

Abstract
Heparan sulfate (HS) is a complex, polyanionic polysaccharide ubiquitously expressed on cell surfaces and in the extracellular matrix. HS interacts with numerous proteins to mediate a vast array of biological and pathological processes. Inhibition of HS‐protein interactions is thus an attractive approach for new therapeutic development for cancer and infectious diseases, including COVID‐19; however, synthesis of well‐defined native HS oligosaccharides remains challenging. This has aroused significant interest in the development of HS mimetics which are more synthetically tractable and have fewer side effects, such as undesired anticoagulant activity. This account provides a perspective on the design and synthesis of different classes of HS mimetics with useful properties, and the development of various assays and molecular modelling tools to progress our understanding of their interactions with HS‐binding proteins.
Keywords: heparan sulfate, heparanase, mimetics, cancer, SARS-CoV-2
This personal account summarizes our work on the design and synthesis of new classes of heparan sulfate (HS) mimetics as potential therapeutics for various diseases including cancer and viral infections. The development of new assays and molecular modelling tools for probing the interactions of HS‐binding proteins with their ligands is also discussed.

1. Introduction
1.1. Heparan Sulfate (HS)
Heparan sulfate (HS) is a highly sulfated linear polysaccharide (a glycosaminoglycan, or GAG) found in the extracellular matrix (ECM) and on the mammalian cell surface. [1] It typically occurs as a HS proteoglycan where two to three HS chains are conjugated to a protein. HS interacts with a vast array of extracellular proteins to regulate their biological activity. [2] These include proteins associated with normal physiological functions, e. g., growth and development, cell to cell communication etc, as well as those that mediate numerous pathological processes such as cancer, inflammation and viral infections. Structurally, HS is a repeating linear copolymer of d‐glucosamine and a uronic acid (Figure 1), either β‐d‐glucuronic acid (GlcA) or α‐l‐iduronic acid (IdoA). [3] HS chains are composed of highly sulfated “S” domains made up of sulfated disaccharide sequences rich in IdoA and spacer domains made up of the non‐sulfated disaccharide sequence. Short mixed sequences of low sulfation also occur between these domains. HS is structurally related to heparin, an anticoagulant drug in clinical use for decades. [4] Heparin contains a specific antithrombin (AT)‐binding pentasaccharide sequence that induces potent anticoagulant activity via inhibition of FXa. Heparin is also more highly sulfated and has a higher proportion of IdoA than HS, but the latter has a more varied structure (Figure 1).
Figure 1.

Structure of heparan sulfate and heparin.
An important structural feature of HS and heparin is the presence of IdoA. This monosaccharide is conformationally flexible and exists in equilibrium between the 1 C 4 and 4 C 1 chair conformations and the 2 S O skew‐boat conformation in solution (Figure 2). [5] The position of the conformational equilibrium is influenced by 2‐O‐sulfation of IdoA and 3‐O‐ and 6‐O‐sulfation on neighbouring residues. [6] The IdoA and IdoA2S residues flip between these conformations to induce kinks or bends in the HS chain to facilitate binding to their target proteins. Due to their important roles in numerous biological and pathological processes, there is great interest in unravelling the molecular details of these HS‐protein interactions to develop new therapeutics. [7] However, the structural heterogeneity of HS and the difficulty in accessing pure, chemically well‐defined HS oligosaccharides means that such structure‐activity relationships are poorly understood. Unfortunately, IdoA and l‐idose both have limited commercial availability. The synthesis of defined HS oligosaccharides is, therefore, the subject of intensive ongoing research (for recent reviews, see refs. [8] ). Despite recent progress, [9] a major challenge in the synthesis of HS oligosaccharides is the efficient gram‐scale preparation of IdoA building blocks and their incorporation into HS oligosaccharides.
Figure 2.

Conformational equilibrium of IdoA2S.
Compounds that mimic the essential structural features of HS and are thus capable of blocking HS‐protein interactions, termed HS (or heparin) mimetics, represent a viable alternative to defined HS oligosaccharides for the development of potential therapeutics. [10] HS mimetics typically do not contain IdoA or other complex building blocks and so are easier to synthesize than native HS oligosaccharides. There is strong interest in HS/heparin mimetics as anticoagulants, [10b] but when targeting other diseases such anticoagulant activity is often an unwanted side effect. However, HS mimetics are often amenable to various modifications to minimize these side effects and/or to improve other properties such as their pharmacokinetics. This account describes our efforts to design and synthesize HS mimetics of different classes for various applications, highlighted by the discovery of amphiphilic HS mimetics, exemplified by pixatimod (PG545), which possess many useful properties. We also discuss new assay development and molecular modelling tools we have developed to study the interactions of HS/heparin and their mimetics with target proteins.
Many of the compounds synthesized, including pixatimod, have been targeted towards heparanase, an enzyme that degrades HS and is intimately involved in cancer, inflammation and viral infections.
1.2. Heparanase
For a comprehensive overview of heparanase, the reader is referred to the recently published Volume 1221 of Advances in Experimental Medicine and Biology entitled “Heparanase: from basic research to clinical applications.” [11] Heparanase is an endo‐β‐glucuronidase responsible for the degradation of HS. It is also the only known mammalian endoglycosidase that can cleave HS chains, whether they are attached to proteoglycans or not. Since the discovery of heparanase in the mid 1970’s, it has been associated with a multitude of cellular functions. [12] Some of the functions dependent on heparanase enzymatic activity include HS turnover, involvement in cell invasion, involvement in the release of ECM‐bound proteins and functioning as the facilitator for the spread of HS‐binding viruses. Heparanase also has non‐enzymatic functions including acting as a cell adhesion molecule, functioning as a promoter of signal transduction and as a transcription factor.
Heparanase is the sole endoglycosidase that degrades HS and is expressed by malignant and normal cells, with activity recognised in numerous tumours, platelets, and tissues. Degradation of HS by heparanase leads to metastasis, which is the spread of cancer cells from the primary site of cancer to a secondary site within the body through neoangiogenesis and tumour cell invasion of the basement membrane and ECM. [12] Heparanase is involved in the regulation and bioavailability of HS‐binding growth factors such as fibroblast growth factor (FGF), vascular endothelial growth factor (VEGF), epidermal growth factor (EGF), hepatocyte growth factor (HGF) and cytokines which promote angiogenesis, a fundamental step in tumour development by stimulating the growth of new blood vessels from the pre‐existing vasculature. The development of heparanase inhibitors [13] or HS mimetics [14] that can block HS‐mediated growth factor activity leading to metastasis, tumour growth and angiogenesis is thus an important strategy for cancer treatment and for inflammatory diseases.
1.3. HS in Viral Infections
The role of HS as a major attachment co‐receptor on the cell surface for many viruses is well established, including herpes simplex virus (HSV), [15] respiratory syncytial virus (RSV), [16] dengue virus (DENV), [17] varicella zoster virus, [18] human immunodeficiency virus (HIV), [19] Epstein‐Barr virus, [20] vaccinia virus, [21] hepatitis C virus [22] and many others. Of particular relevance during the current COVID‐19 pandemic is the direct binding of HS proteoglycans together with co‐receptor Angiotensin‐converting enzyme 2 (ACE2) to facilitate the attachment of spike‐bearing viral particles of SARS‐CoV and SARS‐CoV‐2 to the cell surface to promote viral entry. [23] Molecular studies have shown that the receptor‐binding domain (RBD) in the S1 subunit of the spike glycoprotein mediates binding of SARS‐CoV‐2 to cell surface HS (Figure 3). [23a] Heparin and other HS mimetics have been extensively investigated and have been shown to block infectivity and cell‐to‐cell spread in a multitude of different viruses, [24] including SARS‐CoV‐2 (see below). [25]
Figure 3.
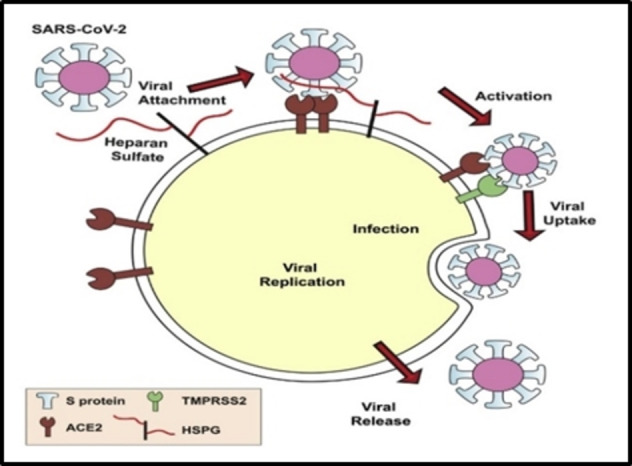
SARS‐CoV‐2 entry mechanism (SARS‐CoV‐2 uses HS and ACE‐2 receptor on the cell surface for entry into the cell). [23a] Reproduced from Ref. [23a] with permission from Cell Press.
Along with HS, heparanase also plays a key role in viral infections. [26] Heparanase expression is upregulated in many viruses and drives the release of progeny virions by cleavage of cell surface HS, although the precise details remain to be elucidated.[ 26 , 27 ]
2. Heparin/Heparan Sulfate Mimetics
The following sections are focussed on our efforts over the years to design and synthesize HS mimetics of different structural classes targeted at a variety of diseases. There have been many contributions from various other labs to develop heparin/HS mimetics of various types, including derivatives of native HS sequences, [28] “unnatural” HS sequences, [29] glycopolymers, [30] and non‐carbohydrate small molecules, [10a] to name a few. It is beyond the scope of this account to discuss these in detail. Instead we direct the reader to some recent pertinent original contributions[ 28 , 29 , 30 ] and reviews. [10]
2.1. Mono‐ and Disaccharide Mimetics Targeted at HS‐Binding Growth Factors
As discussed above, HS is an important player in angiogenesis, which is initiated by the formation of a ternary complex between a growth factor, its receptor and HS. The interactions of FGF‐1 and FGF‐2 with HS and their receptors have been particularly well studied using X‐ray crystallography. [31] Analysis of the bound oligosaccharides in the structures with these growth factors revealed that disaccharide 1 (Figure 4) represents a minimal heparin/HS consensus sequence for FGF binding. [32] We designed simple, non‐HS disaccharides to mimic the essential features of 1, namely the α(1→4) linkage between the two monosaccharide units and the spatial orientation of the two sulfate groups which make contacts with the protein (i. e., GlcN2S and IdoA2S). The first series, [32] exemplified by 2 and 3, also took advantage of the known conformational flexibility of fully sulfated β‐d‐glucose or β‐d‐xylose, [33] using these moieties as surrogates for the flexible IdoA residue. A second series (e. g., 4, 5) utilized a 1,6‐anhydrosugar to mimic the 1 C 4 conformation of bound IdoA. [34] Docking calculations showed that the predicted locations of sulfate groups in the binding site of FGF‐1 were consistent with those observed for co‐crystalized heparin oligosaccharides and correlated with binding affinity (K d) values obtained from SPR binding assays (22 μM to 1.4 mM).
Figure 4.

Structures of minimal HS disaccharide consensus sequence for FGF binding (1) and synthetic disaccharide mimetics (2–5).
We have also investigated combinatorial approaches to small molecule HS mimetics whereby the two key sulfate groups are located on the same monosaccharide scaffold rather than on adjacent monosaccharide moieties in a disaccharide. For example, we utilized the four‐component Ugi condensation to prepare mimetics based on a polysulfated mannose core or two polysulfated mannoses linked by a spacer (e. g., 6–9, Figure 5). [35]
Figure 5.

Structures of HS mimetics. R=SO3Na.
The sulfates serve to anchor the compound to the cationic HS binding site, while the diverse array of functionality introduced by the Ugi condensation probe for adjacent favourable, non‐ionic binding interactions. Similarly, an azide‐functionalized disulfated mannose template was diversified via the Cu(I)‐catalyzed Huisgen azide‐alkyne cycloaddition reaction (e. g., 10), with further extension possible via a Swern oxidation‐Wittig reaction sequence. [36]
The above compounds bind to FGF‐1, ‐2 and VEGF with a range of K d values from μM to mM, with some selectivity observed for the different proteins depending on the type of hydrophobic substitutions. We have also shown that non‐carbohydrate scaffolds can function as HS mimetics. For example, a series of low molecular weight HS mimetics was prepared which comprised a monocyclitol derivatized with a cis‐1,2‐disulfate and various lipophilic and/or heterocyclic groups (e. g., 11–13, Figure 5). [37] The compounds were screened against HSV, a virus that uses HS as an entry receptor, and inhibited infectivity at concentrations approximately 100 times lower than those toxic for cultured cells. The compounds interfered with HSV‐1 attachment to cells and efficiently reduced the cell‐to‐cell spread of the virus. Furthermore, the most potent compounds (11–13) also inactivated the virus particles, perhaps due to their surfactant‐like properties.
2.2. Polyanionic Non‐Carbohydrate HS Mimetics
We have also explored highly sulfated linked cyclitols as non‐carbohydrate HS mimetics. For example, a series of cyclitols with the general structure 14 (Figure 6) were prepared which consisted of two highly sulfated N‐linked bis‐cyclitols coupled by linkers of variable chain length, flexibility, orientation and hydrophobicity, with variations in sulfation also being introduced into some molecules. [38] These compounds were tested against 10 functionally diverse HS‐binding proteins. It was found that there were significant differences in the binding/inhibitory activity for each protein, depending on the nature of the linker and thus the presentation of sulfates.
Figure 6.

Non‐carbohydrate HS mimetics based on sulfated linked cyclitols and RAFT polymers.
Another approach to new HS mimetics we have explored recently utilizes reversible addition‐fragmentation chain transfer (RAFT) polymerization of commercially available, water‐soluble anionic monomers (Figure 6). [39] The RAFT methodology was chosen because reactions can be performed in water and because it offers control of predetermined molecular weight with high monomer conversion and narrow dispersity (Ð). Homopolymers were prepared of sulfonated monomers sodium 4‐styrene sulfonate (SS), sodium‐2‐acrylamido‐2‐methyl‐1‐propane sulfonate (AMPS), potassium‐3‐sulfopropyl acrylate (SPA) and potassium‐3‐sulfopropyl methacrylate (SPMA) with MW range from 5 kDa to 50 kDa. In addition, random copolymers of varying molar ratios of two different sulfonated monomers, [40] or of a sulfonated monomer and acrylic acid (AA) were also prepared, [39] the latter to mimic both types of anionic groups found in HS. Sulfonated homo‐ and copolymers displayed significant anticoagulant activity in APTT, TCT, and thrombin generation assays.
In addition, copolymers of SS and AA, ie., poly(SS‐co‐AA), displayed unprecedented anti‐factor IIa activity. The polymers were subsequently investigated in vitro for their capacities to interfere with tumor cell metastasis. [41] The most promising compounds were poly(SS) and poly(SS‐co‐AA, 1 : 1) (Figure 6), which efficiently attenuated cancer cell‐induced coagulation, and thus platelet activation and degranulation, with similar or slightly better potency than low molecular weight heparin (LMWH), the drug typically used for antithrombotic treatment of cancer patients. Furthermore, they potently inhibited heparanase and bound P‐selectin and the integrin VLA‐4 similar to, or even better than heparin, and thus efficiently blocked melanoma cell binding to endothelium under blood flow conditions. [41]
2.3. Polysulfated Oligosaccharides: PI‐88 (Muparfostat)
Early efforts towards heparanase inhibitors were centred upon the use of modified heparins and sulfated polysaccharides, [42] but due to their high anticoagulant activity they were unsuitable inhibitors for clinical applications. Later, Parish and co‐workers evaluated a library of sulfated (non‐HS) oligosaccharides, which were weaker anticoagulants, for their heparanase inhibition and antiangiogenic activity. [43] It was discovered that chain length and degree of sulfation is an important factor for good inhibitory activity where oligosaccharides with five or more monosaccharide units were found to be most active. PI‐88 (aka muparfostat) displayed antiangiogenic and antimetastatic activity and was thus identified as a potential antitumor drug. [44] PI‐88 was the first of four heparanase inhibitors to progress to the clinic [45] and eventually was evaluated in phase III clinical trials for post‐resection hepatocellular carcinoma, however, it did not meet its primary endpoint and was not approved for use. [46] The other three clinical heparanase inhibitors are pixatimod (PG545, see below) and the chemically modified heparin derivatives roneparstat (SST0001) and necuparanib (M402) (for recent reviews see refs.[ 13 , 14 ]).
PI‐88 is a complex mixture of monophosphorylated polysulfated mannose oligosaccharides, represented in most publications as 15 (Figure 7A), and is prepared by exhaustive sulfation of the oligosaccharide phosphate fraction produced by diploid yeast Pichia holstii NRRL Y‐2448. [47] The major components of PI‐88 are the α‐(1→3)/ α‐(1→2)‐linked penta‐ 16 (∼60 %) and tetrasaccharide 17 (30 %). [48] More recent, detailed NMR structural studies of PI‐88 revealed the presence of minor amounts of all α‐(1→3)‐linked mannoses with terminal α‐(1→3)‐linkages, together with terminal α‐(1→2)‐linked residues and the presence of only α‐(1→3)‐linked disaccharide. [49] Thus, the structure of PI‐88 is best presented as 18 (Figure 7B).
Figure 7.

A. Depiction of structures of PI‐88 mixture (15) and the major components, pentasaccharide 16 and tetrasaccharide 17. B. More complete depiction of the structure of PI‐88 (18) including the presence of minor isomers.
The synthesis of individual PI‐88‐related oligosaccharides (19–22, Figure 8) was achieved by isolation/purification of dephosphorylated α‐(1→3)/α‐(1→2)‐linked manno‐oligosaccharides followed by sulfation. Evaluation of their angiogenic growth factor binding showed that the replacement of the phosphate group with sulfate did not alter the activity. [50] Subsequently these oligosaccharides were synthesized from monosaccharide building blocks using a “1+1” iterative strategy. [51] The tetra‐ and pentasaccharides (21, 22) proved to be potent heparanase inhibitors, whereas tri‐ or disaccharides only displayed partial inhibition.
Figure 8.

Structures of synthetic oligomannosides containing the α‐(1→3)/α‐(1→2)‐linked pentasaccharide backbone of the major component of PI‐88. R=SO3Na.
PI‐88 was evaluated in many preclinical studies to support its progression through the clinic as an anticancer agent (for reviews see refs.[ 44 , 45 ]). PI‐88 also displayed other interesting biological activities associated with its mimicry of HS, e. g., anti‐inflammatory activity and antiviral activity against viruses such as HSV. [27]
The discovery of PI‐88 as a potent heparanase inhibitor and its extensive evaluation in non‐clinical and clinical studies paved the way for discovering next‐generation HS mimetics and heparanase inhibitors with improved properties. A series of fully sulfated PI‐88 analogues (the “PG500” series) were synthesized based on the pentamannoside backbone of PI‐88. [52] Several modifications were made at the reducing end with simple aglycones such as benzyl (23, PG500) and n‐octyl (24, PG501) to alter the pharmacokinetic properties but maintaining the biological activity (Figure 8).
The syntheses were carried out via glycosylation of acetylated pentasaccharide acquired from the PI‐88 manufacturing process followed by deprotection and sulfation. Alternatively, the oligosaccharides could be synthesized from monosaccharide building blocks via a “1+1” iterative strategy. [53]
These analogues bound to FGF‐1, FGF‐2, and VEGF in similar manner to PI‐88 but displayed improved pharmacokinetics. In addition, they were able to inhibit heparanase and cell‐to‐cell spread of HSV‐1 where 23 and 24 showed almost equivalent IC50 values of ∼1–2 μM to PI‐88. In vivo mouse studies of 23 and 24 along with PI‐88 were carried out using the AngioChamber and Angiosponge assays which showed significant inhibition of in vivo angiogenesis. [44a] Subsequently, further analogues were synthesized which introduced lipophilic modifications at the reducing end of the oligosaccharide with different sugar chain length. [54] Pentasaccharide 24 and its tetrasaccharide homologue 25 (PG518) showed good antitumor activity and reduced anticoagulant activity in vivo in a B16 mouse melanoma model which is resistant to PI‐88. In addition, tetrasaccharide 25 displayed better pharmacokinetics in rats in comparison to pentasaccharide 24. [54]
2.4. Discovery of Pixatimod (PG545)
The above modifications of sulfated oligosaccharides which provided compounds with improved pharmacokinetic properties prompted further exploration of lipophilic group modifications. Replacement of small aglycones at the reducing end with significantly more lipophilic groups such as stearamidopropyl and cholestanol resulted in compounds which displayed significantly more potent in vitro and in vivo antiangiogenic and anticancer activity in comparison to the earlier compounds (e. g., 26–27, Figure 8). [55] The carbohydrate backbone was then modified from oligomannoside to oligoglucoside (e. g., 28–30, Figure 9)[ 44b , 55b ] to take advantage of commercially available and inexpensive maltotetraose as the starting material.
Figure 9.

Structures of selected PG500 series compounds with a maltotetraose backbone, including PG545 (28). R=SO3Na.
PG500 series compounds with large lipophilic groups also showed reduced anticoagulant activity, a common side effect of unmodified HS mimetics such as PI‐88. This work ultimately led to the discovery of the clinical candidate PG545 (28), a fully sulfated tetrasaccharide glycoside comprising maltotetraose β‐linked to cholestanol.[ 44b , 55b ] PG545 has been investigated in multiple preclinical studies and has shown potent in vivo efficacy in angiogenesis, solid tumour and metastasis models (for reviews see refs.[ 44b , 56 ]).
These studies also showed that PG545 has a long half‐life compared to other HS mimetics, and thus requires less frequent administration. In addition, it has been discovered that PG545 has immunomodulatory activity [57] which likely contributes to its increased potency. Recently, the results of a phase I study of PG545 for advanced solid tumors were reported. [58] Data from this study support the proposed mechanism of action. Further, its low toxicity made PG545 an attractive candidate for combination clinical trials. PG545 is currently in phase Ib clinical trials in patients with advanced cancer/pancreatic adenocarcinoma in combination with the immune checkpoint inhibitor nivolumab (Opdivo®). [58]
While originally developed for cancer, PG545 has also demonstrated significant antiviral activity against a number of viruses that use HS as an entry receptor with EC50 values ranging from 0.06 to 14 μg/mL, and with low toxicity and high selectivity indices. Activity has been displayed against HSV‐2, [59] HIV, [60] RSV, [61] Ross River virus, [62] Barmah Forest virus, [62] chikungunya virus (CHIKV), [62] and DENV. [63] The antiviral activity against DENV is noteworthy because PG545 inhibits both the Envelope (E) and non‐structural protein 1 (NS1), representing the first identification of a dual targeting drug against DENV infection and NS1 toxicity. The inhibition of cell‐to‐cell spread by PG545 may be linked to its potent inhibition of heparanase, which is known to promote the escape and spread of progeny virions of HS‐binding viruses.[ 29 , 30 ] In addition to blocking virus infectivity, PG545 has also been shown to possess virucidal activity, a unique feature only found in this particular class of amphiphilic HS mimetic. The virucidal activity is due to disruption of the viral lipid envelope [64] by the lipophilic steroid side chain of PG545. In vivo efficacy has been demonstrated in a prophylactic mouse HSV‐2 genital infection model, [64] a prophylactic Ross River virus mouse model, [62] and a therapeutic DENV mouse model. [63] More recently, PG545 has been shown to bind directly to the SARS‐CoV‐2 spike protein receptor binding domain (S1‐RBD), altering its conformation and destabilizing its structure. [65] Molecular modelling identified a binding site overlapping with the ACE2 receptor site (see below). Assays with four different clinical isolates of live SARS‐CoV‐2 virus showed that PG545 potently inhibits infection of Vero cells at doses well within its safe therapeutic dose range. In the transgenic hACE2 mouse model, PG545‐treated animals showed a significant reduction in viral titers in nasal turbinates and in the brain. This demonstration of potent anti‐SARS‐CoV‐2 activity establishes that synthetic HS mimetics can target the HS‐Spike protein‐ACE2 axis. Taken together with other known activities of PG545, these studies provide a strong rationale for further investigation as a potential multimodal therapeutic to address the COVID‐19 pandemic. [65]
3. Tools for Studying HS‐Protein Interactions
3.1. Heparanase Assays
While heparanase has been a recognized therapeutic target since the 1980’s, progress in heparanase research has been relatively slow due to the lack of suitable assays for enzymatic activity. These are largely exacerbated by the complex, heterogeneous nature of the HS substrate. Much effort has been expended over the years in developing a wide variety of assays, however, despite the commercialization of some kits, no single, preferred method has emerged. [66] One of the more common, simpler assays utilizes fondaparinux 31 (Scheme 1), a commercially available anticoagulant drug comprising the methyl glycoside of the specific AT‐binding pentasaccharide sequence of heparin. [67] Fondaparinux has a single point of cleavage which upon heparanase treatment liberates disaccharide 32. This process can be monitored by 1H‐NMR [68] and LC/MS, [69] and can be quantified through reaction with water‐soluble dyes which give UV‐ or fluorescence‐detectable products. [70]
Scheme 1.

Cleavage of fondaparinux (31) by heparanase.
These methods have made assaying heparanase simple and robust, however, they are unsuitable for biological samples due to the presence of endogenous interfering compounds. Thus, synthetic HS disaccharides with fluorogenic leaving groups have been explored as heparanase substrates, e. g., 34 (Figure 10). [71]
Figure 10.

Structures of fluorogenic disaccharide substrates for heparanase.
Recently, we reported the synthesis of disaccharide 35 as a fluorogenic heparanase substrate.[ 66 , 72 ] This compound displayed high affinity for the enzyme but was turned over very slowly, meaning that its usefulness for assaying tight‐binding inhibitors is limited. However, this slow turnover enabled, for the first time, the determination of an X‐ray crystal structure of the substrate bound in the active site with the GlcA residue in an activated 1 S 3 conformation (Figure 11). The structure revealed previously unknown interactions involved in enzymatic processing and suggested using substrates with better leaving groups, e. g., halogenated coumarins. Subsequently, Cui and coworkers [73] reported that difluorocoumarins such as 36 are more efficient aglycone leaving groups for heparanase substrates, in line with our hypotheses.
Figure 11.

Crystal structure of disaccharide substrate occupying the heparanase active site cleft with GlcA in a poised 1 S 3 conformation. Reproduced from Ref. [72] with permission from the Royal Society of Chemistry.
3.2. Biosensor Assays
Determining the binding affinity (K d) of a HS mimetic for its target protein is important for identifying compounds with the potential to block disease processes mediated by that particular protein. Surface plasmon resonance (SPR) spectroscopy is an established method for quantifying such biomolecular interactions and has been successfully used to study HS‐protein interactions. [74] Typical SPR experiments involve the immobilization of one of the binding partners onto a sensor chip surface, and in the case of HS‐protein interactions this is commonly heparin [75] which is used as a proxy for HS.
This more closely mimics natural biological systems where HS on the cell surface binds to target proteins as they flow past. However, HS mimetics screening for drug discovery applications requires a high throughput approach that avoids immobilization of each ligand or of the protein of interest. We developed such an assay whereby immobilised heparin is used to measure binding kinetics in solution.[ 50 , 76 ] The principle of this assay is that a solution, at equilibrium, of the protein and a HS mimetic ligand is passed over the heparin‐coated sensor chip. As unbound protein binds to the heparin, an increase in the SPR response is detected, and its concentration can thus be determined.
A decrease in the free protein concentration as a function of ligand concentration enables the calculation of the K d. Importantly, a decrease in binding of the protein to heparin is observed only if the ligand binds to the protein in the HS (heparin)‐binding site. This assay has been used extensively to study most of the HS mimetics discussed herein.
3.3. Quantification of HS
As discussed above, cell surface HS has important roles in biology via interactions with many proteins. HS is also a marker for a number of lysosomal storage diseases, namely the family of mucopolysaccharidosis (MPS) disorders. [77] MPS disorders are caused by genetic defects in lysosomal HS‐degrading enzymes, which result in the accumulation of undegraded HS. The undegraded HS is associated with multiple pathologies in various organs, including the brain. [78]
The quantification of HS in biological samples, e. g., urine, cerebrospinal fluid, tissue samples etc, is thus of great importance for the diagnosis and prognosis of MPS disorders and for assessing the effectiveness of potential therapeutics aimed at clearing the stored HS in the lysosomes. Quantification of HS is complicated by its heterogeneity and high molecular weight. However, acid‐catalysed methanolysis [79] or butanolysis [80] results in desulfated disaccharide cleavage products which can be detected by LC‐MS/MS. Recently, we synthesized a series of HS‐derived disaccharides as standards for both methanolysis [81] and butanolysis [82] of HS (e. g., 37–41, Figure 12). The disaccharides were prepared by the stereoselective 1,2‐cis glycosylation of suitable GlcA and IdoA acceptors with a 2‐deoxy‐2‐azido thioglucoside donor. The standards enabled the identification of the major products from the LC‐MS/MS chromatograms from both methods.
Figure 12.

Selected synthetic disaccharides derived from methanolysis and butanolysis of HS.
Given the butanolysis method is ∼70‐fold more sensitive than methanolysis, we prepared a deuterium‐labelled version of the major disaccharide butanolysis product 38, namely disaccharide 39, as an internal standard for use in the development of a fully quantitative LC‐MS/MS assay. The utility of this assay was demonstrated by its application to the analysis of brain tissue samples from MPS and control mice which revealed significant differences in the regional accumulation of HS in the brains of affected mice. [82]
3.4. Molecular Modelling
Homogeneous HS oligosaccharides with well‐defined sequences cannot be obtained from natural sources, which limits the molecular determination of HS‐protein interactions using experimental techniques such as X‐ray crystallography or NMR. Oligosaccharides derived from heparin are often used as experimental proxies for HS, but these are limited in their size and structural diversity and may not be representative of the full variety of sequences found naturally in HS. As discussed above, significant progress has been made in the total synthesis of defined HS oligosaccharides, however, the time and resources required is still a significant limitation. Understanding the 3D conformational properties of HS‐derived molecules is complicated by their inherent flexibility, which hinders their crystallization. Furthermore, many protein structures are obtained following crystallization under non‐physiological conditions such as low pH, and in the presence of histidine tags and this could result in false positive or multiple HS binding sites on the protein. [83] Structural characterization by NMR has also proven challenging because of the characteristic overlap of NMR resonances in the spectra of oligosaccharides. Molecular modelling approaches, e. g., docking, molecular dynamics (MD) simulations etc, to study HS‐protein interactions are therefore of great importance. However, these too, have their own challenges due to the high molecular weight, negative charge, flexibility of HS chains, large number of torsional angles between glycosidic bonds, lack of clearly defined pockets on the proteins and multivalent binding to adjacent lattice members, and difficulty defining the impact of solvation/desolvation on GAG/protein structure. The latter are compounded by the conformational flexibility of IdoA residues, as discussed above. The groups of Desai, Imberty, Samsanov, Woods and Pisabarro, among others, have developed and applied various modelling techniques to study GAG‐protein interactions, and these have been extensively reviewed recently. [84]
We have recently attempted to address some of these issues by developing a new docking tool, GlycoTorch Vina (GTV), [85] based on the carbohydrate docking program VinaCarb. GTV contains parameters to model sugars in the 2 S O conformation while also supporting GAG specific glycosidic linkages. GlycoTorch tools can be used to prepare input files for molecular docking of GAGs and tutorials are also provided for running and analysing docking results (http://ericboittier.pythonanywhere.com/Tutorials/).
The small molecule (rotatable bonds less than 10) docking programs like AutoDock Vina and commercial docking programs like Glide and GOLD are trained using a large dataset of high‐resolution protein‐ligand complexes to reproduce poses that are within root mean square deviations of ≤2.0 Å relative to the crystal structures. GAGs differ substantially from small molecules. GAGs contain tens of flexible dihedral torsions and have a limited dataset (only 12 crystal structures) that could be used for benchmarking a program. GTV was able to produce pose predictions with RMSD of 3.1 Å from native, validated X‐ray crystal structures of proteins with bound HS oligosaccharides. Therefore, this represents a statistically significant improvement when compared to AutoDock Vina and Glide where the obtained RMSD were 4.5 Å and 5.9 Å, respectively from the native crystal structures.
Two docking programs, AutoDock Vina and GTV, have been used to study the interaction between keratan sulfate (KS) and its non‐sulfated analogues with human adhesion/growth‐regulatory galectins present in the cornea. [86] The results indicated that compared to AutoDock Vina, the binding free energies from GTV were relatively low. To inhibit virus entry into human cells, targeting the receptor recognition motif of SARS‐CoV‐2 can be considered a compelling approach (Figure 13A). Thus, we used sequence‐based bioinformatics analyses and molecular docking using GTV to elucidate molecular details of binding of heparin oligosaccharides with different lengths to spike SARS‐CoV‐2 RBD. [25a] We have identified three binding sites on the spike RBD surface (Figure 13B). The results for the docking revealed that the heparin octasaccharide (consisting of GlcNS6S‐IdoA2S repeats) has favourable affinity for site I (residues T345, R346, N354, R355, K356, R357, I358, S359, L441, D442, K444, N448, Y449, N450, R466 and R509) whereas the same octasaccharide showed unfavourable preference for site III (residues R403, R408, N409, T415, G416, K417, Y421, N501 and Y505 adjacent to ACE2 interface). Docking of a heparin hexasaccharide indicates that site III residues can accommodate heparin fragments equal to or shorter than a hexasaccharide. The obtained docking results confirmed that the binding of heparin fragments to sites I, II and III is length‐dependent. MD simulations of spike SARS‐CoV‐2 RBD in the presence of oligosaccharides were also carried out to monitor the conformational changes in the protein upon binding of the heparin. The β‐sheets maintain the most stable regions of secondary structure, whereas the α‐ and 310 ‐helices, turns and loops connecting the β‐sheets undergo conformational change and thus prevent binding of the ACE2 receptor. Our modelling data complements circular dichroism data showing that heparin binding induces a conformational change in RBD. We have also used GTV and cosolvent molecular dynamics to identify novel heparin and HS binding capable of bridging the N‐terminal domain and furin cleavage site on the heavily glycosylated spike SARS‐CoV‐2 trimer. [87]
Figure 13.

(A) Three‐dimensional structure of the SARS‐CoV‐2 RBD bound to ACE2 (shown in green ribbon). The SARS‐CoV‐2 RBD core is shown in blue and the receptor binding motif is in red. (B) The binding poses of heparin (shown as ball and stick) at sites I, II and III identified by molecular docking using GTV (protein shown in green). (C) The binding poses of PG545 (shown as ball and stick) at sites I and III as identified through blind molecular docking studies. Molecular graphics were prepared with the Protein Imager [88] and UCSF Chimera 1.14.[89] Not all residues are labelled for clarity.
In the absence of known ligand binding sites, we have carried out blind docking (i. e., docking a ligand to the whole surface of a protein) for prediction of interaction between DENV NS1 and PG545 ligand using Swiss‐dock and PatchDock web‐servers followed by molecular dynamics simulations. [63] It was not surprising that molecular dynamics calculations predicted a PG545 binding site spanning multiple NS1 domains. We have not rigorously tested the performance of GTV for blind docking, thus the number of trials and energy evaluations necessary for a blind docking job for heparin or HS mimetics like PG545 remains unknown. We carried out blind docking of PG545 on the whole surface of SARS‐CoV‐2 RBD (PDB: 6LZG) using GTV parameters, i. e., energy range=12, an exhaustiveness=80, chi_cutoff=1, chi_coeff=2 and the number of modes=100. All sulfate and hydroxyl groups, and glycosidic torsion angles of PG545 were treated as flexible. [65] The top ranked and most populated docked poses predicted the tetrasaccharide of PG545 bound to site I and III similar to heparin binding sites on the spike RBD and the cholestanol occupying the hydrophobic patch on the spike RBD (Figure 13C). The cholestanol binds at the receptor binding motif and thus prevents binding of ACE2 receptor from the host cells. [65]
4. Summary and Outlook
HS is a complex polysaccharide ubiquitously expressed on mammalian cells that interacts with a wide variety of proteins to mediate numerous biological and pathological functions, including many of significant biomedical importance, e. g., cancer, inflammation and viral infections. Blocking these HS‐protein interactions with HS/heparin mimetics is a valid approach for developing new therapeutics because they are generally easier to synthesize than native HS oligosaccharides. These studies have been supported by efforts to develop new assays and molecular modelling tools to study the interactions of HS mimetics with their target proteins. A wide variety of synthetic HS mimetics of different structural classes have been synthesized. A unique feature of this approach is the ability to alter the potency and binding selectivity towards specific proteins involved in the disease of interest by making structural modifications, often with non‐anionic functional groups. Interesting examples are the amphiphilic HS mimetics exemplified by PG545 (pixatimod) which are sulfated oligosaccharides with highly lipophilic side chains, e. g., a steroid. The lipophilic functionality confers a range of unique properties such as reduced anticoagulant activity, improved pharmacokinetics and immunomodulatory activity. This useful combination of activities has supported the clinical development of PG545 as an anticancer agent. HS mimetics also show antiviral activity against a range of viruses that use HS as an entry receptor, including SARS‐CoV‐2, and in the case of amphiphilic HS mimetics many display virucidal activity due to disruption of viral lipid envelopes by their large lipophilic side chains. PG545, in particular, has shown promising in vitro and in vivo activity against SARS‐CoV‐2 at therapeutically relevant concentrations. Together with PG545’s other known activities (e. g., anti‐heparanase, anti‐inflammatory), these recent antiviral data[ 63 , 65 ] provide a strong rationale for its clinical investigation as a potential multimodal therapeutic to address the COVID‐19 pandemic and other coronavirus or flavivirus infections.
Biographical Information
Mohit Chhabra was born in 1991 in Haryana, India. He obtained B.Sc. (Honours) in Chemistry from University of Delhi, India in 2013 and M.Sc. (Pharmaceuticals Chemistry) from VIT University, India in 2015. On completion of his master's degree, he worked as a Research Assistant at the City University of Hong Kong for a short term before moving to Australia in 2017 for his PhD. He is currently a PhD candidate in the School of Chemistry and Molecular Biosciences at the University of Queensland in the lab of A/Prof. Vito Ferro and recently submitted his thesis. He has worked on several projects during his research career involving synthesis of small molecules, carbohydrates and peptides as inhibitors and fluorescent probes. His PhD is focused on the synthesis of pixatimod and its analogues and evaluation of their heparanase inhibitory activity and antiviral activity against DENV and SARS‐CoV‐2.

Biographical Information
Gareth Doherty completed his undergraduate studies at The University of Queensland where be obtained his Bachelor of Science, majoring in chemistry. During this time he took the opportunity to study abroad at Purdue University in the U.S. He then returned to The University of Queensland to complete his honours year in organic chemistry (1 st Class) and commenced his PhD under the supervision of A/Prof. Vito Ferro. He obtained his PhD from The University of Queensland in 2021, where his work focused on drug design and synthesis, with a specific interest in neglected rare diseases, with broader implications for cancer, inflammation and various metabolic disorders.

Biographical Information
Nicholas See was born in Bundaberg, Queensland. After moving to Brisbane in 2016, he graduated with a Bachelor of Advanced Science (Hons I) from the University of Queensland. He is now completing his PhD in theoretical and organic chemistry under the joint supervision of A/Prof. Vito Ferro and A/Prof. Elizabeth Krenske. His research aims to understand the stereochemical outcomes of reactions which deliver rare and valuable l‐hexoses.

Biographical Information
Neha S Gandhi received her B.E. in Computer Sciences from the Maharaja SayajiRao University of Baroda, India in 2000 and worked as department‐in‐charge (Bioinformatics and molecular modelling) at Zydus Research Centre, Ahmedabad, India. Following her MPhil and PhD from Curtin University, Australia (2008–2012), she took a brief post‐doctoral training at the Virginia Commonwealth University, USA. She has been awarded prestigious fellowships from Curtin University and Queensland University of Technology (QUT). She is currently an Advance Queensland Industry Research Fellow and Program Leader in Personalised Medicine of the Centre for Genomics and Personalised Health at QUT. As a computational structural biologist/chemist, her research focuses on investigating the structure‐function of biomolecules in particular structural complexity of glycosaminoglycans, and drug discovery. Using supercomputer, her research team tests millions of drug‐like chemicals against a biological target for treating ageing related diseases to uncover candidate molecules.

Biographical Information
Vito Ferro obtained his PhD from the University of Western Australia in 1992. Following postdoctoral studies at the Carlsberg Laboratory, Denmark, and the University of British Columbia, Canada, he joined Progen Pharmaceuticals Ltd (Brisbane, Australia) where he spent 12 years in various positions, including Director of Drug Discovery. Following a brief period at Queensland University of Technology, he moved to the University of Queensland in 2010 where he is an Associate Professor in the School of Chemistry and Molecular Biosciences and is also a member of the Australian Infectious Diseases Research Centre. His research interests are in carbohydrate and medicinal chemistry with a particular focus on glycosaminoglycans (especially heparan sulfate and heparin) and their mimetics as potential drugs for a range of diseases.

Acknowledgements
V. F. gratefully acknowledges support from the Australian Research Council (DP170104431), the National MPS Society, the Sanfilippo Children's Foundation and the University of Queensland. M. C., G. G. D. and N. W. S. acknowledge the University of Queensland for financial support through the PhD scholarship program. N. S. G. is currently supported by the Advance Queensland Industry Research Fellowship.
M. Chhabra, G. G. Doherty, N. W. See, N. S. Gandhi, V. Ferro, Chem. Rec. 2021, 21, 3087.
References
- 1. Whitelock J. M., Iozzo R. V., Chem. Rev. 2005, 105, 2745–2764. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Xu D., Esko J. D., Annu. Rev. Biochem. 2014, 83, 129–157; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2b. Meneghetti M. C. Z., Hughes A. J., Rudd T. R., Nader H. B., Powell A. K., Yates E. A., Lima M. A., J. R. Soc. Interface 2015, 12, 20150589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rabenstein D. L., Nat. Prod. Rep. 2002, 19, 312–331. [DOI] [PubMed] [Google Scholar]
- 4. Gray E., Mulloy B., Barrowcliffe T. W., Thromb. Haemostasis 2008, 99, 807–818. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Casu B., Choay J., Ferro D. R., Gatti G., Jacquinet J. C., Petitou M., Provasoli A., Ragazzi M., Sinay P., Torri G., Nature 1986, 322, 215–216; [DOI] [PubMed] [Google Scholar]
- 5b. Ferro D. R., Provasoli A., Ragazzi M., Torri G., Casu B., Gatti G., Jacquinet J. C., Sinay P., Petitou M., Choay J., J. Am. Chem. Soc. 1986, 108, 6773–6778; [Google Scholar]
- 5c. Munoz-Garcia J. C., Corzana F., de Paz J. L., Angulo J., Nieto P. M., Glycobiology 2013, 23, 1220–1229. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Ferro D. R., Provasoli A., Ragazzi M., Casu B., Torri G., Bossennec V., Perly B., Sinaÿ P., Petitou M., Choay J., Carbohydr. Res. 1990, 195, 157–167; [DOI] [PubMed] [Google Scholar]
- 6b. Hsieh P.-H., Thieker D. F., Guerrini M., Woods R. J., Liu J., Sci. Rep. 2016, 6, 29602–29602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Xu D., Arnold K., Liu J., Curr. Opin. Struct. Biol. 2018, 50, 155–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.
- 8a. Liu J., Linhardt R. J., Nat. Prod. Rep. 2014, 31, 1676–1685; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8b. Mende M., Bednarek C., Wawryszyn M., Sauter P., Biskup M. B., Schepers U., Bräse S., Chem. Rev. 2016, 116, 8193–8255; [DOI] [PubMed] [Google Scholar]
- 8c. Tsai C.-T., Zulueta M. M. L., Hung S.-C., Curr. Opin. Struct. Biol. 2017, 40, 152–159. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Sun L., Chopra P., Boons G.-J., J. Org. Chem. 2020, 85, 16082–16098; [DOI] [PubMed] [Google Scholar]
- 9b. Anand S., Mardhekar S., Raigawali R., Mohanta N., Jain P., Shanthamurthy C. D., Gnanaprakasam B., Kikkeri R., Org. Lett. 2020, 22, 3402–3406; [DOI] [PubMed] [Google Scholar]
- 9c. Pawar N. J., Wang L., Higo T., Bhattacharya C., Kancharla P. K., Zhang F., Baryal K., Huo C.-X., Liu J., Linhardt R. J., Huang X., Hsieh-Wilson L. C., Angew. Chem. Int. Ed. 2019, 58, 18577–18583; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 18750–18756; [Google Scholar]
- 9d. See N. W., Wimmer N., Krenske E. H., Ferro V., Eur. J. Org. Chem. 2021, 2021, 10, 1575–1584. [Google Scholar]
- 10.For recent reviews see
- 10a. Daniel K. A., Rami A. A.-H., Curr. Med. Chem. 2020, 27, 3412–3447; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10b. Nahain A. A., Ignjatovic V., Monagle P., Tsanaktsidis J., Ferro V., Med. Res. Rev. 2018, 38, 1582–1613; [DOI] [PubMed] [Google Scholar]
- 10c. Mohamed S., Coombe D. R., Pharmaceuticals 2017, 10, 78; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10d. Weiss R. J., Esko J. D., Tor Y., Org. Biomol. Chem. 2017, 15, 5656–5668; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10e. Paluck S. J., Nguyen T. H., Maynard H. D., Biomacromolecules 2016, 17, 3417–3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vlodavsky I., Sanderson R. D., Ilan N. in Heparanase: From Basic Research to Clinical Applications, Vol. 1221 Springer International Publishing, Cham, 2020. [Google Scholar]
- 12.
- 12a. Khanna M., Parish C. R. in Heparanase: Historical Aspects and Future Perspectives, Eds.: Vlodavsky I., Sanderson R. D., Ilan N.), Springer International Publishing, Cham, 2020, pp. 71–96; [DOI] [PubMed] [Google Scholar]
- 12b. Coombe D. R., Gandhi N. S., Front. Oncol. 2019, 9, 1316–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.For recent reviews on heparanase inhibitors see
- 13a. Mohan C. D., Hari S., Preetham H. D., Rangappa S., Barash U., Ilan N., Nayak S. C., Gupta V. K., Basappa, Vlodavsky I., Rangappa K. S., iScience 2019, 15, 360–390; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13b. Fu K., Bai Z., Chen L., Ye W., Wang M., Hu J., Liu C., Zhou W., Eur. J. Med. Chem. 2020, 193, 112221; [DOI] [PubMed] [Google Scholar]
- 13c.See also Ref. [11]. Part V, pp. 473–567.
- 14.For recent reviews on HS mimetics in cancer and inflammation see
- 14a. Lanzi C., Cassinelli G., Molecules 2018, 23, 2915; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14b. Morla S., Int. J. Mol. Sci. 2019, 20, 1963; [Google Scholar]
- 14c. Ferro V., Expert Opin. Ther. Targets 2013, 17, 965–975. [DOI] [PubMed] [Google Scholar]
- 15. Shukla D., Spear P. G., J. Clin. Invest. 2001, 108, 503–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Feldman S. A., Audet S., Beeler J. A., J. Virol. 2000, 74, 6442–6447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen Y., Maguire T., Hileman R. E., Fromm J. R., Esko J. D., Linhardt R. J., Marks R. M., Nat. Med. 1997, 3, 866–871. [DOI] [PubMed] [Google Scholar]
- 18. Jacquet A., Haumont M., Chellun D., Massaer M., Tufaro F., Bollen A., Jacobs P., Virus Res. 1998, 53, 197–207. [DOI] [PubMed] [Google Scholar]
- 19. Roderiquez G., Oravecz T., Yanagishita M., Bou-Habib D. C., Mostowski H., Norcross M. A., J. Virol. 1995, 69, 2233–2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ianelli C. J., DeLellis R., Thorley-Lawson D. A., J. Biol. Chem. 1998, 273, 23367–23375. [DOI] [PubMed] [Google Scholar]
- 21. Chung C. S., Hsiao J. C., Chang Y. S., Chang W., J. Virol. 1998, 72, 1577–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Barth H., Schafer C., Adah M. I., Zhang F., Linhardt R. J., Toyoda H., Kinoshita-Toyoda A., Toida T., Van Kuppevelt T. H., Depla E., Von Weizsacker F., Blum H. E., Baumert T. F., J. Biol. Chem. 2003, 278, 41003–41012. [DOI] [PubMed] [Google Scholar]
- 23.
- 23a. Clausen T. M., Sandoval D. R., Spliid C. B., Pihl J., Perrett H. R., Painter C. D., Narayanan A., Majowicz S. A., Kwong E. M., McVicar R. N., Thacker B. E., Glass C. A., Yang Z., Torres J. L., Golden G. J., Bartels P. L., Porell R. N., Garretson A. F., Laubach L., Feldman J., Yin X., Pu Y., Hauser B. M., Caradonna T. M., Kellman B. P., Martino C., Gordts P. L. S. M., Chanda S. K., Schmidt A. G., Godula K., Leibel S. L., Jose J., Corbett K. D., Ward A. B., Carlin A. F., Esko J. D., Cell 2020, 183, 1043–1057.e1015; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23b. Lang J., Yang N., Deng J., Liu K., Yang P., Zhang G., Jiang C., PLoS One 2011, 6, 23710–23710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nyberg K., Ekblad M., Bergstrom T., Freeman C., Parish C. R., Ferro V., Trybala E., Antiviral Res. 2004, 63, 15–24. [DOI] [PubMed] [Google Scholar]
- 25.
- 25a. Mycroft-West C. J., Su D., Pagani I., Rudd T. R., Elli S., Gandhi N. S., Guimond S. E., Miller G. J., Meneghetti M. C. Z., Nader H. B., Li Y., Nunes Q. M., Procter P., Mancini N., Clementi M., Bisio A., Forsyth N. R., Ferro V., Turnbull J. E., Guerrini M., Fernig D. G., Vicenzi E., Yates E. A., Lima M. A., Skidmore M. A., Thromb. Haemostasis 2020, 120, 1700–1715; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25b. Tree J. A., Turnbull J. E., Buttigieg K. R., Elmore M. J., Coombes N., Hogwood J., Mycroft-West C. J., Lima M. A., Skidmore M. A., Karlsson R., Chen Y. H., Yang Z., Spalluto C. M., Staples K. J., Yates E. A., Gray E., Singh D., Wilkinson T., Page C. P., Carroll M. W., Br. J. Pharmacol. 2021, 178, 626–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Agelidis A., Shukla D. in Heparanase, Heparan Sulfate and Viral Infection, Eds.: Vlodavsky I., Sanderson R. D., Ilan N.), Springer International Publishing, Cham, 2020, pp. 759–770. [DOI] [PubMed] [Google Scholar]
- 27. Koganti R., Suryawanshi R., Shukla D., Cell. Mol. Life Sci. 2020, 77, 5059–5077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.
- 28a. Chopra P., Logun M. T., White E. M., Lu W., Locklin J., Karumbaiah L., Boons G.-J., ACS Chem. Biol. 2019, 14, 1921–1929; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28b. Jain P., Shanthamurthy C. D., Leviatan Ben-Arye S., Yehuda S., Nandikol S. S., Thulasiram H. V., Padler-Karavani V., Kikkeri R., Chem. Commun. 2021, 57, 3516–3519. [DOI] [PubMed] [Google Scholar]
- 29. Shanthamurthy C. D., Leviatan Ben-Arye S., Kumar N. V., Yehuda S., Amon R., Woods R. J., Padler-Karavani V., Kikkeri R., J. Med. Chem. 2021, 64, 3367–3380. [DOI] [PubMed] [Google Scholar]
- 30.
- 30a. Loka R. S., Sletten E. T., Barash U., Vlodavsky I., Nguyen H. M., ACS Appl. Mater. Interfaces 2019, 11, 244–254; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30b. Li J., Cai C., Wang L., Yang C., Jiang H., Li M., Xu D., Li G., Li C., Yu G., ACS Macro Lett. 2019, 8, 1570–1574. [DOI] [PubMed] [Google Scholar]
- 31.
- 31a. DiGabriele A. D., Lax I., Chen D. I., Svahn C. M., Jaye M., Schlessinger J., Hendrickson W. A., Nature 1998, 393, 812–817; [DOI] [PubMed] [Google Scholar]
- 31b. Faham S., Hileman R. E., Fromm J. R., Linhardt R. J., Rees D. C., Science 1996, 271, 1116–1120; [DOI] [PubMed] [Google Scholar]
- 31c. Pellegrini L., Burke D. F., von Delft F., Mulloy B., Blundell T. L., Nature 2000, 407, 1029–1034; [DOI] [PubMed] [Google Scholar]
- 31d. Schlessinger J., Plotnikov A. N., Ibrahimi O. A., Eliseenkova A. V., Yeh B. K., Yayon A., Linhardt R. J., Mohammadi M., Mol. Cell 2000, 6, 743–750. [DOI] [PubMed] [Google Scholar]
- 32. Liu L., Bytheway I., Karoli T., Fairweather J. K., Cochran S., Li C., Ferro V., Bioorg. Med. Chem. Lett. 2008, 18, 344–349. [DOI] [PubMed] [Google Scholar]
- 33.
- 33a. Wessel H. P., Bartsch S., Carbohydr. Res. 1995, 274, 1–9; [DOI] [PubMed] [Google Scholar]
- 33b. Probst K. C., Wessel H. P., J. Carbohydr. Chem. 2001, 20, 549–560. [Google Scholar]
- 34. Fairweather J. K., Karoli T., Liu L., Bytheway I., Ferro V., Carbohydr. Res. 2009, 344, 2394–2398. [DOI] [PubMed] [Google Scholar]
- 35.
- 35a. Liu L., Li C. P., Cochran S., Ferro V., Bioorg. Med. Chem. Lett. 2004, 14, 2221–2226; [DOI] [PubMed] [Google Scholar]
- 35b. Liu L., Li C., Cochran S., Feder D., Guddat L. W., Ferro V., Bioorg. Med. Chem. Lett. 2012, 22, 6190–6194. [DOI] [PubMed] [Google Scholar]
- 36. Liu L., Li C., Cochran S., Jimmink S., Ferro V., ChemMedChem 2012, 7, 1267–1275. [DOI] [PubMed] [Google Scholar]
- 37. Ekblad M., Bergström T., Banwell M. G., Bonnet M., Renner J., Ferro V., Trybala E., Antiviral Chem. Chemother. 2006, 17, 97–106. [DOI] [PubMed] [Google Scholar]
- 38. Freeman C., Liu L., Banwell M. G., Brown K. J., Bezos A., Ferro V., Parish C. R., J. Biol. Chem. 2005, 280, 8842–8849. [DOI] [PubMed] [Google Scholar]
- 39. Nahain A. A., Ignjatovic V., Monagle P., Tsanaktsidis J., Vamvounis G., Ferro V., Biomacromolecules 2020, 21, 1009–1021. [DOI] [PubMed] [Google Scholar]
- 40. Nahain A. A., Ignjatovic V., Monagle P., Tsanaktsidis J., Vamvounis G., Ferro V., Macromol. Biosci. 2020, 20, 2000110. [DOI] [PubMed] [Google Scholar]
- 41. Gockel L. M., Heyes M., Li H., Al Nahain A., Gorzelanny C., Schlesinger M., Holdenrieder S., Li J.-P., Ferro V., Bendas G., ACS Appl. Mater. Interfaces 2021, 13, 7080–7093. [DOI] [PubMed] [Google Scholar]
- 42. Parish C. R., Coombe D. R., Jakobsen K. B., Bennett F. A., Underwood P. A., Int. J. Cancer 1987, 40, 511–518. [DOI] [PubMed] [Google Scholar]
- 43. Parish C. R., Freeman C., Brown K. J., Francis D. J., Cowden W. B., Cancer Res. 1999, 59, 3433–3441. [PubMed] [Google Scholar]
- 44.
- 44a. Ferro V., Dredge K., Liu L., Hammond E., Bytheway I., Li C., Johnstone K., Karoli T., Davis K., Copeman E., Gautam A., Semin. Thromb. Hemostasis 2007, 33, 557–568; [DOI] [PubMed] [Google Scholar]
- 44b. Chhabra M., Ferro V., Adv. Exp. Med. Biol. 2020, 1221, 473–491. [DOI] [PubMed] [Google Scholar]
- 45. Kudchadkar R., Gonzalez R., Lewis K. D., Expert Opin. Invest. Drugs 2008, 17, 1769–1776. [DOI] [PubMed] [Google Scholar]
- 46. Chen P. J., Lee P. H., Han K. H., Fan J., Cheung T. T., Hu R. H., Paik S. W., Lee W. C., Chau G. Y., Jeng L. B., Wang H. J., Choi J. Y., Chen C. L., Cho M., Ho M. C., Wu C. C., Lee K. S., Mao Y., Hu F. C., Lai K. L., Ann. Oncol. 2017, 28, 213. [Google Scholar]
- 47.
- 47a. Ferro V., Fewings K., Palermo M. C., Li C., Carbohydr. Res. 2001, 332, 183–189; [DOI] [PubMed] [Google Scholar]
- 47b. Yu G., Gunay N. S., Linhardt R. J., Toida T., Fareed J., Hoppensteadt D. A., Shadid H., Ferro V., Li C., Fewings K., Palermo M. C., Podger D., Eur. J. Med. Chem. 2002, 37, 783–791. [DOI] [PubMed] [Google Scholar]
- 48. Ferro V., Li C., Fewings K., Palermo M. C., Linhardt R. J., Toida T., Carbohydr. Res. 2002, 337, 139–146. [DOI] [PubMed] [Google Scholar]
- 49.
- 49a. Handley P. N., Carroll A., Ferro V., Carbohydr. Res. 2017, 446–447, 68–75; [DOI] [PubMed] [Google Scholar]
- 49b. Elli S., Stancanelli E., Handley P. N., Carroll A., Urso E., Guerrini M., Ferro V., Glycobiology 2018, 28, 731–740. [DOI] [PubMed] [Google Scholar]
- 50. Cochran S., Li C., Fairweather J. K., Kett W. C., Coombe D. R., Ferro V., J. Med. Chem. 2003, 46, 4601–4608. [DOI] [PubMed] [Google Scholar]
- 51. Fairweather J. K., Hammond E., Johnstone K. D., Ferro V., Bioorg. Med. Chem. 2008, 16, 699–709. [DOI] [PubMed] [Google Scholar]
- 52. Karoli T., Liu L., Fairweather J. K., Hammond E., Li C. P., Cochran S., Bergefall K., Trybala E., Addison R. S., Ferro V., J. Med. Chem. 2005, 48, 8229–8236. [DOI] [PubMed] [Google Scholar]
- 53. Liu L. G., Johnstone K. D., Fairweather J. K., Dredge K., Ferro V., Aust. J. Chem. 2009, 62, 546–552. [Google Scholar]
- 54. Johnstone K. D., Karoli T., Liu L., Dredge K., Copeman E., Li C. P., Davis K., Hammond E., Bytheway I., Kostewicz E., Chiu F. C., Shackleford D. M., Charman S. A., Charman W. N., Harenberg J., Gonda T. J., Ferro V., J. Med. Chem. 2010, 53, 1686–1699. [DOI] [PubMed] [Google Scholar]
- 55.
- 55a. Dredge K., Hammond E., Davis K., Li C. P., Liu L., Johnstone K., Handley P., Wimmer N., Gonda T. J., Gautam A., Ferro V., Bytheway I., Invest. New Drugs 2010, 28, 276–283; [DOI] [PubMed] [Google Scholar]
- 55b. Ferro V., Liu L., Johnstone K. D., Wimmer N., Karoli T., Handley P., Rowley J., Dredge K., Li C. P., Hammond E., Davis K., Sarimaa L., Harenberg J., Bytheway I., J. Med. Chem. 2012, 55, 3804–3813. [DOI] [PubMed] [Google Scholar]
- 56. Hammond E., Dredge K., Adv. Exp. Med. Biol. 2020, 1221, 539–565. [DOI] [PubMed] [Google Scholar]
- 57. Hammond E., Haynes N. M., Cullinane C., Brennan T. V., Bampton D., Handley P., Karoli T., Lanksheer F., Lin L., Yang Y., Dredge K., J. Immunother. Cancer 2018, 6, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Dredge K., Brennan T. V., Hammond E., Lickliter J. D., Lin L., Bampton D., Handley P., Lankesheer F., Morrish G., Yang Y., Brown M. P., Millward M., Br. J. Cancer 2018, 118, 1035–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ekblad M., Adamiak B., Bergstrom T., Johnstone K. D., Karoli T., Liu L., Ferro V., Trybala E., Antiviral Res. 2010, 86, 196–203. [DOI] [PubMed] [Google Scholar]
- 60. Said J., Trybala E., Andersson E., Johnstone K., Liu L., Wimmer N., Ferro V., Bergstrom T., Antiviral Res. 2010, 86, 286–295. [DOI] [PubMed] [Google Scholar]
- 61. Lundin A., Bergstrom T., Andrighetti-Frohner C. R., Bendrioua L., Ferro V., Trybala E., Antiviral Res. 2012, 93, 101–109. [DOI] [PubMed] [Google Scholar]
- 62. Supramaniam A., Liu X., Ferro V., Herrero L. J., Antimicrob. Agents Chemother. 2018, 62, 01959–01917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Modhiran N., Gandhi N. S., Wimmer N., Cheung S., Stacey K., Young P. R., Ferro V., Watterson D., Antiviral Res. 2019, 168, 121–127. [DOI] [PubMed] [Google Scholar]
- 64. Said J. S., Trybala E., Görander S., Ekblad M., Liljeqvist J.-Å., Jennische E., Lange S., Bergström T., Antimicrob. Agents Chemother. 2016, 60, 1049–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Guimond S. E., Mycroft-West C. J., Gandhi N. S., Tree J. A., Le T. T., Spalluto C. M., Humbert M. V., Buttigieg K. R., Coombes N., Elmore M. J., Nyström K., Said J., Setoh Y. X., Amarilla A. A., Modhiran N., Sng J. D. J., Chhabra M., Young P. R., Lima M. A., Yates E. A., Karlsson R., Miller R. L., Chen Y.-H., Bagdonaite I., Yang Z., Stewart J., Hammond E., Dredge K., Wilkinson T. M. A., Watterson D., Khromykh A. A., Suhrbier A., Carroll M. W., Trybala E., Bergström T., Ferro V., Skidmore M. A., Turnbull J. E., bioRxiv 2021, 2020.2006.2024.169334. [Google Scholar]
- 66. Chhabra M., Ferro V., Molecules 2018, 23, 2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Petitou M., van Boeckel C. A., Angew. Chem. Int. Ed. 2004, 43, 3118–3133; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 3180–3196. [Google Scholar]
- 68. Wilson J. C., Laloo A. E., Singh S., Ferro V., Biochem. Biophys. Res. Commun. 2014, 443, 185–188. [DOI] [PubMed] [Google Scholar]
- 69. Bisio A., Mantegazza A., Urso E., Naggi A., Torri G., Viskov C., Casu B., Semin. Thromb. Hemostasis 2007, 33, 488–495. [DOI] [PubMed] [Google Scholar]
- 70.
- 70a. Hammond E., Li C. P., Ferro V., Anal. Biochem. 2010, 396, 112–116; [DOI] [PubMed] [Google Scholar]
- 70b. Melo C. M., Tersariol I. L., Nader H. B., Pinhal M. A., Lima M. A., Carbohydr. Res. 2015, 412, 66–70. [DOI] [PubMed] [Google Scholar]
- 71. Pearson A. G., Kiefel M. J., Ferro V., von Itzstein M., Org. Biomol. Chem. 2011, 9, 4614–4625. [DOI] [PubMed] [Google Scholar]
- 72. Wu L., Wimmer N., Davies G. J., Ferro V., Chem. Commun. (Cambridge, U. K.) 2020, 56, 13780–13783. [DOI] [PubMed] [Google Scholar]
- 73. Liu J., Schleyer K. A., Bryan T. L., Xie C., Seabra G., Xu Y., Kafle A., Cui C., Wang Y., Yin K., Fetrow B., Henderson P. K. P., Fatland P. Z., Liu J., Li C., Guo H., Cui L., Chem. Sci. 2021, 12, 239–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.
- 74a. Zhang F., Datta P., Dordick J. S., Linhardt R. J., J. Pharm. Sci. 2020, 109, 975–980; [DOI] [PubMed] [Google Scholar]
- 74b. Zhao J., Kong Y., Zhang F., Linhardt R. J., Biochem. Physiol. 2018, 7, 241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Osmond R. I. W., Kett W. C., Skett S. E., Coombe D. R., Anal. Biochem. 2002, 310, 199–207. [DOI] [PubMed] [Google Scholar]
- 76. Cochran S., Li C. P., Ferro V., Glycoconjugate J. 2009, 26, 577–587. [DOI] [PubMed] [Google Scholar]
- 77.
- 77a. Muenzer J., Rheumatol. 2011, 50, 4–12; [Google Scholar]
- 77b. Neufeld E. F., Muenzer J. in The mucopolysaccharidoses, Eds.: Scriver C. R., Beaudet A. L., Sly W. S., Valle D., McGraw-Hill, New York, 2001, pp. 3421–3452. [Google Scholar]
- 78. Viana G. M., Priestman D. A., Platt F. M., Khan S., Tomatsu S., Pshezhetsky A. V., J. Clin. Med. 2020, 9, 396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Trim P. J., Lau A. A., Hopwood J. J., Snel M. F., Rapid Commun. Mass Spectrom. 2014, 28, 933–938. [DOI] [PubMed] [Google Scholar]
- 80. Trim P. J., Hopwood J. J., Snel M. F., Anal. Chem. 2015, 87, 9243–9250. [DOI] [PubMed] [Google Scholar]
- 81. He Q. Q., Trim P. J., Snel M. F., Hopwood J. J., Ferro V., Org. Biomol. Chem. 2018, 16, 8791–8803. [DOI] [PubMed] [Google Scholar]
- 82. He Q. Q., Trim P. J., Lau A. A., King B. M., Hopwood J. J., Hemsley K. M., Snel M. F., Ferro V., ACS Chem. Neurosci. 2019, 10, 3847–3858. [DOI] [PubMed] [Google Scholar]
- 83.
- 83a. Singh A., Kett W. C., Severin I. C., Agyekum I., Duan J., Amster I. J., Proudfoot A. E. I., Coombe D. R., Woods R. J., J. Biol. Chem. 2015, 290, 15421–15436; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83b. Cui J. Y., Zhang F., Nierzwicki L., Palermo G., Linhardt R. J., Lisi G. P., Biochem. 2020, 59, 3541–3553; [DOI] [PubMed] [Google Scholar]
- 83c. Ziarek J. J., Veldkamp C. T., Zhang F., Murray N. J., Kartz G. A., Liang X., Su J., Baker J. E., Linhardt R. J., Volkman B. F., J. Biol. Chem. 2013, 288, 737–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Paiardi G., Milanesi M., Wade R. C., D'Ursi P., Rusnati M., Biomol. Eng. 2021, 11, 739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Boittier E. D., Burns J. M., Gandhi N. S., Ferro V., J. Chem. Inf. Model. 2020, 60, 6328–6343. [DOI] [PubMed] [Google Scholar]
- 86. Miller M. C., Cai C., Wichapong K., Bhaduri S., Pohl N. L. B., Linhardt R. J., Gabius H.-J., Mayo K. H., Sci. Rep. 2020, 10, 15708–15708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Schuurs Z. P., Hammond E., Elli S., Rudd T. R., Mycroft-West C. J., Lima M. A., Skidmore M. A., Karlsson R., Chen Y.-H., Bagdonaite I., Yang Z., Ahmed Y. A., Richard D. J., Turnbull J., Ferro V., Coombe D. R., Gandhi N. S., Comput. Struct. Biotechnol. J. 2021, 19, 2806–2818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Tomasello G., Armenia I., Molla G., Bioinformatics 2020, 36, 2909–2911. [DOI] [PubMed] [Google Scholar]
- 89. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E., J. Comput. Chem. 2004, 25, 1605–1612. [DOI] [PubMed] [Google Scholar]