

Abstract
Intrinsically disordered proteins (IDPs) regularly constitute components of larger protein assemblies contributing to architectural stability. Two small, highly acidic IDPs have been linked to the so‐called PCI complexes carrying PCI‐domain subunits, including the proteasome lid and the COP9 signalosome. These two IDPs, DSS1 and CSNAP, have been proposed to have similar structural propensities and functions, but they display differences in their interactions and interactome sizes. Here we characterized the structural properties of human DSS1 and CSNAP at the residue level using NMR spectroscopy and probed their propensities to bind ubiquitin. We find that distinct structural features present in DSS1 are completely absent in CSNAP, and vice versa, with lack of relevant ubiquitin binding to CSNAP, suggesting the two proteins to have diverged in both structure and function. Our work additionally highlights that different local features of seemingly similar IDPs, even subtle sequence variance, may endow them with different functional traits. Such traits may underlie their potential to engage in multiple interactions thereby impacting their interactome sizes.
Keywords: IDP, interactomes, NMR, proteasome, signalosome, ubiquitin
1. INTRODUCTION
The abundance and relevance of proteins and protein regions without well‐defined three‐dimensional structures, the intrinsically disordered proteins (IDPs)—or regions (IDRs), have been well established in past years.1, 2, 3, 4, 5 This is especially evident in eukaryotes where an estimated 30–40% of the proteome is predicted to be disordered or contain disordered regions.3, 6, 7 IDPs populate ensembles of dynamically interchangeable, disordered conformations, but they still remain fully functional performing various crucial cellular tasks, in for example, regulatory and signaling functions,8 and they are implicated in various diseases like cancers and neurodegenerative disorders.9, 10 Protein–protein interactions are essential for such processes, involving intricate interaction networks where the ability to associate with multiple targets is important. Because IDPs encompass a limited amount of stabilizing hydrophobic interactions, they present a larger exposed surface area with a high rotational freedom resulting in greater conformational flexibility and binding propensity. These properties render them malleable and confer functional advantages, one of which is the ability to bind multiple binding partners.11, 12 Such binding often occurs by exploitation of minor, limited binding sites constituted by small linear motifs (SLiMs).4, 13 The prevalence of disordered proteins in the eukaryotic proteome3, 6, 7 is evidence of their importance in biological processes, but due to their low sequence conservation compared to folded counterparts,14, 15 evolutionary links between different IDPs can be difficult to track and thus often relies on functional analyses.
Deletion of Split‐hand/Split‐foot 1 (DSS1), identified from a gene connected to split‐hand‐split‐foot malformation,16 is a small IDP enriched in acidic residues and highly conserved among eukaryotic species.9, 17 In the yeast Saccharomyces cerevisiae, it was identified as a suppressor of mutants in the exocyst complex and named Suppressor of Exocyst Mutation 1 (Sem1)18; yet this function is not conserved in its fission yeast Schizosaccharomyces pombe (S. pombe) orthologue DSS1/SEM119; from here on only named DSS1. It is an integral subunit of the 26S proteasome, where it functions as a molecular tether promoting assembly of the proteasome (Figure 1a).31, 32, 33 The proteasome lid is one of several homologous complexes that share similar overall structural architecture, harboring the so‐called PCI complexes—named so from the 26S proteasome lid, the COP9 signalosome (CSN), and the eukaryotic translation initiation factor‐3 (eIF3).25 These multiprotein complexes contain subunits with PCI folds, which display a bipartite structure consisting of a structurally conserved C‐terminal winged‐helix domain, and a more divergent N‐terminal domain based on a stack of α‐helices, the number and arrangement of which varies.26 The arrangement of and interactions between PCI‐domains are the structural hallmark of the PCI complex26, 34 (see an overview of the PCI complex subunit composition in Figure 1a).
FIGURE 1.
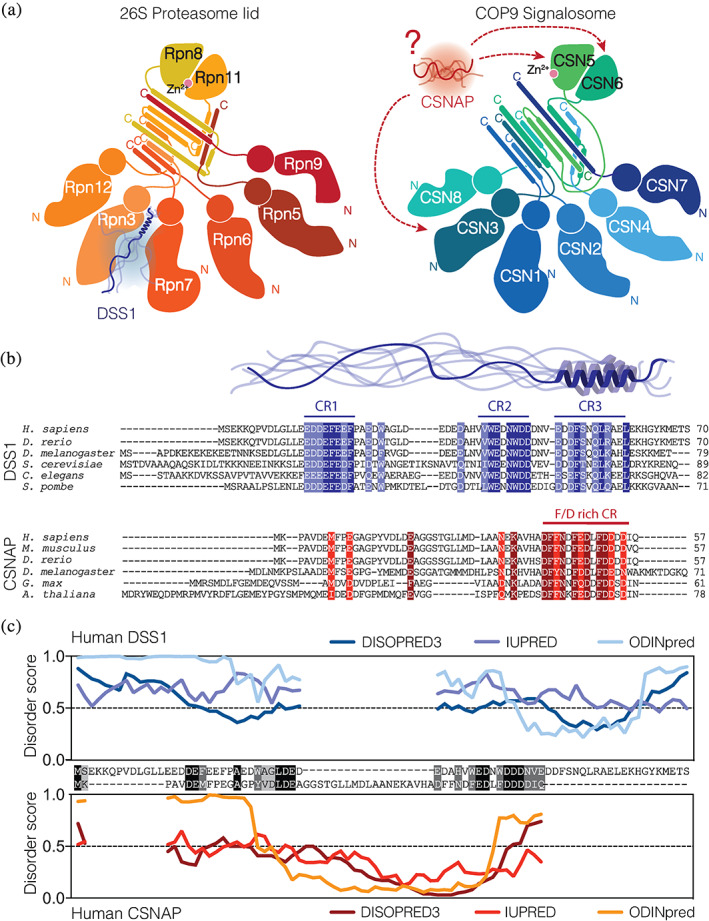
Comparison of the functional propensities of DSS1 and CSNAP. (a) Schematic illustration of the 26S proteasome lid (left) and the COP9 signalosome (right). The subunit composition of the 26S proteasome lid is based on information on the helical bundle20 and cryo‐EM structures.21, 22 The representation of the COP9 signalosome is based on a crystal structure.23 The localization of CSNAP in the PCI complex still remains to be defined, but the known interactions of CSNAP with subunits are indicated with dashed arrows.24 PCI complexes contain six PCI domain subunits that fold into an α‐helical structure topped with winged helix domains (circles) characteristic of the PCI “horseshoe” fold, and by two MNP domains (rounded triangles). The subunits are connected by a helical bundle.25, 26 (b) Multiple sequence alignment of DSS1/SEM1 homologues (blue) and of CSNAP homologues (red) from different eukaryotic organisms. Conserved residues have been shaded (dark, identical; light, conserved) and the conserved regions (CR) indicated in both proteins. (c) Pairwise sequence alignment of human DSS1 and human CSNAP (black, identical; grey, conserved) with a sequence identity and similarity of 17.6% and 26.4%, respectively, as well as the predicted disorder of DSS1 (blue) and CSNAP(red) using IUPRED,27, 28 DISOPRED329 and OdiNPred30
Within the proteasome lid, DSS1 interacts with the two PCI‐domain subunits, RPN3 and RPN7.26, 33 In doing so, it stabilizes the complex.35 In addition, DSS1 interacts with several other PCI‐domain‐containing complexes, including the TREX‐2 complex involved in mRNA export36, 37 and the Csn12‐Thp3 complex, implicated in mRNA splicing.37, 38 Although these binding partners share the presence of PCI folds and therefore are structurally similar, structural data have demonstrated distinct conformations of DSS1 in different PCI domain‐containing protein complexes.17, 33, 36, 39 DSS1 also engages with other proteins and protein complexes that do not have PCI domains, including BRCA239, 40 and the single‐strand DNA binding complex RPA,41, 42 involved in DNA repair. Further, S. pombe DSS1 interacts with ubiquitin independently of its association with the proteasome lid subunits and may serve as a ubiquitin receptor for the proteasome.43
Deduced from sequence analysis,33 and experimentally validated for the S. pombe orthologue by nuclear magnetic resonance (NMR) spectroscopy, free DSS1 is an IDP, with a ~50% transiently populated α‐helix in its C‐terminus.43 Multiple sequence alignments of DSS1 homologues (Figure 1b, top panel) highlight two additional conserved regions characterized by aromatic and hydrophobic residues flanked by several acidic residues. These two regions form extended transient structures in the unbound state in S. pombe DSS1 and both interact with ubiquitin, either with both sites engaging mono‐ubiquitin, or with higher affinity to di‐ubiquitin; correspondingly these sites were dubbed ubiquitin binding site (UBS) 1 and 2.43 The three conserved regions of DSS1 are differentially involved in partner binding. The α‐helix and a region including UBS2 are important for binding Thp1 in the TREX‐2 complex,37, 38 while the stretch containing both UBS1 and the α‐helix is crucial for association with the RPA complex.41 Interestingly, deletion of the α‐helix does not eliminate its multi‐specificity, but modulates ligand preference.42
The COP9 signalosome (CSN) is conserved in eukaryotic cells and acts as a regulator of a large family of E3 ligases called Cullin‐RING ubiquitin ligases (CRL).44, 45 Like the proteasome lid, several CSN subunits contain PCI domains. But, rather than incorporating DSS1, the CSN appears to rely on the DSS1‐like CSN Acidic Protein (CSNAP).24 CSNAP is a 57‐residues paralogue to DSS1, phylogenetically more recent, and only found in metazoans. Multiple alignments of homologous CSNAP sequences from different species (Figure 1b) demonstrate a conserved C‐terminus enriched in Phe and Asp, likely to be important for function. Notably, this region, and especially the Phe residues, were necessary for the incorporation of CSNAP into the CSN through interactions with subunits CSN3, CSN5 and CSN624 (Figure 1a). The absence of CSNAP has global effects on the cell cycle, cell viability and DNA damage response,46 although the exact molecular mechanisms behind these observations remain elusive.
Thus, shown from solved structures by cryo‐EM and by chemical cross‐linking coupled with mass spectrometry, both DSS1 and CSNAP bind PCI domains in various protein complexes.21, 33 However, despite their similarities, DSS1 and CSNAP each interact with different complexes and different PCI‐containing subunits within the respective PCI‐complexes (Figure 1a), suggesting some functional divergence. It therefore remains to be shown whether CSNAP is only a CSN binding protein, or whether its interactome includes PCI‐domain proteins outside of the CSN or even other proteins not adopting the PCI‐fold as observed for the multifunctional DSS1. Both the proteasome and the CSN regulate processes related to ubiquitylation, which might suggest a potential conserved ubiquitin binding ability for CSNAP and DSS1. However, structural details of these human paralogues are lacking. Therefore, to address why two small, very similar, and acidic disordered proteins can have such divergent interactomes, we have undertaken a comparative structural approach. We ask whether the similar architectural role played by CSNAP and DSS1 in PCI complexes is rooted in shared structural properties. We also tested the conservation of ubiquitin binding in human DSS1 and CSNAP independently of their association with respective PCI complexes, and compared this to ubiquitin binding of S. pombe DSS1 previously characterized by NMR spectroscopy.43 Using NMR and circular dichroism (CD) spectroscopy combined with ubiquitin binding assays, we find distinct features present in DSS1 to be completely absent in CSNAP, and vice versa, suggesting that they have diverged in both structure and function. Our work highlights how seemingly similar IDPs have subtle, but important local features that endow them with different functional traits that can be deterministic for their interactomes.
2. RESULTS
2.1. Sequence analyses predict DSS1 and CSNAP as related IDPs
As a first step in our comparison of DSS1 and CSNAP, we noted that both sequences are enriched in acidic and aromatic residues (Figure 1b), but pairwise sequence alignment of the human sequences demonstrated a sequence identity of just 17.6%24 (Figure 1c). The sequences share only one region, which corresponds to the UBS1 mapped in S. pombe DSS143 and which maps to the conserved C‐terminus of CSNAP. The sequence transiently populating an α‐helix in DSS1 appears not to be conserved in CSNAP (Figure 1b,c). However, based on the sequence pattern of Phe and Asp separated by three and four residues, the conserved C‐terminal region in CSNAP was previously suggested to form an amphipathic α‐helix.24
We next generated sequence‐based disorder predictions of human CSNAP and human DSS1 and compared these to their pairwise sequence alignment (Figure 1c). In agreement with previous structural characterization of S. pombe DSS1,43 disorder predictions and published chemical shifts of a longer construct including human DSS1 (BMRB:27475),47 human DSS1 was predicted to be largely disordered with some order in the conserved region from residues D52‐K62 matching to where S. pombe DSS1 has its transient helix (Figure 1c). CSNAP was predicted to be highly disordered except for residues L32‐F51, for which the IUPred and DISOPRED3 scores were lower compared to DSS1. This region contains the sequence motif conserved in DSS1 (Figure 1c). The disorder prediction performed using ODiNPred, a deep neural network predictor based on NMR chemical shifts,30 indicated high levels of disorder outside of the region Y16‐L50, which also includes the sequence stretch disparate from DSS1. Overall, the disorder predictions suggest that CSNAP contains more order than DSS1.
2.2. DSS1 and CSNAP are structurally distinct IDPs
To probe these differences and similarities further, we addressed the disorder experimentally using CD and NMR spectroscopy in both DSS1 and CSNAP. First, to map the average secondary structure content of the two proteins, we recorded far‐UV CD spectra on recombinant human DSS1 and CSNAP. Both showed an intense negative ellipticity with a global minimum just under 200 nm, characteristic of highly unstructured proteins (Figure 2a). Negative ellipticity was also visible as a broad minimum around 220 nm suggesting the presence of transient structures, likely from α‐helices or poly‐proline II (PPII)/extended structures.48, 49, 50, 51 The local minimum of DSS1 was slightly shifted to higher wavelength than CSNAP, and it had a higher residual ellipticity at 190 nm; an indication of a larger content of α‐helix structure in DSS1 than in CSNAP.52, 53, 54
FIGURE 2.
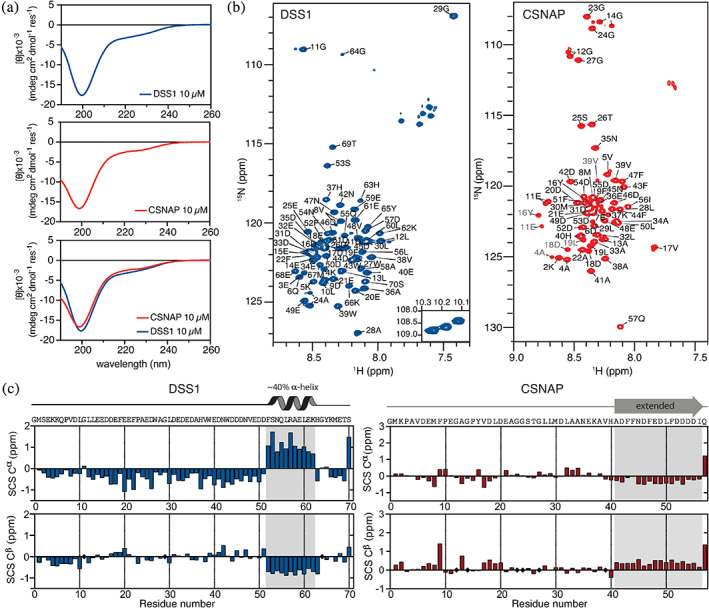
Structural properties of DSS1 and CSNAP. (a) Far‐UV CD spectra of 10 μM DSS1 and CSNAP in 20 mM Na2HPO4/NaH2PO4, pH 7.4, 25°C. The minimum ellipticities are at 199 nm (DSS1) and 198 nm (CSNAP). (b) Assigned 1H‐15N HSQC spectra of 140 μM DSS1 (left, blue) and 210 μM CSNAP (right, red) in 20 mM Na2HPO4/NaH2PO4, 150 mM NaCl, pH 7.4 at 10°C. Assigned residues indicated in grey are from minor populations caused by cis‐trans proline isomerization. (c) Secondary chemical shift analysis of the Cα and Cβ nuclei for DSS1 (left, blue) and CSNAP (right, red). Residues without Cβ are marked with black diamonds. The unassigned non‐native Gly is set to zero in the sequence numeration. Dominant transiently populated structures are indicated above the plots
To further decompose these populations to individual residues, we generated 13C‐15N‐stable isotope‐labelled proteins and assigned the backbone resonance of Cα, Cβ, and carbonyl C′ from sets of triple resonance NMR spectra. For both proteins, the assigned 1H‐15N HSQC spectra (Figure 2b) demonstrated low dispersion in the proton dimension characteristic of disordered proteins, in full accordance with the CD spectra. The peaks were well separated and allowed for assignment of 97% of the expected backbone resonances of DSS1 and 96% for CSNAP. In addition, both DSS1 and CSNAP displayed additional peaks in the 1H‐15N HSQC spectrum, located around prolines (P7–P24 in DSS1 and P3–V6 and P10–L19 in CSNAP), and which were found to arise from minor populations of prolines in cis‐conformations (see Figure S1). The cis populations of P7 and P23 in DSS1 were 10.8% and 13.9%, while for P3, P10, and P15 in CSNAP they were 8.0%, 11.8%, and 22.5%, respectively.
From the assigned backbone resonances recorded at identical conditions, the secondary chemical shifts (SCS) of the Cα and Cβ nuclei of DSS1 and CSNAP, respectively, were calculated using recent random coil chemical shift values55(Figure 2c). The chemical shifts of human DSS1 were recently assigned, but at different conditions and using a longer construct (including 17 N‐terminal non‐native residues).47 However, at pH 7.4, the SCSs were comparable to those previously determined for S. pombe DSS143 as well as to those reported for human DSS1.47 Positive Cα SCS in combination with negative Cβ SCS indicated that human DSS1 had a transiently formed α‐helix, populated to ~40% in the region D52‐K62. This region coincides well with the α‐helix defined in S. pombe DSS1 (F55‐K66), although the helix in human DSS1 is three residues shorter and with smaller SCS, suggesting it to be less populated. Outside the helical region, the Cα SCS of DSS1 were consistently negative throughout the remainder of the sequence, with two minima at around E20 and N42; each one close to the two aromatic and acidic motifs. The lack of corresponding upshifts in the Cβ SCS indicates no β‐strand propensity, but rather the presence of extended structure or PPII.56
The calculated SCS confirmed that CSNAP is largely disordered, as only small fluctuations around zero were observable for the first 45 residues (Figure 2c, right panel). A slight downshift in the Cα SCSs combined with a concomitant upshift of the same magnitude in the Cβ SCS were evident in the remaining C‐terminal stretch. This suggests the presence of a lowly populated β‐strand or extended structure and coincides with the region predicted to be less disordered (Figure 1c). Most importantly, the SCS analysis clearly established that CSNAP does not populate any α‐helical structures. Instead, the conserved F/D‐region in the C‐terminus of CSNAP adopts an extended structure, which distinctly differentiates itself from the rest of the sequence, where no other transiently populated secondary structures could be detected. With aromatic residues flanked by acidic residues, this conserved region has a similar amino acid composition as the UBS1‐region in DSS1. However, they do not show the same SCS profiles, with DSS1 lacking a general upshift in Cβ SCSs. Thus, despite similar overall sequence properties and similar CD spectra profiles, DSS1 and CSNAP have different structural properties, with DSS1 harboring a ~40% populated α‐helix in its C‐terminus and CSNAP a ~25% populated strand‐like, extended structure. The latter structure has similar, but not identical sequence properties to the UBS1 in S. pombe DSS1, which could suggest that CSNAP is able to bind ubiquitin.
Finally, to address their chain dimensions, we used 1H pulse field gradient NMR spectroscopy to measure diffusion coefficients and calculated from this their radius of hydration (R h) using similar data from an internal reference, 1,4‐dioxane. The experiments were conducted at three different temperatures (10, 20, and 37°C) chosen to compare across the triple resonance NMR and far‐UV CD measurements to the physiologically relevant temperature. The R h of DSS1 and CSNAP were 27.4 ± 0.8 Å and 25 ± 3 Å at 10°C, respectively (see Table 1 and Supporting Information). The R h values for DSS1 were largely temperature independent, whereas CSNAP became more compact at higher temperature. Compared to theoretically derived values for random coil chains and for IDPs (Table 1), the measured R h of DSS1 reflected a more expanded chain than for an average IDP. For CSNAP, the R h was at 10°C only slightly larger than the theoretically estimated value, but smaller than the estimated R h IDP at 37°C. Thus, relative to their chain lengths, DSS1 is generally more expanded than CSNAP, with the dimensions of CSNAP being more temperature dependent. The differences in compaction as well as secondary structure elements thus clearly differentiate the two evolutionary related IDPs.
TABLE 1.
Experimentally determined R h values at different temperatures and derived theoretical R h values57 for human DSS1 and CSNAP
| Temperature(°C) | DSS1 R h (Å) | CSNAP R h (Å) |
|---|---|---|
| 10 | 27.4 ± 0.8 | 25 ± 3 |
| 20 | 30 ± 1 | 22 ± 2 |
| 37 | 27.0 ± 0.4 | 19.0 ± 0.7 |
| R h Denat | 24.2 | 21.6 |
| R h IDP | 23.1 | 20.4 |
2.3. Only DSS1 binds ubiquitin
S. pombe DSS1 binds ubiquitin through UBS1 and UBS2, but it remains to be established if human DSS1 and CSNAP also have relevant affinities for ubiquitin. To address this, we first tested ubiquitin binding of human DSS1 and CSNAP by co‐precipitation assays using GST‐tagged DSS1 and CSNAP with 6His‐tagged human ubiquitin. In these assays only DSS1 was able to co‐precipitate ubiquitin (Figure 3a), highlighting that ubiquitin interaction is conserved between DSS1 orthologues and suggesting that any interaction between CSNAP and ubiquitin is weak.
FIGURE 3.
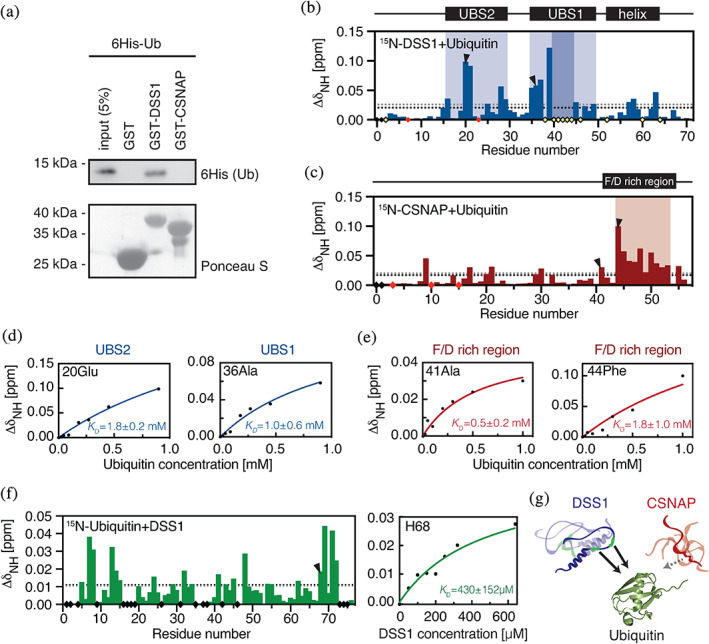
Ubiquitin binding to DSS1 and CSNAP. (a) Ubiquitin co‐precipitation assays. Purified GST‐DSS1, GST‐CSNAP or, as a control, GST alone, was incubated with 6His‐tagged human ubiquitin. After precipitation with glutathione Sepharose beads, the material was analyzed by SDS‐PAGE and western blotting using antibodies to the 6His‐tag on ubiquitin (Ub). Ponceau S staining was used to assess loading. CSP of (b) DSS1 and (c) CSNAP in the presence of 10 times molar excess of ubiquitin with UBS1 and UBS2 in DSS1 indicated in light shaded blue (disappearing residues in darker color) and the weak ubiquitin interaction region in CSNAP in light shaded red. Disappearing signals are displayed as yellow diamonds, unassigned residues as black diamonds and prolines as red diamonds. The black and grey dotted lines correspond to the mean CSP and the standard deviation, respectively. (d) Changes in chemical shift by increasing ubiquitin concentrations for DSS1‐Glu20 (top right) located in the proposed UBS2 and DSS1‐Ala36 (bottom right) in UBS1. The blue lines are global fits of residues within the individual regions. (e) Changes in chemical shifts of CSNAP‐Ala41 and CSNAP‐Phe44 upon increasing ubiquitin concentrations, each demonstrating different binding behavior. The red line corresponds to a global fit of the residues with similar binding saturation (Group1: 41Ala, 45 Asn and 47Phe; Group 2: 44Phe, 46Asp, 48Glu, 50 Leu). (f) CSP of 15N‐ubiquitin in the presence of 8 times molar excess of DSS1 at 25°C. Right panel: CSP of ubiquitin‐His68 at 10°C with different DSS1 concentrations, green line shows the fitted binding curve. (g) Summarizing illustration showing DSS1 interacting with ubiquitin (PDB:1D3Z) through two regions, whereas CSNAP only has very weak affinity for ubiquitin, not detectable in co‐precipitation assay and likely not physiologically relevant
For more details and quantitative assessments of affinity, increasing concentrations of human mono‐ubiquitin were titrated into human DSS1 and CSNAP, and the chemical shift changes followed in a series of 1H‐15N‐HSQC‐based NMR spectra. Chemical shift perturbation (CSP) analyses comparing the peak positions in the free and bound states, showed two regions in human DSS1 to be affected, indicating two ubiquitin binding sites (Figure 3b). The region binding ubiquitin with the highest affinity, UBS1, was identified from a set of disappearing peaks originating from residues V38‐D46, combined with large CSPs from residues D35‐H37 (Figure 3b).43 The conserved UBS1 form an extended structure in the unbound state, known to be further structured upon binding different partners.17 Due to line broadening of the resonances within the key binding site, an accurate K D could not be determined.58 Instead, a global fit of the CSPs of the non‐disappearing neighboring residues at the edges were used to determine a minimum affinity. The residues demonstrated linear behavior upon ubiquitin addition and resulted in an apparent affinity of K Dapp = 1 ± 0.6 mM for UBS1; an affinity comparable to another disordered UBS present in the acidic tail of Cdc34 and determined by NMR spectroscopy to ~1 mM.59 This apparent affinity is weaker than for UBS1 in S. pombe,43 but was extracted from residues on the edge of the binding site. To circumvent this, we instead used 15N‐labelled ubiquitin and titrated in unlabeled human DSS1. Here, binding between 15N‐ubiquitin and DSS1 reached saturation, but slower tumbling at 10°C resulted in significant line broadening of residues involved in the interaction, which made them unsuited for global fit. However, the CSP profile of the bound state of ubiquitin at 25°C (Figure 3f, left panel) directly revealed the main residues involved in DSS1 binding to be Thr7, Leu8, Ile13, Lys48, His68, Leu69, Leu71, and Arg73, which constitute the surface‐exposed, common hydrophobic binding site of ubiquitin. All of these except His68 displayed considerable signal loss at 10°C with increasing DSS1 concentrations, therefore only His68 was used to estimate the binding affinity. Fitting the CSPs of His68 gave a K D of 430 ± 152 μM (Figure 3f, right panel); an affinity similar to many other ubiquitin binding domains.8, 59, 60, 61
In S. pombe, the second site, UBS2 binds ubiquitin much weaker, and in human DSS1, UBS2 was even less well‐defined, with inconsistently dispersed CSPs over the region D16‐G29. Fitting resulted in a very low apparent affinity estimate of K D,USB2 = 1.8 ± 0.2 mM. Upon increasing the ubiquitin concentration further, additional effects became visible outside of the UBS regions. Three residues (Arg57, Ala58, and His63) preceding or within the helical region of DSS1 displayed CSPs above one standard deviation and three others had disappearing signals (Phe52, Leu60, and Gly54). This was not observed in S. pombe 43 and suggests that human DSS1 may engage more surface than UBS1 in ubiquitin binding, although this may be dependent on the used concentrations and conditions.
Titration of CSNAP with ubiquitin resulted in much smaller CSPs and fewer perturbed residues (Figure 3c). Moreover, none of the NMR signals broadened beyond detection and no significant signal intensity loss was observed, supporting the much weaker affinity observed in the pull‐down assay. A larger fraction of the NMR signals from CSNAP appeared completely unaffected by the presence of ubiquitin. The most affected residues involving A41‐D55 were located in the C‐terminus, which formed an extended structure (Figure 2c), and a few residues with CSP > 0.05 ppm were seen dispersed throughout the sequence. Plotting the CSPs as a function of concentration revealed a linear relationship confirming a very weak ubiquitin affinity, and an inaccurate affinity estimation. The most affected residues demonstrated two different behaviors as exemplified by Ala41 and Phe44, respectively (right panel of Figure 3b), further complicating curve fitting. Although binding did not reach saturation, estimated affinities were K D(app) = 0.8 ± 0.2 mM and K D(app) = 2 ± 1 mM (Figure 3e). Combined with the results of the pull‐down assays, the weak affinity for ubiquitin is likely not of biological relevance for CSNAP.
3. DISCUSSION
Two disordered paralogues, the highly acidic proteins DSS1 and CSNAP, have been linked and suggested to have structural and functional similarities. Still, their known interactome sizes differ considerably and data have been lacking to validate their connection. Thus, to gain insight into the evolutionary link between these two disordered PCI‐binding proteins and understand the structural properties that facilitate a vast interactome for a small IDP, a structural and functional comparative study of human DSS1 and the paralogue CSNAP was performed. The sequence conservation of the two eukaryotic IDPs (Figure 1) demonstrated DSS1 to be more conserved than CSNAP, with three highly conserved regions compared to only one in CSNAP. Both proteins are disordered, but have distinct structure propensities that segregate them. In the C‐terminus, human DSS1 adopts an α‐helix coinciding with the α‐helix in S. pombe DSS1, although slightly less populated (~40% compared to ~50% in S. pombe DSS1)43 and shorter. Further, outside this region, a consistent downshift in Cα SCSs combined with Cβ SCSs close to zero in human DSS1 indicated extended structure formation. As this pattern was also detected in S. pombe DSS143 and in a different human DSS1 construct,47 it supports this to arise from extended structure elements in DSS1 homologues. The negative Cα SCSs could indicate the formation of polyproline type II (PPII) structures, a frequently occurring extended structure in IDPs that is hard to detect and quantify.50, 56 In contrast, the C‐terminus of CSNAP encompassing A41‐I56 transiently populates extended structure, likely of a β‐strand nature. Even though DSS1 has negative Cα SCSs, the lack of positive Cβ SCS suggests that the structural properties of DSS1 are different from the ones contributing to the observed SCSs in the C‐terminus of CSNAP. The differences in extended structure might be manifested in the overall protein chain dimension, a notion addressed by measuring their R h values. For IDPs, high net charge may result in more expanded chains due to charge repulsion.57 The obtained R h values (Table 1) indicated that DSS1 is generally more expanded than CSNAP and this coincides with the higher number of net charges per residue (CSNAP:0.25; DSS1:0.31). Further, CSNAP demonstrated more compaction with increasing temperature, not detected for DSS1, but previously observed for other IDPs.62, 63, 64 The structural characteristics of CSNAP are mostly random‐coil with transiently formed extended structure in the C‐terminus, leaving the helix in DSS1 unique and not recapitulated in CSNAP, despite previous suggestions.24 These structural differences hint to distinct behaviors of the two disordered paralogues.
The observed differences could give insight into the structural requirements for the multi‐specificity of DSS1. Until now, CSNAP has only been associated with the subunits of the CSN,24 whereas DSS1 is well known for engaging in several different types of protein complexes.17, 42 The overall structural differences that have been revealed here could be attributes defining why one of these two acidic and disordered, apparently similar, PCI‐binding IDPs, is highly multivalent, while the other appears exclusive to its CSN‐related PCI complex. As the conserved C‐terminus of CSNAP is enriched in aromatic and acidic residues in a pattern similar to the known UBS1 of DSS1, and due to the regulatory function of the CSN in ubiquitylation pathways, we addressed whether CSNAP could interact with ubiquitin. CSPs demonstrated effects in regions of DSS1 similar to the known UBS1 and UBS2 in S. pombe DSS1 (see Figure 3a). The apparent affinities of the UBSs in human DSS1 were lower compared to S. pombe DSS1,43 but in a similar range as other ubiquitin binding motifs.59, 60, 61 In the case of UBS2, the affinity was too low to be accurately determined.58 Because the UBS1 of DSS1 shares sequence similarities with the conserved C‐terminus of CSNAP, this was expected to interact with ubiquitin. Indeed, the NMR titration of CSNAP with ubiquitin confirmed a very weak interaction with the conserved C‐terminus, and much weaker than UBS1 in DSS1. Using pull‐down assays, we could not demonstrate binding of ubiquitin to CSNAP, thus questioning a relevant biological role for the observed low affinity ubiquitin binding.
In spite of the generally lower sequence conservation of IDRs,65 substitution rates of amino acids can vary in IDPs/IDRs due to SLiMs and posttranslational sites like phosphorylation sites.66 In both the UBS1 of DSS1 and the Asp/Phe‐rich C‐terminus of CSNAP, the positions of the aromatics with their adjacent acidic residues are conserved, indicating a shared functional importance. Nevertheless, the aromatic residues clearly distinguish the conserved regions of the two paralogues. The Trp residues in DSS1 are in CSNAP replaced by Phe, which in turn are crucial for its association with the CSN.24 This could indicate that the Trp residues are important for stronger ubiquitin binding, a conclusion supported by our pull‐down assays.
Since the CSN catalyzes de‐neddylation of CRLs, it is possible that CSNAP instead functions as a NEDD8 receptor for the CSN. NEDD8 and ubiquitin have distinct biological functions, but their close homology allows many ubiquitin binding domains to bind both proteins via a conserved hydrophobic patch (Leu8‐Ile44‐His68‐Val70).67, 68, 69 However, due to Arg72 in ubiquitin, and Ala72 in NEDD8, some UBDs can discriminate between ubiquitin and NEDD8.70, 71 The stronger cation‐π electron interaction between Arg and Trp compared to Arg and Phe,72, 73 further supports our suggestion of selectivity in binding between CSNAP and DSS1. In DSS1, acidic and hydrophobic residues interact with the hydrophobic patch on ubiquitin formed by Ile13, Ile44, and Leu69, flanked by two basic regions.43 This region is conserved in NEDD8 and recent work furthermore demonstrate that the catalytic activity of the CSN is not significantly affected by the absence of CSNAP,46 which indicates that NEDD8 interaction is likely irrelevant for CSNAP function in the CSN.
From sequence alignments and the analogous engagement of disordered DSS1 and CSNAP in their respective PCI complexes, it is clear that there is a structural and functional link between the two proteins. Several studies have demonstrated the association of DSS1 in a great number of functionally and structurally distinct protein complexes.17, 42 As CSNAP was only recently discovered and described, its implication in the CSN is not fully understood and CSNAP has not yet been identified in any other protein complexes.24, 46 Nevertheless, the sequence and structural similarities lead to the question of whether CSNAP is multi‐specific like DSS1. The multivalency of DSS1 is related to its disordered characteristics, the presence of several conserved regions and the ability to form a transiently populated α‐helix that can regulate the different interactions and do so additionally by dynamically folding back and shielding the UBS1 region.42 The structural characteristics of the human DSS1 and CSNAP uncovered in this study showed that CSNAP is more featureless, being more randomly disordered and lacking the α‐helix of DSS1, while adopting extended structure only in its C‐terminus. This could indicate that CSNAP may not undergo the same regulation mechanism as DSS1 and indeed be less multi‐specific. Further, as several binding regions in an IDP can facilitate multivalency, the presence of several conserved regions in DSS1 and a large structural versatility versus the single conserved region identified in CSNAP, could suggest its multivalency to be rooted in these. Finally, our work also indicates that commonly conserved regions with slightly different structural propensities and content, still can deviate so much in the strengths of their interactions, that it can affect biological relevance. Here, the change from Phe to Trp leads to interaction rewiring. The implications of this seemingly small change infer that the UBS1 of DSS1 shows propensity for being extended, whereas the same region in CSNAP adopts a strand‐like structure leading also to a much weaker affinity for ubiquitin.
Discerning homology between DSS1 and CSNAP from the primary structure is challenging due to the general low sequence conservation of IDPs compared to folded proteins65 and with a sequence identity of only 17.6%. However, the similar IDP properties and their incorporation into PCI‐complexes, suggest that they likely derived from a common ancestor. In unicellular eukaryotes like yeasts that do not contain CSNAP, the CSN also in general appears simpler and contains fewer subunits.74 Despite apparent similarities between DSS1 and CSNAP, our work highlights clear evolutionary variations distinguishing conformation and interaction specificity, a phenomenon previously studied in folded homologues.75, 76 Among the notable structural differences, the lack of helix structure in CSNAP, and the difference in aromaticity as well as compaction, likely underlie the poor ubiquitin interaction of the conserved C‐terminus of CSNAP. Thus, the ability to bind ubiquitin has been lost in CSNAP. These subtle, but effectual difference suggest that CSNAP might not retain the same multivalent properties featured by DSS1.
4. MATERIALS AND METHODS
4.1. Disorder prediction and sequence alignments
Homologous sequences of DSS1 from H. sapiens (NP_006295.1), D. rerio (NP_955887.1), D. melanogaster (NP_652555.1), S. cerevisiae (NP_010651.3), C. elegans (Q95Y72.2), and S. pombe (NP_594968.1) were used in a multiple sequence alignment using CLUSTAL OMEGA77 which shows the conserved positions and semi‐conserved positions with similar chemical properties. The same was performed for CSNAP with homologous sequences identified using the HMMER software from H. sapiens (NP_001156896.1), M. musculus (NP_001156897.1), D. rerio (NP_957141.2t), D. melanogaster (NP_652383.1), G. max (XP_003555729.1), and A. thaliana (NP_189071.1). Disorder predictions of the human DSS1 and CSNAP sequences were obtained using online predictors: IUPred2,27, 28 DISOPRED329, 78 and ODiNPred.30 The computed disorder scores were displayed in the reading frame of the pairwise sequence alignment obtained with the EMBOSS NEEDLE algorithm.77
4.2. Purification of recombinant human CSNAP and DSS1
Human CSNAP (UniProt: J3QT29) and human DSS1 (UniProt P60896) were expressed from a pD454‐GST vector in E. coli BL21 DE3 using unlabeled as well as 13C,15N labelled M9 media. Expression was induced using 1 mM β‐d‐1‐thiogalactopyranoside (IPTG) for 3 hr at 37°C in M9 media containing 15NH4Cl and 13C6‐glucose. Harvested cells were lysed by French press (American Instrument Company) in 1×PBS buffer pH 7.4 with a Complete Mini Protease Inhibitor Tablets (Roche). Following centrifugation at 20,000g for 20 min at 4°C, the lysate supernatants containing DSS1 or CSNAP were incubated with equilibrated glutathione sepharose 4B fast flow resin (GE healthcare) for 45 min at room temperature. After collecting the flow‐through and washing the resin with 4–5 CV of washing buffer (1×PBS), DSS1 or CSNAP was eluted in two to three fractions of 5 ml with elution buffer (50 mM Tris, 10 mM reduced l‐glutathione, pH 8). This was followed by another wash and the resin was re‐equilibrated with 1×PBS before re‐binding the flow‐through. Fractions containing DSS1 or CSNAP were collected, adjusted to the cleavage buffer (50 mM Tris, 10 mM reduced l‐glutathione, 150 mM NaCl, 1 mM DTT, pH 8), and digested with TEV protease (1:70 substrate to protease ratio) overnight at 4°C on a tilting table. Protease and GST‐tag were removed by temperature denaturation at 72°C for 10 min, followed by centrifugation for 15 min at 4°C 20,000g. Further purification was performed by reversed‐phase HPLC. DSS1 was purified using a SOURCE 15RPC ST 4.6/100 (GE Healthcare) column equilibrated in 50 mM NH4HCO3 and eluted with 50 mM NH4HCO3 70% acetonitrile. The final purification of CSNAP was performed on a Zorbax 300 Å StableBond C18 column (Agilent) equilibrated with Milli Q (MQ) water containing 0.1% TFA and eluted with 70% acetonitrile, 0.8% TFA.
4.3. Purification of ubiquitin
N‐terminally 6×‐His tagged human ubiquitin was expressed from a pET30b vector in E. coli BL21 DE3 cells grown in LB medium. Cells were harvested and lysed by French press (American Instrument Company) in 20 mM Na2HPO4/NaH2PO4, 10 mM imidazole, pH 7.5, and the lysate was poured onto a gravity flow column containing 5 ml equilibrated Ni‐NTA Sepharose 6 Fast Flow resin (Ge Healthcare). The resin was washed with 5 CV 20 mM Na2HPO4/NaH2PO4, 150 mM NaCl 20 mM imidazole pH = 7.5, followed by elution in four fractions of 5 mL with 20 mM Na2HPO4/NaH2PO4, 150 mM NaCl 250 mM imidazole pH = 7.5. Ubiquitin was further purified by size exclusion chromatography on a HPLC Äkta Purifier system (GE Healthcare) using a HiLoad 16 60 Superdex 75 Prep Grade (bed volume 120 ml) column in 20 mM Na2HPO4/NaH2PO4150 mM NaCl pH = 7.4.
4.4. Circular dichroism Sp
The lyophilized DSS1 and CSNAP were resuspended in 20 mM Na2HPO4/NaH2PO4, pH 7.4, and transferred to a rectangular 1 mm quartz SUPRASIL cell (Hellma). Far‐UV CD spectra from 190 nm to 260 nm were recorded on a Jasco J‐810 CD spectropolarimeter at 25°C equipped with a Peltier control. All spectra were acquired with a scan accumulation of 15, data pitch of 0.5 nm, 10 nm/min scanning speed, response time of 2 s and band width of 1 nm. Background spectra of the buffer were recorded identically and subtracted. The spectra were smoothed using the Jasco software.
4.5. NMR spectroscopy
For backbone resonance assignments of DSS1 and CSNAP, triple resonance experiments of 140 μM 15N,13C DSS1 or 210 μM 15N,13C CSNAP, in 20 mM Na2HPO4/NaH2PO4, pH = 7.4, 150 NaCl, 10% (v/v) D2O, 1% (v/v) NaN3, and 1% (v/v) 4,4‐dimethyl‐4‐silapentane‐1‐sulfonic acid (DSS) in a 5 mm Shigemi BMS tube (Bruker). The backbone resonances were assigned from 1H,15N HSQC, and 3D HN(CA)CO, HNCACB, CBCA(CO)NH, and HN(CA)NNH spectra. All 3D spectra were recorded using non‐uniform sampling79, 80 with a 25% data reduction at 10°C on a 600 MHz Bruker spectrometer for DSS1 and an 800 MHz spectrometer for CSNAP. The spectra were reconstructed, and Fourier transformed with qMDD81 and data analysis was performed in CcprNmr analysis.82
4.5.1. Titration experiments
1H,15N HSQC spectra of 90 μM 15N DSS1 or 100 μM 15N CSNAP in 20 mM Na2HPO4/NaH2PO4, 150 mM NaCl, pH 7.2 10% (v/v) D2O, 1% (v/v) NaN3, and 1% (v/v) DSS with increasing ubiquitin concentrations ranging from 0 to 10 times molar excess, were recorded at 10°C on a 600 MHz Bruker spectrometer. 1H,15N HSQC spectra of 40 μM 15N‐labelled human monoubiquitin were recorded in 20 mM Na2HPO4/NaH2PO4, 150 mM NaCl, pH 7.2 10% (v/v) D2O, 1% (v/v) NaN3, and 1% (v/v) DSS at 10°C on a 800 MHz Bruker spectrometer and titrated with increasing Human DSS1 concentrations up to 16 times molar excess and recorded. Due to slower protein tumbling at 10°C and therefore significant line broadening, additional spectra of 40 μM 15N‐ubiquitin alone and in presence of 8 times molar excess of DSS1 were recorded at 25°C and the chemical shifts used to make the CSP plot over the ubiquitin sequence.
4.5.2. Diffusion/Rh measurements
Unlabeled CSNAP or DSS1 in 20 mM Na2HPO4/NaH2PO4, 150 NaCl pH 7.4. 10% D2O 1% NaN3, 0.25 mM DSS. An internal reference 1, 4‐dioxane was added to a concentration of 1 mg/ml. A 1H PFG NMR diffusion experiments (standard Bruker PFG‐LED) were recorded on a 600 MHz magnet at 10, 20, and 37°C. A total of 32 spectra were recorded with gradient strengths varying from 2% to 98% of maximum strength. The used gradient pulse length λ was 3 ms and the diffusion time set to 200 ms. The pseudo‐2D data was Fourier transformed, phased and baseline corrected in Topspin (Bruker), and the peak intensities further analyzed in Dynamics Center (Bruker). Only peaks from 1,4‐dioxane at 3.75 ppm and the region from 2.5 ppm to 0.5 ppm corresponding to the resonances of aliphatic side chains of the protein were included in the analysis. The diffusion constant was obtained by fitting the peak intensities to the Stejskal‐Tanner equation:
where is the intensity, the gradient strength, the gyromagnetic ratio of 1H, is the gradient length, the diffusion time and the diffusion constant. All the fitted diffusion constants for the protein were frequency distributed in a histogram and fitted to a Gaussian function:
where A is the amplitude, μ the mean and the standard deviation. In order to assess the robustness of the extracted R h, different bin sizes were used. The internal reference 1, 4‐dioxane with the known R h of 2.12 Å was used to calculate the R h of the protein with the equation according to Wilkins et al.83
The R h of the sequences was determined theoretically based on the simple power‐law scaling relationship:
where R 0 and v are defined constants, and N correspond to the number of amino acids. Theoretical R h values were calculated based on this relationship using constants fitted to describe proteins under denaturing conditions (R 0 = 2.33 and v = 5.04957) and IDPs. The equation using the equation corrected for net charge and proline contributions to estimate the R h of IDPs:
where P Pro is the fraction of proline residues, |Q| is the net charge of the protein, and S His* is a scaling factor applied if the protein has a poly‐histidine tag and is set to 1 if no tag is present. A–D are constants from the slopes (A and C) and offsets (B and D) of the linear fits between real R h and P Pro or |Q| of different IDPs. They were set to A = 1.24, B = 0.904, C = 0.00759, and D = 0.963, which were the best‐fit parameters for a data set of 32 IDPs.57
4.5.3. Data analysis
Chemical shift of assigned peaks in all the 1H‐15N HSQC spectra were extracted from CcpNmr Analysis, and CSPs of assigned residues for each titration were calculated using the equation:
where and are the chemical shift changes of a residue specific peak in the 1H and 15N dimensions respectively. The value 0.154 is the scaling factor on the N nuclei that balances the changes in chemical shifts between the two nuclei and has been experimentally determined.84
Signals demonstrating a CSP above average plus one standard deviation were taken into consideration for fitting of K D. Peaks in crowded spectral regions, or peaks disappearing were not included in the fit. For the remaining signals K D was obtained from a global fit to the following equation.58
where and are the observed and maximum chemical shift changes in the 1H and 15N dimensions, [P] 0 is the total protein concentration, and [L] 0 the total ligand concentration.
4.5.4. Co‐precipitation assays
GST‐tagged human DSS1 and CSNAP were produced in E. coli BL21(DE3) cells from pGEX‐6P1 (Genscript). Cell lysis, purification, and co‐precipitation assays were performed as described previously.43 The 6His‐tagged human mono‐ubiquitin was purchased from Boston Biochemicals. Following precipitation, the protein was eluted with SDS sample buffer. The samples were resolved by SDS‐PAGE on 15% acrylamide gels and analyzed by Western blotting using penta‐His antibodies (Qiagen, ID: 34660). The secondary antibody was from Dako Cytomation.
4.6. Data deposition
The backbone chemical shifts of human DSS1 and human CSNAP have been deposited in the BioMagResBank under the accession codes 50906 and 50908, respectively.
AUTHOR CONTRIBUTIONS
Sarah F. Ruidiaz: Conceptualization; data curation; formal analysis; investigation; visualization; writing ‐ original draft; writing‐review & editing. Jesper E. Dreier: Formal analysis; investigation. Rasmus Hartmann‐Petersen: Conceptualization; funding acquisition; investigation; resources; supervision; writing‐review & editing. Birthe B. Kragelund: Conceptualization; funding acquisition; investigation; project administration; resources; supervision; validation; writing ‐ original draft; writing‐review & editing.
Supporting information
Appendix S1: Supporting Information.
ACKNOWLEDGMENTS
The authors thank Anne‐Marie Lauridsen and Signe A. Sjørup for expert technical assistance and Andreas Prestel for assistance with NMR. This work was supported by grants from the Danish Council for Independent Research (to R.H.P.: Technology and Production Sciences, Grant No.: 9041‐00062B) and the Novo Nordisk Foundation Challenge program REPIN (to B.B.K. and R.H.P.: #NNF18OC0033926). We thank Villumfonden and Novo Nordisk Fonden for supporting the NMR infrastructure. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Ruidiaz SF, Dreier JE, Hartmann‐Petersen R, Kragelund BB. The disordered PCI‐binding human proteins CSNAP and DSS1 have diverged in structure and function. Protein Science. 2021;30:2069–2082. 10.1002/pro.4159
Funding information Novo Nordisk Fonden, Grant/Award Number: #NNF18OC0033926; Teknologi og Produktion, Det Frie Forskningsråd, Grant/Award Number: 9041‐00062B; Villum Fonden
REFERENCES
- 1.Wright PE, Dyson HJ. Intrinsically unstructured proteins: Re‐assessing the protein structure‐function paradigm. J Mol Biol. 1999;293:321–331. [DOI] [PubMed] [Google Scholar]
- 2.Uversky VN. Natively unfolded proteins: A point where biology waits for physics. Protein Sci. 2002;11:739–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dunker AK, Lawson JD, Brown CJ, et al. Intrinsically disordered protein. J Mol Graph Model. 2001;19:26–59. [DOI] [PubMed] [Google Scholar]
- 4.Van Der Lee R, Buljan M, Lang B, et al. Classification of intrinsically disordered regions and proteins. Chem Rev. 2014;114:6589–6631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dunker AK, Cortese MS, Romero P, Iakoucheva LM, Uversky VN. Flexible nets: The roles of intrinsic disorder in protein interaction networks. FEBS J. 2005;272(20):5129–5148. [DOI] [PubMed] [Google Scholar]
- 6.Ward J, Sodhi J, McGu L, Buxton B, Jones D. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J Mol Biol. 2004;337:35–645. [DOI] [PubMed] [Google Scholar]
- 7.Xue B, Dunker AK, Uversky VN. Orderly order in protein intrinsic disorder distribution: Disorder in 3500 proteomes from viruses and the three domains of life. J Biomol Struct Dyn. 2012;30:137–149. [DOI] [PubMed] [Google Scholar]
- 8.Hicke L, Schubert HL, Hill CP. Ubiquitin‐binding domains. Nat Rev Mol Cell Biol. 2005;6:610–621. [DOI] [PubMed] [Google Scholar]
- 9.Babu MM, van der Lee R, de Groot NS, Gsponer J. Intrinsically disordered proteins: Regulation and disease. Curr Opin Struct Biol. 2011;21:432–440. [DOI] [PubMed] [Google Scholar]
- 10.Gsponer J, Futschik ME, Teichmann SA, Babu MM. Tight regulation of unstructured proteins: From transcript synthesis to protein degradation. Science. 2008;322:1365–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Perovic V, Sumonja N, Marsh LA, et al. IDPpi: Protein‐protein interaction analyses of human intrinsically disordered proteins. Sci Rep. 2018;8:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Teilum K, Olsen JG, Kragelund BB. On the specificity of protein‐protein interactions in the context of disorder. Biochem J. 2021;478:2035–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Olsen JG, Teilum K, Kragelund BB. Behaviour of intrinsically disordered proteins in protein–protein complexes with an emphasis on fuzziness. Cell Mol Life Sci. 2017;74:3175–3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dunker AK, Oldfield CJ, Meng J, et al. The unfoldomics decade: An update on intrinsically disordered proteins. BMC Genomics. 2008;9:S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brown CJ, Johnson AK, Dunker AK, Daughdrill GW. Evolution and disorder. Curr Opin Struct Biol. 2011;21:441–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crackower MA, Scherer SW, Rommens JM, et al. Characterization of the split hand/split foot malformation locus SHFM1 at 7q21.3‐q22.1 and analysis of a candidate gene for its expression during limb development. Hum Mol Genet. 1996;5:571–579. [DOI] [PubMed] [Google Scholar]
- 17.Kragelund BB, Schenstrøm SM, Rebula CA, Panse VG, Hartmann‐Petersen R. DSS1/Sem1, a Multifunctional and Intrinsically Disordered Protein. Trends Biochem Sci. 2016;41:446–459. [DOI] [PubMed] [Google Scholar]
- 18.Jäntti J, Lahdenranta J, Olkkonen VM, Söderlund H, Keränen S. SEM1, a homologue of the split hand/split foot malformation candidate gene Dss1, regulates exocytosis and pseudohyphal differentiation in yeast. Proc Natl Acad Sci U S A. 1999;96:909–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kampmeyer C, Karakostova A, Schenstrøm SM, et al. The exocyst subunit Sec3 is regulated by a protein quality control pathway. J Biol Chem. 2017;292:15240–15253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Estrin E, Lopez‐Blanco JR, Chacón P, Martin A. Formation of an intricate helical bundle dictates the assembly of the 26S proteasome lid. Structure. 2013;21:1624–1635. [DOI] [PubMed] [Google Scholar]
- 21.Schweitzer A, Aufderheide A, Rudack T, et al. Structure of the human 26S proteasome at a resolution of 3.9 Å. Proc Natl Acad Sci U S A. 2016;113:7816–7821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dambacher CM, Worden EJ, Herzik MA, Martin A, Lander GC. Atomic structure of the 26S proteasome lid reveals the mechanism of deubiquitinase inhibition. Elife. 2016;5:e13027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lingaraju GM, Bunker RD, Cavadini S, et al. Crystal structure of the human COP9 signalosome. Nature. 2014;512:161–165. [DOI] [PubMed] [Google Scholar]
- 24.Rozen S, Ben‐dor S, Sharon M, et al. CSNAP is a stoichiometric subunit of the COP9 signalosome. Cell Rep. 2015;13:585–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hofmann K, Bucher P. The PCI domain: A common theme in three multiprotein complexes. Trends Biochem Sci. 1998;23:204–205. [DOI] [PubMed] [Google Scholar]
- 26.Pick E, Hofmann K, Glickman MH. PCI complexes: Beyond the proteasome, CSN, and eIF3 troika. Mol Cell. 2009;35:260–264. [DOI] [PubMed] [Google Scholar]
- 27.Erdős G, Dosztányi Z. Analyzing protein disorder with IUPred2A. Curr Protoc Bioinformatics. 2020;70:e99. [DOI] [PubMed] [Google Scholar]
- 28.Mészáros B, Erdős G, Dosztányi Z. IUPred2A: context‐dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res. 2018;46:329–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jones DT, Cozzetto D. DISOPRED3: precise disordered region predictions with annotated protein‐binding activity. Bioinformatics. 2015;31:857–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dass R, Mulder FAA, Nielsen JT. ODiNPred: comprehensive prediction of protein order and disorder. Sci Rep. 2020;10:14780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bohn S, Sakata E, Beck F, et al. Localization of the regulatory particle subunit Sem1 in the 26S proteasome. Biochem Biophys Res Commun. 2013;435:250–254. [DOI] [PubMed] [Google Scholar]
- 32.Jossé L, Harley ME, Pires IMS, Hughes DA. Fission yeast Dss1 associates with the proteasome and is required for efficient ubiquitin‐dependent proteolysis. Biochem J. 2006;393:303–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tomko RJ, Hochstrasser M. The intrinsically disordered Sem1 protein functions as a molecular tether during proteasome lid biogenesis. Mol Cell. 2014;53:433–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ellisdon AM, Stewart M. Structural biology of the PCI‐protein fold. Bioarchitecture. 2012;2:118–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Funakoshi M, Li X, Velichutina I, Hochstrasser M, Kobayashi H. Sem1, the yeast ortholog of a human BRCA2‐binding protein, is a component of the proteasome regulatory particle that enhances proteasome stability. J Cell Sci. 2004;117:6447–6457. [DOI] [PubMed] [Google Scholar]
- 36.Ellisdon AM, Dimitrova L, Hurt E, Stewart M. Structural basis for the assembly and nucleic acid binding of the TREX‐2 transcription‐export complex. Nat Struct Mol Biol. 2012;19:328–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Faza MB, Kemmler S, Jimeno S, et al. Sem1 is a functional component of the nuclear pore complex‐associated messenger RNA export machinery. J Cell Biol. 2009;184:833–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wilmes GM, Bergkessel M, Bandyopadhyay S, et al. A genetic interaction map of RNA‐processing factors reveals links between Sem1/Dss1‐containing complexes and mRNA export and splicing. Mol Cell. 2008;32:735–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marston NJ, Richards WJ, Hughes D, Bertwistle D, Marshall CJ, Ashworth A. Interaction between the product of the breast cancer susceptibility gene BRCA2 and DSS1, a protein functionally conserved from yeast to mammals. Mol Cell Biol. 1999;19:4633–4642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang H, Jeffrey PD, Miller J, et al. BRCA2 function in DNA binding and recombination from a BRCA2‐DSS1‐ssDNA structure. Science. 2002;297:1837–1849. [DOI] [PubMed] [Google Scholar]
- 41.Zhao W, Vaithiyalingam S, San Filippo J, et al. Promotion of BRCA2‐dependent homologous recombination by DSS1 via RPA targeting and DNA mimicry. Mol Cell. 2015;59:176–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schenstrøm SM, Rebula CA, Tatham MH, et al. Expanded interactome of the intrinsically disordered protein Dss1. Cell Rep. 2018;25:862–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Paraskevopoulos K, Kriegenburg F, Tatham MH, et al. Dss1 is a 26S proteasome ubiquitin receptor. Mol Cell. 2014;56:453–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barth E, Hu R, Baniahmad A, Marz M. The evolution of COP9 signalosome in unicellular and multicellular organisms. Genome Biol Evol. 2016;8:1279–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dubiel W, Chaithongyot S, Dubiel D, Naumann M. The COP9 signalosome: A multi‐DUB complex. Biomolecules. 2020;10:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Füzesi‐Levi MG, Fainer I, Ivanov Enchev R, et al. CSNAP, the smallest CSN subunit, modulates proteostasis through cullin‐RING ubiquitin ligases. Cell Death Differ. 2020;27:984–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stefanovie B, Hengel SR, Mlcouskova J, et al. DSS1 interacts with and stimulates RAD52 to promote the repair of DSBs. Nucleic Acids Res. 2020;48:694–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rucker AL, Creamer TP. Polyproline II helical structure in protein unfolded states: Lysine peptides revisited. Protein Sci. 2002;11:980–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Makowska J, Rodziewicz‐Motowidło S, Bagińska K, et al. Polyproline II conformation is one of many local conformational states and is not an overall conformation of unfolded peptides and proteins. Proc Natl Acad Sci U S A. 2006;103:1744–1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Adzhubei AA, Sternberg MJE, Makarov AA. Polyproline‐II helix in proteins: Structure and function. J Mol Biol. 2013;425:2100–2132. [DOI] [PubMed] [Google Scholar]
- 51.Woody RW. Circular dichroism and conformation of unordered peptides. Adv Biophys Chem. 1992;2:37–79. [Google Scholar]
- 52.Kelly SM, Jess TJ, Price NC. How to study proteins by circular dichroism. Biochim Biophys Acta. 2005;1751:119–139. [DOI] [PubMed] [Google Scholar]
- 53.Greenfield NJ. Using circular dichroism spectra to estimate protein secondary structure. Nat Protoc. 2007;1:2876–2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Drake AF, Siligardi G, Gibbons WA. Reassessment of the electronic circular dichroism criteria for random coil conformations of poly(L‐lysine) and the implications for protein folding and denaturation studies. Biophys Chem. 1988;31:143–146. [DOI] [PubMed] [Google Scholar]
- 55.Kjaergaard M, Poulsen FM. Sequence correction of random coil chemical shifts: correlation between neighbor correction factors and changes in the Ramachandran distribution J Biomol NMR. 2011;50(2):157–165. [DOI] [PubMed] [Google Scholar]
- 56.Treviño MÁ, Pantoja‐Uceda D, Menéndez M, Gomez MV, Mompeán M, Laurents DV. The singular NMR fingerprint of a polyproline II helical bundle. J Am Chem Soc. 2018;140:16988–17000. [DOI] [PubMed] [Google Scholar]
- 57.Marsh JA, Forman‐Kay JD. Sequence determinants of compaction in intrinsically disordered proteins. Biophys J. 2010;98:2383–2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Teilum K, Ben M, Kunze A, Erlendsson S, Kragelund BB. ( S ) Pinning down protein interactions by NMR. Protein Sci. 2017;26:436–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Choi YS, Wu K, Jeong K, et al. The human Cdc34 carboxyl terminus contains a non‐covalent ubiquitin binding activity that contributes to SCF‐dependent ubiquitination. J Biol Chem. 2010;285:17754–17762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cohen A, Rosenthal E, Shifman JM. Analysis of structural features contributing to weak affinities of ubiquitin/protein interactions. J Mol Biol. 2017;429:3353–3362. [DOI] [PubMed] [Google Scholar]
- 61.Hurley JH, Lee S, Prag G. Ubiquitin‐binding domains. Biochemistry. 2006;399:361–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Aznauryan M, Nettels D, Holla A, Hofmann H, Schuler B. Single‐molecule spectroscopy of cold denaturation and the temperature‐induced collapse of unfolded proteins. J Am Chem Soc. 2013;135:14040–14043. [DOI] [PubMed] [Google Scholar]
- 63.Kjærgaard M, Nørholm AB, Hendus‐Altenburger R, Pedersen SF, Poulsen FM, Kragelund BB. Temperature‐dependent structural changes in intrinsically disordered proteins: Formation of α‐helices or loss of polyproline II? Protein Sci. 2010;19(8):1555–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wuttke R, Hofmann H, Nettels D, et al. Temperature‐dependent solvation solvation modulates modulates the the dimensions dimensions of of disordered disordered proteins proteins. Proc Natl Acad Sci U S A. 2019;111:5213–5218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Brown CJ, Takayama S, Campen AM, et al. Evolutionary rate heterogeneity in proteins with long disordered regions. J Mol Evol. 2002;55:104–110. [DOI] [PubMed] [Google Scholar]
- 66.Fahmi M, Ito M. Evolutionary approach of intrinsically disordered CIP/KIP proteins. Sci Rep. 2019;9:1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bandau S, Knebel A, Gage ZO, Wood NT, Alexandru G. UBXN7 docks on neddylated cullin complexes using its UIM motif and causes HIF1α accumulation. BMC Biol. 2012;10:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.den Besten W, Verma R, Kleiger G, Oania RS, Deshaies RJ. NEDD8 links Cullin–Ring ubiquitin ligase function to the p97 pathway. Nat Struct Mol Biol. 2012;19:511–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Oved S, Mosesson Y, Zwang Y, et al. Conjugation to Nedd8 instigates ubiquitylation and down‐regulation of activated receptor tyrosine kinases. J Biol Chem. 2006;281:21640–21651. [DOI] [PubMed] [Google Scholar]
- 70.Shen LN, Liu H, Dong C, Xirodimas D, Naismith JH, Hay RT. Structural basis of NEDD8 ubiquitin discrimination by the deNEDDylating enzyme NEDP1. EMBO J. 2005;24:1341–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shin YC, Chen JH, Chang SC. The molecular determinants for distinguishing between ubiquitin and NEDD8 by USP2. Sci Rep. 2017;7:2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dougherty DA. Cation‐π interactions in chemistry and biology: A new view of benzene, phe, tyr and trp. Science. 1996;271(5246):163–168. [DOI] [PubMed] [Google Scholar]
- 73.Gallivan JP, Dougherty DA. Cation‐π interactions in structural biology. Proc Natl Acad Sci U S A. 1999;96:9459–9464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Qin N, Xu D, Li J, Deng XW. COP9 signalosome: Discovery, conservation, activity, and function. J Integr Plant Biol. 2020;62:90–103. [DOI] [PubMed] [Google Scholar]
- 75.Lite TLV, Grant RA, Nocedal I, Littlehale ML, Guo MS, Laub MT. Uncovering the basis of protein‐protein interaction specificity with a combinatorially complete library. Elife. 2020;9:e60924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Batchelor JD, Doucleff M, Lee CJ, et al. Structure and regulatory mechanism of Aquifex aeolicus NtrC4: Variability and evolution in bacterial transcriptional regulation. J Mol Biol. 2008;384:1058–1075. [DOI] [PubMed] [Google Scholar]
- 77.Madeira F, Park YM, Lee J, et al. The EMBL‐EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019;47:W636–W641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ward JJ, McGuffin LJ, Bryson K, Buxton BF, Jones DT. The DISOPRED server for the prediction of protein disorder. Bioinformatics. 2004;20:2138–2139. [DOI] [PubMed] [Google Scholar]
- 79.Kumar A, Brown SC, Donlan ME, Meier BU, Jeffs PW. Optimization of two‐dimensional NMR by matched accumulation. J Magn Reson. 1991;95:1–9. [Google Scholar]
- 80.Rovnyak D, Sarcone M, Jiang Z. Sensitivity enhancement for maximally resolved two‐dimensional NMR by nonuniform sampling. Magn Reson Chem. 2011;49:483–491. [DOI] [PubMed] [Google Scholar]
- 81.Orekhov VY, Jaravine VA. Analysis of non‐uniformly sampled spectra with multi‐dimensional decomposition. Prog Nucl Magn Reson Spectrosc. 2011;59:271–292. [DOI] [PubMed] [Google Scholar]
- 82.Vranken WF, Boucher W, Stevens TJ, et al. The CCPN data model for NMR spectroscopy: Development of a software pipeline. Proteins. 2005;59:687–696. [DOI] [PubMed] [Google Scholar]
- 83.Wilkins DK, Grimshaw SB, Receveur V, Dobson CM, Jones JA, Smith LJ. Hydrodynamic radii of native and denatured proteins measured by pulse field gradient NMR techniques. Biochemistry. 1999;38:16424–16431. [DOI] [PubMed] [Google Scholar]
- 84.Mulder FA, Schipper D, Bott R, Boelens R. Altered flexibility in the substrate‐binding site of related native and engineered high‐alkaline Bacillus subtilisins. J Mol Biol. 1999;292:111–123. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supporting Information.