

Abstract
Metabolomics is unique among omics technologies in being applicable to metabolism and toxicity studies broadly across organisms (e.g., humans, other mammals, model organisms and even bacteria) and across biological materials (e.g., blood, urine, saliva, biopsy, stool), including cultured cells and subcellular fractions. Metabolomics can be used to characterize biologic response patterns in humans as well as to support mechanistic studies in model systems and ex vivo studies. A broad range of resources are available, including publicly accessible data repositories (e.g., Metabolomics Workbench), tools for biostatistics and bioinformatics (e.g., MetaboAnalyst), metabolite identification (e.g., Metlin) and pathway analysis (e.g., Kyoto Encyclopedia of Genes and Genomes). Thus, metabolomics is more than a promise of the future, metabolomics is already available as a translational approach to facilitate precision medicine. This ACT Symposium review will contain an introduction to metabolomics in toxicity studies followed by sections on translational metabolic networks, translational metabolite biomarkers of acetaminophen-induced acute liver injury, translational framework using high-resolution metabolomics for integrated pharmacokinetics and pharmacodynamics, and precision medicine applications: extracting actionable targets from untargeted metabolomics data following one year in space.
Introduction
The typical metabolomics workflow includes sample collection, sample analysis, bioinformatic data analysis and application.1,2 Study designs can vary; in some cases, for instance, there may be interest in surveillance of use of a specific drug and potential toxic metabolite in a population. Alternatively, sample collection may consist of a baseline pre-treatment sample, followed by samples collected during and possibly after dosing. The samples collected and analyzed in toxicity studies are usually blood or urine, but can also include feces, salvia, breath and tissue. Samples should be collected, stored, and prepared according to standard operating procedures (SOPs) to minimize sample (and analyte) degradation that can lead to experimental variance and false discovery, the latter of which may obscure true biological variance. This preanalytical precision can begin by taking a structured approach to addressing the three major domains where experimental variance is typically introduced: (a) patient/participant, (b) physician/clinician, and (c) laboratory. Each domain has multiple potential contributors to preanalytical variance for which methodical controls must be established. Those elements that cannot be controlled should be annotated, so that they can be modeled if necessary. The next step in the workflow is data acquisition, and many laboratories use non-targeted (global profiling) methods of nuclear magnetic resonance (NMR) spectroscopy or mass spectrometry (MS) to collect the data. Alternatively, targeted metabolic profiling is used when pre-selected chemical classes (e.g., amino acids, bile acids, steroids, or lipids) are evaluated. In either case, standards and quality control measures are required during NMR, LC/MS, or GC/MS data acquisition. The third step is bioinformatic data analyses of NMR or MS data that are used to build models of patient response to a drug or toxicant. Biostatistical methods are used when there is a need to test specific hypotheses. After processing and normalizing the metabolomics data, unsupervised methods such as Principal Component Analysis (PCA) can be used to look for general patterns and outliers in the data. Supervised methods like partial least squares-discriminate analysis (PLS-DA) and artificial neural networks (ANN) can be employed for further data mining. These may be used to formulate a relationship between metabolomic data and other data types including clinical chemistry, histopathology, imaging results and other omics analyses. The application step generally involves some sort of pathway analysis for biological interpretation and discovery of potential absorption, distribution, metabolism and excretion (ADME), predictive or biomarkers of drug or toxicity response. In the use of metabolomics for biomarker discovery, a subsequent study is needed to validate pathways or biomarkers and test hypotheses. Once validated, these pathways or biomarkers can be used in precision medicine.
Response to toxicants
Toxicity is a temporal process that follows the toxicokinetics. In most toxicity studies, the subject is initially in homeostasis before being exposed to the toxicant. Once the toxicant has been added to a system, it enters ADME (absorption, metabolism, distribution and excretion) and the toxin or its reactive metabolites can cause cellular and organ damage via interactions with biomolecules (i.e. proteins, DNA. RNA, lipids, and/or metabolites where it causes a toxicological impact).3 We call this phase 1 of the response to the toxicant. In phase 2, the toxicant initiates a cellular injury, where the cell can try to recover from the injury before overt injury signals occur. If that happens there is adaptation and recovery towards homeostasis. When this happens perturbations in biochemical pathways that are altered without observable injury could be associated mechanisms associated with protection such as glutathione depletion for toxicants that cause production of reactive oxygen species (ROS). If recovery does not occur, phase 3 is organ injury where clinical chemistry biomarkers such as alanine aminotransferase (ALT) for liver toxicity and histopathology are observed. If the organ can repair and protect itself from dysfunction, recovery and adaptation towards healthy homeostatic status can occur. Phase 4 is organ failure for which biomarkers of organ failure and organ recovery from organ injury are needed.
Metabolomics Sample Collection and at Different Phases of Toxicity
Blood or urine samples can be collected from all four phases of the toxicity response of a subject.4 The predose sample can be used to discover biomarkers of susceptibility/risk and prediction of pharmacokinetics. Predose samples can act as personalized control samples. In a longitudinal study, one can collect and analyze samples from all 4 phases of toxicity depending on toxicokinetics and when samples are collected. Longitudinal samples offer the opportunity for personalized medicine by being able to track patient response to therapies in real time. Analysis of phase I samples provide information and biomarkers associated with ADME processes. Analysis of phase 2 samples provide information before organ injury, protection processes and early predictive biomarkers of toxicity. Samples from phase 3 are related to organ Injury and can be compared to clinical chemistry and histopathology biomarkers. Analysis of phase 4 samples provide biomarkers associated with organ failure and recovery from organ failure.
Translational Metabolic Networks
Mechanistic models can help to translate experimental findings between species.5 Large amounts of data are routinely collected during the experimental process of multi-scale omics studies (metabolomics, transcriptomics, proteomics, fluxomics, etc.). In the context of toxicology, experimental data can be combined with mechanistic models to better understand both the response to a compound and the potential species-specific responses. Because mechanistic models serve as a collection of what we know about the underlying biology for species, these models can help interpret the complex changes and provide mechanistic hypotheses for observed changes in diverse data types. For example, a recently published review highlights the utility of large data sets, such as transcriptomics, coupled with mechanistic models to yield unique insights into species-specific differences.5 This coupling aids in the formation of hypotheses that can begin to illuminate the differences seen between rat and human responses to various compounds.
One type of mechanistic model that can be used to yield insight into mechanistic differences between species are genome-scale metabolic network reconstructions (GENREs), which are mathematical representations of a species’ metabolic reactions.6 These models provide a mechanistic representation of metabolism through stoichiometrically balanced reactions and gene-protein-reaction rules governing each metabolic reaction, allowing for the integration of different types of data. For example, transcriptomics and metabolomics data can be integrated with GENREs to generate mechanistic predictions for experimentally observed differences between species.
GENREs Connect Genotype to Phenotype through Metabolic Reactions and Gene Protein Reaction Rules
Genome-scale metabolic network reconstruction (GENRE) connects the genotype to the phenotype through metabolic reactions and gene protein reaction rules.6,7 These models represent the known metabolic reactions of an organism, the metabolites that are converted in those reactions, and the enzymes that catalyze each of these reactions into a mathematical matrix. For example, central carbon metabolism can be used to build a simplified GENRE by representing ATP and glycolysis as shown in Figure 2. The upper middle panel shows a simplified schematic breakdown of glucose to ATP, including glycolysis, the tricarboxylic acid cycle, and the electron transport chain. The resulting mathematical matrix is shown at the bottom of the figure, where each column represents a reaction and each row represents a metabolite in the network. Models capture metabolic reactions in cellular compartments such as in the cytosol ‘[c]’ or the within the mitochondria ‘[m].’ The values within the matrix are indicative of whether the metabolite is being consumed (negative, blue) or produced (positive, red). Overall, constraints can be applied to specific genes, proteins, metabolic reactions, and metabolites of glycolysis, such as ATP production, or protein synthesis, to generate context-specific predictions (lower middle panel).6
Figure 2.
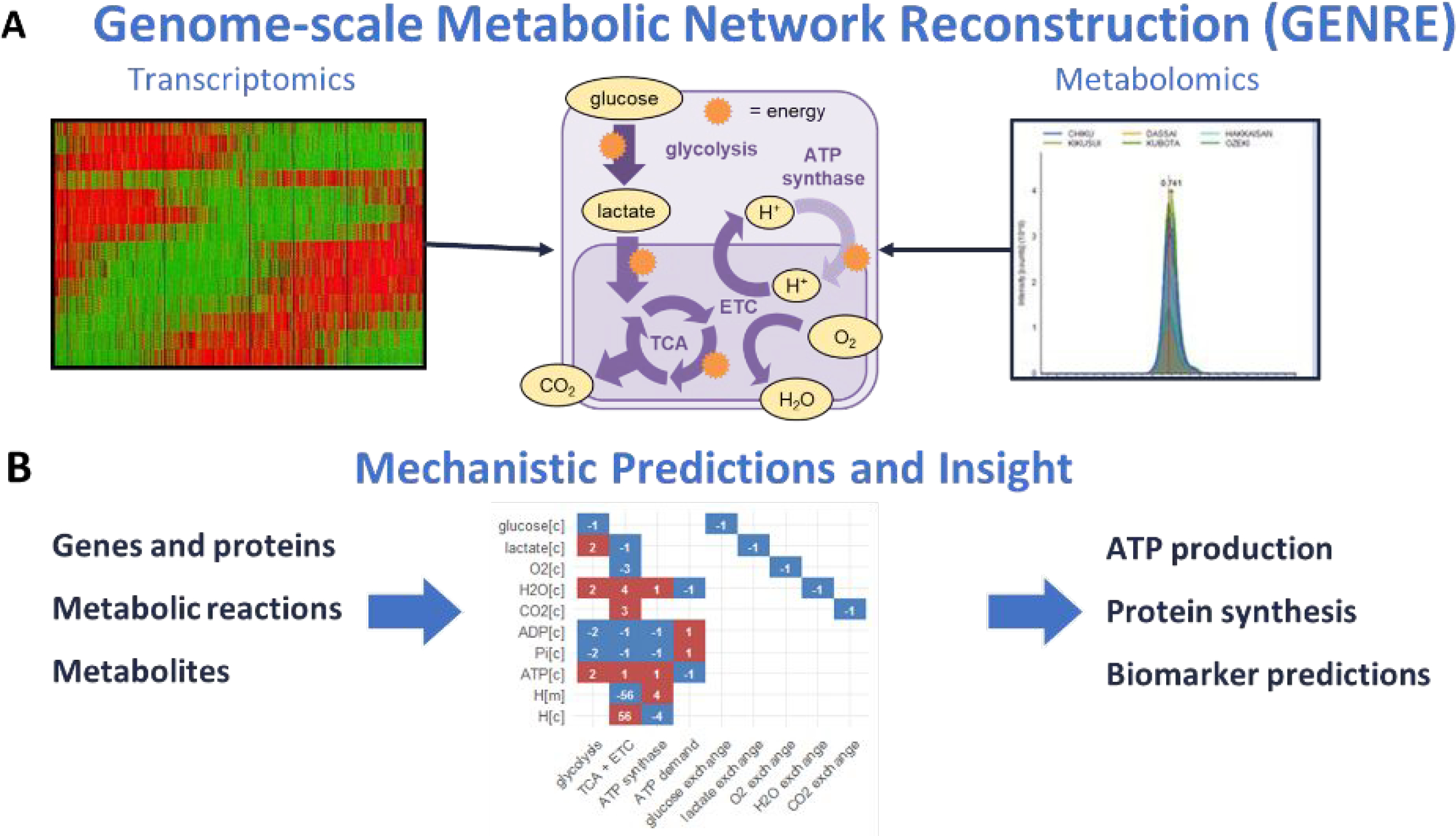
Genome-scale Metabolic Network Reconstruction (GENRE).6,7 Transcriptomics can be integrated with metabolic network reconstructions to turn off reactions which do not have genes expressed or to constrain the amount of flux these reactions can carry based on levels of transcription. Extracellular metabolomics data can be integrated as constraints on metabolites that are brought into the network while intracellular metabolomics data can be used to calculate thermodynamic constraints or place constraints on reactions that consume or produce the metabolite.
Paired Models of Human and Rat Metabolism
Given that rats are commonly used as a surrogate for humans in toxicity studies, we have previously published paired GENREs of human and rat metabolism to allow for species-specific comparisons. To our knowledge, these models represent the first attempt to allow for systems-level metabolic comparisons between rats and humans. As discussed above, GENREs allow for comparisons to be made by integrating context-specific data, such as cells exposed to a variety of compounds in vitro. Using these models, the context-specific data can be integrated and generate context specific predictions for how different species respond.7
The Models Capture Known Functional Differences between rats and Humans
In the process of building these models, it was necessary to incorporate species-specific metabolism, which allowed these models to capture known functional differences between rats and humans, such as differences in vitamin C metabolism, purine metabolism, and sialic acid metabolism. Some metabolic reactions occur only in humans while others occur only in rats. For example, humans do not have the Gulo gene, a gene that is necessary to catalyze the last step of vitamin C synthesis, making vitamin C an essential nutrient for humans. Rats, on the other hand, do have this gene and are able to synthesize vitamin C. This makes rats immune to developing scurvy, while humans can develop scurvy within insufficient dietary vitamin C.
Another example of species-specific metabolism is in the purine degradation pathway, in which inosine and urate are broken down into allantoin. Rats can degrade urate into allantoin which makes rats resistant to the development of Gout. Humans, on the other hand, are unable to metabolize urate, leading to the accumulation of urate and an increased susceptibility to Gout.
Similarly, the model captures species-specific differences in the synthesis of sialic acid. Both rats and humans are able to produce Neu5Ac, but humans cannot synthesize Neu5Gc due to the lack of Cmah gene.7
Models Allow for Integration of Data to Predict Differences between Species
Although these models were developed in parallel, each model still retains species-specific metabolism through not only unique metabolic reactions but also unique gene-protein reactions for each species. The complex relationships between genes and their metabolic reactions were matched between species by using homologous genes, thus, capturing known differences in the number and type of enzymes that catalyze each metabolic reaction. These paired models can then be used to integrate species-specific data for cells exposed to a variety of compounds in vitro, capturing both known differences in metabolism and differences in gene expression Further, the Japanese Toxicogenomic database has species-specific transcriptomic data for approximately 70 compounds which can be integrated with each species-specific model to make predictions for metabolites produced each network.7
Model Predictions Can be Validated Using Targeted Metabolomics Data
By using the Transcriptionally Inferred Metabolic Biomarker Response (TIMBR) algorithm,7 species-specific data can be integrated with the GENREs to make context-specific predictions. This algorithm integrates transcriptomics data as weights on each reaction in the network and then solves for the most likely pathway that produces each metabolite in the network based on the transcriptomics data. These pathways are summarized into production scores. Based on the scores, comparisons between treated and control conditions can be used to identify metabolites with either higher or lower scores when compared to a control model.
For example, TIMBR can be used to integrate human-specific transcriptomics data with the human metabolic network (iHsa) to make context specific predictions (Fig. 3). In this example, metabolite production scores in response to two structurally similar compounds, theophylline (y-axis) and caffeine (x-axis), are compared. Integration of the species-specific data with the species-specific model allows for these computational predictions. The graph shows us that human cells in vitro respond differently to theophylline and caffeine, despite their structural similarities. In rats, similar responses are seen for both compounds, which is demonstrated by the high R2 value as shown in Figure 3.7 Finally, specific metabolites can be selected to experimentally validate the computational predictions using targeted metabolite assays. The metabolites glucose and urea, which were predicted to be differentially produced between rats and humans using the presented model and algorithm, were chosen for validation.
Figure 3.
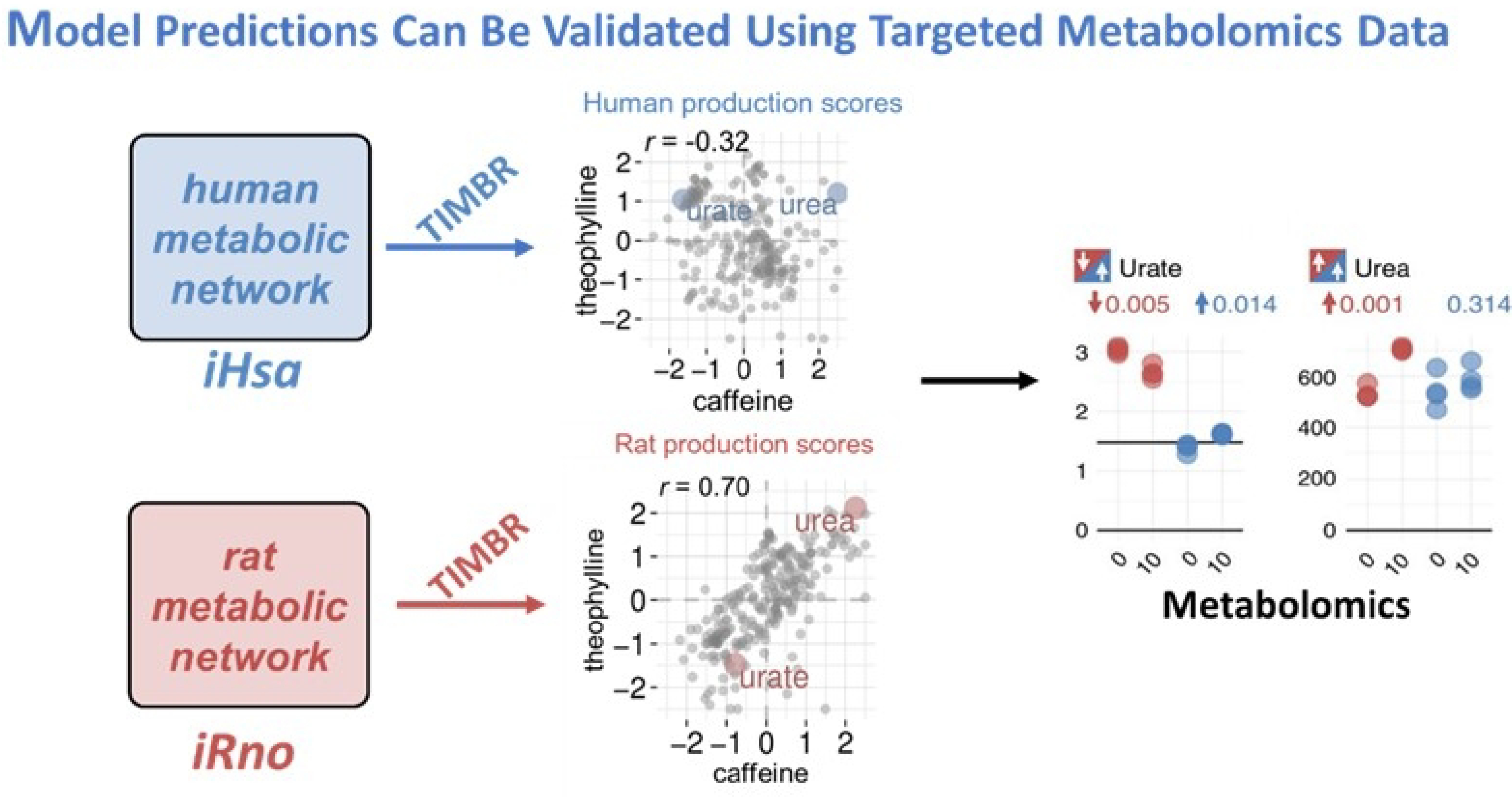
Model Predictions Can be Validated Using Targeted Metabolomics Data. Gene expression data for human hepatocytes treated in vitro with either caffeine or theophylline were integrated with iHsa or iRno to predict changes in the production of metabolites in the network. Although these compounds are structurally similar, we see little correlation between production of metabolites. We can validate select metabolite predictions using metabolomics data and then compare species-specific responses (blue – human, red – rat) to identify metabolites that are differentially produced.7).
It is important to note the layers of complexity that these models can help interpret. First, there are differences in the transcriptomic data. It is well known that differentially expressed genes cannot directly be translated to another species using homologous genes. Second, the complex relationships between changes in gene expression and metabolic function can be captured using GENREs and the presented TIMBR algorithm.7 These approaches were extended to both the liver and kidneys using experimentally collected data on acetaminophen.6,8
Translational Metabolite Biomarkers of Acetaminophen-Induced Acute Liver Injury
Acetaminophen (N-acetyl-para-aminophenol, paracetamol, APAP) is a non-steroidal anti-inflammatory drug (NSAID) because it inhibits cyclooxygenase (COX) pathways but it is mechanistically different from most other NSAIDs in that it does not have anti-inflammatory properties. At therapeutic levels, the safety profile of acetaminophen is good. However, if taken in large amounts APAP can cause severe liver toxicity. In the United States, APAP toxicity is the most common cause of liver transplantation and is the second most common cause worldwide. Further, in 2004 acetaminophen toxicity was responsible for 56,000 emergency department visits, 2600 hospitalizations, and 500 deaths per year in the United States.9
Acetaminophen Metabolism
Initially, acetaminophen (APAP) goes through phase 2 conjugations. Sulfation attaches a sulfate group to the para hydroxyl group, creating APAP-sulfate. The sulfation phase conjugation occurs at a frequency of approximately 25%. Similarly, APAP can undergo glucuronidation, which replaces the hydrogen atom of the para-hydroxyl group with a glucuronide moiety and creates APAP-glucuronide. APAP undergoes glucuronidation with a frequency of 50–70%. Both, APAP-sulfate and APAP-glucuronide are excreted in the urine. Alternatively, if these two pathways are saturated, APAP can go through phase 1 metabolism via the enzymatic action of cytochrome P4502E1. This leads to the formation of the toxic metabolite NAPQI. If NAPQI is not deactivated it can form covalent linkages with proteins and other molecules known as APAP-adducts. NAPQI is deactivated through the action of glutathione (GSH), which covalently bonds to NAPQI at carbon 3 of the benzene ring, forming APAP-GSH. Further processing leads to the truncation and excretion of APAP-GSH as APAP-N-Acetylcysteine (NAC).10
Acetaminophen Toxicity
APAP-induced toxicity causes oxidative stress, reactive oxygen species (ROS) production, glutathione depletion, mitochondrial dysfunction, disruption of energy metabolism, liver necrosis, and alters immune response. APAP overdose is frequently the cause of acute liver failure in the U.S. and is associated with hepatic necrosis. Previous nonclinical reports have illuminated the mechanisms behind acetaminophen toxicity. In 2003, Coen et al. reported disruption of energy metabolism during APAP toxicity.11 Chen and colleagues utilized LC/MS-based metabolomics to profile serum samples from control, APAP-treated wild-type and Cyp2e-1 null mice. They observed the accumulation of long-chain acylcarnitines in the serum of APAP-treated wild-type mice after APAP treatment, consistent with disruption of fatty acid β-oxidation in the mitochondria.12
β-oxidation of Fatty Acids in the Mitochondria
Acylcarnitines are important biomolecules in β-oxidation and assessing overall mitochondrial health. Initially, fatty acids are combined with CoA via fatty acid synthetase creating acyl-CoA. In order to cross the outer mitochondrial membrane into the intermembrane space, fatty acids must first be converted to acylcarnitine. This reaction is catalyzed by carnitine-palmitoyl transferase I (CPT1). Acylcarnitine enters the inner mitochondrial matrix through the action of CPT2. This step is followed by the conversion of acylcarnitine to acyl-CoA via carnitine/acylcarnitine carrier (CAC). When acyl-CoA has been transported to the inner mitochondrial matrix, β-oxidation can commence. If the mitochondria are dysfunctional or injured, acylcarnitine accumulates in the blood. Therefore, increased concentrations of acylcarnitines in circulation, due to the impairment of the mitochondria, can be a biomarker of mitochondrial dysfunction and disruption in energy metabolism.
PLS of Serum Acylcarnitines from Mice Dosed APAP
To highlight the relationship of several serum acylcarnitines following APAP dosing in mice, Bhattacharyya and colleagues utilized a partial least squares regression model. The data showed that long-chain acylcarnitines (i.e., palmitoyl-, oleoyl-, myristoyl-carnitine) are increased at early timepoints and decreased at later timepoints after APAP dosing. In contrast, glutathione and liver weight could be increased at late timepoints and decreased at earlier timepoints. Overall, their data is indicative of these biomarkers being time- and phase-dependent in APAP-induced toxicity.13
ALT and Serum Acylcarnitines Level Changes in APAP-Dosed Mice
Further exploration of the metabolic responses of several biomarkers of toxicity following APAP administration in mice (200 mg/kg) reveal levels of alanine aminotransferase (ALT), an indicator of hepatic injury, begin at lower concentrations and increase significantly above control levels from 4–8 hours. From 8–48 hours, ALT concentrations began to return to baseline levels. In contrast, APAP-adducts in serum increase quickly within the first hour and remain significantly increased compared to controls through 1, 2, and 4 hours. APAP-adduct concentrations begin decreasing after 4 hours, yet remain significantly higher than controls through the entire 48-hour period. Acylcarnitines fall in between ALT and APAP-adducts, slowly rising and reaching their max between 2–4 hours and returning to their normal concentrations by 8 hours post-APAP administration.13
PLS-DA of APAP Overdose in Children
A Partial least squares discriminant analysis model was used to discern the relationship between acylcarnitines, age, and gender by assessing toxicity status of APAP overdose in children via LC/MS. In this study, there were three dose groups, the therapeutic dose, control, and overdose groups. It was found that the area of the loadings associated with the PLS of the overdose group corresponded to long-chain acylcarnitines (i.e., palmitoyl-, oleoyl-, myristoyl-carnitine), age, and gender,14 which is consistent with previous preclinical study.13
Effects of N-acetylcysteine Treatment in Children
The clinical remedy for APAP overdose is N-acetylcysteine (NAC) treatment, which affects the toxicity and dynamics. Bhattacharyya et al.14 assessed how the time to treatment with NAC affected serum ALT and acylcarnitine levels. If NAC treatment was given before 24 hours after APAP overdose, the APAP-adducts, ALT, carnitine, acetyl, and long-chain acylcarnitines have a time-to-max between 24 and 48 hours. Alternatively, if NAC treatment is received after 24 hours, the maximum levels of these biomarkers in serum occurred over a much longer period and in several cases approached 5 days.14 This demonstrates how the administration of NAC has a significant impact on the toxicity cascade of APAP.
Time Responses of Palmitoyl Carnitine vs ALT
Palmitoyl carnitine peaks before ALT peaks in rodents and humans when NAC treatment is delayed. In mice, palmitoyl carnitine (shown in blue in Fig. 4a) increases to a maximum concentration by 4 hours and falls below control levels by 8 hours. ALT (in red) reaches its maximum concentration around 8 hours and returns to baseline by 48 hours. In rats (Fig. 4b), palmitoyl carnitine peaks at 6 hours and returns to baseline by 72 hours. ALT peaks at 24 hours and returns to baseline by 72 hours. In human APAP overdose, with late NAC treatment, palmitoyl carnitine reaches its maximum concentration by 24 hours and returns to normal around 72 hours. The maximum concentration of ALT occurs at 72 hours before returning below the control levels by day 6. This data helps to establish a signature for palmitoyl carnitine, initially rising quickly, then dropping below normal, and ultimately recovering to normal levels. This palmitoyl carnitine signature is seen across species.10
Figure 4.
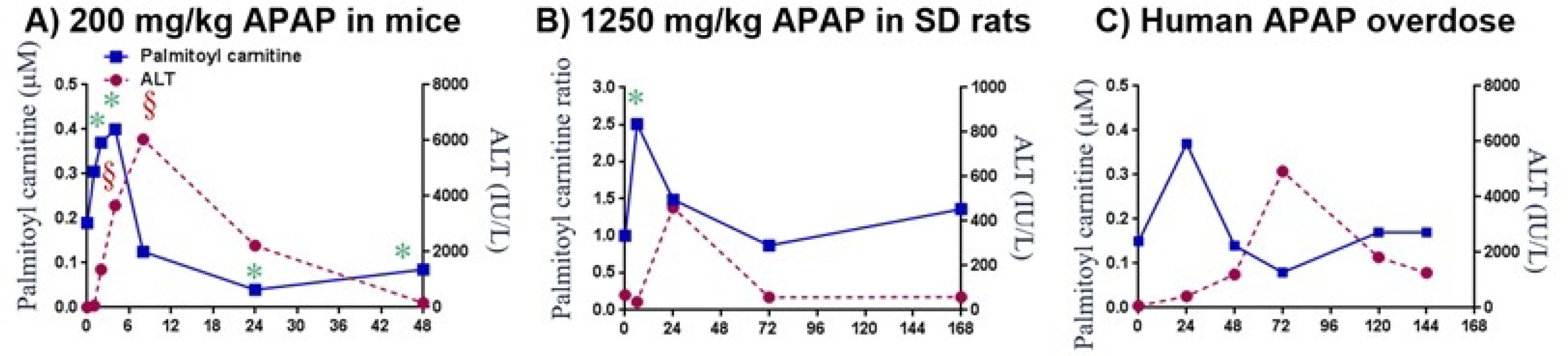
Acetaminophen Hepatotoxicity: Translational Time Response of Palmitoyl Carnitine vs ALT. Plots showing time response of blood palmitoyl carnitine versus ALT in A) mice dosed with 200 mg/kg;13 B) SD rats dosed with 1250 mg/kg APAP;15 C) human APAP overdose patient.10,14
Acylcarnitines Biomarker Conclusions
Several important findings have come out of this research on the responses of rodents and humans to APAP overdose and NAC treatment. (1) Long chain acylcarnitines were increased in rodents and children after APAP overdose, (2) NAC treatment before 24 hours after APAP overdose had shorter time to peak acylcarnitine levels, and (3) increased long chain acylcarnitines, that remain in circulation for a short period of time, are early indicators of reduced β-oxidation and mitochondrial dysfunction.
Previous Reports of Bile Acids in APAP Overdose
Previously reported findings on bile acids have demonstrated their importance as biomarkers of APAP toxicity. Bile acids are metabolized from cholesterol and are involved in many processes in the liver and gastrointestinal tract including maintenance of energy homeostasis, activation of nuclear receptors and cell signaling pathways, cell proliferation and inflammation.16,17 In the 1970s, total bile acids were discovered to be altered in APAP toxicity in the clinical setting. At the time, total bile acid levels were found to be below the sensitivity of available clinical measurement.18–20 Since then, bile acids have been reported as biomarkers of liver injury in many rodent and human clinical studies.21–23
Bile Acids in SD Rat APAP Study
Using Sprague-Dawley rats given a low dose of 100 mg APAP/kg body weight or a high dose of 1250 mg APAP/kg body weight and data from three different MS methods, Sun et al. analyzed the transcriptomic and specific bile acid profiles that participate in the metabolic pathway of bile acids in response to APAP drug-induced hepatotoxicity. Data from all three techniques showed the serum concentration of cholic acid was decreased at 6 hours, significantly elevated at 24 hours, and decreased slightly from its concentration at 24 hours but remained elevated at 72 hours. Deoxycholic acid was only measured by two of the MS methods and showed a similar pattern with a decrease at 6 hours and a significant increase at 24 hours. Glycocholic acid was also significantly elevated at 24 hours across the three MS methods. Increases in levels of bile acids along with bile acid transporter genes, Mdr and Mrp, are consistent with damage-initiated secretion of bile acids.15
Mean Data for Peak ALT and Bile Acids by Therapeutic Group
In a study comparing APAP-adducts and bile acids in children and adolescents with APAP toxicity, James et al. performed three-way and pairwise comparisons of peak serum ALT, APAP-adducts, and bile acid concentrations of the therapeutic exposure group, the healthy control group, and the overdose group. Peak ALT differed significantly between the three groups (p = 0.003). There was a greater difference seen between the therapeutic dose and the overdose groups (p = 0.003) than between the therapeutic group and the control (p = 0.001). The authors noted that peak ALT for the overdose group was not higher than it was for the therapeutic group, which was likely attributable to a large number of patients in the overdose group receiving NAC treatment before developing toxicity.
Utilizing the three-way comparison, significant differences for serum bile acid levels were found for six of the nine bile acids. The largest difference in bile acids between groups was found for the overdose group compared to the healthy control group, shown by the pairwise comparison. When comparing the overdose group to the healthy group, taurodeoxycholic acid (TDC), glycodeoxycholic acid (GDCA), and glycochenodeoxycholic acid (GCDCA) were elevated and taurocholic acid (TCA) and cholic acid (CA) were lower than controls. Alternatively, when the overdose group was compared to the therapeutic group, only chenodeoxycholic acid (CDCA) was significantly higher and TCA and glycocholic acid (GCA) were lower in the overdose group.24
Comparison of Time to APAP-adducts vs. Bile Acids
Next, James et al. compared the time to reach peak bile acid measurements as a function of APAP protein adduct concentration. Liver injury is considered to be fairly severe at concentrations of 1000 ALT IU/I, which equates to approximately 1.0 nmol/mL APAP-adducts. For patients with < 1.0 nmol/mL of APAP-adducts, the time to reach maximal peak measures was shorter (2 days) compared to patients whose APAP-adducts were greater than or equal to 1.0 nmol/mL (time to peak was around 4 days). Correlation analysis revealed that the highest correlation among APAP-adducts and bile acids was for TDCA (R = 0.604; p<0.001), GDCA (R = 0.581; p<0.001), and GCDCA (R = 0.571; p<0.001).24 Using ROC analysis, James et al. found bile acid cut points that allow for the maximal sum of sensitivity and specificity between patients with peak APAP-adducts either greater than or less than 1.0 nmol/mL. The TDCA cut point was 0.56 pM with an area under the curve of 0.92 and a sensitivity/specificity of 0.89/0.84. The GDCA cut point was 2.37 μM with an area under the curve of 0.80 and a sensitivity/specificity of 0.67/0.87.24
Urine – Significant Urine Metabolites correlated to ALT and APAP Adducts
Using NMR and QToF-MS, Schnackenberg et al. found significant urine metabolites correlated to ALT and APAP-induced hepatotoxicity. Early on, APAP-adducts are associated with energy metabolism in the blood. This shows that the disruption of energy metabolism in response to APAP-adducts occurs early. ALT is associated with more markers and may be associated with the gastrointestinal system or possibly the urea cycle. The indoxyl marker is significantly associated with both the ALT and APAP adduct markers. It has been suggested by several groups that indoxyl sulfate is a biomarker of liver injury.25 The MetaboAnalyst pathway analysis of urine metabolites revealed that liver injury is associated with energy and oxidative stress pathways. Urine metabolomics in children with APAP overdose is consistent with reports in non-clinical APAP urine studies that energy and oxidative stress pathways are associated with injury.25
Precision Medicine Applications: Extracting Actionable Targets from Untargeted Metabolomics Data Following One Year in Space
Humans entering the spaceflight environment present a complex set of health, safety, and performance concerns. Current problems encountered in orbital space habitation include space-associated neuro-ocular syndrome, jugular vein stasis, altered bone metabolism, altered muscle metabolism, immune deficits, cognitive changes, and many other side effects. Precision medicine approaches derived from understanding molecular complexity may represent one way to address such concerns.
Translating Molecular Complexity into Actionable Countermeasures
The objective of precision medicine in spaceflight is to identify individual patterns of variance, and with appropriate development, translate that molecular complexity into actionable countermeasures for individual astronauts. This is accomplished via a progression that attempts to 1) describe molecular patterns of variance, 2) describe features (analytes) associated with that variance, 3) describe molecular pathways and network changes, 4) describe biological meaning, 5) identify potential actionable targets (assessment, intervention), and 6) develop countermeasures (or generate new hypothesis for future investigation).
The NASA Twins Study of one year in space was the first multi-scale omics study conducted in space, which was intended to address a gap in our understanding of the molecular dynamics of humans during prolonged space habitation. In this study, one astronaut spent 340 days aboard the International Space Station (ISS), while his identical twin (also an astronaut) remained on Earth. Multiple blood samples were collected (flight = 19; ground = 12) from both subjects beginning approximately 6 months prior to the launch date, during the 340 days aboard the ISS, and 6 months after return. Longitudinal assessments included the genome, epigenome, transcriptome, proteome, metabolome, microbiome, and immunome.26
Untargeted Metabolome Feature Selection: p-Cresol Trajectory after One Year in Space
From the untargeted metabolome data, we identified an elevation of the uremic toxin p-cresol, which is produced by gut microbial fermentation of dietary tyrosine. In addition, we examined some of the associated molecular networks. p-Cresol has adverse metabolic effects via depletion of the hepatic sulfur pool, which impacts metabolism of drugs (acetaminophen, levothyroxine, amoxicillin, morphine, hydrocortisone, etc.), endogenous compounds (estrogens, androgens, catecholamines, bile acids, thyroid hormones, etc.), and xenobiotics. We investigated the potential relevance of these findings to spaceflight) today and in the future.26,27
A comprehensive pre-mission, in-flight, and post-flight analysis of p-cresol glucuronide and p-cresol sulfate was performed (Fig. 5). We found that there are significant elevations in p-cresol glucuronide and p-cresol sulfate concentrations during the first half of the one year in space. Both measures neared control levels by flight day 180.
Figure 5.
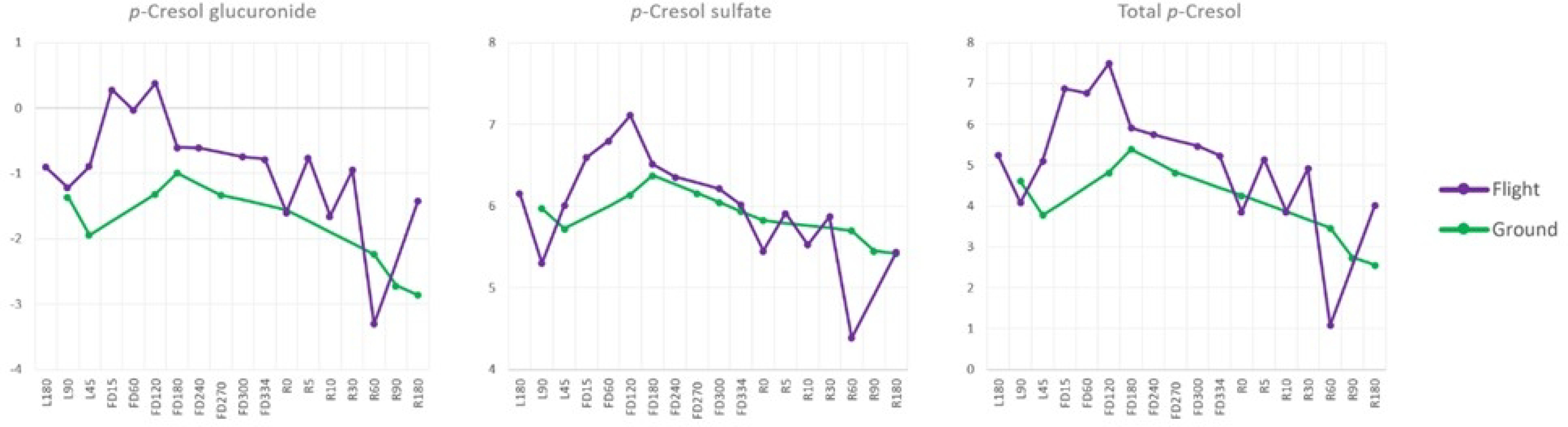
NASA Twins Study: p-Cresol Glucuronide and Sulfate Trajectories. Measurements for p-Cresol glucuronide, p-cresol sulfate, and total p-cresol from 180 days pre-launch (L), in-mission flight days (FD), and 180 days post-mission (return to Earth, R) are shown. In-flight p-cresol measurements (purple) are shown relative to ground control measurements (green). The data shows that both measures, as well as total p-cresol, increased significantly during the initial portion of the in-flight mission when compared to control measures on the ground.
p-Cresol Origins, Metabolism, and Relevance to Drug Metabolism
p-Cresol is generated through a metabolic pathway in which dietary tyrosine is acted upon by particular gut microbes (e.g., clostridium species) and converted into p-cresol. Once p-cresol is created in the gut, it enters the liver and is converted to p-cresol glucuronide and p-cresol sulfate. The relevance of p-cresol to drug metabolism is demonstrated via the metabolic actions following oral APAP loading. The absorption of oral APAP takes place in the small intestine. Once absorbed, APAP is metabolized in the following ways. It can undergo processing via CYP450 2E1, as previously described, or it can undergo sulfonation or glucuronidation.
A classic study conducted by Clayton et al. is instructive, as we work to understand the potential importance of p-cresol production in space. They examined APAP-sulfate, APAP-glucuronide, p-cresol sulfate conjugate, and p-cresol glucuronide conjugate following oral loading of 1000 mg of APAP in humans. It was discovered that individuals with high pre-dose urinary levels of p-cresol had lower post-dose urinary ratios of APAP-sulfate to APAP-glucuronide.28 These reduced levels of APAP-sulfate suggest that p-cresol competes with APAP for the hepatic sulfur groups needed for sulfur conjugation. This further demonstrates the importance of sulfur compounds for the safe metabolism of both p-cresol and APAP. Therefore, adequate sulfur compounds must be made available through dietary sources to replete the sulfur pool when p-cresol and APAP are concomitantly present.
Phase 1 and Phase II Drug Metabolism: From General to Specific (General Biotransformation of APAP)
In phase I drug metabolism, the drug undergoes enzymatic biotransformation to create either electrophiles through oxidation, or nucleophiles through hydrolysis or reduction reactions. The drug can be further metabolized via phase II enzymatic conjugation reactions. For electrophilic drug metabolites, this phase II metabolism occurs though glutathione conjugation that leads to a soluble excretion product. Nucleophilic intermediates can undergo phase II processes, such as sulfation, acetylation, and glucuronidation.
As previously described, APAP can undergo oxidation through CYP450 2E1, generating NAPQI as its primary product. This can lead to covalent adducts and liver injury. However, phase II metabolism leads to modification of NAPQI via glutathione conjugation, which leads to the generation of the safe excretion product NAPQI-GSH. APAP metabolism may also proceed through glucuronidation (UGT; UDP-glycosyltransferase) and conversion to p-cresol sulfate through SULT (sulfotransferase).
Toxicological Concerns in the Spaceflight Environment
A critical feature of the p-cresol-APAP problem in space arises due to the competition of the two metabolic pathways for sulfur groups. For example, when the sulfur pool is low in the presence of APAP, this may lead to the generation of protein adducts, nucleic acid adducts, cell apoptosis, cell necrosis, impaired oxidative phosphorylation, and liver injury. Essentially, the increase in p-cresol that we observed in the space environment may increase the susceptibility to APAP toxicity by reducing available hepatic sulfur.
In addition, metabolism of other drugs and endogenous metabolites may also become compromised in the spaceflight environment. The extent to which elevated p-cresol in space may reduce sulfur groups necessary for metabolism of endogenous compounds has not been studied. Beyond this, p-cresol may produce notable physiologic effects independent of its interaction or competition with drug or endogenous metabolite metabolism.29
Countermeasure Considerations
It must be noted that using untargeted metabolome data from an N=2 study to discuss spaceflight countermeasures related to p-cresol may seem premature. The natural approach would be to use this signal from untargeted data to generate a new hypothesis, which could then be followed up by using targeted methods on future space missions. While this is clearly warranted, we must also understand that spaceflight does not easily provide sufficient cohort numbers to render optimum decision support. Moreover, astronauts have pressing needs to have matters that affect their health, safety, and performance addressed more quickly than is easily achieved in some of the lengthy spaceflight mission architectures.
Given these limitations, it is useful to contemplate potential countermeasures that might be considered for mission application and, further, where such applications might be individualized, based on laboratory analytics (i.e., precision medicine). Several candidates arise as potential countermeasures where precision medicine applications might be used and where such measures could be employed in a pre-mission, in-mission, or post-mission environment.
Among the primary considerations is ensuring the sulfur pool can adequately compensate for the elevations in p-cresol resulting from any medication that may impact it. This might be accomplished through the use of NAC, which is the clinical treatment for APAP toxicity, as it quickly replenishes the sulfur pool. In addition to upregulation of the sulfur pool, NAC also restores glutathione synthesis, allowing NAPQI to undergo glutathione conjugation and be safely excreted following APAP ingestion.30 Administration of oral or intravenous glutathione could potentially also be an effective pre-mission countermeasure.
One mechanism by which elevated p-cresol levels can be effectively reduced is by limiting its production, which can be accomplished through selective dietary modification. For instance, dietary restriction of foods rich in tyrosine may reduce p-cresol levels, as tyrosine is directly converted to p-cresol via microbes in the gut. However, dietary restriction of tyrosine may present an entirely new set of problems in space, due to the effect that reduced protein intake would have on muscle (e.g., leucine, isoleucine, valine), as well as further reducing the intake of sulfur-containing amino acids (e.g., methionine). Additional methods of modification of p-cresol include probiotics and prebiotics (e.g., resistant starch), as these have been shown to modify microbial populations that lead to reduced p-cresol production in humans and animals.27
Translational Framework Using High-Resolution Metabolomics for Integrated Pharmacokinetics and Pharmacodynamics
Metabolome-wide Association study of anti-epileptic drug treatment during pregnancy
Use of metabolic changes associated with anti-epileptic drug use in pregnancy provides examples of additional ways metabolomics can be used for precision medicine. The integration of targeted and non-targeted pharmacokinetic and pharmacodynamic analysis linked to specific external exposures can be utilized to develop outcome measures, such as biomarkers of risk and health outcomes. Walker et al. used metabolome-wide association to study anti-epileptic drug treatment during pregnancy. Women with epilepsy are especially vulnerable during pregnancy and require continuous anti-epileptic drug treatment to avoid injury to themselves and the developing embryo and fetus. However, the reality of prolonged treatment of pregnant women with anti-epileptic drugs presents a risk to the fetus as well. Understanding the drug-metabolome associations can be useful to improve drug selection and dosing to maximize the benefits and minimize the risks. 31
MWAS study of lamotrigine
In this study, plasma blood samples were collected for pregnant women with epilepsy. Of the samples collected, 39 samples were from women receiving Lamotrigine monotherapy and 10 samples were collected from women receiving no treatment. As can be seen in Figure 6A, there are a large number of mass spectral signals significantly associated with lamotrigine dose measured values in plasma. Hierarchical cluster analysis effectively separates individuals taking lamotrigine monotherapy from individuals who were not receiving any treatment (Fig. 6B). Metabolic associations show very significant associations with lamotrigine metabolites, as well as a variety of other significant metabolic associations.31 Although correlations do not establish causation, observed metabolic pathway associations with antiepilepsy drugs are supported by prior clinical research and animal model studies for changes in neurosteroids (progesterone and 3β-androstanediol), metabolites related to neurotransmitters (tyrosine, tryptophan, biopterin), folate metabolism [5-methyltetrahydrofolate (5-MTHF) and tetrahydrofolate (THF)], and purine metabolism [inosine, uric acid, 1-(5-phospho-D-ribosyl)-5-amino-4-imidazolecarboxylate, deoxyadenosine, deoxyguanosine 5’-triphosphate].
Figure 6.
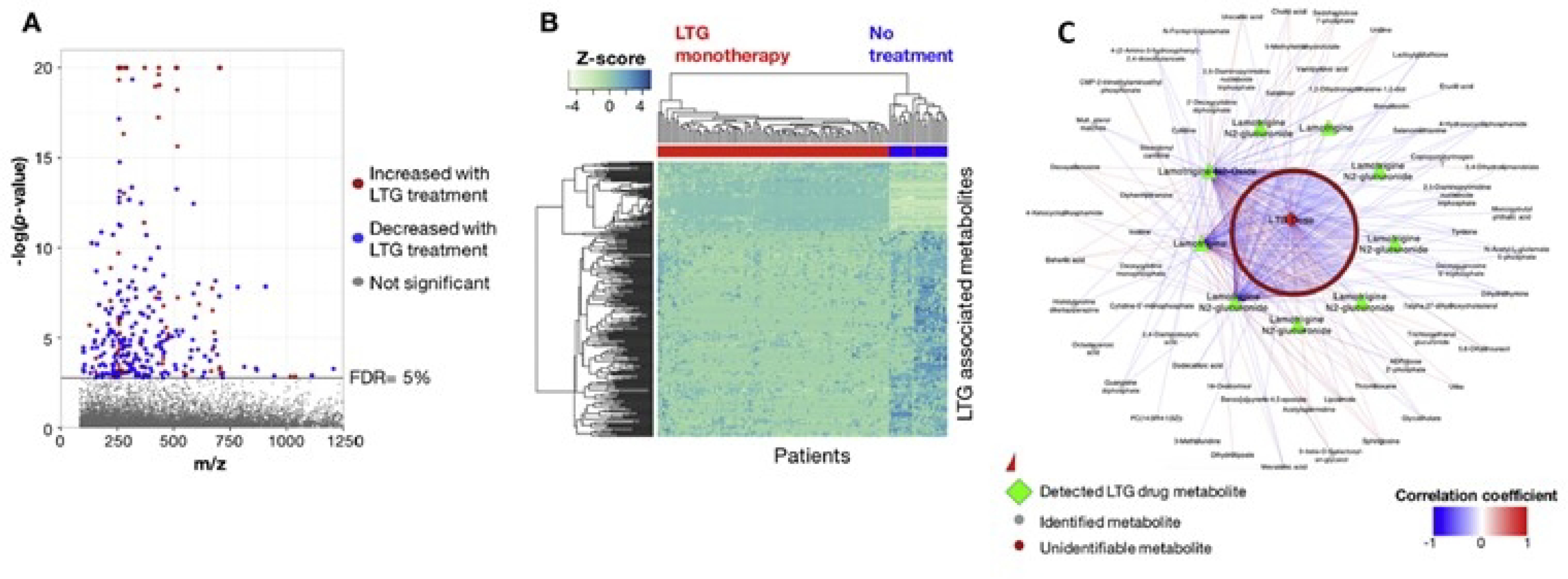
Metabolome-Wide Association Study (MWAS) of Lamotrigine. A) Manhattan plot of −log p for m/z features associated with Lamotrigine therapy; B) Unsupervised hierarchical clustering of m/z features from Manhattan plot clustered in both the metabolite and patient dimensions; C) Both identified metabolites and unidentified m/z features associated with Lamotrigine.31
Comparisons of Drug Responses can help guide therapeutics
Comparing responses of various similar drugs can help guide therapeutics. In this same study, lamotrigine-associated pathways and levetiracetam-associated pathways were compared. The results showed that some of the pathways are the same. However, it is not currently understood whether this is good or bad because these shared pathways could be indicators of common beneficial effects or common adverse effects. If a large enough population has been studied and associations with adverse outcomes have been found, such an approach may reveal associations with specific pathways. Therefore, discriminatory pathways that differ between drugs can potentially be used for diagnostic methods to identify adverse outcomes.31
If the pathway effects are linked to birth defects, then metabolic patterns could be used to adjust dose or switch therapies, possibly before or during pregnancy. When lamotrigine is given as a therapeutic, folate metabolism and methionine salvage pathways are altered in a dose-dependent manner. When levetiracetam is given, it alters butyrate lipoate and mevalonate metabolism. Although this type of analysis has not yet been developed for any therapeutic applications, understanding the metabolic effects associated with drug responses can help guide their therapeutic approach.31
Lowest hanging fruit for precision therapeutics involves dose optimization
Dose optimization is an essential aspect involved in precision therapeutics. This principle is illustrated by an in vitro cell study in which cells in culture were exposed to increasing manganese doses from a physiologically important range up through a toxic range. Results showed that adaptive responses were separated from toxic responses in a variety of different ways. By measuring cell death several days after treatment, we were able to show that early metabolic changes could be used to classify toxicity based on subsequent cell death as an outcome. By using an interaction of the metabolome and transcriptome, clear separation of adaptive and toxic responses was obtained. Therefore, from a basic scientific standpoint, it is appropriate to apply this type of approach at the human level to separate adaptive metabolic responses from toxic metabolic responses. Furthermore, once the pathways are defined, this creates additional levels of understanding. It is possible to use this approach starting in pre-treatment and increasing the dose, then taking sequential measurements of the individual to determine which dose is producing the desired metabolic outcome. In parallel, sequential measurements will show at which doses adverse metabolic responses begin to occur.32
Mechanistic links through integrated omics
This integrated omics approach can be extended to include redox proteomics along with metabolomics and transcriptomics and further separate physiologic network responses from toxicological network responses. In an SH-SY5Y cell culture study, transcriptomic, metabolomic, and redox proteomic measurements were taken to assess the differences between the physiologic network responses and the toxicological network responses in mitochondria. Cells were incubated for 5 hours at physiological manganese (Mn) concentrations (≤10 μM) or toxicological Mn concentrations (50 and 100 μM) and returned to normal culture medium and measured again at 24 and 48 hours. Exposure to higher concentrations of Mn resulted in higher cell death than physiological concentrations. The network structures at physiologic levels of Mn included > 280 metabolites and higher numbers of transcripts as well as redox proteins. Cells exposed to toxicological Mn only produced 80 metabolites and relatively few transcripts and redox proteins. This study showed cells were able to adapt under physiologic Mn concentrations but toxicological concentrations disrupted metabolic connectivity. Further detailed analysis of these network structures provided insights into associated mechanisms and metabolic pathways. It is evident that this multiscale omics approach could yield enhanced capabilities of evaluation and provide beneficial mechanistic knowledge in human studies in response to interventions and altered physiologic environments. 32–24
Combining metabolomics with other omics and phenotypic data to define dose optimization, therapeutic need or adverse outcome
Further, the combination of omics and phenotypic data can help define dose optimization, therapeutic need, or adverse outcomes. For example, in a study of microbiome-metabolome interactions of human lung bronchoalveolar lavage (BAL) in healthy individuals with HIV-1 infection, results showed that the healthy HIV-1 infected individuals had four main hubs in the microbiome-metabolome interaction. These hubs included Staphylococcaceae, Nocardiodaceae, Caulobacteraceae, and Streptococcus, the organisms commonly causing opportunistic infection in this population. The results suggest that this untargeted data-driven approach can be used to identify hubs, dose optimization, therapeutic needs and adverse outcomes,35 thereby suggesting substantial potential for precision medicine.
Useful information may be obtained from G × M × E (2:16:35)
Additional useful information may be obtained from examining the Genome × Metabolome × Exposome (G × M × E). The “exposome” includes the full range of daily exposures encountered in an environment, such as drug dose e.g. (Fig. 7). External exposures can be harmful exposures or beneficial therapeutic exposures. In a pilot study of US Military Personnel with occupational exposures, results showed that combined genomic, metabolomic, and external exposures were linked to health outcomes. Metabolome data were combined with environmental exposures and ICD-9 codes (health data of military personnel) using data-driven integration tools to identify central communities of interactions. Dioxins were used as an environmental exposure, cotinine was used as a behavioral measure (smoking), and benzo(a)pyrene diol epoxide (BPDE) was used as an occupational exposure. Health outcomes included allergies, respiratory conditions, and cardiovascular health. Results showed that with this approach, it becomes feasible to link exposures to health outcomes and precision medicine. 36
Figure 7.
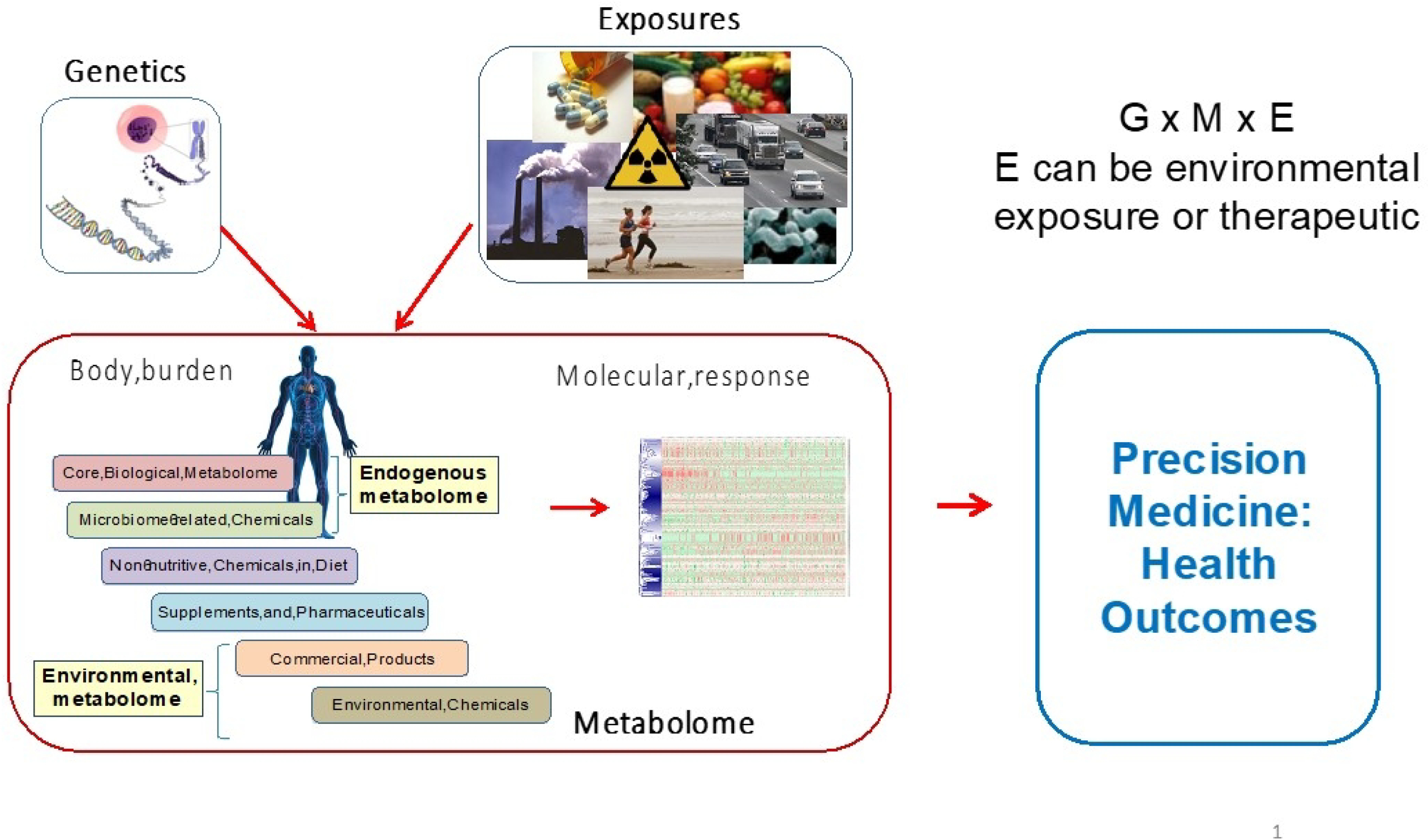
Useful Information obtained from G × M × E for Precision Medicine. Genetics (G), exposure (E) (environmental and direct exposure to diet and drugs) and metabolome response (M) related to Precision Health outcomes.36 Composite illustration by DI Walker, S Li and DP Jones used genetics image from Pixabay and exposures composite of drugs (Tom Varco), smokestacks (Alfred Palmer), food (Peggy Greb, USDA); runners: Mike Baird (http://www.flickr.com/photos/mikebaird/3539161615/) [CC BY2.0 (http://creativecommons.org/licenses/by/2.0)], and Campylobacter jejuni (De Wood modified by Chris Poole).
Discussion/Conclusion
The examples shown here illustrate a broad range of success stories and the extensive potential of metabolomics for precision medicine. The capability to translate toxicologic studies across species and to mechanistically link differences to specific genetic variants is critically important to advance the goals of precision medicine. Additionally, the ability to scale research from individuals to populations and to compare metabolites across biological sample types create important opportunities for new diagnostics and assessment of therapeutic efficacy. Metabolomics also allows us to query the complex biological response to various occupational, environmental, or operating environments. Identification of patterns of variance in these circumstances can provide insights into novel assessments, prophylactics, treatments, or hypotheses for future study. Global untargeted metabolomics has long-term potential for further development as part of routine healthcare for dose optimization and therapeutic follow-up to simultaneously measure pharmacokinetics, pharmacodynamics and potential adverse responses. Together these examples demonstrate that metabolomics is emerging as a truly translational tool for precision medicine.
Figure 1.
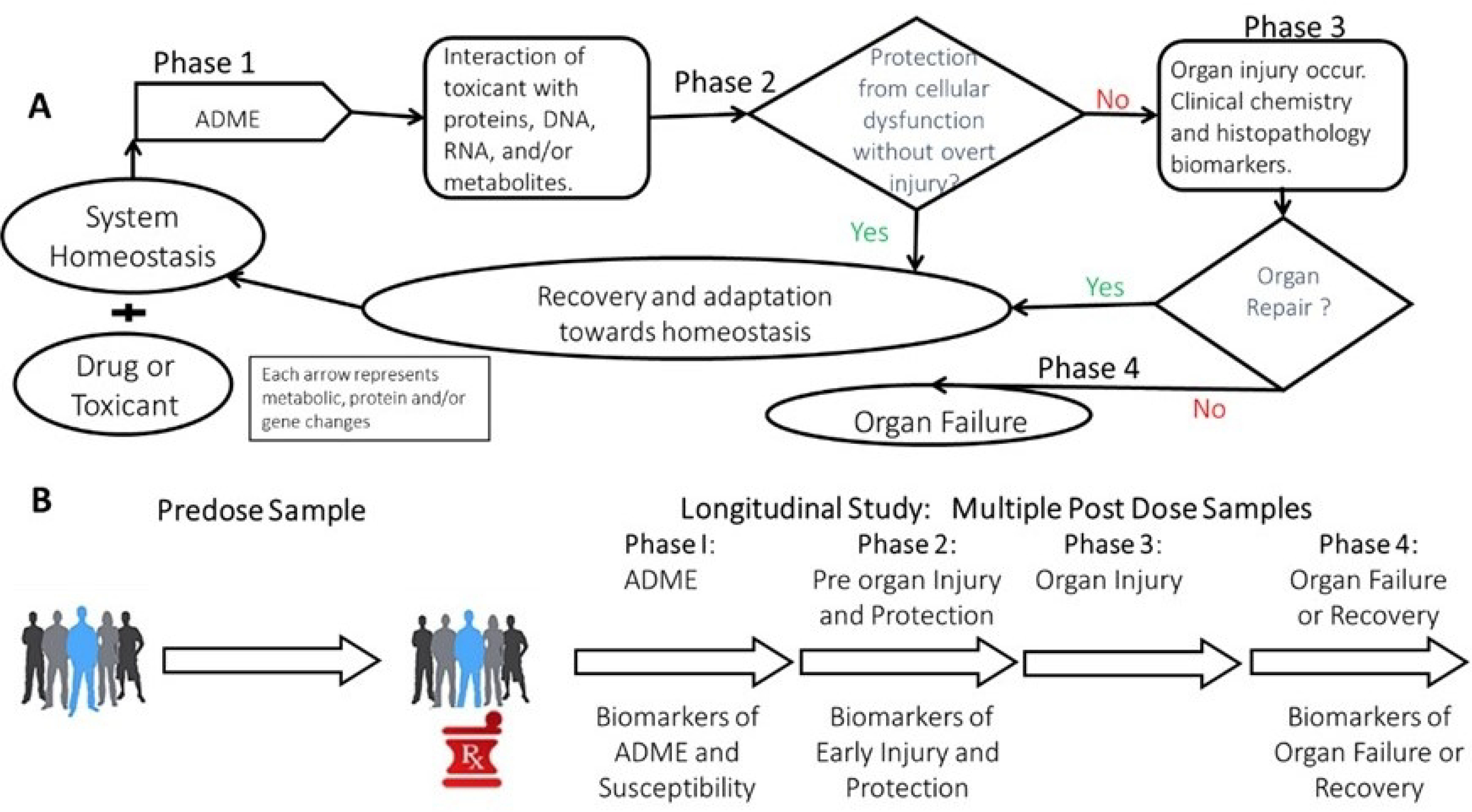
General Metabolomics Flow. A) Scheme of the four phases of responses to drug/toxicants exposure in a system.3 B) Metabolomics samples collection pre dose and longitudinal samples post dosing that relate to the 4 phases of responses to a toxicant show in panel A.2
Acknowledgements and Disclaimer
DPJ was supported in part by NIH grants ES023485, ES031824, ES019776, ES030163, ES029490, ES032189 and DoD grant W81XWH2010103. The opinions expressed in this manuscript do not necessarily represent those of the US the Food and Drug Administration. The NASA Twins Study of one year in space was supported by NASA: NNX14AH51G (All Twins PIs); TRISH: NNX16AO69A:0107 and NNX16AO69A:0061; the Bert L and N Kuggie Vallee Foundation, the WorldQuant Foundation, The Pershing Square Sohn Cancer Research Alliance, and the Bill and Melinda Gates Foundation (OPP1151054) for funding. Our thanks to Michael Snyder and Tejas Mishra of Stanford University for untargeted metabolomics. A special thanks to Christopher E. Mason of Weill Cornell Medicine for ongoing research collaboration in the post-hoc analysis of the NASA Twins Study data and contribution to the findings reported in this paper.
Footnotes
Conflict(s) of interest
DPJ: None
References
- 1.Beger RD, Sun J, Schnackenberg LK. Metabolomics approaches for discovering biomarkers of drug-induced hepatotoxicity and nephrotoxicity. Toxicol Appl Pharmacol. 2010;243(2):154–166. doi: 10.1016/j.taap.2009.11.019 [DOI] [PubMed] [Google Scholar]
- 2.Beger RD, Schmidt MA, Kaddurah-Daouk R. Current concepts in pharmacometabolomics, biomarker discovery, and precision medicine. Metabolites. 2020; 10(4). doi: 10.3390/metabo10040129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sun J, Slavov S, Schnackenberg LK, et al. Identification of a metabolic biomarker panel in rats for prediction of acute and idiosyncratic hepatotoxicity. Comput Struct Biotechnol J. 2014;10(17):78–89. doi: 10.1016/j.csbj.2014.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beger RD, Flynn TJ. Pharmacometabolomics in drug safety and drug-exposome interactions. Metabolomics. 2016;12(7). doi: 10.1007/s11306-016-1061-2 [DOI] [Google Scholar]
- 5.Dougherty BV, Papin JA. Systems biology approaches help to facilitate interpretation of cross-species comparisons. Curr Opin Toxicol. 2020;23–24:74–79. doi: 10.1016/j.cotox.2020.06.002 [DOI] [Google Scholar]
- 6.Rawls KD, Dougherty BV., Blais EM, et al. A simplified metabolic network reconstruction to promote understanding and development of flux balance analysis tools. Comput Biol Med. 2019;105:64–71. doi: 10.1016/j.compbiomed.2018.12.010 [DOI] [PubMed] [Google Scholar]
- 7.Blais EM, Rawls KD, Dougherty B V., et al. Reconciled rat and human metabolic networks for comparative toxicogenomics and biomarker predictions. Nat Commun. 2017;8:1–15. doi: 10.1038/ncomms14250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rawls KD, Dougherty BV., Vinnakota KC, et al. Predicting changes in renal metabolism after compound exposure with a genome-scale metabolic model. Toxicol Appl Pharmacol. 2021;412. doi: 10.1016/j.taap.2020.115390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee WM. Acetaminophen and the U.S. acute liver failure study group: Lowering the risks of hepatic failure. Hepatology. 2004;40(1):6–9. doi: 10.1002/hep.20293 [DOI] [PubMed] [Google Scholar]
- 10.Beger RD, Bhattacharyya S, Yang X, et al. Translational biomarkers of acetaminophen-induced acute liver injury. Arch Toxicol. 2015;89(9):1497–1522. doi: 10.1007/s00204-015-1519-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coen M, Lenz EM, Nicholson JK, Wilson ID, Pognan F, Lindon JC. An integrated metabonomic investigation of acetaminophen toxicity in the mouse using NMR spectroscopy. Chem Res Toxicol. 2003;16(3):295–303. doi: 10.1021/tx0256127 [DOI] [PubMed] [Google Scholar]
- 12.Chen C, Krausz KW, Shah YM, Idle JR, Gonzalez FJ. Serum metabolomics reveals irreversible inhibition of fatty acid β-oxidation through the suppression of PPARα activation as a contributing mechanism of acetaminophen-induced hepatotoxicity. Chem Res Toxicol. 2009;22(4):699–707. doi: 10.1021/tx800464q [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bhattacharyya S, Pence L, Beger R, et al. Acylcarnitine Profiles in Acetaminophen Toxicity in the Mouse: Comparison to Toxicity, Metabolism and Hepatocyte Regeneration. Metabolites. 2013;3(3):606–622. doi: 10.3390/metabo3030606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bhattacharyya S, Yan K, Pence L, et al. Targeted liquid chromatography – mass spectrometry analysis of serum acylcarnitines in acetaminophen toxicity in children. Biomark Med. 2014;8(2):147–159. doi: 10.2217/bmm.13.150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sun J, Ando Y, Ahlbory-dieker D, et al. Systems Biology Investigation to Discover Metabolic Biomarkers of Acetaminophen-Induced Hepatic Injury Using Integrated Transcriptomics and Metabolomics. J Mol Biomark Diagn. 2013;s1(002):1–11. doi: 10.4172/2155-9929.s1-002 [DOI] [Google Scholar]
- 16.Hylemon PB, Zhou H, Pandak WM, Ren S, Gil G, Dent P. Bile acids as regulatory molecules. J Lipid Res. 2009;50(8):1509–1520. doi: 10.1194/jlr.R900007-JLR200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Trauner M, Claudel T, Fickert P, Moustafa T, Wagner M. Bile acids as regulators of hepatic lipid and glucose metabolism. Dig Dis. 2010;28(1):220–224. doi: 10.1159/000282091 [DOI] [PubMed] [Google Scholar]
- 18.James O, Douglas AP, Watson AJ, Infirmary RV. Liver Damage After Paracetamol. Lancet. 1975;2:579–581. doi: 10.1016/s0140-6736(75)90170-1 [DOI] [PubMed] [Google Scholar]
- 19.Kaplowitz N, Kok E, Javitt NB. Postprandial Serum Bile Acid for the Detection of Hepatobiliary Disease. JAMA J Am Med Assoc. 1973;225(3):292–293. doi: 10.1001/jama.1973.03220300048011 [DOI] [PubMed] [Google Scholar]
- 20.Korman MG, Hofmann AF, Summerskill WHJ. Assessment of Activity in Chronic Active Liver Disease: Serum Bile Acids Compared with Conventional Tests and Histology. N Engl J Med. 1974;290(25):1399–1402. doi: 10.1056/NEJM197406202902503 [DOI] [PubMed] [Google Scholar]
- 21.Luo L, Schomaker S, Houle C, Aubrecht J, Colangelo JL. Evaluation of serum bile acid profiles as biomarkers of liver injury in rodents. ToxicolSci. 2014;137(1):12–25. doi: 10.1093/toxsci/kft221 [DOI] [PubMed] [Google Scholar]
- 22.Yamazaki M, Miyake M, Sato H, et al. Perturbation of bile acid homeostasis is an early pathogenesis event of drug induced liver injury in rats. Toxicol Appl Pharmacol. 2013;268(1):79–89. doi: 10.1016/j.taap.2013.01.018 [DOI] [PubMed] [Google Scholar]
- 23.Woolbright BL, McGill MR, Staggs VS, et al. Glycodeoxycholic acid levels as prognostic biomarker in acetaminophen-induced acute liver failure patients. Toxicol Sci. 2014;142(2):436–444. doi: 10.1093/toxsci/kfu195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.James L, Yan K, Pence L, et al. Comparison of bile acids and acetaminophen protein adducts in children and adolescents with acetaminophen toxicity. PLoS One. 2015;10(7):1–13. doi: 10.1371/journal.pone.0131010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schnackenberg LK, Sun J, Bhattacharyya S, Gill P, James LP, Beger RD. Metabolomics analysis of urine samples from children after acetaminophen overdose. Metabolites. 2017;7(3). doi: 10.3390/metabo7030046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garrett-Bakelman FE, Darshi M, Green SJ, et al. The NASA twins study: A multidimensional analysis of a year-long human spaceflight. Science (80-). 2019;364(6436). doi: 10.1126/science.aau8650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schmidt MA, Meydan C, Schmidt CM, Afshinnekoo E, Mason CE, BioRxiv. Elevation of Gut-Derived p-Cresol During Spaceflight and its Effect on Drug Metabolism and Performance in Astronauts. bioRxiv. Published online 2020:1–10. doi: 10.1101/2020.11.10.374645 [DOI] [Google Scholar]
- 28.Clayton TA, Baker D, Lindon JC, Everett JR, Nicholson JK. Pharmacometabonomic identification of a significant host-microbiome metabolic interaction affecting human drug metabolism. Proc Natl AcadSci U S A. 2009;106(34):14728–14733. doi: 10.1073/pnas.0904489106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vanholder R, Schepers E, Pletinck A, Nagler E V., Glorieux G. The uremic toxicity of indoxyl sulfate and p-cresyl sulfate: A systematic review. J Am Soc Nephrol. 2014;25(9):1897–1907. doi: 10.1681/ASN.2013101062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ben-Shachar R, Chen Y, Luo S, Hartman C, Reed M, Nijhout HF. The biochemistry of acetaminophen hepatotoxicity and rescue: A mathematical model. Theor Biol Med Model. 2012;9(1). doi: 10.1186/1742-4682-9-55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Walker DI, Perry-Walker K, Finnell RH, et al. Metabolome-wide association study of anti-epileptic drug treatment during pregnancy. Toxicol Appl Pharmacol. 2019;363:122–130. doi: 10.1016/j.taap.2018.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fernandes J, Chandler JD, Lili LN, et al. Transcriptome analysis reveals distinct responses to physiologic versus toxic manganese exposure in human neuroblastoma cells. Front Genet. 2019;10:1–15. doi: 10.3389/fgene.2019.00676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Go YM, Fernandes J, Hu X, Uppal K, Jones DP. Mitochondrial network responses in oxidative physiology and disease. Free Radic Biol Med. 2018;116:31–40. doi: 10.1016/j.freeradbiomed.2018.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Uppal K, Ma C, Go YM, Jones DP. XMWAS: A data-driven integration and differential network analysis tool. Bioinformatics. 2018;34(4):701–702. doi: 10.1093/bioinformatics/btx656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cribbs SK, Uppal K, Li S, et al. Correlation of the lung microbiota with metabolic profiles in bronchoalveolar lavage fluid in HIV infection. Microbiome. 2016;4(3):1–11. doi: 10.1186/s40168-016-0147-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thakar J, Thatcher TH, Smith MR, et al. Integrative Network Analysis Linking Clinical Outcomes with Environmental Exposures and Molecular Variations in Service Personnel Deployed to Balad and Bagram. J Occup Environ Med. 2019;61(12):S65–S72. doi: 10.1097/JOM.0000000000001710 [DOI] [PMC free article] [PubMed] [Google Scholar]