

Abstract
Ferredoxin‐NADP+ reductases (FNRs) are ubiquitous flavoenzymes involved in redox metabolisms. FNRs catalyze the reversible electron transfer between NADP(H) and ferredoxin or flavodoxin. They are classified as plant‐ and mitochondrial‐type FNR. Plant‐type FNRs are divided into plastidic and bacterial classes. The plastidic FNRs show turnover numbers between 20 and 100 times higher than bacterial enzymes and these differences have been related to their physiological functions. We demonstrated that purified Escherichia coli FPR (EcFPR) contains tightly bound NADP+, which does not occur in plastidic type FNRs. The three‐dimensional structure of EcFPR evidenced that NADP+ interacts with three arginines (R144, R174, and R184) which could generate a very high affinity and structured site. These arginines are conserved in other bacterial FNRs but not in the plastidic enzymes. We have cross‐substituted EcFPR arginines with residues present in analogous positions in the Pisum sativum FNR (PsFNR) and replaced these amino acids by arginines in PsFNR. We analyzed all proteins by structural, kinetic, and stability studies. We found that EcFPR mutants do not contain bound NADP+ and showed increased K m for this nucleotide. The EcFPR activity was inhibited by NADP+ but this behavior disappeared as arginines were removed. A NADP+ analog of the nicotinamide portion produced an activating effect on EcFPR and promoted the NADP+ release. Our results give evidence for a new model of NADP+ binding and catalysis in bacterial FNRs.We propose that this tight NADP+ binding constitutes an essential catalytic and regulatory mechanism of bacterial FNRs involved in redox homeostasis.
Keywords: catalytic mechanism, catalytic site, Escherichia coli, ferredoxin‐NADP+ reductase, NADP+ binding
Abbreviations
- DMAP
4‐Dimethylaminopyridine
- FNR
ferredoxin‐NADP+ reductase
- FPR
bacterial type ferredoxin‐NADP+ reductase
- G6P
glucose‐6‐phosphate
- G6PD
glucose‐6‐phosphate dehydrogenase
- PeaFNR, EcFPR, PcFPR, BaFPR, LepFNR
ferredoxin‐NADP+ reductase from Pisum sativum, Escherichia coli, Pectobacterium carotovorum, Brucella abortus, and Leptospira interrogans, respectively
1. INTRODUCTION
Ferredoxin‐NADP+ reductases (FNRs) constitute a family of monomeric hydrophilic proteins that contain non‐covalently bound FAD as a prosthetic group. These enzymes are widely distributed among organisms and are involved in the electron transfer of biologically important processes. FNRs are classified as plant and mitochondrial‐type. Plant‐type FNRs are grouped into plastidic and bacterial classes. Members of these two classes differ not only in sequence alignment but also in the environment of the active site, FAD conformation and catalytic efficiency.1, 2
Both plastidic and bacterial FNRs fold in two domains. The N‐terminal and C‐terminal domains are responsible for the binding of the prosthetic group FAD and the substrate NADP(H), respectively. In plastidic FNRs, the FAD binds in an extended conformation while it is in a folded form in the bacterial enzymes (named FPRs). The extended conformation in plastidic FNRs is favored by a β‐hairpin structure that is replaced by a short loop in FPRs. The folded FAD in FPRs is stabilized by the stacking of its adenosine to an aromatic residue found in a C‐terminal tail present in FPRs but absent in FNRs. Another difference between FNRs and FPRs also occurs at the active site. The FAD isoalloxazine ring stacks between two aromatic residues, being on the re‐face a conserved C‐terminal tyrosine in plastidic FNRs. Subclass II FPRs conserve this aromatic residue, but subclass I members lack it.1, 3
Subclass I FPRs was subdivided into two new groups: subclass IA and subclass IB.4 The main structural differences between these subclasses are located in the C‐terminal region. While enzymes from subclass IA have a conserved lysine, the subclass IB FPRs have a glutamate or an aspartate at the equivalent position and a more extended C‐terminal region. Consequently, subclass IA is defined by the C‐terminal sequence VEK and the subclass IB by the sequence (V/A)G(E/D)G(I/V).1
The C‐terminal tyrosine of plastidic FNRs would be displaced to allow the entry of NADP+ during catalysis. These FNRs show turnover numbers between 20 and 100 times greater than bacterial enzymes. This suggests that FPRs have catalytic limitations, which were overcome by the functional counterpart present in photosynthetic organisms and tissues. Moreover, the FPRs contain a structured variable carboxyl terminus that has not allowed to propose logical models that justify how the substrate reaches the active site.1, 5, 6 Escherichia coli FPR (EcFPR) can exchange reduction equivalents with both ferredoxin and flavodoxin.7, 8 The direction of the reaction in bacteria is optimized to favor the consumption of NADPH.9 The enzyme participates in several metabolic pathways, and its deletion produces bacteria that are highly sensitive to oxidative stress.7, 10, 11 Different studies suggest that bacteria need to finely regulate NADP(H) pool homeostasis to generate an appropriate stress response. This regulatory function would be carried out with the direct participation of FPR, as was observed in E. coli.12, 13
We have previously obtained crystals of a mutant EcFPR with bound NADP+, although the nucleotide substrate had not been added in the crystallization procedure. The three‐dimensional structure showed that the NADP+ molecule interacts with three arginines (R144, R174, and R184).14 These residues could be responsible for generating a structured site with a very high affinity for NADP+, as was proposed for the Pseudomonas aeruginosa FPR.15 These arginine residues are conserved in other FPRs of different subclasses, but not in the plastidic type enzymes. Pisum sativum FNR contains an arginine analogous to R174 (R229), but R144 corresponds to a proline (P199) and R184 to a tyrosine (Y240). In this study, we demonstrated that purified E. coli FPR contains tightly bound NADP+. The nucleotide remains attached to the enzyme after extensive dialysis and gel filtration. To elucidate if this also occurs with other FPRs or FNRs, we analyzed another subclass II FPR from Pectobacterium carotovorum, a subclass IA FPR from Brucella abortus and two different plastidic type FNRs, from P. sativum and Leptospira interrogans.16 We observed that althought with different extent, all FPRs contain the nucleotide bound. In contrast, the plastidic type FNRs did not. The NADP+ binding may exert some enzyme regulation since we observed a correlation between the amounts of bound NADP+ and the catalytic rates of bacterial and plastidic type FNRs. The presence of NADP+ considerably reduced the catalytic activity of EcFPR, which was recovered when the nucleotide was removed from the enzyme. Recently, it was shown that oxido‐reductase activity of FPR from B. ovis depends on the NADPH concentration with inhibition by its excess.3 This may potentially occur as a consequence of the reaction product (NADP+) accumulation. The NADP+ binding also produced a structural stabilization of subclass II FPRs, but no effect was observed on the plastidic counterparts. In order to further investigate the participation of some specific residues, we constructed different EcFPR and P. sativum FNR mutant enzymes. The structural and kinetic characterization of these proteins allowed us to confirm that R144 and R184 in EcFPR are involved in a structured site with a very high affinity for NADP+.
Our results suggest that FPRs show differential NADP+ binding mode and catalytic mechanism from the ones previously described for the plastidic type FNRs.5, 6, 17 A novel hypothetical model of the NADP+ binding and catalysis mechanism in FPRs is proposed, as well as the physiological implications that it may have for the bacteria.
2. RESULTS
2.1. Determination of the NADP + content tightly bound to the enzyme
The amount of NADP+ that remained bound to purified E. coli FPR (EcFPR) was analyzed. We developed an enzymatic method based on the reaction of glucose‐6‐phosphate dehydrogenase (G6PD) to measure the nucleotide. The NADP+ content was determined as the μmoles of NADPH produced by the reduction of NADP+ by the dehydrogenase in the presence of glucose‐6‐phosphate (G6P) using a standard NADP+curve. The same analyses were performed with other bacterial reductases as FPR from P. carotovorum (PcFPR) and B. abortus (BaFPR) and with the plastidic type FNR from L. interrogans (LepFNR)16 and P.sativum (PeaFNR). These FPRs and FNRs were obtained as recombinant proteins in E. coli with N‐terminal polyhistidine tags. The purification procedures involved two steps of immobilized‐metal affinity chromatography. The histidine tags were removed by specific proteolytic digestion and wild type or mutant enzymes without N‐terminal extensions were obtained. Then, samples were subjected to extensive dialysis (see Material and Methods). As shown in Table 1, after this purification process the NADP+ remained bound to EcFPR at a ratio of 0.99 mol/mol. The same trend was observed for PcFPR and although to a lesser extent, for BaFPR. The analyzed plastidic type FNRs did not contain NADP+ bound. These results were confirmed by High‐performance liquid chromatography, where the NADP+ content bound to the reductases was determined by the peak of nucleotide eluted from previously heat‐denatured samples (Table S1).
TABLE 1.
NADP+ content bound to purified FNR enzymes from different organisms
| Enzyme | NADP+/FNR (mol/mol) |
|---|---|
| EcFPR | 0.99 ± 0.05 |
| PcFPR | 0.65 ± 0.05 |
| BaFPR | 0.30 ± 0.02 |
| LepFNR | 0.16 ± 0.03 |
| PeaFNR | 0.08 ± 0.03 |
Note: The NADP+ content was determined by the reaction of the G6PD activity in 50 mM Tris–HCl, pH 8.0, at 25°C. Values are mean ± SD of at least three independent measurements.
EcFPR and PcFPR belong to the subclass II FPRs group meanwhile, BaFPR corresponds to the subclass IA.1 As was previously reported, the subclass IA FPR showed higher turnover numbers than the subclass II enzymes, and plastidic FNRs were more catalytically active than the bacterial reductases (Table 2).1 Therefore, we found a correlation between the amounts of NADP+ bound to the FNRs and their catalytic rate constants. This may suggest a regulatory function for NADP+ on these flavoenzymes.
TABLE 2.
Kinetic parameters of the FNR as obtained for the diaphorase activity
| Enzyme | NADPH | |
|---|---|---|
| Km (μM) | kcat (s−1) | |
| EcFPR | 5 ± 1 | 49 ± 2 |
| PcFPR | 9 ± 1 | 50 ± 1 |
| BaFPR | 13 ± 1 | 146 ± 3 |
| LepFNR | 22 ± 1 | 280 ± 4 |
| PeaFNR | 29 ± 5 | 349 ± 20 |
Note: NADPH‐ferricyanide diaphorase activity in 50 mM Tris–HCl, pH 8.0, at 25°C. Values are mean ± SD of at least three independent measurements.
2.2. The effect of NADP + on the structural stability of FPR
The protein melting point (T m), the temperature at which a protein denatures, can be measured by differential scanning fluorometry. The thermal unfolding of the proteins in the presence of a hydrophobic fluorescent dye18 was analyzed using a real‐time thermocycler. Then, a simple adjustment procedure with the OriginPro Software (OriginLab Corporation, Massachusetts) allowed the determination of T m (Figure S1).19 The difference between these temperatures in the presence and absence of a ligand can be related to the protein's affinity for this molecule. In this study, we used this approach to analyze the effect of the bound NADP+ on the structural stability of FPRs.
As shown in Figure 1a, the removal of NADP+ using a column of Cibacron Blue‐Agarose resin produced a decrease of T m values in EcFPR and PcFPR. However, no differences were observed in the T m determined before and after treatment with Cibacron Blue‐Agarose with BaFPR and the plastidic type FNRs analyzed (Table S1). Similar to BaFPR, no change in T m has been reported for B. ovis FPR in a recent study.3 This is expected considering that FPRs from both Brucella species share 99% protein sequence identity. EcFPR or PcFPR titration with a NADP+ solution increased T m until reaching a ratio of 1:1 (protein: ligand). The T m of PeaFNR remained unchanged with the addition of increasing concentrations of NADP+ (Figure 1b). These observations indicate that the NADP+ binding results in structural stabilization of the subclass II FPRs. Our results suggest that the NADP+ binding mode is different in the plastidic type FNRs.
FIGURE 1.
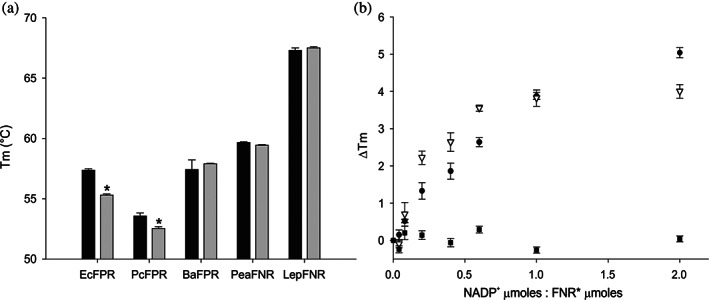
Stability analysis of the FNR. T m determination by adjusting the real‐time denaturation curves. (a) Comparison of the stability of the different FNRs un‐eluted (FNR, black bars) and eluted (FNR*, grey bars) from the Cibacron Blue column. (b) Comparison of the thermal stabilities of EcFPR (circles), PcFPR (triangles), and PeaFNR (squares) with increasing concentrations of NADP+. Bars indicate mean ± SD of three independent experiments
2.3. Design and construction of different FPR/FNR variants
Crystals of a mutant EcFPR with NADP+ bound have been previously obtained without the addition of the nucleotide during the crystallization process.14 The three‐dimensional structure evidenced that NADP+ interacts with three arginines (R144, R174 and R184 in Figure 2a) which could generate a very high affinity and structured site, as was proposed for the P. aeruginosa enzyme.15 These three amino acids are conserved in other FPRs, but not in the plastidic enzymes found in plant chloroplasts and cyanobacteria (Figure 2b). The structural alignment of EcFPR with the plastidic P. sativum FNR showed that of these three arginines, only R174 is present in PeaFNR (R229); R144 corresponds to a proline (P199) and R184 to a tyrosine (Y240) (Figure 2b, c). Based on this, we have cross‐substituted EcFPR arginines with proline and tyrosine residues, which are present in analogous positions in PeaFNR (P199 and Y240) and replaced both amino acids by arginines in PeaFNR. Therefore, we generated the R144P, R184Y, and R144PR184Y variants of EcFPR and P199R, Y240R, and P199RY240R variants of PeaFNR. The mutant forms were expressed and purified following the same protocols used with the wild type enzymes. In all cases, we obtained active soluble proteins with similar yields.
FIGURE 2.
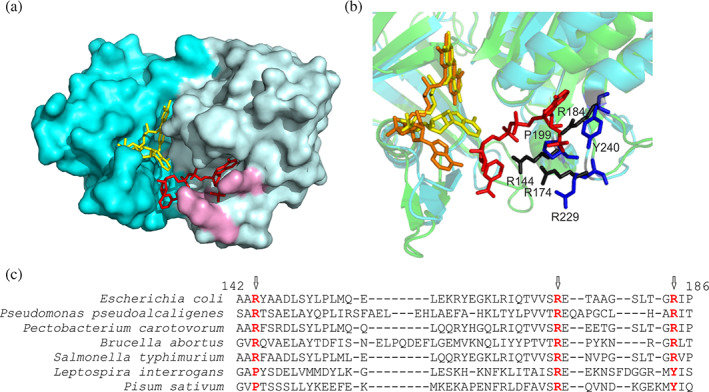
The nucleotide binding site of FNR. (a) Surface model of EcFPR (PDB 2XNJ) containing bound NADP+. FAD is represented in yellow and NADP+ in red. The three arginine residues that interact with NADP+ are shown in purple. (b) Crystal structure of EcFPR (light blue) superimposed to PeaFNR (PDB 1QG0) (green). Arginines present in EcFPR are shown in black and amino acids corresponding to PeaFNR in blue. The NADP+ molecule of EcFPR is shown in red. The FAD molecules have different conformation: folded (yellow) in EcFPR and extended (orange) in PeaFNR. (c) Structural alignment of different FPRs and of the plastidic type LepFNR and PeaFNR. The three arginine residues would be conserved in the FPRs, but only one would be present in the plastidic type (colored in red). Panels A and B were designed from the structures of the proteins obtained by X‐ray diffraction, using the Pymol version 1.8.0.0. program. Panel C alignment was performed with the T‐Coffee Software. The results are consistent with the Pymol alignment function
2.4. Characterization of the FPR/FNR variants
All EcFPR and PeaFNR variants showed a similar absorption spectrum between 300 and 600 nm, with absorption peaks at approximately 385 and 456 nm corresponding to the cofactor FAD. Also, the circular dichroism (CD) spectra in the far‐UV region were registered and no substantial differences were observed between the wild type and mutant reductases (Figure S2). Thus, we assumed that no significant structural changes were produced by the arginine residues replacements.
The content of bound NADP+ in each purified protein variant was determined by the reaction of G6PD, as previously described. The wild type EcFPR showed a ratio of 0.99 mol NADP+/mol FPR while none of its mutants contained bound NADP+ (Figure 3a). Consequently, we confirmed that R144 and R184 are involved in the tight binding of NADP+ to EcFPR. The replacement of the analogous residues in PeaFNR by arginines did not produce an increase in the content of bound NADP+, suggesting again that the NADP+ binding mode would be different in the plastidic FNR. Then, the melting point of each purified protein was estimated by differential scanning fluorometry, before and after removing the bound NADP+ with a Cibacron‐Blue Agarose resin. Only wild type EcFPR showed a decrease in T m value after this procedure. The T m of EcFPR and PeaFNR mutants were not affected by the treatment (Figure 3b and Table S3).
FIGURE 3.
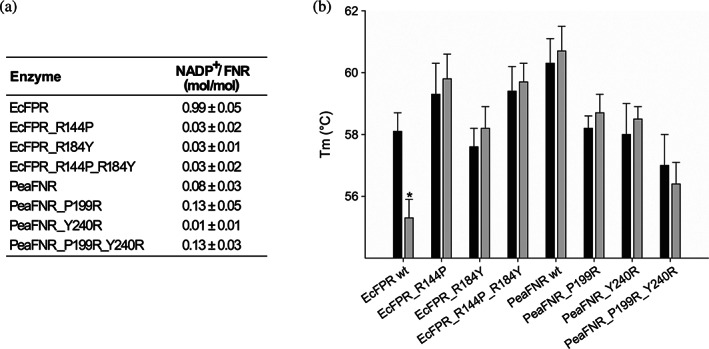
Relationship between NADP+ content and thermal stability of wild type and mutant EcFPR and PeaFNR. (a) Estimation of the NADP+ content bound to each reductase based on the NADPH generated by the reaction of G6PD. Values are mean ± SD of at least three independent measurements. (b) Comparison of the thermal stability of wild type and mutant EcFPR and PeaFNR un‐eluted (black bars) and eluted (grey bars) from the Cibacron Blue column. Bars indicate mean ± SD of three independent experiments
To kinetically characterize the proteins under study, we assayed the diaphorase activity with NADPH using a regenerating system with G6P and G6PD, NADP+ and potassium ferricyanide as electron acceptor substrate (see Section 4). The catalytic constants were determined from the hyperbolic curves obtained (Table 3). In the case of EcFPR, we observed that the replacement of R144 or R184 produced an increase of K m value for NADPH being more pronounced for R184. Furthermore, the EcFPR variant with both arginine residues replaced by proline and tyrosine showed a 260 times greater K m than the wild type enzyme. Consequently, EcFPR_R144P, EcFPR_R184Y and EcFPR_R144P_R184Y were 1‐, 2‐, and 3‐orders of magnitude less catalytically efficient than EcFPR, respectively. These catalytic alterations were not observed when we compared wild type and the different PeaFNR variants.
TABLE 3.
Kinetic parameters of wild type and mutant EcFPR and PeaFNR as obtained for the diaphorase activity
| Enzyme | NADPH | ||
|---|---|---|---|
| Km (μM) | kcat (s−1) | Catalytic efficiency (μM−1 s−1) | |
| EcFPR | 5 ± 1 | 49 ± 2 | 10 ± 4 |
| EcFPR_R144P | 88 ± 10 | 83 ± 3 | 0.9 ± 0.3 |
| EcFPR_R184Y | 480 ± 112 | 20 ± 1 | 0.04 ± 0.02 |
| EcFPR_R144P_R184Y | 1289 ± 210 | 4.9 ± 0.3 | 0.004 ± 0.001 |
| PeaFNR | 29 ± 5 | 349 ± 20 | 12 ± 6 |
| PeaFNR_P199R | 28 ± 5 | 381 ± 20 | 14 ± 6 |
| PeaFNR_Y240R | 33 ± 4 | 283 ± 7 | 9 ± 3 |
| PeaFNR_P199_Y240R | 24 ± 1 | 225 ± 1 | 9 ± 2 |
Note: NADPH‐ferricyanide diaphorase activity in 50 mM Tris–HCl, pH 8.0, at 25°C. Values are mean ± SD of at least three independent measurements.
We also analyzed the effect produced by the presence of NADP+ in the reaction medium on the catalytic properties of EcFPR. The diaphorase activity was assayed using NADPH as the electron donor substrate with the regenerating system (NADP+ absence condition) or without it (NADP+ presence condition) in the cuvette. The regenerating system was used to deplete NADP+ from the reaction medium. From the data shown in Table 4, we concluded that NADP+ produced an inhibition on EcFPR as the wild type enzyme was 10 times more catalytically efficient when the oxidized nucleotide generated was depleted from the reaction medium. This inhibition was not observed with the different EcFPR variants, thus indicating that R144 and R184 are involved in the inhibition site.
TABLE 4.
Kinetic parameters of wild type and mutant EcFPR and PeaFNR in the presence and absence of NADP+ as obtained for the diaphorase activity
| Enzyme | NADPH | ||||
|---|---|---|---|---|---|
| Km (μM) | Km fold change (+/− NADP+) | kcat (s−1) | Catalytic efficiency (μM−1 s−1) | Catalytic efficiency fold change (−/+ NADP+) | |
| EcFPR* | 6.8 ± 0.5 | 11.6 | 54 ± 1 | 8 ± 2 | 10 |
| 79 ± 23 | 69 ± 6 | 0.8 ± .05 | |||
| EcFPR_R184Y* | 90 ± 2 | 1.6 | 60 ± 1 | 0.7 ± 0.1 | 1.4 |
| 144 ± 15 | 69 ± 3 | 0.5 ± 0.2 | |||
| EcFPR_R144P* | 442 ± 63 | 1.1 | 26 ± 1 | 0.06 ± 0.02 | 1.2 |
| 471 ± 55 | 23 ± 1 | 0.05 ± 0.01 | |||
| EcFPR_R144P_R184Y* | 2672 ± 685 | 0.9 | 15 ± 1 | 0.006 ± 0.003 | 1 |
| 2455 ± 704 | 16 ± 1 | 0.006 ± 0.003 | |||
| PeaFNR* | 20 ± 2 | 1.8 | 294 ± 7 | 15 ± 5 | 1.8 |
| 37 ± 7 | 314 ± 19 | 8 ± 4 | |||
| PeaFNR_P199R* | 25 ± 1 | 3.3 | 286 ± 5 | 11 ± 2 | 3.6 |
| 83 ± 17 | 279 ± 42 | 3 ± 2 | |||
| PeaFNR_Y240R* | 15 ± 1 | 1.7 | 259 ± 6 | 17 ± 5 | 1.5 |
| 26 ± 6 | 288 ± 18 | 11 ± 5 | |||
| PeaFNR_P199R_Y240R* | 19 ± 2 | 4 | 226 ± 6 | 12 ± 4 | 3 |
| 77 ± 26 | 280 ± 34 | 4 ± 2 | |||
Note: NADPH‐ferricyanide diaphorase activity in 50 mM Tris–HCl, pH 8.0, at 25°C. Values are mean ± SD of at least three independent measurements. *FNR eluted from the Cibacron Blue column. White: NADP+ absence, gray: NADP+ presence.
2.5. Analysis of the binding mode of NADP + to the active site
It has been postulated that NADP(H) binds to FNR through a bipartite site. The nucleotide is initially bound to the enzyme in an unproductive conformation, where the 2′‐P‐AMP portion first interacts with a preformed binding site (P hemi‐site), but with the nicotinamide far away from the catalytic site. The nicotinamide moiety then moves into proximity with the isoalloxazine (N hemi‐site) and establishes a productive conformation.1, 5
The 4‐Dimethylaminopyridine (DMAP) is a derivative of the pyridine molecule, and it can be considered as a NADP+ analog for the nicotinamide portion (Figure 4a). Paladini et al. reported that DMAP influences the nicotinamide interaction at the N hemi‐site but not the interaction of the nucleotide at P hemi‐site.20 We observed that purified EcFPR and PcFPR, both subclass II FPRs, contain tightly bound NADP+. Moreover, they showed less catalytic efficiency in the presence of NADP+. Thus, we decided to determine the content of bound NADP+ in a sample of wild type EcFPR previously incubated with DMAP, and then filtered by size exclusion chromatography using a Sephadex G‐25 column in order to separate the released NADP+ from the protein sample. We estimated a ratio of 0.54 ± 0.06 mol NADP+/mol FPR. A control sample treated under the same conditions but without the addition of DMAP, showed a ratio of 0.99 ± 0.05 mol NADP+/mol FPR. When the diaphorase activity of EcFPR was evaluated in the absence or presence of 20 mM DMAP, we observed an activating effect exerted by DMAP on the enzyme activity (Figure 4b). However, the presence of this compound produced a ~ 4‐fold increase in the K m for the nucleotide substrate and, consequently, a 2.5‐fold decrease in the catalytic efficiency of EcFPR. These alterations were not observed with the mutant enzymes (Table 5). Taken these results together, it can be argued that in the subclass II FPRs, the NADP+ would be constitutively interacting at the P hemi‐site with high affinity through its 2′‐P‐AMP portion. The occupancy of the N hemi‐site produces a decrease in the P hemi‐site affinity and subsequently, the NADP+ release from it. R144 and R184 are both part of the P hemi‐site in EcFPR since the replacement of these residues does not affect the kinetic parameters of the mutant enzymes in the presence of DMAP.
FIGURE 4.
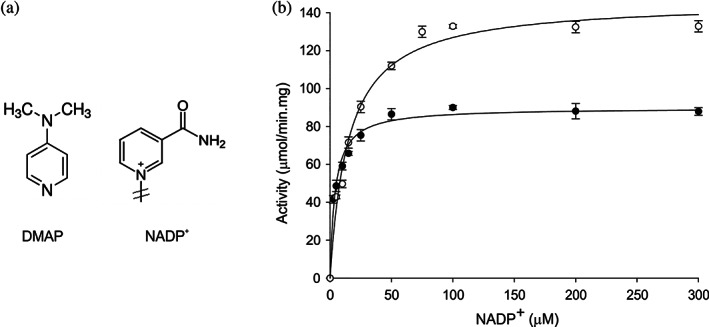
Effect of DMAP on the diaphorase activity of wild type EcFPR. (a) Structural analogies between NADP+ and DMAP in the nicotinamide region. (b) Changes in diaphorase activity in the presence (white circles) and absence (black circles) of DMAP as a function of NADP+ concentration. Bars indicate mean ± SD of three independent experiments
TABLE 5.
Kinetic parameters of wild type and mutant EcFPR in the presence and absence of DMAP as obtained for the diaphorase activity
| Enzyme | NADPH | ||||
|---|---|---|---|---|---|
| Km (μM) | Km fold change (+/− DMAP) | kcat(s−1) | Catalytic efficiency (μM−1.s−1) | Catalytic efficiency fold change (−/+ DMAP) | |
| EcFPR | 4.1 ± 0.5 | 3.7 | 41 ± 1 | 10 ± 3 | 2.5 |
| EcFPR + DMAP | 15 ± 2 | 67 ± 2 | 4 ± 1 | ||
| EcFPR_R184Y | 85 ± 4 | 0.7 | 64 ± 2 | 0.8 ± 2 | 0.7 |
| EcFPR_R184Y + DMAP | 60 ± 2 | 67 ± 1 | 1.1 ± 0.2 | ||
| EcFPR_R144P | 408 ± 25 | 0.9 | 15.7 ± 0.5 | 0.04 ± 0.01 | 0.8 |
| EcFPR_R144P + DMAP | 360 ± 13 | 17.6 ± 0.4 | 0.05 ± 0.01 | ||
| EcFPR_R144P_R184Y | 1285 ± 308 | 1.1 | 2.9 ± 0.1 | 0.002 0 ± 0.001 | 1 |
| EcFPR_R144P_R184Y + DMAP | 1420 ± 411 | 3.5 ± 0.2 | 0.002 0 ± 0.001 | ||
Note: NADPH‐ferricyanide diaphorase activity in 50 mM Tris–HCl, 20 mM DMAP, pH 8.0, at 25°C. Values are mean ± SD of at least three independent measurements.
In order to confirm our hypothesis, we performed an experiment in which the reduction of purified wild type EcFPR and PeaFNR, and EcFPR_R144P_R184Y was monitored by absorption spectroscopy. The protein samples were incubated in a cuvette with G6PD or, with this enzyme and its substrate G6P. Then absorbance spectra were registered between 300 and 600 nm. In the case of the wild type EcFPR, we observed the FAD reduction only when both G6PD and G6P were present in the cuvette, thus indicating the presence of NADP+ in the wild type EcFPR sample (Figure 5a). The oxidized nucleotide bound to wild type EcFPR was reduced by the G6PD reaction and, consequently, NADPH was able to reduce the FAD of the reductase. The FAD reduction did not occur either with the wild type PeaFNR or the EcFPR_R144P_R184Y samples because these proteins do not contain tightly bound NADP+ (Figure 5b,c). Finally, the content of NADP+ bound to the purified wild type EcFPR was determined after the G6PD reaction proceeded. The sample was filtered through a centrifugal concentrator and the NADP+ present in the retained volume was quantified. After this procedure, we observed a significant decrease in the NADP+/FPR ratio. A control sample showed that EcFPR was entirely retained by the filter (Figure 5d). This confirmed that the presence of NADPH produces the release of NADP+ from EcFPR.
FIGURE 5.
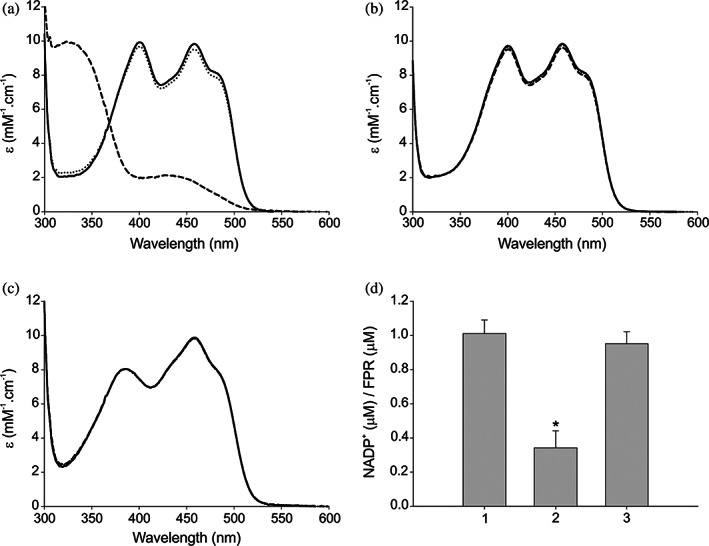
Analysis of the binding mode of NADP+. (a‐c) Spectroscopic visualization of the reduction reaction with EcFPR, EcFPR_R144P_R184Y, and PeaFNR, respectively. Solid line, control FNR; dotted line, FNR + G6PD; dash line, FNR + G6PD + G6P. (d) Determination of the NADP+ content in EcFPR: 1, unfiltered EcFPR; 2, retained EcFPR in the centrifugal concentrator after reaction with G6PD and G6P; 3, retained EcFPR in the centrifugal concentrator without adding G6PD and G6P
3. DISCUSSION
Previous studies suggested that EcFPR co‐purified with its nucleotide substrate.14 Here, our results firmly demonstrate that purified EcFPR contains tightly bound NADP+ at a 1:1 M ratio. This was also observed with PcFPR, another subclass II FPR and, although with a smaller ratio, with BaFPR, a subclass IA FPR. In contrast, we determined that plastidic type FNRs do not contain bound NADP+.
The tight binding of NADP+ has been reported for other enzymes. The native form of bovine lens aldose reductase is characterized by a tightly bound pyridine coenzyme, which is retained during the purification steps.21 The bound nucleotide appears to be related to the redox state of cysteinyl residues of the enzyme. Kirkman et al. found that the catalase of human erythrocytes is a major NADPH‐binding protein within those cells.22 In the same way, bovine liver and canine catalases also contain tightly bound NADPH.23 These catalases, which are tetramers, contain one tightly bound NADPH per subunit. Gaetani et al.24 suggested that the bound NADPH may participate in preventing the formation of compound II, the reversibly inactivated state of catalase. Human glucose 6‐phosphate dehydrogenase, purified after overexpression in E. coli, contained one molecule/subunit of acid‐extractable “structural” NADP+ and no NADPH. This tightly bound NADP+ was reduced by glucose‐6‐phosphate, presumably following migration to the catalytic site due to conformational changes of the enzyme induced on the binding of G6P. The Kd for the structural NADP+ was estimated as 37 nM, 200‐fold lower than for catalytic NADP+.25 In the case of EcFPR, many attempts for measuring the Kd for NADP+ were unsuccessful because we could not reach equilibrium conditions between the bound and unbound nucleotide fractions at a detectable protein concentration. However, we observed that the addition of 0.5 μM NADP+ pulses to 5 μM EcFPR produced a practically total binding of the nucleotide (Figure S3). This would indicate that the EcFPR affinity for NADP+ is very high, probably with a K d in the nM range. These observations are consistent with the fact that the enzyme retains the nucleotide after extensive dialysis.
The tightly bound NADP+ to G6PD seems crucial, not for activity, but rather for the long‐term stability of the enzyme at the physiological temperature of 37°C.25 Recently, Morales‐Luna et al.26 performed a functional and structural analysis of fused glucose‐6‐phosphate dehydrogenase‐phosphogluconolactonase protein from the protozoan Trichomonas vaginalis. They found that the protein becomes more stable in the presence of the NADP+. We determined that the removal of NADP+ produced a decrease of T m values in EcFPR and PcFPR. Moreover, when these enzymes were titrated with a NADP+ solution their T m increased until reaching a ratio of 1:1 (protein: NADP+), showing a structural stabilization of FPR. These FPRs probably became more stable by NADP+ binding due to the new interactions formed between the protein and NADP+. However, we think that the EcFPR structural stabilization by the tight NADP+ binding is a consequence of the main role, which would be the regulation of the enzyme catalytic activity. This argument is supported by the fact that in EcFPR crystals, the NADP+ was bound to its catalytic site.14
FNR·NADP(H) complex formation was interpreted as a two‐step binding of the nucleotide to a bipartite site.27 The first step involves a strong interaction of FNR with the adenosine part of NADP(H) (at P hemi‐site), followed by isomerization leading to nicotinamide docking (at N hemi‐site) and, eventually, hydride transfer.5 The nicotinamide docking is energetically costly and weakens the entire interaction to a remarkable extent. Measurements of complex formation in plastidic type FNR from P. sativum showed that under saturating conditions <20% of the nicotinamide is placed in contact with the flavin and is therefore available for hydride transfer.17 This model explains why FNR·NADP(H) crystals were only obtained with engineered plastidic type FNR proteins in which the C‐terminal tyrosine had been replaced by nonaromatic residues such as serine.27 In these FNR variants, the displacement of the phenol group is no longer required, and both pockets of the binding site would be readily accessible.
The bipartite nature of NADP+ binding highlights an ambiguity of spectroscopically based assessments of structural interactions of extended ligands such as NADP+. The weak difference spectrum observed for NADP+ binding to wild type P. sativum FNR is not due to a weak electronic interaction between flavin and the nicotinamide portion of NADP+. It instead reflects an interaction that displays a significant intrinsic signal in the difference spectrum but only occurs for a low percentage of the molecules on a certain moment. At a more general level, this result illustrates that it is not always correct to assume that when an extended ligand is present at saturating concentrations, the entire binding site will be interacting with all portions of the bound ligand.28
The crystal structure of FPR from P. aeruginosa (paFPR)15 is almost identical to the structure of EcFPR.8 In the structure of paFPR the terminal Tyr conserved in the plastidic FNRs, is replaced by Ala254, which is followed by a C‐terminal extension (AFVEK258). Considering the NADP+ binding mechanism proposed for the FNR, this extension can be expected to slow the rate of conformational rearrangements needed to access the “productive” conformation of cofactors thought to facilitate direct hydride transfer.25
The structure of the paFPR·NADP+ complex reported by Wang et al. provided the first view of the interactions between NADP+ and paFPR.25 In this study, the authors reported that the structure of paFPR has a preformed binding pocket, where residues involved in important contacts with the 2′P group of NADP+ require little or no rearrangement to accommodate the nucleotide. Among these residues, R182 and R190 are conserved in subclass II bacterial reductases. They also determined similar values of Kd for the binding of NADP+ and the competitive inhibitor ADP, which indicates that stabilization of the paFPR·NADP+ complex is derived mainly from interactions between the protein and the adenosine 2′, 5′‐diphosphate moiety in NADP+. In contrast to observations made with spinach FNR, paFPR binding affinity for NADP+ is slightly larger than its affinity for ADP, which indicates that the nicotinamide nucleotide moiety does not undergo destabilizing interactions with the enzyme, as is thought to be the case in the binding of NADP+ to plastidic FNRs. It is therefore likely that NADP+ binding to paFPR occurs in one step, rather than two, as was thought for plastidic FNRs.25
Purified EcFPR showed NADPH‐ferricyanide diaphorase activity. Thus, NADPH was able to reduce the enzyme‐containing tightly bound NADP+. Accordingly, N hemi‐site would be available to allow the NADPH entrance to the catalytic site. The kinetic experiments using DMAP, a NADP+ analog of the nicotinamide portion, showed that the presence of this compound produced a ~ 4‐fold increase in the K m for the NADPH and an activating effect on the EcFPR activity. Moreover, DMAP, which is assumed to interact at the N hemi‐site, promoted the NADP+ release from the enzyme. This observation may explain how the catalysis proceeds and gives evidence for a novel catalytic model. In this sense, the interaction of the incoming substrate nicotinamide at the N hemi‐site would produce the release of the tightly bound NADP+. Consequently, the nicotinamide entrance to the N hemi‐site facilitates the proper substrate binding and completion of the catalytic turnover. This interconnection between both hemi‐sites was previously observed in PeaFNR.20 Finally, we confirmed that the reduction of the EcFPR causes the release of NADP+ from the enzyme. Taken all these findings together, we postulated a new model of the NADP+ binding and catalysis mechanism for the subclass II reductases, which can probably be extended to all FPRs (Figure 6). In this model, the NADP+ would bind to the reductase through a bipartite site (N and P hemi‐sites) with different affinities. The release of the nucleotide tightly bound to the P hemi‐site (with the highest affinity) would be promoted by the binding of an incoming NADPH molecule to the N hemi‐site, through its nicotinamide moiety.
FIGURE 6.
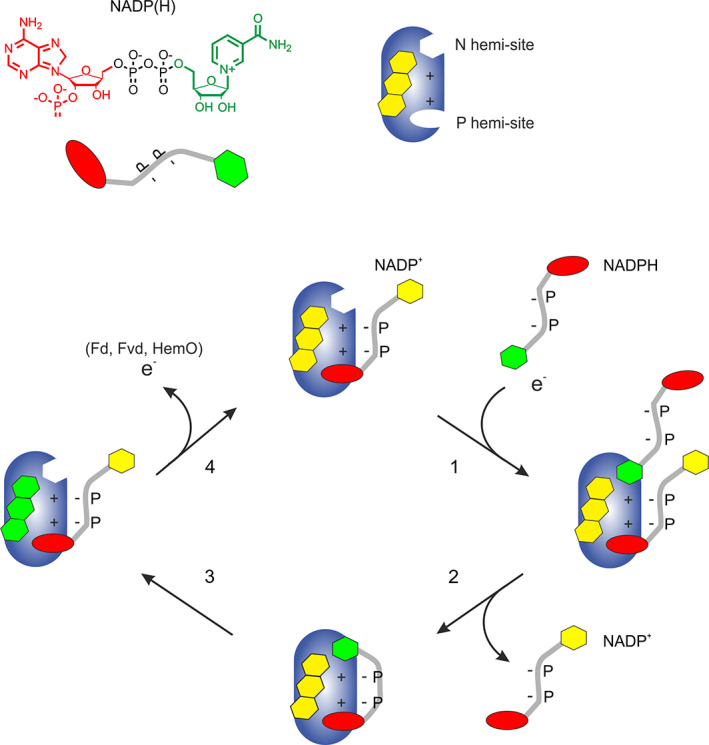
Hypothetical model of the mechanism of NADP+ binding and catalysis in subclass II FPRs. The natural state of FPR is depicted with the NADP+ molecule attached by 2′‐P‐AMP portion to P hemi‐site. In the presence of NADPH, an approximation of the nicotinamide portion towards the isoalloxazine ring of FAD occurs, as outlined in (1). This approximation results in the flavoprotein reduction and simultaneous displacement of the NADP+ molecule bound to P hemi‐site. Next, the P hemi‐site is occupied by the 2′‐P‐AMP portion of the incoming NADPH (2). This leads to the electron transfer process (3). Finally, the nicotinamide portion separates from the enzyme, preserving the binding of the NADP+ molecule through the P hemi‐site (4). Nucleotides states: oxidized, in yellow; reduced, in green
Consistent with these results are our observations using NADH. The FAD of EcFPR can be reduced by either NADPH or NADH, indicating that the N hemi‐site is available and that the reduced nicotinamide from these nucleotides is able to transfer electrons to the flavin. However, the diaphorase activity of EcFPR is negligible with NADH as an electron donor substrate (Figure S4). NADH can not properly bind to the enzyme P hemi‐site, thus preventing it from performing a complete turnover.
EcFPR can reduce both ferredoxin and flavodoxin. The reaction direction is optimized to favor the oxidation of NADPH. Physiologically, EcFPR participates in the response mechanism to oxidative stress produced by reactive oxygen species. This reductase has also been related to the activation of enzymes associated with anaerobic metabolism through the flavodoxin reduction.12, 28 Evidence has been obtained that bacteria need to finely regulate the homeostasis of the NADP(H) pool, which enables them to respond to stress conditions appropiately. This regulatory function would be carried out with the direct involvement of EcFPR.13 Many considerations have been made regarding the catalytic difference between plastidic and bacterial type FNRs. Some studies have proposed that the NADP+ binding mode could be an important feature in the catalytic difference observed between these enzymes.29
Our studies showed that EcFPR is purified with the NADP+ tightly bound and that this binding produces an inhibition of the catalytic activity, which disappears when the nucleotide is released from the enzyme. We propose that this tight binding of NADP+ could be involved in the NADPH/NADP+ regulation. At low cellular NADPH concentration, the enzyme displays a basal activity as it is inhibited by the tightly bound NADP+. When the NADPH concentration increases, the oxidized nucleotide is released and the enzyme activated, thus accelerating the removal of the reduced nucleotide excess. In other words, this control valve opens at times when the NADPH levels become dangerous for cell life.
The dynamics of the FNRs catalytic sites has always been an exciting topic not yet fully elucidated. Several pieces of evidence suggested the movement of C‐terminal tyrosine entering and leaving the N hemi‐site in plastidic type FNRs.5, 17, 30, 31, 32 A connection between the two hemi‐sites was also observed. Paladini et al. showed that the interaction of the 2′‐P‐AMP moiety from NADP+ induces a change that favors the interaction of the nicotinamide, thereby facilitating the catalytic process.20 In the bacterial enzymes a displacement of the C‐terminal extension is also expected to obtain a catalytically competent conformation. However, the interaction between the aromatic residue located in the C‐terminal extension and the adenosine of FAD suggests that an important rearrangement is necessary to allow nicotinamide to reach the active site. It has been reported that the 2′‐P‐AMP binding region does not suffer major conformational changes upon coenzyme binding in FPRs since the cavity is already preformed.15 Accordingly, similar binding affinities were reported for NADP+ and 2′‐P‐AMP.33 These observations allowed to conclude that the entrance of nicotinamide to the catalytic site is not promoted by NADP+ binding at the P hemi‐site. The model proposed in this study raises the possibility that nicotinamide leads the initial entrance of NADPH to the active site. This partial binding produces a subsequent protein rearrangement facilitating the release of the tightly bound NADP+, allowing catalysis to occur. This would imply that the nicotinamide binding in the N hemi‐site should promote a conformational change, albeit a transitory one, that alters the P hemi‐site affinity. Our model does not predict whether the affinities for NADP+ and NADPH are different, but these could change transiently according to the presence of substrate molecules in each hemi‐site. The development of new experimental strategies in the future will allow to capture these internal movements in FPRs. A crucial evolutionary difference between plastidic and bacterial type FNRs may exist. The catalytic mechanism of the formers has been optimized for NADPH production. In contrast, FPRs have been adapted to regulate and control their activity according to the role they must fulfill in nature.
Concluding, the tight binding of NADP+ identified and characterized in this study, may be an essential catalytic and regulatory mechanism of subclass II FPRs. This phenomenon could be used as a differential target for the inactivation of metabolic pathways in which FPR participates in pathogenic bacteria. These pathways are especially appropriate for the development of antimicrobial agents because they are not present in humans.
4. MATERIALS AND METHODS
4.1. Plasmid constructions, protein expression, and purification
Wild type and mutant EcFPR and PeaFNR were expressed in Escherichia coli using the pET205 vector,14, 34 containing an ampicillin resistance marker cassette. All plasmids constructed express a Trx‐His‐Tag‐FNR fusion protein with a thrombin recognition site between the His‐Tag and the mature FNR.
PcFPR, BaFPR and LepFNR were expressed in E. coli using pET‐TEV.35 This plasmid is derived from the pET28a vector in which the thrombin cleavage site was replaced by a TEV cleavage site along with an extension of six histidine residues. The T7 affinity tag was removed from this vector.
All FNR variants were expressed in E. coli BL21(DE3)pLysS by induction with 250 μM IPTG at 20°C for 16 h obtaining similar yields in the soluble fraction in all cases. Then, cells were collected, and the recombinant enzymes were purified from cell extracts by affinity chromatography using Ni‐NTA agarose. Proteins were eluted with 100 mM imidazole and dialyzed against 50 mM Tris–HCl (pH 8), 150 mM NaCl. Subsequently, proteins were digested with thrombin or TEV protease, and the His‐Tag removed by a second Ni‐NTA affinity chromatographic procedure. The purity of all protein preparations was assessed by SDS‐PAGE, and protein concentrations were determined by UV–visible spectroscopy.
4.2. Construction of the mutant enzymes
Primers were designed in order to obtain mutant enzymes by site‐directed mutagenesis. The appropriate plasmids were used as templates to create single mutants EcFPR_R144P, EcFPR_R184Y, and the double mutant EcFPR_R144P_R184Y. Likewise, plasmids coding for PeaFNR_P199R and PeaFNR_Y240R were generated.
These mutations were incorporated using the QuikChange Lightning Site‐Directed Mutagenesis Kit (Agilent Technologies, Inc. USA) following the supplier instructions.
Double mutant PeaFNR (PeaFNR_P199R_Y240R) could not be obtained by the aforementioned method, so new oligos were designed to incorporate the Y240R mutation into PeaFNR_P199R using a modified site‐directed mutagenesis technique.36 The high efficiency of this method allowed us to incorporate the desired mutation.
4.3. Spectral analysis
Absorption spectra were recorded on a spectrophotometer Shimadzu UV‐2450 at 25°C. Samples were filtered through a G25 Sephadex (Sigma) spin‐column equilibrated with 50 mM Tris–HCl (pH 8), 150 mM NaCl before measurements to eliminate any non‐protein bound FAD traces. FNR extinction coefficients (ε) were determined based on FAD absorbance changes observed after enzyme denaturation by treatment with 0.2% (w/v) SDS. The absorption spectra were measured again between 300 and 600 nm, corresponding to the free FAD in solution. Based on the data obtained, the ε were calculated according to the following equation:
with εFAD 450 nm = 11.3 mM−1 cm−1.
4.4. Circular dichroism spectra of enzymes
CD spectra in the UV‐far region were recorded for wild type and mutant EcFPR and PeaFNR in order to assess the impact of the amino acid changes on the structural integrity of the reductases. A JASCO J‐1500 spectropolarimeter at 25°C was used. The protein concentration was 5 μM. The spectra were reported in units of molar ellipticities [θ] (degrees.cm2.dmol−1) according to the following conversion:
where θobs is the ellipticity given by the polar spectrum; MRW is the protein molecular weight in Daltons and the number of amino acids ratio; l is the cuvette pathlength; and c is the protein concentration in g/ml.
4.5. Determination of the NADP + content in the FNR by enzymatic activity
The amount of NADP+ bound to FPR/FNR was quantified by determining the NADPH generated by the reaction of G6PD. Samples were heat‐treated (5 min at 90°C) to release the nucleotide into the medium. Then they were centrifuged at maximum speed and supernatants were analyzed. The reaction medium (700 μl) contained 50 mM Tris–HCl (pH 8), 3 mM G6P, and 1 U of G6PD. After adding the sample, the absorbance at 340 nm was recorded, and the μmoles of NADPH generated by NADP+ reduction were calculated using an ε340 NADPH = 6.22 mM−1. Solutions of NADP+ between 0 and 500 μM, heated as the protein samples, were used as controls for the construction of a calibration curve. The NADP+ content bound to wild type EcFPR was also determined in the presence of 4‐dimethylaminopyridine (DMAP). In this case, a 30 μM wild type EcFPR sample was previously incubated with 20 mM DMAP for 15 min, at 25°C, and then filtered by size exclusion chromatography with a Sephadex G‐25 column in order to separate the released NADP+ from the protein sample.
4.6. Enzymatic assays
FNR‐dependent diaphorase activity was determined using published methods.37 The reaction medium contained 50 mM Tris–HCl (pH 8), 1 mM K3Fe(CN)6, 1–500 μM NADP(H) and 100–500 nM FNR. In the determinations in which a regenerating system was used, the reaction medium also contained 3 mM G6P and 1 U G6PD. The reaction was followed by a decrease in the absorbance at 420 nm by K3Fe(CN)6 reduction. For the measurements that were carried out in the presence of the NADP+ analog DMAP, 20 mM was added to the reaction medium. All kinetic experiments were performed at 25°C. Experimental data were fitted to theoretical curves using SIGMAPLOT (Systat Software Inc., Point Richmond, California).
In the experiment in which the reduction of wild type EcFPR and PeaFNR, and EcFPR_R144P_R184Y was spectroscopically monitored, the absorption spectra of 75 μM samples of these proteins were scanned between 300 and 600 nm. Then, they were incubated with 1 U G6PD or, with this enzyme and its substrate, 3 mM G6P, for 5 min at 25°C and the absorption spectra were registered again.
4.7. Separation of the NADP + bound to the enzymes
The NADP+ bound to FPR/FNR was separated by affinity chromatography with a Cibacron Blue‐Agarose resin (Cibacron Blue 3GA Fast flow agarose, SIGMA). 2 mg of protein were added to 800 μl of the matrix. The reductase binds to the matrix with high affinity, with the concomitant release of the protein‐bound NADP+. Then proteins were eluted from the column with 750 mM NaCl. The samples obtained were analyzed by microplate absorbance measurements. Although the nucleotide and protein spectra partially overlap, it was possible to select certain wavelengths to differentiate the compounds eluted from the column as follows: NADP+, at 260 nm; FAD, at 456 nm, and the aromatic residues, at 280 nm. Aliquots were collected from both fractions not fixed to the matrix and those eluted with NaCl and the absorbance profiles were obtained for each protein.
4.8. Enzyme stability analysis
Protein stability was evaluated in a Real‐time Thermal Cycler with Realplex software. The sample volume was 20 μl, and the protein concentration of 0.5 mg/ml. Sypro Orange was used as a fluorescent probe in a final concentration of ×10. The excitation wavelength was 520 nm, and the emission was at 550 nm. Each sample was evaluated in triplicate. The program was initialized with a temperature of 20°C for 10 min, and then the temperature was increased 1°C every 1 min for 70 cycles until reaching a final temperature of 90°C. Denaturation curves were obtained, and T m calculated using OriginPro software. By plotting the first derivative of the fluorescence emission (−dF/dT) as a function of temperature, a negative Gaussian curve was obtained where the melting temperature (T m) is represented by the minimum value of the curve (Figure S1). The existence of significant differences with respect to the control condition was determined by t‐Student test (p < 0.05) and they were denoted with an asterisk.
AUTHOR CONTRIBUTIONS
Paula Monchietti: Investigation; methodology; writing ‐ original draft; writing‐review & editing. Arleth S. López Rivero: Investigation; methodology. Eduardo A. Ceccarelli: Conceptualization; funding acquisition; supervision; writing‐review & editing. Daniela L. Catalano‐Dupuy: Conceptualization; funding acquisition; investigation; methodology; supervision; writing ‐ original draft; writing‐review & editing.
Supporting information
TABLE S1 Determination of the content of NADP+ bound to purified FNR enzymes from different organisms by HPLC
FIGURE S1: Thermal stability of EcFPR* in the presence or absence of NADP+
TABLE S2: Melting points of thermal unfolding transitions of the FNR
FIGURE S2: Spectroscopic characterization of wild type and mutant EcFPR and PeaFNR
FIGURE S3: Isothermal titration calorimetry (ITC) for the binding of NADP+ to EcFPR
TABLE S3: Melting points of thermal unfolding transitions of wild type and mutant EcFPR and PeaFNR
FIGURE S4: Electron transfer between EcFPR and NADPH or NADH
ACKNOWLEDGMENTS
This study was supported by grants from PICT 2017‐1789 and PCT 2018‐4494, ANCYPT, Ministerio de Ciencia, Tecnología e Innovación Productiva, Argentina (www.agencia.mincyt.gov.ar); PIP 112‐201201‐00345‐CO from CONICET, Consejo Nacional de Investigaciones Científicas y Técnicas, Argentina (www.conicet.gov.ar) and, 800201180300113UR and BIO497 from the University of Rosario, Argentina (www.unr.edu.ar.). Eduardo A. Ceccarelli and Daniela L. Catalano‐Dupuy are staff members and Paula Monchietti is a fellow of CONICET.
Monchietti P, López Rivero AS, Ceccarelli EA, Catalano‐Dupuy DL. A new catalytic mechanism of bacterial ferredoxin‐NADP+ reductases due to a particular NADP+ binding mode. Protein Science. 2021;30:2106–2120. 10.1002/pro.4166
Funding information ANCYPT, Ministerio de Ciencia, Tecnología e Innovación Productiva Argentina, Grant/Award Numbers: PICT 2017‐1789, PCT 2018‐4494; University of Rosario Argentina, Grant/Award Numbers: BIO497, 800201180300113UR; CONICET, Consejo Nacional de Investigaciones Científicas y Técnicas, Argentina, Grant/Award Number: PIP 112‐201201‐00345‐CO
REFERENCES
- 1.Catalano‐Dupuy D, Rial D, Ceccarelli E. Ferredoxin‐NADP+ reductases. In: Gruyter DE, editor. Handbook of Flavoproteins. Vol. 1, Oxidases, dehydrogenases and related systems. Berlin/Boston: Hille, Russ; Miller, Susan and Palfey, Bruce, 2013; p. 313–335. [Google Scholar]
- 2.Koskela MM, Dahlstrom KM, Goni G, et al. Arabidopsis FNRL protein is an NADPH‐dependent chloroplast oxidoreductase resembling bacterial ferredoxin‐NADP(+) reductases. Physiol Plant. 2018;162, 177–190. [DOI] [PubMed] [Google Scholar]
- 3.Perez‐Amigot D, Taleb V, Boneta S, et al. Towards the competent conformation for catalysis in the ferredoxin‐NADP(+) reductase from the Brucella ovis pathogen. Biochim Biophys Acta. 2019;1860:148058. [DOI] [PubMed] [Google Scholar]
- 4.Tondo M, Musumeci M, Delprato M, Ceccarelli E, Orellano E. Structural‐functional characterization and physiological significance of ferredoxin‐NADP+ reductase from Xanthomonas axonopodis pv. Citri. PLoS One. 2011;6:e27124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carrillo N, Ceccarelli E. Open questions in ferredoxin‐NADP+ reductase catalytic mechanism. Eur J Biochem. 2003;270:1900–1915. [DOI] [PubMed] [Google Scholar]
- 6.Mulo P, Medina M. Interaction and electron transfer between ferredoxin‐NADP(+) oxidoreductase and its partners: structural, functional, and physiological implications. Photosynth Res. 2017;134:265–280. [DOI] [PubMed] [Google Scholar]
- 7.Bianchi V, Reichard P, Eliasson R, et al. Escherichia coli ferredoxin NADP+ reductase: activation of E. coli anaerobic ribonucleotide reduction, cloning of the gene (fpr), and overexpression of the protein. J Bacteriol. 1993;175:1590–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ingelman M, Bianchi V, Eklund H. The three‐dimensional structure of flavodoxin reductase from Escherichia coli at 1.7 A resolution. J Mol Biol. 1997;268:147–157. [DOI] [PubMed] [Google Scholar]
- 9.Medina M, Gomez‐Moreno C. Interaction of ferredoxin–NADP+ reductase with its substrates: optimal interaction for efficient electron transfer. Photosynth Res. 2004;79:113–131. [DOI] [PubMed] [Google Scholar]
- 10.Krapp A, Tognetti V, Carrillo N, Acevedo A. The role of ferredoxin‐NADP+ reductase in the concerted cell defense against oxidative damage. Studies using Escherichia coli mutants and cloned plant genes. J Biochem. 1997;249:556–563. [DOI] [PubMed] [Google Scholar]
- 11.Giro M, Carrillo N, Krapp A. Glucose‐6‐phosphate dehydrogenase and ferredoxin‐NADP(H) reductase contribute to damage repair during the soxRS response of Escherichia coli . Microbiology. 2006;152, 1119–1128. [DOI] [PubMed] [Google Scholar]
- 12.Krapp A, Rodriguez R, Poli H, Paladini D, Palatnik J, Carrillo N. The flavoenzyme ferredoxin (flavodoxin)‐NADP(H) reductase modulates NADP(H) homeostasis during the soxRS response of Escherichia coli . J Bacteriol. 2002;184:1474–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krapp A, Humbert M, Carrillo N. The soxRS response of Escherichia coli can be induced in the absence of oxidative stress and oxygen by modulation of NADPH content. Microbiology. 2011;157:957–965. [DOI] [PubMed] [Google Scholar]
- 14.Musumeci M, Botti H, Buschiazzo A, Ceccarelli E. Swapping FAD binding motifs between plastidic and bacterial ferredoxin‐NADP(H) reductases. Biochemistry. 2011;50:2111–2122. [DOI] [PubMed] [Google Scholar]
- 15.Wang A, Zeng Y, Han H, et al. Biochemical and structural characterization of Pseudomonas aeruginosa Bfd and FPR: ferredoxin NADP+ reductase and not ferredoxin is the redox partner of heme oxygenase under iron‐starvation conditions. Biochemistry. 2007;46, 12198–12211. [DOI] [PubMed] [Google Scholar]
- 16.Catalano‐Dupuy D, Musumeci M, Lopez‐Rivero A, Ceccarelli E. A highly stable plastidic‐type ferredoxin‐NADP(H) reductase in the pathogenic bacterium Leptospira interrogans . PLoS One. 2011;6:e26736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Piubelli L, Aliverti A, Arakaki A, et al. Competition between C‐terminal tyrosine and nicotinamide modulates pyridine nucleotide affinity and specificity in plant ferredoxin‐NADP+ reductase. J Biol Chem. 2000;275:10472–10476. [DOI] [PubMed] [Google Scholar]
- 18.Biggar KK, Dawson NJ, Storey KB. Real‐time protein unfolding: a method for determining the kinetics of native protein denaturation using a quantitative real‐time thermocycler. Biotechniques. 2012;53:231–238. [DOI] [PubMed] [Google Scholar]
- 19.Huynh K, Partch CL. Analysis of protein stability and ligand interactions by thermal shift assay. Curr Protoc Protein Sci. 2015;79: 28.29.21–28.29.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Paladini D, Musumeci M, Carrillo N, Ceccarelli E. Induced‐fit and equilibrium dynamics for high catalytic efficiency in ferredoxin‐NADP(H) reductases. Biochemistry. 2009;48:5760–5768. [DOI] [PubMed] [Google Scholar]
- 21.Del Corso A, Barsacchi D, Giannessi M, et al. Bovine lens aldose reductase: tight binding of the pyridine coenzyme. Arch Biochem Biophys. 1990;283:512–518. [DOI] [PubMed] [Google Scholar]
- 22.Kirkman HN, Gaetani GF, Clemons EH. NADP+‐binding proteins causing reduced availability and sigmoid release of NADP+ in human erythrocytes. J Biol Chem. 1986;261:4039–4045. [PubMed] [Google Scholar]
- 23.Kirkman HN, Gaetani GF. Catalase: a tetrameric enzyme with four tightly bound molecules of NADPH. Proc Natl Acad Sci U S A. 1984;81:4343–4347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gaetani GF, Ferraris AM, Sanna P, Kirkman HN. A novel NADPH:(bound) NADP+ reductase and NADH:(bound) NADP+ transhydrogenase function in bovine liver catalase. Biochem J. 2005;385:763–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang X, Chan T, Lam V, Engel P. What is the role of the second “structural” NADP+‐binding site in human glucose 6‐phosphate dehydrogenase? Protein Sci. 2008;17, 1403–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morales‐Luna L, Hernandez‐Ochoa B, Ramirez‐Nava EJ, et al. Characterizing the fused TvG6PD::6PGL protein from the protozoan Trichomonas vaginalis, and effects of the NADP(+) molecule on enzyme stability. Int J Mol Sci. 2020;21:4831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Deng Z, Aliverti A, Zanetti G, et al. A productive NADP+ binding mode of ferredoxin–NADP+ reductase revealed by protein engineering and crystallographic studies. Nat Struct Biol. 1999;6:847–853. [DOI] [PubMed] [Google Scholar]
- 28.Bianchi V, Haggård‐ljungquist E, Pontis E, Reichard P. Interruption of the ferredoxin (flavodoxin)‐NADP+ oxidoreductase gene of Escherichia coli does not affect anaerobic growth but increases sensitivity to paraquat. J Bacteriol. 1995;177:4528–4531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Catalano‐Dupuy D, López‐Rivero A, Soldano A, Ceccarelli EA. Redox proteins as targets for drugs development against pathogens. Curr Pharm Des. 2013;19:2594–2605. [DOI] [PubMed] [Google Scholar]
- 30.Catalano‐Dupuy DL, Orecchia M, Rial DV, Ceccarelli EA. Reduction of the pea ferredoxin‐NADP(H) reductase catalytic efficiency by the structuring of a carboxyl‐terminal artificial metal binding site. Biochemistry. 2006;45:13899–13909. [DOI] [PubMed] [Google Scholar]
- 31.Peregrina JR, Lans I, Medina M. The transient catalytically competent coenzyme allocation into the active site of Anabaena ferredoxin‐NADP+reductase. Eur Biophys J. 2012;41:117–128. [DOI] [PubMed] [Google Scholar]
- 32.Lans I, Medina M, Rosta E, et al. Theoretical study of the mechanism of the hydride transfer between ferredoxin‐NADP+ reductase and NADP+: the role of Tyr303. J Am Chem Soc. 2012;134, 20544–20553. [DOI] [PubMed] [Google Scholar]
- 33.Nogues I, Hervas M, Peregrina JR, et al. Anabaena flavodoxin as an electron carrier from photosystem I to ferredoxin‐NADP+ reductase. Role of flavodoxin residues in protein‐protein interaction and electron transfer. Biochemistry. 2005;44, 97–104. [DOI] [PubMed] [Google Scholar]
- 34.Rial DV, Arakaki AK, Ceccarelli EA. Interaction of the targeting sequence of chloroplast precursors with Hsp70 molecular chaperones. Eur J Biochem. 2000;267:6239–6248. [DOI] [PubMed] [Google Scholar]
- 35.Liu H, Naismith JH. A simple and efficient expression and purification system using two newly constructed vectors. Protein Expr Purif. 2009;63:102–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu H, Naismith JH. An efficient one‐step site‐directed deletion, insertion, single and multiple‐site plasmid mutagenesis protocol. BMC Biotechnol. 2008;8:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zanetti G. A lysyl residue at the NADP+ binding site of ferredoxin‐NADP+ reductase. Biochim Biophys Acta. 1976;445:14–24. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
TABLE S1 Determination of the content of NADP+ bound to purified FNR enzymes from different organisms by HPLC
FIGURE S1: Thermal stability of EcFPR* in the presence or absence of NADP+
TABLE S2: Melting points of thermal unfolding transitions of the FNR
FIGURE S2: Spectroscopic characterization of wild type and mutant EcFPR and PeaFNR
FIGURE S3: Isothermal titration calorimetry (ITC) for the binding of NADP+ to EcFPR
TABLE S3: Melting points of thermal unfolding transitions of wild type and mutant EcFPR and PeaFNR
FIGURE S4: Electron transfer between EcFPR and NADPH or NADH