

Abstract
The novel coronavirus disease 2019 (COVID‐19) caused by severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) still has serious negative effects on health, social life, and economics. Recently, vaccines from various companies have been urgently approved to control SARS‐CoV‐2 infections. However, any specific antiviral drug has not been confirmed so far for regular treatment. An important target is the main protease (Mpro), which plays a major role in replication of the virus. In this study, Gaussian and residue network models are employed to reveal two distinct potential allosteric sites on Mpro that can be evaluated as drug targets besides the active site. Then, Food and Drug Administration (FDA)‐approved drugs are docked to three distinct sites with flexible docking using AutoDock Vina to identify potential drug candidates. Fourteen best molecule hits for the active site of Mpro are determined. Six of these also exhibit high docking scores for the potential allosteric regions. Full‐atom molecular dynamics simulations with MM‐GBSA method indicate that compounds docked to active and potential allosteric sites form stable interactions with high binding free energy (∆G bind ) values. ∆G bind values reach −52.06 kcal/mol for the active site, −51.08 kcal/mol for the potential allosteric site 1, and − 42.93 kcal/mol for the potential allosteric site 2. Energy decomposition calculations per residue elucidate key binding residues stabilizing the ligands that can further serve to design pharmacophores. This systematic and efficient computational analysis successfully determines ivermectine, diosmin, and selinexor currently subjected to clinical trials, and further proposes bromocriptine, elbasvir as Mpro inhibitor candidates to be evaluated against SARS‐CoV‐2 infections.
Keywords: allostery, drug repurposing, main protease, SARS‐CoV‐2
1. INTRODUCTION
Coronaviruses (CoV) belong to the family of Coronaviridae, single‐stranded RNA viruses that spread widely among humans and other mammals, causing serious upper respiratory tract diseases. 1 Over the past 17 years, two novel CoVs, namely severe acute respiratory syndrome CoV (SARS‐ CoV) 2 and Middle East respiratory syndrome CoV (MERS‐CoV) 3 have emerged and infected humans, resulting in high fatality rates and large economic losses. The new CoVs, designated as SARS‐CoV‐2, is first identified in Wuhan, China, during December 2019 4 and spread widely around the world, and so declared as a pandemic on March 2020 by the World Health Organization (WHO). 5
The viral life cycle involves entry, replication of genetic material, protein translation, assembly, and release from the host cell. Strategies for drug development target viral proteins and host receptors to interfere with different stages of the CoV life cycle. 6 Spike (S) protein plays a vital role in viral entry into the host cell, hence it is an attractive target for blocking SARS‐CoV‐2 infection. 7 Several theoretical studies have been performed to identify inhibitors against SARS‐CoV‐2 spike protein. 8 , 9 , 10 RNA‐dependent RNA polymerase (RdRp) is another important target for SARS‐CoV‐2 due to its crucial role in replicating the positive sense viral RNA. 6 Promising drug candidates against SARS‐CoV‐2 RdRp have been reported in several in silico drug repurposing studies. 11 , 12 , 13 , 14 One of the best‐characterized drug targets among CoV is the Mpro (also called 3CLpro) that is functional as a homodimer, where each monomer contains the catalytic dyad defined by H41 and C145 residues. 15 This enzyme is essential for processing the polyproteins that are translated from the viral RNA 16 and mediating the maturation of the non‐structural proteins, which is the main step in the replication of the virus. As a vital enzyme of SARS‐CoV‐2, inhibiting the activity of Mpro would block viral replication and transcription. 17 In addition to kinetic studies indicating that the active form of the Mpro corresponds to a homodimer, 18 significant conformational differences between the monomer and dimeric states have been reported in recent studies. 19 , 20 To analyze structural and dynamic properties of Mpro, all‐atom MD simulations of various Mpro mutants have been performed by Amamuddy et al 21 emphasizing that mutations located near the active site control the bending motions needed for catalysis, so they may influence enzymatic activity. All these studies suggest that drug discovery combining docking and MD simulations should be performed using the homodimeric conformation instead of the monomer. Previous molecular docking and MD simulation studies targeting SARS‐CoV‐2 Mpro also reported plausible molecules to test experimentally. 22 , 23 , 24 , 25 , 26
To tackle this pandemic, overall efforts have been made to develop effective and safe therapies (including vaccines) for COVID‐19. In the earlier studies, crystal structure of Mpro in complex with peptide‐like irreversible inhibitors (Michael acceptor N3 and its carbonyl derivative α‐ketoamide) are used to design N3‐analogues as potential inhibitors against Mpro. 27 , 28 Covalent inhibitors provide many pharmacological advantages such as enhanced biochemical and cellular potency, selectivity, and prolonged duration of action. 29 , 30 However, their potential for nonspecific reactivity, toxicity, and undesired side‐effects, rapid in vivo metabolism and reduced oral bioavailability make the irreversible inhibitors less efficient as therapeutic agents. 31 , 32 , 33 , 34 On the other hand, drug repositioning or repurposing is a faster and less costly solution to propose potential effective drugs useful to control emerged infectious outbreaks immediately, as new drug development takes more than 10 years. 35 , 36 Currently, there are no clinically approved therapeutics available. At this point, already known and FDA‐approved potential candidate drugs can be screened and re‐evaluated based on antiviral effects for alternative treatments. 37 In this regard, enzyme kinetics and cellular antiviral assays reveal that boceprevir, 38 GC‐376, 39 calpain inhibitors II, XII, 40 and bepridil 41 inhibit SARS‐CoV‐2 Mpro in the micromolar and nanomolar range. Several drugs employed for various diseases are also being tested in numerous clinical trials, including remdesivir, 42 favipiravir, 43 chloroquine, 44 dexamethasone, 45 nafamostat, 46 and ivermectin. 47 Furthermore, screening of approved and clinical drugs 48 and covalent and non‐covalent fragments 49 have experimentally confirmed two allosteric binding sites on Mpro, suggesting a novel multi‐cyclin‐dependent kinase inhibitor with moderate antiviral activity.
In the light of the reports stated above, it is evident that a search for effective drugs, having the potential to inhibit SARS‐CoV‐2, has become a global pursuit. In this study, we investigate the repurposing of the existing FDA‐approved drugs to target COVID‐19 virus Mpro using a systematic computational approach combining different methods, namely elastic network theory, molecular docking, and MD simulations. We apply residue network and Gaussian network models (GNM) to identify potential allosteric sites on Mpro, which may be evaluated to regulate the enzymatic activity of this protease. Proteins are all considered to use allostery to accomplish their function, whether or not they undergo a large conformational change. 50 Allosteric sites provide alternative drug binding regions on the same proteins, which improves the likelihood of effective drugs with greater specificity and regulates the activity of proteins by remotely affecting their active sites. 51 , 52 Molecular docking is one of the most common computational approaches to determine potential drugs regarding their binding pose as well as scoring their binding affinity. 53 We then employ molecular docking of FDA‐approved drugs against active site of Mpro and its potential allosteric sites. We use AutoDock Vina for molecular docking studies of the constructed library comprising over 2400 molecules. We rank compounds based on their binding scores and consider promising hit compounds from docking experiments with good vina scores in 50‐ns MD simulations using AMBER16 54 to validate the stability of docked binding modes. MD simulations are performed for ligand‐Mpro complexes, where whether both catalytic sites or dimer interfaces (ie, potential allosteric sites) accommodate hit compounds. MD simulation trajectories are then analyzed with the Molecular Mechanics Generalized Born Surface Area (MM/GBSA) method 55 to evaluate binding characteristics of dimeric Mpro to 14 promising hit compounds at the active site and eight promising drug candidates at the potential allosteric sites.
This study reveals unknown potential allosteric sites of Mpro, suggests FDA‐approved drugs to target Mpro from its critical sites, and describes a systematic methodology that can be also used for drug repurposing for other diseases that urgently wait efficient therapeutics.
2. METHODOLOGY
In the present study, 2447 FDA‐approved drugs are screened with a molecular docking approach against the active site and potential allosteric sites of Mpro. We employ GNM and residue network model to identify potential allosteric sites of Mpro that can serve as drug‐binding regions, besides the active sites. Based on the vina docking score rankings and the active site interaction patterns, MD simulations of the most promising hit compounds bound to active site and potential allosteric sites of Mpro are performed. In order to understand the behavior of ligand molecules and their interactions with the active site and potential allosteric sites of the enzyme, 50 ns MD simulations are carried. The binding free energy analysis of Mpro complexed with the most promising hit compounds is performed using the MM/GBSA method. 55 All findings are evaluated together to propose FDA‐approved drugs to target the Mpro. The schematic representation of the approach followed for repurposing FDA‐approved drugs is provided in Figure 1.
FIGURE 1.
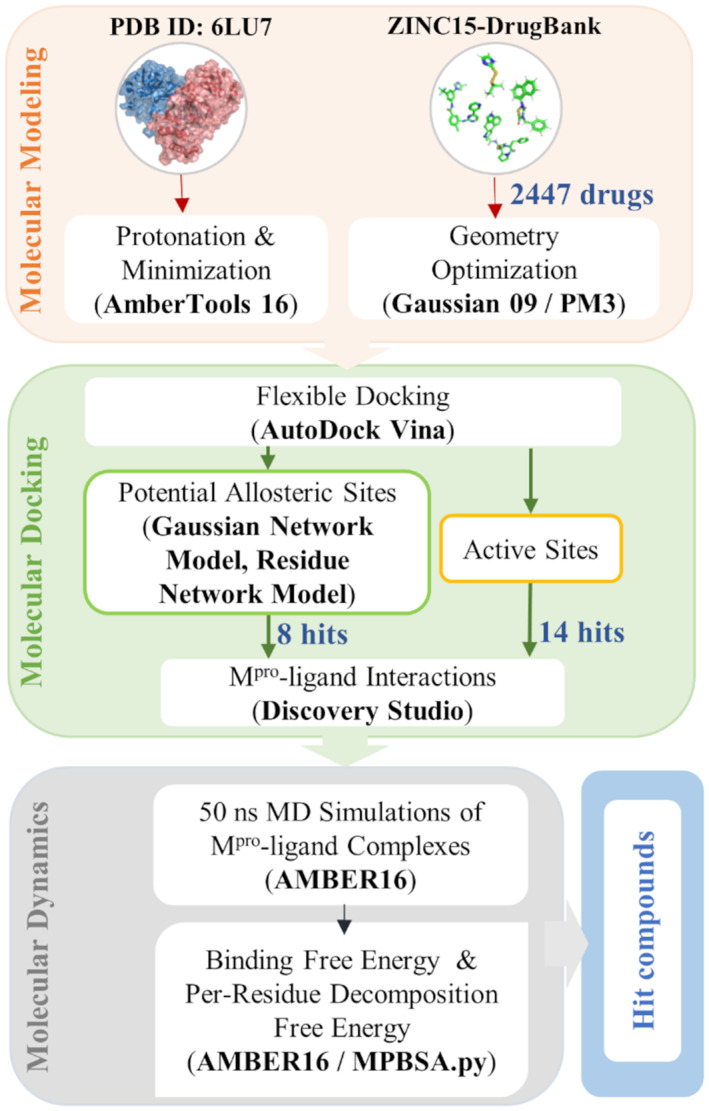
The computational workflow followed for the repurposing of FDA‐approved drugs
2.1. Determination of potential allosteric sites
GNM is an elastic network model (ENM), which describes the protein structure as a network of connected nodes. Nodes are usually placed at Cα positions of the amino acids. Neighboring amino acid pairs within a cut‐off distance of 7 Å are then linked by uniform elastic springs. 56
The total potential energy of the constructed network of N nodes is given as,
| (1) |
where γ is the spring constant, R ij and R ij 0 are the instantaneous and equilibrium distances between the i th and j th nodes, respectively. Γ ij is the ij th element of the Kirchhoff matrix Γ (N × N) containing the connectivity information of the nodes. The singular decomposition of Γ = UΛU T gives the eigenvalues and eigenvectors corresponding to coupled motions of the nodes. Here, U is the orthogonal eigenvector matrix with elements u k indicating the k th eigenvector. Λ is the diagonal matrix of eigenvalues λ k . N − 1 eigenvalues give the vibrational frequencies of N − 1 modes, while one eigenvalue is zero indicating the rigid body motion.
Cross‐correlation between i th and j th nodes is defined by,
| (2) |
and their relative fluctuations are calculated as,
| (3) |
Cross‐correlations calculated for the low‐frequency motions (slow modes) reveal dynamic domains of proteins where groups of amino acids fluctuate in the same direction. Low‐frequency motions of the proteins correspond to their functional globular motions. Therefore, they are highly useful to understand the functional mechanisms of the protein. 57 On the other hand, ⟨∆R ij 2 ⟩ describes the relative mobility of i th and jth nodes. The high‐frequency motions in fast modes calculated with ⟨∆R ij 2⟩ serve to find critical residues related to folding core 58 , 59 or binding, such as protein, DNA, or drug binding sites. 59 , 60 Here, we analyze the six slowest modes that give information about collective functional motions of the main protease with the highest contribution. Also, we consider the 20 fastest modes to detect the residues with high‐frequency fluctuations. These residues have a high capacity to alter the energy landscape such as after binding a drug molecule, thus, highlight plausible drug target regions besides the active site of the main protease.
The residue network model is similar to GNM; a network of connected nodes is constructed based on the protein structure. Nodes are placed at C α atoms, and two nodes (amino acids) are linked if they have atom‐atom neighboring within a cut‐off distance of 4.5 Å. This cut‐off distance includes van der Waals and electrostatic interactions. The local interaction strength of a (i, j) node‐pair is calculated as,
| (4) |
Here, N ij is the total number of heavy atom pairs of ı th and ȷ th nodes. The biasing effect of the amino acid size is eliminated by weighting N ij using their total number of atoms N i and N j . 61 In this approach, the node pairs with a high interaction strength can be considered closer to each other. Thus, the length of edges between two neighboring nodes is calculated by 1/a ij . This formulation suppresses the bias towards covalently bonded interactions and covers both covalent and long‐range interactions. 62
The centrality measure of betweenness reveals the frequently visited nodes or “hubs” located on the shortest paths that are calculated for the network. The betweenness (C B ) value is determined as 63 ;
| (5) |
Here, σ ij is the shortest number of routes between nodes i and j, σ ij (l) is the shortest number of routes between nodes i and j passing through node l. In this line, the nodes with high (C B ) values in the residue network have a high potential to reside on the allosteric communication paths 62 , 64 that can be evaluated as novel drug targets.
2.2. Molecular docking
The crystal structure of Mpro in complex with N3 at 2.1 Å resolution is retrieved from the Protein Data Bank (PDB ID:6lu7). 16 The dimeric Mpro structure is hydrogenated at pH 7.0 using AMBER16 54 following a validation of the protonation states of the charged side chains with PROPKA3.1. 65 Then, the dimer is subjected to energy minimization using AMBER16. FDA‐approved drugs are downloaded from ZINC15 66 and DrugBank 67 databases. Molecules are geometrically optimized using Gaussian 09 68 at PM3 level of theory. 69 The sdf files of the molecules are converted into Gaussian input files (.gjf) using OpenBabel‐3.0. 70 A library composed of 2447 optimized drugs is created for molecular docking studies.
Molecular docking of Mpro against the FDA‐approved drugs is performed using AutoDock Vina software. 71 The grid is centered within the active site cavity of subunit A and subunit B with a size of 20 Å × 20 Å × 20 Å in the x‐, y‐, z‐axes, respectively. On the other hand, the grid sizes are set as 20 Å × 20 Å × 25 Å and 26 Å × 20 Å × 20 Å for potential allosteric sites 1 and 2, respectively. To validate our docking protocol, rigid docking of N3 ligand from PDB ID:6lu7 into the active site of Mpro is performed. From this calculation, H41, C145, H163, E166, Q189 residues on subunit A, and S1 residue of subunit B are determined to make critical contributions to the vina docking score (kcal/mol); therefore, they are kept flexible during flexible docking. Similarly, for the potential allosteric site 1, K12, C16, and K97 residues on both subunits are kept flexible, whereas E288, D289, E290, F291 residues on subunit A are kept flexible in docking studies for the potential allosteric site 2. In docking runs, we collect the best 10 poses for each ligand in the active site cavity and potential allosteric regions. The ligand‐protein interactions are analyzed with Discovery Studio Free Visualizer 2020 72 and the results are reported as vina docking scores (kcal/mol).
2.3. Molecular dynamics simulations
The parameters, coordinate, and topology input files are generated for all compounds, the crystal structure of the protein (PDB ID:6lu7), and protein‐ligand complexes using tLeaP module implemented in AMBER16. For the ligand parameterization, the general Amber force field (GAFF), 73 and the antechamber and parmchk2 modules are used with AM1‐BCC charges. 74 The parameters for the protein are described using the AMBER ff14SB force field. 75 TIP3P water model 76 is used to solvate each system with a 12 Å padding resulting in a cubic box dimension of 112 Å × 112 Å × 112 Å. The Na+ ions are added to neutralize the system electrostatically, yielding approximately 128 000 atoms in the simulation systems without considering ligand molecules.
The Particle Mesh Ewald summation technique 77 is used with the default 8 Å cutoff. A time step of 2.0 fs is used for the implementation of SHAKE algorithm 78 to fix the bonds involving hydrogens. The system is minimized with 50 000 iterations of the steepest‐descent method, while the positions of all heavy atoms are fixed by imposing harmonic restraints. The equilibration processes of the samples are carried out in four consecutive steps to maintain the interactions for the docking pose and to assess the relative stability of the pose before continuing further. (a) 100 ps of MD is performed in NPT ensemble at a temperature of 10 K with a strong temperature coupling while the harmonic restraints of 10 kcal/mol/Å2 are maintained for all heavy atoms. (b) a 100 ps long NPT MD simulation is performed while only the protein‐heavy atoms are restrained. These two steps are carried out to generate a proper geometry for the hydrogen atoms, and to allow H‐bond interactions between the water molecules and the enzyme complex. (c) a 2 ns NPT MD simulation is performed to increase system temperature from 10 to 300 K while the harmonic restraints are maintained only for all heavy atoms of the protein and the ligand(s). (d) a total of 25 ns MD in NVT ensemble at a temperature of 300 K is performed. During this run, restrictions on the main atoms are gradually removed in the first 10 ns by decreasing the harmonic restraint value on the heavy atoms by 0.5 kcal/mol/Å2 after each 500 ps. Finally, 50 ns production MD simulations are performed in NVT ensemble using the Langevin thermostat. 79
MD simulations are performed for the complexes, where ligands are docked to both active sites of the Mpro. In this way, better sampling of the ligand‐active site interactions is achieved. For the other ligand‐Mpro complexes, there is only one ligand in one potential allosteric site. For all cases, ligands in their binding sites are monitored to ensure that they do not diffuse to the solvent. Therefore, the binding free energy calculations for the ligands enable a reliable comparison to evaluate their binding affinities.
2.4. Binding free energy calculations
The values of the free energy of binding (∆G bind ) for a ligand can be calculated according to the equation 55 :
| (6) |
| (7) |
| (8) |
where ΔE MM is the molecular mechanics energy of the molecule expressed as the sum of the internal energy of the molecule and the electrostatic and van der Waals interactions. ΔG sol denotes the solvation energy which is composed of the polar (ΔG PB/GB) and nonpolar contributions (ΔG SA). The entropic contribution can be neglected because of similar types of ligands bind to the receptor, and enthalpic contribution is sufficient to compare different ligands. The protein‐ligand interactions and the binding free energies are obtained using the MMPBSA.py 80 and Sander modules of AMBER16, respectively.
2.5. Determining the most promising compounds
The most promising compounds are selected following these steps:
Compounds with the highest vina docking scores down to 15% less of the best binding score are noted;
Molecular interactions critical in ligand stabilization and binding affinity, such as H‐bonds, sulfur bridges, and so on. within the substrate‐binding pocket of the protein‐ligand complexes from (1) are analyzed in detail;
For the active site calculations, we select compounds interacting with the catalytic dyad residues H41 and C145 similar to the binding pose of N3 inhibitor. This indicates a better fit as well as the good binding affinity of the compounds to the active site;
For the potential allosteric sites, we especially consider compounds having a high binding affinity towards both active site and potential allosteric regions.
Selected compounds in complex with the dimeric enzyme are then subjected to 50‐ns long MD simulations coupled with MM/GBSA calculations. Protein‐ligand complexes with binding free energies higher than approximately 25% of the best binding free energy value are considered as promising hit compounds to be further evaluated in vitro and in vivo studies.
3. RESULTS AND DISCUSSION
Since there is an urgent need to alleviate the COVID‐19 pandemic, the repurposing of existing FDA‐approved drugs is a highly effective strategy that reduces time cost, investment, and risks compared to traditional drug development strategies. 81 , 82 Herein, we aim to identify potential inhibitors against Mpro by employing docking and MD simulations combined with MM/GBSA studies to calculate the binding energies. All calculations are performed with the Mpro dimer, which represents its functional state. A total of 2447 FDA‐approved drugs are screened using molecular docking methodology against the active site of Mpro on both subunits and potential allosteric sites (Figure 2). Initially, the molecular docking protocol is validated by re‐docking the crystallographic N3 ligand (PDB ID:6lu7) to the active sites of Mpro. Both the crystallized and docked structures for N3 indicate similar poses (Figure S1) with a root‐mean‐square deviation (RMSD) value of 1.9 Å, which is considered sufficient.
FIGURE 2.
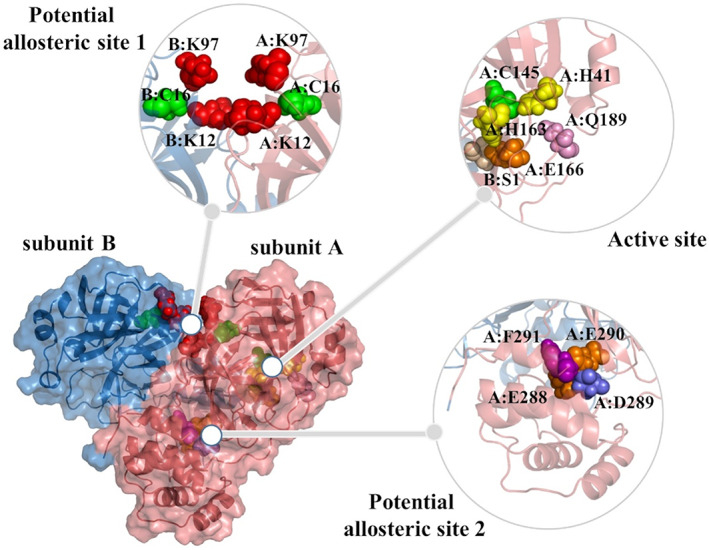
Surface representation of dimeric Mpro, where two subunits are in salmon and skyblue. One active site and two potential allosteric sites are explicitly shown. The key residues in the flexible docking experiments are in spheres
3.1. Docking to the active sites of Mpro
We perform flexible docking experiments of over 2400 FDA‐approved drugs with AutoDock Vina against the active site on both monomers of the Mpro. The active site residues making critical contributions to the binding score (H41, C145, H163, E166, Q189 on subunit A, and S1 on subunit B) are kept flexible during docking experiments (Figure 2). Molecular docking studies reveal 14 promising Mpro inhibitor candidates that belong to different chemical classes, namely ergot alkaloids, flavonoids, antivirals, anti‐parasitics, antibiotics, anti‐cancer agents, analgesics, and cardiac glycosides. Vina docking scores of these drugs range from −11.6 kcal/mol (dihydroergotamine) to −8.4 kcal/mol (elbasvir) for the active site on both subunits (A/B). The detailed results on the docking score (kcal/mol), DrugBank identification, and chemical classification of each compound are given in Table 1.
TABLE 1.
Vina docking scores (kcal/mol) of the hit compounds among FDA‐approved drugs against Mpro
| Classification | Drug name DrugbankID | Chemical formula structure | Vina Docking Score (kcal/mol) | ||
|---|---|---|---|---|---|
| Active site subunit A/B | Allosteric site 1 | Allosteric site 2 | |||
| Ergot‐alkaloids |
Dihydroergotamine DB00320 |
C 33 H 37 N 5 O 5
|
−11.6/−11.3 | −10.6 | −12.1 |
| Bromocriptine DB01200 |
C 32 H 40 BrN 5 O 5
|
−9.9/−9.9 | −9.7 | −11.2 | |
| Anti−cancer agent |
Nilotinib DB04868 |
C 28 H 22 F 3 N 7 O
|
−10.8/−10.8 | −9.5 | −11.1 |
|
Entrectinib DB11986 |
C 31 H 34 F 2 N 6 O 2
|
−10.2/−10.3 | −9.6 | −10.9 | |
|
Selinexor DB11942 |
C 17 H 11 F 6 N 7 O
|
−10.1/−10.1 | −8.2 | −9.5 | |
| Anti‐parasitic |
Ivermectin DB00602 |
C 95 H 146 O 28
|
−10.2/−9.7 | −10.0 | −7.5 |
| Antiviral |
Paritaprevir DB09297 |
C 40 H 43 N 7 O 7 S
|
−10.2/−10.2 | −10.1 | −10.0 |
|
Elbasvir DB11574 |
C 49 H 55 N 9 O 7
|
−8.8/−8.4 | −10.8 | −11.1 | |
| Antibiotic | Quinupristin DB01369 |
C 53 H 67 N 9 O 10 S
|
−10.0/−10.0 | −9.8 | 2.0 |
|
Rifapentine DB01201 |
C 47 H 64 N 4 O 12
|
−9.8/−9.9 | −10.4 | −0.4 | |
| Flavanoids |
Rutin DB01698 |
C 27 H 30 O 16
|
−10.6/−10.5 | −8.1 | −9.5 |
|
Diosmin DB08995 |
C 28 H 32 O 15
|
−9.8/−9.4 | −9.4 | −10.6 | |
| Cardiac glycoside |
Digitoxin DB01396 |
C 41 H 64 O 13
|
−10.0/−9.8 | −10.3 | −11.1 |
| Analgesic |
Antrafenine DB01419 |
C 30 H 26 F 6 N 4 O 2
|
−9.9/−9.7 | −7.3 | −10.9 |
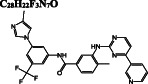
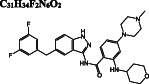

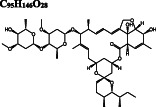
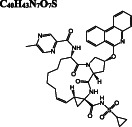
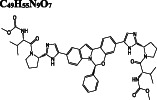
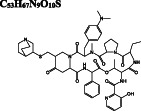
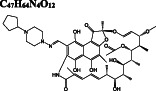
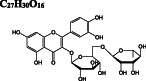
Dihydroergotamine is predicted as the compound with the highest binding affinity towards the active site of dimeric Mpro. It interacts with the residues typically dispersed in domain 2 (residues 102‐184) (Figure 3A) predominantly in the form of hydrogen bond interactions. Another ergot alkaloid derivative in our list is bromocriptine exhibiting H‐bond interactions with H41 and C145 residues of the catalytic dyad, respectively (Figure 3B). Anti‐cancer agents, nilotinib, entrectinib, and selinexor are ranked among the top best molecules in our results, displaying H‐bond, π‐alkyl, π‐sulfur, alkyl, and halogen interactions with the active site residues, especially with the catalytically active C145 residue (Figure 3C‐E).
FIGURE 3.
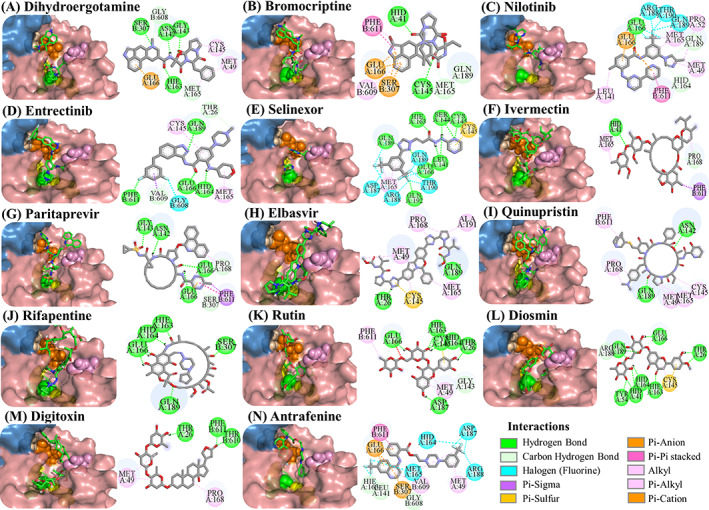
Docking poses of potential Mpro inhibitors into the substrate‐binding site. The docking results for (A) dihydroergotamine, (B) bromocriptine, (C) nilotinib, (D) entrectinib, (E) selinexor, (F) ivermectin, (G) paritaprevir, (H) elbasvir, (I) quinupristin, (J) rifapentine, (K) rutin, (L) diosmin, (M) digitoxin, (N) antrafenine are shown as salmon (subunit A) and skyblue (subunit B) surface. Ligands are depicted in green and flexible residues are represented in spheres. Their two‐dimensional interaction maps are also shown
Ivermectin and paritaprevir exhibit relatively higher binding affinity. The hydrogen bond interaction with H41 residue (Figure 3F) for ivermectin, and several H‐bond, π‐π stacking, π‐sulfur π‐alkyl, and alkyl interactions (Figure S2) with domain two residues especially with C145 residue of the catalytic dyad (Figure 3G‐H) observed for paritaprevir. Although elbasvir displays a relatively lower binding affinity towards the active site with −8.8 kcal/mol compared to other FDA‐approved drugs, it shows several H‐bond, π‐sulfur, π‐alkyl, and alkyl interactions (Figure S2) with domain 2 residues especially with C145 residue of catalytic dyad (Figure 3G‐H). Besides several H‐bond interactions with residues typically dispersed in domain 1 (residue 1‐101) and domain 2 (residue 102‐184), just like paritaprevir and rifapentine, quinupristin displays alkyl interaction with C145 residue of the active site (Figure 3I‐J). We also identify two flavonoid compounds, rutin, and diosmin, displaying high‐binding affinity towards Mpro. Besides a vast number of interactions with residues of domain 2 (Figure S2), rutin exhibits H‐bond and π‐sulfur interactions with C145 residue of the catalytic dyad (Figure 3K), whereas diosmin displays H‐bond interaction with H41 residue and π‐alkyl and π‐sulfur interactions with C145 residue of catalytic dyad (Figure 3). Digitoxin and antrafenine interact with the active site by forming several H‐bond, π‐π stacked, π‐cation, π‐anion, π‐alkyl, alkyl, and halogen interactions (Figure 3). Considering all analyses, ligands mostly interact with T26, H41, H163, H164, E166, R188, Q189, and T190 residues by forming H‐bond and halogen interactions, with F305 residue of subunit B by π − π stacked interaction and with M49, C145, M165, and P168 residues by π‐alkyl, π‐sulfur, and alkyl interactions, in accordance with those stabilizing N3 inhibitor inside the substrate‐binding pocket. 16 These residues are also reported in previous docking studies targeting Mpro. 22 , 23 , 24
The number of interactions and interaction types of all compounds mentioned above are given in Figure S2. We posit that these 14 hit compounds are good candidates as Mpro inhibitors targeting the active site of Mpro and therefore the complex stability of these compounds are further evaluated in 50‐ns MD simulations and binding free energy calculations.
3.2. Docking to potential allosteric sites of Mpro
Figure 4A displays the low‐frequency (slow modes) and the high‐frequency (fast modes) dynamics of Mpro calculated by GNM. The cross‐correlations of residue pairs are determined for the six slowest modes that contribute to 16% of the overall dynamics, and they are projected on the structure. Same colored regions point to groups of amino acids with positive cross‐correlation moving in the same direction, and different colored regions have anti‐correlated motions. Positively correlated amino acids form dynamic domains moving as rigid bodies for the functional dynamics of the protein. Hinge regions located between the dynamic domains consist of amino acids with low mobility and provide them rotational freedom. For instance, in slow mode 1, each monomer is a distinct dynamic domain having anti‐correlated motions such as to open and close the cleft at the subunit interface. This motion is coordinated by a hinge region including the whole subunit interface. We also note that the dynamic domains of Mpro at the other slowest modes consist of its building blocks, that is, its structural domains I (amino acids 10‐99), II (100‐184), and III (201‐303), separated by hinge regions.
FIGURE 4.
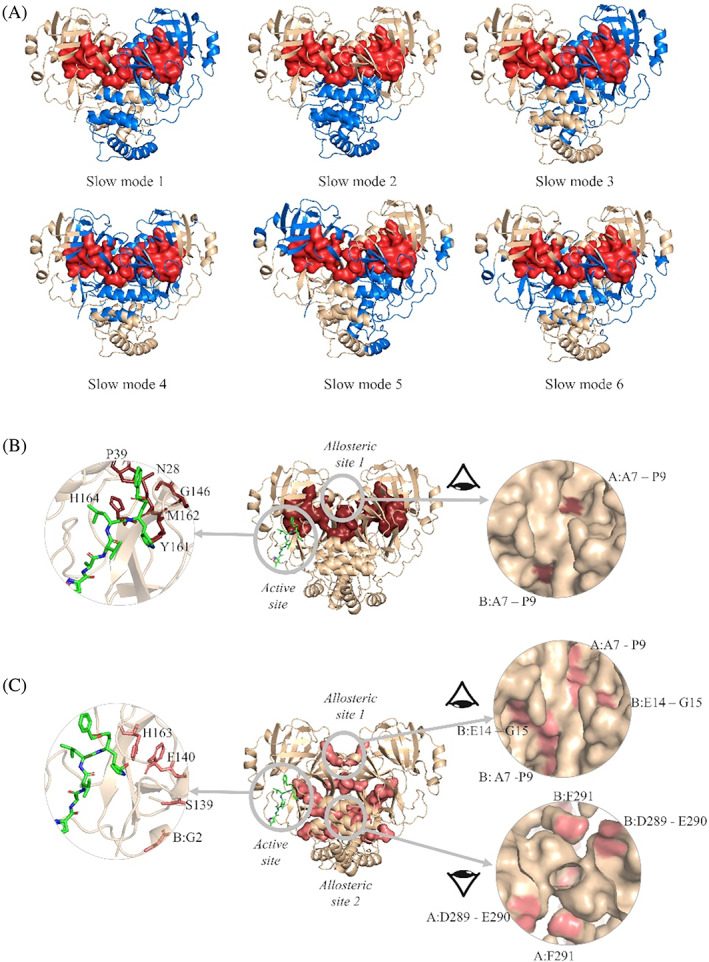
(A) Dynamic domains from the six slowest modes of Mpro calculated by GNM. Distinct dynamic domains are colored in blue and wheat. Amino acids with high relative mobility in the 20 fastest modes are shown in red surface representation. Potential drug binding sites predicted by (B) GNM and (C) the residue network model. In (B), amino acids with high relative mobility in 20 fastest modes of GNM are shown in brown. In (C), the hub residues predicted with residue network model are in pink. The ligand N3 (in green) at one active site is also shown with the predicted amino acids in the left panel, and potential allosteric sites are shown in the right panel
Figure 4A also shows amino acids with high relative mobility ⟨∆R ij 2 ⟩ in the 20 fastest modes. They have a high potential to change the conformational energy landscape of the protein upon ligand binding. Therefore, the amino acids both having high relative mobility in the fastest modes and involving in the hinge regions of the slowest modes can be evaluated as potential drug binding sites, as was recently shown for the bacterial ribosome. 83 At the same time, they plausibly highlight allosteric regions that can affect the activity of the protein upon perturbation. Accordingly, M6‐P9, V13, V18, N28, G29, C38, P39, A116, C117, G146, S147, Y161, M162, and H164 are the residues with high relative mobility located at the hinge regions. Indeed, P39, G146, S147, Y161, M162, H164 are located next to the active sites, as shown in Figure 4B. Recent MD simulations also indicated that mutations at residues A7 and A116 increase the proximal interactions, which in turn affect the dynamics and the dimer stability. 21 We propose the region pinned by A7‐P9 as a potential allosteric site, since it is located on a cavity at the subunit interface, and is solvent accessible.
Then, we employ the residue network model and centrality measurement of betweenness to determine hub residues with high potential to assist the flow of information in the structure. 62 We use the crystal structure of Mpro (PDB ID:6lu7), and calculate betweenness values CB. The top 0.05 quantile (CB > 0.0485) is designated to hub residues as in 62 (Figure 4C). All findings are given in Table S4. Residue network model predicts the active site residues F140 and H163 as hub residues, while it also reveals critical amino acids potentially taking part in allosteric communication of the active sites with distant regions. Supporting our results, mutation studies using MD simulations 21 proposed A7 and G15 as critical residues impairing the functional motions of Mpro. Among the other hub residues listed in Table S1, E14, and G15 are solvent‐accessible and are located at the potential allosteric region predicted by GNM (Figure 4B and C, right panel). GNM and residue network model calculations both indicate the same region as a drug target, called allosteric site 1 including residues P9, E14, and G15. In addition, E290 and F291 mark a second potential allosteric region (Figure 4C, right panel) located at the interface of domains III on both subunits, thus, has a high potential to affect the functional activity of Mpro. The potential allosteric site 2 is next to the previously proposed allosteric site, revealed by crystallographic screening of covalent and noncovalent fragments. 49 Very recently, two allosteric sites are determined by X‐ray high‐throughput screening on Mpro 48 ; one site is close to the catalytic region within the C‐terminal dimerization domain and the other is between the catalytic and the dimerization domain. The latter is in proximity of the potential allosteric site 2, suggested by our calculations.
After we predict potential drug binding sites on Mpro, we perform flexible docking studies of over 2400 FDA‐approved drugs against these potential allosteric sites of Mpro with AutoDock Vina. Dihydroergotamine (DB00320), nilotinib (DB04868), entrectinib (DB11986), digitoxin (DB01396), bromocriptine (DB01200), and diosmin (DB08995) exhibit good binding affinities not only to the active site but also to the predicted allosteric sites of Mpro with vina docking scores up to −12.1 to kcal/mol (Table 1). These drugs form H‐bond interactions with K97 of subunit A, and K12 of both subunits, which are kept flexible during docking experiments for potential allosteric site 1. They also make π‐alkyl, alkyl, and halogen interactions with domain 1 (residue 1‐101) of both subunits (Figure S3). On potential allosteric site 2, domain III (W207, L282 G283, L286, E288, E290, F291, R310, K311) and domain I (R4, K5, K137) residues are highlighted due to their H‐bond, π‐cation, π‐anion, π‐alkyl, and alkyl interactions with the docked ligands (Figure S4). In addition, we determine two other hits for the potential allosteric sites. Interestingly, elbasvir has a higher binding affinity for the potential allosteric sites 1 (−10.8 kcal/mol) and 2 (−11.1 kcal/mol) compared to both active sites of Mpro (−8.8 and − 8.4 kcal/mol). Selinexor has a slightly lower binding affinity for the potential allosteric sites (−8.2 and − 9.5 kcal/mol) as compared to the active sites (−10.1 kcal/mol). Evidently, the potential allosteric sites on Mpro can be indeed novel target sites, which motivates us to further investigate the stability of these 8 hit compound—Mpro complexes with 50‐ns long MD simulations.
3.3. MD simulations of Mpro‐ligand complexes
The main purpose of this study is to determine FDA‐approved drugs that exhibit high binding affinity to active and/or potential allosteric sites of Mpro. We apply a comprehensive docking protocol and select a total of 14 ligands complexed with the Mpro active site to perform MD simulations. Among these, eight drugs namely bromocriptine, diosmin, dihydroergotamine, nilotinib, entrectinib, digitoxin, selinexor, and elbasvir have also high docking scores for the potential allosteric sites 1 and 2. Therefore, MD simulations are also carried for these ligands docked to the potential allosteric sites. MD simulations are performed for the functional dimer, where the hit compounds are either bound to both active sites or the allosteric sites.
The stability of the ligand‐complex systems is monitored by the RMSD of the backbone Cα atoms and root‐mean‐square fluctuations (RMSF) of the residues, where the initial frame is taken as the reference. RMSD and RMSF graphs reveal that systems reached equilibrium at the end of 25‐ns equilibration run. The stability of each system in 50‐ns production run with detailed analysis of RMSD and RMSF graphs are presented in Figures S5‐S34. The RMSD values for the protein are around 2 Å, indicating convergence of the simulations. We then focus on the stability of ligands that are docked to both active sites, that is, the same ligand both in subunits A and B. Here, ligands can be mobile at both active sites without leaving the pocket, such as nilotinib (Figure S8), entrectinib (Figure S12), and elbasvir (Figure S18). On the other hand, the mobility of ligands on each subunit may differ; such as antrafenine (Figure S9), dihydroergotamine (Figure S15), and quinipristine (Figure S10) have greater mobility at one active site and stable in the other. Similar observations are made for the potential allosteric sites. Digitoxin (Figure S22) and elbasvir (Figures S25 and S33) are mobile in their binding pockets. Nilotinib and entrectinib diffuse from their first binding pockets (allosteric site 1) at the equilibration period and towards the end of the simulations, respectively. They bind neighboring sites (subunit A), where they remain during the simulations. Nilotinib and entrectinib interact with A70 and N72 residues by forming H‐bond interactions (Table S3) and remain stable in these neighboring sites. It should be noted that these two residues are on the second shell of the predicted allosteric site. Other than indicated, ligands are stable in their initial docking sites, that is, active sites, potential allosteric sites 1 and 2.
The binding free energy values are calculated using the MM/GBSA approach and reported separately for each ligand‐binding site pair. For all investigated ligands, the binding is thermodynamically favorable and Tables S2‐S4 tabulates the energetic components of the binding free energy, namely van der Waals energy (∆E vdw ), electrostatic energy (∆E ele ), General Born solvation (∆E GB ), and surface area energies (∆E sur ). In addition, Table S2 lists the H‐bond interactions between the active site residues of Mpro and ligands that are established throughout the simulation. Here, residues H41, N142, E166, Q189, T190, and Q192 on both subunits are taking a role to stabilize the ligands via H‐bond interactions. This finding indicates the reliability of the flexible docking protocol followed in this study, where H41, E166, and Q189 are let flexible. The detailed information on the % occupancy of H bonds formed between active site residues and ligands are given in Tables S5 and S6 for subunits A and B, respectively.
Vina docking scores and ∆G bind values of hit compounds at both subunits are displayed in Figure 5A. Vina score ranking is in agreement with MM/GBSA binding energy rankings for most of the ligands. Dihydroergotamine has the highest vina docking score and also has a high binding free energy in the active site. On the other hand, elbasvir exhibits a lower binding affinity towards the active sites, which is consistent with vina score values. Even though ivermectin, bromocriptine, nilotinib, digitoxin, and diosmin exhibit moderate vina scores, they show high‐binding affinities to the active site of Mpro. There may be differences in the binding affinities of the ligands in different subunits of dimeric Mpro. This is plausibly due to the collective dynamics of the enzyme that can modulate the cooperativity of distant active sites. 84
FIGURE 5.
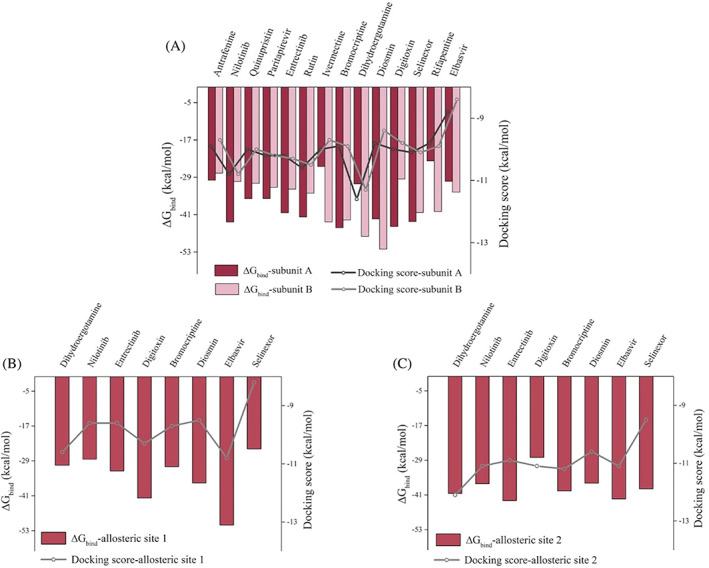
∆G bind and docking score values of potential hit compounds bound to (A) active site, (B) potential allosteric site 1, and (C) potential allosteric site 2 of Mpro. Red bars correspond to MM/GBSA calculations of subunit A, pink bars correspond to the same calculations of subunit B, and salmon bars correspond to the same calculations of potential allosteric sites (interface) (primary y‐axis). Dark gray line corresponds to docking score of subunit A, whereas light gray line corresponds to docking score of subunit B, and gray line corresponds to docking score of potential allosteric sites (secondary y‐axis)
For the potential allosteric sites 1 and 2, elbasvir is highlighted both by vina docking scores and MM/GBSA calculations (Figure 5B, C). Elbasvir is followed by digitoxin and diosmin with promising binding free‐energy values for the allosteric site 1. On the other hand, entrectinib, dihydroergotamine, and bromocriptine show a high‐binding affinity towards the allosteric site 2. Selinexor has a high affinity for the active site; MM/GBSA calculations are also performed for selinexor docked to allosteric regions. Its binding free energy for the allosteric site 1 is worth noting (Figure 5B).
H‐bond interactions between the potential allosteric site 1 and 2 residues are detailed in Table S3 and Table S4, respectively. The potential allosteric site 1 and 2 are located at the subunit interface. In the first site, the ligands are mostly stabilized via H bond interactions involving K12, E14, N72, K97, and S121 residues of both subunits and Y154 residue on subunit B. The stabilization of ligands at the potential allosteric site 2 is maintained by K5, K137, W207, G283, and S284 residues on both subunits and E288 residue on subunit A. The % occupancies of H‐bond interactions at these sites are detailed in Tables S7 and S8.
Consequently, selinexor, bromocriptine, and diosmin exhibit high‐binding affinity on both active sites and show relatively higher docking scores for both potential allosteric sites. Ivermectin also has a high affinity for the active site and it is currently used in clinical trials to treat COVID‐19 according to WHO (WHO/2019‐nCoV/therapeutics/2021.1). In addition, elbasvir has a high affinity for both potential allosteric sites. Therefore, these compounds are further subjected to energy decomposition calculations per residue basis. In this way, we aim to determine which residues are mainly responsible for the stabilization of the ligands, hence one can propose a pharmacophore for rational drug design.
3.3.1. Per‐residue free‐energy decomposition
Figure 6 displays the per‐residue free‐energy decomposition for selinexor, bromocriptine, diosmin, and ivermectin where their stabilizations are predominantly maintained by 12 to 19 residues. Complex formation between active site of dimeric Mpro and selinexor involves mostly hydrophobic, positively charged, and polar residues of domains 1 and 2 at both subunits (Figure 6A). Same residues at both subunits energetically contribute to the binding of selinexor to similar extents that are reflecting on their binding free energies at subunits A and B (Figure 5A). Similarly, for bromocriptine, the interactions on both active sites involve the same hydrophobic and polar residues that stabilize the ligand in the binding sites (Figure 6B). For the diosmin‐Mpro complex, interacting residues with the ligand differ (Figure 6C). Hydrophobic residues L50, L167, P168, and A191 contribute to stabilizing diosmin at subunit A, whereas polar residues T25, N142, S144, C145, Q189 of domains 1 and 2 mostly facilitate its subunit B binding. This dramatic difference in the type of binding residues is the reason of the difference in binding free‐energy values for subunit A (−42.39 kcal/mol) and B (−52.06 kcal/mol). The binding free energy of ivermectin at the active sites is less than diosmin (approximately 40 kcal/mol), where polar (S46, Q189, Q192) and positively charged (H41, R188) residues of subunit A, and hydrophobic residues (L27, M49, G143, M165, L167, and P168) of subunit B stabilize the ligand (Figure 6D).
FIGURE 6.
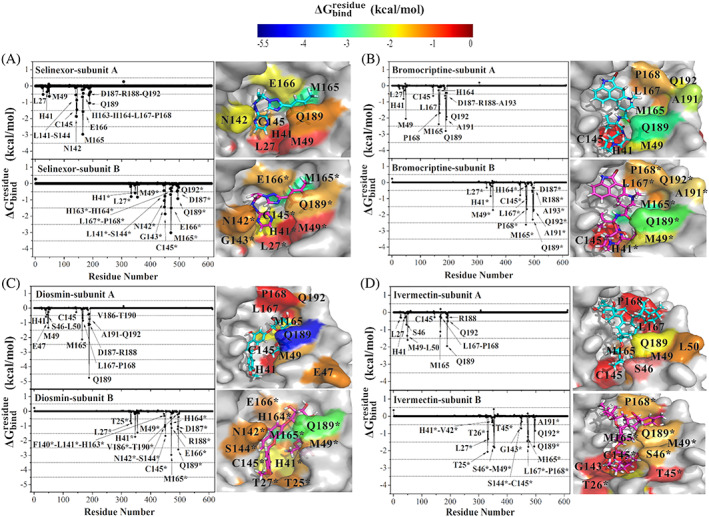
values of (A) selinexor, (B) bromocriptine, (C) diosmin, and (D) ivermectin in complex with the active site of subunits A and B of Mpro. The contributing residues involved in ligand binding are colored according to their values, where the highest to lowest free energies are shaded from blue to red, respectively
For these four selected ligands, hydrophobic (L27, M49, G143, M165), polar (T25, T26, S46, N142, S144, C145, Q189, T190, Q192), positively charged (H41, H163, H164, R188), and negatively charged (E166, D187) residues significantly contribute to ∆G bind values of ligand‐enzyme complexes (Figure 7). Notably, catalytic residues H41 and C145, and M49, M165, Q189 consistently involve in binding of the ligands, as also reported by previous studies on Mpro. 16 , 17 , 84 , 85 , 86 These residues are also reported for N3 16 complexed with Mpro crystal structure, and for the proteolysis reaction catalyzed by Mpro investigated using QM/MM molecular dynamics simulations. 84 , 85 , 86 , 87
FIGURE 7.
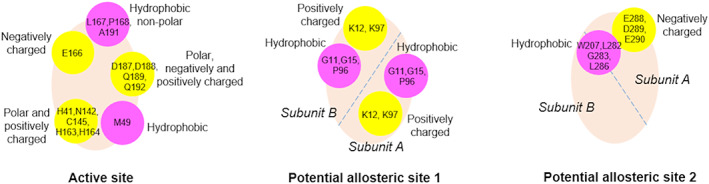
Characteristics of the residues interacting with the hit compounds at the target sites of Mpro. The approximate locations of the hydrophobic (in magenta) and polar/charged (in yellow) residue groups are shown on the binding cavities (in wheat)
The same analysis is carried for elbasvir docked to potential allosteric sites 1 and 2. Elbasvir has a high‐binding affinity for both potential allosteric sites; −51.08 and − 42.37 kcal/mol for allosteric sites 1 and 2, respectively. Potential allosteric sites are located at the subunit interfaces, therefore, residues of both subunits simultaneously contribute to ligand stabilization. Elbasvir binds to potential allosteric site 1 via the energetic contributions of hydrophobic G11, G15, P96, and positively charged K12, K97 residues of both subunits, whereas hydrophobic residues W207, L282, G283, L286 of both subunits stabilize its binding at potential allosteric site 2 (Figure 8A). We also investigate the per‐residue free‐energy decomposition of elbasvir on the active site. Its binding free energy at the active sites is less than that at the potential allosteric sites (approximately −32 kcal/mol), where hydrophobic and polar residues of subunit A, and polar residues (C44, T45, S46, N142, S144, and Q189) of subunit B stabilize the ligand (Figure 8B). Moreover, S301, G302, and V303 residues of subunit A are noted to take a role in the stabilization of elbasvir at subunit B.
FIGURE 8.
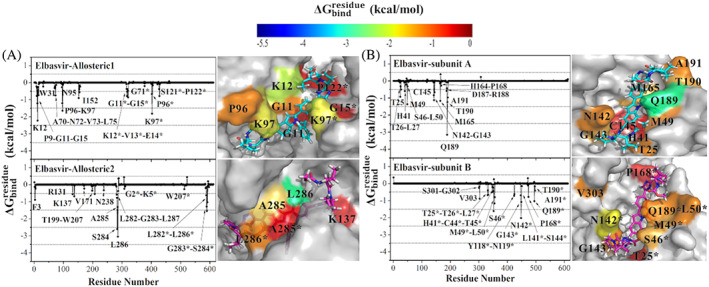
values of (A) elbasvir in complex with the potential allosteric sites, and (B) elbasvir in complex with the active site of Mpro. The contributing residues involved in ligand binding are colored according to their values, where the highest to lowest free energies are shaded from blue to red, respectively
In general, hydrophobic residues of both subunits contribute to ∆G bind values, but G11, K12, G15, and K97 residues of both subunits are highlighted at the potential allosteric site 1. For the potential allosteric site 2, positively charged K5 residue and hydrophobic F3, W207, A285, F291 residues of both subunits seem to help ligand binding, where L282, G283, S284, and L286 of both subunits significantly contribute to ∆G bind values (Figure 7). Here, K12, K97, and F291 are kept flexible during docking studies to obtain more accurate docking poses as initial ligand‐protein complex structure in MD simulations.
Among the hit 14 compounds that we identify in this study, high‐binding free energies and a large number of molecular interactions of dihydroergotamine, 88 rutin, 89 , 90 paritaprevir, 91 ivermectin, 92 antrafenine 93 with the substrate‐binding pocket of Mpro are also reported in other in silico studies, thus supporting our approach. In vitro studies have recently reported the antiviral activity of digitoxin and nilotinib against SARS‐CoV‐2. 94 , 95 However, inhibitory effects of these drugs against Mpro have not been clinically tested yet. In addition to these studies, we here identify diosmin, selinexor, bromocriptine, ivermectin, and elbasvir with an inhibitory potential against Mpro. Diosmin (NCT04452799) mixture with hesperidin 96 and selinexor (NCT04349098) have already been under clinical trials against SARS‐CoV‐2. Selinexor, a novel class of anti‐cancer agent, is evaluated in clinical trials for hospitalized patients with SARS‐CoV‐2 infection, resulting in both anti‐viral and anti‐inflammatory activities in patients. Being a member of the flavonoid family, diosmin is used in the treatment of venous disease and displays anti‐inflammatory action. 97 Considering the importance of natural products as antiviral agents recently, 98 , 99 diosmin could be a promising drug to treat COVID‐19. Worth to note that selinexor and diosmin have not been tested against Mpro; yet, they can be evaluated as potent Mpro inhibitors. The inhibitory effects of bromocriptine, an ergot‐derived dopamine receptor agonist, on zika virus and dengue virus replication have been reported, 100 , 101 , 102 so it has also a high potential for the treatment of SARS‐CoV‐2 infected patients. Ivermectin is an anti‐parasitic drug the usage of which extended from veterinary medicine to humans. 103 In addition to in silico studies 104 involving the interactions between ivermectin and SARS‐CoV‐2 3CLpro, an in vitro study is performed by Caly et al. 92 indicating ivermectin's capability to reduce viral RNA. Ivermectin (NCT04668469) has recently been evaluated for the clinical trials. 47 Elbasvir is another antiviral drug in our list that displays a higher binding affinity towards potential allosteric sites than the active site of Mpro, agreeing with previous in silico studies for Mpro. 105 , 106 Hit compounds identified as potent Mpro inhibitors in computational studies should be experimentally tested whether they are promiscuous inhibitors or not. 107 , 108 Regarding the immediate need for therapeutics against SARS‐CoV‐2, bromocriptine and elbasvir should be evaluated for advanced experimental research to cure COVID‐19.
4. CONCLUSION
In this study, we conduct a systematic approach to suggest potent inhibitory compounds to inhibit the activity of Mpro by combining different computational methods. First, we predict two potential allosteric sites by GNM and residue network model as alternative drug binding sites on the Mpro native structure. These residues have a high capacity to alter the conformational energy landscape of the enzyme upon ligand binding, thus affecting its functional motions. Then, flexible docking runs are performed for active sites on both subunits and two potential allosteric sites of the dimeric enzyme using FDA‐approved compound library containing over 2400 FDA‐approved drugs. Taking N3, the original inhibitor in the crystal structure as a reference, we determine the compounds interacting with catalytic dyad residues (H41, C145), establishing a large number of H‐bond and having high vina docking scores for the target sites.
Accordingly, 14 hits are determined for the active site, while six of these have also high docking scores for the allosteric sites. These promising drugs are further investigated with 50‐ns‐long MD simulations coupled with the MM/GBSA calculations to calculate free‐binding energy values and their binding characteristics. Results for molecular interactions and binding free‐energy values for the majority of ligands indicate that vina docking score ranking is in harmony with MM/GBSA free‐binding energy rankings. In fact, the analysis of docking poses reveals that flexible residues are mainly involved in the stabilization of ligands on the binding regions, leading to plausible complex structures for the MD simulations. The energy decomposition per residue analyses highlight the catalytic residues H41, C145, and substrate binding site residues M49, M165, and Q189, which are involved in ligand‐Mpro complex formation. The hits found from the calculations establish various interactions, such as H‐bond, π‐alkyl, π‐sulfur, and alkyl interactions with the binding pockets, which traces a clear template for pharmacophore design (Figure 7).
Bromocriptine, diosmin, selinexor, ivermectin, elbasvir, nilotinib, entrectinib, rutin, dihydroergotamine, and digitoxin are determined to have a high affinity for Mpro. These FDA‐approved drugs are suggested as anti‐COVID‐19 therapeutics to be further evaluated in vitro and in vivo testing for viral activity. Indeed, our calculations successfully determine diosmin, ivermectin, and selinexor that have already been subjected to clinical trials. Therefore, our systematic approach followed in this study serves as a guideline to propose effective compounds that can be rapidly tested in clinical trials for the treatment of various diseases, including COVID‐19.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.
DATA AVAILABILITY
The authors confirm that the data supporting the findings of this study are available within the article or its supplementary material.
5.
PEER REVIEW
The peer review history for this article is available at https://publons.com/publon/10.1002/prot.26164.
Supporting information
Appendix S1: Supporting Information
Video S1
Video S2
ACKNOWLEDGEMENTS
The authors thank the National Center of High‐Performance Computing (UHeM) at ITU (1007692020) for computational time. National Center of High‐Performance Computing (UHeM) at ITU, Grant/Award Number: 1007692020.
Yuce M, Cicek E, Inan T, Dag AB, Kurkcuoglu O, Sungur FA. Repurposing of FDA‐approved drugs against active site and potential allosteric drug‐binding sites of COVID‐19 main protease. Proteins. 2021;89(11):1425-1441. 10.1002/prot.26164
Funding information National Center of High Performance Computing (UHeM) at ITU, Grant/Award Number: 1007692020
Contributor Information
Ozge Kurkcuoglu, Email: olevitas@itu.edu.tr.
Fethiye Aylin Sungur, Email: aylin.sungur@itu.edu.tr.
REFERENCES
- 1. Zumla A, Chan JFW, Azhar EI, Hui DSC, Yuen KY. Coronaviruses‐drug discovery and therapeutic options. Nat Rev Drug Discov. 2016;15:327‐347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cheng VCC, Lau SKP, Woo PCY, Yuen KY. Severe acute respiratory syndrome coronavirus as an agent of emerging and reemerging infection. Clin Microbiol Rev. 2007;20(4):660‐694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chan JFW, Lau SKP, To KKW , Cheng VCC, Woo PCY, Yuen K. Middle East respiratory syndrome coronavirus: another zoonotic betacoronavirus causing SARS‐like disease. Clin Microbiol Rev. 2015;28(2):465‐522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhu N, Zhang D, Wang W, Li X. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. 2020;382:727‐733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cucinotta D, Vanelli M. WHO declares COVID‐19 a pandemic. Acta Biomed. 2020;91(6):157‐160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Joshi S, Joshi M, Degani MS. Tackling SARS‐CoV‐2: proposed targets and repurposed drugs. Future Med Chem. 2020;12(17):1579‐1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Singh E, Khan RJ, Jha RK, et al. A comprehensive review on promising anti‐viral therapeutic candidates identified against main protease from SARS‐CoV‐2 through various computational methods. J Genet Eng Biotechnol. 2020;18(1):69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Basu A, Sarkar A, Maulik U. Molecular docking study of potential phytochemicals and their effects on the complex of SARS‐CoV2 spike protein and human ACE2. Sci Rep. 2020;10(1):1‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ali A, Vijayan R. Dynamics of the ACE2–SARS‐CoV‐2/SARS‐CoV spike protein interface reveal unique mechanisms. Sci Rep. 2020;10(1):1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Deganutti G, Prischi F, Reynolds CA. Supervised molecular dynamics for exploring the druggability of the SARS‐CoV‐2 spike protein. J Comput Aided Mol des. 2020;35:195‐207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gul S, Ozcan O, Asar S, Okyar A, Barıs I, Kavakli IH. In silico identification of widely used and well‐tolerated drugs as potential SARS‐CoV‐2 3C‐like protease and viral RNA‐dependent RNA polymerase inhibitors for direct use in clinical trials. J Biomol Struct Dyn. 2020;1‐20. 10.1080/07391102.2020.1802346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pokhrel R, Chapagain P, Siltberg‐Liberles J. Potential RNA‐dependent RNA polymerase inhibitors as prospective therapeutics against SARS‐CoV‐2. J Med Microbiol. 2020;69:864‐873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cozac R, Medzhidov N, Yuki S. Predicting inhibitors for SARS‐CoV‐2 RNA‐dependent RNA polymerase using machine learning and virtual screening. arXiv (preprint). 2020. https://arxiv.org/abs/2006.06523v1. [Google Scholar]
- 14. Parvez MSA, Karim MA, Hasan M, et al. Prediction of potential inhibitors for RNA‐dependent RNA polymerase of SARS‐CoV‐2 using comprehensive drug repurposing and molecular docking approach. Int J Biol Macromol. 2020;163:1787‐1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Xia B, Kang X. Activation and maturation of SARS‐CoV main protease. Protein Cell. 2011;2(4):282‐290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jin Z, Du X, Xu Y, et al. Structure of Mpro from SARS‐CoV‐2 and discovery of its inhibitors. Nature. 2020;582(7811):289‐293. [DOI] [PubMed] [Google Scholar]
- 17. Zhang L, Lin D, Sun X, et al. Crystal structure of SARS‐CoV‐2 main protease provides a basis for design of improved a‐ketoamide inhibitors. Science. 2020;368(6489):409‐412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Graziano V, McGrath WJ, Yang L, Mangel WF. SARS CoV main proteinase: the monomer‐dimer equilibrium dissociation constant. Biochemistry. 2006;45(49):14632‐14641. [DOI] [PubMed] [Google Scholar]
- 19. Bello M. Prediction of potential inhibitors of the dimeric SARS‐CoV2 main proteinase through the MM/GBSA approach. J Mol Graph Model. 2020;101:107762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Grottesi A, Bešker N, Emerson A, et al. Computational studies of SARS‐CoV‐2 3clpro: insights from md simulations. Int J Mol Sci. 2020;21(15):5346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Amamuddy OS, Verkhivker GM, Bishop ÖT. Impact of early pandemic stage mutations on molecular dynamics of SARS‐CoV‐2 MPro . J Chem Inf Model. 2020;60(10):5080‐5102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ngo ST, Quynh N, Pham A, Le LT, Pham D, Vu VV. Computational determination of potential inhibitors of SARS‐CoV—2 Main protease. J Chem Inf Model. 2020;60(12):5771‐5780. [DOI] [PubMed] [Google Scholar]
- 23. Gahlawat A, Kumar N, Kumar R, Sandhu H, Singh IP, Singh S. Structure‐based virtual screening to discover potential Lead molecules for the SARS‐CoV—2 Main protease. J Chem Inf Model. 2020;60(12):5781‐5793. [DOI] [PubMed] [Google Scholar]
- 24. Kumar S, Sharma PP, Shankar U, et al. Discovery of new Hydroxyethylamine analogs against 3CLproProtein target of SARS‐CoV‐2: molecular docking, molecular dynamics simulation, and structure‐activity relationship studies. J Chem Inf Model. 2020;60(12):5754‐5770. [DOI] [PubMed] [Google Scholar]
- 25. Deeks HM, Walters RK, Barnoud J, Glowacki DR, Mulholland AJ. Interactive molecular dynamics in virtual reality is an effective tool for flexible substrate and inhibitor docking to the SARS‐CoV‐2 Main protease. J Chem Inf Model. 2020;60(12):5803‐5814. [DOI] [PubMed] [Google Scholar]
- 26. Sacco MD, Ma C, Lagarias P, et al. Structure and inhibition of the SARS‐CoV‐2 main protease reveal strategy for developing dual inhibitors against Mpro and cathepsin L. Sci Adv. 2020;6(50):eabe0751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Frecer V, Miertus S. Antiviral agents against COVID‐19: structure‐based design of specific peptidomimetic inhibitors of SARS‐CoV‐2 main protease. RSC Adv. 2020;10(66):40244‐40263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Arafet K, Serrano‐Aparicio N, Lodola A, et al. Mechanism of inhibition of SARS‐CoV‐2 Mpro by N3 peptidyl Michael acceptor explained by QM/MM simulations and design of new derivatives with tunable chemical reactivity. Chem Sci. 2021;12(4):1433‐1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Singh J, Petter RC, Baillie TA, Whitty A. The resurgence of covalent drugs. Nat Rev Drug Discov. 2011;10(4):307‐317. [DOI] [PubMed] [Google Scholar]
- 30. Kalgutkar AS, Dalvie DK. Drug discovery for a new generation of covalent drugs. Expert Opin Drug Discov. 2012;7(7):561‐581. [DOI] [PubMed] [Google Scholar]
- 31. González‐Bello C. Designing irreversible inhibitors ‐ worth the effort? ChemMedChem. 2016;11(1):22‐30. [DOI] [PubMed] [Google Scholar]
- 32. Pillaiyar T, Manickam M, Namasivayam V, Hayashi Y, Jung SH. An overview of severe acute respiratory syndrome‐coronavirus (SARS‐CoV) 3CL protease inhibitors: Peptidomimetics and small molecule chemotherapy. J Med Chem. 2016;59(14):6595‐6628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Aljoundi A, Bjij I, El Rashedy A, Soliman MES. Covalent versus non‐covalent enzyme inhibition: which route should we take? A justification of the good and bad from molecular Modelling perspective. Protein J. 2020;39(2):97‐105. [DOI] [PubMed] [Google Scholar]
- 34. Jena NR. Drug targets, mechanisms of drug action, and therapeutics against SARS‐CoV‐2. Chem Phys Impact. 2021;2:100011. [Google Scholar]
- 35. Dyall J, Coleman CM, Hart BJ, et al. Repurposing of clinically developed drugs for treatment of Middle East respiratory syndrome coronavirus infection. Antimicrob Agents Chemother. 2014;58(8):4885‐4893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Parvathaneni V, Gupta V. Utilizing drug repurposing against COVID‐19—efficacy, limitations, and challenges. Life Sci. 2020;259:118275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Song Y, Zhang M, Yin L, et al. COVID‐19 treatment: close to a cure? A rapid review of pharmacotherapies for the novel coronavirus (SARS‐CoV‐2). Int J Antimicrob Agents. 2020;56(2):106080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fu L, Ye F, Feng Y, et al. Both Boceprevir and GC376 efficaciously inhibit SARS‐CoV‐2 by targeting its main protease. Nat Commun. 2020;11(1):4417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Vuong W, Khan MB, Fischer C, et al. Feline coronavirus drug inhibits the main protease of SARS‐CoV‐2 and blocks virus replication. Nat Commun. 2020;11(1):4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ma C, Sacco MD, Hurst B, et al. Boceprevir, GC‐376, and calpain inhibitors II, XII inhibit SARS‐CoV‐2 viral replication by targeting the viral main protease. Cell Res. 2020;30(8):678‐692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Vatansever EC, Yang KS, Drelich AK, et al. Bepridil is potent against SARS‐CoV‐2 in vitro. PNAS. 2021;118(10):e2012201118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Beigel JH, Tomashek KM, Dodd LE, et al. Remdesivir for the treatment of Covid‐19 — final report. N Engl J Med. 2020. 2020;383:1813–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chen C, Zhang Y, Huang J, et al. Favipiravir versus Arbidol for COVID‐19: a randomized clinical trial. medRxiv. 2020. 10.1101/2020.03.17.20037432. [DOI] [Google Scholar]
- 44. Nicastri E, Petrosillo N, Bartoli TA, et al. National Institute for the infectious diseases “L. Spallanzani”, IRCCS. Recommendations for COVID‐19 clinical management. Infect Dis Rep. 2020;12(1):3‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Horby P, Lim WS, Emberson JR, et al. Dexamethasone in hospitalized patients with Covid‐19. N Engl J Med. 2021;384(8):693‐704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hoffmann M, Schroeder S, Kleine‐Weber H, Müller MA, Drosten C, Pöhlmann S. Nafamostat mesylate blocks activation of SARS‐CoV‐2: new treatment option for COVID‐19. Antimicrob Agents Chemother. 2020;64(6):e00754‐e00720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. López‐Medina E, López P, Hurtado IC, et al. Effect of Ivermectin on time to resolution of symptoms among adults with mild COVID‐19: a randomized clinical trial. JAMA. 2021;325(14):1426‐1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Günther S, Reinke PYA, Fernández‐Garciá Y, et al. X‐ray screening identifies active site and allosteric inhibitors of SARS‐CoV‐2 main protease. Science. 2021;372(6542):642‐646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Douangamath A, Fearon D, Gehrtz P, et al. Crystallographic and electrophilic fragment screening of the SARS‐CoV‐2 main protease. Nat Commun. 2020;11(1):5047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Del Sol A, Tsai CJ, Ma B, Nussinov R. The origin of allosteric functional modulation: multiple pre‐existing pathways. Structure. 2009;17(8):1042‐1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ramirez UD, Myachina F, Stith L, Jaffe EK. Docking to large allosteric binding sites on protein surfaces. Adv Exp Med Biol. 2010;680:481‐488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Guarnera E, Berezovsky IN. Allosteric sites: remote control in regulation of protein activity. Curr Opin Struct Biol. 2016;37:1‐8. [DOI] [PubMed] [Google Scholar]
- 53. Meng X‐Y, Zhang H‐X, Mezei M, Cui M. Molecular docking: a powerful approach for structure‐based drug discovery. Curr Comput Aided‐Drug des. 2011;7(2):146‐157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Case DA, Betz RM, Cerutti DS, et al. AMBER 2016. San Francisco: University of California; 2016. [Google Scholar]
- 55. Hou T, Wang J, Li Y, Wang W. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J Chem Inf Model. 2011;51(1):69‐82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Haliloglu T, Bahar I, Erman B. Gaussian dynamics of folded proteins. Phys Rev Lett. 1997;79(16):3090‐3093. [Google Scholar]
- 57. Bahar I, Lezon TR, Yang LW, Eyal E. Global dynamics of proteins: bridging between structure and function. Annu Rev Biophys. 2010;39:23‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ozkan SB, Dalgýn GS, Haliloglu T. Unfolding events of chymotrypsin inhibitor 2 (CI2) revealed by Monte Carlo (MC) simulations and their consistency from structure‐based analysis of conformations. Polymer. 2004;45(2):581‐595. [Google Scholar]
- 59. Ozbek P, Soner S, Haliloglu T. Hot spots in a network of functional sites. PLoS One. 2013;8(9):e74320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ozbek P, Soner S, Erman B, Haliloglu T. DNABINDPROT: fluctuation‐based predictor of DNA‐binding residues within a network of interacting residues. Nucleic Acids Res. 2010;38:W417‐W423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Brinda KV, Vishveshwara S. A network representation of protein structures: implications for protein stability. Biophys J. 2005;89(6):4159‐4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kürkçüoğlu Ö. Exploring allosteric communication in multiple states of the bacterial ribosome using residue network analysis. Turkish J Biol. 2018;42(5):392‐404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Fokas AS, Cole DJ, Ahnert SE, Chin AW. Residue geometry networks: a rigidity‐based approach to the amino acid network and evolutionary rate analysis. Sci Rep. 2016;6(1):1‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Guzel P, Kurkcuoglu O. Identification of potential allosteric communication pathways between functional sites of the bacterial ribosome by graph and elastic network models. Biochim Biophys Acta ‐ Gen Subj. 2017;1861(12):3131‐3141. [DOI] [PubMed] [Google Scholar]
- 65. Olsson MHM, SØndergaard CR, Rostkowski M, Jensen JH. PROPKA3: consistent treatment of internal and surface residues in empirical pKa predictions. J Chem Theory Comput. 2011;7(2):525‐537. [DOI] [PubMed] [Google Scholar]
- 66. Irwin JJ, Shoichet BK. ZINC ‐ a free database of commercially available compounds for virtual screening. J Chem Inf Model. 2005;45(1):177‐182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wishart DS, Knox C, Guo AC, et al. DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006;34:D668‐D672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Frisch MJ, Trucks GW, Schlegel HB, et al. Gaussian 09, Revision B.01. 2009. [Google Scholar]
- 69. Stewart JJP. Optimization of parameters for semiempirical methods I Method. J Comput Chem. 1989;10(2):221‐264. [Google Scholar]
- 70. O'Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. Open babel: an open chemical toolbox. J Chem. 2011;3(33):1‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2009;31(2):174‐182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Discovery Studio Visualizer . Dassault Systèmes BIOVIA, San Diego; 2020.
- 73. Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general Amber force field. J Comput Chem. 2004;25(9):1157‐1174. [DOI] [PubMed] [Google Scholar]
- 74. Jakalian A, Jack DB, Bayly CI. Fast, efficient generation of high‐quality atomic charges. AM1‐BCC model: II. Parameterization and validation. J Comput Chem. 2002;23(16):1623‐1641. [DOI] [PubMed] [Google Scholar]
- 75. Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, Simmerling C. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J Chem Theory Comput. 2015;11(8):3696‐3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79(2):926‐935. [Google Scholar]
- 77. Darden T, York D, Pedersen L. Particle mesh Ewald: an N·log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98(12):10089‐10092. [Google Scholar]
- 78. Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n‐alkanes. J Comput Phys. 1977;23(3):327‐341. [Google Scholar]
- 79. Uberuaga BP, Anghel M, Voter AF. Synchronization of trajectories in canonical molecular‐dynamics simulations: observation, explanation, and exploitation. J Chem Phys. 2004;120(14):6363‐6374. [DOI] [PubMed] [Google Scholar]
- 80. Miller BR, McGee TD, Swails JM, Homeyer N, Gohlke H, Roitberg AE. MMPBSA.Py: an efficient program for end‐state free energy calculations. J Chem Theory Comput. 2012;8(9):3314‐3321. [DOI] [PubMed] [Google Scholar]
- 81. Cheng YS, Williamson PR, Zheng W. Improving therapy of severe infections through drug repurposing of synergistic combinations. Curr Opin Pharmacol. 2019;48:92‐98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Pandey A, Nikam AN, Shreya AB, et al. Potential therapeutic targets for combating SARS‐CoV‐2: drug repurposing, clinical trials and recent advancements. Life Sci. 2020;256:117883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Kurkcuoglu O, Gunes MU, Haliloglu T. Local and global motions underlying antibiotic binding in bacterial ribosome. J Chem Inf Model. 2020;60(12):6447‐6461. [DOI] [PubMed] [Google Scholar]
- 84. Bello M, Martínez‐Muñoz A, Balbuena‐Rebolledo I. Identification of saquinavir as a potent inhibitor of dimeric SARS‐CoV2 main protease through MM/GBSA. J Mol Model. 2020;26(12):1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Arun KG, Sharanya CS, Abhithaj J, Francis D, Sadasivan C. Drug repurposing against SARS‐CoV‐2 using E‐pharmacophore based virtual screening, molecular docking and molecular dynamics with main protease as the target drug repurposing against SARS‐CoV‐2 using E‐pharmacophore based virtual screening, molecular doc. J Biomol Struct Dyn. 2020;1‐12. 10.1080/07391102.2020.1779819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Nutho B, Hengphasatporn K, Pattaranggoon NC. Why are Lopinavir and ritonavir effective against the newly emerged coronavirus 2019? Atomistic insights into the inhibitory mechanisms. Biochemistry. 2020;59(18):1769‐1779. [DOI] [PubMed] [Google Scholar]
- 87. Świderek K, Moliner V. Revealing the molecular mechanisms of proteolysis of SARS‐CoV‐2 Mproby QM/MM computational methods. Chem Sci. 2020;11(39):10626‐10630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Kadioglu O, Saeed M, Greten HJ, Efferth T. Identification of novel compounds against three targets of SARS CoV2 coronavirus by combined virtual screening and supervised machine learning. Comput Biol Med. 2021;133:104359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Coppola M, Mondola R. Phytotherapeutics and SARS‐CoV‐2 infection: potential role of bioflavonoids. Med Hypotheses. 2020;140:109766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Hu X, Cai X, Song X, et al. Possible SARS‐coronavirus 2 inhibitor revealed by simulated molecular docking to viral main protease and host toll‐like receptor. Future Virol. 2020;15(6):359‐368. [Google Scholar]
- 91. Alamri MA, Tahir Ul Qamar M, Mirza MU, et al. Pharmacoinformatics and molecular dynamics simulation studies reveal potential covalent and FDA‐approved inhibitors of SARS‐CoV‐2 main protease 3CLpro . J Biomol Struct Dyn. 2020;1‐13. 10.1080/07391102.2020.1782768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Caly L, Druce JD, Catton MG, Jans DA, Wagstaff KM. The FDA‐approved drug ivermectin inhibits the replication of SARS‐CoV‐2 in vitro. Antiviral Res. 2020;178:104787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Vishal M, Pravin D, Himani G, Nilam V, Urvisha B, Rajesh P. Drug repurposing of approved drugs Elbasvir, Ledipasvir, Paritaprevir, Velpatasvir, Antrafenine and ergotamine for combating COVID19. Cambridge Open Engage (preprint). 2020. 10.26434/chemrxiv.12115251.v2. [DOI] [Google Scholar]
- 94. Jeon S, Ko M, Lee J, et al. Identification of antiviral drug candidates against SARS‐CoV‐2 from FDA‐approved drugs. Antimicrob Agents Chemother. 2020;64(7). https://doi.org/10.1128/AAC.00819‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Cagno V, Magliocco G, Tapparel C, Daali Y. The tyrosine kinase inhibitor nilotinib inhibits SARS‐CoV‐2 in vitro. Basic Clin Pharmacol Toxicol. 2021;128(4):621‐624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Haggag YA, El‐Ashmawy NE, Okasha KM. Is hesperidin essential for prophylaxis and treatment of COVID‐19 infection? Med Hypotheses. 2020;144:109957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Aboul‐Enein HY. A spectrophotometric method for Diosmin determination. Open Chem Biomed Methods J. 2011;3(1):123‐127. [Google Scholar]
- 98. Arakawa T, Yamasaki H, Ikeda K, Ejima D, Naito T, Koyama A. Antiviral and Virucidal activities of natural products. Curr Med Chem. 2009;16(20):2485‐2497. [DOI] [PubMed] [Google Scholar]
- 99. Lin MV, Chung R. Recent FDA approval of sofosbuvir and simeprevir. Implications for current HCV treatment. Clin Liver Dis. 2014;3(3):65‐68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Chan JFW, Chik KKH, Yuan S, et al. Novel antiviral activity and mechanism of bromocriptine as a Zika virus NS2B‐NS3 protease inhibitor. Antiviral Res. 2017;141:29‐37. [DOI] [PubMed] [Google Scholar]
- 101. Kato F, Ishida Y, Oishi S, et al. Novel antiviral activity of bromocriptine against dengue virus replication. Antiviral Res. 2016;131:141‐147. [DOI] [PubMed] [Google Scholar]
- 102. Seo EJ, Sugimoto Y, Greten HJ, Efferth T. Repurposing of bromocriptine for cancer therapy. Front Pharmacol. 2018;9:1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Canga AG, Prieto AMS, Diez Liébana MJ, Martínez NF, Sierra Vega M, García Vieitez JJ. The pharmacokinetics and interactions of ivermectin in humans ‐ a mini‐review. AAPS J. 2008;10(1):42‐46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. González‐paz LA, Lossada CA, Moncayo LS, et al. Molecular docking and molecular dynamic study of two viral proteins associated with SARS‐CoV‐2 with Ivermectin. Preprint. 10.20944/preprints202004.0334.v1. [DOI] [Google Scholar]
- 105. Tiwari S, Jain R, Banerjee I. Repurposing of FDA approved drugs targeting Main protease MPro for SARS‐CoV‐2. Preprint. 10.21203/rs.3.rs-72576/v1. [DOI] [Google Scholar]
- 106. Nguyenla X, Wehri E, van Dis E, et al. Discovery of SARS‐CoV‐2 antiviral synergy between remdesivir and approved drugs in human lung cells. bioRxiv. 2020. 10.1101/2020.09.18.302398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Ma C, Hu Y, Townsend JA, et al. Ebselen, Disulfiram, Carmofur, PX‐12, Tideglusib, and Shikonin are nonspecific promiscuous SARS‐CoV‐2 Main protease inhibitors. ACS Pharmacol Transl Sci. 2020;3(6):1265‐1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Ma C, Wang J. Dipyridamole, chloroquine, montelukast sodium, candesartan, oxytetracycline, and atazanavir are not SARS‐CoV‐2 main protease inhibitors. PNAS. 2021;118(8):e2024420118. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supporting Information
Video S1
Video S2
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article or its supplementary material.