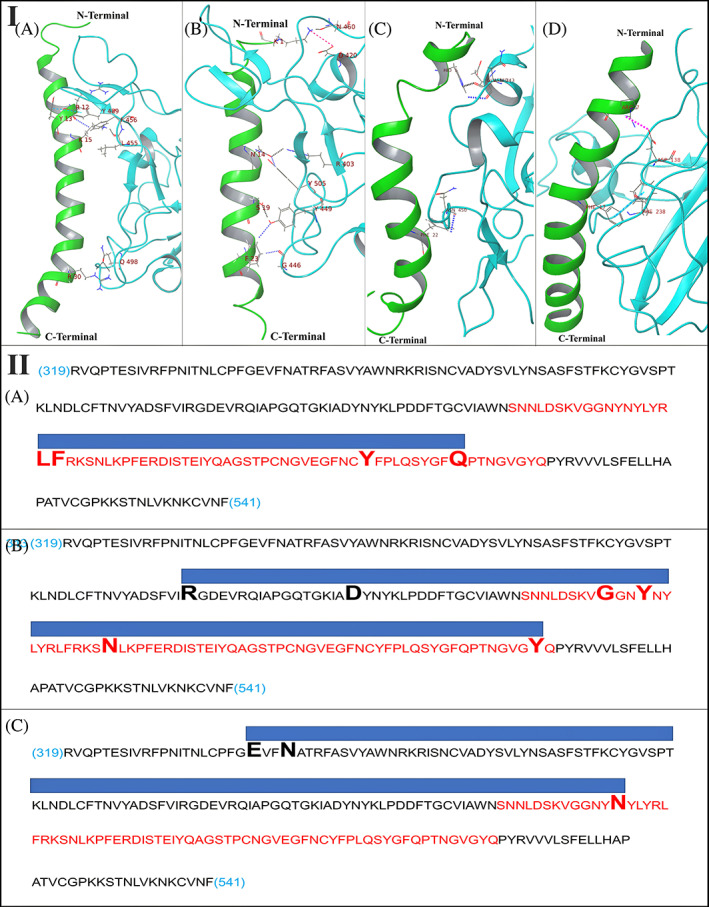
Panel I: Intermolecular interactions and binding mode of (A) PACAP‐38 with SARS‐CoV‐2 RBD, (B) Amylin with SARS‐CoV‐2 RBD, (C) GLP‐2 with SARS‐CoV‐2 RBD, and (D) LL‐37 with SARS‐CoV‐2 region (96–318) which is adjacent to RBD. Hydrogen bonds, aromatic hydrogen bonds, Pi‐Pi stacking and salt bridges are represented by dashed black, blue, sky blue and pink colored lines respectively. Panel II: Diagrammatic representation of molecular docking of control peptides to RBD. Sequence: RBD sequence (319 to 541 of UniProt ID PODTC2 common to 6MOJ and 6LZG); Red sequence: RBM sequence; Large font bold residues: Contacted by the peptide; Blue bar: Contact region covered by the peptide. (A) PACAP‐38 binding to RBD OF 6LZG (Control 1); (B) Amylin binding to RBD OF 6LZG (Control 2); (C) GLP‐2 binding to RBD of 6LZG (Control 3)
From these docking studies, we can conclude that, in silico, LL‐37 shows a binding affinity and relative stability of binding toward the RBD of SARS‐CoV‐2. Now, with a lower structural alignment (with NTH) TM‐score, visually poorer alignment with a bifurcation, and lesser number of aligned residue pairs with d < 5.0 Å; 2 KB8 has shown almost comparable docking as LL‐37. This would be attributed to the amino acid sequence of 2 KB8 which must have amino acids at appropriate positions so that they could dock with RBD. But, even so, the chances of LL‐37 binding to RBD are greater than 2 KB8, because LL‐37, with its better structural attributes, would first more easily enter and fit into the binding region, and then only the amino acid interactions would take place.
3.5. Conformational changes (RMSD) and free energy calculations (MM/GBSA)
The binding stability of protein‐ligand (RBD‐Peptides) complexes in a fully solvated system were studied using RMSD in the C‐α atoms of the RBD after binding with NTH, LL‐37 and control peptides in this study. The RMSD calculations were performed on 1000 trajectories/conformations retrieved during the 100 ns MD simulations and were compared with the initial confirmation (docked pose) of their respective complex.
Figure 5 shows the RMSD plot for SARS‐CoV‐2 RBD upon binding of peptides. The values indicated that the RMSD values for RBD from 6LZG and 6M0J upon binding to LL‐37 deviate <5.00 Å with mean RMSDs of 3.75 ± 0.56 Å and 3.74 ± 0.82 Å, respectively, and the RMSD is <4.00 Å during the first 40 ns. The control peptides viz. 2D2P and 2L63 complexed with RBD6LZG, also LL‐37 complexed with region adjacent to the RBD6LZG shows RMSD values ranging from >5 Å to 8 Å during 100 ns MD simulation with mean RMSDs of 6.10 ± 1.30 Å, 5.64 ± 1.26 Å, and 5.95 ± 1.39 Å, respectively. Whereas the 2KB8_RBD6LZG complex shows larger deviation during 50 ns to 84 ns with mean RMSD of 4.27 ± 1.00 Å. Only in the initial phase (0‐40 ns) and last phase of 100 ns (84–100 ns) simulation time the complex equilibrates within 4 Å and shows complex stability. In the case of the NTH_RBD6LZG complex, the equilibration was seen during initial 63 ns, later this complex deviates up to 6 Å with mean RMSD of 4.12 ± 1.32 Å.
FIGURE 5.
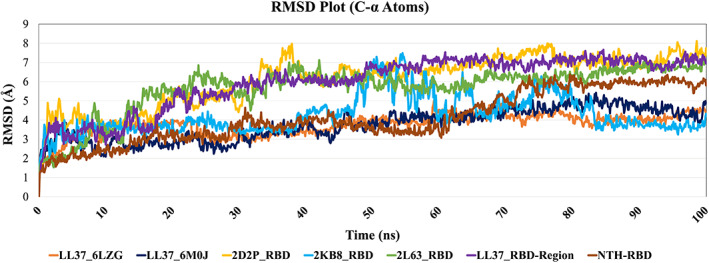
The SARS‐CoV‐2 RBD Root Mean Square Deviation (RMSD) upon binding with NTH and selected peptides in this study
Likewise, the Prime MM/GBSA energies for all the complexes were computed to reveal the strength of the docked complexes during 100 ns MD simulation and the results are provided in Table 4. Remarkably, the peptide LL37 used in this study gives a stronger binding affinity toward the RBD6LZG of SARS‐CoV‐2. The complexes LL37_RBD6LZG and LL37_RBD6M0J exhibited −144.50 kcal/mol and −118.98 kcal/mol binding free energies, respectively. Also, NTH extracted from the ACE2 of 6LZG complexed with RBD6LZG shows −112.26 kcal/mol binding free energy, which is also stronger when compared with the other peptides used as a control in this study. The control peptides binding to RBD, viz., 2D2P_RBD6LZG, 2 KB8_RBD6LZG, and 2 L63_RBD6LZG give −91.80 kcal/mol, −82.14 kcal/mol, and −88.12 kcal/mol binding energies, respectively. LL‐37 complexed with the region adjacent to RBD6LZG had an energy of −93.38 kcal/mol. Collectively, from the MD simulation and MM/GBSA calculations, we can consider that the LL‐37 shows stronger binding affinity toward RBD of SARS‐CoV‐2 as it shows the minimum deviation and favorable binding free energies compared to other peptides (controls) 2D2P, 2KB8 and 2L63 and validates the macromolecular docking protocol.
TABLE 4.
Binding free energies (Prime MM/GBSA) for RBD‐peptide complexes during 100 ns MD simulation
| SARS‐CoV‐2 RBD_Peptide complex | ΔG bind (kcal/mol) |
|---|---|
| LL37_RBD (PDB ID: 6LZG) | −144.50 |
| LL37_RBD (PDB ID: 6M0J) | −118.98 |
| 2D2P_RBD (PDB ID: 6LZG) | −91.80 |
| 2 KB8_RBD (PDB ID: 6LZG) | −82.14 |
| 2 L63_RBD (PDB ID: 6LZG) | −88.12 |
| LL37_Region Adjacent to RBD (PDB ID: 6LZG) | −93.38 |
| NTH_RBD (PDB ID: 6LZG) | −112.26 |
3.6. Sequence variation in geographical isolates of SAR‐CoV‐2
For analyzing the variations in the sequence of SARS‐CoV‐2 isolated from different geographical regions we used the “Latest Global Analysis” tool of the “nextstrain.org” database, 43 wherein we downloaded the analyzed sequences of 3564 genomes of different geographical isolates of SARS‐CoV‐2 reported from December 2019 to 9th November 2020. Subsequently we analyzed the mutations of amino acid residues which are observed to participate in the interaction with LL‐37 in our docking. As shown in Figure 3 (Panel II), the number of mutations of these residues in 3564 SARS‐CoV‐2 genomes is only 1 or 0 and hence these amino acids are well conserved, which is a plus point for LL‐37 as a therapeutic.
3.7. Cell‐penetrating peptide activity of LL‐37
Two prediction servers were used. The SkipCPP‐Pred tool 44 (http://server.malab.cn/SkipCPP-Pred/Index.html) predicted LL‐37 to belong to the cell‐penetrating class with a prediction confidence of 0.806. The MLCPP algorithm 45 (http://www.thegleelab.org/MLCPP/) predicted LL‐37 to be a CPP with a probability score of 0.68. The uptake efficiency was predicted as Low with a probability score of 0.45. Additionally, LL‐37 is found deposited as a CPP in the database CPPsite 2.0. 46 , 47
3.8. Safety analyses of LL‐37
It is recognized that peptides, even including LL‐37, could have adverse effects when used as therapeutics. 48 For possible delivery of LL‐37 through the inhalatory route, LL‐37 was assessed for allergenicity. Taking an example of a human polypeptide which is used by inhalation in clinical practice, namely insulin, it has been reported that allergic reactions can occur although insulin is an innate protein. 49 , 50 The algorithm AllergenFP v.1.0 51 predicted LL‐37 to be a “probable non‐allergen.” Using the server AllerCatPro 52 LL‐37 as a query gave the result “No Evidence” for allergenicity. AllergenFP v.1.0 predicted human Insulin (UniProt sequence ID P01308) to be a “probable allergen” and AllerCatPro gave the result “Strong Evidence” for insulin as an allergen. The software program ToxinPred 53 predicted LL‐37 to be a Non‐Toxin by all Support Vector Machine methods available on the server. For prediction of hemolytic activity, the tool HAPPENN, which employs neural networks method was used. 54 LL‐37 was predicted to have very low hemolytic scores of 0.073, 0.089, and 0.09 by the three methods available in the tool, degree of hemolytic activity increasing from 0 to 1.
4. DISCUSSION
In summary, based on our in silico findings, we propose that LL‐37 could be used as a therapeutic for Covid‐19. Besides the known action of LL‐37 against enveloped viruses, we provide in silico data for the binding of LL‐37 to the RBD of SARS‐CoV‐2 itself. The very good structural alignment of LL‐37 with the NTH suggests that LL‐37 could stop RBD from binding ACE2 even by simply occupying the space intended by the virus for NTH. Also, from analysis of the docking studies (Figure 3, Panel II), it can be seen that LL‐37 spans a considerable stretch of RBD, from Glu407 to Tyr506 in both the Test dockings which includes the entire Receptor Binding Motif. This again is indicative of the potential for effective blocking of the RBD. Although the docking energy of LL‐37 is not comparable to that of ACE2, if present at sufficient concentration in the body, due to its structural similarity to NTH, LL‐37 could also simply create a physical hindrance for the virus binding to ACE2. It may be noted that the interactions of the peptide PACAP‐38 with RBD span a much shorter region of RBD than LL‐37. Also, although the interactions of GLP‐2 span a long region of RBD, the RBM is almost completely missed out by this peptide. Amylin spans the RBD well, but as explained above, NTH is more likely to bind to RBD than Amylin. Besides, PACAP‐38, Amylin, and GLP‐2, based on their normal function in the body, are unsuitable to use as therapeutic peptides, unlike LL‐37. Now, it is of noteworthy importance that, following our previous report of in silico binding of LL‐37 to RBD, 55 it has been demonstrated in vitro that LL‐37 does bind to RBD. 56 This corroborates our prediction as sound, and also highlights the value of in silico studies. In the pages of scientific literature, ours was the very first study describing the binding of LL‐37 to the RBD of the virus causing an epic pandemic, and the value this could have. From the RMSD plots (Figure 5), a clear difference can be seen between the profiles of LL‐37 and those of the negative control peptides, which strengthens the validity of the docking scores. It should be noted that LL‐37 has been put through a very stringent test, because peptides as similar to LL‐37 as possible have been selected as the controls, in spite of which there is a difference between LL‐37 and the controls. The plot of LL‐37 is close to that of NTH. With respect to binding free energies, the percentage difference between LL‐37:RBD binding and Other peptide:RBD binding ranges from 25.78% to 55.03%, which are most conclusive figures. The binding energy of LL‐37 is close to that of NTH. The possible binding of LL‐37 to RBD and consequent inhibition of viral binding to cells echoes other such examples mentioned in the Introduction.
This finding of LL‐37 binding to RBD also provides a boost for the use of Vitamin D at an effective, medically prescribed dose for protection against SARS‐CoV‐2, especially as LL‐37 is expressed in the respiratory epithelial cells also, and Vitamin D elevates LL‐37 levels. The European Food and Safety Authority advise 4000 IU/day and the Endocrine Society, 10 000 IU/day of Vitamin D, as an upper limit for safety. 57 A randomized controlled trial involving 373 healthy adults having normal Vitamin D levels, reported similar safety data for Vitamin D doses up to 400, 4000, and 10 000 IU/day. 58 In the future, it would be helpful to design a water‐soluble derivative of Vitamin D to avoid the adverse effect of hypervitaminosis.
Even when the RBD is in the UP state, it might be difficult for the immune surveillance mechanism of neutralizing antibodies to reach the RBD, and hence the efficient immune evasion by the virus. Also, it is reasonable to conjecture that a small peptide like LL‐37 would more easily access the RBD in its UP state, than large molecules like neutralizing antibodies. The prediction that LL‐37 is a cell‐penetrating peptide raises the interesting possibility of LL‐37 intracellularly binding to the Spike protein of SARS‐CoV‐2 as the protein is produced in the infected cells and then, following viral particle assembly, release of virions from the cells which already have the RBD blocked by LL‐37 and hence cannot infect new cells.
Regarding the safety of using LL‐37 as a therapeutic, it has been cautioned that there could be adverse effects including cell membrane‐destabilization above critical concentrations of LL‐37. 48 A clinical trial testing the safety of LL‐37 in the treatment of non‐healing leg ulcers demonstrated that topical LL‐37 application was safe. 59 Now, LL‐37 has been approved for Phase II clinical trials (https://clinicaltrials.gov; ClinicalTrials.gov Identifier: NCT04098562) for its anti‐microbial action in the management of diabetic foot ulcers. 48 There is also approval for a clinical study of intratumoral injection of LL‐37 for the condition of melanoma (ClinicalTrials.gov Identifier: NCT02225366). At high concentrations in vitro, LL‐37 has been found cytotoxic to different eukaryotic cells, 60 but when human serum is present, the cytotoxic activity of LL‐37 is inhibited, and this could be the reason why LL‐37 is not cytotoxic in the body. Indeed, the cytotoxicity of LL‐37 secreted into the blood is negated by the binding of LL‐37 to plasma proteins such as apolipoprotein A‐I. 61 It is well elucidated how the cytotoxic effects of LL‐37 are counteracted by its binding to apolipoprotein A‐I. 62 The KD of the binding is such that apolipoprotein A‐I at its physiological concentration in plasma, can render inactive (including cytotoxic activity) 90% of the LL‐37 molecules present at concentrations which would be toxic to the body's cells. This still leaves sufficient free LL‐37 molecules for antimicrobial action. Data is also indicative that eukaryotic cells are more resistant (than prokaryotic cells) to the cytotoxicity of LL‐37 due to the expression of heparan sulfate by eukaryotic cells and the structure and composition of their membranes. 11 A very recent report describes a cooperation between LL‐37 and Human Neutrophil Peptide‐1 that shields mammalian plasma membranes from lysis. 63 Now, it has been reported that excision of the N‐terminal hydrophobic residues from LL‐37 reduces its cytotoxicity. 61 From Figure 2(Panel I,B), it can be seen that the first three N‐terminal amino acids (LLG) of LL‐37 do not align with the NTH residues. Also, from the docking data, these three amino acids do not form interactions with RBD. This means that, for the development of LL‐37 as a therapeutic, the option of removing two‐three amino acids from the N‐terminus is available.
The predicted non‐allergenicity of LL‐37 can mean that LL‐37 could be administered to patients through the respiratory tract route, which would have more immediate efficacy in treating the disease. Also, this would greatly improve the deliverability to large populations in a shorter time, due to ease of administration compared to an injectable. If administered intravenously, being a peptide, LL‐37 could migrate from the circulation to the infected regions in the lungs. Peptide therapeutics enter the tissues from the vasculature by diffusion or convective extravasation, and transfer from the circulation to the tissues also depends on the properties of specific peptides. 64 In relation to the lungs, as respiratory epithelial cells express hCap‐18, LL‐37 has been found to be present in the bronchial alveolar lavage fluid, 65 and increased levels have been detected in tracheal aspirates during infection. In the lungs, the normal role of LL‐37 is to provide innate immunity against bacterial infections. In the case of Covid‐19, SARS‐CoV‐2 infects the respiratory epithelial cells, 66 the hCap‐18 expression system of which could hence be adversely affected, which anyway is upregulated in response to bacterial infection. Thus, as we have shown that there could be a binding connection between LL‐37 and RBD, it could be advocated that external administration of LL‐37 directly into the lungs would be beneficial in the treatment of Covid‐19. Besides, the epithelial cells yet unaffected by the virus, can be stimulated to produce LL‐37 by the use of Vitamin D. Caution would have to be exercised if LL‐37 is to be used as a therapeutic, because it is reported to contribute to inflammatory processes in the lungs. At the same time, LL‐37 is also involved in wound repair involving skin epithelial cells. 65 This role of LL‐37 may have (positive) implications for Covid‐19. It has been reviewed that Vitamin D could have a protective effect against Covid‐19 severity through its upregulation of cathelicidin, which in turn results in decrease in the levels of pro‐inflammatory cytokines and increase in anti‐inflammatory cytokines. 67 Consequently, the cytokine storm associated with Covid‐19 could be controlled. The development of LL‐37 as a therapeutic for Covid‐19 will have to be in the context of the various roles and effects of the molecule. But the fact that LL‐37 is a molecule already present in the human circulation, and LL‐37 as a therapeutic has received approval for clinical trials could help in paving the way for its use in the case of Covid‐19. LL‐37 as a therapeutic, which would need only peptide synthesis, would also be relatively cost‐effective.
Further experimental studies, in addition to the in vitro work mentioned above, 55 would be required to evaluate in detail the binding of LL‐37 with the RBD. If such studies confirm the finding of our in silico studies, then LL‐37 has a good potential to be developed as a therapeutic. As it is a molecule native to the human body, its development up the regulatory pathway could be of shorter duration. To minimize the morbidity until the developed/upcoming vaccines become available to all people in the world, Vitamin D administration in a safe range to the entire population could be advocated by authorities. Our findings add significant weight to a similar recommendation made, 68 based on the association of Vitamin D deficiency with heightened risk of getting Covid‐19 infection. It could be the simplest prophylaxis to implement, and worldwide Vitamin D supplementation could be advocated, as only possible benefit could come out of it, when consumed within a safe limit as advised by authorities.
CONFLICT OF INTEREST
The authors declare no potential conflict of interest.
AUTHOR CONTRIBUTIONS
Manisha Deshpande conceived, designed and led the project and carried out the Structural Alignment study, the Safety studies, CPP prediction, and data analyses. She has also written most of the Introduction and Discussion of the manuscript. Kiran Lokhande performed all the Molecular Docking, MD simulation, and MM/GBSA free energy calculations and analyses, analyzed the Structural Alignment results and has written the details about the docking, MD simulation and MM/GBSA in the manuscript. Tanushree Banerjee gave the advice to carry out structural studies, performed the mutation study, and contributed to the writing. Kakumani Venkateswara Swamy mentored Kiran Lokhande, did analysis of the data and critical reading of the manuscript and gave valuable suggestions. Payel Ghosh provided the facility to perform MM/GBSA calculations using Prime (Schrodinger) software, and she has also contributed to performing the MM/GBSA calculations, and critically revised the manuscript. All authors have critically read and approved the final manuscript.
ACKNOWLEDGMENTS
The authors are grateful for the valued support of Dr. D. Y. Patil Vidyapeeth (DPU), Pune, India, and of Prof. Jayanta K. Pal, Director, Dr. D. Y. Patil Biotechnology and Bioinformatics Institute (DYPBBI, a constituent of DPU), Pune, for carrying out this work. They are also thankful for the valued support of Director and Head of MIT School of Bioengineering Sciences & Research, Pune. Senior Research Fellowship awarded to Mr. Kiran Bharat Lokhande (Project ID: 2019‐3458; File No.: ISRM/11(54)/2019) by the Indian Council of Medical Research, New Delhi is also acknowledged. TB would like to acknowledge the intramural research grant (Project No. DPU/559(1)/2020 dated 25th Sep 2020) from Dr. D. Y. Patil Vidyapeeth, Pune, “Developing therapeutics and diagnostics for Covid‐19.” The authors acknowledge the support of the Bioinformatics Centre, Savitribai Phule Pune University, Pune, for providing the Schrodinger software for free energy calculations.
Lokhande KB, Banerjee T, Swamy KV, Ghosh P, Deshpande M. An in silico scientific basis for LL‐37 as a therapeutic for Covid‐19. Proteins. 2022;90(5):1029‐1043. 10.1002/prot.26198
DATA AVAILABILITY STATEMENT
Data are available on request from the authors.
REFERENCES
- 1. Vabret N, Britton GJ, Gruber C, et al. Immunology of COVID‐19: current state of the science. Immunity. 2020;52(6):910‐941. doi: 10.1016/j.immuni.2020.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Calina D, Docea AO, Petrakis D, et al. Towards effective COVID‐19 vaccines: updates, perspectives and challenges (review). Int J Mol Med. 2020;46(1):3‐16. doi: 10.3892/ijmm.2020.4596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Abduljalil JM, Abduljalil BM. Epidemiology, genome, and clinical features of the pandemic SARS‐CoV‐2: a recent view. New Microbes New Infect. 2020;35:100672. doi: 10.1016/j.nmni.2020.100672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lan J, Ge J, Yu J, et al. Structure of the SARS‐CoV‐2 spike receptor‐binding domain bound to the ACE2 receptor. Nature. 2020;581(7807):215‐220. doi: 10.1038/s41586-020-2180-5 [DOI] [PubMed] [Google Scholar]
- 5. Chowdhury R, Boorla VS, Maranas CD. Computational biophysical characterization of the SARS‐CoV‐2 spike protein binding with the ACE2 receptor and implications for infectivity. Comput Struct Biotechnol J. 2020;18:2573‐2582. doi: 10.1016/j.csbj.2020.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Agier J, Efenberger M, Brzezińska‐Błaszczyk E. Cathelicidin impact on inflammatory cells. Cent Eur J Immunol. 2015;40(2):225‐235. doi: 10.5114/ceji.2015.51359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mansour SC, Pena OM, Hancock RE. Host defense peptides: front‐line immunomodulators. Trends Immunol. 2014;35(9):443‐450. doi: 10.1016/j.it.2014.07.004 [DOI] [PubMed] [Google Scholar]
- 8. Sørensen OE, Follin P, Johnsen AH, et al. Human cathelicidin, hCAP‐18, is processed to the antimicrobial peptide LL‐37 by extracellular cleavage with proteinase 3. Blood. 2001;97(12):3951‐3959. doi: 10.1182/blood.v97.12.3951 [DOI] [PubMed] [Google Scholar]
- 9. Tang X, Basavarajappa D, Haeggström JZ, Wan M. P2X7 receptor regulates internalization of antimicrobial peptide LL‐37 by human macrophages that promotes intracellular pathogen clearance. J Immunol. 2015;195(3):1191‐1201. doi: 10.4049/jimmunol.1402845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dahl S, Anders E, Gidlöf O, Svensson D, Nilsson BO. The host defense peptide LL‐37 triggers release of nucleic acids from human mast cells. Peptides. 2018;109:39‐45. doi: 10.1016/j.peptides.2018.10.001 [DOI] [PubMed] [Google Scholar]
- 11. Zhang X, Oglęcka K, Sandgren S, et al. Dual functions of the human antimicrobial peptide LL‐37‐target membrane perturbation and host cell cargo delivery. Biochim Biophys Acta. 2010;1798(12):2201‐2208. doi: 10.1016/j.bbamem.2009.12.011 [DOI] [PubMed] [Google Scholar]
- 12. Tripathi S, Wang G, White M, Qi L, Taubenberger J, Hartshorn KL. Antiviral activity of the human cathelicidin, LL‐37, and derived peptides on seasonal and pandemic influenza a viruses. PLoS One. 2015;10(4):e0124706. doi: 10.1371/journal.pone.0124706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ahmed A, Siman‐Tov G, Hall G, Bhalla N, Narayanan A. Human antimicrobial peptides as therapeutics for viral infections. Viruses. 2019;11(8):704. doi: 10.3390/v11080704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Crane‐Godreau MA, Clem KJ, Payne P, Fiering S. Vitamin D deficiency and air pollution exacerbate COVID‐19 through suppression of antiviral peptide LL37. Front Public Health. 2020;8:232. doi: 10.3389/fpubh.2020.00232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Alagarasu K, Patil PS, Shil P, et al. In‐vitro effect of human cathelicidin antimicrobial peptide LL‐37 on dengue virus type 2. Peptides. 2017;92:23‐30. doi: 10.1016/j.peptides.2017.04.002 [DOI] [PubMed] [Google Scholar]
- 16. Currie SM, Gwyer Findlay E, McFarlane AJ, et al. Cathelicidins have direct antiviral activity against respiratory syncytial virus in vitro and protective function in vivo in mice and humans. J Immunol. 2016;196(6):2699‐2710. doi: 10.4049/jimmunol.1502478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yu J, Dai Y, Fu Y, et al. Cathelicidin antimicrobial peptides suppress EV71 infection via regulating antiviral response and inhibiting viral binding. Antiviral Res. 2021;187:105021. doi: 10.1016/j.antiviral.2021.105021 [DOI] [PubMed] [Google Scholar]
- 18. Gurlek A, Pittelkow MR, Kumar R. Modulation of growth factor/cytokine synthesis and signaling by 1alpha,25‐dihydroxyvitamin D(3): implications in cell growth and differentiation. Endocr Rev. 2002;23(6):763‐786. doi: 10.1210/er.2001-0044 [DOI] [PubMed] [Google Scholar]
- 19. Svensson D, Nebel D, Nilsson BO. Vitamin D3 modulates the innate immune response through regulation of the hCAP‐18/LL‐37 gene expression and cytokine production. Inflamm Res. 2016;65(1):25‐32. doi: 10.1007/s00011-015-0884-z [DOI] [PubMed] [Google Scholar]
- 20. Merzon E, Tworowski D, Gorohovski A, et al. Low plasma 25(OH) vitamin D level is associated with increased risk of COVID‐19 infection: an Israeli population‐based study. FEBS J. 2020;287(17):3693‐3702. doi: 10.1111/febs.15495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Meltzer DO, Best TJ, Zhang H, Vokes T, Arora V, Solway J. Association of vitamin D status and other clinical characteristics with COVID‐19 test results. JAMA Netw Open. 2020;3(9):e2019722. doi: 10.1001/jamanetworkopen.2020.19722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Greiller CL, Martineau AR. Modulation of the immune response to respiratory viruses by vitamin D. Nutrients. 2015;7(6):4240‐4270. doi: 10.3390/nu7064240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhao L, Cao Z, Bian Y, Hu G, Wang J, Zhou Y. Molecular dynamics simulations of human antimicrobial peptide LL‐37 in model POPC and POPG lipid bilayers. Int J Mol Sci. 2018;19(4):1186. doi: 10.3390/ijms19041186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shajahan A, Supekar NT, Gleinich AS, Azadi P. Deducing the N‐ and O‐glycosylation profile of the spike protein of novel coronavirus SARS‐CoV‐2. Glycobiology. 2020;30(12):981‐988. doi: 10.1093/glycob/cwaa042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cai Y, Zhang J, Xiao T, et al. Distinct conformational states of SARS‐CoV‐2 spike protein. Science. 2020;369(6511):1586‐1592. doi: 10.1126/science.abd4251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mercurio I, Tragni V, Busto F, De Grassi A, Pierri CL. Protein structure analysis of the interactions between SARS‐CoV‐2 spike protein and the human ACE2 receptor: from conformational changes to novel neutralizing antibodies. Cell Mol Life Sci. 2020;78(4):1‐22. doi: 10.1007/s00018-020-03580-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang G. Structures of human host defense cathelicidin LL‐37 and its smallest antimicrobial peptide KR‐12 in lipid micelles. J Biol Chem. 2008;283(47):32637‐32643. doi: 10.1074/jbc.M805533200 [DOI] [PubMed] [Google Scholar]
- 28. Wang Q, Zhang Y, Wu L, et al. Structural and functional basis of SARS‐CoV‐2 entry by using human ACE2. Cell. 2020;181(4):894‐904.e9. doi: 10.1016/j.cell.2020.03.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tateishi Y, Jee JG, Inooka H, Tochio H, Hiroaki H, Shirakawa M. The solution structure of micelle‐bound peptide. 2006. doi: 10.2210/pdb2D2P/pdb [DOI]
- 30. Patil SM, Xu S, Sheftic SR, Alexandrescu AT. Dynamic alpha‐helix structure of micelle‐bound human amylin. J Biol Chem. 2009;284(18):11982‐11991. doi: 10.1074/jbc.M809085200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Venneti KC, Hewage CM. Conformational and molecular interaction studies of glucagon‐like peptide‐2 with its N‐terminal extracellular receptor domain. FEBS Lett. 2011;585(2):346‐352. doi: 10.1016/j.febslet.2010.12.011 [DOI] [PubMed] [Google Scholar]
- 32. Sastry GM, Adzhigirey M, Day T, Annabhimoju R, Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des. 2013;27(3):221‐234. doi: 10.1007/s10822-013-9644-8 [DOI] [PubMed] [Google Scholar]
- 33. Zhang Y, Skolnick J. TM‐align: a protein structure alignment algorithm based on the TM‐score. Nucleic Acids Res. 2005;33(7):2302‐2309. doi: 10.1093/nar/gki524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xu J, Zhang Y. How significant is a protein structure similarity with TM‐score = 0.5? Bioinformatics. 2010;26(7):889‐895. doi: 10.1093/bioinformatics/btq066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Csaba G, Birzele F, Zimmer R. Systematic comparison of SCOP and CATH: a new gold standard for protein structure analysis. BMC Struct Biol. 2009;9:23. doi: 10.1186/1472-6807-9-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yan Y, Tao H, He J, Huang SY. The HDOCK server for integrated protein‐protein docking. Nat Protoc. 2020;15(5):1829‐1852. doi: 10.1038/s41596-020-0312-x [DOI] [PubMed] [Google Scholar]
- 37. Huo J, Zhao Y, Ren J, et al. Neutralization of SARS‐CoV‐2 by destruction of the Prefusion spike [published correction appears in Cell Host Microbe. 2020 Sep 9;28(3):497]. Cell Host Microbe. 2020;28(3):445‐454.e6. doi: 10.1016/j.chom.2020.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bowers KJ, Chow E, Xu H, et al. Scalable algorithms for molecular dynamics simulations on commodity clusters. Proceedings of the ACM/IEEE Conference on Supercomputing (SC06), Tampa, Florida; New York City, IEEE; 2006:11‐17. [Google Scholar]
- 39. Kollar J, Frecer V. How accurate is the description of ligand‐protein interactions by a hybrid QM/MM approach? J Mol Model. 2017;24(1):11. doi: 10.1007/s00894-017-3537-z [DOI] [PubMed] [Google Scholar]
- 40. Jacobson MP, Pincus DL, Rapp CS, et al. A hierarchical approach to all‐atom protein loop prediction. Proteins. 2004;55(2):351‐367. doi: 10.1002/prot.10613 [DOI] [PubMed] [Google Scholar]
- 41. Shang J, Ye G, Shi K, et al. Structural basis of receptor recognition by SARS‐CoV‐2. Nature. 2020;581(7807):221‐224. doi: 10.1038/s41586-020-2179-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yan R, Zhang Y, Li Y, Xia L, Guo Y, Zhou Q. Structural basis for the recognition of SARS‐CoV‐2 by full‐length human ACE2. Science. 2020;367(6485):1444‐1448. doi: 10.1126/science.abb2762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hadfield J, Megill C, Bell SM, et al. Nextstrain: real‐time tracking of pathogen evolution. Bioinformatics. 2018;34(23):4121‐4123. doi: 10.1093/bioinformatics/bty407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wei L, Tang J, Zou Q. SkipCPP‐Pred: an improved and promising sequence‐based predictor for predicting cell‐penetrating peptides. BMC Genomics. 2017;18(Suppl 7):742. doi: 10.1186/s12864-017-4128-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Manavalan B, Subramaniyam S, Shin TH, Kim MO, Lee G. Machine‐learning‐based prediction of cell‐penetrating peptides and their uptake efficiency with improved accuracy. J Proteome Res. 2018;17(8):2715‐2726. doi: 10.1021/acs.jproteome.8b00148 [DOI] [PubMed] [Google Scholar]
- 46. Agrawal P, Bhalla S, Usmani SS, et al. CPPsite 2.0: a repository of experimentally validated cell‐penetrating peptides. Nucleic Acids Res. 2016;44(D1):D1098‐D1103. doi: 10.1093/nar/gkv1266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gautam A, Singh H, Tyagi A, et al. CPPsite: a curated database of cell penetrating peptides. Database (Oxford). 2012;2012:bas015. doi: 10.1093/database/bas015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chen CH, Lu TK. Development and challenges of antimicrobial peptides for therapeutic applications. Antibiotics (Basel). 2020;9(1):24. doi: 10.3390/antibiotics9010024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gatto NM, Bracken MB, Kolitsopoulos F, et al. Pulmonary and cardiovascular safety of inhaled insulin in routine practice: the Exubera large simple trial (VOLUME). Contemp Clin Trials Commun. 2019;18:100427. doi: 10.1016/j.conctc.2019.100427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mastrandrea LD. Inhaled insulin: overview of a novel route of insulin administration. Vasc Health Risk Manag. 2010;6:47‐58. doi: 10.2147/vhrm.s6098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Dimitrov I, Naneva L, Doytchinova I, Bangov I. AllergenFP: allergenicity prediction by descriptor fingerprints. Bioinformatics. 2014;30(6):846‐851. doi: 10.1093/bioinformatics/btt619 [DOI] [PubMed] [Google Scholar]
- 52. Maurer‐Stroh S, Krutz NL, Kern PS, et al. AllerCatPro‐prediction of protein allergenicity potential from the protein sequence. Bioinformatics. 2019;35(17):3020‐3027. doi: 10.1093/bioinformatics/btz029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gupta S, Kapoor P, Chaudhary K, et al. In silico approach for predicting toxicity of peptides and proteins. PLoS One. 2013;8(9):e73957. doi: 10.1371/journal.pone.0073957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Timmons PB, Hewage CM. HAPPENN is a novel tool for hemolytic activity prediction for therapeutic peptides which employs neural networks. Sci Rep. 2020;10(1):10869. doi: 10.1038/s41598-020-67701-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lokhande KB, Banerjee T, Swamy KV, Deshpande M. An in silico scientific basis for LL‐37 as a therapeutic and vitamin D as preventive for Covid‐19. (Version 1). ChemRxiv. 2020. doi: 10.26434/chemrxiv.12928202.v1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Roth A, Lütke S, Meinberger D, et al. LL‐37 fights SARS‐CoV‐2: the vitamin D‐inducible peptide LL‐37 inhibits binding of SARS‐CoV‐2 spike protein to its cellular receptor angiotensin converting enzyme 2 in vitro. bioRxiv. 2020. doi: 10.1101/2020.12.02.408153 [DOI] [Google Scholar]
- 57. Amrein K, Scherkl M, Hoffmann M, et al. Vitamin D deficiency 2.0: an update on the current status worldwide. Eur J Clin Nutr. 2020;74(11):1498‐1513. doi: 10.1038/s41430-020-0558-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Billington EO, Burt LA, Rose MS, et al. Safety of high‐dose vitamin D supplementation: secondary analysis of a randomized controlled trial. J Clin Endocrinol Metab. 2020;105(4):dgz212. doi: 10.1210/clinem/dgz212 [DOI] [PubMed] [Google Scholar]
- 59. Grönberg A, Mahlapuu M, Ståhle M, Whately‐Smith C, Rollman O. Treatment with LL‐37 is safe and effective in enhancing healing of hard‐to‐heal venous leg ulcers: a randomized, placebo‐controlled clinical trial. Wound Repair Regen. 2014;22(5):613‐621. doi: 10.1111/wrr.12211 [DOI] [PubMed] [Google Scholar]
- 60. Johansson J, Gudmundsson GH, Rottenberg ME, Berndt KD, Agerberth B. Conformation‐dependent antibacterial activity of the naturally occurring human peptide LL‐37. J Biol Chem. 1998;273(6):3718‐3724. doi: 10.1074/jbc.273.6.3718 [DOI] [PubMed] [Google Scholar]
- 61. Ciornei CD, Sigurdardóttir T, Schmidtchen A, Bodelsson M. Antimicrobial and chemoattractant activity, lipopolysaccharide neutralization, cytotoxicity, and inhibition by serum of analogs of human cathelicidin LL‐37. Antimicrob Agents Chemother. 2005;49(7):2845‐2850. doi: 10.1128/AAC.49.7.2845-2850.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Xhindoli D, Pacor S, Benincasa M, Scocchi M, Gennaro R, Tossi A. The human cathelicidin LL‐37—a pore‐forming antibacterial peptide and host‐cell modulator. Biochim Biophys Acta. 2016;1858(3):546‐566. doi: 10.1016/j.bbamem.2015.11.003 [DOI] [PubMed] [Google Scholar]
- 63. Drab E, Sugihara K. Cooperative function of LL‐37 and HNP1 protects mammalian cell membranes from lysis. Biophys J. 2020;119(12):2440‐2450. doi: 10.1016/j.bpj.2020.10.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Diao L, Meibohm B. Pharmacokinetics and pharmacokinetic‐pharmacodynamic correlations of therapeutic peptides. Clin Pharmacokinet. 2013;52(10):855‐868. doi: 10.1007/s40262-013-0079-0 [DOI] [PubMed] [Google Scholar]
- 65. Tjabringa GS, Rabe KF, Hiemstra PS. The human cathelicidin LL‐37: a multifunctional peptide involved in infection and inflammation in the lung. Pulm Pharmacol Ther. 2005;18(5):321‐327. doi: 10.1016/j.pupt.2005.01.001 [DOI] [PubMed] [Google Scholar]
- 66. Mason RJ. Pathogenesis of COVID‐19 from a cell biology perspective. Eur Respir J. 2020;55(4):2000607. doi: 10.1183/13993003.00607-2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Grant WB, Lahore H, McDonnell SL, et al. Evidence that vitamin D supplementation could reduce risk of influenza and COVID‐19 infections and deaths. Nutrients. 2020;12(4):988. doi: 10.3390/nu12040988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Griffin G, Hewison M, Hopkin J, et al. Preventing vitamin D deficiency during the COVID‐19 pandemic: UK definitions of vitamin D sufficiency and recommended supplement dose are set too low. Clin Med (Lond). 2020;21(1):e48‐e51. doi: 10.7861/clinmed.2020-0858 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data are available on request from the authors.