

Abstract
Introduction
Naïve T cells activated by cognate antigens and co-stimulation proliferate and differentiate to effector T cells. The shift from a resting to a proliferative state entails profound changes in cellular metabolism, in particular increases in glycolysis, glutaminolysis, and mitochondrial metabolism, to produce high levels of ATP. T cells depend on a translational burst to produce the metabolic enzymes that support an increase in metabolism, and to produce the protein components of clonal T cell progeny and their cytokines. Paradoxically, the metabolic processes that provide energy for growth and expansion also produce reactive oxygen species (ROS), which are capable of inducing oxidative stress that leads to translation repression. On the other hand, ROS function as second messengers in T cell receptor (TCR) signaling and are essential for proliferation and development of effector function. This suggests that in order to preserve the signaling activities of ROS, protective mechanisms against oxidative stress may occur at multiple levels beyond simply reducing ROS levels in T cells.
Rationale
From a mouse forward genetic screen for mutations affecting immunity, we previously identified a recessive mutation in Slfn2, leading to elevated susceptibility to bacterial and viral infections, and diminished numbers of T cells that failed to proliferate in response to infection and diverse proliferative stimuli. Here, we aimed to investigate the molecular function of SLFN2 in T cells by generating mice with a T cell-specific deletion of Slfn2
Results
T cell-specific SLFN2-deficient mice displayed compromised humoral and cellular immune responses to immunization with a T cell-dependent antigen and to infection with mouse cytomegalovirus, respectively. These defects stemmed from impaired CD4+ and CD8+ T cell proliferative responses to TCR stimulation, despite normal induction of TCR signaling events in SLFN2-deficient T cells. Whereas IL-2 production by SLFN2-deficient T cells was normal after TCR stimulation, the cells failed to proliferate in response to exogenous IL-2. IL-2R signaling was defective. Abrogation of the mitogenic effects of IL-2 was due to a failure to translationally upregulate the β and γ chains of the IL-2 receptor. Indeed, there was a globally dampened translational response to TCR activation in SLFN2-deficient T cells in vitro and in vivo.
The cellular oxidative stress response includes translation repression by tRNA fragments generated by angiogenin (ANG), a stress-induced tRNA-directed RNase. ANG cleaves tRNAs within their anticodon loops, yielding 30–40-nucleotide tRNA fragments (tiRNA). In response to TCR activation, SLFN2-deficient T cells accumulated tiRNA, which could be reduced by antioxidant treatment or knockdown or inhibition of ANG. Moreover, global translation rates in activated SLFN2-deficient T cells could be rescued by antioxidant treatment or by ANG knockdown. SLFN2 directly bound to tRNAs, but exerted no nucleolytic activity towards them, unlike other SLFN proteins. Binding of SLFN2 to tRNAs blocked tRNA cleavage by ANG, thereby averting tiRNA accumulation and tiRNA-mediated translation repression.
Conclusion
We describe a protective mechanism by which SLFN2 shields tRNA from oxidative stress-induced cleavage, thereby preventing the translation inhibitory effects of ROS produced in response to T cell activation. Importantly, SLFN2 acts downstream of ROS production itself, leaving ROS functions in T cell metabolism and signaling intact. We identify angiogenin as a stress-activated RNase whose effects are opposed by SLFN2 in T cells. Our data provide further support for a key role of SLFN family members in the regulation of RNA and translation.
Reactive oxygen species (ROS) increase in activated T cells because of metabolic activity induced to support T cell proliferation and differentiation. We show that these ROS trigger an oxidative stress response leading to translation repression. This response is countered by Schlafen 2 (SLFN2), which directly binds tRNAs to protect them from cleavage by the RNase angiogenin (ANG). T cell-specific SLFN2 deficiency results in the accumulation of tRNA fragments, which inhibit translation and promote stress granule formation. IL-2Rβ and IL-2Rγ fail to be translationally upregulated following TCR stimulation, rendering SLFN2-deficient T cells insensitive to IL-2’s mitogenic effects. SLFN2 confers resistance against the ROS-mediated translation-inhibitory effects of oxidative stress normally induced by T cell activation, permitting robust protein synthesis necessary for T cell expansion and immunity.
One-Sentence Summary:
SLFN2 upholds translation in activated T cells by protecting tRNA from cleavage by ANG in response to oxidative stress.
Graphical Abstract
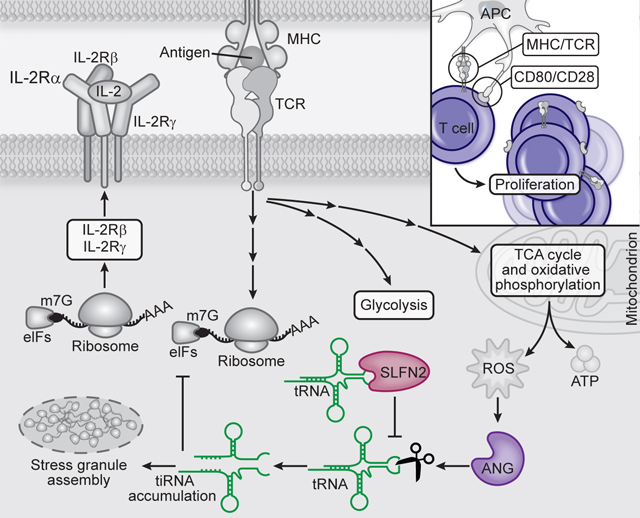
In response to antigen engagement and costimulation, T cells reprogram their metabolism to produce high levels of energy and biosynthetic precursors necessary for the growth and clonal expansion of activated T cells and their subsequent differentiation to effectors (1). This metabolic reprogramming occurs within 24 hours after stimulation, a period when T cells increase in size (2), and depends critically upon translational induction of metabolic enzymes (3). During the subsequent 24 to 72 hours when T cells are actively proliferating, translation of mRNAs encoding ribosome proteins is dramatically upregulated, followed about 3 days later by an abrupt downregulation of translation as T cells stop dividing prior to the contraction phase (4).
Reactive oxygen species (ROS) are byproducts of the activated T cell metabolic processes that produce ATP (5, 6). ROS act as a second messenger in TCR signaling necessary to promote T cell proliferation (7). However, in the absence of antioxidant production, ROS accumulate to compromise metabolic reprogramming and impair T cell proliferation (8). The cellular oxidative stress response includes translation repression, the increased expression of stress response genes, and the formation of stress granules (9, 10). Although translation repression by phospho-eIF2α is well characterized (11), a role for tRNA cleavage in this process has only recently been established (12–14). Oxidative stress-induced tRNA cleavage is mediated by angiogenin (ANG) (15–17), a member of the RNase superfamily (18). ANG cleaves tRNAs primarily within their anticodon loops, yielding 30–40-nucleotide (nt) tRNA-derived stress-induced fragments (tiRNA), which may displace eIF4G from the m7G cap or silence translation similarly to microRNAs (19, 20).
Here, we provide evidence for a T cell oxidative stress response resulting from TCR and costimulatory receptor engagement, which is characterized by the induction of tiRNAs, translation repression, and stress granule formation. This response is suppressed by Schlafen 2 (SLFN2). Thus, T cells require SLFN2 to maintain their capacity for rapid, abundant protein translation necessary for T cell-mediated immunity.
Results
Impaired T cell-mediated immunity in T cell-specific Slfn2-deficient mice
Using N-ethyl-N-nitrosourea, we previously created mice with a missense mutation in Slfn2, leading to lymphoid and myeloid immunodeficiency (21). To examine the T cell-intrinsic role of SLFN2, we generated mice in which Slfn2 was conditionally deleted in CD4+ and CD8+ T cells (fig. S1, A to C). Slfn2 mRNA was barely detectable in splenic T cells from CD4-Cre; Slfn2f/f mice (fig. S1D). The frequencies of CD4+ and CD8+ T cells in the thymus, spleen, lymph node, and blood were similar for control (Slfn2f/f) and CD4-Cre; Slfn2f/f mice, suggesting there was no cell-autonomous requirement for SLFN2 in T cell survival (fig. S1E). However, T cell-dependent antibody responses were reduced in CD4-Cre; Slfn2f/f mice compared to Slfn2f/f mice after immunization with a replication-defective recombinant Semliki Forest virus vector encoding β-galactosidase (rSFV-β-gal) (22) (Fig. 1A). By contrast, T cell-independent antibody responses to NP-Ficoll immunization were comparable between Slfn2f/f and CD4-Cre; Slfn2f/f mice (Fig. 1A). Strikingly, the function of CD4-Cre; Slfn2f/f CD8+ T cells was impaired, as indicated by the diminished frequency of antigen-specific IFNγ+ CD8+ T cells induced by restimulation with either MCMV-derived peptides or PMA and ionomycin 10 days after acute MCMV infection (Fig. 1, B and C). Thus, SLFN2 function in T cells is necessary for T cell-mediated immunity.
Fig. 1. Impaired T cell-mediated immunity in T cell-specific SLFN2-deficient mice.

(A) Serum β-gal-specific IgG on day 14 after immunization (left) and NP-specific IgM on day 7 after immunization (right) of Slfn2f/f and CD4-Cre; Slfn2f/f mice (n=10 per genotype) with rSFV-encoded β-gal and NP-Ficoll, respectively. Each symbol represents an individual mouse. (B) Representative flow cytometry analysis of IFN-γ+ CD8+ T cells isolated from the spleens of Slfn2f/f and CD4-Cre; Slfn2f/f mice on day 10 after MCMV infection and then stimulated in vitro with MCMV peptides m139 or M45 or PMA–ionomycin. Numbers indicate percent cells in boxed area. (C) Quantification of the percentage of IFN-γ+ CD8+ T cells among 1×106 spleen cells assessed by flow cytometry as in B (n=4 per genotype). (D) Mean EAE clinical scores of age-matched Slfn2f/f and CD4-Cre; Slfn2f/f mice (n=10 per genotype) on the indicated days after immunization with MOG35–55. (E) IL-17 (left) or IFN-γ (right) concentration in the culture medium of spleen cells harvested from Slfn2f/f and CD4-Cre; Slfn2f/f mice (n=10 per genotype) on day 10 after MOG35–55 immunization and stimulated with MOG35–55 in vitro for the indicated time. (F) Immunoblot analysis of GFP-SLFN2 expression in splenic T cells isolated from BAC transgenic mice expressing GFP-tagged SLFN2 and stimulated with anti-CD3/CD28 for the indicated time (left). Quantification of the average intensity of the GFP-SLFN2 protein bands normalized to time 0 hour from three independent experiments (right). (G) Representative flow cytometry analysis of CFSE intensity in splenic Slfn2f/f or Slfn2−/− (from CD4-Cre; Slfn2f/f mice) CD4+ and CD8+ T cells left unstimulated or stimulated with anti-CD3/CD28 for 48 hours and 72 hours (left). Numbers indicate percent dye-positive cells in bracketed region. Frequency of CFSElo cells among dye-positive CD4+ and CD8+ T cells stimulated with anti-CD3/CD28 antibodies for 48 hours and 72 hours (n=6 per genotype) (right). (H) Percentage of Slfn2f/f and Slfn2−/− splenic T cells that incorporated BrdU or expressed annexin V (apoptotic cells) 48 hours and 72 hours after anti-CD3/CD28 stimulation, assessed by flow cytometry (n=6 per genotype). (I) Representative flow cytometry analysis of intracellular cyclin D1 and Ki67 expression in splenic T cells isolated from Slfn2f/f and CD4-Cre; Slfn2f/f mice (n=6 per genotype) and stimulated with anti-CD3/CD28 for 48 hours. Isotype, Slfn2f/f T cells stained with isotype-matched control antibody. (J) CFSE-labeled OT-1 T cells from Slfn2f/f and CD4-Cre; Slfn2f/f mice (n=3 per genotype) were adoptively transferred into Rag1−/− recipients. Representative flow cytometry analysis of CFSE intensity in OT-1 T cells isolated from the spleens of Rag1−/− recipients 72 hours after OVA immunization (n=3 per genotype). Numbers indicate percent dye-positive cells in bracketed region. P-values were determined by Student’s t test and error bars indicate SD except that mean clinical EAE scores were analyzed by the Mann–Whitney U test (n.s., not significant, *P<0.5, **P <0.01, ***P <0.001). Data are representative of two (I) or three (A to H, J) independent experiments.
CD4+ T cells are the primary mediators of experimental autoimmune encephalomyelitis (EAE), a mouse model of multiple sclerosis, by producing Th1 and Th17 cytokines. We tested whether loss of Slfn2 in T cells was sufficient to impair EAE development. Immunization with MOG35–55/CFA emulsion and pertussis toxin resulted in much less severe EAE in CD4-Cre; Slfn2f/f mice compared to Slfn2f/f control mice (Fig. 1D). Spleen cells isolated from CD4-Cre; Slfn2f/f mice on day 10 post-EAE induction produced dramatically reduced levels of IL-17A and IFNγ relative to Slfn2f/f control cells upon stimulation with MOG35–55 autoantigen (Fig. 1E).Thus, SLFN2 is also critical for T cell function in this autoimmune context.
Impaired proliferation by SLFN2-deficient T cells in response to TCR and costimulatory receptor stimulation
SLFN2 expression was upregulated after anti-CD3/CD28 activation of T cells isolated from bacterial artificial chromosome (BAC) transgenic mice expressing GFP-tagged SLFN2 (Fig. 1F). Slfn2 mRNA levels were also upregulated in wild-type T cells upon TCR stimulation (fig. S1F). To examine the proliferative response of primary T cells isolated from CD4-Cre; Slfn2f/f mice (hereafter SLFN2-deficient or Slfn2−/− T cells) to engagement of TCR plus CD28, we measured cell divisions of dye-labeled splenic T cells. After stimulation with anti-CD3/CD28 for 48 hours, few SLFN2-deficient CD4+ or CD8+ T cells had divided, whereas control cells expanded as expected. After stimulation for 72 hours, a significantly lower percentage of SLFN2-deficient T cells had divided compared with control T cells (Fig. 1G). The frequency of Slfn2−/− T cells that had incorporated BrdU was also reduced compared to that of Slfn2f/f T cells 48 and 72 hours after stimulation (Fig. 1H). By contrast, the frequency of annexin V-expressing cells (Fig. 1H) was similar at both time points suggesting that apoptosis was not responsible for the functional defects in SLFN2-deficient T cells. Consistent with these data, cell proliferation markers Ki67 and cyclin D1 showed reduced expression in SLFN2-deficient T cells than in control cells after anti-CD3/CD28 stimulation (Fig. 1I). To test proliferation in response to antigen-specific stimulation in vivo, CD4-Cre; Slfn2f/f and Slfnf/f mice were crossed with OT-1 TCR transgenic mice. OT-1 CD8+ T cells were then dye-labeled and adoptively transferred into Rag1−/− mice, which were subsequently immunized with ovalbumin (OVA). Fewer SLFN2-deficient T cells underwent proliferation compared to control T cells in response to OVA immunization (Fig. 1J). Thus, SLFN2-deficient T cells have an impaired proliferative response to TCR and costimulatory receptor activation.
We examined canonical signaling events induced by anti-CD3/CD28 stimulation in T cells. Phosphorylation of PLC-γ, Zap70, LAT, ERK, and Akt, calcium influx, and NF-κB p65 and NFAT nuclear translocation were normal after anti-CD3/CD28 stimulation of naïve Slfn2−/− T cells (fig. S2A–C). Cell surface expression of the T cell activation markers CD25 (IL-2Rα) and CD44 was similar between Slfn2−/− T cells and control Slfn2f/f T cells 48 hours after stimulation (fig. S2D). Thus, the defective proliferative response of Slfn2−/− T cells occurred despite intact proximal TCR signaling.
Abrogation of responses to IL-2 is due to diminished IL-2 receptor expression in SLFN2-deficient T cells
T cell stimulation through the TCR induces both IL-2 secretion and responsiveness to IL-2 through transcriptional and translational upregulation of the high affinity IL-2 receptor, leading to autocrine/paracrine signaling and T cell proliferation (23). Although we detected similar IL-2 production by naïve Slfn2−/− and Slfn2f/f T cells in response to anti-CD3/CD28 stimulation (Fig. 2A), both CD4+ and CD8+ SLFN2-deficient T cells failed to proliferate in response to exogenous IL-2 stimulation (Fig. 2B). In addition, control T cells displayed greatly enhanced phosphorylation of Stat1 and Stat5 in response to stimulation, whereas Slfn2−/− T cells did not. This was true even after the addition of exogenous IL-2, which further elevated phosphorylated Stat1 and Stat5 levels in control cells (Fig. 2C). Thus, although IL-2 production is normal, IL-2 receptor signaling is impaired in SLFN2-deficient T cells.
Fig. 2. Abrogation of responses to IL-2 is due to diminished IL-2 receptor expression in SLFN2-deficient T cells.

(A) IL-2 concentration in the culture medium of Slfn2f/f or Slfn2−/− (from CD4-Cre; Slfn2f/f mice) CD4+ and CD8+ T cells left unstimulated or stimulated with anti-CD3/CD28 for 24 hours and 48 hours (n=4 per genotype). (B) Representative flow cytometry analysis of CFSE intensity in Slfn2f/f or Slfn2−/− splenic CD4+ and CD8+ T cells that were pre-activated with anti-CD3/CD28 for 24 hours, then labeled with CFSE, and treated with IL-2 for 48 hours in the absence of anti-CD3/CD28 antibody (n=3 per genotype). (C) Immunoblot analysis of total and phosphorylated Stat1 and Stat5 in Slfn2f/f or Slfn2−/− splenic T cells pre-activated with anti-CD3/CD28 for 24 hours followed by IL-2 stimulation for 30 min with cells pooled from 4–8 mice per genotype. (D) Representative flow cytometry analysis (left) and mean fluorescence intensity (right) of surface expression levels of IL-2Rβ and IL-2Rγ on Slfn2f/f or Slfn2−/− splenic T cells stimulated with anti-CD3/CD28 for the indicated time (n=4 per genotype). Isotype, Slfn2f/f T cells stained with isotype-matched control antibody. (E) Immunoblot analysis of total and phosphorylated Stat5 and Erk in Slfn2f/f or Slfn2−/− splenic T cells pre-activated with anti-CD3/CD28 for 24 hours followed by IL-2, IL-7, or IL-15 stimulation for 30 min, and cells were pooled from 4–8 mice per genotype. (F) qRT-PCR analysis of IL-2Rβ and IL-2Rγ transcripts in total mRNA isolated from Slfn2f/f or Slfn2−/− splenic T cells stimulated with anti-CD3/CD28 for the indicated time (n= 4 per genotype). Data were normalized to β-actin mRNA expression. (G) Immunoblot analysis of IL-2Rβ and IL-2Rγ in Slfn2f/f or Slfn2−/− splenic T cells stimulated with anti-CD3/CD28 for the indicated time with cells pooled from 4–8 mice per genotype. P-values were determined by Student’s t test (n.s., not significant, *P<0.5, **P<0.01, ***P<0.001). Data are representative of two (D) or three (A to C, E to G) independent experiments, and error bars indicate SD.
The high-affinity IL-2 receptor is a heterotrimer consisting of IL-2Rα (CD25), IL-2Rβ (CD122), and IL-2Rγ (γc or CD132) subunits (23). Cell surface expression of IL-2Rβ and IL-2Rγ was significantly lower in SLFN2–deficient T cells than in Slfn2f/f T cells as early as 8 hours (IL-2Rβ) and 4 hours (IL-2Rγ) after stimulation with anti-CD3/CD28 antibodies. This difference was amplified at later time points as Slfn2f/f cells further increased cell surface receptor expression (Fig. 2D). The phosphorylation of Stat5 and ERK was also reduced in Slfn2−/− T cells in response to stimulation of other γc-containing cytokine receptors, such as IL-7 and IL-15 receptors (Fig. 2E). qRT-PCR analysis showed that the relative levels of Il2rb and Il2rg mRNA in Slfn2−/− T cells were comparable to those in control cells before and after anti-CD3/CD28 stimulation (Fig. 2F). By contrast, IL-2Rβ and IL-2Rγ protein levels were similar in Slfn2−/− T cells and control cells before anti-CD3/CD28 stimulation, but the upregulation of total IL-2Rβ and IL-2Rγ protein in response to stimulation was only observed in control T cells (Fig. 2G). Thus, SLFN2 is necessary for the upregulation of IL-2Rβ and IL-2Rγ total protein levels, but not their transcripts, in activated T cells.
Attenuated translational burst in activated SLFN2-deficient T cells
Given that similar expression levels of Il2rb and Il2rg mRNA were induced in Slfn2f/f and Slfn2−/− T cells by TCR activation, we investigated whether the reduced expression of IL-2Rγ protein in activated SLFN2-deficient T cells could be due to aberrant protein translation and/or degradation. Although total IL-2Rγ protein amounts were initially lower in activated Slfn2−/− T cells than in Slfn2f/f T cells, they decreased at a similar rate in T cells of both genotypes treated with the translation inhibitor cycloheximide (Fig. 3A), suggesting that protein degradation is normal in SLFN2-deficient T cells. By contrast, inhibiting either lysosome-mediated or proteasome-mediated protein degradation in activated SLFN2-deficient T cells using NH4Cl and MG132, respectively, failed to significantly increase total IL-2Rγ levels, as seen in control T cells (Fig. 3B). Thus, translation of IL-2Rγ is impaired in SLFN2-deficient T cells.
Fig. 3. Attenuated translational burst in activated SLFN2-deficient T cells.

(A) Immunoblot analysis of IL-2Rγ in Slfn2f/f or Slfn2−/− (from CD4-Cre; Slfn2f/f mice) splenic T cells stimulated with anti-CD3/CD28 for 24 hours and then treated with cycloheximide for the indicated time with cells pooled from 4–8 mice per genotype (right). Quantification of the intensity of the IL-2Rγ protein bands normalized to time 0 hour (left). (B) Immunoblot analysis of IL-2Rγ in Slfn2f/f or Slfn2−/− splenic T cells stimulated with anti-CD3/CD28 for 24 hours and then treated with NH4Cl (10 mM) or MG132 (10 μM) for the indicated time, and cells were pooled from 4–8 mice per genotype. (C) OP-puro mean fluorescence intensity in Slfn2f/f or Slfn2−/− splenic T cells (n=5 per genotype) stimulated with anti-CD3/CD28 for the indicated time and then labeled with OP-puro for 30 min. Each symbol represents an individual mouse. (D) Representative flow cytometry analysis of OP-puro fluorescence in Slfn2f/f or Slfn2−/− splenic T cells stimulated with anti-CD3/CD28 for 24 hours, followed by OP-puro or PBS incubation (with Slfn2f/f cells as a control) for 30 min. (E) Representative flow cytometry analysis of intracellular Ki67 levels (left) and OP-puro fluorescence (right) in OT-1 cells from Slfn2f/f or CD4-Cre; Slfn2f/f mice 48 hours after i.m. OVA immunization followed by a 1-hour OP-puro treatment in vivo (n=3 per genotype). Control, antibody isotype (left) or PBS treatment (right) of Slfn2f/f cells. P-values were determined by Student’s t test (*P<0.5, **P<0.01, ***P<0.001). Data are representative of two (B to E) or three (A) independent experiments, and error bars indicate SD.
We examined global protein translation in SLFN2-deficient and control T cells or mice using O-propargyl-puromycin (OP-puro), an aminoacyl-tRNA mimic, which forms covalent conjugates with nascent polypeptide chains (24). Basal translation was slightly but significantly reduced in Slfn2−/− T cells compared to Slfn2f/f T cells (Fig. 3C). Both SLFN2-deficient and control T cells increased their translation rate in response to anti-CD3/CD28 stimulation, but the rate of OP-puro incorporation in SLFN2-deficient T cells was reduced relative to that in control T cells (Fig. 3, C and D). To determine whether reduced protein translation also occurred in response to antigen-specific stimulation in vivo, OT-1; CD4-Cre; Slfn2f/f and OT-1 Slfn2f/f mice were used to assess protein synthesis in OT-1 T cells after intramuscular OVA immunization. OT-1 SLFN2-deficient T cells exhibited reduced OP-puro fluorescence intensity relative to control OT-1 T cells, and this reduction correlated with decreased Ki67 expression levels (Fig. 3E). Thus, translation is impaired in SLFN2-deficient T cells. Control T cells initiate a burst of translation between 8 and 24 hours after TCR activation, but this response is attenuated in SLFN2-deficient T cells, presumably resulting in a failure to proliferate.
SLFN2 directly binds to tRNAs
SLFN family proteins share a conserved N-terminal domain. In human (h) and rat (r) SLFN13 and hSLFN11, the N-terminal domain harbors tRNA/rRNA-targeted endoribonuclease activity (25, 26). However, using sequence comparison (fig. S3A) and structural homology modeling (Fig. 4A), we found that SLFN2 lacks two of three acidic residues reported to form the nucleolytic active site of rSLFN13. We directly measured endoribonuclease activity of purified recombinant SLFN2 protein, finding no cleavage of tRNA or rRNA by SLFN2 in two different assays (fig. S3, B to D). Thus, SLFN2 is not a tRNA- or rRNA-directed endoribonuclease.
Fig. 4. SLFN2 is not an RNase but directly binds to tRNAs.

(A) Comparison of the putative catalytic site of SLFN2 to the catalytic site of rSLFN13N in a homology model generated using SWISS-MODEL. Catalytic residues identified in rSLFN13 and the corresponding residues in SLFN2 are shown in orange. (B) qRT-PCR analysis of 20 tRNAs that were immunoprecipitated with FLAG antibody from lysates of EL4 cells stably expressing either GFP-Fg or SLFN2-Fg. Data were normalized to β-actin mRNA expression. Inset shows immunoblot analysis of immunoprecipitated GFP-Fg and SLFN2-Fg (n=4 independent cultures per construct). (C) qRT-PCR analysis of tRNAGly that was immunoprecipitated with FLAG antibody from lysates of Neuro-2a cells transfected with Slfn2-WT-Fg or Fg-tagged SLFN2 variants (n=4 independent cultures per construct). Data are expressed relative to GFP-Fg and normalized to Slfn2-WT-Fg, which was set to a value of 1. Immunoprecipitated SLFN2-Fg and Fg-tagged SLFN2 variants were detected by immunoblot (below). (D) RNA blot analysis of tRNAGly and tRNASer immunoprecipitated with FLAG antibody from lysates of Neuro-2a cells transfected with Slfn2-WT-Fg or FG-tagged SLFN2 variants. IP, immunoprecipitation. NB, Northern (RNA) blot. (E) EMSA analysis of in vitro transcribed, biotin-labeled tRNAGly and tRNASer after incubation with recombinant wild-type SLFN2 or SLFN2 variants purified from 293F cells. (F) Gel filtration chromatography analysis of in vitro complex formation between recombinant SLFN2 purified from 293F cells and in vitro transcribed tRNAGly. The indicated fractions were run on either SDS-PAGE gel or 15% denaturing urea PAGE gel for analysis of SLFN2 and tRNAGly in each fraction by Coomassie blue staining or SYBR Gold staining. Data are representative of two (B and C) or three (D to F) independent experiments, and error bars indicate SD.
Two conserved tRNA binding patches identified in rSLFN13 are present in SLFN2 and consist of R67, K68, N71, K242, K249, and Q301 (fig. S3A). We therefore tested whether SLFN2 binds to tRNAs. C-terminally Flag-tagged SLFN2 (SLFN2-Fg) or GFP-Fg were immunoprecipitated from mouse cell lines expressing each of these proteins and the immunoprecipitates were used for qRT-PCR or RNA blot analysis of bound tRNAs. Compared to GFP-Fg, SLFN2-Fg immunoprecipitated with high levels of multiple tRNAs (Fig. 4B). Mutation of any one of the six binding-patch residues significantly reduced the quantity of tRNAGly that immunoprecipitated with SLFN2-Fg (Fig. 4C). A triple mutation of either binding patch (T1: R67A, K68A, and N71A; T2: K242A, K249A, and Q301A) almost completely abolished binding to prototypical tRNAs tRNAGly and tRNASer (Fig. 4, C and D). Electrophoretic mobility shift assays (EMSAs) demonstrated that recombinant wild-type SLFN2 protein, but not SLFN2T1 or SLFN2T2, directly bound to in vitro transcribed tRNAGly and tRNASer (Fig. 4E) with dissociation constants (Kd) of 167.3 ± 15.44 nM and 101.8 ± 10.24 nM, respectively (fig. S3E). Finally, using gel filtration, we showed that recombinant SLFN2 and tRNAGly coeluted in fractions distinct from those fractions containing only tRNAGly or SLFN2, indicating that SLFN2 and tRNAGly form a stable complex in vitro (Fig. 4F). Thus, SLFN2 binds tRNAs directly and forms a stable complex with them.
SLFN2 protects tRNAs from oxidative stress-induced cleavage by ANG after T cell activation
Strikingly, we observed significant accumulation of short RNA fragments, mainly 30–40 nt in length, in unstimulated splenic Slfn2−/− T cells but not in Slfn2f/f T cells (Fig. 5A). The quantity of these fragments increased while full-length tRNAs decreased after anti-CD3/CD28 stimulation of SLFN2-deficient T cells (Fig. 5B and fig. S4A). Such abnormalities occurred in the cytoplasm but were undetectable in the nucleus, consistent with SLFN2 localization specifically in the cytoplasm as previously reported (27) (Fig. 5B). No reduction of 5S, 5.8S (Fig. 5B), 18S, or 28S rRNAs (fig. S3F) from resting or activated Slfn2−/− T cells was observed. To determine whether the 30–40-nt RNA fragments found in SLFN2-deficient T cells were derived from tRNAs, the region of the gel containing them was excised and fragments were eluted and cloned. Notably, 91.3% of the clones contained tRNA-derived fragments, with the remainder containing either rRNA- (6.93%) or mRNA-derived (1.73%) fragments (table S1). Using RNA sequencing (RNA-seq), we found that unstimulated SLFN2-deficient T cells contained 3.1-fold more tRNA fragments than Slfn2f/f T cells (Fig. 5C). tRNA fragments (31–40 nt) comprised 92.6% of total tRNA fragments in SLFN2-deficient T cells and were fivefold more abundant than those in Slfn2f/f T cells (Fig. 5C and data S1). Activated SLFN2-deficient T cells accumulated even more tRNA fragments compared to Slfn2f/f T cells (8.7-fold more, 91.9% 31–40 nt) accompanied by a 22.13% reduction of full-length tRNAs not observed in unstimulated cells (Fig. 5D). RNA blot analysis (fig. S4B) confirmed these findings. Thus, the aberrant accumulation of tRNA fragments and reduction of the parental full-length tRNAs occur in SLFN2-deficient T cells and become more pronounced after T cell activation.
Fig. 5. Antioxidant inhibition of ROS prevents accumulation of tRNA fragments, suppression of translation, and stress granule assembly, permitting activation-induced proliferation of SLFN2-deficient T cells.

(A) Total RNA extracted from unstimulated Slfn2f/f or Slfn2−/− (from CD4-Cre; Slfn2f/f mice) splenic T cells, separated by 15% denaturing urea PAGE, and visualized by SYBR Gold staining. Positions of tRNA and 30–40-nt small RNA fragments are indicated. (B) Slfn2f/f or Slfn2−/− T cells were left unstimulated or stimulated with anti-CD3/CD28 antibodies for 24 hours. Cell lysates were fractionated into nuclear and cytoplasmic fractions before total RNA was isolated from each fraction, separated by 15% denaturing urea PAGE, and visualized by SYBR Gold staining. Positions of 5.8S RNA, 5S RNA, tRNA, and 30–40-nt RNA fragments are indicated. See also fig. S4 for longer exposure. (C and D) RNA-seq analysis of relative levels of full-length tRNAs (left) and relative levels of tRNA-derived fragments with indicated size ranges (right) from (C) resting Slfn2f/f or Slfn2−/− T cells left untreated or treated with 1 mM H2O2 for 2 hours or (D) Slfn2f/f or Slfn2−/− T cells stimulated with anti-CD3/CD28 for 24 hours in the presence or absence of 10 mM NAC (n=3 mice per genotype and treatment). (E) ROS levels in Slfn2f/f or Slfn2−/− splenic T cells (n=6 mice per genotype) as assessed by staining with DCF-DA. T cells were stimulated for 24 hours with anti-CD3/CD28 in the presence or absence of NAC prior to DCF-DA staining. (F) Total RNA from Slfn2f/f or Slfn2−/− splenic T cells stimulated with anti-CD3/CD28 for 24 hours in the presence or absence of NAC was separated by 15% denaturing urea PAGE and visualized by SYBR Gold staining. (G) Total RNA from unstimulated Slfn2f/f or Slfn2−/−splenic T cells treated with 1 mM H2O2 for indicated time was separated by 15% denaturing urea PAGE and visualized by SYBR Gold staining. Positions of 5.8S RNA, 5S RNA, tRNA, and 30–40-nt RNA fragments are indicated. (H) Representative flow cytometry analysis of OP-puro fluorescence in Slfn2f/f or Slfn2−/− splenic T cells stimulated with anti-CD3/CD28 in the presence or absence of NAC for 24 hours and then labeled with OP-puro for 30 min. PBS was used as a negative control. (I) OP-puro mean fluorescence intensity in Slfn2f/f or Slfn2−/− splenic T cells (n=4 mice per genotype), stimulated with anti-CD3/CD28 in the presence or absence of NAC for 24 hours and then labeled with OP-puro for 30 min. (J) Confocal immunofluorescence microscopy images of G3BP1 (green) and eIF4G (red) in Slfn2f/f or Slfn2−/− splenic T cells stimulated with anti-CD3/CD28 in the presence or absence of NAC for 24 hours. Cell nuclei were stained with SYTOX Deep Red (blue). Scale bar: 10 μm. (K) Quantification of stress granules (G3BP1+eIF4G+) in Slfn2f/f or Slfn2−/− splenic T cells stimulated as in J (n=100 cells per genotype). (L) Representative flow cytometry analysis of CFSE intensity in Slfn2f/f or Slfn2−/− splenic T cells stimulated with anti-CD3/CD28 in the presence or absence of NAC for 72 hours. Numbers indicate percent dye-positive cells in bracketed region. P-values were determined by Student’s t test (n.s., not significant, *P<0.5, **P<0.01, ***P<0.001). Data are representative of three independent experiments (A and B, E to L), and error bars indicate SD.
tRNA cleavage yielding tiRNA is a cellular response to oxidative stress (14, 17) and ROS are known to be generated during increased mitochondrial metabolism associated with T cell activation(6). We found that Slfn2f/f and Slfn2−/− T cells produced similar amounts of ROS in response to anti-CD3/CD28 stimulation (Fig. 5E). However, the accumulation of tiRNA in stimulated SLFN2-deficient T cells could be reduced by the antioxidant N-acetylcysteine (NAC) (Fig. 5, D and F). Conversely, directly treating unstimulated T cells with a low concentration of H2O2 resulted in excessive accumulation of tiRNA in Slfn2−/− T cells compared to Slfn2f/f T cells (Fig. 5, C and G), suggesting that tRNAs in SLFN2-deficient T cells are more susceptible to cleavage in response to oxidative stress. We found that ROS and tiRNA accumulation correlated with decreased translation in SLFN2-deficient T cells by measuring their translation rate using OP-puro after anti-CD3/CD28 stimulation for 24 hours. The rate of OP-puro incorporation was reduced in activated Slfn2−/− T cells compared to Slfn2f/f T cells but was substantially rescued when anti-CD3/CD28 stimulation was performed in the presence of NAC (Fig. 5, H and I). The formation of stress granules—assemblies of non-translating messenger ribonucleoproteins formed from mRNAs stalled in ribosomes (10)—after anti-CD3/CD28 stimulation of Slfn2−/− T cells could be inhibited by NAC (Fig. 5, J and K). Moreover, NAC treatment during anti-CD3/CD28 stimulation markedly rescued the cell proliferation defect of Slfn2−/− T cells (Fig. 5L). Thus, antioxidant inhibition of ROS in activated SLFN2-deficient T cells is sufficient to greatly reduce abnormal accumulation of tiRNA and subsequent translation suppression and stress granule assembly. This then increases the activation-induced proliferation of SLFN2-deficient T cells.
tiRNAs have been previously shown to be generated in mammalian cells by the RNase ANG in response to oxidative stress (12, 15–17) and their role in translation inhibition is well established (13, 28). We hypothesized that direct binding of SLFN2 to tRNA is necessary to protect tRNA from cleavage and thereby support translation during T cell activation. ANG mRNA and protein expression increased in both Slfn2f/f and Slfn2−/− mouse T cells after anti-CD3/CD28 stimulation in agreement with findings from activated human T cells (29) (fig. S5, A and B). No significant change in subcellular localization of either ribonuclease/angiogenin inhibitor 1 (RNH1) or ANG was observed after T cell activation (fig. S5B) compared with previous observations in HeLa cells treated with sodium arsenite (30), suggesting a different mechanism for activation of ANG in activated T cells. RNH1 protein levels remained stable under conditions of elevated oxidative stress after T cell activation, resulting in an excess of cytosolic ANG that is not associated with RNH1 and may actively cleave tRNA (fig. S5C). Notably, NAC treatment reduced the level of ANG protein expression after T cell activation (fig. S5, A to C), indicating that oxidative stress is necessary for induction of ANG expression in activated T cells.
To test whether SLFN2 directly inhibits tRNA cleavage by ANG, we performed in vitro ANG cleavage assays using yeast tRNAs as a substrate. Incubation with ANG resulted in the accumulation of tRNA fragments, but this effect was reduced by preincubation of recombinant SLFN2 with tRNAs (Fig. 6, A and B, and data S2). By contrast, SLFN2T1, a mutant protein impaired in tRNA binding (Fig. 4, C and D), failed to block the ANG-dependent production of tRNA fragments (Fig. 6, A and B). Using primary mouse T cells, we showed that the amount of tiRNA in SLFN2-deficient T cells could be reduced by knockdown of ANG or overexpression of the ANG inhibitory protein RNH1 (30) (Fig. 6, C and D, and fig. S6, A and B) and this correlated with improved cellular protein translation rates (Fig. 6E). In addition, knockdown of ANG or overexpression of RNH1 led to restored IL-2Rβ and IL-2Rγ protein levels (Fig. 6F), as well as improved cell proliferation and BrdU incorporation in activated SLFN2-deficient T cells (Fig. 6, G and H). Overexpression of SLFN2, but not SLFNT1, had effects similar to ANG knockdown or inhibition in SLFN2-deficient T cells (Fig. 6, C to H, and fig. S6C), indicating that the interaction between SLFN2 and tRNAs is necessary for these effects. Thus, ANG mediates tRNA cleavage in SLFN2-deficient T cells resulting in impaired translation and cell proliferation in response to TCR stimulation.
Fig. 6. SLFN2 protects tRNAs from ANG-mediated cleavage in T cells.

(A) tRNA cleavage activity of mouse ANG towards 2 μM yeast total tRNA in the presence of 2 μM recombinant MBP (negative control), SLFN2, or SLFN2 T1, as measured by the release of perchloric acid-soluble fragments detected by absorbance at 260 nm. (B) RNA-seq analysis of relative levels of full-length tRNAs (left) and relative levels of tRNA-derived fragments with indicated size ranges (right). Total tRNA and tRNA-derived fragments were isolated from in vitro yeast tRNA cleavage assays performed with 1 μM mouse ANG and 2 μM indicated other proteins (n=3 assays per condition). (C) Total RNA extracted from activated Slfn2f/f or Slfn2−/− splenic T cells expressing the indicated proteins or shRNAs for 72 hours, separated by 15% denaturing urea PAGE, and visualized by SYBR Gold staining. Positions of tRNA and 30–40-nt tiRNA are indicated. (D) RNA-seq analysis of relative levels of full-length tRNAs (left) and relative levels of tRNA-derived fragments with indicated size ranges (right) isolated from activated Slfn2f/f or Slfn2−/− splenic T cells expressing the indicated proteins or shRNAs (n=3 per genotype) for 72 hours. (E) Representative flow cytometry analysis of OP-puro fluorescence in activated Slfn2−/− splenic T cells expressing the indicated proteins or shRNAs for 48 hours, followed by OP-Puro or PBS incubation (with “Slfn2−/−(SLFN2)” as a control) for 30 min (left). Quantified OP-puro mean fluorescence intensity is shown (n=5 per genotype) (right). (F) Immunoblot analysis of IL-2Rβ and IL-2Rγ in activated Slfn2f/f or Slfn2−/− splenic T cells expressing the indicated proteins or shRNAs for 48 hours. (G) Representative flow cytometry analysis of CellTrace Violet intensity in activated Slfn2−/− splenic T cells expressing the indicated proteins or shRNAs (left) for 72 hours. Numbers indicate percent dye-positive cells in bracketed region. Frequency of CellTrace Violetlo cells among dye-positive T cells (n=6 per genotype) (right). (H) Percentage of BrdU positive, activated Slfn2−/− splenic T cells expressing the indicated proteins or shRNAs for 48 hours, assessed by flow cytometry (n=6 per genotype). P-values were determined by Student’s t test (n.s., not significant, *P<0.5, **P<0.01, ***P<0.001). Data are representative of two (A and E) or three (C, F to H) independent experiments, and error bars indicate SD.
ANG is a secreted ribonuclease reported to act non-cell-autonomously (18, 31) through binding to the plexin-B2 receptor (32). We knocked down plexin-B2 expression in SLFN2-deficient T cells and examined tiRNA levels. In contrast to the effect of ANG knockdown, plexin-B2 knockdown failed to reduce the amount of tiRNA in SLFN2-deficient T cells (fig. S7, A and B). Thus, plexin-B2 is not required for ANG-mediated tRNA cleavage in these cells.
Discussion
Both CD4+ and CD8+ T cells depend on a translational burst to support the rapid switch to glycolysis and glutaminolysis induced by TCR engagement and later to produce the translation machinery (i.e. ribosomal proteins) that will generate the protein components of clonal T cell progeny and their cytokines (1, 3, 4). Paradoxically, ROS generated in activated T cells as a byproduct of electron transport chain reactions activate the tRNA-directed RNase activity of ANG, resulting in production of tiRNAs which suppress translation. Given the necessity of ROS for the proliferative response to TCR and costimulatory receptor activation (8), T cells apparently developed a means, dependent on SLFN2, to specifically circumvent the translation inhibitory effects of ROS while maintaining their necessary functions in supporting metabolic reprogramming and signaling. Interestingly, T cells from Slfn2eka/eka mice express markers of ER stress (33), another cellular stress that may activate ANG (34). These findings suggest that SLFN2 may play a similar role during ER stress, potentially mitigating the translation-inhibitory effects of tiRNA on proteins necessary to resolve ER stress.
We found that SLFN2 is rapidly upregulated after TCR stimulation, suggesting that SLFN2 exerts its protective effect during the metabolic reprogramming phase. SLFN2 levels continue to increase until at least 48 hours after TCR stimulation and may therefore also be important for upholding translation potential during the phase of T cell expansion. In contrast to IL-2 and IL-2Rα, which are known to be strongly upregulated at the transcriptional level upon T cell activation (35, 36), IL-2Rβ and IL-2Rγ are among the translation targets affected by SLFN2 deficiency in T cells. This is probably due to weaker TCR-dependent upregulation of their transcription and their rapid consumption during clonal expansion. Identification of other proteins dependent upon SLFN2 for translation may reveal novel therapeutic targets for the manipulation of T cell expansion.
Together with several recent publications (25, 26, 37–39), our findings support the idea that SLFN family proteins interact with and regulate RNA in a variety of capacities with ensuing control of translation. hSLFN11 and hSLFN13 have been reported to regulate translation of cellular and viral proteins by nucleolytic cleavage of tRNAs. By contrast, our data suggest a different mechanism for SLFN2, which lacks nucleolytic activity and instead confers protection from it. This is perhaps not surprising given the evolutionary distance between SLFN2 and SLFN11/13, which are classified into distinct phylogenetic groups (40). Whether SLFN2 protects tRNAs from cleavage by other T cell-expressed SLFN proteins (e.g. SLFN3/4/8/9/10 (41, 42)) is an interesting possibility to be explored.
ANG affects multiple tissues and cellular processes through interactions with RNA (31, 32). ANG may act endogenously within the cell expressing it (43), or may be secreted to affect neighboring or distant cells (18, 32). The latter situation was reported to apply to hematopoietic stem cells (HSC), in which ANG promotes stemness and quiescence (31, 32, 44). Our findings raise the question of whether ANG function in T cells is cell-autonomous or not. Plexin-B2 has recently been implicated as a receptor for ANG on multiple cell types including HSC (32). Notably, plexin-B2 is also expressed on T cells where it promotes mTORC1 activation necessary for T cell activation and differentiation (45). However, no reduction of tiRNA was observed when plexin-B2 was knocked down in SLFN2-deficient T cells. SLFN2 is most highly expressed in the thymus and spleen, but also in other tissues, with moderate expression in intestine, bladder, embryonic liver, and mammary gland (46). Further studies are needed to investigate whether SLFN2 counters the function of ANG in cell types other than T cells.
Materials and Methods
Mice
Mus musculus C57BL/6J (#000664), 129 ROSA26Flpo (#007844), and OT-1 transgenic (#003831) mice were from The Jackson Laboratory. All experiments used age- and sex-matched mice (6–12 weeks old). All mouse studies were conducted according to protocols (2017–102166 and 2017–102357) approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Texas Southwestern Medical Center.
Generation of a conditional Slfn2 mutant allele
To generate a conditional Slfn2 mutant allele, the Slfn2-targeting vector was constructed with pGKneoF2L2DTA containing two loxP sites harboring a neomycin resistance cassette flanked by two FRT sites as previously described (47). Targeting arms were amplified from 129SvEv genomic DNA by PCR and verified by sequencing. The Slfn2 5′ targeting arm (4 kb), 3′ targeting arm (2 kb), and knock-in arm (2.6 kb) were cloned into the vector with SacII/NotI, HindIII/EcoRV, and SmaI, respectively. The targeting vector was linearized and electroporated into 129SvEv-derived ES cells. Using Southern blot analysis with a 0.7-kb 5′ external probe and PCR amplification with one primer outside of the 3′ targeting construct in conjunction with another primer in the selectable marker, positive ES cells were identified and used for blastocyst injection (see Figure S1A). The resulting chimeric mice were bred with mice expressing FLPe recombinase to remove the neomycin resistance cassette, and obtain germline transmission of the Slfn2floxP (Slfn2f) allele. Slfn2f/f mice were crossed with CD4-Cre mice (JAX stock #017336) obtained from The Jackson Laboratory (48). Primer sequences for PCR genotyping of the floxP allele are as follows: F, 5′-CACATATGCAGGCAAAACACCAAT-3′; R, 5′-TAATCCTTGTTCATCTCTTTTCTTGTCCTT-3′.
Transgenic mice
The BAC clone RP23–246H3, containing the Slfn2 locus, was obtained from the BACPAC Resources Center at the Children’s Hospital Oakland Research Institute. The eGFP cassette was inserted at the initiation codon of the Slfn2 coding sequence, which is located within exon 1, by homologous recombination using a targeting vector designed for BAC recombineering. After electroporation into the recombinogenic Escherichia coli, homologous recombination was confirmed by PCR and sequencing the BAC using primers that flanked the upstream and downstream of eGFP. The modified BAC DNA was extracted, and circular intact DNA was microinjected into fertilized embryos by standard pronuclear injection techniques. Genomic DNA isolated from mouse tails was extracted, and founder mice carrying the BAC transgene were identified by PCR. Primer sequences for PCR genotyping are as follows: F, 5′-AGCAAATGGACTTTGCTGCTTG-3′; R, 5′-GAACTTGTGGCCGTTTACGTC-3′.
Flow cytometry
For surface markers, cells were stained in PBS with 2% BSA (w/v). For intracellular staining, cells were fixed and permeabilized using BD Cytofix/Cytoperm Fixation/Permeabilization Solution Kit (BD Biosciences) before staining with the proper antibodies. For BrdU incorporation, after activation of cells, BrdU (BD Biosciences) was added for 90 min. Cells were stained with anti-BrdU (3D4; BD Biosciences) as described in BrdU Flow Kit (BD Biosciences). Apoptotic T cells were stained using Annexin V Apoptosis Detection Kit (BD Biosciences) according to the manufacturer’s instructions. For intracellular ROS measurement, T cells were stimulated with anti-CD3 plus anti-CD28 antibody for the indicated time before incubation with freshly prepared 10 μM 2′,7′-dichlorodihydrofluorescein diacetate (DCF-DA, D399, Invitrogen) for 30 min (8). Cells were then analyzed by flow cytometry as described. For calcium influx measurements, splenocytes at a density of 107 cells/ml were surface-stained with anti-CD4 and anti-CD8 mAbs, washed, and loaded with 2 μg/ml of Indo-1 (Invitrogen) at 37°C for 45 min. Cells were washed and stimulated with 10 μg/ml of biotin-labeled anti-CD3 (145–2C11) plus anti-CD28 (37.51) antibody (#13–0031-85 and #13–0281-85, eBioscience) at 37°C for 30 min. Baseline fluorescence (ratio of 405 nm/485 nm) was measured, and calcium flux was determined in the presence of 10 μg/ml of streptavidin (#85878, Sigma) by flow cytometry. Flow cytometry data were acquired on a LSR II Fortessa cell analyzer (BD Biosciences) and analyzed with FlowJo software (TreeStar), as described previously (49).
Immunization and ELISA
Immunization and ELISA was performed as previously described (50). On day 0, mice were i.p. immunized with either 2×106 infectious units (IU) of rSFV vector encoding β-galactosidase (rSFV-β-gal) or 40 μg of NP-AECM-Ficoll (Biosearch Technologies). On days 7 and 14, blood was collected for analysis of the antibody responses to NP-Ficoll (NP-specific IgM) and rSFV-β-gal (β-galactosidase-specific IgG) by ELISA.
ELISA analysis of IL-2 was performed according to the manufacturer’s instructions (#88–7024-22, eBioscience).
Protein expression and purification
Recombinant proteins MBP (maltose binding protein), and MBP-SLFN2 containing an N terminal MBP-tag were expressed in E. coli Rosetta (DE3). Transformed bacteria were cultured at 37°C in rich broth medium with glucose and ampicillin before induction with 0.1 mM isopropyl-1-thio-β-d-galactopyranoside at an OD600 nm of 0.5 and grown overnight at 16°C. Cells were lysed in 20 mM Tris-HCl, pH 8, 200 mM NaCl, 1 mM EDTA, and 1mM DTT with cOmplete Protease Inhibitor Cocktail (Roche). The proteins were purified over amylose resin and FPLC columns. The purity of the proteins was estimated by Coomassie blue staining. Recombinant mouse angiogenin 2 (ANG) was generated as described (31) for the in vitro cleavage assay, considering the dramatic upregulated expression of mouse angiogenin 2 in T cells after TCR activation, and its higher ribonucleolytic activity compared to angiogenin 1 (51). Recombinant SLFN2, SLFN2T1 and SLFN2T2 proteins were expressed heterologously in HEK293F cells (Life Technologies) using the BacMam system (Thermo Fisher Scientific) by using the protocol previously described (52, 53). GST-SLFN2, GST-SLFN2T1 and GST-SLFN2T2 containing an N-terminal GST tag followed by a cleavage site for PreScission protease was cloned into a pEZT-BM vector. The baculovirus was generated in Sf9 cells (Life Technologies) and used to infect HEK293F cells at a ratio of 1:20 (virus:HEK293F, v/v) and supplemented with 5 mM sodium butyrate to increase protein expression. Cells were cultured in suspension at 37°C for 48 hours and collected by centrifugation at 3000g. The cell pellet was resuspended in lysis buffer (20 mM Tris, pH 8.0, 400 mM NaCl) with cOmplete Protease Inhibitor Cocktail (Roche) and homogenized by sonication on ice, followed by centrifugation at 20,000g for 50 min. The GST-fusion proteins were captured by Glutathione Sepharose 4B resin. After extensive washing with lysis buffer to remove unbound proteins, SLFN2, SLFN2T1 and SLFN2T2 were cleaved from GST affinity tag by treatment with the PreScission protease on resin at 4°C for overnight. The eluted protein was further purified on a Superose 6 10/300 GL increase 10/300 column (GE healthcare) in buffer (20 mM HEPES pH 8.0, 150 mM KCL, 5 mM MgCl2, 1 mM DTT, and 5% Glycerol). Peak fractions were collected, concentrated and kept at −80°C before use. To analyze SLFN2-tRNA complex formation, purified SLFN2 and tRNAGly were mixed at 1:2 molar ratio in 20 mM HEPES pH 8.0, 150 mM KCL, 5 mM MgCl2, 1 mM DTT, and 5% glycerol, and incubated on ice for 30 min before analysis by Superose 6 size exclusion chromatography.
tRNA substrate preparation
Preparation of tDNA transcription template and in vitro transcription of tRNAs was performed as previously described (26, 54). To prepare tRNAGly and tRNASer, 50 ng/μl of linearized plasmid containing tDNA transcription template was mixed with 20 mM of each NTP, 20 mM Tris-HCl, pH 8.0, 150 mM NaCl, 20 mM MgCl2, 5 mM DTT, 1 mM spermidine, and 0.3 μM T7 RNA polymerase at 37°C overnight. The tRNA product was initially purified with phenol chloroform extraction and isopropanol precipitation, and further purified by 8 M urea polyacrylamide gel electrophoresis. The tRNA of the correct size was cut out and then passively diffused into DEPC-treated water overnight at 4°C and concentrated by using a spin concentrator with a 10-kDa cutoff (Millipore). Sequences of the tRNA transcripts used in this study are listed in table S2. To label the 3′ end of RNA probes used in EMSA, the Pierce RNA 3′ End Biotinylation Kit (Thermo Fisher Scientific) was used to generate biotin-labeled tRNA probes.
tRNA electrophoretic mobility shift assay (EMSA)
EMSA was performed following the protocols described in LightShift Chemiluminescent RNA EMSA Kit (Thermo Fisher Scientific). Briefly, 2 μM MBP, SLFN2, SLFN2T1, and SLFN2T2 were incubated with 5 μM in vitro-transcribed and biotin-labeled tRNAGly(CCC) or tRNASer(UAG) in buffer containing 10 mM HEPES, pH 7.3, 20 mM KCl, 1 mM MgCl2, 1 mM DTT, and 5% glycerol with 1 U/μl Rnasin Plus Rnase Inhibitor for 30 min at 25°C. After the incubation, each sample was mixed with loading buffer containing 3% (v/v) glycerol and 0.05% (w/v) bromophenol blue before loading onto a 6% DNA retardation gel (Invitrogen). The gel was pre-run for 45 min at 15 V/cm with running buffer containing 0.5X TBE. Samples were electrophoresed until the bromophenol blue dye migrated approximately 2/3 down the length of the gel, followed by electrophoretic transfer and crosslinking of transferred tRNAs to a nylon membrane. Biotin-labeled tRNAs were detected by chemiluminescence with CCD camera. To estimate of the binding affinity between tRNA and SLFN2, 50 nM Biotin-labeled type I tRNAGly(CCC) or type II tRNASer(UAG) was incubated with various concentrations of SLFN2 in EMSA binding buffer for 30 min at room temperature, and the dissociation constant for tRNAs was determined by using EMSAs and ImageJ to quantify the signal intensity of the bound fraction as SLFN2 protein was titrated. The values for the Kd are obtained from the curve-fitting.
In vitro rRNA and tRNA cleavage assays
The cleavage assay on rRNAs and tRNAs was performed as described before (26). Briefly, total RNA was isolated from EL4 cell lysates using TRIzol reagent (Thermo Fisher Scientific). Two micromolar proteins were incubated with 5 μg total RNA in cleavage buffer containing 40 mM Tris-HCl, pH 8.0, 20 mM KCl, 4 mM MgCl2 and 2 mM DTT for 30 min at 37°C. The cleaved products were resolved by either 1% denaturing agarose gel electrophoresis or 10% denaturing urea PAGE, and visualized by SYBR Gold (# S11494, Thermo Fisher Scientific).
The cleavage assay on total yeast tRNA with mouse ANG was carried out as described (55, 56). Briefly, assays were performed by preincubating 2 μM yeast tRNAs (Sigma, St Louis, MO, USA) with 2 μM SLFN2 or SLFN2T1 protein purified from 293F cells for 30 min, then adding indicated concentrations of mouse ANG for another 4 hours at 37°C. A260 was plotted against the protein concentration as a measure of Rnase activity. To further validate the cleavage results, total tRNA and tRNA-derived fragments were extracted by using TRIzol reagent for RNA sequencing to quantify the levels of the total tRNA and the range of tRNA-derived fragments after the cleavage assay.
To directly measure endoribonuclease activity of SLFN2, recombinant SLFN2 protein was expressed and purified from either BL21 (DE3) E. coli or mammalian 293 Freestyle (293F) cells by affinity purification and gel filtration. No cleavage of tRNAs was observed after incubating MBP-tagged SLFN2 (MBP-SLFN2) purified from bacteria or non-tagged SLFN2 purified from 293F cells with either total RNA isolated from the mouse lymphoma cell line EL4 in the same cleavage conditions that were used to assay the endoribonuclease activity of human and rat SLFN13 N-terminal domains (26), or with yeast tRNA, whereas the tRNA-specific ribonuclease ANG (57) displayed robust cleavage activity.
Cell lines, plasmids, and transfections
Mouse EL4 (ATCC TIB-39) and Neuro-2a (ATCC CCL-131) cell lines were acquired from ATCC, and were maintained at 37°C, 5% CO2 in high glucose DMEM supplemented with 10% heat-inactivated fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mM L-glutamine, 1X MEM non-essential amino acids, 1 mM sodium pyruvate, and 50 μM 2-mercaptoethanol. EL4 cell lines with stable expression of either GFP-Fg or SLFN2-Fg were generated using lentivirus-based vectors as previously described. DNA constructs expressing GFP-Fg, Slfn2-Fg and Fg-tagged Slfn2 variants were created by subcloning the corresponding cDNA coding sequence into pSin vector containing C-terminal FLAG (Fg), and PCR-based site-directed mutagenesis was used to generate corresponding variants. DNA constructs expressing Slfn2, Slfn2-Fg, and RNH1 were created by subcloning the corresponding cDNA coding sequence into pLVX vector. Ctrl, ANG, and Plexin-B2 shRNA were cloned into pXY-IRES, which was a gift from Dr. Z. Chen (University of Texas Southwestern Medical Center). Neuro-2a cells were seeded in six-well plates and left to adhere for 24 hours so that they reached 70% to 90% confluence at the time of transfection. PolyJet in vitro transfection reagent (SignaGen Laboratories) was used to transfect pSin-Slfn2-Fg and its variants according to the manufacturer’s instructions. After 16 hours, medium was replaced with fresh medium and cells were lysed 48 hours after transfection to analyze protein expression. Human freestyle HEK293F cells were acquired from Life Technologies and maintained in FreeStyle 293 Expression Medium.
For mouse primary T cell electroporation, mouse T cell Nucleofector kit (Lonza) was used to introduce plasmids containing indicated cDNAs or shRNAs. 24 hours after anti-CD3/CD28 antibody stimulation, T cells were electroporated with the indicated plasmids mixed with 1/10 pmax-GFP plasmid, to identify cells expressing proteins or shRNAs of interest. Cells were cultured for an additional 48 hours in the presence of anti-CD3/CD28 antibodies and then GFP+ T cells were sorted for further analysis. For ANG mRNA knockdown, the mixture of ANG1 and ANG2 shRNA was used to obtain desired knockdown efficiency for inactivation of ANG-mediated ribonuclease activity.
Primary cell isolation, in vitro activation, and measurement of proliferation
CD4+ and CD8+ T cells were isolated from spleens of male or female mice with the indicated genotypes by using mouse Pan T Cell Isolation Kit II or Naive T Cell Isolation Kit (Miltenyi Biotec) in combination with magnetic bead sorting. T cells were stimulated in vitro with Dynabeads Mouse T-Activator CD3/CD28 (Thermo Fisher Scientific), or with plate-bound anti-CD3 antibody (BD Biosciences, 5 μg/ml) plus anti-CD28 antibody (BD Biosciences, 5 μg/ml), or with PMA–ionomycin (50 ng/ml PMA and 1 μg/ml ionomycin) for the indicated time. For cytokine stimulation of pre-activated pan T cells, pan T cells were activated with anti-CD3/CD28 antibodies for 24 hours, rested for 4 hours without antibody stimulation, and then stimulated with IL-2 (10 ng/ml), IL-7 (100 ng/ml), or IL-15 (100 ng/ml) for 30 min. For the stimulation of splenocytes, 1×106 splenocytes isolated from MCMV-infected mice on day 10 post-infection were seeded in 96-well flat-bottom plates and stimulated with either 2 μg/ml MCMV-derived peptide (M45: HGIRNASFI; m139: TVYGFCLL) or PMA–ionomycin in the presence of brefeldin A for 6 hours, followed with surface staining and intracellular staining. For examination of IL-2Rγ degradation and synthesis, T cells were treated after anti-CD3/CD28 stimulation with cycloheximide (10 μg/ml), NH4Cl (10 mM), or MG132 (10 μM). Quantification of band intensities was performed with ImageJ64. For analysis of RNAs in splenic T cells, total RNA was isolated using TRIzol reagent and subjected to 15% denaturing urea PAGE followed by SYBR Gold staining.
For proliferation assays using CFSE or CellTrace Violet, CFSE or CellTrace Violet Cell Proliferation Kits (Thermo Fisher Scientific) were used. Briefly, T cells were labeled for 10 min with CFSE (carboxyfluorescein diacetate succinimidyl ester) or CellTrace Violet, followed by two washing steps. Cells were stimulated with anti-CD3 plus anti-CD28 antibody for the indicated time, and CFSE or CellTrace Violet were analyzed by flow cytometry.
Immunofluorescence staining and imaging
Briefly, T cells were grown for 24 hours on glass chambered coverslips (μ-Slide 8-Well, ibidi) which were pre-coated with anti-CD3 and anti-CD28, followed by fixation and permeabilization using BD Cytofix/Cytoperm Fixation/Permeabilization Solution Kit (BD Biosciences). Primary antibody incubations were performed in 1X perm/wash buffer overnight at 4°C. Secondary antibodies (Thermo Fisher Scientific) were tagged with Alexa 488 or Alexa 546, and incubated with cells for 1 hour at room temperature. Following washes with PBS, cells were mounted with a nuclei dye (SYTOX Deep Red), and viewed at room temperature using a Spinning Disk Field Scanning Confocal System (Yokogawa CSU-W1; Zeiss) with a 63X /1.4 OilDICIII objective lens and a Prime 95B back-illuminated sCMOS camera. Cells were manually scored for SGs using fluorescence microscopy with eIF4G, and G3BP1 as SG markers. Only granules costained for both eIF4G and G3BP1 were considered SGs (58).
RNA immunoprecipitation and tRNA-derived fragment cloning
For RNA immunoprecipitation, EL4 stable cell lines and transiently transfected Neuro-2a cells were lysed on ice in immunoprecipitation buffer (25 mM Tris-HCl pH 7.4, 150 mM NaCl, 1% (v/v) NP-40, 1 mM EDTA, 5% (v/v) glycerol, and protease inhibitor cocktail [Roche] with SUPERase In Rnase Inhibitor [Invitrogen]). Immunoprecipitation was performed using anti-FLAG M2 agarose beads (Sigma) at 4°C with rotation overnight in immunoprecipitation buffer, followed by four washes with immunoprecipitation buffer containing SUPERaseIn Rnase Inhibitor. Each sample was divided into two aliquots, one aliquot for immunoblot analysis using anti-FLAG M2 antibody (Sigma), and the other aliquot for qRT-PCR analysis was resuspended in TRIzol reagent (Thermo Fisher Scientific) to recover immunoprecipitated tRNAs.
For tRNA-derived fragment cloning, ZR small-RNA PAGE Recovery Kit (Zymo Research) and TruSeq Small RNA Library Preparation Kits (Illumina) were used. Briefly, total RNA from Slfn2f/f or CD4-Cre; Slfn2f/f T cells was resolved by 15% denaturing urea PAGE, and small RNA fragments of approximately 20–50 nt were excised and eluted from the gels. Eluted RNA fragments were ligated with 3′ and 5′ adapters (Illumina), followed by reverse transcription and linear PCR amplification as described in the Library Preparation kit. PCR products were cloned into pCR2.1 Vector by using the TA cloning kit (Thermo Fisher Scientific) and analyzed by capillary sequencing. A total of 173 clones containing inserts of 20–50 nt were analyzed.
Immunoprecipitation and immunoblot analysis
Cells were lysed on ice in immunoprecipitation buffer (25 mM Tris-HCl pH 7.4, 150 mM NaCl, 1% (v/v) NP-40, 1 mM EDTA, 5% (v/v) glycerol, and protease inhibitor cocktail [Roche]) for 30 min. The lysates were cleared by centrifugation and quantified by Bradford protein assay (Bio-Rad) or BCA assay (Pierce). For immunoprecipitation, equivalent amounts of cell lysates were incubated at 4°C with rotation overnight in immunoprecipitation buffer with the indicated antibodies. Protein A/G magnetic beads (Thermo Scientific) were added to the extracts and mixed by rotation for an additional 2 hours at 4°C. For immunoblot analysis, whole-cell lysates and immunoprecipitates were separated by SDS-PAGE, transferred to nitrocellulose membranes, and analyzed using the indicated primary antibodies and horseradish peroxidase-conjugated secondary antibody. Signals were detected using Super Signal West Dura Chemiluminescent Substrate (Thermo Scientific) and a CCD camera. When needed, the image was quantified with ImageJ64.
To assess NF-kB and NFAT1 nuclear translocation, purified T cells were left unstimulated or stimulated with plate-bound anti-CD3 plus anti-CD28 antibodies for 4 hours, and fractionated according to the manufacturer’s protocols (NE-PER Nuclear and Cytoplasmic Extraction Reagents, Thermo Scientific).
Antibodies, cytokines, and inhibitors
Antibodies used for flow cytometry, immunoblotting, and immunostaining analysis are listed in table S3. To generate SLFN2 antibody, N-terminally His-tagged SLFN2 (amino acids 1 to 246) was expressed in BL21 E. coli (Invitrogen) by induction with 0.1 mM IPTG. His-SLFN2-N protein was purified on SDS-PAGE and used to immunize rabbits for SLFN2 antibody generation (Cocalico Biologicals).
Recombinant mouse IL-2 protein (#402-ML-020), recombinant mouse IL-7 protein (#407-ML-005), recombinant mouse IL-15 protein (447-ML-010) were from R&D Systems. Hydrogen peroxide solution (#349887), N-acetyl-cysteine (#A9165), cycloheximide (C4859), MG132 (#474790), and ammonium chloride (#254134) were from Sigma.
Quantitative RT-PCR and primers
RNA was isolated with TRIzol reagent or as specified in the figures, and cleaned with the TURBO DNA-free Kit (Invitrogen Ambion). Reverse transcription was performed using the High Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific). The qPCR reactions were carried out using a StepOne Plus Real-Time PCR System using Powerup SYBR Green Master Mix according to the manufacturer’s protocols (Thermo Fisher Scientific). The relative levels of RNAs of interest were calculated based on ΔCt values and subsequent normalization to β-actin mRNA levels, or others indicated in the figures. Primer sequences are listed in table S2.
tRNA demethylation and qPCR-based tRNA assay
After RNA was TRIzol extracted, the tRNA demethylation and reverse transcription were carried out as described (25). Primer sequences are listed in table S2.
RNA sequencing and analysis
Total yeast tRNAs from the in vitro cleavage assay, and tRNAs and small RNAs isolated from primary mouse T cells by using mirVana miRNA Isolation Kit (Life Technologies), were deacylated and demethylated for subsequent cDNA library preparation using TruSeq Small RNA Library Prep kit (Illumina). Deacylation was performed by incubating RNA in 20 mM Tris HCl (pH 9.0) at 37°C for 40 min. Each cDNA library with index was size-selected on 8% TBE-PAGE gel to isolate cDNAs corresponding to full-length tRNAs (a prominent band at ~70 bp) and tRNA fragments (<70 bp). Individual purified libraries were pooled and sequenced by using the NSQ 500/550 Mid Output KT v2.5 (Illumina) with three flowcells for mouse full-length tRNAs, mouse tRNA fragments, and yeast tRNAs including both full-length and fragments. For RNA-seq data analysis of total tRNAs and tRNA-derived fragments, mapping and processing of RNA-seq reads was performed as previously described (59–61) and alignments were performed against the yeast and the mouse reference genomes, and the predicted tRNA genes were downloaded from the GtRNAdb v2.0 (January 2016). Briefly, adaptors were removed using cutadapt 1.9.dev4 with a minimum read length remaining of 10 bps. Reads were then mapped to the SacCer or mm10 genomes (https://genome.ucsc.edu/) using bowtie2 version 2.2.3 using local alignment and default options. Mapped reads were then compared to bedfiles containing tRNA regions (http://gtrnadb.ucsc.edu/) using bedtools version 2.26.0. For tRNA fragment analysis, counts were binned according to size ranges indicated in the figures (ranging from 10 to 70 nt) and their percentages were calculated among counted tRNA fragments of all sizes.
RNA blot analysis
TRIzol-purified total RNA was resolved on 10% denaturing urea PAGE gels, and subsequently transferred for 1 hour onto Biodyne B Nylon Membrane in 0.5X TBE at 25 V at room temperature. After transfer, membranes were crosslinked in a UV Stratalinker 2400 cross-linker (Stratagene), prehybridized with 10 ml ULTRAhyb-Oligo hybridization buffer (Thermo Fisher Scientific) for 1 hour at 42°C and then subjected to hybridization with 10 pmol biotin-labeled DNA oligo probes (Integrated DNA Technologies) at 42°C for at least 14 hours. Membranes were then rinsed and washed twice with wash buffer (2X saline sodium citrate with 0.5% (w/v) SDS) at 42°C for 1 hour and analyzed using Chemiluminescent Nucleic Acid Detection Module (89880) (Thermo Fisher Scientific). The probe sequences (table S2) were chosen based on predictions from the GtRNAdb database (tRNAscan-SE analysis of complete genomes (62)) and previously validated mouse tRNA gene families (63).
Measurement of nascent protein synthesis
For measuring protein synthesis in vitro, Click-iT Plus OPP Alexa Fluor 647 Protein Synthesis Assay Kit (Thermo Fisher Scientific) was used as indicated. T cells were incubated with OP-puro in culture for 30 min before or at various time points after anti-CD3/CD28 stimulation, and then cells were fixed and permeabilized using BD Cytofix/Cytoperm Fixation/Permeabilization Solution Kit (BD Biosciences). The azide-alkyne cycloaddition was performed according to the manufacturer’s instructions. After the 30-min reaction, the cells were washed and analyzed by flow cytometry.
For in vivo analysis, OP-Puro (MedchemSource; 50 mg per kg of body mass; pH 6.4–6.6 in PBS) was injected intraperitoneally into mice that were immunized with 100 μg of freshly prepared EndoFit ovalbumin (OVA, InvivoGen) intramuscularly 48 hours earlier. One hour later mice were euthanized, unless indicated otherwise. Splenocytes were collected and 2×106 cells were stained with combinations of antibodies against cell-surface markers as described in the figures. After washing, the cells were fixed, permeabilized, and the azide-alkyne cycloaddition was performed according to the manufacturer’s instructions. Mean OP-Puro fluorescence reflected absolute fluorescence values for each cell population from multiple independent experiments.
In vivo antigen-specific T cell proliferation
CD4-Cre; Slfn2f/f mice were crossed with OT-1 transgenic mice to produce OT-1; Slfn2f/f and OT-1; CD4-Cre; Slfn2f/f mice. CD8+ T cells were isolated from the spleens of OT-1; Slfn2f/f and OT-1; CD4-Cre; Slfn2f/f mice using the CD8a+ T Cell Isolation Kit (Miltenyi Biotec). These cells were then labeled with CFSE. Labeled cells were injected into individual CD45.1 Rag1−/− mice (2×106 cells per mouse). Four hours later, mice were i.p. injected with 100 μg of freshly prepared EndoFit ovalbumin (OVA, InvivoGen). Seventy-two hours after OVA immunization, CD45.1 recipient mice were euthanized and spleen cells were harvested for analysis of OVA-specific CD8+ T cell proliferation by flow cytometry.
MCMV infection
For MCMV infection, MCMV stock (Smith strain) was prepared as described (21). Mice were infected with MCMV (1.5×105 pfu per 20 g of body weight) by intraperitoneal injection.
EAE induction and measurement of cytokine production
For the induction of EAE, Slfn2f/f and CD4-Cre; Slfn2f/f mice were immunized subcutaneously with 150 μg of myelin oligodendrocyte glycoprotein (MOG) peptide (MOG35–55) (AnaSpec) emulsified in CFA on day 0. After immunization, the mice were i.p. injected with 200 ng of pertussis toxin (List Biological Laboratories) on days 0 and 2. Animals were scored for clinical signs of EAE according to the following criteria: 0, no clinical signs; 1, complete tail paralysis; 2, abnormal gait; 3, partial hind limb paralysis; 4, complete hind limb paralysis; 5, moribund or death, as described before (64).
Splenocytes isolated from the immunized mice on day 10 post-EAE induction, were seeded onto 96-well plates (1×106 cells per well), and then cultured for 24 and 48 hours with 50 μg/ml MOG35–55 peptide in the culture medium (RPMI 1640 medium). Supernatants were collected and assayed for IL-17A and IFN-γ by ELISA according to the manufacturer’s instructions (eBioscience).
SLFN2 structural homology modeling
Homology modeling of SLFN2 protein was performed using SWISS-MODEL based on the crystal structure of rat SLFN13 (Protein Data Bank (PDB) 5YD0) with which SLFN2 protein has 38.9% sequence identity (65). The absolute quality of the homology model in comparison to X-ray crystallography reference structures was estimated by qualitative model energy analysis (QMEA) norm score and z score. The SLFN2 model had QMEAN z score of −2.24 and QMEAN norm score of 0.67, indicating that the predicted model is well placed within the range of scores for similarly sized resolved structures in the reference set.
Statistical analysis
All comparisons of differences were between two unpaired experimental groups. An unpaired t test (Student’s t test) is appropriate and was used for such comparisons, except that mean clinical EAE score differences were analyzed by Mann–Whitney U test. Statistical analysis was performed using GraphPad Prism software. Differences with P≥0.05 were considered to be not significant (n.s.). Differences with P<0.05 were considered significant. P-values are denoted by *P<0.05, **P<0.01, and ***P<0.001. No pre-specified effect size was assumed, and in general, three or more mice or independent cell cultures of each genotype or condition were used in experiments. This sample size was sufficient to demonstrate statistically significant differences in comparisons between two unpaired experimental groups by an unpaired t test. The investigator was not blinded to genotypes or group allocations during any experiment. The allocation of samples or animals to experimental groups was random. No data were excluded from any analyses. For immunoblots and RNA blots, a representative of at least three independent experiments is shown.
Supplementary Material
Acknowledgments:
We thank Drs. J. Erzberger, Y. Liu, and X. Bai for technical help and comments on this study, Dr. G. Karlsson Hedestam for the gift of rSFV-encoded β-gal, and D. La Vine for assistance with the summary figure.
Funding: This work was supported by National Institutes of Health grants U19 AI100627 and R01 AI125581 to B.B. and by the Lyda Hill Foundation.
Footnotes
Competing interests: Authors declare no competing interests.
Data and materials availability: RNA-seq raw data have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE167919 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE167919). All other data are available in the main text or the supplementary materials. The Materials Design Analysis Reporting (MDAR) reproducibility checklist is available in Supplementary Materials.
References and Notes:
- 1.Chang CH et al. , Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 153, 1239–1251 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang R. et al. , The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 35, 871–882 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ricciardi S. et al. , The Translational Machinery of Human CD4(+) T Cells Is Poised for Activation and Controls the Switch from Quiescence to Metabolic Remodeling. Cell Metab 28, 895–906 e895 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Araki K. et al. , Translation is actively regulated during the differentiation of CD8(+) effector T cells. Nat Immunol 18, 1046–1057 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lian G. et al. , Glutathione de novo synthesis but not recycling process coordinates with glutamine catabolism to control redox homeostasis and directs murine T cell differentiation. Elife 7, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Franchina DG, Dostert C, Brenner D, Reactive Oxygen Species: Involvement in T Cell Signaling and Metabolism. Trends Immunol 39, 489–502 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Sena LA et al. , Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity 38, 225–236 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mak TW et al. , Glutathione Primes T Cell Metabolism for Inflammation. Immunity 46, 675–689 (2017). [DOI] [PubMed] [Google Scholar]
- 9.Holcik M, Sonenberg N, Translational control in stress and apoptosis. Nat Rev Mol Cell Biol 6, 318–327 (2005). [DOI] [PubMed] [Google Scholar]
- 10.Protter DSW, Parker R, Principles and Properties of Stress Granules. Trends Cell Biol 26, 668–679 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wek RC, Jiang HY, Anthony TG, Coping with stress: eIF2 kinases and translational control. Biochem Soc Trans 34, 7–11 (2006). [DOI] [PubMed] [Google Scholar]
- 12.Ivanov P, Emara MM, Villen J, Gygi SP, Anderson P, Angiogenin-induced tRNA fragments inhibit translation initiation. Mol Cell 43, 613–623 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thompson DM, Parker R, Stressing out over tRNA cleavage. Cell 138, 215–219 (2009). [DOI] [PubMed] [Google Scholar]
- 14.Thompson DM, Lu C, Green PJ, Parker R, tRNA cleavage is a conserved response to oxidative stress in eukaryotes. RNA 14, 2095–2103 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fu H. et al. , Stress induces tRNA cleavage by angiogenin in mammalian cells. FEBS Lett 583, 437–442 (2009). [DOI] [PubMed] [Google Scholar]
- 16.Emara MM et al. , Angiogenin-induced tRNA-derived stress-induced RNAs promote stress-induced stress granule assembly. J Biol Chem 285, 10959–10968 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yamasaki S, Ivanov P, Hu GF, Anderson P, Angiogenin cleaves tRNA and promotes stress-induced translational repression. J Cell Biol 185, 35–42 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fett JW et al. , Isolation and characterization of angiogenin, an angiogenic protein from human carcinoma cells. Biochemistry 24, 5480–5486 (1985). [DOI] [PubMed] [Google Scholar]
- 19.Schimmel P, The emerging complexity of the tRNA world: mammalian tRNAs beyond protein synthesis. Nat Rev Mol Cell Biol 19, 45–58 (2018). [DOI] [PubMed] [Google Scholar]
- 20.Haussecker D. et al. , Human tRNA-derived small RNAs in the global regulation of RNA silencing. RNA 16, 673–695 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Berger M. et al. , An Slfn2 mutation causes lymphoid and myeloid immunodeficiency due to loss of immune cell quiescence. Nat Immunol 11, 335–343 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hidmark AS et al. , Humoral responses against coimmunized protein antigen but not against alphavirus-encoded antigens require alpha/beta interferon signaling. J Virol 80, 7100–7110 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ross SH, Cantrell DA, Signaling and Function of Interleukin-2 in T Lymphocytes. Annu Rev Immunol 36, 411–433 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu J, Xu Y, Stoleru D, Salic A, Imaging protein synthesis in cells and tissues with an alkyne analog of puromycin. Proc Natl Acad Sci U S A 109, 413–418 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li M. et al. , DNA damage-induced cell death relies on SLFN11-dependent cleavage of distinct type II tRNAs. Nat Struct Mol Biol 25, 1047–1058 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang JY et al. , Structure of Schlafen13 reveals a new class of tRNA/rRNA- targeting RNase engaged in translational control. Nat Commun 9, 1165 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Katsoulidis E. et al. , Role of Schlafen 2 (SLFN2) in the generation of interferon alpha-induced growth inhibitory responses. J Biol Chem 284, 25051–25064 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saikia M, Hatzoglou M, The Many Virtues of tRNA-derived Stress-induced RNAs (tiRNAs): Discovering Novel Mechanisms of Stress Response and Effect on Human Health. J Biol Chem 290, 29761–29768 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eleftheriadis T. et al. , Angiogenin is upregulated during the alloreactive immune response and has no effect on the T-cell expansion phase, whereas it affects the contraction phase by inhibiting CD4(+) T-cell apoptosis. Exp Ther Med 12, 3471–3475 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pizzo E. et al. , Ribonuclease/angiogenin inhibitor 1 regulates stress-induced subcellular localization of angiogenin to control growth and survival. J Cell Sci 126, 4308–4319 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goncalves KA et al. , Angiogenin Promotes Hematopoietic Regeneration by Dichotomously Regulating Quiescence of Stem and Progenitor Cells. Cell 166, 894–906 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu W. et al. , Plexin-B2 Mediates Physiologic and Pathologic Functions of Angiogenin. Cell 171, 849–864 e825 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Omar I, Lapenna A, Cohen-Daniel L, Tirosh B, Berger M, Schlafen2 mutation unravels a role for chronic ER stress in the loss of T cell quiescence. Oncotarget 7, 39396–39407 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mami I. et al. , Angiogenin Mediates Cell-Autonomous Translational Control under Endoplasmic Reticulum Stress and Attenuates Kidney Injury. J Am Soc Nephrol 27, 863–876 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rothenberg EV, Ward SB, A dynamic assembly of diverse transcription factors integrates activation and cell-type information for interleukin 2 gene regulation. Proc Natl Acad Sci U S A 93, 9358–9365 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim HP, Leonard WJ, The basis for TCR-mediated regulation of the IL-2 receptor alpha chain gene: role of widely separated regulatory elements. EMBO J 21, 3051–3059 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li L. et al. , The translin-TRAX complex (C3PO) is a ribonuclease in tRNA processing. Nat Struct Mol Biol 19, 824–830 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fletcher SJ et al. , Role of the novel endoribonuclease SLFN14 and its disease-causing mutations in ribosomal degradation. RNA 24, 939–949 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lin YZ et al. , Equine schlafen 11 restricts the production of equine infectious anemia virus via a codon usage-dependent mechanism. Virology 495, 112–121 (2016). [DOI] [PubMed] [Google Scholar]
- 40.Bustos O. et al. , Evolution of the Schlafen genes, a gene family associated with embryonic lethality, meiotic drive, immune processes and orthopoxvirus virulence. Gene 447, 1–11 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Geserick P, Kaiser F, Klemm U, Kaufmann SH, Zerrahn J, Modulation of T cell development and activation by novel members of the Schlafen (slfn) gene family harbouring an RNA helicase-like motif. Int Immunol 16, 1535–1548 (2004). [DOI] [PubMed] [Google Scholar]
- 42.Mavrommatis E, Fish EN, Platanias LC, The schlafen family of proteins and their regulation by interferons. J Interferon Cytokine Res 33, 206–210 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kishimoto K, Liu S, Tsuji T, Olson KA, Hu GF, Endogenous angiogenin in endothelial cells is a general requirement for cell proliferation and angiogenesis. Oncogene 24, 445–456 (2005). [DOI] [PubMed] [Google Scholar]
- 44.Silberstein L. et al. , Proximity-Based Differential Single-Cell Analysis of the Niche to Identify Stem/Progenitor Cell Regulators. Cell Stem Cell 19, 530–543 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ito D. et al. , mTOR Complex Signaling through the SEMA4A-Plexin B2 Axis Is Required for Optimal Activation and Differentiation of CD8+ T Cells. J Immunol 195, 934–943 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yue F. et al. , A comparative encyclopedia of DNA elements in the mouse genome. Nature 515, 355–364 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xin M. et al. , Regulation of insulin-like growth factor signaling by Yap governs cardiomyocyte proliferation and embryonic heart size. Sci Signal 4, ra70 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee PP et al. , A critical role for Dnmt1 and DNA methylation in T cell development, function, and survival. Immunity 15, 763–774 (2001). [DOI] [PubMed] [Google Scholar]
- 49.Wang KW et al. , Enhanced susceptibility to chemically induced colitis caused by excessive endosomal TLR signaling in LRBA-deficient mice. Proc Natl Acad Sci U S A, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Choi JH et al. , IgD class switching is initiated by microbiota and limited to mucosa-associated lymphoid tissue in mice. Proc Natl Acad Sci U S A 114, E1196–E1204 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nobile V, Vallee BL, Shapiro R, Characterization of mouse angiogenin-related protein: implications for functional studies on angiogenin. Proc Natl Acad Sci U S A 93, 4331–4335 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Morales-Perez CL, Noviello CM, Hibbs RE, Manipulation of Subunit Stoichiometry in Heteromeric Membrane Proteins. Structure 24, 797–805 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.She J. et al. , Structural insights into the voltage and phospholipid activation of the mammalian TPC1 channel. Nature 556, 130–134 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jiang F. et al. , Structures of a CRISPR-Cas9 R-loop complex primed for DNA cleavage. Science 351, 867–871 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shapiro R, Riordan JF, Vallee BL, Characteristic ribonucleolytic activity of human angiogenin. Biochemistry 25, 3527–3532 (1986). [DOI] [PubMed] [Google Scholar]
- 56.Iyer S, Holloway DE, Acharya KR, Crystal structures of murine angiogenin-2 and −3-probing ‘structure--function’ relationships amongst angiogenin homologues. FEBS J 280, 302–318 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Saxena SK, Rybak SM, Davey RT Jr., Youle RJ, Ackerman EJ, Angiogenin is a cytotoxic, tRNA-specific ribonuclease in the RNase A superfamily. J Biol Chem 267, 21982–21986 (1992). [PubMed] [Google Scholar]
- 58.Kedersha N, Anderson P, Mammalian stress granules and processing bodies. Methods Enzymol 431, 61–81 (2007). [DOI] [PubMed] [Google Scholar]
- 59.Torres AG, Reina O, Stephan-Otto Attolini C, Ribas de Pouplana L, Differential expression of human tRNA genes drives the abundance of tRNA-derived fragments. Proc Natl Acad Sci U S A 116, 8451–8456 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Su Z, Kuscu C, Malik A, Shibata E, Dutta A, Angiogenin generates specific stress-induced tRNA halves and is not involved in tRF-3-mediated gene silencing. J Biol Chem 294, 16930–16941 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Thomas SP, Hoang TT, Ressler VT, Raines RT, Human angiogenin is a potent cytotoxin in the absence of ribonuclease inhibitor. RNA 24, 1018–1027 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chan PP, Lowe TM, GtRNAdb 2.0: an expanded database of transfer RNA genes identified in complete and draft genomes. Nucleic Acids Res 44, D184–189 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Coughlin DJ, Babak T, Nihranz C, Hughes TR, Engelke DR, Prediction and verification of mouse tRNA gene families. RNA Biol 6, 195–202 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nakagawa K. et al. , Schlafen-8 is essential for lymphatic endothelial cell activation in experimental autoimmune encephalomyelitis. Int Immunol 30, 69–78 (2018). [DOI] [PubMed] [Google Scholar]
- 65.Waterhouse A. et al. , SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Research 46, W296–W303 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.