

Abstract
Background
The protozoan parasites in the Cryptosporidium genus cause both acute diarrheal disease and subclinical (ie, nondiarrheal) disease. It is unclear if the microbiota can influence the manifestation of diarrhea during a Cryptosporidium infection.
Methods
To characterize the role of the gut microbiota in diarrheal cryptosporidiosis, the microbiome composition of both diarrheal and surveillance Cryptosporidium-positive fecal samples from 72 infants was evaluated using 16S ribosomal RNA gene sequencing. Additionally, the microbiome composition prior to infection was examined to test whether a preexisting microbiome profile could influence the Cryptosporidium infection phenotype.
Results
Fecal microbiome composition was associated with diarrheal symptoms at 2 timepoints. Megasphaera was significantly less abundant in diarrheal samples compared with subclinical samples at the time of Cryptosporidium detection (log2 [fold change] = –4.3; P = 10–10) and prior to infection (log2 [fold change] = –2.0; P = 10–4); this assigned sequence variant was detected in 8 children who had diarrhea and 30 children without diarrhea. Random forest classification also identified Megasphaera abundance in the pre- and postexposure microbiota as predictive of a subclinical infection.
Conclusions
Microbiome composition broadly, and specifically low Megasphaera abundance, was associated with diarrheal symptoms prior to and at the time of Cryptosporidium detection. This observation suggests that the gut microenvironment may play a role in determining the severity of a Cryptosporidium infection.
Clinical Trials Registration. NCT02764918.
Keywords: Cryptosporidium, microbiome/microbiota, parasite, diarrhea, Bangladesh
Megasphaera abundance in the stool of Bangladeshi infants is associated with the development of diarrhea upon infection with the Cryptosporidium parasite.
Protozoan parasites in the Cryptosporidium genus cause both acute diarrhea and subclinical (ie, nondiarrheal) disease, and both clinical outcomes are associated with poor physical and neurocognitive growth in infants [1–6]. These parasites are the fifth leading cause of diarrhea in young children [7], and recent studies have estimated the global burden of Cryptosporidium diarrhea mortality to be as high as 50 000 deaths annually [8]. This burden is disproportionately borne by young children [9]. Importantly, no therapies exist to treat Cryptosporidium infection in children or immunocompromised individuals [10]. Thus, there is a pressing need to prevent cryptosporidiosis mortality.
Understanding the difference in the host, parasite, and environment during acute diarrheal and subclinical infections may reveal new therapeutic solutions. Human polymorphisms are associated with an increased host susceptibility to cryptosporidiosis; however, these mutations do not completely explain the differences in infection outcomes [11, 12]. Parasite genetics (within and across species) have been associated with differences in their host range [13–16]. The role of the microbiome upon infection by Cryptosporidium has been examined in healthy adults [17] and animals [18, 19]; however, its role in differentiating diarrheal and subclinical infections is not known, nor is the impact of any differences in the microbiome composition occurring during infant cryptosporidiosis.
Here, we interrogate the association between diarrheal status during cryptosporidiosis and a child’s microbiome using fecal samples from infants living in Mirpur and Mirzapur, Bangladesh. In Mirpur, Cryptosporidium diarrhea was frequent (24% of infections); detected Cryptosporidium species included Cryptosporidium hominis, Cryptosporidium parvum, and Cryptosporidium meleagridis, with C. hominis as the most common. In contrast, most infections in Mirzapur were subclinical (98%), and C. meleagridis was the most common detected species [1]. Because Cryptosporidium-associated diarrhea was infrequent in Mirzapur and most infections involved C. meleagridis rather than C. hominis or C. parvum, the association between diarrheal status and microbiome composition in infants in Mirzapur could not be decoupled from an alternative infection phenotype caused by C. meleagridis. We therefore focused our analysis on Mirpur due to the variation in diarrheal status and the dominance of the C. hominis species in this population. We found that the microbiota demonstrated high variability between children but, despite this observation, microbiota composition and a low abundance of Megasphaera were associated with diarrheal symptoms both at the time of Cryptosporidium detection and prior to infection. Thus, we propose that Megasphaera may prevent acute diarrhea during parasite infection or at least can serve as a biomarker for other unknown protective factors.
MATERIALS AND METHODS
Cohort
Children were enrolled into a community-based prospective cohort study of enteric infections that was established at the urban and rural Bangladesh sites, Mirpur and Mirzapur, respectively (ClinicalTrials.gov identifier NCT02764918) (Figure 1A) [1, 14]. Stool samples were collected monthly and during diarrheal episodes. Diarrhea was defined as ≥3 loose stools within 24 hours, as reported by the child’s caregiver. Both pan-species and species-specific quantitative polymerase chain reaction assays were used to identify the Cryptosporidium species infecting the children (Steiner et al 2018) [1]. If positive samples were collected within an interval of ≤65 days, they were regarded as derived from 1 infection event [1, 14]. In addition to the collection of stool samples, a study database was created containing clinical information on each episode of diarrhea a child experienced, antibiotic consumption, and anthropometric measurements as well as data on the household demographics [1]. A subset of the Cryptosporidium-positive and corresponding “predetection” Cryptosporidium-negative surveillance samples were analyzed. The data from Mirzapur (Figure 1A) were only included in the post hoc analysis due to the limited amount of information on the antibiotic history of these children, the rarity of diarrheal cases at this site, and the high prevalence of C. meleagridis at the site relative to the more common C. hominis species detected in Mirpur.
Figure 1.

Study design. A, Overall cohort design and sample collection. For more information, see [1, 14]. Samples from Mirzapur were only used in post hoc analysis in Figure 4F. B, Paired samples were selected to assess Cryptosporidium-positive samples (time of detection) and the preceding surveillance sample (predetection). Cryptosporidium-positive samples were identified from both monthly surveillance and diarrheal stool samples, generating our subclinical and diarrheal sample groups.
The study was approved by the Ethical and Research Review Committees of the International Centre for Diarrhoeal Disease Research, Bangladesh, and by the Institutional Review Board of the University of Virginia. For each child, informed written consent was obtained from their parent or guardian.
DNA Extraction
On the day of collection, stool samples were brought to the study clinic and transported to our laboratory at 4°C, where they were aliquoted in DNase- and Rnase-free cryovials for storage at –80°C. For DNA extraction, samples were thawed and 200 mg removed for total nucleic acid extraction (see details in [1]). To verify the extraction protocol, phocine herpesvirus (European Virus Archive Global, Department of Virology, Erasmus Medical Center, Rotterdam, the Netherlands) and bacteriophage MS2 (ATCC 15597B; American Type Culture Collection, Manassas, Virginia) were added into each sample as positive controls.
16S Ribosomal Sequencing and Processing
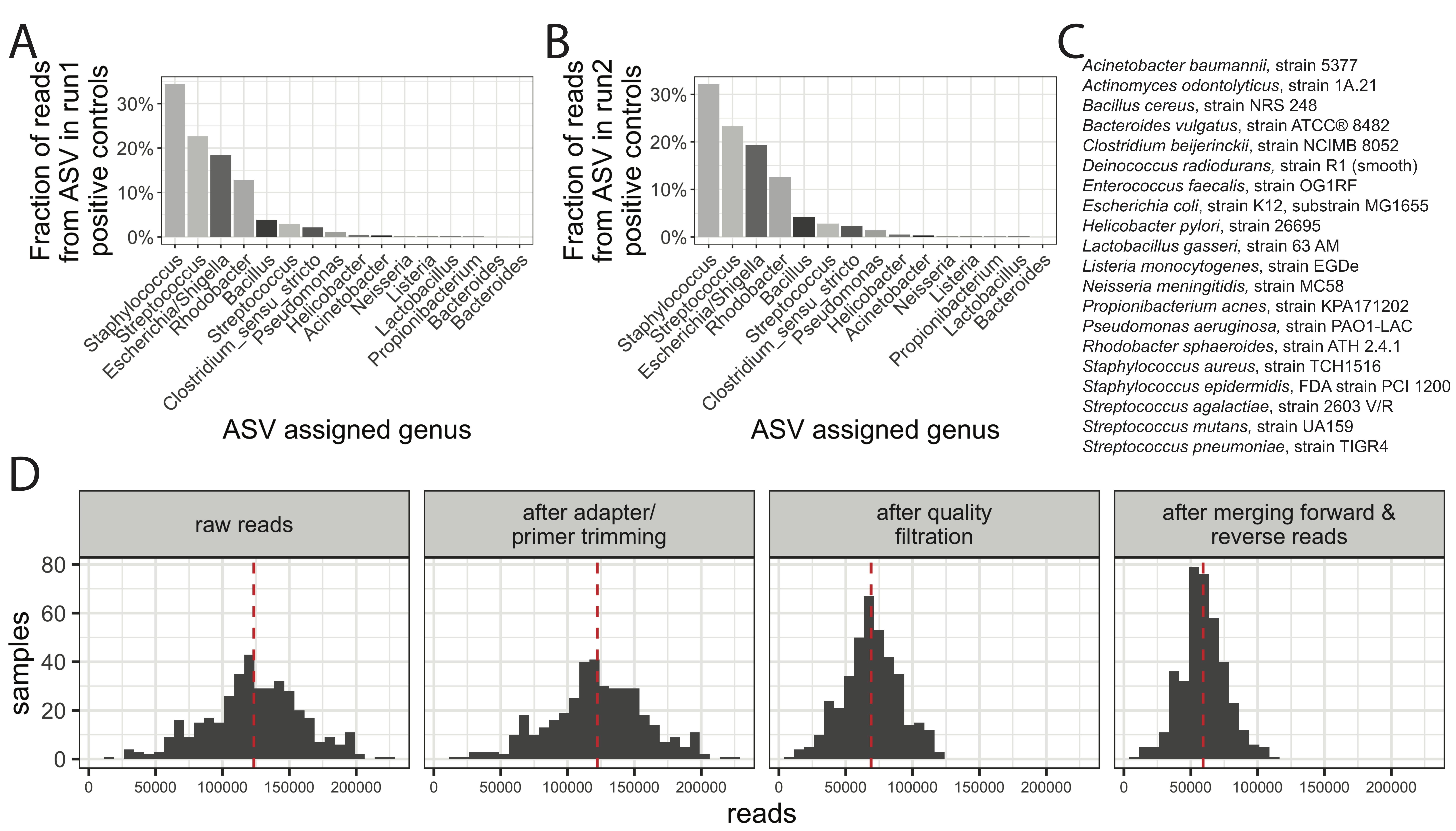
The V4 region of the 16S ribosomal (rRNA) gene was amplified using the previously described phased Illumina-eubacteria primers and protocol from [20, 21] with the minor modification that the Illumina MiSeq version 3 chemistry was used to generate 300-bp paired-end reads. Sequencing was performed by the University of Virginia’s Genome Analysis and Technology Core. Negative controls included extraction blanks throughout the amplification and sequencing process. As positive controls, DNA was extracted from the HM-782D Mock Bacteria Community (ATCC through BEI Resources) and analyzed on each sequencing run (Supplementary Figure 1A–C). Additionally, a PhiX DNA library was added at 20% into each sequencing run to increase genetic diversity prior to parallel sequencing in both forward and reverse directions using the Miseq version 3 kit and machine (per the manufacturer’s protocol).
Sequencing produced 48 146 401 reads with a mean of 118 295.8 and median of 121 519 reads per sample (raw reads from Supplementary Figure 1D). Sequence adaptors were then removed using Bbtools [22] and primers were removed using CutAdapt [23]; quality-based filtering was performed with DADA2 [24]. Quality filtration reduced the total number of reads to a mean of 59 202.2 reads per sample (Supplementary Figure 1D). In brief, reads were removed and trimmed based on overall read quality and base pair quality: forward and reverse reads were trimmed to 250 or 200 bp and removed if there were more than 3 or 6 expected errors, respectively. Reads were also truncated at the first instance of a quality score (Phred or Q score) of ≤2. Next, forward and reverse reads were merged with only 1 mismatch permitted. Last, taxa assignments were made using DADA2’s naive Bayesian classifier method and the Ribosomal Database Project’s Training Set 16 (release 11.5) reference database [24] and reads that did not map to bacteria (including human contaminants, archaea, and mitochondrial or chloroplast DNA) were removed, resulting in a mean of 27 809 reads per sample.
Samples with <10 000 reads and unpaired samples (those with no predetection or time-of-detection sample within 42 days) were removed from consideration; all were subsampled to a uniform depth of 10 000 reads per sample to correct for differences in sequencing depth across samples and to enable the comparison (rather than cataloging) of sequenced taxa among samples [25]. Following these filtration and processing steps, 2953 amplicon sequence variants (ASVs) and 182 stool samples remained in the dataset.
The 182 paired predetection and time-of-detection samples (91 pre- and 91 postdetection), as well as additional positive and negative control samples (amplification blanks) and additional samples that did not pass our selection criteria, were split into 2 sequencing runs to increase the sequencing depth. The first sequencing run included all predetection samples and the second sequencing run included all time-of-detection samples. As an unintentional result of this choice, sequencing batch effects may result in spurious differences between predetection and time-of-detection samples; thus, analyses are focused on symptomatic vs subclinical samples within each time point (ie, within the same sequencing batch).
Statistical and Machine Learning Analyses
All of the following data processing and statistical analyses were performed in R software [24, 26–29] (see Supplementary Materials for code and software versions). Appropriate statistical tests were selected and are described as introduced throughout the Results.
For machine learning analyses, random forest analysis was used to classify subclinical or diarrheal samples using associated metadata and/or ASV abundances, and the trained models (ie, classifiers) were used to identify individual variables that were important for prediction accuracy [30]. Within a random forest classifier, individual trees were built from subsets of the data and model performance was evaluated by predicting the class of each sample using only the trees in the random forest that were not constructed using that sample (ie, out-of-bag performance). Here, variables were ranked by their effect on classifier certainty, which influenced overall accuracy, using the mean decrease in node impurity (via the Gini coefficient). Variables that maximally split samples by classification group yielded a larger forest-wide node impurity (or evenness of the split); thus, more important variables had a higher mean decrease in node impurity. Analytic code is provided in the Supplementary Materials; analyses and figure generation were performed in R software [29, 31–43].
RESULTS
Prevalence of Diarrhea and Antibiotic Use
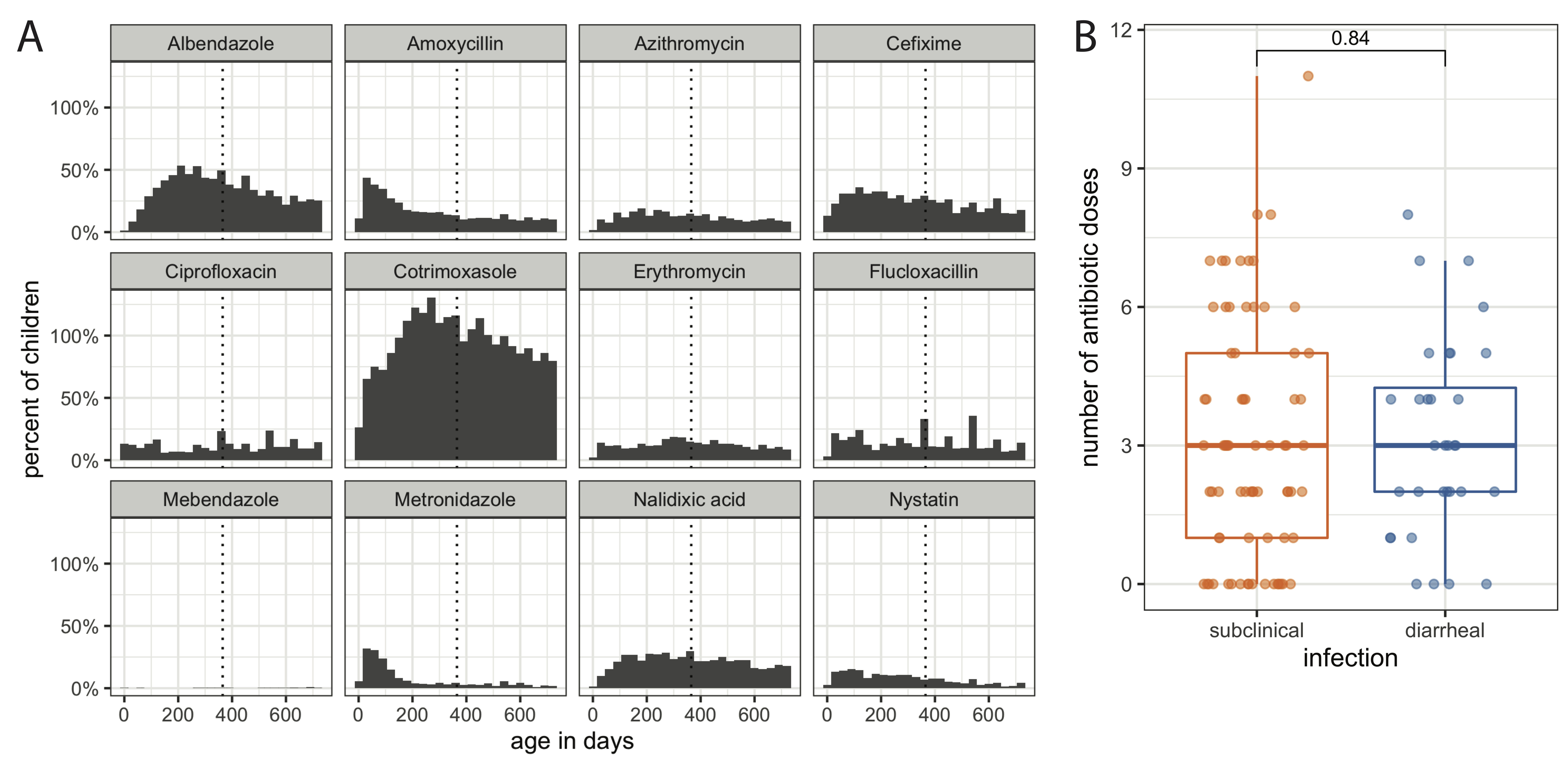
Infants were enrolled into a prospective cohort from Mirpur, Dhaka, Bangladesh to study enteric infections (Figure 1A); this cohort was part of a larger assessment of diarrhea in Bangladesh, published previously (Figure 1A) [1]). Each child was monitored by community health workers for enteric disease, including collection of monthly surveillance and diarrheal stool samples during the first 2 years of life. Diarrhea and antibiotic use were common in this cohort (Figure 2A and Supplementary Figure 2), and Cryptosporidium species, including C. hominis and C. meleagridis, were frequently detected during diarrhea (Table 1). These parasites cause both subclinical and overt diarrheal infections [1].
Figure 2.

Diarrheal infection and antibiotic treatment were common and heterogenous in infants from Mirpur. A, Prevalence of diarrhea. Frequency of diarrheal episodes per child. Full Mirpur cohort is shown in in gray; red subset indicates the children whose samples were used in the microbiome study (and all subsequent figures). B, All-cause diarrhea was heterogenous among children with divergent Cryptosporidium outcomes. Number of diarrheal events per child based on cumulative Cryptosporidium status, both over the first 2 years of life. C, Antibiotic usage was heterogenous among children with divergent Cryptosporidium outcomes. Number of antibiotic events per child based on cumulative Cryptosporidium status, both over the first 2 years of life. Combination therapies were treated as separate doses. For B and C, the full cohort was used and statistics are shown if significant. For B and C, each box represents the median (inner line), 25th percentile, and 75th percentile. Upper whiskers extend from the top of the box to the largest value within 1.5 times the interquartile range (distance between 25th and 75th percentile), and the lower whisker extends to the smallest value within 1.5 times the interquartile range. P values were generated from a t test without multiple testing correction.
Table 1.
Sample Summary Statistics for Samples From Mirpur
| Subclinical Infections | Diarrheal Infections | Total | ||
|---|---|---|---|---|
| Children | 72 | |||
| Male/female sex | 28/44 | |||
| Children with repeat infections in dataset | 19 | |||
| Samples | 182 | |||
| No. of PD samples | 60 | 31 | 91 | |
| No. of TOD samples | 60 | 31 | 91 | |
| Age at collection, d, mean (SD) | 362.5 (128.8) | 321.3 (136.3) | 348.7 (132.1) | |
| Days between PD and TOD samplea, mean (SD) | 31.1 (4.6) | 19.2 (9.1) | 27.0 (8.6) | |
| Parasite burden at TOD (pan-Cryptosporidium qPCR Ct), mean (SD) | 28.6 (6.2) | 29.9 (7.3) | 29.0 (6.6) | |
| Positive qPCR (for positive samples) | Pan-Cryptosporidium: 100% | Pan-Cryptosporidium: 100% | Pan-Cryptosporidium: 100% | |
| C. hominis: 60% | C. hominis: 58% | C. hominis: 59% | ||
| C. meleagridis: 7% | C. meleagridis: 6% | C. meleagridis: 7% | ||
| First infection/repeat infection, No. | 42/18 | 28/3 | 70/21 |
Abbreviations: Ct, cycle threshold; PD, predetection; qPCR, quantitative polymerase chain reaction; SD, standard deviation; TOD, time of detection.
aStatistically different between subclinical and diarrheal infections via t test (P = 4 × 10–8). All other comparisons between clinical types were not significantly different using a t test (all comparisons except first vs subsequent infection) or χ 2 test (first vs subsequent infection).
Children who had at least 1 symptomatic episode of cryptosporidiosis had more cumulative episodes of diarrhea than children with exclusively subclinical infections or no Cryptosporidium-positive stool samples (Figure 2B). Additionally, children with only diarrheal episodes (ie, no observed subclinical cryptosporidiosis) had more frequent exposure to antibiotics than children who had never tested positive for Cryptosporidium (Figure 2C). Frequent antibiotic use occurred (Supplementary Figure 2A), but there was no difference in antibiotic use during the month prior to infection between children with subclinical or diarrheal infections (Supplementary Figure 2B).
Microbiota Sequencing
Given the difference in all-cause diarrheal frequency between children with subclinical and diarrheal cryptosporidiosis (Figure 2B), we hypothesized that microbiome composition may influence the development of acute symptoms during cryptosporidiosis. 16S rRNA gene sequencing was performed on both the time-of-detection stool samples (Cryptosporidium positive, including subclinical and diarrheal) and the corresponding surveillance stool collected immediately prior to the Cryptosporidium-positive sample (predetection; Figure 1B) for a subset of children who tested positive for Cryptosporidium (Table 1 and Figure 2A). Predetection samples were collected within approximately 1 month of the time-of-detection samples (Table 1).
Sequencing produced 48 146 401 reads with a mean of 118 295.8 and median of 121 519 reads per sample (raw reads from Supplementary Figure 1D). Following quality filtration and taxonomy assignment, a mean of 27 809 reads per sample remained, permitting us to subsample reads to a uniform depth of 10 000 reads per sample to correct for differences in sequencing depth across samples.
Microbiota Diversity
Following sequencing, taxonomy was assigned to reads using DADA2. Nearly 25% of reads were assigned to an ASV belonging to the genus Bifidobacterium (Figure 3A) that represents a number of functionally diverse species which colonize the infant gastrointestinal tract soon after birth. Microbiota α-diversity measures (richness and evenness) were not statistically significantly different between sample groups (2-way analysis of variance, post hoc testing via Tukey honest significant difference method; significance cutoff of P < .05; Figure 3B and 3C). Despite this lack of significance (P > .21 for all comparisons), the microbiota of infants who had diarrheal infection was, on average, less diverse than infants with subclinical infection, both prior to and at the time of infection (Figure 3B and 3C). Moreover, this cohort exhibited high interindividual variation as many ASVs were specific to just a few children. Only a few ASVs were found in >50% of samples (Figure 3D).
Figure 3.

Microbiome samples were highly variable. A, Most abundant amplicon sequence variants (ASVs) in the study. Only the top 10 most abundant ASVs are shown; the abundance of these common ASVs per sample is also represented in Supplementary Figure 3. Nearly 25% of all reads were assigned to an ASV in the Bifidobacterium genus. B, Richness of each sample, or the number of ASVs present in a sample, was not significantly different across sample groups. B and C, Each box represents the median (inner line), 25th percentile, and 75th percentile. Upper whiskers extend from the top of the box to the largest value within 1.5 times the interquartile range (distance between 25th and 75th percentile), and the lower whisker extends to the smallest value within 1.5 times the interquartile range. C, Evenness was also minimally different across sample groups. Evenness is a diversity metric calculated to represent how many different species are present and how well distributed those species are across samples; it is calculated using the inverse Simpson index. No significant differences in evenness was observed among any comparisons of clinical type (2-way analysis of variance with multiple testing correction via Tukey honest significant difference). D, Fraction of all samples containing a particular ASV, ordered by from highest to lowest. Very few ASVs were detected in many samples; however, almost all samples contain the most common Bifidobacterium ASVs.
Associations Between Diarrheal Symptoms and the Microbiota
To identify compositional differences in the microbiome among sample groups, principal coordinate analysis was performed using the Euclidean distance between samples. Predetection samples overlapped substantially with Cryptosporidium-positive samples and, among positive samples, subclinical and diarrheal samples did not separate (permutational multivariate analysis of variance using distance matrices [PERMANOVA]; P > .05; Figure 4A). Alternative distance metrics, such as Unifrac, also failed to separate sample groups (Supplementary Figure 4A). The change in microbiota from predetection to time of detection for each child was similarly variable for both diarrheal and subclinical infections (PERMANOVA; P > .05; Supplementary Figure 4B).
Figure 4.

Identifying associations between diarrheal symptoms and the microbiota. A, Predetection (PD) and time-of-detection (TOD) sample microbiota were indistinguishable via principal coordinate analysis using a permutational multivariate analysis of variance using distance matrices and a significance cutoff of P < .05, as were subclinical and diarrheal Cryptosporidium-positive samples. Principal coordinate analysis of amplicon sequence variants (ASV) quantification across all samples using Euclidean distance. B, Univariate statistics identifies ASVs associated with symptoms in the PD samples and TOD samples. Statistically significant differential expressed ASVs are colored, whereas gray points represent ASVs that were not different or not significantly different, using DESeq2. Large points indicate ASVs that were also identified as important using random forest classification, whereas small points were not among the top 15 most important variables. Random forest classifiers were built to predict the presence of diarrhea upon Cryptosporidium infection. Importantly, purple points represent statistically significant ASVs that were also among the most important variables for classifiers made at both timepoints. C, Random forest classifiers were built from the TOD microbiota (blue) or predetection microbiota (red). Area under the curve (AUC), a metric of classifier accuracy, is listed for each classifier. D, Most important variables, as ranked by mean decrease in node impurity (or Gini importance), from the PD and TOD classifiers. Important variables were similarly important, within and across models. Of note, age was not an important variable in the TOD classifier. E, One ASV assigned to the Megasphaera genus was significantly less abundant in diarrheal cases via univariate analyses (at both timepoints) and was among the top 15 most important variables for the classifiers for both timepoints. Relative abundance of each ASV is plotted for each sample, with each box representing the median (inner line), 25th percentile, and 75th percentile. Upper whiskers extend from the top of the box to the largest value within 1.5 times the interquartile range (distance between 25th and 75th percentile), and the lower whisker extends to the smallest value within 1.5 times the interquartile range. F, The Megasphaera ASV was also more likely to be high-abundance (above dashed line) in samples at the second study site, Mirzapur, where diarrheal cryptosporidiosis was less common when compared to Mirpur; however, environmental factors, including the causal Cryptosporidium species, were also different in Mirzapur [1]. Increased Megasphaera abundance in Mirzapur may partially explain reduced diarrhea associated with cryptosporidiosis in that community.
Given the lack of separation between samples when considering overall microbiome composition, univariate analyses were used to identify individual ASVs that were significantly different between subclinical and diarrheal samples prior to and at the time of infection (Figure 4B). However, univariate statistics rely on assumptions of independence and, thus, may perform poorly with microbiome datasets due to correlations between and statistical interactions among members of the microbiota [44]. To make robust inferences of the importance of individual ASVs, we utilized a univariate approach designed specifically for sparse count data [45], as well as random forest classification to consider interactions among ASVs. Interpreting the results of these 2 approaches together provided a more stringent assessment of ASV importance.
Thus, classification using the random forest models was performed to determine if specific members of the microbiota were predictive of the development of diarrheal symptoms; important variables from the random forest models are highlighted on the volcano plots, which also show the results of univariate statistical tests (Figure 4B and 4C). This machine learning approach was used to prioritize the results generated from univariate statistics. Classifier performance using the predetection or time-of-detection microbiome separately yielded predictive models (area under the curve >0.6 for both prior to and at the time of infection Figure 4C); this performance was similar to the highest-performing classification models across a metanalysis of case-control clinical microbiome studies [46, 47].
Both classifiers supported conclusions drawn by univariate analyses and identified several additional ASVs as important to classify subclinical and diarrheal samples (Figure 4B and 4C). Some important microbes for each classifier were not enriched in either sample group (Figure 4B), suggesting that these ASVs are only important when analyzed in combination with others. Despite the effect of antibiotic treatment on the microbiota [48], the addition of a child’s antibiotic history did not significantly augment classifier performance (Supplementary Figure 5), indicating that there was no interaction between the important ASVs and antibiotic use. The infecting Cryptosporidium species (C. hominis or C. meleagridis) were not important variables in the random forest models, and child age was not an important variable in the time-of-detection model (Figure 4D).
We focused on ASVs that were identified via both the univariate statistics and machine learning approaches. For the predetection timepoint, these prioritized ASVs were assigned to the Megasphaera, Flavonifractor, Morganella, Collinsella, and Lactobacillus genera; for the time-of-detection timepoint, these included the same Megasphaera ASV, as well as ASVs assigned to Parabacteroides, Enterococcus, Prevotella, Bifidobacterium, Sutterella, Veillonella, Megamonas, and Faecalibacterium (Figure 4B and 4D). Combinations of ASVs were more predictive of diarrhea than any individual ASV, as evident by the similar Gini importance for all important variables (Figure 3D).
One Megasphaera ASV in particular was identified at both timepoints and both analytic approaches (Figure 4B and 4E). This Megasphaera ASV also accounted for at least 1% of reads across the entire study (Figure 2A), and was present in 25% of samples (Supplementary Figure 6A). This bile acid–resistant species colonizes the small intestines [49], among other sites on the human body [50, 51]. It can therefore be a major component of the microbiome at the site of Cryptosporidium parasite colonization. The other ASVs that contributed to model performance were either less abundant or resided predominantly in the large bowel. Interestingly, Megasphaera ASVs broadly did not show the same trend as the important individual ASV (Supplementary Figure 6B) and were present in 54.9% of samples.
Although there were many environmental differences between the study sites, this ASV was also more likely to be detected at high abundance in our second study site, rural Mirzapur (Figure 4F), despite the observation that Megasphaera ASV did not vary with Cryptosporidium species (Supplementary Materials). The most common Cryptosporidium species at Mirzapur was C. meleagridis rather than the C. hominis in Mirpur, but C. meleagridis has been associated with gastrointestinal disease in other studies and has also been shown to cause diarrhea in a human challenge experiment [52, 53]. Children in Mirzapur were, however, less likely to develop diarrhea upon Cryptosporidium infection; 3% of Cryptosporidium-positive stools in Mirzapur were diarrheal, compared to 32% in Mirpur [1].
DISCUSSION
Here, we identified differences in the microbiota composition and in the abundance of an individual ASV, Megasphaera, in infants who had either a subclinical or a diarrheal Cryptosporidium infection. Fecal samples from 72 Cryptosporidium-infected children in Mirpur, Bangladesh, were used to profile the human microbiota during cryptosporidiosis (Table 1 and Figure 1) with 16S rRNA gene sequencing (Figure 3). It is well established that the microbiome shifts with child development [54–56] and that it is highly variable in infants aged <2 years [57–59]. There was also universally frequent antibiotic use and enteric infections in this young population (Table 1, Figure 2C, and Supplementary Figure 2). It was therefore unsurprising that there was a high degree of intersample variability among these infants’ samples (Figure 3A and 3D).
Despite this variation, microbiome composition was predictive of diarrheal symptoms at the time of infection and up to a month prior (Figure 4C). Although individual members of the microbiome were associated with diarrhea (Figure 4B), no single ASV completely explained the clinical type of infection (Figure 4D). This observation is consistent with animal models of infection that have highlighted a complex relationship between the microbiota, host, and parasite [60–62]. For example, previous work found that antibiotics alone did not sensitize immunocompetent mice to infection [18], although certain probiotics [63], antibiotics [64], and deprivation of prebiotics [65] could exacerbate disease severity.
Higher abundance of 1 ASV, Megasphaera (class: Clostridia), was associated with subclinical Cryptosporidium infection whereas its absence or low abundance was more common in cases of Cryptosporidium-associated diarrhea (Figure 4B and 4D). This Megasphaera ASV was not associated with antibiotic use in this cohort (Supplementary Figure 5) or all-cause diarrhea (ie, total number of diarrheal episodes; Supplementary Materials). Megasphaera species can collocate in the small intestines [49] with Cryptosporidium, and they were more frequently observed at high abundance in a community in which diarrhea was rarely seen during cryptosporidiosis (Figure 4F) [1]. Megasphaera are known to synthesize short-chain fatty acids [66], compounds that regulate the intestinal homeostasis [67], impact the host immune response [68], and modulate osmotic diarrhea [69]. Interestingly, Megasphaera elsdenii is used as a probiotic in veterinary medicine to treat diet-induced metabolic acidosis because of the bacteria’s ability to convert lactate (a key acidic metabolite responsible for acidosis) to short-chain fatty acids [70]. This ability of Megasphaera to produce short-chain fatty acids or to modulate the host’s immune system through other mechanisms may be protective in attenuating disease outcome during Cryptosporidium infection. Alternatively, Megasphaera may be a biomarker for another microbiome- or immune-mediated mechanism of protection from diarrhea.
Limitations of this study include the wide age range of children enrolled in this study, the microbial diversity of samples, widespread antibiotic use and infections, and the unknown generalizability to global populations. In addition, technical limitations include moderate sample size, the fact that time-of-detection and predetection samples were sequenced separately, and the need for read count normalization due to the variable sequencing depth across samples.
In sum, the microbiome was predictive of Cryptosporidium diarrhea both prior to and at the time of infection. Low abundance of 1 member of the microbiome, Megasphaera, was associated with diarrheal symptoms. There is currently no effective drug for treating Cryptosporidium diarrhea in children, and modulating members of the microbiota such as Megasphaera may be an appealing prevention strategy.
Supplementary Data
Supplementary materials are available at Clinical Infectious Diseases online. Consisting of data provided by the authors to benefit the reader, the posted materials are not copyedited and are the sole responsibility of the authors, so questions or comments should be addressed to the corresponding author.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Notes
Author contributions. M. A. C. and C. A. G. conceived of the study and W. A. P. led the study. R. H. and A. S. G. F. founded the birth cohort and directed the Bangladesh studies. G. L. M. and M. A. C. conceived of the analyses. Field work and data collection at the International Centre for Diarrhoeal Disease Research, Bangladesh (icddr,b) were performed by M. A. and M. K., with A. S. G. F. and R. H. providing supervision. DNA extraction and 16S rRNA library preparation were performed by M. J. U., and U. N. oversaw the clinical database. G. L. M. performed some preliminary microbiome sequence processing and machine learning analyses, with supervision by J. P. Final analyses and drafting of the manuscript were performed by M. A. C. All authors edited and approved the final manuscript.
Acknowledgments. The authors thank the children and parents from the icddr,b study sites as well as the field workers, physicians, scientists, and staff of the Emerging Infectious Diseases Division of icddr,b for their key contributions to this research. The authors also thank the members of the Petri lab group for their feedback, especially Jeff Donowitz, Kevin Steiner, Chelsea Marie, and Barbara Mann, and the University of Virginia Genome Analysis and Technology Core for conducting the sequencing for this study.
Disclaimer. The funders had no role in study design, data collection and analysis, or decision to submit for publication.
Financial support. This work was supported by the National Institutes of Health (grant numbers R01 AI043596 to W. A. P. and C. A. G.; T32 LM012416 to J. P. and G. L. M.; and R21 142656 to C. A. G.); the University of Virginia (Engineering-in-Medicine Seed funding to M. A. C., J. P., and W. A. P.); the Bill & Melinda Gates Foundation (grant number OPP1100514); and the PhRMA Foundation (Postdoctoral Fellowship in Translational Medicine and Therapeutics to M. A. C.). The governments of Bangladesh, Canada, Sweden, and the United Kingdom provide core support to icddr,b.
Potential conflicts of interest. W. A. P. acts as a consultant for TechLab Inc, which produces diagnostics for cryptosporidiosis. All other authors report no potential conflicts of interest. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
REFERENCES
- 1.Steiner KL, Ahmed S, Gilchrist CA, et al. . Species of cryptosporidia causing subclinical infection associated with growth faltering in rural and urban Bangladesh—a birth cohort study. Clin Infect Dis 2018; 67:1347–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Checkley W, Epstein LD, Gilman RH, Black RE, Cabrera L, Sterling CR. Effects of Cryptosporidium parvum infection in Peruvian children: growth faltering and subsequent catch-up growth. Am J Epidemiol 1998; 148:497–506. [DOI] [PubMed] [Google Scholar]
- 3.Korpe PS, Haque R, Gilchrist C, et al. . Natural history of cryptosporidiosis in a longitudinal study of slum-dwelling Bangladeshi children: association with severe malnutrition. PLoS Negl Trop Dis 2016; 10:e0004564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lima AA, Fang G, Schorling JB, et al. . Persistent diarrhea in northeast Brazil: etiologies and interactions with malnutrition. Acta Paediatr Suppl 1992; 381:39–44. [DOI] [PubMed] [Google Scholar]
- 5.Guerrant DI, Moore SR, Lima AA, Patrick PD, Schorling JB, Guerrant RL. Association of early childhood diarrhea and cryptosporidiosis with impaired physical fitness and cognitive function four-seven years later in a poor urban community in northeast Brazil. Am J Trop Med Hyg 1999; 61:707–13. [DOI] [PubMed] [Google Scholar]
- 6.Sallon S, Deckelbaum RJ, Schmid II, Harlap S, Baras M, Spira DT. Cryptosporidium, malnutrition, and chronic diarrhea in children. Am J Dis Child 1988; 142:312–5. [DOI] [PubMed] [Google Scholar]
- 7.Platts-Mills JA, Babji S, Bodhidatta L, et al. ; MAL-ED Network Investigators . Pathogen-specific burdens of community diarrhoea in developing countries: a multisite birth cohort study (MAL-ED). Lancet Glob Health 2015; 3:e564–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Khalil IA, Troeger C, Rao PC, et al. . Morbidity, mortality, and long-term consequences associated with diarrhoea from Cryptosporidium infection in children younger than 5 years: a meta-analyses study. Lancet Glob Health 2018; 6:e758–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.GBD 2016 Diarrhoeal Disease Collaborators. Estimates of the global, regional, and national morbidity, mortality, and aetiologies of diarrhoea in 195 countries: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Infect Dis 2018; 18:1211–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Manjunatha UH, Vinayak S, Zambriski JA, et al. . A Cryptosporidium PI(4)K inhibitor is a drug candidate for cryptosporidiosis. Nature 2017; 546:376–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wojcik GL, Korpe P, Marie C, et al. . Genome-wide association study of cryptosporidiosis in infants implicates PRKCA. mBio 2020; 11:e.03343-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kirkpatrick BD, Haque R, Duggal P, et al. . Association between Cryptosporidium infection and human leukocyte antigen class I and class II alleles. J Infect Dis 2008; 197:474–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nader JL, Mathers TC, Ward BJ, et al. . Evolutionary genomics of anthroponosis in Cryptosporidium. Nat Microbiol 2019; 4:826–36. [DOI] [PubMed] [Google Scholar]
- 14.Gilchrist CA, Cotton JA, Burkey C, et al. . Genetic diversity of Cryptosporidium hominis in a Bangladeshi community as revealed by whole-genome sequencing. J Infect Dis 2018; 218:259–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heiges M, Wang H, Robinson E, et al. . CryptoDB: a Cryptosporidium bioinformatics resource update. Nucleic Acids Res 2006; 34:D419–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sateriale A, Šlapeta J, Baptista R, et al. . A genetically tractable, natural mouse model of cryptosporidiosis offers insights into host protective immunity. Cell Host Microbe 2019; 26:135–46.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chappell CL, Darkoh C, Shimmin L, et al. . Fecal indole as a biomarker of susceptibility to Cryptosporidium infection. Infect Immun 2016; 84:2299–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harp JA, Wannemuehler MW, Woodmansee DB, Moon HW. Susceptibility of germfree or antibiotic-treated adult mice to Cryptosporidium parvum. Infect Immun 1988; 56:2006–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harp JA, Chen W, Harmsen AG. Resistance of severe combined immunodeficient mice to infection with Cryptosporidium parvum: the importance of intestinal microflora. Infect Immun 1992; 60:3509–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Faith JJ, Guruge JL, Charbonneau M, et al. . The long-term stability of the human gut microbiota. Science 2013; 341:1237439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Caporaso JG, Lauber CL, Walters WA, et al. . Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J 2012; 6:1621–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bushnell B;Joint Genome Institute. BBTools: a suite of fast, multithreaded bioinformatics tools designed for analysis of DNA and RNA sequence data.2018. Available at: https://jgidoegov/data-and-tools/bbtools. Accessed 24 July 2020. [Google Scholar]
- 23.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J 2011; 17:10–2. [Google Scholar]
- 24.Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, Holmes SP. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods 2016; 13:581–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weiss S, Xu ZZ, Peddada S, et al. . Normalization and microbial differential abundance strategies depend upon data characteristics. Microbiome 2017; 5:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.R Computing Team. A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing, 2015. [Google Scholar]
- 27.R Computing Team. RStudio: integrated development for R. Boston, MA: RStudio, Inc,2015. [Google Scholar]
- 28.Morgan M, Anders S, Lawrence M, Aboyoun P, Pagès H, Gentleman R. ShortRead: a bioconductor package for input, quality assessment and exploration of high-throughput sequence data. Bioinformatics 2009; 25:2607–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McMurdie PJ, Holmes S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 2013; 8:e61217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Breiman L. Random forests. Mach Learn 2001; 45:5–32. [Google Scholar]
- 31.Wickham H. reshape2: Flexibly reshape data: a reboot of the reshape package.R package version 2012:1. Available at: http://cran.ms.unimelb.edu.au/web/packages/reshape2/. Accessed 1 October 2018.
- 32.Wickham H, Chang W, Others. ggplot2: an implementation of the grammar of graphics. R package version 0 7.2008. Available at: http://ftp.auckland.ac.nz/software/CRAN/src/contrib/Descriptions/ggplot.html. Accessed 1 October 2018.
- 33.Attali D, Baker C. ggExtra: add marginal histograms to “ggplot2,” and more “ggplot2”enhancements. R package version 0 3. 2016:4. [Google Scholar]
- 34.Kassambara A. ggpubr":ggplot2” based publication ready plots. R package version 0 1.2017:6. [Google Scholar]
- 35.Allaire JJ, Xie Y, McPherson J, et al. . rmarkdown: dynamic documents for R. 2018. Available at: https://CRAN.R-project.org/package=rmarkdown. Accessed 1 October 2018.
- 36.Xie Y. knitr: a comprehensive tool for reproducible research in R. Implement Reprod Res 2014. Available at: https://books.google.com/books?hl=en&lr=&id=WVTSBQAAQBAJ&oi=fnd&pg=PA3&ots=qSxw89GmV3&sig=FRrY5j5zcadovLD4VH5P7ZZZYCA. Accessed 1 October 2018. [Google Scholar]
- 37.Liaw A, Wiener M, Others. Classification and regression by randomForest. R News 2002; 2:18–22. [Google Scholar]
- 38.Paluszynska A, Biecek P. randomForestExplainer: explaining and visualizing random forests in terms of variable importance.2017. Available at: https://CRAN.R-project.org/package=randomForestExplainer. Accessed 1 October 2018.
- 39.Urrea V, Calle M. AUCRF: variable selection with random forest and the area under the curve. R package version 1 1. 2012. [Google Scholar]
- 40.Robin X, Turck N, Hainard A, et al. . pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics 2011; 12:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wickham H, Francois R, Henry L, Müller K. dplyr: a grammar of data manipulation. R package version 0 4. 2015:3. [Google Scholar]
- 42.Wagner H. Vegan: community ecology package. R package. 2015. [Google Scholar]
- 43.Wickham H, Henry L, Others. tidyr: Easily Tidy Data with “spread ()”and “gather ()”Functions. R package version 0 8. 2018:2. [Google Scholar]
- 44.Calle ML. Statistical analysis of metagenomics data. Genomics Inform 2019; 17:e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014; 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Duvallet C, Gibbons SM, Gurry T, Irizarry RA, Alm EJ. Meta-analysis of gut microbiome studies identifies disease-specific and shared responses. Nat Commun 2017; 8:1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Topçuoğlu BD, Lesniak NA, Ruffin MT 4th, Wiens J, Schloss PD. A framework for effective application of machine learning to microbiome-based classification problems. mBio 2020; 11:e.00434-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bokulich NA, Chung J, Battaglia T, et al. . Antibiotics, birth mode, and diet shape microbiome maturation during early life. Sci Transl Med 2016; 8:343ra82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen RY, Kung VL, Das S, et al. . Duodenal microbiota in stunted undernourished children with enteropathy. N Engl J Med 2020; 383:321–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zozaya-Hinchliffe M, Martin DH, Ferris MJ. Prevalence and abundance of uncultivated Megasphaera-like bacteria in the human vaginal environment. Appl Environ Microbiol 2008; 74:1656–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guo C, Li Y, Wang P, et al. . Alterations of gut microbiota in cholestatic infants and their correlation with hepatic function. Front Microbiol 2018; 9:2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chappell CL, Okhuysen PC, Langer-Curry RC, Akiyoshi DE, Widmer G, Tzipori S. Cryptosporidium meleagridis: infectivity in healthy adult volunteers. Am J Trop Med Hyg 2011; 85:238–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cama VA, Bern C, Roberts J, et al. . Cryptosporidium species and subtypes and clinical manifestations in children, Peru. Emerg Infect Dis 2008; 14:1567–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Odamaki T, Kato K, Sugahara H, et al. . Age-related changes in gut microbiota composition from newborn to centenarian: a cross-sectional study. BMC Microbiol 2016; 16:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yatsunenko T, Rey FE, Manary MJ, et al. . Human gut microbiome viewed across age and geography. Nature 2012; 486:222–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Costello EK, Carlisle EM, Bik EM, Morowitz MJ, Relman DA. Microbiome assembly across multiple body sites in low-birthweight infants. mBio 2013; 4:e00782–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Avershina E, Storrø O, Øien T, Johnsen R, Pope P, Rudi K. Major faecal microbiota shifts in composition and diversity with age in a geographically restricted cohort of mothers and their children. FEMS Microbiol Ecol 2014; 87:280–90. [DOI] [PubMed] [Google Scholar]
- 58.Kurokawa K, Itoh T, Kuwahara T, et al. . Comparative metagenomics revealed commonly enriched gene sets in human gut microbiomes. DNA Res 2007; 14:169–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Koenig JE, Spor A, Scalfone N, et al. . Succession of microbial consortia in the developing infant gut microbiome. Proc Natl Acad Sci U S A 2011; 108:4578–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.McKenney EA, Greene LK, Drea CM, Yoder AD. Down for the count: Cryptosporidium infection depletes the gut microbiome in Coquerel’s sifakas. Microb Ecol Health Dis 2017; 28:1335165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ichikawa-Seki M, Motooka D, Kinami A, et al. . Specific increase of Fusobacterium in the faecal microbiota of neonatal calves infected with Cryptosporidium parvum. Sci Rep 2019; 9:12517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ras R, Huynh K, Desoky E, Badawy A, Widmer G. Perturbation of the intestinal microbiota of mice infected with Cryptosporidium parvum. Int J Parasitol 2015; 45:567–73. [DOI] [PubMed] [Google Scholar]
- 63.Oliveira BCM, Widmer G. Probiotic product enhances susceptibility of mice to cryptosporidiosis. Appl Environ Microbiol 2018; 84:e.01408-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Charania R, Wade BE, McNair NN, Mead JR. Changes in the microbiome of Cryptosporidium-infected mice correlate to differences in susceptibility and infection levels. Microorganisms 2020; 8:879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Oliveira BCM, Bresciani KDS, Widmer G. Deprivation of dietary fiber enhances susceptibility of mice to cryptosporidiosis. PLoS Negl Trop Dis 2019; 13:e0007411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shetty SA, Marathe NP, Lanjekar V, Ranade D, Shouche YS. Comparative genome analysis of Megasphaera sp. reveals niche specialization and its potential role in the human gut. PLoS One 2013; 8:e79353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Parada Venegas D, De la Fuente MK, Landskron G, et al. . Short chain fatty acids (SCFAs)-mediated gut epithelial and immune regulation and its relevance for inflammatory bowel diseases. Front Immunol 2019; 10:277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bachem A, Makhlouf C, Binger KJ, et al. . Microbiota-derived short-chain fatty acids promote the memory potential of antigen-activated CD8+ T cells. Immunity 2019; 51:285–97.e5. [DOI] [PubMed] [Google Scholar]
- 69.Binder HJ. Role of colonic short-chain fatty acid transport in diarrhea. Annu Rev Physiol 2010; 72:297–313. [DOI] [PubMed] [Google Scholar]
- 70.Sedighi R, Alipour D. Assessment of probiotic effects of isolated Megasphaera elsdenii strains in Mehraban sheep and Holstein lactating cows. Anim Feed Sci Technol 2019; 248:126–31. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.