

Graphical abstract
Keywords: SARS-CoV2, Spike trimers, ACE2 dimer, Super-complex, Peptidase domain, Prefusion conformation, Post-fusion conformation
Highlights
-
•
Formation of super-complexes of the spike protein trimer with three ACE-2 dimers.
-
•
Formation of super-complexes of ACE-2 dimer with two spike protein trimers.
-
•
Cooperative interactions within formed super-complexes.
-
•
Membrane geometry on ACE-2 activity within super-complexes.
Abstract
The membrane fusion mechanism of SARS-CoV-2 offers an attractive target for the development of small molecule antiviral inhibitors. Fusion involves an initial binding of the crown-like trimeric spike glycoproteins of SARS-CoV-2 to the receptor angiotensin II-converting enzyme 2 (ACE2) on the permissive host cellular membrane and a prefusion to post-fusion conversion of the spike trimer. During this conversion, the fusion peptides of the spike trimer are inserted into the host membrane to bring together the host and viral membranes for membrane fusion in highly choreographic events. However, geometric constraints due to interactions with the membranes remain poorly understood. In this study, we build structural models of super-complexes of spike trimer/ACE2 dimers based on the molecular structures of the ACE2/neutral amino acid transporter B(0)AT heterodimer. We determine the conformational constraints due to the membrane geometry on the enzymatic activity of ACE2 and on the viral fusion process. Furthermore, we find that binding three ACE2 dimers per spike trimer is essential to open the central pore as necessary for triggering productive membrane fusion through an elongation of the central stalk. The reported findings thus provide valuable insights for targeting the membrane fusion mechanism for drug design at the molecular level.
1. Introduction
SARS-CoV-2 (Severe Acute Respiratory Syndrome Coronavirus-2) is responsible for the current ongoing COVID-19 pandemic that has already infected over 206 million people and killed more than 4.3 million patients worldwide [1], [2], [3], [4]. Global efforts are underway to find potential treatments of this virus and its variants using fundamental understanding at the molecular level, resulting in many crystallographic and cryogenic electron microscopic (cryo-EM) structures [5], [6], [7], [8], [9]. However, the viral membrane fusion process that could offer an attractive target for antiviral small molecule inhibitors has remained largely unexplored [10], [11]. Here, we focus on structural models of spike trimer/ACE2 dimer super-complexes that provide valuabe insights on conformational arrangements required for membrane fusion.
SARS-CoV-1 and SARS-CoV-2 are RNA viruses with a protective membrane decorated with densely packed trimeric spike glycoproteins (S proteins). A wealth of structural information has rapidly accumulated, including characterization of the supra-organization of SARS-CoV viral particles based on in situ atomic force electron microscopy, transmission electron microscopy, and electron tomography [12], [13], [14], [15], [16], [17], [18]. SARS-CoV-2 is a non-homogeneous, non-symmetric, spherical particle of about 900 Å in diameter with ∼ 48 S trimers on the surface. Contrary to SARS-CoV-1 where the S trimers exhibit a regular P2-symmetry-like lattice [13], SARS-CoV-2 exhibits a more disordered arrangement.
The S trimers bind to the human ACE2 (Agiotensin-II Converting Enzyme-2) receptors on the host cell membrane and initiate the viral membrane fusion process, enabling the viral content including the genomic RNA to enter the host cell for rapid replication [5], [6], [7], [19], [20], [21], [22], [23], [24], [25], [26]. Recent studies have reported molecular structures of individual S protein trimers of SARS-CoV-2 in both prefusion and post-fusion states as well as of the ACE2 dimer in open and closed states, sub-complexes of S trimer with the peptidase domain (PD) of ACE2, and the receptor-binding domain (RBD) of the S protein with intact ACE2 [19], [20], [5], [6], [7]. Despite the wealth of piecemeal structural information, the structure of the super-complex that leads to membrane fusion upon bringing together the viral and host cellular membranes remains to be established and is the subject of this study. In addition, the interactions that are essential to modulate the physiological enzymatic activity of ACE2 for converting the vasoconstrictor 8-residue peptide hormone of angiotensin-2 to the vasodilator 7-residue peptide hormone of angiotensin remain poorly understood [27], [28] and are addressed through our modeling of super-complexes of the spike trimer with ACE2 dimers.
Membrane fusion is a thermodynamically unfavorable process that requires activation and is often coupled to other highly exergonic mechanisms that help to overcome significant kinetic barriers [29], [30], [31], [32], [33], [34], [35]. Coronaviruses exhibit a conserved type I membrane fusion mechanism in which HR-1 (Heptad Repeat-1) and HR-2 (Heptad Repeat-2) of the S protein are folded into two separated helices in the meso-stable prefusion state that pack against each other like a ‘loaded spring’ [35], [36]. The two units become a single elongated continuous helix in the post-fusion state in which the three elongated helices from the three subunits of the S trimer form an ultra-stable core structure. Prior to the conversion, the S protein is proteolytically cleaved into fragments S1 and S2 [20]. The S1 fragment contains the receptor-binding domain (RBD) and S2 has the fusion peptide (FP). However, the post-fusion state of the S2 fragment differs from an initially formed extended intermediate state. In the post-fusion state, viral and host membranes are merged after the FP migrates next to the trans-membrane domain (TMD). In the prefusion state, FP and TMD are located on the opposite ends of the S2 fragment, and the viral and host membranes remain separated by ∼200 Å or more. The length of the extended helices and central stalk vary among the various coronaviruses as they interact with different host receptors and coreceptors for viral entry [37]. In general, membrane fusion involves five states: (i) prefusion, (ii) extended intermediate, (iii) collapsed intermediate, (iv) hemi-fusion intermediate, and (v) the ultra-stable post-fusion product, which makes the fusion process irreversible. Non-specific dissociation of S1 from S2 could lead to the rapid conversion of S2 from prefusion to post-fusion. However, such dissociation would not necessarily cause membrane fusion if the ACE-2 bound RBD of S1 plays an active role in bringing together the membranes of the host and the virus. Therefore, it is necessary to elucidate the extended intermediates critical for membrane fusion, based on known prefusion and post-fusion S protein structures.
The important influence of the membrane on the assembly of macromolecular supercomplexes is often underestimated. High-resolution in vitro structural studies are typically carried out using truncated soluble forms of transmembrane proteins. However, the binding energies can be quite different, as for example in the spike protein-ACE2 subcomplexes mentioned above. Beyond the spike protein, perhaps the most classic example is the large difference in binding affinity of insulin and insulin-like growth factor-I (IGF1) when comparing the full-length and truncated soluble forms of the human insulin receptor [38]. Not only does the binding affinity differ by 3 orders of magnitude, but also the nature of binding is altered [38]. In fact, the full-length insulin receptor dimer unexpectedly binds four insulins, not just two as previously observed in the truncated insulin receptor [39], [40]. Therefore, the TMD as well as the geometry of the membrane could dramatically alter quaternary interactions of the proteins involved. We computationally address the importance of the membrane in the viral-host membrane fusion in this study.
Extensive studies have already been carried out on how FPs might be folded and inserted into the different phospholipid membrane, both experimentally and computationally [41], [42], [43], [44], [45], [46], [47], [48]. Those studies focused on a step after the FPs have been repositioned from a location near the viral membrane buried inside the pre-fusion state to a new location near the host membrane in an exposed position, pointing away in the post-fusion state. Thus, our study fundamentally differs from the prior work because we focus on an intermediate step from the pre-fusion to postfusion conversion. Moreover, even after the FP has been inserted into the host membrane and after the S2 peptide in the proposed hemifusion state, there is still a large energy barrier for transition of the two membranes that are in contact by a single line (through the S2 fragment in a lying-down position) to those with a pore generated to connect them. A minimal requirement for pore formation is that three contact lines between the two membranes from three S2 fragments are needed to be arranged in a closed triangle, a topological requirement that is essential in the three dimension and has often been overlooked. A simple analogy of this requirement in the membrane fusion process via a pore formation can be viewed in the reverse process of cell division through contractile ring [49], [50].
2. Computational methods and atomic models
Least-square alignment of equivalent structures was carried out initially using secondary-structure based alignment with the graphics Coot and then using the CCP4 suite for further alignment [51], [52]. The corresponding transformation matrices were used for docking and making movies. Figures were made using the graphics application PyMOL [53].
To address the feasibility of the proposed formation of spiker trimers with ACE2 dimers, we used the Molecular Mechanics Generalized Born Solvationn Area Model [54] as implemented in the AmberTools20 package to assess the free energy of complex formation after energy minimization with the PMEMD (also part the Amber package) [55]. Starting coordinates were taken from the trimeric spike protein with one RBD in an up position and two RBDs in a down position (PDB accession number of 6vsb) and from the dimeric ACE2 in complex with B0AT and one RBD bound (6m17) [7], [20]. For the simplicity of calculations, we removed the membrane phospholipids, transmembrane helical domains, and glycans because none of them was directly involved in complex formation. This omission would have some effects on solvation energy, but we estimated this effect to be relatively small and should not affect the main conclusion on our computional results because they are far away from the site of complex formation. However, the validation of this approximation needs to be further confirmed experimentally. Nevertheless, membranes must play an important geometrical role on the fusion reaction because a single contact line between two membranes will not open a pore between them.
Three complexes were examined computationally: (i) complex-1 with one ACE2/B0AT dimer bound to one spike trimer, (ii) complex-2 with two ACE2/B0AT dimers bound to the spike trimer after the second RBD of the spike protein underwent from the down-to-up transition, and (iii) complex-3 with three ACE2/B0AT dimers bound to the spike trimer after the second and third RDBs of the spike protein underwent from the down-to-up transition. Conformation of the spike trimer in its supercomplex with three ACE2/B0AT dimers done this stuy was essentially identical to one predicted computationally and independently [56]. Schrödinger Maestro 2020–2 (Schrödinger, LLC, New York, NY) was used to visualize and reconstruct the protein structures through a pre-processing step with its Protein Preparation Wizard. This interface GUI included proper assignment of bond order, ionization and protonation state, rebuilding missing side chain atoms and H atoms, fixing disoriented side chains, and capping both N and C termini. Without the transmembrane helical domain, the sequence of spike protein included residues 11 to 1140.
3. Results
3.1. Models of membrane-bound spike trimers and dimers of ACE2/B(0)AT heterodimer
Viral membrane fusion requires significant conformational changes of both viral and host cellular membranes. Here, we analyze geometric constraints essential for membrane fusion using atomistic models of the fully glycosylated, full-length S trimer of SARS-CoV-2 in the prefusion state embedded in a viral membrane. The models have been generated from the structures of ACE2 complexed with the membrane-bound neutral amino acid transporter, B(0)AT, in a host cellular membrane (Fig. 1) [55]. In the S model, there is an extended central stalk of about 90 Å before the TM is anchored to the viral membrane, with models for HR2 and the C-terminal domains of S2 recently confirmed by experiments [36]. While most of the S trimers have rigid crown-like structures, some exhibit a flexible extension [16], [17]. We address whether the joined HR1/HR2 continuous helix of S2 (formed in the first step of viral membrane fusion from the two single helices) is long enough and necessary to insert its FP into the host cellular membrane.
Fig. 1.

Full atomistic models of S trimer within the viral membrane and ACE2 complex within the host cellular membrane. (a) Asymmetric 6vyb S trimer. One subunit is in blue-to-red rainbow color, the other two subunits are in silver and grey. Glycans are CPK sphere representation. (b, c) Two orthogonal views of two symmetric 6vxx models with glycans omitted. Computationally modeled extension of about 90 Å is indicated. (d) Symmetric 6m17 dimer of heterodimeric ACE2/B(0)AT complex. (e, f) Two orthogonal views of three dimers of ACE2/B(0)AT complex with the ACE2 in a closed conformation. See Fig. 1 for comparison with the closed and open-1 structures and Video 1A, 1B, and 1C for motions between them. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
We have used these fully atomistic models of the membrane portion to generate two S trimers (starting with PDB ID of 6vxx), including three copies of the atomistic model for three dimers of ACE2/B(0)AT complex (6m17) to form S trimer/ACE2 dimer super-complexes (Fig. 1b, 1c, 1e, 1f) [7], [20]. The two S trimers on the SARS-CoV-2 viral surface are more flexible, with geometry based on the S-to-S distribution on SARS-CoV-1 viral particles [13], [16]. The geometry of the three ACE2 dimers was assumed on the basis of the symmetrized open-2 conformation (6crz) of the SARS-CoV-1 S trimer, as discussed below [25]. The relationship of the three conformations observed for the S protein trimers is described in online Supporting materials (OSM, Fig. S1; Video 1A, 1B, 1C).
3.2. Symmetrized open conformations of spike trimers
The central pore of the S1 trimer must open up to accommodate an elongated S2 central stalk formed in an extended helix intermediate during the first step of viral membrane fusion. In either the symmetric closed S trimer or the asymmetric open conformations, two or three RBDs are in the “down” position, completely blocking the extension of S2 through the S1 central pore. In the presence of some neutralizing antibodies, all three S subunits in an “up” position have been observed in which antibodies block the binding of ACE2 [9]. For other antibodies, the up-down equilibrium of the S protein is shifted to the “down” position [9]. Therefore, the symmetric S trimer ought to exist in both open-1 and open-2 conformations as would asymmetric mixed-open conformations. The symmetric open conformation defines the minimum pore size because the mixed-open conformation could still obstruct the central pore by a single “down” conformation of the RBD regardless of “open” conformations of the remaining two RBDs. We describe the nature of symmetric open conformations before discussing their implication on viral membrane fusion.
We applied 3-fold symmetry for generating open-1 and open-2 conformations (Fig. 2a-c). For the symmetric closed conformation, three RBDs of each S trimer strongly interact with each other (Fig. 2a). In the symmetric open-1 conformation, each RBD is displaced away from the 3-fold axis by 21.2 Å with an open central pore (Fig. 2b; Video 1C). In the symmetric open-2 conformation, the displacement of RBD increases by an additional 2.5 Å with further opening of the central pore, which is large enough for the passage of an extended S2 central stalk (Fig. 2c). However, the orientation of the RBD is altered by 16.7° between the open-1 and open-2 conformations, which would result in a large displacement of the ACE2 dimer on the host membrane away from each other, thus thinning the host cellular membrane. Given the known amount of rotation between the closed and open-1 conformations, 18 intermediate structures were computationally generated by linear extrapolation of rotation angles. A series of overlaid structures demonstrate motion of the resulting conversion (Fig. 2d). The rotation does not significantly change the interaction of the RBD with the N-terminal domain (NTD) of a neighboring monomer. Likewise, three intermediate structures are generated for visualization of the open-1-to-open-2 conversion (Fig. 2e). In the second set of displacements, the distance between the RBD and its interacting NTD increases slightly. In the 6crz structure [25], the NTD follows the movement of the RBD with the rotation axis closer to the center of the NTD. In both sets of motions, the RBD surface of S1 that binds the PD of ACE2 rotates outward for easy accessibility to the PD. At the same time, the NTD of S1 moves downward toward the viral membrane, bringing the two membranes closer together.
Fig. 2.

Comparison of three spike conformations in context of trimer. (a) Two orthogonal views of the closed conformation (6vxx). (b) Two orthogonal views of the open-1 conformation (modeled after 6vyb). (c) Two orthogonal views of the open-2 conformation (modeled after PDB ID 6crz). (c,d) Two orthogonal views of the closed-to-open-1 movies with starting model in green and ending model in red, and intermediates in partial transparency. (e) Two orthogonal views of the open-1-to-open-2 movies. (f) Surface model for end-on view of model of the closed-to-open-1 movies. (g) Surface model for end-on view of model of the open-1-to-open-2 movies. See Video 1C for motions of symmetrized opening and closing of the central pore of the spike trimer associated with motions of the RBDs. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
The importance of the symmetrized spike trimer has recently been confirmed in the complex of soluble spike trimer with an engineered ACE2 trimer [57]. The binding affinity of the spike protein from a monomerized ACE2 to the trimerized ACE2 has improved by 3 orders of magnitude (with apparent IC50 values of 27 nM and 30 pM, respectively). Yet, there is very little difference in the binding of soluble spike trimer between the monomeric ACE2 and physiologically relevant ACE2 dimer. This highlights strong cooperativity and allostery within the spike protein-multiple ACE2 complexes, and suggests that the geometry of dimeric ACE2 is not suitable for binding two RBDs of each spike trimer even though the mean binding affinity of ACE2 to the subsequential RBDs significantly increases upon an initial binding. Thus, additional ACE2 dimers would be required for formation of larger super-complex as a basis for our modeling.
3.3. Symmetric dimer of full-length ACE2 complexed with B(0)AT
The full-length ACE2 consists of three domains: the peptidase domain (PD), collectrin domain (CD), and type I helical TMD (Fig. 4a) [7]. ACE2 belongs to the type I membrane protein family and forms a rigid dimer mainly through the CD and its surrounding residues as well as indirectly through its TMD with the neutral amino acid transporter B(0)AT (or B0AT) (Fig. 3a). The high-affinity binding of the RBD to the ACE2 dimer, with one RBD interacting with the PD of dimeric ACE2, does not alter the conformation of the ACE2 dimer in that structure (Fig. 3b). A bi-lobal structure of PD has a substrate-binding cleft that opens and closes for the extracellular processing of angiotensin-2 to generate angiotensin (residues 1–7) and remains in the open position in the absence of its substrate [27], [28], [58]. Evidence exists that PD can be locked down in a closed conformation or inhibited conformation with an inhibitor. For instance, human testicular ACE2 PD is observed in the inhibited conformation when inhibitor MLN-4760 with a Zn2+ ion is bound in its peptidase active site, involving a subdomain rotation of about 17° from the open to closed conformations (Video 2A, 2B) [58].
Fig. 4.

A fusion-active extended intermediate of the S2 central stalk. (a-c) Views of docked three ACE2 dimers onto the extended stalk. The viral membrane is shown in small circles, and the middle plane of the host membrane is shown in ellipses around each ACE2 dimer. They are approximately parallel to one another. The top end of the EMD-9597 is close to the middle plane of the host membrane. (a) Same view as (h) of Fig. S2. (b) 60° rotated view. (c) Host end view. See Fig. S2 for piecewise assembly. See Figs. S2 and S3 for conformational changes of spike subunit and trimer during the prefusion to post-fusion conversion, and Video 3A, 3B, and 3C for motions of ACE2 for continued opening of the central pore of the spike trimer and for resulting the post-fusion S2 in super-complexes.
Fig. 3.

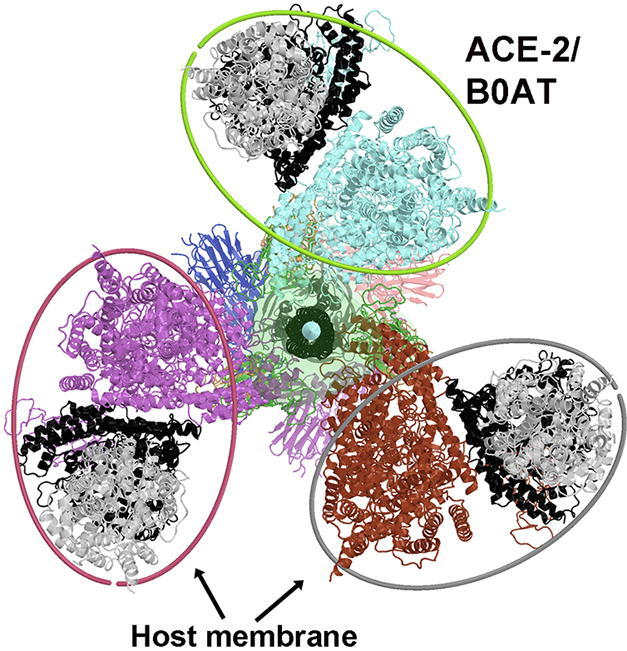
The full-length ACE2 dimers in complex with the two RBDs of spike proteins and with two neutral amino-acid transporter B0AT for modeling super-complexes. (a) Dimer of ACE2/B0AT heterodimer (PDB ID/6m17). One ACE2 is rainbow colored from N-to-C termini, and the other is salmon. One B0AT is in cyan, and the other is in yellow. Two RBDs of spike are in magenta and cyan. (b) Docking two asymmetric spike trimers (6vyb) onto the 6m17 ACE2 dimer by aligning their RBD-PD interaction. The dyad is in cyan with a length of 400 Å, and the 3-fold axes are in green with a length of 250 Å. Middle planes of the host membrane are indicated with ellipses surrounding the TMDs of the ACE2/B0AT dimer; locations of the viral membrane are shown red circles, which differ between the two spike trimers, and between the spike trimers and the ACE2 dimer. Distance between the two spike trimers at the viral membrane is about 260 Å. (c) A close-up view of the PD of ACE2 in 6m17. (d) A close-up view of the testicular ACE2 PD with the inhibitor MLN-4760 (red spheres) bound in the closed conformation (1r4l). (e) Modeled closed ACE2 on the basis of 6 m17 and 1r4l. (f) Docking two asymmetric spike trimers (6vyb) onto the closed ACE2 dimer. Spacing between the two spike trimers is about 140 Å. (g) Three additional views of (f), tilted view, viral end view, and host end view. (h) One symmetrized 6crz spike trimer with three closed ACE2 dimers. (i) The 3-fold arrangement of the three closed ACE2 dimers viewed from the viral end with the 6crz trimer removed. (j-l) Three views of the smallest function of spike/ACE2 super-complex, containing three ACE2 dimers and three spike trimers surrounding the central spike trimer. (j) Viral end view. (k) Side view with viral membrane the bottom and host membrane on the top (l) Host end view. See Video 2A and 2B for opening and closing of the ACE2 cleft in complexes with the spike trimers with and without an inhibitor of ACE2 bound. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
The spacing between two RBDs bound within the ACE2 dimer in either open or closed conformation is far greater than the distance between any two RBDs within each S trimer and therefore this would exclude the possibility that these two RBDs belong to a single S trimer (Fig. 3a, e). Thus, two RBDs bound to ACE2 dimer would have to come from two adjacent S trimers on the surface of coronavirus. Upon superposition of the common RBD-PD interactions present in both the ACE2/RBD complex and the spike protein/PD complex [19], [20], [22], [24], [25], we dock two S trimers to each ACE2 dimer, and three ACE2 dimers to each S trimer. This process can be repeated to form larger super-complexes having multiple ACE2 dimers and multiple S trimers.
3.4. Symmetric super-complex of one ACE2 dimer with two SARS-CoV-2 S trimers
When two S trimers are docked onto the inhibited conformation of the ACE2 dimer, the 3-fold axes of the S trimers are approximately parallel to the 2-fold dyad of ACE2 dimer. The viral membrane and host membrane are also approximately parallel with each other (Fig. 3f, g). The estimated distance between the two membranes is ∼ 250 Å, which is approximately the length of a fusion-active extended intermediate. The spacing between the two S trimers bound to the single ACE2 dimer is about 140 Å, matching the spacing of two adjacent S trimers on the viral surface [13] to represent an initial binding mode. Given that two adjacent S trimers are likely arrange with P2 symmetry on the surface of coronaviruses, the dyad of two closest S trimers should be aligned with that of the ACE2 dimer [13]. Therefore, it is likely that coronaviruses have exploited the symmetry and dimension of the ACE2/B(0)AT complex for simultaneously binding two adjacent S trimers on their surface.
When two S trimers are docked onto the open conformation of the ACE2 dimer, the PD of ACE2 rotates outward by about 17° from its closed conformation as does the bound RBD (Fig. 3; Video 2). As a result, the 3-fold axis of the S trimer and the 2-fold dyad of ACE2 dimer have a tilt angle of about 20° (and the spacing between the two S trimers at the viral membrane surface increases to about 260 Å). However, the tilted configuration is not consistent with the convex surface of the virus particle, consequently the ACE2 open conformation cannot simultaneously bind two S trimers on the surface of the same virus. This observation suggests that the open conformation of the ACE2 dimer can bind only one S trimer and that the open-to-closed transition must occur before it binds the second S trimer. Given the nanomolar binding affinity between the RBD of the S protein and the PD of ACE2, this binding energy is sufficient to drive the open-to-closed transition of ACE2 dimer, and likely lock down the PD of ACE2 in the inhibited conformation. In this conformation, the peptidase active site of ACE2 should no longer be accessible for processing its substrate. Therefore, our analysis suggests that before the viral membrane process, SARS-CoV-2 S proteins appear to have an ability to disable ACE2′s normal extracellular peptidase activity.
3.5. Symmetric super-complex of three ACE2 dimers with SARS-CoV-2 S trimers
Our modeling analysis shows that binding the two S trimers of SARS-CoV-2 to the membrane-bound ACE2 dimer likely abolishes the normal function of ACE2 activity because the density of S trimers on the surface of coronaviruses is relatively high, resulting in each ACE2 dimer binding to two adjacent S trimers. However, favorable binding alone may not be sufficient for SARS-CoV-2 to enter the host cell for replication because the central pore of the S trimer remains obstructed, and the S2 central stalk cannot be extended. A single ACE2 attached SARS-CoV-2 could not sufficiently open the central pore of the S trimer when its two RBDs remain in “down” conformations. There are two possible mechanisms for how the S1 central pore may open: (i) cooperatively opening the S central pore upon simultaneous binding three ACE2 dimers to one S trimer or (ii) non-specific dissociation of the S1 trimer. However, any dissociation of the S1 trimer before the viral and host cellular membranes are aligned would rapidly trigger the prefusion to post-fusion conversion of S2 and abort the viral membrane fusion reaction.
We docked three ACE2 dimers with B(0)AT onto the symmetric open-1 and open-2 conformations of the S1 trimer and observed that only the open-2 form could allow for simultaneous binding of three ACE2 dimers without physical overlapping (Fig. 4a-c). The binding of one ACE2 dimer to one RBD in the open-1 conformation has physically excluded the binding of the second and third ACE2 dimers to the other two RBDs. Without ACE2 dimers, these two RBDs may prefer to adopt the “down” configuration, establishing interactions with each other as in the observed asymmetric structure [5], [20]. Upon the binding of ACE2 to the first RBD of S1 trimer, the equilibirum of the remaining two RBDs is shifted to the high-affinity state for binding additional ACE2 [57]. Thus, with increasing cellular ACE2 level, binding additional ACE2 dimers may be possible. This would accelerate the closed to open-1 transition as well as the open-1 to open-2 transition, stabilizing the symmetric open-2 conformation. The central pore of the S trimer in the symmetric open-2 conformation is large enough for the extended S2 central stalk to pass through the pore (Fig. 2, and see below).
In the presence of potent neutralizing antibodies, the equilibrium of the S protein conformations of SARS-CoV-2 has been altered, and the population of three RBDs in an all “up” conformation increases [9]. This all-up conformation would accelerate the conversion of the S2 fragment in the loaded spring state to an extended state before the virus is attached to the host membrane via binding to ACE2. In fact, these antibodies may disable this fusion apparatus by causing it to misfire. In situ studies have shown that the virus has a mixed population of both prefusion and post-fusion structures on its surface [36], [16], [17], [18].
3.6. Viral membrane fusion mechanism at the cellular level
Viral membrane fusion requries an extension of its S2 central stalk that uses the sticky FP to insert itself into the host cellular membrane and then pulls the membrane toward the viral membrane for fusion. This process is irreversible so that it has a one-time capacity as a loaded spring for viral membrane fusion. When it occurs before alignment of viral and host membranes, it becomes abortive for fusion reaction. Thus, this process could be a target of anti-viral drug design. Having structures of only the prefusion and post-fusion states is not sufficient for understanding this entire process. Thus, it is essential to have snapshots of all key intermediates to fully understand this process.
Since an initial proposal of the viral membrane fusion mechanism has been published [29], [30], transient fusion-active intermediates have yet to be captured structurally. Once a spike protein is in an extended fusion-active conformation, it is rapidly converted to the most stable post-fusion state via a hemi-fusion state with or without membrane. In the hemi-fusion state, the central stalk lies horizontally to the membranes, becoming a single contact line between the two membranes while all S1 fragments are dissociated from the S2 central stalk. Before and after the hemi-fusion state, the orientation of the S2 central stalk is inverted along with a migration of FP from one end to the other. In this case, the relative orientation of the elongated S2 central stalk to that of the S1 trimer is difficult to model precisely. In this study, we remove all moving parts associated with the S1 trimer and keep only the central 3-helical coiled-coils of the S2 trimer to measure the dimension of the S2 trimer within the S1 trimer.
We started this modeling using the murine hepatitis viral (MHV) S trimer in both its prefusion and post-fusion states. Our modeling results have been fully confirmed after the post-fusion cryo-EM structure of the SARS-CoV-2 S2 fragment became available (see OSM, Figs. S2, S3) [22], [36], [59]. This modeling provides geometric constraints for understanding the viral membrane fusion mechanism at the cellular level. In the early steps of this process, the S2 trimer remains tightly associated with the S1 trimer, which is bound to ACE2 in a tripodal configuration. The three ACE2 dimers pull the three legs of the tripod by moving away from each other (Fig. 1, Fig. 4; Video 3A, 3B, 3C). This causes thinning of the host cellular membrane at the midpoint of the three ACE2 dimers because a fixed number of lipid molecules occupies an enlarged membrane surface (Video 3C). This destabilizes the host cellular membrane for insertion of FPs. This motion also brings the host cellular and the viral membranes closer together (Video 3C). Once the FPs of the S2 fusion trimer are inserted into the host cellular membrane, it would be converted rapidly to a hemi-fusion state where the location of the 3-fold axis of the host cellular membrane has depleted lipid molecules at the midpoint of the super-complexes (Video 3C). Topologically, the formation of three-dimensional hemi-fusion state between a host cell and virus would require the cooperation of multiple copies of the S trimer-ACE2 dimer because a single spike trimer in the hemi-fusion state could only make a single contact line between the two membranes and would likely not be sufficient to open up a large pore connecting the two membranes. A minimal requirement for a pore formation is via three contact lines that are arranged in a closed triangle. The membrane fusion process is thus a series of highly choreographic events, requiring cooperative interactions between the ACE2-bound S1 and S2 during the conversion of S2. One can accelerate the prefusion to post-fusion conversion of the spike protein in vitro by simply treating with protease followed by heating denaturation [21]. However, such conversion is non-productive for membrane fusion.
3.7. Free energy calculations for formation of proposed supercomplexes
Free energy calculated for formation of one, two, and three ACE2/B0AT dimers with one spike trimer in solution as well as in vacuum shows a complicated pattern of allosteric regulation during complex formation (Fig. 5, Table 1). In vacuum, the binding of one ACE2/B0AT dimer with one spike trimer is strongly favored energetically. The binding of a second or a third ACE2/B0AT dimer has a binding energy almost 10- or 3-times larger, respectively, relative to the already bound ACE2/B0AT dimer. Solvation effects apparently modulate this pattern with very limited positive cooperativity in ACE2/B0AT dimer binding. In the solution state, the binding of the first ACE2/B0AT dimer is found to be slightly unfavorable, whereas the binding of a second or third ACE2B9AT dimer is favored by an additive 45–50 kcal/mol.
Fig. 5.

Models for free energy calculation. (a) spike trimer with one RBD in an up position and ACE2/B0AT with one RBD (green) bound, which RBD would be replaced with the trimeric spike. (b) One ACE2/B0AT dimer to one spike trimer. (c) Two ACE2/B0AT dimers bound to one spike trimer. (d) Three ACE2/B0AT dimers bound to one spike trimer. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Table 1.
Free energies calculated for formation of trimeric spike with one, two, and three ACE-2/B0AT dimers (without transmembrane domains and without glycans because neither is directly involved in complex formation).a.
| Spike trimer: ACE-2 dimer | Binding Free energy (ΔG0, kcal/mole) |
||
|---|---|---|---|
| Vacuum | Solvation | Overall | |
| 1:1 as in Complex-1 | −171.5 | 177.1 | 5.6 |
| 1:2 as in Complex-2 | −1845.9 | 1790.8 | −55.2 |
| 1:3 as in Complex-3 | −2395.3 | 2295.1 | −100.2 |
a.Omission of transmembrane domains and glycans can affect solvent energy to some extent, which would be likely canceled out because they are far away from the site of complex formation.
Part of this regulation is related to free energy of an up-to-down conversion of the RBD of the trimeric spike protein. When two RBDs are in the down position, they interact with each other favorable so that the conversion of the down-to-up position is energetically unfavorable [19], [20]. The binding of two additional ACE2/B0AT dimers can overcome this energy barrier but not the binding of one additional ACE2/B0AT dimer. Our computations therefore reveal how SARS-CoV-2 have maximized its capacity for the membrane fusion reaction. It always has one RBD of the spike trimer in an “up” position for an initial attachment to the host membrane. When the host cell membrane has a low level of ACE2/B0AT dimers, the virus is evolutionarily compliant because the fusion process cannot be completed for the spike trimer with single ACE2/B0AT dimer. When the level of ACE2/B0AT dimer is upregulated, the virus begins to form supercomplexes and initates the membrane fusion reaction.
4. Discussion
The step-by-step visualization of viral membrane fusion is extremely challenging because the reaction intermediates are short-lived. Moreover, the fusion process can be studied only in pseudo viruses or live viruses. However, the resolution of electron tomographic structures in live viruses is still very limited and thus these studies would not provide atomic details of this process. The FP of the spike protein is initially located near the viral membrane in the prefusion state, transiently extending over 130–190 Å to insert into the host membrane in the first fusion intermediate, and then pulls the viral and host membranes towards each other to merge them – like the sticky tongue of a cameleon lizard catching a prey. So far, only two of the five predicted spike conformations of any virus have been structurally characterized, corresponding only to the beginning and ending points of the fusion process. The reason why it is difficult to capture the three other fusion intermediates is that they are extremely unstable and rapidly convert to the ultra-stable, post-fusion endpoint. Even the prefusion state of the spike protein of SARS-CoV-2 is very unstable such that its wild-type protein is extremely difficult to purify in sufficient amount for structural studies. Current structural information on that state is obtained from laboratory-engineered mutant spike proteins with multiple mutations that are designed to specifically stabilize it [20].
Similar stabilizing mutational approaches may be applicable to other unstable fusion intermediates for structural studies. Our computational models provide structural information on the membrane fusion process that should be particularly valuable to design mutants of detectable intermediates for structural studies. In this study, we have focused on symmetric spike trimers to provide information on the dimensions of its central pore, and how to keep the S2 fragment in the prefusion state. We find that when the S1 fragment falls off the complex prematurely, or when its central pore opens asymmetrically (which is known to occur), nothing could prevent the rapid conversion of the S2 fragment. When the conversion occurs away from the host cells, the spike protein would undergo an abortive fusion process that would make the virus non-infectious. The computationally modeled structure of the first fusion intermediate suggests that it will rapidly converge to the post-fusion conformation due to a large ‘downhill’ free energy gradient. One could target the conversion from the prefusion state to the first intermediate state for drug design. This could have two orthogonal approaches. The first approach is to design small molecular inhibitors that suppress this conversion, and the second approach is to rapidly trigger this conversion before the virus approaches the host cells. The second approach can be effective because it could permanently disable the infectiousness of the virus while the first approach only slows down the viral infection.
We anticipate our analysis of membrane geometric constraints upon formation of super-complexes between dimeric ACE2 with trimeric spike protein should provide valuable insights into the observed correlation of SARS-CoV-2 infection and organ failure by disabling the normal function of the ACE2 that produces the vasodilator 7-residue peptide hormone of angiotensin [60]. Thus, the reported results could inspire the exploration of a new treatment of COVID-19 with the peptidase domain of human recombinant ACE2 for neutralizing the spike protein while maintaining a sufficient level of the vasodilator. Infection of SARS-CoV in cell lines appears to increase the level of angiotensin-II (the substrate of ACE2) and decrease the level of the ACE2 expression, which is likely responsible for organ failure [61], [62]. Currently, treatment with soluble human recombinant ACE2 (not just its peptidase domain as we propose) is underway [63], [64].
Declaration of Competing Interest
The authors declare that there is no competing interest in this study.
Acknowledgements
This work was supported by GM106121 (V.S.B) and supercomputer resources from NERSC. We thank Drs. Joan A. Steitz and Walther Mothes for discussion during the course of this work, and Angela Miccinello for editorial work.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.csbj.2021.08.053.
Contributor Information
Jimin Wang, Email: jimin.wang@yale.edu.
Victor S. Batista, Email: victor.batista@yale.edu.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Zhou P., Yang X.-L., Wang X.-G., Hu B., Zhang L., Zhang W. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu F., Zhao S.u., Yu B., Chen Y.-M., Wang W., Song Z.-G. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lu R., Zhao X., Li J., Niu P., Yang B.o., Wu H. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395:565–574. doi: 10.1016/S0140-6736(20)30251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhu N.a., Zhang D., Wang W., Li X., Yang B.o., Song J. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. 2020;382:727–733. doi: 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lan J., Ge J., Yu J., Shan S., Zhou H., Fan S. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020;581:215–220. doi: 10.1038/s41586-020-2180-5. [DOI] [PubMed] [Google Scholar]
- 6.Shang J., Ye G., Shi K.e., Wan Y., Luo C., Aihara H. Structural basis of receptor recognition by SARS-CoV-2. Nature. 2020;581:221–224. doi: 10.1038/s41586-020-2179-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yan R., Zhang Y., Li Y., Xia L.u., Guo Y., Zhou Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science. 2020;367:1444–1448. doi: 10.1126/science.abb2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cao Y., Su B., Guo X., Sun W., Deng Y., Bao L. Potent neutralizing antibodies against SARS-CoV-2 identified by high-throughput single-cell sequencing of convalescent patients' B cells. Cell. 2020 doi: 10.1016/j.cell.2020.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barnes C.O., West A.P., Huey-Tubman K.E., Hoffmann M.A.G., Sharaf N.G., Hoffman P.R. Structures of human antibodies bound to SARS-CoV-2 spike reveal common epitopes and recurrent features of antibodies. Cell. 2020;182:828–842.e16. doi: 10.1016/j.cell.2020.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xia S., Liu M., Wang C., Xu W., Lan Q., Feng S. Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 2020;30:343–355. doi: 10.1038/s41422-020-0305-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tang T., Bidon M., Jaimes J.A., Whittaker G.R., Daniel S. Coronavirus membrane fusion mechanism offers a potential target for antiviral development. Antiviral Res. 2020;178:104792. doi: 10.1016/j.antiviral.2020.104792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ng M.L., Lee J.W.M., Leong M.L.N., Ling A.-E., Tan H.-C., Ooi E.E. Topographic changes in SARS coronavirus-infected cells at late stages of infection. Emerg Infect Dis. 2004;10:1907–1914. doi: 10.3201/eid1011.040195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Neuman B.W., Adair B.D., Yoshioka C., Quispe J.D., Orca G., Kuhn P. Supramolecular architecture of severe acute respiratory syndrome coronavirus revealed by electron cryomicroscopy. J Virol. 2006;80:7918–7928. doi: 10.1128/JVI.00645-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barcena M., Oostergetel G.T., Bartelink W., Faas F.G.A., Verkleij A., Rottier P.J.M. Cryo-electron tomography of mouse hepatitis virus: insights into the structure of the coronavirion. Proc Natl Acad Sci U S A. 2009;106:582–587. doi: 10.1073/pnas.0805270106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ertel KJ, Benefield D, Castano-Diez D, Pennington JG, Horswill M, den Boon JA, Otegui MS, Ahlquist P. 2017. Cryo-electron tomography reveals novel features of a viral RNA replication compartment. Elife 6:e25940. [DOI] [PMC free article] [PubMed]
- 16.Klein S., Cortese M., Winter S.L., Wachsmuth-Melm M., Neufeldt C.J., Cerikan B. SARS-CoV-2 structure and replicaiton characrterized by in situ cryo-electron tomography. Nature Comm. 2020;11:5885. doi: 10.1038/s41467-020-19619-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Turoňová B., Sikora M., Schürmann C., Hagen W.J.H., Welsch S., Blanc F.E.C. In situ structural analysis of SARS-CoV-2 spike reveals flexibility mediated by three hinges. Science. 2020 doi: 10.1126/science:abd5223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ke Z., Oton J., Qu K., Cortese M., Zila V., McKeane L. Structures and distributions of SARS-CoV-2 spike proteins on intact virions. Nature. 2020 doi: 10.1038/s41586-020-2665-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wrapp D., Wang N., Corbett K.S., Goldsmith J.A., Hsieh C.-L., Abiona O. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367:1260–1263. doi: 10.1126/science.abb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walls A.C., Park Y.J., Tortorici M.A., Wall A., McGuire A.T., Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. 2020;181(281–292) doi: 10.1016/j.cell.2020.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li F. Structure, function, and evolution of coronavirus spike proteins. Annu Rev Virol. 2016;3:237–261. doi: 10.1146/annurev-virology-110615-042301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Song W., Gui M., Wang X., Xiang Y.e., Heise M.T. Cryo-EM structure of the SARS coronavirus spike glycoprotein in complex with its host cell receptor ACE2. PLoS Pathog. 2018;14:e1007236. doi: 10.1371/journal.ppat.1007236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yuan Y., Cao D., Zhang Y., Ma J., Qi J., Wang Q. Cryo-EM structures of MERS-CoV and SARS-CoV spike glycoproteins reveal the dynamic receptor binding domains. Nat Commun. 2017;8:15092. doi: 10.1038/ncomms15092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gui M., Song W., Zhou H., Xu J., Chen S., Xiang Y.e. Cryo-electron microscopy structures of the SARS-CoV spike glycoprotein reveal a prerequisite conformational state for receptor binding. Cell Res. 2017;27:119–129. doi: 10.1038/cr.2016.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kirchdoerfer R.N., Wang N., Pallesen J., Wrapp D., Turner H.L., Cottrell C.A. Stabilized coronavirus spikes are resistant to conformational changes induced by receptor recognition or proteolysis. Sci Rep. 2018;8:15701. doi: 10.1038/s41598-018-34171-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li F., Li W., Farzan M., Harrison S.C. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science. 2005;309:1864–1868. doi: 10.1126/science.1116480. [DOI] [PubMed] [Google Scholar]
- 27.Tipnis S.R., Hooper N.M., Hyde R., Karran E., Christie G., Turner A.J. A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J Biol Chem. 2000;275:33238–33243. doi: 10.1074/jbc.M002615200. [DOI] [PubMed] [Google Scholar]
- 28.Donoghue M., Hsieh F., Baronas E., Godbout K., Gosselin M., Stagliano N. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1–9. Circ Res. 2000;87 doi: 10.1161/01.res.87.5.e1. E1-9. [DOI] [PubMed] [Google Scholar]
- 29.Harrison S.C. Mechanism of membrane fusion by viral envelope proteins. Adv Virus Res. 2005;64:231–261. doi: 10.1016/S0065-3527(05)64007-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harrison S.C. Viral membrane fusion. Nat Struct Mol Biol. 2008;15:690–698. doi: 10.1038/nsmb.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Harrison S.C. Viral membrane fusion. Virology. 2015;479-480:498–507. doi: 10.1016/j.virol.2015.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.White J.M., Delos S.E., Brecher M., Schornberg K. Structures and mechanisms of viral membrane fusion proteins: multiple variations on a common theme. Crit Rev Biochem Mol Biol. 2008;43:189–219. doi: 10.1080/10409230802058320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kielian M. Mechanisms of virus membrane fusion proteins. Annu Rev Virol. 2014;1:171–189. doi: 10.1146/annurev-virology-031413-085521. [DOI] [PubMed] [Google Scholar]
- 34.Rey F.A., Lok S.-M. Common features of enveloped viruses and implications for immunogen design for next-generation vaccines. Cell. 2018;172:1319–1334. doi: 10.1016/j.cell.2018.02.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Colman P.M., Lawrence M.C. The structural biology of type I viral membrane fusion. Nat Rev Mol Cell Biol. 2003;4:309–319. doi: 10.1038/nrm1076. [DOI] [PubMed] [Google Scholar]
- 36.Cai Y., Zhang J., Xiao T., Peng H., Sterling S.M., Walsh R.M. Distinct conformational states of SARS-CoV-2 spike protein. Science. 2020 doi: 10.1126/science:abd4251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pancera M., Zhou T., Druz A., Georgiev I.S., Soto C., Gorman J. Structure and immune recognition of trimeric pre-fusion HIV-1 Env. Nature. 2014;514:455–461. doi: 10.1038/nature13808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schaffer L. A model for insulin binding to the insulin receptor. Eur J Biochem. 1994;221:1127–1132. doi: 10.1111/j.1432-1033.1994.tb18833.x. [DOI] [PubMed] [Google Scholar]
- 39.Li J., Choi E., Yu H., Bai X.C. Structural basis of the activation of type 1 insulin-like growth factor receptor. Nat Commun. 2019;10:4567. doi: 10.1038/s41467-019-12564-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Uchikawa E, Choi E, Shang G, Yu H, Bai XC. 2019. Activation mechanism of the insulin receptor revealed by cryo-EM structure of the fully liganded receptor-ligand complex. Elife 8:48630. [DOI] [PMC free article] [PubMed]
- 41.Sainz B., Jr., Rausch J.M., Gallaher W.R., Garry R.F., Wimley W.C. The aromatic domain of the coronavirus class I viral fusion protein induces membrane permeabilization: putative role during viral entry. Biochemistry. 2005;44:947–958. doi: 10.1021/bi048515g. [DOI] [PubMed] [Google Scholar]
- 42.Guillén J., Kinnunen P.K.J., Villalaín J. Membrane insertion of the three main membranotropic sequences from SARS-CoV S2 glycoprotein. Biochim Biophys Acta. 2008;1778:2765–2774. doi: 10.1016/j.bbamem.2008.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mahajan M., Bhattacharjya S. NMR structures and localization of the potential fusion peptides and the pre-transmembrane region of SARS-CoV: implications in membrane fusion. Biochim Biophys Acta. 2015;1848:721–730. doi: 10.1016/j.bbamem.2014.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mahajan M., Chatterjee D., Bhuvaneswari K., Pillay S., Bhattacharjya S. NMR structure and localization of a large fragment of the SARS-CoV fusion protein: implications in viral cell fusion. Biochim Biophys Acta Biomembr. 2018;1860:407–415. doi: 10.1016/j.bbamem.2017.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Meher G., Bhattacharjya S., Chakraborty H. Membrane cholesterol modulates oligomeric status and peptide-membrane interaction of severe acute respiratory syndrome coronavirus fusion peptide. J Phys Chem B. 2019;123:10654–10662. doi: 10.1021/acs.jpcb.9b08455. [DOI] [PubMed] [Google Scholar]
- 46.Chakraborty H., Bhattacharjya S. Mechanistic insights of host cell fusion of SARS-CoV-1 and SARS-CoV-2 from atomic resolution structure and membrane dynamics. Biophys Chem. 2020;265:106438. doi: 10.1016/j.bpc.2020.106438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pattnaik G.P., Bhattacharjya S., Chakraborty H. Enhanced cholesterol-dependent hemifusion by internal fusion peptide 1 of SARS coronavirus-2 compared to Its N-terminal counterpart. Biochemistry. 2021;60:559–562. doi: 10.1021/acs.biochem.1c00046. [DOI] [PubMed] [Google Scholar]
- 48.Borkotoky S., Dey D., Banerjee M. Computational insight into the mechanism of SARS-CoV-2 membrane fusion. J Chem Inf Model. 2021;61:423–431. doi: 10.1021/acs.jcim.0c01231. [DOI] [PubMed] [Google Scholar]
- 49.Lutkenhaus and J., Addinall S.G. Bacterial cell division and the Z ring. Annu Rev Biochem. 1997;66:93–116. doi: 10.1146/annurev.biochem.66.1.93. [DOI] [PubMed] [Google Scholar]
- 50.Miller A.L. The contractile ring. Curr Biol. 2011;21:R976–R978. doi: 10.1016/j.cub.2011.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Emsley P., Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 52.Winn M.D., Ballard C.C., Cowtan K.D., Dodson E.J., Emsley P., Evans P.R. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Delano W.L. Delano Scientific; San Carlos, CA: 2009. The Pymol Molecular Graphics System. [Google Scholar]
- 54.Jo S, Vargyas M, Vasko-Szedlar J, Roux B, Im W. 2008. PBEQ-Solver for online visualization of electrostatic potential of biomolecules. Nucleic Acids Res 36:W270-5. [DOI] [PMC free article] [PubMed]
- 55.Woo H., Park S.-J., Choi Y.K., Park T., Tanveer M., Cao Y. Developing a fully-glycosylated full-length SARS-CoV-2 spike protein model in a viral membrane. J Phys Chem B. 2020 doi: 10.1021/acs.jpcb.0c0455310.1021/acs.jpcb.0c04553.s001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sztain T, Ahn SH, Bogetti AT, Casalino L, Goldsmith JA, McCool RS, Kearns FL, McCammon JA, McLellan JS, Chong LT, Amaro RE. 2021. A glycan gate controls opening of the SARS-CoV-2 spike protein. bioRxiv doi:10.1101/2021.02.15.431212. [DOI] [PMC free article] [PubMed]
- 57.Guo L., Bi W., Wang X., Xu W., Yan R., Zhang Y. Engineered trimeric ACE2 binds viral spike protein and locks it in “Three-up” conformation to potently inhibit SARS-CoV-2 infection. Cell Res. 2021;31:98–100. doi: 10.1038/s41422-020-00438-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Towler P., Staker B., Prasad S.G., Menon S., Tang J., Parsons T. ACE2 X-ray structures reveal a large hinge-bending motion important for inhibitor binding and catalysis. J Biol Chem. 2004;279:17996–18007. doi: 10.1074/jbc.M311191200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Walls A.C., Tortorici M.A., Snijder J., Xiong X., Bosch B.-J., Rey F.A. Tectonic conformational changes of a coronavirus spike glycoprotein promote membrane fusion. Proc Natl Acad Sci U S A. 2017;114:11157–11162. doi: 10.1073/pnas.1708727114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Verdecchia P., Cavallini C., Spanevello A., Angeli F. The pivotal link between ACE2 deficiency and SARS-CoV-2 infection. Eur J Intern Med. 2020;76:14–20. doi: 10.1016/j.ejim.2020.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kuba K., Imai Y., Rao S., Gao H., Guo F., Guan B. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat Med. 2005;11:875–879. doi: 10.1038/nm1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Imai Y., Kuba K., Rao S., Huan Y.i., Guo F., Guan B. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature. 2005;436:112–116. doi: 10.1038/nature03712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang K., Gheblawi M., Oudit G.Y. Angiotensin Converting Enzyme 2: a double-edged sword. Circulation. 2020;142:426–428. doi: 10.1161/CIRCULATIONAHA.120.047049. [DOI] [PubMed] [Google Scholar]
- 64.Batlle D, Wysocki J, Satchell K. 2020. Soluble angiotensin-converting enzyme 2: a potential approach for coronavirus infection therapy? Clin Sci (Lond) 134:543-545. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.