

Abstract
CAD is a 1.5 MDa particle formed by hexameric association of a 250 kDa protein divided into different enzymatic domains, each catalyzing one of the initial reactions for de novo biosynthesis of pyrimidine nucleotides: glutaminase‐dependent Carbamoyl phosphate synthetase, Aspartate transcarbamoylase, and Dihydroorotase. The pathway for de novo pyrimidine synthesis is essential for cell proliferation and is conserved in all living organisms, but the covalent linkage of the first enzymatic activities into a multienzymatic CAD particle is unique to animals. In other organisms, these enzymatic activities are encoded as monofunctional proteins for which there is abundant structural and biochemical information. However, the knowledge about CAD is scarce and fragmented. Understanding CAD requires not only to determine the three‐dimensional structures and define the catalytic and regulatory mechanisms of the different enzymatic domains, but also to comprehend how these domains entangle and work in a coordinated and regulated manner. This review summarizes significant progress over the past 10 years toward the characterization of CAD's architecture, function, regulatory mechanisms, and cellular compartmentalization, as well as the recent finding of a new and rare neurometabolic disorder caused by defects in CAD activities.
Keywords: aspartate transcarbamoylase, carbamoyl phosphate synthetase, de novo pyrimidine biosynthesis, dihydroorotase, multienzymatic protein, nucleotide metabolism, rare diseases
1. INTRODUCTION
Pyrimidine nucleotides are building blocks of DNA and RNA and key activators of intermediate metabolites in a number of cellular processes, including glycosylation and carbohydrate and phospholipid synthesis.1, 2 The cells obtain the supply of pyrimidines through two different metabolic pathways (Figure 1). Slowly dividing or differentiated cells use preferentially salvage pathways to uptake preformed nucleosides (e.g., uridine) from the diet or from the breakdown of nucleic acids, whereas, proliferating cells, whether normal (e.g., activated lymphocytes) or tumoral, depend on de novo (“of new”) synthesis of pyrimidine nucleotides. The de novo pathway consists of six enzymatic steps yielding uridine 5‐monophosphate (UMP), from which all other pyrimidine nucleotides are made (Figure 1).3 In a first step, carbamoyl phosphate synthetase (CPS; EC 6.3.5.5) makes carbamoyl phosphate (CP) from bicarbonate, ammonia (released from glutamine) and two ATP molecules. Then, aspartate transcarbamoylase (ATC; EC 2.1.3.2) uses CP and aspartate (Asp) to form carbamoyl aspartate. Next, dihydroorotase (DHO; EC 3.5.2.3) catalyzes the condensation of carbamoyl aspartate to dihydroorotate, which is oxidized to orotate by the dihydroorotate dehydrogenase (DHODH; EC 1.3.5.2). Orotate, a pyrimidine base, is attached to ribose by the enzyme orotate phosphoribosyltransferase (OPRT; EC 2.4.2.10), producing the nucleotide orotidine 5‐monophosphate (OMP) that is subsequently decarboxylated by OMP decarboxylase (OMPDC; EC 4.1.1.23) to make UMP.
FIGURE 1.
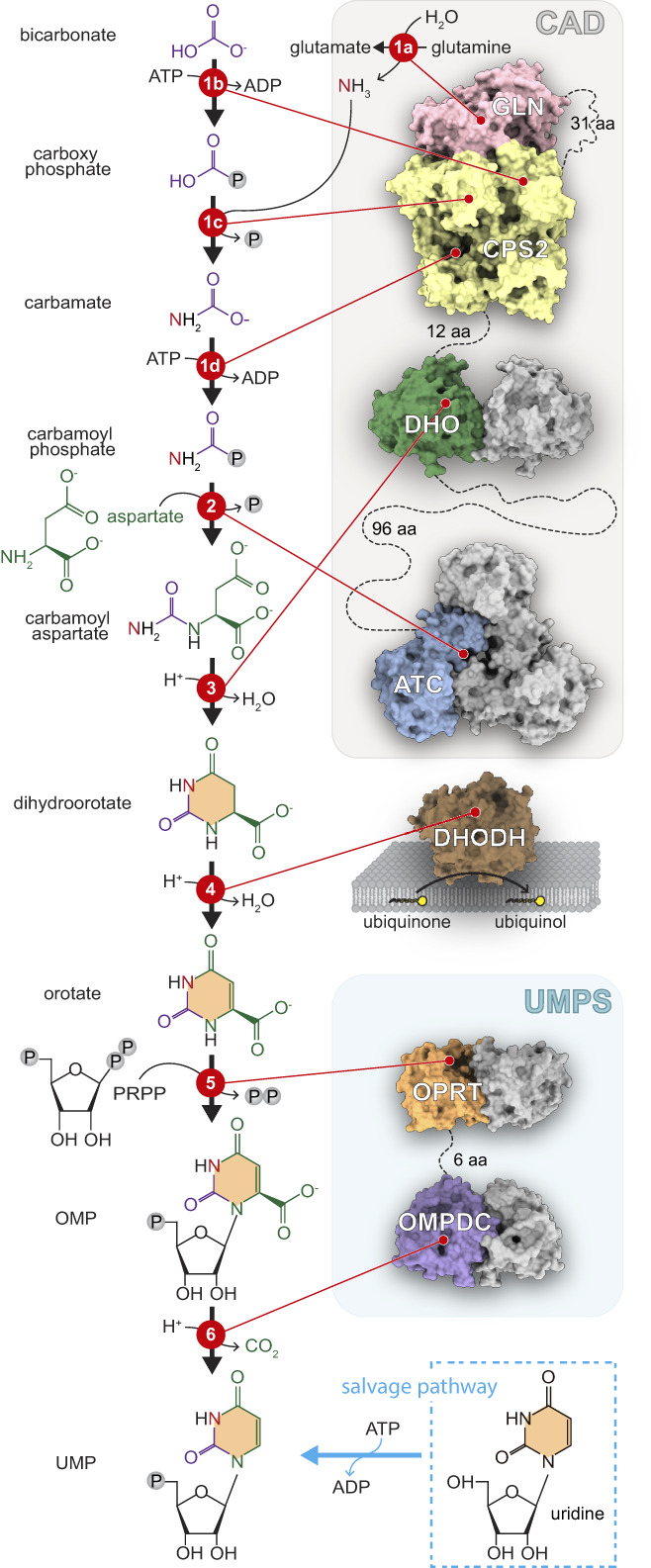
Scheme of de novo pathway for UMP synthesis. In animals, the first three enzymes of the pathway (CPS, ATC, and DHO) are fused into a single multifunctional protein named CAD, and the last two enzymes (OPRT and OMPDC) are fused into the bifunctional protein UMPS. DHODH is attached to the inner mitochondrial membrane and uses ubiquinone as co‐factor. The structures of the human enzymes are shown in surface representation and covalent linkages are indicated with dashed lines. The structure of CAD's CPS‐2 is unknown, and has been represented by the related arginine‐specific human CPS‐1 structure (PDB ID 5DOU and 5DOT). Some enzymatic domains are represented in its oligomeric form with only one of the subunit colored
Although the six steps for de novo UMP synthesis are preserved in all living organisms, the organization of the enzymatic machinery varies greatly (Figure 2a).3, 4, 5 Whereas most prokaryotes encode individual enzymes for each step, in humans and other animals, the glutamine‐dependent CPS (CPS‐2), ATC and DHO activities are fused as distinct functional domains into a single ~250 kDa polypeptide named CAD (Figures 1 and 2a).6 This multifunctional protein further self‐assembles into hexamers, forming a megaenzyme nearly half the size of a ribosome.7 OPRT and OMPDC are also fused into a single polypeptide named UMP synthetase (UMPS), while the remaining enzyme, DHODH, is anchored to the inner mitochondrial membrane, coupling the synthesis of pyrimidines to the respiratory chain (Figure 1). Fungi, on the other hand, have a CAD‐like protein (named ura2 in S. cerevisiae) with an inactive DHO‐like domain,8 and the DHO, DHODH, OPRT and OMPDC activities are performed by separated monofunctional proteins (Figure 2a). Most of these enzymes in plants and protists are also monofunctional, except for a bifunctional UMPS that in some protists (e.g., Trypanosoma) fuses the domains in reversed order (Figure 2a).5, 9
FIGURE 2.
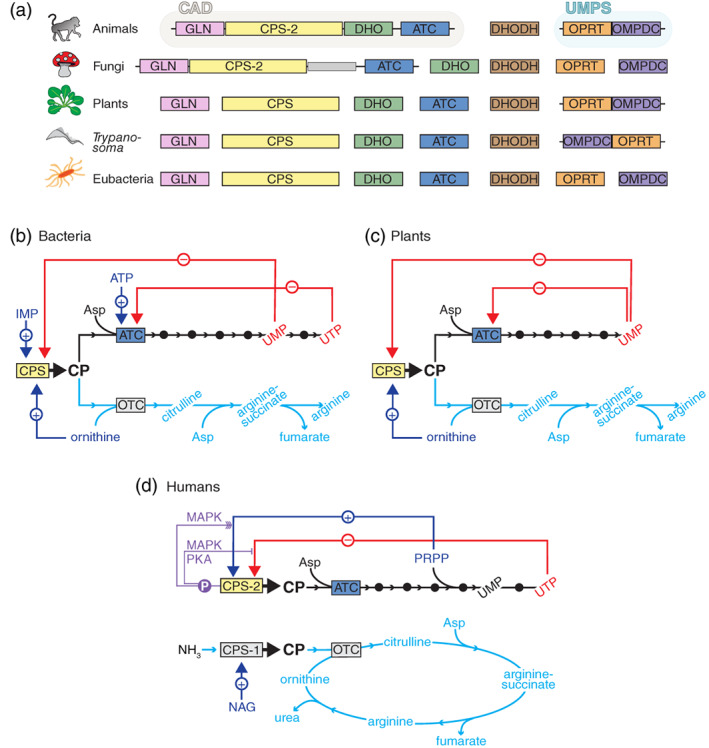
Organization and regulation of the enzymes involved in the de novo synthesis of pyrimidine nucleotides. (a) Scheme of the six enzymatic activities for de novo UMP synthesis, as domains within multifunctional proteins or as independent enzymes. (b–d) Regulation of the pathways for the biosynthesis of UMP (black lines) and arginine (cyan lines) in bacteria (b), plants (c) and humans and most terrestrial animals (d). Red lines indicate allosteric inhibition, and blue lines allosteric activation. Purple lines indicate how phosphorylations inhibit or enhance (multiple arrows) the allosteric regulation of CAD's CPS‐2 activity. OTC, ornithine transcarbamoylase; NAG, N‐acetyl‐l‐glutamate
These differences in the enzyme organization are accompanied by differences in the regulation of the pathway. In prokaryotes and plants, CPS makes CP both for the biosynthesis of UMP and arginine (Figure 2b,c).3 Thus, ATC becomes the first committed enzyme for pyrimidine synthesis and is the control point of the pathway, being feedback inhibited by UTP (and CTP) and activated by ATP in the case of bacteria,10 or inhibited by UMP in plants (Figure 2b,c).11 In these organisms, CPS is also inhibited by pyrimidines and activated by ornithine (a substrate for arginine synthesis). Then, when the pyrimidine pool builds up, both ATC and CPS are inhibited, and ornithine counteracts the CPS inhibition, diverting CP synthesis to the arginine pathway (Figure 2b,c). In animals and fungi, on the other hand, there are separated CP pools for pyrimidines and arginine biosynthesis (Figure 2d).3 The arginine‐specific CP is produced by a CPS (CPS‐1) localized in the mitochondrial matrix, and in most terrestrial animals, this enzyme lacks glutaminase activity and funnels the circulating ammonia for its detoxification through the urea cycle (Figure 2d). In turn, the CP pool for pyrimidine synthesis is produced by CAD's CPS‐2, which is the rate‐limiting and control point of the de novo pathway, being allosterically inhibited by UTP and activated by phosphoribosyl pyrophosphate (PRPP) (Figure 2d). This allosteric regulation is further modulated through phosphorylation of the CPS‐2 domain. Phosphorylation by the cyclic AMP‐dependent protein kinase (PKA) overcomes the inhibition by UTP,12 whereas phosphorylation by the mitogen‐activated protein kinase (MAPK) converts UTP from an inhibitor to a modest activator and promotes the activation by PRPP (Figure 2d).13 Both phosphorylations are proposed to occur sequentially and to regulate the activity of CAD throughout the cell cycle.14
Whereas significant progress has been made in the structural and biochemical characterization of the enzymes initiating the de novo biosynthesis of pyrimidines in bacteria, CAD, the multienzymatic protein leading this essential metabolic pathway in animals, remains surrounded by mystery. What are the structures and functional mechanisms of CAD's enzymatic domains? What are the evolutionary advantages for their fusion? How they assemble? What is the architecture of the megaenzyme? Where is CAD within the cell? Why there are no diseases associated with defects of any of CAD's essential activities? The last 10 years provided important answers to these questions, and we believe it is a good timing to review the efforts toward understanding CAD and its function in health and disease.
2. GETTING CAD IN SHAPE
2.1. Starting from the end: The ATC domain
The discovery of CAD is ligated to the rational design of PALA (N‐phosphonacetyl‐l‐aspartate), an ATC inhibitor with structural features of the substrates CP and Asp that mimics the transition‐state of the reaction.15 PALA proved to be a potent inhibitor of Escherichia coli ATC (ecATC), and also penetrated easily in mammalian cells, arresting the growth of mouse tumors and proving that de novo pyrimidine synthesis is mandatory for cell proliferation.16, 17 However, a fraction of PALA‐treated cells overcame the inhibitory effect by producing large amounts of ATC,18 and the attempt to purify the enzyme led to the striking discovery that it was covalently attached to CPS and DHO forming a single multienzymatic protein.6
Forty years later, the first detailed structural information about CAD, that of the isolated human ATC domain free and bound to PALA, came to light (Table 1).19, 24 The structure of the ATC domain of the CAD‐like protein from Chaetomium thermophilum was also determined.23 The structures reveal a homotrimer of triangular appearance and with high similarity to bacterial ATCs (Figure 3a). Each subunit of 33 kDa is divided into two domains of comparable size (Figure 3b): an N‐terminal domain that provides most contacts with the other subunits and holds the site for CP, and a C‐terminal domain bearing the site for Asp. Both domains are structured by a central β‐sheet of five parallel strands flanked by α‐helices. The active site is formed at the cleft between the N‐ and C‐domains and includes a loop (CP‐loop) from the adjacent subunit (Figure 3a–c, colored in magenta). CP is the substrate binding in the first place, inducing the repositioning of the CP‐loop from the adjacent subunit and the closure of the C‐domain, which create the pocket for Asp. Upon Asp binding, the subunit closes even more, with a large movement of a loop at the C‐domain (Asp‐loop) that traps and brings the substrates in close contact to favor the reaction (Figure 3b,c).19, 25
TABLE 1.
Crystal structures of CAD and CAD‐like domains deposited in the Protein Data Bank
| Protein | ID | Comments |
|---|---|---|
| Human ATC | 5G1O | Apo form19 |
| 5G1P | Bound to CP19 | |
| 5G1N | Bound to PALA19 | |
| Human DHO | 4C6C, 4BY3 | Apo form with complete active site20 |
| 4C6B | Apo form with non‐carboxylated K155620 | |
| 5YNZ | Mutant K1556A21 | |
| 4C6D, 4C6E, 4C6F, 4C6I, 4C6J, 4C6K, 4C6L, 4C6M, 4C6N, 4C6O, 4C6P, 4C6Q | WT and mutants bound to substrates at pH 5.5–8.020 | |
| 6HFD, 6HFE, 6HFF, 6HFH, 6HFI, 6HFJ, 6HFK, 6HFL, 6HFN, 6HFP, 6HFQ, 6HFR, 6HFS, 6HFU, 6HG1, 6HG2, 6HG3 | F1563 mutants in apo form or bound to substrates at pH 6.5–7.522 | |
| C. thermophilum ATC | 5NNN | Apo form23 |
| 5NNQ | Bound to CP23 | |
| C. thermophilum inactive DHO‐like | 5NNL | Apo form23 |
Abbreviations: ATC, aspartate transcarbamoylase; DHO, dihydroorotase.
FIGURE 3.
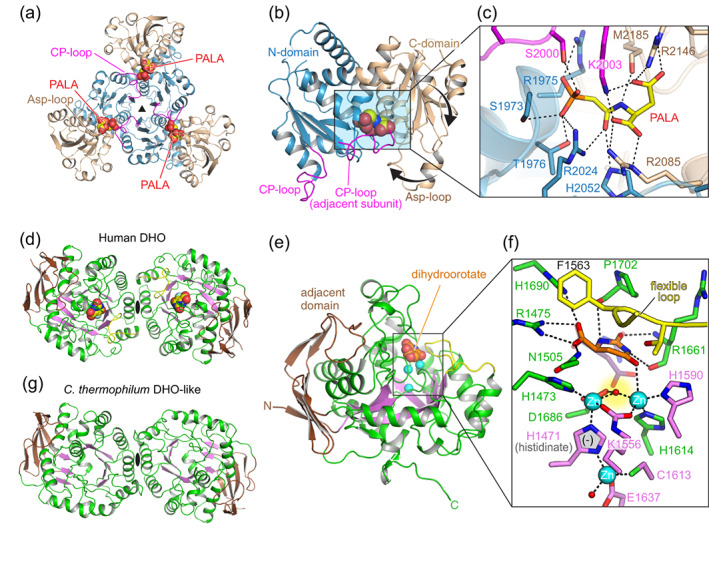
Crystal structures of CAD's ATC and DHO domains. (a) Cartoon representation of human ATC trimer bound to PALA with each subunit colored with the N‐ and C‐domain in blue and brown, respectively. (b) Detail of the ATC subunit with the CP‐loop colored in magenta and the movement of the C‐domain and the Asp‐loop indicated by thick arrows. (c) Detail of the active site with PALA bound. (d) Dimeric structure of the DHO domain of human CAD, with the central β‐barrel colored magenta and the adjacent domain in brown. The product of the reaction, dihydroorotate, is shown within the active site and represented as spheres. (e,f) Detail of the DHO subunit and of the active site, with Zn2+ ions represented as cyan spheres and the catalytic flexible loop colored in yellow. In panel (f), the substrate carbamoyl aspartate is shown superimposed in semitransparent stick representation. (g) Crystal structure of the dimeric and inactive DHO‐like domain of Chaetomium thermophilum CAD‐like protein
Surprisingly, the human ATC trimer was shown to bind PALA with high affinity to the first two subunits, but with 100‐fold lower affinity to the third one.19 Since PALA is a transition‐state analog, this suggested that despite being a trimer, only two ATC subunits catalyze the reaction at a time. The idea, although bizarre, received support from the first structure of an ATC from plants. The Arabidopsis ATC was shown to bind only one PALA molecule per trimer, and thus to catalyze the reaction only in one subunit at a time.26 The mechanistic explanation resides in the CP‐loop, that upon reaching the catalytic state, blocks the movement of the other CP‐loops to a similar active conformation.26
In sum, CAD's ATC domain depends on its oligomerization as a trimer to be active, and within this association there must be a communication between subunits that coordinates the sequential firing of the active sites.
2.2. DHO, a structural element with replaceable enzymatic activity
DHO is a metalloenzyme catalyzing the reversible interconversion of carbamoyl aspartate to dihydroorotate (Figure 1). The isolated 43 kDa DHO domain of human CAD was the first eukaryotic DHO to be structurally determined (Table 1).20, 27 The protein is a homodimer in solution, with each subunit folded in a barrel of eight β‐strands flanked on the outer surface by α‐helices and an additional subdomain formed by the N‐ and the C‐terminal regions (Figure 3d,e). The active site locates in a cavity shaped by the loops connecting the β‐barrel with the outer α‐helices and contains two catalytic Zn2+ ions coordinated by the side chain of a carboxylated Lys, four His and one Asp (Figure 3e,f). The active site is superimposable to that of E. coli DHO,28 including a catalytic flexible loop that orients the substrate in the active site, stabilizes the transition state of the reaction and excludes water from the active site.20, 22, 29 CAD's DHO also has a third Zn2+ ion at approximately the center of the β‐barrel that interacts with one of the catalytic metals through the side chain of a rare histidine with negative charge (histidinate anion) (Figure 3f). This third Zn2+ ion has not been described in bacterial DHOs and is proposed to increase stability and to influence the electrostatic environment of the active site.20, 30
The crystal structure of the inactive DHO‐like domain of the CAD‐like protein from C. thermophilum has also been determined (Figures 2a and 3g; Table 1).23 The structure shows a protein devoid of active site but assembled in identical dimers to those observed in human DHO (Figure 3g). The dimerization involves lateral contacts of the α/β‐barrel opposite to the adjacent domain, with both subunits oriented in the same direction. Interestingly, the construction of a monomeric human DHO mutant, proved that dimerization is not required for the enzyme to be active.20
Thus, in addition to the enzymatic activity, which is replaceable in the case of fungi, the DHO and DHO‐like domains preserve an oligomeric organization that is conserved from yeast to humans, and that could be required for the assembly of the multienzymatic particle.
2.3. Known unknowns of CPS‐2 structure
The smallest intermediate in the de novo pathway, CP, is synthesized by the largest enzyme (Figures 1 and 2a). CPS‐2 covers the ~1.000 aa N‐terminal half of CAD and is divided in two parts: a ~40 kDa GLN domain that obtains ammonia from glutamine (Figure 1, reaction 1a), and a ~120 kDa synthetase (or SYN) domain that (a) phosphorylates bicarbonate to form carboxyphosphate (1b), (b) condensates carboxyphosphate and ammonia into carbamate (1c), and (c) phosphorylates carbamate to make CP (1d).3 So far, there is no direct structural information of CAD's CPS‐2, and only the structures of E. coli CPS and the arginine‐specific human CPS‐1 (huCPS‐1) have been determined.31, 32 These evolutionary distant enzymes have a very similar structure, which is also expected to be shared by CAD's CPS‐2.
CPS is organized into multiple domains responsible for the partial reactions and connected by an internal tunnel to shuttle the unstable reaction intermediates (Figure 4a).31, 32 The GLN moiety, whether encoded as a different protein (ecCPS) or covalently attached to the SYN domain (huCPS‐1, CPS‐2), folds in a globular shape divided into a ~16 kDa N‐terminal lobe and a ~25 kDa C‐terminal lobe, both in close contact with the SYN domain (Figure 4a). The release of ammonia from glutamine occurs at the interface between both lobes and involves an activating His and a catalytic Cys that forms of a glutamyl‐thioester intermediate (Figure 4b).33 huCPS‐1 lacks glutaminase activity and replaces the catalytic Cys by a serine residue. In human CAD, the predicted catalytic residues are C252 and H336 (Figure 4b). The ammonia, whether hydrolyzed from glutamine or directly captured by the GLN domain is released to the SYN domain, which is organized into a pseudo‐homodimer formed by two homologous phosphorylation domains, an integration domain interconnecting the phosphorylation domains and the GLN moiety, and a C‐terminal regulatory domain (Figure 4a). The phosphorylation domains fold into three lobes that “grasp” the ATP (ATP‐grasp fold). The most N‐terminal domain phosphorylates bicarbonate and contacts the GLN moiety, whereas the most C‐terminal domain phosphorylates carbamate and interacts with the regulatory region. The catalytic residues and divalent cations occupy similar positions in the phosphorylation sites of ecCPS and huCPS‐1 and are also conserved in CPS‐2 (Figure 4c).
FIGURE 4.

The structures of E. coli CPS and human CPS‐1 shed light on the structure of CAD's CPS‐2. (a) Cartoon representation of E. coli CPS heterodimer with the small glutaminase (GLN) subunit colored magenta and the large synthetase (SYN) subunit colored in yellow (phosphorylation domains), blue (integration domain) and green (regulatory domain). The image was composed from different structures of the protein in complex with the indicated compounds that are shown as spheres (PDB ID 1T36, 1C3O, 1JBD). The internal tunnel connecting the active sites is represented as a semitransparent tube. (b–d) Detail of the glutaminase active site (b), the carbamate phosphorylation site (c) and the regulation site (d) in E. coli CPS. The equivalent regions of human CPS‐1 have been superimposed and colored in white. Some important residues are numbered in pink (b), green (d) or yellow (c) for E. coli CPS and gray for human CPS‐1. The equivalent conserved residue in CAD's CPS‐2 are indicated in between parenthesis
As mentioned, CPS‐2, ecCPS and huCPS‐1 are regulated by different allosteric effectors (Figure 2b–d), which bind to the ~20 kDa C‐terminal regulatory domain (Figure 4a,d).34 This region exhibits a similar Rossmann‐fold in ecCPS and huCPS‐1, with a five‐stranded parallel β‐sheet flanked on both side by α‐helices. Interestingly, and despite their different nature, the ecCPS effectors UMP (inhibitor), IMP (activator) and ornithine (activator) and the huCPS‐1 effector NAG bind in a similar position at the carboxy‐edge of the central β‐sheet (Figure 4d), and interact with residues also conserved in CPS‐2 (Figure 4d).
2.4. The CAD puzzle: How to put the pieces together
Knowing that DHO and ATC formed dimers and trimers, respectively, a model was proposed for the architecture of CAD, where the hexamers would actually be “dimers of trimers.”35 According to the model, three CAD polypeptides could engage through their ATC domains forming a trimer, and two of these trimers could further dimerize by association of the DHO domains. A similar model had also been suggested for the Neurospora CAD‐like protein, although dimerization was proposed to occur through the CPS domains.36 The central role of the ATC domain was proven by mutations in the predicted trimer interface, which impede the formation of the hexamers.37 Then, to further validate the “dimer of trimers” model, a construct spanning the DHO and ATC domains of human CAD, including the 96 aa linker, was made and shown to form hexamers in solution.23 In turn, similar DHO‐ATC constructs bearing mutations at the ATC or DHO oligomerization interfaces were shown to assemble into dimers or trimers, respectively. Similar results were obtained for a construct spanning the DHO‐like and ATC domains of C. thermophilum CAD‐like protein.23 Based on these results, we constructed a model for a CAD hexameric particle with D3 symmetry—a threefold axis with three perpendicular twofold axes—where two ATC trimers oriented face‐to‐face occupy apical positions separated by three DHO dimers (Figure 5a). The CPS‐2 could arrange as three dimers surrounding the central DHO‐ATC scaffold. Both ecCPS and huCPS‐1 are known to form dimers in solution, although this does not affect their activity.40, 41 Indeed, both proteins crystallized as dimers, and the dimerization interface was shown to be adjacent to the regulatory domain, as represented in Figure 5a for the putative CPS‐2 dimer.
FIGURE 5.
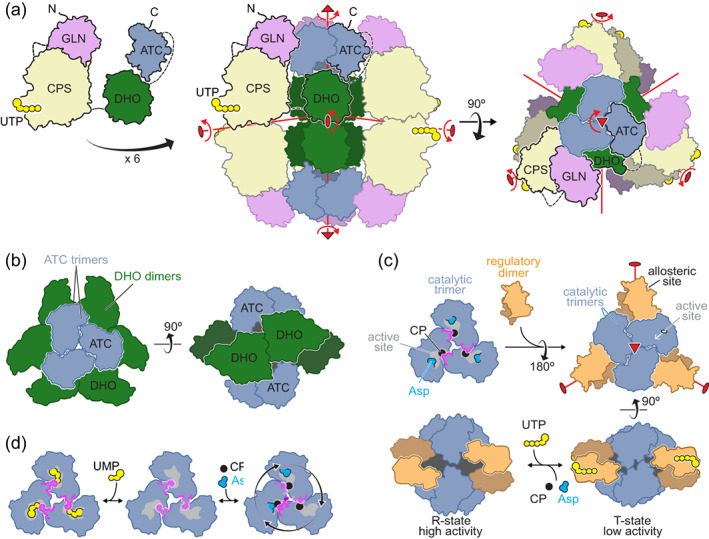
A possible model of CAD hexameric particle. (a) Model proposed for the organization of CAD into a hexameric particle with D3 symmetry. The symmetry axes are indicated in red. (b) Schematic drawing of the dodecameric complex formed between two ATC trimers and three DHO dimers in the bacteria Aquifex aeolicus (PDB ID 3D6N).38 (c) Drawing of the association of the catalytic trimers and regulatory dimers in E. coli ATC (PDB IDs 3CSU, 1D09 and 4FYX).39 (d) Drawing of Arabidopsis ATC trimer with UMP bound at the active site or catalyzing the reaction only in one subunit at a time (PDB IDs 6YPO, 6YY1 and 6YSP).26
While experimental evidence is still awaiting, one can consider how this CAD model agrees with biochemical data, what could be the mechanistic advantages and how it compares to the organization of these enzymatic activities in other organisms.
3. THE ASSEMBLY AND REGULATION OF PYRIMIDINE FACTORIES
The covalent linkage of the three enzymes leading the de novo UMP synthesis might improve the metabolic efficiency of the pathway in a number of ways.6, 42 The production of a single multienzymatic protein grants the coordinated and stoichiometric production of the activities acting consecutively in the pathway and favors their co‐localization within the cell, minimizing the diffusion of metabolic intermediates and the interference with other pathways. Also, proximity might favor the association between the enzymes, perhaps enhancing their stability, and allowing the shuttling of intermediates between active sites. CP is very unstable43 and is partially channeled in CAD and CAD‐like proteins between the CPS‐2 and ATC active sites.44, 45, 46 In the CAD model, the exit of the CPS‐2 tunnel faces the gap between DHO dimers, so that CP could be delivered into the inner space and diffuse a short distance to the ATC trimers (Figure 5a),23, 47 where the high affinity for CP would facilitate its capture.19, 37
The tight association of the enzymatic domains could also favor more complex mechanisms of regulation. It is known that binding of UTP or PRPP to the CPS‐2 regulatory domain affects ATC activity; whereas, binding of PALA to ATC changes the substrate affinity of CPS‐2.48 This “reciprocal allostery” could be explained as the result of concerted movements of the CAD model around the symmetry axes (Figure 5a, red arrows). Similar to the changes induced by NAG in huCPS‐1,31 the binding of effectors to the regulatory domain could induce a reorientation in the CPS‐2 dimer, with concomitant movements of DHO dimers and ATC trimers around and along their symmetry axes; reciprocally, the closure of the ATC subunits upon substrates or PALA binding could translate into movements of the CPS‐2 subunits. These concerted movements could control the affinity for the substrates, the rate and coupling of the reactions and the channeling of reaction intermediates. The equilibrium between different conformational states of the CAD particle could be further modulated by posttranslational modifications (e.g., phosphorylations12, 13, 49, 50) or interactions with other proteins.51
The proposed CAD model shares some features with the association of ATC and DHO in some bacteria. In Aquifex aeolicus, two ATC trimers assemble face‐to‐face with three DHO dimers in a complex with D3 symmetry needed for both enzymes to achieve maximal activity (Figure 5b).38, 52 The DHO subunits dimerize through lateral contacts of the β‐barrel like human DHO, but the subunits are oriented toward opposite ATC trimers favoring the communication between their active sites.53 In other bacteria (e.g., Pseudomonas aeruginosa), a similar association is proposed between ATC trimers and dimers of an inactive DHO that would play a structural role similar to the DHO‐like domain of fungal CAD‐like protein.54
The interaction between ATC and DHO is not mandatory. In some bacteria, like E. coli, two catalytic ATC trimers associate through three dimers of regulatory subunits (Figure 5c), forming a dodecameric holoenzyme with no interaction with DHO.10, 39 Within this complex, the substrate affinity and activity of the ATC trimers is modulated by binding of ATP (activator) or UTP and CTP (inhibitors) to the regulatory dimers (Figures 2b and 5c). The allosteric mechanism involves the rotation and translation of the catalytic trimers and regulatory dimers around and along their symmetry axes, resulting in a conformational equilibrium between active (R‐state) and inactive (T‐state) forms (Figure 5c).39, 55 The E. coli DHO, on the other hand, is an independent homodimer formed by subunits interacting through the loops above the active sites and not by lateral contacts as in human or A. aeolicus DHOs.28, 29 The E. coli DHO structure and dimeric association is shared by the active yeast DHO, which replaces the inactive DHO‐like domain of the CAD‐like protein,56 and is also probably shared by the DHO in plants.20 Intriguingly, in plants, ATC was long known to be a trimer without regulatory subunits but yet still inhibited by UMP (Figure 2c).57 This observation was puzzling, since the isolated bacterial ATC or CAD's ATC domains lack regulatory properties.19 But recently, the crystal structure of Arabidopsis ATC bound to UMP revealed a unique mechanism of regulation, where the nucleotide binds to the active sites rather than to an allosteric pocket, and directly competes with the binding of the substrates (Figure 5d).26
In sum, it is fascinating how homologous enzymes with overall structural similarity and virtually identical active sites combine into assemblies endowed with different regulatory properties (Figure 2b–d). It is even more fascinating to observe how macromolecular complexity builds up from small—sometimes subtle—changes in protein sequence.
4. LOCALIZATION OF CAD IN THE CELL
The architecture is not the only elusive aspect of CAD. Its cellular location has been controversial and attracted much attention. CAD is ubiquitously expressed in all cells (Figure 6a). Early studies using differential centrifugation, located the CPS‐2, ATC and DHO activities exclusively in the microsomal fraction of rat tissues,60 whereas others found a significant activity (20–30%) in crude nuclear fractions of rat ascites cellular carcinoma.61 The use of immunolocalization techniques showed CAD exclusively at the cytoplasm of baby hamster kidney (BHK) cells,62 but other study using antibodies against synthetic CAD peptides found the protein in the nucleus and cytoplasm of BHK cells, and also reported its localization throughout the nuclear matrix and outside the mitochondria of spermatozoa.63 CAD has also been reported in the nuclear matrix of adenovirus‐infected HeLa cells, at sites of active viral replication.64
FIGURE 6.
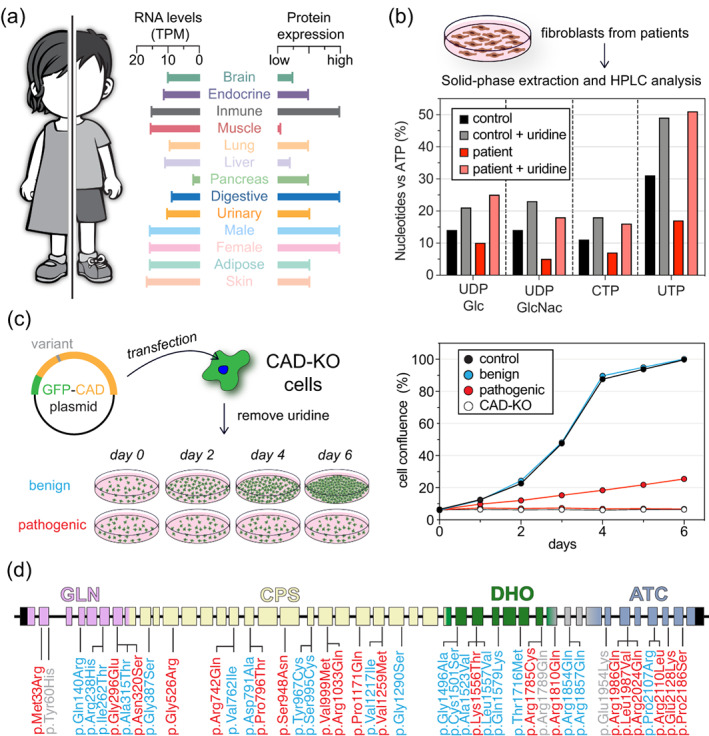
CAD function in health and disease. (a) Expression of CAD in human tissues based on RNA levels (in transcripts per million, TPM) and protein expression data obtained from the Human Protein Atlas database (www.proteinatlas.org). (b) Functional assay to validate the pathogenicity of CAD variants using patient‐derived fibroblast and HPLC analysis of nucleotide levels. The graph is adapted from Koch et al.58 (c) Growth complementation assay using CAD‐knockout (CAD‐KO) cells unable to grow in absence of uridine. Transfection with a functional CAD variant, rescues the growth phenotype in absence of uridine. If transfected cells do not grow, the variant is identified as pathogenic. Graph adapted from del Caño‐Ochoa et al.59 (d) Representation of the 44 exons in the CAD gene, indicating all the clinical variants described up today, colored according to the result of the cellular complementation assay: pathogenic (red), benign (cyan) or not tested (grey)
Parallel studies in yeast, proved by immunocytochemistry and using GFP‐fused constructs, that yeast CAD‐like protein (Ura2) is cytoplasmic and excluded from the nucleus.65 Nonetheless, using a similar GFP‐hybrid, Sigoillot et al. reported the translocation of a significant fraction (20–30%) of hamster CAD to the nucleus during S phase.14, 66 Attracted by the possible function of CAD in the nucleus, our group used GFP‐ and Cherry‐tagged human and hamster CAD hybrids, antibodies against the endogenous protein, life imaging and subcellular fractionation, but repeatedly failed to detect the nuclear translocation of CAD throughout the entire cell cycle.67 Even the addition of a strong nuclear trafficking signal failed to change the cytosolic distribution of the protein. Instead, we found that the antibody used by others recognized at least one nuclear protein other than CAD,67 a result that was confirmed using CAD‐knockout human cells (unpublished), and that reassured our conclusions that CAD localizes exclusively in the cytoplasm.
But the location of CAD is not an ended question. Different groups reported an intriguing punctuated patterns of CAD in the cytoplasm that does not correlate with mitochondria, cytoskeleton, lysosomes or other subcellular structures.14, 50, 62, 67, 68, 69 Remarkably, this dotted distribution is enhanced upon activation of de novo pyrimidine synthesis. In addition to MAPK and PKA phosphorylation, CAD is phosphorylated at the DHO‐ATC linker by the S6 kinase in the downstream pathway of mTORC1.49, 50 The phosphorylation activates CAD and enhances its punctuated distribution. These enlarge CAD foci have been related to protein oligomerization,50 but other factors could be involved, including the interactions with other proteins of the mTORC pathways, like Rheb or mLST8.69, 70 This punctuated CAD pattern resembles the foci observed upon activation of the de novo biosynthesis of purine nucleotides. Under conditions of high purine demand, the six enzymes involved in this pathway (three are multienzymatic like CAD) cluster into a dynamic multienzyme complex named “purinosome,” where they might find a favorable microenvironment for enhanced stability and transfer of intermediates without interference of other metabolic pathways.71 The possibility that CAD assembles into a supramolecular complex or “pyrimidinosome” is of great relevance to understand the regulation of de novo UMP synthesis and deserves further investigation.
5. DEFECTS IN CAD CAUSE A RARE NEUROMETABOLIC DISEASE
Partial or total loss of function of any of the three enzymes involved in de novo pyrimidine biosynthesis cause severe congenital metabolic disorders. UMPS deficiency or orotic aciduria (OMIM #258900) is a rare autosomal recessive disease characterized by excretion of orotic acid, anemia, seizures and intellectual and motor impairment. DHODH defects, on the other hand, cause Miller syndrome (OMIM #263750), a rare malformation disorder with no intellectual damage. However, the fact that no diseases were associated with defects in CAD led to the belief that mutations compromising any of its enzymatic activities would be lethal. Then, in 2015, the first case of a child with a rare inborn error of metabolism and biallelic mutations in CAD was reported,72 and was shortly followed by a second study describing four similar cases from three unrelated families.58 This CAD‐deficiency (OMIM #616457), also known as early infantile epileptic encephalopathy‐50 (EIEE‐50) or CAD congenital disorder of glycosylation, typically manifests at an early age as a progressive refractory epilepsy with loss of skills, global developmental delay, intellectual disability and movement disorders, anemia and abnormal red blood cells.58, 72, 73, 74, 75, 76 The course of the disease can be lethal, with most untreated cases not surviving beyond 4.5–9 years of age.73 However, oral supplementation with uridine or UMP replenish the pyrimidine levels through the salvage pathway (Figure 1) and leads to significant improvement, with immediate cessation of seizures, and progression from a minimally conscious state to communicate and move without assistance after several weeks of treatment.58, 73, 74, 75, 76
An early treatment can prevent irreversible damage,58 but the diagnosis is difficult since there are no biomarkers for screening and symptoms are easily confused or masked by nutritional factors. So far, all 31 CAD‐deficient patients reported worldwide were diagnosed by exome sequencing and validation of CAD variants. However, the large size of CAD and our still limited knowledge about how it functions impose great difficulties to interpret the pathogenic potential of clinical missense mutations.59 To test the functionality of these variants it is possible to use patient‐derived fibroblast and measure the levels of nucleotides and UDP‐sugars (Figure 5b), as well as the incorporation of labelled Asp into DNA and RNA.58, 72 A complementary cellular assay has been developed using human CAD‐knockout cells that only proliferate in media supplemented with uridine or upon transfection with a functional CAD variant (Figure 5c).59 This assay is used to distinguish between “benign” variants that restore cell proliferation in absence of uridine from those pathogenic variants that damage CAD activity and impair de novo pyrimidine synthesis and cell proliferation (Figure 5d).
In the future, the advent of affordable next generation sequencing for genetic testing, the automation of functional assays and an in‐depth knowledge of CAD structure and function will improve the diagnosis and treatment of children affected by this rare neurometabolic disease.
CONFLICT OF INTEREST
The authors declare no competing financial interests.
AUTHOR CONTRIBUTIONS
Francisco del Caño‐Ochoa: Writing ‐ original draft; writing‐review & editing. Santiago Ramón‐Maiques: Funding acquisition; visualization; writing ‐ original draft; writing‐review & editing.
ACKNOWLEDGMENTS
This work was supported by grants from the Spanish Ministry of Science and Innovation (RTI2018‐098084‐B‐100; AEI/FEDER, UE) and from the Fundación Ramón Areces (XX National Call, area of Rare Diseases).
del Caño‐Ochoa F, Ramón‐Maiques S. Deciphering CAD: Structure and function of a mega‐enzymatic pyrimidine factory in health and disease. Protein Science. 2021;30:1995–2008. 10.1002/pro.4158
Funding information Fundación Ramón Areces, Grant/Award Number: XX National Call; area of Rare Diseases; Ministerio de Ciencia e Innovación, Grant/Award Number: RTI2018‐098084‐B‐100; AEI/FEDER; UE
REFERENCES
- 1.Evans DR, Guy HI. Mammalian pyrimidine biosynthesis: Fresh insights into an ancient pathway. J Biol Chem. 2004;279:33035–33038. [DOI] [PubMed] [Google Scholar]
- 2.Löffler M, Fairbanks LD, Zameitat E, Marinaki AM, Simmonds HA. Pyrimidine pathways in health and disease. Trends Mol Med. 2005;11:430–437. [DOI] [PubMed] [Google Scholar]
- 3.Jones ME. Pyrimidine nucleotide biosynthesis in animals: Genes, enzymes, and regulation of UMP biosynthesis. Annu Rev Biochem. 1980;49:253–279. [DOI] [PubMed] [Google Scholar]
- 4.Del Cano‐Ochoa F, Moreno‐Morcillo M, Ramon‐Maiques S. CAD, a multienzymatic protein at the head of de novo pyrimidine biosynthesis. Subcell Biochem. 2019;93:505–538. [DOI] [PubMed] [Google Scholar]
- 5.Zrenner R, Stitt M, Sonnewald U, Boldt R. Pyrimidine and purine biosynthesis and degradation in plants. Annu Rev Plant Biol. 2006;57:805–836. [DOI] [PubMed] [Google Scholar]
- 6.Coleman PF, Suttle DP, Stark GR. Purification from hamster cells of the multifunctional protein that initiates de novo synthesis of pyrimidine nucleotides. J Biol Chem. 1977;252:6379–6385. [PubMed] [Google Scholar]
- 7.Lee L, Kelly RE, Pastra‐Landis SC, Evans DR. Oligomeric structure of the multifunctional protein CAD that initiates pyrimidine biosynthesis in mammalian cells. Proc Natl Acad Sci U S A. 1985;82:6802–6806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Souciet JL, Nagy M, Le Gouar M, Lacroute F, Potier S. Organization of the yeast URA2 gene: Identification of a defective dihydroorotase‐like domain in the multifunctional carbamoylphosphate synthetase‐aspartate transcarbamylase complex. Gene. 1989;79:59–70. [DOI] [PubMed] [Google Scholar]
- 9.French JB, Yates PA, Soysa DR, et al. The Leishmania donovani UMP synthase is essential for promastigote viability and has an unusual tetrameric structure that exhibits substrate‐controlled oligomerization. J Biol Chem. 2011;286:20930–20941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Allewell NM. Escherichia coli aspartate transcarbamoylase: Structure, energetics, and catalytic and regulatory mechanisms. Annu Rev Biophys Biophys Chem. 1989;18:71–92. [DOI] [PubMed] [Google Scholar]
- 11.Yon RJ. End‐product inhibition of aspartate carbamoyltransferase from wheat germ. Biochem J. 1971;121:18P–19P. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carrey EA, Campbell DG, Hardie DG. Phosphorylation and activation of hamster carbamyl phosphate synthetase II by cAMP‐dependent protein kinase. A novel mechanism for regulation of pyrimidine nucleotide biosynthesis. EMBO J. 1985;4:3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Graves LM, Guy HI, Kozlowski P, et al. Regulation of carbamoyl phosphate synthetase by MAP kinase. Nature. 2000;403:328–332. [DOI] [PubMed] [Google Scholar]
- 14.Sigoillot FD, Berkowski JA, Sigoillot SM, Kotsis DH, Guy HI. Cell cycle‐dependent regulation of pyrimidine biosynthesis. J Biol Chem. 2003;278:3403–3409. [DOI] [PubMed] [Google Scholar]
- 15.Collins KD, Stark GR. Aspartate transcarbamylase interaction with the transition state analogue N‐(phosphonacetyl)‐L‐aspartate. J Biol Chem. 1971;246:6599–6605. [PubMed] [Google Scholar]
- 16.Swyryd EA, Seaver SS, Stark GR. N‐(phosphonacetyl)‐L‐aspartate, a potent transition state analog inhibitor of aspartate transcarbamylase, blocks proliferation of mammalian cells in culture. J Biol Chem. 1974;249:6945–6950. [PubMed] [Google Scholar]
- 17.Yoshida T, Stark GR, Hoogenraad J. Inhibition by N‐(phosphonacetyl)‐L‐aspartate of aspartate transcarbamylase activity and drug‐induced cell proliferation in mice. J Biol Chem. 1974;249:6951–6955. [PubMed] [Google Scholar]
- 18.Kempe TD, Swyryd EA, Bruist M, Stark GR. Stable mutants of mammalian cells that overproduce the first three enzymes of pyrimidine nucleotide biosynthesis. Cell. 1976;9:541–550. [DOI] [PubMed] [Google Scholar]
- 19.Ruiz‐Ramos A, Velazquez‐Campoy A, Grande‐Garcia A, Moreno‐Morcillo M, Ramon‐Maiques S. Structure and functional characterization of human aspartate transcarbamoylase, the target of the anti‐tumoral drug PALA. Structure. 2016;24:1081–1094. [DOI] [PubMed] [Google Scholar]
- 20.Grande‐Garcia A, Lallous N, Diaz‐Tejada C, Ramon‐Maiques S. Structure, functional characterization, and evolution of the dihydroorotase domain of human CAD. Structure. 2014;22:185–198. [DOI] [PubMed] [Google Scholar]
- 21.Cheng J‐H, Huang Y‐H, Lin J‐J, Huang C‐Y. Crystal structures of monometallic dihydropyrimidinase and the human dihydroorotase domain K1556A mutant reveal no lysine carbamylation within the active site. Biochem Biophys Res Commun. 2018;505:439–444. [DOI] [PubMed] [Google Scholar]
- 22.Del Cano‐Ochoa F, Grande‐Garcia A, Reverte‐Lopez M, D'Abramo M, Ramon‐Maiques S. Characterization of the catalytic flexible loop in the dihydroorotase domain of the human multi‐enzymatic protein CAD. J Biol Chem. 2018;293:18903–18913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moreno‐Morcillo M, Grande‐Garcia A, Ruiz‐Ramos A, Del Cano‐Ochoa F, Boskovic J, Ramon‐Maiques S. Structural insight into the core of CAD, the multifunctional protein leading de novo pyrimidine biosynthesis. Structure. 2017;25:912–923. [DOI] [PubMed] [Google Scholar]
- 24.Ruiz‐Ramos A, Lallous N, Grande‐Garcia A, Ramon‐Maiques S. Expression, purification, crystallization and preliminary X‐ray diffraction analysis of the aspartate transcarbamoylase domain of human CAD. Acta Cryst F. 2013;69:1425–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Collins KD, Stark GR. Aspartate transcarbamylase. Studies of the catalytic subunit by ultraviolet difference spectroscopy. J Biol Chem. 1969;244:1869–1877. [PubMed] [Google Scholar]
- 26.Bellin L, Del Caño‐Ochoa F, Velázquez‐Campoy A, Möhlmann T, Ramón‐Maiques S. Mechanisms of feedback inhibition and sequential firing of active sites in plant aspartate transcarbamoylase. Nat Commun. 2021;12:947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lallous N, Grande‐Garcia A, Molina R, Ramon‐Maiques S. Expression, purification, crystallization and preliminary X‐ray diffraction analysis of the dihydroorotase domain of human CAD. Acta Cryst F. 2012;68:1341–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thoden JB, Phillips GN, Neal TM, Raushel FM, Holden HM. Molecular structure of dihydroorotase: A paradigm for catalysis through the use of a binuclear metal center. Biochemistry. 2001;40:6989–6997. [DOI] [PubMed] [Google Scholar]
- 29.Lee M, Chan CW, Mitchell Guss J, Christopherson RI, Maher MJ. Dihydroorotase from Escherichia coli: Loop movement and cooperativity between subunits. J Mol Biol. 2005;348:523–533. [DOI] [PubMed] [Google Scholar]
- 30.Huang YH, Huang CY. Creation of a putative third metal binding site in type II dihydroorotases significantly enhances enzyme activity. Protein Pept Lett. 2015;22:1117–1122. [DOI] [PubMed] [Google Scholar]
- 31.de Cima S, Polo LM, Diez‐Fernandez C, et al. Structure of human carbamoyl phosphate synthetase: Deciphering the on/off switch of human ureagenesis. Sci Rep. 2015;5:16950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thoden JB, Holden HM, Wesenberg G, Raushel FM, Rayment I. Structure of carbamoyl phosphate synthetase: A journey of 96 A from substrate to product. Biochemistry. 1997;36:6305–6316. [DOI] [PubMed] [Google Scholar]
- 33.Chaparian MG, Evans DR. The catalytic mechanism of the amidotransferase domain of the Syrian hamster multifunctional protein CAD. Evidence for a CAD‐glutamyl covalent intermediate in the formation of carbamyl phosphate. J Biol Chem. 1991;266:3387–3395. [PubMed] [Google Scholar]
- 34.Liu X, Guy HI, Evans DR. Identification of the regulatory domain of the mammalian multifunctional protein CAD by the construction of an Escherichia coli hamster hybrid carbamyl‐phosphate synthetase. J Biol Chem. 1994;269:27747–27755. [PubMed] [Google Scholar]
- 35.Carrey EA. The shape of CAD. In: Davidson JN, editor. Paths to pyrimidines—an international newsletter. Volume 3. University of Kentucky, 1995; p. 68–72. [Google Scholar]
- 36.Makoff AJ, Buxton FP, Radford A. A possible model for the structure of the Neurospora carbamoyl phosphate synthase‐aspartate carbamoyl transferase complex enzyme. Mol Gen Genet. 1978;161:297–304. [DOI] [PubMed] [Google Scholar]
- 37.Qiu Y, Davidson JN. Substitutions in the aspartate transcarbamoylase domain of hamster CAD disrupt oligomeric structure. Proc Natl Acad Sci U S A. 2000;97:97–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang P, Martin PD, Purcarea C, et al. Dihydroorotase from the hyperthermophile Aquifex aeolicus is activated by stoichiometric association with aspartate transcarbamoylase and forms a one‐pot reactor for pyrimidine biosynthesis. Biochemistry. 2009;48:766–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lipscomb WN, Kantrowitz ER. Structure and mechanisms of Escherichia coli aspartate transcarbamoylase. Acc Chem Res. 2011;45:444–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Anderson PM. Carbamoyl‐phosphate synthetase: An example of effects on enzyme properties of shifting an equilibrium between active monomer and active oligomer. Biochemistry. 1986;25:5576–5582. [DOI] [PubMed] [Google Scholar]
- 41.Lusty CJ. Catalytically active monomer and dimer forms of rat liver carbamoyl‐phosphate synthetase. Biochemistry. 1981;20:3665–3674. [DOI] [PubMed] [Google Scholar]
- 42.Davidson JN, Chen KC, Jamison RS, Musmanno LA, Kern CB. The evolutionary history of the first three enzymes in pyrimidine biosynthesis. Bioessays. 1993;15:157–164. [DOI] [PubMed] [Google Scholar]
- 43.Shi D, Caldovic L, Tuchman M. Sources and fates of carbamyl phosphate: A labile energy‐rich molecule with multiple facets. Biology (Basel). 2018;7:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Christopherson RI, Jones ME. The overall synthesis of L‐5, 6‐dihydroorotate by multienzymatic protein pyr1‐3 from hamster cells. Kinetic studies, substrate channeling, and the effects of inhibitors. J Biol Chem. 1980;255:11381–11395. [PubMed] [Google Scholar]
- 45.Mally MI, Grayson DR, Evans DR. Catalytic synergy in the multifunctional protein that initiates pyrimidine biosynthesis in Syrian hamster cells. J Biol Chem. 1980;255:11372–11380. [PubMed] [Google Scholar]
- 46.Penverne B, Belkaid M, Herve G. In situ behavior of the pyrimidine pathway enzymes in Saccharomyces cerevisiae. 4. The channeling of carbamylphosphate to aspartate transcarbamylase and its partition in the pyrimidine and arginine pathways. Arch Biochem Biophys. 1994;309:85–93. [DOI] [PubMed] [Google Scholar]
- 47.Moreno‐Morcillo M, Ramon‐Maiques S. CAD: A multifunctional protein leading de novo pyrimidine biosynthesis. Encyclopedia of life sciences. John Wiley and Sons, 2017. [Google Scholar]
- 48.Irvine HS, Shaw SM, Paton A, Carrey EA. A reciprocal allosteric mechanism for efficient transfer of labile intermediates between active sites in CAD, the mammalian pyrimidine‐biosynthetic multienzyme polypeptide. Eur J Biochem/FEBS. 1997;247:1063–1073. [DOI] [PubMed] [Google Scholar]
- 49.Ben‐Sahra I, Howell JJ, Asara JM, Manning BD. Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science. 2013;339:1323–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Robitaille AM, Christen S, Shimobayashi M, et al. Quantitative phosphoproteomics reveal mTORC1 activates de novo pyrimidine synthesis. Science. 2013;339:1320–1323. [DOI] [PubMed] [Google Scholar]
- 51.Lindsey‐Boltz LA, Wauson EM, Graves LM, Sancar A. The human Rad9 checkpoint protein stimulates the carbamoyl phosphate synthetase activity of the multifunctional protein CAD. Nucleic Acids Res. 2004;32:4524–4530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Prange T, Girard E, Fourme R, et al. Pressure‐induced activation of latent Dihydroorotase from Aquifex aeolicus as revealed by high pressure protein crystallography. FEBS J. 2019;286:1204–1213. [DOI] [PubMed] [Google Scholar]
- 53.Evans HG, Fernando R, Vaishnav A, et al. Intersubunit communication in the dihydroorotase‐aspartate transcarbamoylase complex of Aquifex aeolicus. Protein Sci. 2014;23:100–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schurr MJ, Vickrey JF, Kumar AP, et al. Aspartate transcarbamoylase genes of pseudomonas putida: Requirement for an inactive dihydroorotase for assembly into the dodecameric holoenzyme. J Bacteriol. 1995;177:1751–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schachman HK. Can a simple model account for the allosteric transition of aspartate transcarbamoylase? J Biol Chem. 1988;263:18583–18586. [PubMed] [Google Scholar]
- 56.Guan H‐H, Huang Y‐H, Lin E‐S, Chen C‐J, Huang C‐Y. Structural basis for the interaction modes of dihydroorotase with the anticancer drugs 5‐fluorouracil and 5‐aminouracil. Biochem Biophys Res Commun. 2021;551:33–37. [DOI] [PubMed] [Google Scholar]
- 57.Yon RJ, Grayson JE, Chawda A. The quaternary structure of wheat‐germ aspartate transcarbamoylase. Biochem J. 1982;203:413–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Koch J, Mayr JA, Alhaddad B, et al. CAD mutations and uridine‐responsive epileptic encephalopathy. Brain. 2017;140:279–286. [DOI] [PubMed] [Google Scholar]
- 59.del Caño‐Ochoa F, Ng BG, Abedalthagafi M, et al. Cell‐based analysis of CAD variants identifies individuals likely to benefit from uridine therapy. Genet Med. 2020;22:1598–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bottomley RH, Lovig CA. Subcellular distribution of rat aspartate carbamoyltransferase. Biochim Biophys Acta. 1967;148:588–590. [DOI] [PubMed] [Google Scholar]
- 61.Shoaf WT, Jones ME. Uridylic acid synthesis in Ehrlich ascites carcinoma. Properties, subcellular distribution, and nature of enzyme complexes of the six biosynthetic enzymes. Biochemistry. 1973;12:4039–4051. [DOI] [PubMed] [Google Scholar]
- 62.Chaparian MG, Evans DR. Intracellular location of the multidomain protein CAD in mammalian cells. FASEB J. 1988;2:2982–2989. [DOI] [PubMed] [Google Scholar]
- 63.Carrey EA, Dietz C, Glubb DM, Loffler M, Lucocq JM, Watson PF. Detection and location of the enzymes of de novo pyrimidine biosynthesis in mammalian spermatozoa. Reproduction. 2002;123:757–768. [PubMed] [Google Scholar]
- 64.Angeletti PC, Engler JA. Adenovirus preterminal protein binds to the CAD enzyme at active sites of viral DNA replication on the nuclear matrix. J Virol. 1998;72:2896–2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Benoist P, Feau P, Pliss A, et al. The yeast Ura2 protein that catalyses the first two steps of pyrimidines biosynthesis accumulates not in the nucleus but in the cytoplasm, as shown by immunocytochemistry and Ura2‐green fluorescent protein mapping. Yeast. 2000;16:1299–1312. [DOI] [PubMed] [Google Scholar]
- 66.Sigoillot FD, Kotsis DH, Serre V, Sigoillot SM, Evans DR, Guy HI. Nuclear localization and mitogen‐activated protein kinase phosphorylation of the multifunctional protein CAD. J Biol Chem. 2005;280:25611–25620. [DOI] [PubMed] [Google Scholar]
- 67.Del Caño‐Ochoa F, Ramón‐Maiques S. The multienzymatic protein CAD leading the de novo biosynthesis of pyrimidines localizes exclusively in the cytoplasm and does not translocate to the nucleus. Nucleosides Nucleotides Nucleic Acids. 2020;39:1320–1334. [DOI] [PubMed] [Google Scholar]
- 68.Brandt J, Wendt L, Bodmer BS, Mettenleiter TC, Hoenen T. The cellular protein CAD is recruited into ebola virus inclusion bodies by the nucleoprotein NP to facilitate genome replication and transcription. Cell. 2020;9:1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sato T, Akasu H, Shimono W, et al. Rheb protein binds CAD (carbamoyl‐phosphate synthetase 2, aspartate transcarbamoylase, and dihydroorotase) protein in a GTP‐ and effector domain‐dependent manner and influences its cellular localization and carbamoyl‐phosphate synthetase (CPSase) activity. J Biol Chem. 2015;290:1096–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nakashima A, Kawanishi I, Eguchi S, et al. Association of CAD, a multifunctional protein involved in pyrimidine synthesis, with mLST8, a component of the mTOR complexes. J Biomed Sci. 2013;20:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pedley AM, Benkovic SJ. A new view into the regulation of purine metabolism: The purinosome. Trends Biochem Sci. 2017;42:141–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ng BG, Wolfe LA, Ichikawa M, et al. Biallelic mutations in CAD, impair de novo pyrimidine biosynthesis and decrease glycosylation precursors. Hum Mol Genet. 2015;24:3050–3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rymen D, Lindhout M, Spanou M, et al. Expanding the clinical and genetic spectrum of CAD deficiency: An epileptic encephalopathy treatable with uridine supplementation. Genet Med. 2020;22:1589–1597. [DOI] [PubMed] [Google Scholar]
- 74.Frederick A, Sherer K, Nguyen L, et al. Triacetyluridine treats epileptic encephalopathy from CAD mutations: A case report and review. Ann Clin Transl Neurol. 2021;8:284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Russo R, Marra R, Andolfo I, et al. Uridine treatment normalizes the congenital dyserythropoietic anemia type II‐like hematological phenotype in a patient with homozygous mutation in the CAD gene. Am J Hematol. 2020;95:1423–1426. [DOI] [PubMed] [Google Scholar]
- 76.Zhou L, Xu H, Wang T, Wu Y. A patient with CAD deficiency responsive to uridine and lietrature review. Front Neurol. 2020;11:5. [DOI] [PMC free article] [PubMed] [Google Scholar]