

Summary
Fusion-transcription factors (fusion-TFs) represent a class of driver oncoproteins that are difficult to therapeutically target. Recently, protein degradation has emerged as a strategy to target these challenging oncoproteins. The mechanisms that regulate fusion-TF stability, however, are generally unknown. Using CRISPR-Cas9 screening, we discovered tripartite motif-containing 8 (TRIM8) as an E3 ubiquitin ligase that ubiquitinates and degrades EWS/FLI, a driver fusion-TF in Ewing sarcoma. Moreover, we identified TRIM8 as a selective dependency in Ewing sarcoma compared with >700 other cancer cell lines. Mechanistically, TRIM8 knockout led to an increase in EWS/FLI protein levels that was not tolerated. EWS/FLI acts as a neomorphic substrate for TRIM8, defining the selective nature of the dependency. Our results demonstrate that fusion-TF protein stability is tightly regulated and highlight fusion oncoprotein-specific regulators as selective therapeutic targets. This study provides a tractable strategy to therapeutically exploit oncogene overdose in Ewing sarcoma and potentially other fusion-TF-driven cancers.
Keywords: fusion oncoproteins, EWS/FLI, Ewing sarcoma, TRIM8, E3 ligases, ubiquitination, protein degradation, neomorphic substrate, tumor dependency, oncogene overdose
Graphical abstract
Highlights
-
•
The E3 ligase TRIM8 is an exquisitely selective dependency in Ewing sarcoma
-
•
TRIM8 regulates EWS/FLI expression but not its wild-type counterparts
-
•
K334 is critical for TRIM8-mediated degradation of EWS/FLI
-
•
Increased EWS/FLI levels induce cell death in Ewing sarcoma cells
Using CRISPR-Cas9 screens, Seong et al. demonstrate that the E3 ligase TRIM8 degrades the EWS/FLI fusion oncoprotein in Ewing sarcoma cells. Knockout of TRIM8 is selectively lethal in Ewing sarcoma compared with >700 non-Ewing cancer models. TRIM8 loss increases EWS/FLI protein levels, which is toxic to Ewing sarcoma cells.
Introduction
Balanced chromosomal rearrangements often lead to gene fusions, some of which act as dominant oncoproteins that drive tumorigenesis (Mitelman et al., 2007). Two major fusion oncogene classes include fusion kinases, such as BCR/ABL1 in chronic myeloid leukemia, and fusion-transcription factors (fusion-TFs), such as EWS/ETS in Ewing sarcoma. Although inhibition of fusion kinases can be achieved for clinical efficacy (Cocco et al., 2018; Kantarjian et al., 2002; Shaw et al., 2014), therapeutic targeting of fusion-TFs has remained challenging due to the well-established difficulties in developing small-molecule inhibitors for transcription factors. Successful targeting, however, can lead to cures as exemplified by arsenic and retinoic acid treatment for patients with acute promyelocytic leukemia, both of which lead to degradation of the driver fusion-TF PML/RARα (Lo-Coco et al., 2013; Zhu et al., 2001).
The ubiquitin (Ub)-proteasome system (UPS) is the main mechanism by which cells regulate protein degradation to maintain protein homeostasis. In UPS, Ub-ligase (E3) enzymes, in conjunction with Ub-activating (E1) and Ub-conjugating (E2) enzymes, polyubiquitinate substrate proteins for proteasomal degradation. Cancer cells depend on protein quality control to cope with excess and abnormal proteins (Deshaies, 2014). This vulnerability led to the clinical development of proteasome inhibitors, such as bortezomib, as a cancer therapy. In addition, the protein stability of oncogenes and tumor suppressors is highly regulated and offers an opportunity for therapeutic intervention, such as the development of MDM2 inhibitors (Shangary and Wang, 2009). More recently, lenalidomide was shown to selectively target the transcription factors IKZF1 and IKZF3 (Ikaros family zinc-finger proteins 1 and 3), for proteasomal degradation by co-opting the cereblon E3 ligase complex for its anti-tumor effects in multiple myeloma (Kronke et al., 2014; Lu et al., 2014).
Ewing sarcoma is characterized by a chromosomal rearrangement that fuses the transactivation domain of EWS with the DNA binding domain of ETS transcription factors, most commonly FLI1, leading to expression of the EWS/FLI fusion oncoprotein (Delattre et al., 1992). EWS/FLI acts as a pioneering transcription factor that opens closed chromatin by recruiting chromatin remodeling complexes, such as the BAF complex (Boulay et al., 2017). Consequently, an aberrant transcriptional program is activated that promotes tumorigenesis and maintenance of Ewing sarcoma tumors. Despite EWS/FLI acting as a driver oncogene in Ewing sarcoma, there are no therapies that directly and specifically target it. Moreover, genome-wide sequencing studies have demonstrated that Ewing sarcoma tumors have quiet genomes with few recurrent mutations (Brohl et al., 2014; Crompton et al., 2014; Tirode et al., 2014), posing a challenge to identifying alternative therapeutic targets.
Here, we identify TRIM8 as an E3 ligase that regulates EWS/FLI protein degradation. We show that TRIM8 is a strong selective dependency in Ewing sarcoma and that TRIM8 degradation disrupts the fusion oncoprotein rheostat leading to oncogene overdose. Moreover, we demonstrate that EWS/FLI acts as a neomorphic substrate for TRIM8 and that the regulation is specific to the fusion oncoprotein and not the wild-type (WT) counterparts. Our findings suggest that EWS/FLI can be indirectly, but selectively, targeted and that other fusion oncoprotein-specific regulators may provide new therapeutic targets with exquisite selectivity for fusion-driven cancers.
Results
Flow cytometry-based CRISPR screen identifies TRIM8 as a regulator of EWS/FLI stability
To determine whether protein degradation can be exploited for therapeutic targeting of fusion-TFs, we studied the mechanisms that regulate the degradation/stability of EWS/FLI in Ewing sarcoma. To this end, we generated a 293T cell line model expressing EWS/FLI-GFP-IRES-mCherry that can report on EWS/FLI protein stability (Figure S1A). Concurrent examination of protein levels for EWS/FLI-GFP and the mCherry control allows us to investigate mechanisms specific to EWS/FLI compared with mechanisms regulating general transcription or protein homeostasis. We observed that high expression of the full-length EWS/FLI in 293T cells was not well tolerated, leading to the rise of two distinct subpopulations: (1) cells with low expression of full-length EWS/FLI-GFP in the nucleus and (2) cells with high expression of degraded EWS/FLI-GFP in both nucleus and cytoplasm (Figures 1A and 1B). To select cells that only express full-length EWS/FLI, we sorted GFPlow cells and selected a single-cell clone (EWS/FLI-GFP-1) with moderate expression of EWS/FLI-GFP in the nucleus (Figures 1A, 1B, and S1B). CRISPR-mediated EWS/FLI knockout in these reporter cells gave rise to a GFP-mCherry+ subpopulation; thus, faithfully reporting on EWS/FLI protein levels (Figures S1C and S1D). Using this reporter line, we performed a flow cytometry-based CRISPR-Cas9 screen (Figure 1C) to identify regulators of EWS/FLI protein stability. Cas9-positive EWS/FLI reporter cells were infected with the genome-scale CRISPR Avana library (Doench et al., 2016), passaged for 7 days, and sorted into GFPlow and GFPhigh populations (Figure 1D). Massively parallel sequencing of single guide RNAs (sgRNAs) from sorted populations revealed an enrichment of sgRNAs targeting EWSR1 and FLI1 in the GFPlow subpopulation compared with the pre-sort population, supporting the validity of the screen (Figure S1E). Strikingly, we observed an enrichment of sgRNAs targeting TRIM8 as the only significant enrichment in both replicates in the GFPhigh subpopulations (Figure 1E) (Table S1). TRIM8 encodes for tripartite motif-containing 8 (TRIM8), an E3 ligase that ubiquitinates target proteins for proteasomal degradation (Okumura et al., 2010; Ye et al., 2017). Unexpectedly, sgRNAs targeting TRIM8 were also enriched in the GFPlow cells (Figure S1E). We examined the TRIM8 gRNA distribution and observed greater enrichment of TRIM8 targeting sgRNAs in GFPhigh-sorted population compared with the GFPlow-sorted population (Figure S1F). Validation experiments confirmed that TRIM8 knockout predominantly results in increased EWS/FLI levels; however, minor subpopulations of cells that are unable to tolerate increased EWS/FLI can abrogate EWS/FLI protein expression consistent with the TRIM8 sgRNAs observed in the GFPlow-sorted subpopulation in the screen (Figures S1G and S1H). To further support this hypothesis, we performed a time course experiment with EWS/FLI overexpression in WT or TRIM8 knockout (TRIM8−/−) 293T cells. We detected higher EWS/FLI levels in the GFP+mCherry+ population in TRIM8 knockout cells compared with WT cells as expected, but also observed an increase in the GFP−mCherry+ subpopulation in TRIM8 knockout cells over time as compared with WT control cells (Figures S1I–S1L).
Figure 1.

CRISPR screens identify TRIM8 as a regulator of EWS/FLI protein stability and a selective dependency in Ewing sarcoma
(A and B) Immunoblot and images showing the expression level and localization of EWS/FLI-GFP in the reporter cell line and subpopulations. ∗Indicates a non-specific band.
(C and D) Schematic of flow cytometry-based CRISPR screening pipeline and the gating strategy used in the screen.
(E) Scatterplot showing average log2 fold changes in sgRNA abundance in replicates in the GFPhigh-sorted population. Negative control guides are highlighted in gray. sgRNAs targeting TRIM8 are highlighted in red. Each dot represents an average of log2 fold changes for four independent sgRNAs per gene.
(F and G) Scatterplots showing Ewing sarcoma relative dependency on TRIM8 in screens with the Avana (F) and GecKO (G) libraries. The x axis shows the gene’s dependency score in each cell line. The y axis shows the gene’s dependency rank in an individual cell line.
(H and I) Comparison of 14 Ewing sarcoma with 724 other cancer cell lines (H) and 11 Ewing sarcoma with 32 other cancer cell lines (I) demonstrates enrichment of TRIM8 dependency in Ewing sarcoma. Each circle represents a single gene. The x axis shows the effect size, which is the mean difference of dependency scores in Ewing sarcoma cell lines compared with other lines screened. Negative effect size indicates that Ewing sarcoma cells are more dependent on that gene compared with other cancer cell lines screened. The y axis shows the significance calculated as –log10(q value) from empirical-Bayes-moderated t statistics with Benjamini-Hochberg correction.
(J) A scatterplot showing ranked disease-enriched dependency in the Avana library (n = 738). The x axis shows the t statistics and the y axis shows the significance calculated as –log10(q value) from empirical Bayes-moderated t statistics with Benjamini-Hochberg correction. PubMed hits represent the number of papers retrieved when searched on PubMed.
TRIM8 is a strong selective dependency in Ewing sarcoma
Genome-scale CRISPR-Cas9 screens have revealed genetic dependencies in multiple cancers (Behan et al., 2019; Tsherniak et al., 2017) offering inroads into identifying therapeutic targets for diseases with a paucity of recurrently mutated genes, such as pediatric cancers. Strikingly, we identified TRIM8 as a top enriched dependency in Ewing sarcoma (Dharia et al., 2021) in two independent CRISPR screens using the GeCKO library (Sanjana et al., 2014), where 43 cancer cell lines were screened, and the Avana library (Doench et al., 2016), where over 700 cancer cell lines were screened (Figures 1F–1I). The TRIM8 dependency was independent of the ETS fusion partner; EWS/FLI-, EWS/ERG-, and EWS/FEV1-positive Ewing sarcoma cell lines were all dependent on TRIM8 (Figures 1F and 1G). Moreover, the TRIM8 dependency in Ewing sarcoma is the strongest, previously unreported enriched dependency identified within the Avana Dependency Map screen (Figure 1J). We validated the TRIM8 dependency in multiple Ewing sarcoma models in vitro and observed decreased proliferation and induction of apoptosis with CRISPR-mediated TRIM8 knockout (Figures 2A–2C and S2A–S2D). Importantly, we observed suppression of tumor growth with TRIM8 knockout in vivo using multiple Ewing sarcoma xenograft models (Figures 2D, S2E, and S2F). Immunoblot analysis of delayed tumors progressing in the TRIM8 knockout group showed no evidence of sustained TRIM8 knockout, suggesting that cells cannot tolerate TRIM8 knockout in vivo (Figure S2G). Furthermore, we failed to select single-cell clones of TRIM8 knockout in vitro (Figure S2H). Taken together, our data suggest that TRIM8 knockout cannot be tolerated by Ewing sarcoma cells in vitro or in vivo.
Figure 2.

TRIM8 regulates EWS/FLI protein levels and Ewing sarcoma cell growth
(A) TC32 cells infected with non-targeting (sgNT) or TRIM8-directed sgRNAs (sgTRIM8) were assessed for growth using CellTiter-Glo. Mean of eight technical replicates ± SD of relative growth are shown. Data representative of three independent experiments.
(B and C) Proliferation (B) and induction of apoptosis (C) in TC32 cells infected with either sgNT or sgTRIM8 were assessed by measuring EdU incorporation (FC, fold change) and cleaved PARP and cleaved caspase-3 levels, respectively. Mean FC of three biological replicates ± SEM are shown for (B). Statistical significance calculated using unpaired two-tailed Student's t test. ∗p < 0.05, ∗∗p < 0.01.
(D) Tumor growth of TC32 cells infected with sgNT or two different sgTRIM8s that were implanted subcutaneously: sgNT (n = 6), sgTRIM8-1 (n = 7), and sgTRIM8-2 (n = 7). Mean tumor volume ± SEM are shown. Statistical significance calculated using unpaired two-tailed Student's t test. ∗∗p < 0.01.
(E) Ewing sarcoma cell lines were infected with either sgNT or sgTRIM8s and immunoblotted with the indicated antibodies. Data representative of three independent experiments.
(F) Schematic of the TRIM8 dTAG system.
(G) Immunoblot showing expression of N- or C-terminally FKBP12F36V-2XHA-tagged (N- or C-dTAG) TRIM8 (anti-HA) and endogenous TRIM8 (anti-TRIM8) in TC32 TRIM8 dTAG cells.
(H and I) TC32 TRIM8 dTAG cells treated with 1 μM dTAGV-1 molecule for 24 h immunoblotted with the indicated antibodies (H) and assessed for cell growth using CellTiter-Glo (I). Mean of eight technical replicates ± SD of relative growth are shown. Data representative of three independent experiments.
(J) TC71 C-dTAG TRIM8 cells were treated with the indicated concentrations of dTAGV-1 for 48 h and immunoblotted with the indicated antibodies.
(K) TC71 C-dTAG TRIM8 cells were treated with dTAGV-1 (1 μM) for the indicated time-points and immunoblotted with the indicated antibodies.
(L and M) TC32 (L) and TC71 (M) cells infected with lentivirus encoding TRIM8 and an E3 ligase domain deletion mutant (TRIM8ΔRING) were immunoblotted with the indicated antibodies (left) and assessed for cell growth using CellTiter-Glo (right). Mean of eight technical replicates ± SD of relative growth are shown. Data representative of three independent experiments.
(N) EdU incorporation of indicated Ewing sarcoma cells overexpressing either control vector or TRIM8-V5 for 4 days. Data represents the fold change (FC) in percent of EdU+ cells ± SEM from two independent experiments with technical duplicates. Statistical significance calculated using unpaired two-tailed Student's t test. ∗∗∗p < 0.001.
(O) TC32 cells infected with varying amounts of lentivirus encoding TRIM8-V5 for 48 h and immunoblotted with the indicated antibodies.
(P) TC32 cells infected with lentivirus encoding TRIM8-V5, TRIM8ΔRING-V5, or control vector were subcutaneously injected into nude mice: vector (n = 10), TRIM8-V5 (n = 10), and TRIM8ΔRING-V5 (n = 7). Mean tumor volume ± SEM are shown. Statistical significance calculated using unpaired two-tailed Student's t test. ∗p < 0.05.
TRIM8 regulates EWS/FLI protein expression to regulate cell growth in Ewing sarcoma
Given that we identified TRIM8 as a regulator of EWS/FLI protein stability in 293T cells, we asked if TRIM8 can regulate EWS/FLI in Ewing sarcoma cells. We knocked out TRIM8 and suppressed TRIM8 expression using CRISPR and siRNA, respectively, and observed increased EWS/FLI expression, and NKX2-2 expression, a known EWS/FLI target gene (Figures 2E and S2I–S2K). We also observed increased EWS/ERG expression and decreased growth with TRIM8 knockout (Figure S2L), consistent with our finding that EWS/ERG-driven Ewing sarcoma cells are dependent on TRIM8 (Figures 1F and 1G). To further validate the relationship between TRIM8 and EWS/FLI, we generated Ewing sarcoma cells expressing degradable TRIM8 using the dTAG system (Nabet et al., 2018) (Figure 2F). The dTAG system utilizes a heterobifunctional small molecule that specifically binds and brings in close proximity a FKBP12F36V-tagged target protein and the E3 ligase complex, leading to ubiquitination and proteasome-mediated degradation of the target protein. Here, we concurrently expressed FKBP12F36V-2XHA-tagged TRIM8 and knocked out the endogenous TRIM8 (Figures 2G and S2M). Then, we used a highly selective dTAG molecule that co-opts the VHL E3 ligase (dTAGV-1) (Nabet et al., 2020) to induce degradation of TRIM8. dTAGV-1-mediated degradation of N- or C-terminally FKBP12F36V-2XHA-tagged TRIM8 (N/C-dTAG TRIM8) led to a suppression of cell growth and increased EWS/FLI levels (Figures 2H, 2I, and S2N). Leveraging the dTAG system, we observed a dose-dependent increase in EWS/FLI upon TRIM8 degradation (Figures 2J and S2O). Moreover, we examined the kinetics of EWS/FLI upregulation and observed a significant increase in EWS/FLI levels 16 h post-TRIM8 degradation by western immunoblot (Figure 2K).
To test the hypothesis that TRIM8 is an E3 ligase for EWS/FLI, we overexpressed TRIM8 and observed decreased EWS/FLI levels, associated with suppression of growth and proliferation (Figures 2L–2N and S2P–S2S). As predicted, we observed that TRIM8 overexpression led to a decrease in EWS/FLI levels in a concentration-dependent manner (Figure 2O). We also demonstrated that the E3 ligase activity is required for the regulation of EWS/FLI as overexpression of a TRIM8 RING domain deletion mutant (TRIM8ΔRING) failed to downregulate EWS/FLI expression. In fact, the TRIM8ΔRING mutant acts as a dominant negative as its overexpression led to increased EWS/FLI levels and suppression of growth (Figures 2L, 2M, and S2P–S2S). Importantly, we observed that overexpression of TRIM8 or TRIM8ΔRING mutant decreased the tumor growth in vivo (Figure 2P). Finally, TRIM8-mediated downregulation of EWS/FLI was rescued with a proteasome inhibitor, confirming regulation at the protein level (Figures S2T and S2U).
“Goldilocks principle” of EWS/FLI
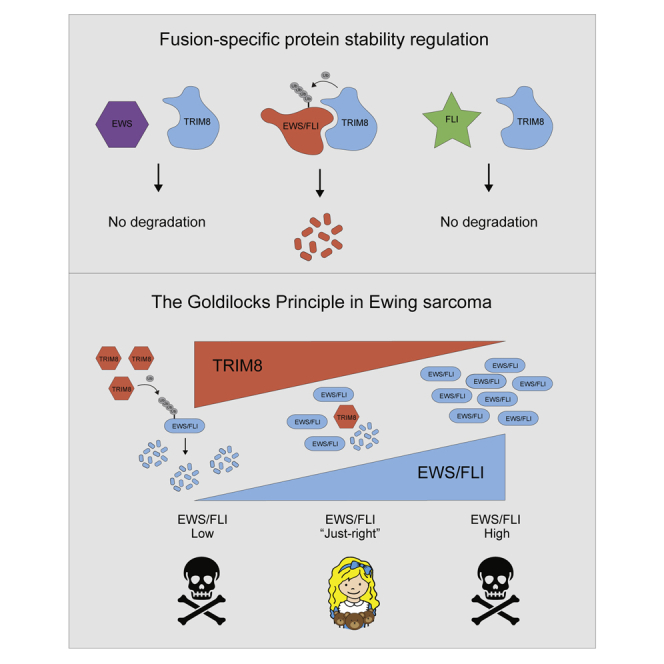
Oncogenes are regulated at multiple levels, including transcriptional and post-translational, to enable oncogenic functions (Schuijers et al., 2018; Willumsen et al., 1984). Although oncogenes can promote cancer cell survival, they can also induce senescence and apoptosis (McMahon, 2014; Serrano et al., 1997). Given that TRIM8 knockout increased EWS/FLI levels, which was associated with a growth suppression, we examined whether enhanced EWS/FLI expression is deleterious to Ewing sarcoma. To address this hypothesis, we engineered multiple Ewing sarcoma cell lines with a doxycycline (dox)-inducible EWS/FLI. We observed that dox-induced overexpression of EWS/FLI suppressed growth and induced apoptosis in vitro (Figures 3A–3C and S3A–D). Importantly, a Ewing sarcoma cell line and a minimally passaged Ewing sarcoma patient-derived xenograft (PDX) (ES-PDX-001) (Garcia-Dominguez et al., 2018) overexpressing EWS/FLI displayed delayed tumor growth in vivo (Figures 3D and 3E). Furthermore, we overexpressed a degradable EWS/FLI (FKBP12F36V-2XHA-EWS/FLI; N-dTAG EWS/FLI) and induced degradation, which partially rescued the growth suppression observed with EWS/FLI overexpression (Figure 3F). These data suggest that Ewing sarcoma cells cannot tolerate excessive EWS/FLI levels.
Figure 3.

TRIM8 fine-tunes EWS/FLI expression to promote survival
(A and B) A673, TC71, and TC32 cells expressing doxycycline (dox)-inducible EWS/FLI-HA were treated with dox (500 ng/mL) for 2, 3, and 5 days, respectively. Cells were immunoblotted with the indicated antibodies (A) and assessed for growth using CellTiter-Glo (B). Mean of eight technical replicates ±SD of relative growth are shown. Data representative of two independent experiments.
(C) Bar graph depicts percentage of annexin V+ cells in Ewing cell lines after 24 h (RDES) or 48 h (TC71, SKNEP1, EW8, A673) of dox-induction (500 ng/mL). Statistical significance calculated using unpaired two-tailed Student's t test. ∗∗p < 0.01.
(D and E) TC32 (D) and ES-PDX-001 (E) cells were infected with either EWS/FLI or a vector control and subcutaneously injected into mice. Tumor outgrowth was monitored by measuring tumor volume. Mice for vector (n = 10) and N-dTAG EWS/FLI (n = 10) for TC32 (D) and vector (n = 10) and EWS/FLI-V5 (n = 10) for ES-PDX001 (E). Mean tumor volume ±SEM are shown. Statistical significance calculated using unpaired two-tailed Student's t test. ∗∗p < 0.01, ∗∗∗p < 0.001. Note: tumors from TC32 vector control mice are equivalent to those in Figure 2P.
(F) TC32 were infected with vector or N-dTAG EWS/FLI for 3 days, treated with DMSO or dTAGV-1 (1 μM) for 48 h, and then lysed and immunoblotted with the indicated antibodies (left) or assessed for growth (right) using CellTiter-Glo. Mean of eight technical replicates ±SD of relative growth are shown. Statistical significance calculated using unpaired two-tailed Student's t test. ∗∗∗p < 0.001. Data representative of three independent experiments.
(G) Scatterplot of gene set enrichment analysis (GSEA) for significantly upregulated genes (adj. p < 0.05) after TRIM8 degradation (24 h). Gene sets significantly enriched (adj. p < 10−20) are highlighted in red. GSEA was performed with mSigDB C2 collection v.7.0.
(H and I) EWS502 N-dTAG EWS/FLI cells were infected with either vector or TRIM8ΔRING for 48 h and then treated with either DMSO or dTAGV-1 (1 μM) for 24 h and immunoblotted with the indicated antibodies (H) or assessed for cell growth by cell counting (I). Mean of two technical replicates ± SD are shown. Statistical significance calculated using unpaired two-tailed Student's t test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. Data representative of three independent experiments.
(J–M) EWS502 N-dTAG EWS/FLI cells were either transfected with control (siCtrl) or TRIM8-targeting (siTRIM8) siRNAs (J) or infected with either non-targeting (sgNT) or TRIM8-targeting sgRNA (sgTRIM8) (L) for 48 h. Cells were then treated with either DMSO or indicated concentrations of dTAGV-1 for 48 h and were assessed for growth using CellTiter-Glo (K and M). Mean of eight technical replicates ± SD of relative growth are shown. Statistical significance calculated using unpaired two-tailed Student's t test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. Data representative of two independent experiments.
(N) Conceptual model of the Goldilocks principle with EWS/FLI dosage.
To examine the early transcriptional consequences of EWS/FLI overexpression, we performed RNA sequencing 8 h after dox-induction of EWS/FLI. We primarily observed upregulation of genes suggesting transcriptional activation (Figure S3E), and gene set enrichment analysis showed an enrichment of EWS/FLI UP signatures (composed of genes activated by EWS/FLI) (adj. p < 0.05) (Figure S3F). Similarly, we observed transcriptional activation after 24 h TRIM8 degradation (Figure S3G) and enrichment of the same EWS/FLI UP signatures as the only significantly (adj. p < 10−20) enriched signatures (Figure 3G). We did not observe changes in EWS/FLI transcript level upon TRIM8 degradation, further confirming that TRIM8-mediated regulation of EWS/FLI occurs post-transcriptionally (Figure S3H). These data suggest that the major transcriptional consequence of TRIM8 degradation is activation of the EWS/FLI transcriptional program.
Next, we asked whether the impaired growth observed with TRIM8 knockout is mediated by increased EWS/FLI expression. To this end, we used a Ewing sarcoma cell line engineered to express N-dTAG EWS/FLI and in which endogenous EWS/FLI has been knocked out by CRISPR (Nabet et al., 2020). In this model, EWS/FLI can be degraded in a concentration-dependent manner upon dTAGV-1 treatment (Figure 3H). We overexpressed the dominant negative TRIM8ΔRING mutant and then induced partial degradation of EWS/FLI with dTAGV-1 to near control protein levels, which rescued the diminished growth phenotype (Figures 3H and 3I). In addition, we observed that CRISPR-mediated TRIM8 knockout and siRNA-mediated TRIM8 knockdown phenotypes can also be rescued by degrading EWS/FLI to control levels (Figures 3J–3M). These rescue experiments confirm that increased EWS/FLI is, in part, responsible for the impaired growth phenotype observed with TRIM8 inhibition. We next asked whether EWS/FLI levels are anti-correlated with TRIM8 levels in Ewing sarcoma cells. We observed a trend toward an inverse correlation between EWS/FLI and TRIM8 protein levels in Ewing sarcoma cell lines (Figures S3I and S3J). Based on our findings, we propose a model in which TRIM8 tightly regulates and maintains EWS/FLI at “just-the-right” protein level to promote cell survival (Figure 3N).
Increased levels of EWS/FLI exacerbate DNA damage in Ewing sarcoma cells
Ewing sarcoma tumors harbor copy-number loss of CDKN2A (∼30%) and TP53 mutations (∼15%) (Brohl et al., 2014; Crompton et al., 2014; Tirode et al., 2014), both which have been shown to regulate senescence (Lin et al., 1998). We, however, observed no correlation between TRIM8 dependency and either TP53 or CDKN2A status (Figures S4A and S4B). Moreover, we did not observe a senescence phenotype with TRIM8 knockout/degradation in Ewing sarcoma cells (Figure S4C). TRIM8 previously has been shown to regulate tumor necrosis factor (TNF) and interferon (IFN) signaling to modulate immune responses to pathogens (Li et al., 2011; Maarifi et al., 2019). We thus examined whether TNF-α and IFNs can regulate TRIM8 levels in Ewing sarcoma and vice versa. We observed that neither TNF-α nor IFN γ/β treatment regulates TRIM8 levels in Ewing sarcoma cells. Similarly, neither TRIM8 knockout nor overexpression regulates TNF or IFN signaling in Ewing sarcoma cell lines, as measured by altered pSTAT1 or altered levels of nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha (IkBα) (Figures S4D–S4H).
EWS/FLI has been associated with R loop formation (Gorthi et al., 2018) and impaired DNA damage repair response, which confer increased sensitivity of Ewing sarcoma cells to DNA-damaging agents (Brenner et al., 2012; Iniguez et al., 2018). We observed that EWS/FLI overexpression, TRIM8 degradation, and overexpression of the TRIM8ΔRING mutant can exacerbate DNA damage (Figures 4A–4C and S3A–S3C), leading to increased sensitivity to olaparib, a PARP inhibitor (Figures 4D and 4E). Moreover, TRIM8 degradation sensitized Ewing sarcoma cells to DNA-damaging agents, such as cisplatin and etoposide, in a cell line-dependent manner (Figures 4F–4I). We next asked whether TRIM8 inhibition correlates with sensitivity to DNA damage repair inhibitors, such as PARP inhibitors, in Ewing sarcoma cells. Ewing sarcoma cell lines show increased sensitivity to PARP inhibitors that possess stronger PARP trapping potency (Figure 4J), and TRIM8 dependency is significantly correlated with response to PARP inhibitors that show strongest PARP trapping potencies (Figures 4K–4O). These data suggest that PARP trapping may be a potential mechanism of DNA damage upon TRIM8 inhibition in Ewing sarcoma cells.
Figure 4.

EWS/FLI overexpression and TRIM8 degradation increase DNA damage and sensitize cells to PARP inhibitors
(A) TC71 and TC32 cells expressing dox-inducible EWS/FLI-HA were treated with doxycycline (dox) (500 ng/mL) for 48 h and immunoblotted with the indicated antibodies.
(B) TC71 and TC32 C-dTAG TRIM8 cells were treated with either DMSO control or dTAGV-1 (1 μM) for 48 h and immunoblotted with the indicated antibodies.
(C) TC32 and TC71 cells were infected with lentivirus encoding either vector control, TRIM8-V5, or TRIM8ΔRING-V5 and immunoblotted with the indicated antibodies.
(D–I) TC32 and TC71 C-dTAG TRIM8 cells were pre-treated with either DMSO control or dTAGV-1 (1 μM) for 48 h and assessed for sensitivity to olaparib (D and E), cisplatin (F and G), and etoposide (H and I). Mean of eight technical replicates ±SD of relative viability are shown. Statistical significance calculated using unpaired two-tailed Student's t test. Data in (A–I) representative of three independent experiments.
(J) Heatmap showing area under the curve values of PARP inhibitor sensitivity of Ewing sarcoma cells in the PRISM Repurposing Secondary Screen at the Broad Institute (https://depmap.org/portal/).
(K–O) Scatterplots showing the correlation between TRIM8 dependency and sensitivity to PARP inhibitors: talazoparib (K), niraparib (L), olaparib (M), rucaparib (N), and veliparib (O). Spearman correlation (R) and p value from one-sided exact t test are shown.
EWS/FLI acts as a neomorphic substrate
The striking specificity of TRIM8 dependency prompted us to characterize the molecular basis for TRIM8-mediated regulation of EWS/FLI. First, we demonstrated that TRIM8 interacts with and polyubiquitinates EWS/FLI in 293T overexpression experiments (Figures 5A and 5B). Moreover, we observed that TRIM8ΔRING mutant retained binding to EWS/FLI but failed to ubiquitinate EWS/FLI, as expected (Figures 5A and 5B). Similarly, we observed that TRIM8 interacts with and ubiquitinates EWS/ERG (Figures 5C and 5D). To assess the specificity of TRIM8 for the fusion oncoproteins, we examined whether TRIM8 can degrade WT EWS, FLI, or ERG proteins and observed that TRIM8 failed to degrade these WT proteins (Figure 5E). In addition, we observed no consistent changes in WT FLI expression upon overexpression of TRIM8 in two acute myeloid leukemia (AML) cell lines that express WT FLI (Figure S5). We then asked whether TRIM8 can bind to these WT proteins and observed decreased TRIM8 binding for WT EWS and FLI proteins, but not ERG protein (Figure 5C). Moreover, we observed that TRIM8 overexpression led to polyubiquitination of EWS/FLI and EWS/ERG fusions but not WT FLI or ERG proteins (Figure 5D). Interestingly, EWS was poly-ubiquitinated upon TRIM8 overexpression; however, this did not lead to degradation. Taken together, our data suggest that the TRIM8-mediated degradation is specific to EWS/FLI and not WT counterpart proteins, which may, in part, explain the selective TRIM8 dependency in Ewing sarcoma.
Figure 5.

EWF/FLI is a neomorphic substrate of TRIM8 for degradation
(A) 293T cells were transfected with the indicated constructs for 48 h and then treated with 500 nM carfilzomib for 6 h, lysed, immunoprecipitated, and immunoblotted with the indicated antibodies. Data representative of three independent experiments.
(B) 293T cells were transfected with the indicated constructs for 48 h and treated with DMSO or 500 nM carfilzomib for 6 h. Cells were then lysed, immunoprecipitated, and immunoblotted with the indicated antibodies. Data representative of three independent experiments.
(C) 293T cells transfected with either fusion oncoproteins or WT counterparts for 48 h were treated with 1 μM carfilzomib for 6 h, lysed, immunoprecipitated, and immunoblotted with the indicated antibodies. Data representative of three independent experiments.
(D) 293T cells were transfected with the indicated constructs for 48 h then lysed, immunoprecipitated, and immunoblotted with the indicated antibodies. Data representative of three independent experiments.
(E) 293T cells were transfected with either FLAG-tagged fusion oncoproteins or WT counterparts for 48 h, lysed, and immunoblotted with the indicated antibodies. Data representative of three independent experiments.
EWS/FLI and TRIM8 interaction in Ewing sarcoma cells
Given that we characterized the EWS/FLI and TRIM8 interaction in overexpression settings, we next sought to define their interaction in Ewing sarcoma cells. E3 ligase-substrate interactions are typically weak and transient, posing detection challenges in the native setting. Adding to the problem of detecting endogenous interactions in Ewing cells, despite relatively high TRIM8 expression in mesenchymal stem cells, the putative cell of origin of Ewing sarcoma, TRIM8 expression is among the lowest in Ewing sarcoma cell lines in the Cancer Cell Line Encyclopedia and in primary tumors in the Treehouse dataset. Moreover, EWS/FLI does not regulate TRIM8 expression (Figures S6A–I). We thus sought to detect the interaction between EWS/FLI and TRIM8 using the proximity ligation assay (PLA), which allows in situ detection of protein-protein interactions at single-molecule resolution (Figure 6A). We took advantage of our TRIM8 dTAG Ewing sarcoma cells that allow us to utilize HA and FLI antibodies to detect TRIM8 and EWS/FLI, respectively. The PLA showed interaction between EWS/FLI and TRIM8, which was abrogated upon TRIM8 degradation (Figures 6B and 6C).
Figure 6.

TRIM8 interacts with and ubiquitinates EWS/FLI in Ewing sarcoma cells
(A) Schematic of the proximity ligation assay (PLA).
(B) Histogram showing fluorescence signal from the PLA in TC71 C-dTAG TRIM8 cells. No antibody (neg. ctrl), anti-FLI, and anti-HA alone were used as negative controls. Anti-FLI and anti-HA combination were used to detect interaction between TRIM8 and EWS/FLI.
(C) PLA signal TC71 C-dTAG TRIM8 cells treated with either DMSO or dTAGV-1 (1 μM) for 48 h. Data representative of three independent experiments.
(D) Schematic of fluorescent proteins expressed in A673-E/F-Halo cell line is shown above. Airyscan super-resolution images of the nucleus (Hoechst stained, blue), endogenous EWS/FLI-Halo (JF646 labeled, red), and transiently expressed TRIM8-EGFP (green) in an A673-E/F-Halo cell.
(E) Two-color image showing that endogenous EWS/FLI-Halo hubs enrich for TRIM8-EGFP. The nuclear fluorescence background is subtracted in both channels of the average image (see the STAR Methods for details).
(F) Average background-free radial profiles of EWS/FLI-Halo and TRIM8-EGFP at EWS/FLI-Halo hubs (averaged from 6,872 hubs in 15 cells).
(G and H) Poly-ubiquitinated proteins in TC71 cells infected with either non-targeting or TRIM8-targeting sgRNA were enriched with tandem ubiquitin binding entities (TUBE). Both input and TUBE-enriched proteins were immunoblotted with the indicated antibodies. EWS/FLI levels were quantified using ImageJ and the poly(ub)-E/F:total-E/F ratio was used to calculate poly-ubiquitinated EWS/FLI levels. Fold change (FC) in poly-ubiquitinated EWS/FLI ±SEM from three independent experiments is shown in (H). ∗∗∗p < 0.001.
(I) TC71 C-dTAG TRIM8 cells treated with 1 μM of dTAGV-1 for 6 h in triplicate were lysed and immunoblotted with the indicated antibodies. Six hour pomalidomide (1 μM) treatment served as a positive control for TMT mass spectrometry.
(J) Total proteome of cells in (I) was analyzed using TMT quantification mass spectrometry. Non-EWS/FLI proteins that significantly changed in abundance are highlighted in blue and EWS/FLI in red. Significance cutoffs used were p < 0.0001 and fold change (FC) > 1.25 and noted with dotted lines. Note: ERG1 (Q14534) is a squalene monooxygenase and not the ETS family member ERG (P11308).
EWS/FLI possesses a low complexity domain and has previously been shown to form local high-concentration hubs at its endogenous target genes via multivalent homotypic interactions, which are essential for the transcriptional activation function of EWS/FLI (Chong et al., 2018). To further support the EWS/FLI and TRIM8 interaction and assess whether TRIM8 co-localizes to EWS/FLI transcriptional hubs, we utilized super-resolution imaging. Specifically, we exploited a knockin A673 cell line where the endogenous EWS/FLI is fused to HaloTag (A673-E/F-Halo), which can be fluorescently labeled to enable visualization of endogenous EWS/FLI at its normal expression levels (Chong et al., 2018). We transiently expressed EGFP-tagged TRIM8 in the A673-E/F-Halo cells and performed simultaneous imaging of EWS/FLI-Halo and TRIM8-EGFP with Airyscan confocal super-resolution microscopy (Figure 6D). We observed that endogenous EWS/FLI-Halo forms numerous small local high-concentration hubs in the cell nucleus (Figures 6D), consistent with previous findings (Chong et al., 2018). TRIM8-EGFP at low expression levels is distributed throughout the cell nucleus with a modest degree of heterogeneity (Figure 6D). We detected the enrichment of TRIM8 at many EWS/FLI hubs; however, the crowded distribution of nuclear EWS/FLI hubs makes it difficult to clearly demonstrate TRIM8 enrichment at individual hubs. By averaging the images of nearly 7,000 individual EWS/FLI hubs in multiple cells, the signal-to-noise ratio is markedly improved to reveal TRIM8 enrichment at EWS/FLI hubs (Figures 6E and 6F). This result suggests that TRIM8 interacts with EWS/FLI in Ewing sarcoma cells.
We next asked whether TRIM8 can facilitate ubiquitination of EWS/FLI in Ewing sarcoma cells. To this end, we performed the tandem ubiquitin binding entity (TUBE) enrichment assay, which allows for enrichment of poly-ubiquitinated proteins (Hjerpe et al., 2009). We observed a decrease in the proportion of poly-ubiquitinated EWS/FLI levels in TRIM8 knockout cells compared with control cells (Figures 6G and 6H), suggesting that TRIM8 regulates ubiquitination of EWS/FLI in Ewing sarcoma.
While E3 ligases confer substrate specificity in the ubiquitin cascade, they often ubiquitinate multiple substrates. To examine the target repertoire of TRIM8, we performed unbiased global proteomics with dTAGV-1-mediated TRIM8 degradation. To identify direct substrates of TRIM8, we chose a time point 6 h post-degradation for mass spectrometry. Upon TRIM8 degradation, we observed EWS/FLI as one of the most significantly increased proteins (p < 0.0001, fold change > 1.25) among a small subset of the TRIM8 substrate candidates identified (Figures 6I and 6J; Table S2). Of note, EWS/FLI protein levels did not increase in a pomalidomide treatment control (Table S2), suggesting that upregulated EWS/FLI expression is specific to TRIM8 degradation and not an experimental artifact of degradation per se.
C1 and C5 domains of EWS/FLI are critical for TRIM8-mediated degradation
E3 ligases typically recognize a specific “degron” on their substrates for interaction. These degrons can exist in many forms including a short peptide sequence or a specific structural motif. To investigate whether we can identify the degron in EWS/FLI, we first examined whether the N-terminal EWS (N-EWS) or C-terminal FLI (C-FLI) portion of the fusion is regulated by TRIM8 (Figure 7A). We did not detect a change in expression of N-EWS or C-FLI with TRIM8 overexpression (Figure 7B). Moreover, we observed a loss of N-EWS mutant binding and a significant decrease in the C-FLI mutant binding to TRIM8, as well as a loss of ubiquitination with both mutants (Figures 7C and 7D). Given that the C-FLI mutant retained partial binding to TRIM8, we generated C-terminal deletion mutants (ΔC1-5) and observed that ΔC1 and ΔC5 mutants were resistant to TRIM8-mediated degradation (Figures S7A and S7B), suggesting the presence of a possible degron in these domains. Notably, we observed a significant decrease in the stability of the ΔC2-4 mutants, which may have an impact on interpreting TRIM8-mediated regulation of these mutants. Further biochemical experiments showed that C1 and C5 domains are required, but not sufficient, for TRIM8-mediated ubiquitination and subsequent degradation (Figures S7C–S7E). These data suggest that C1 and C5 domains likely promote a proper structural conformation of EWS/FLI that allows for efficient ubiquitination and subsequent degradation by TRIM8.
Figure 7.

Identification of domains critical for TRIM8-mediated degradation of EWS/FLI
(A) Schematics of EWS/FLI deletion mutants.
(B) EWS/FLI, N-EWS, and C-FLI mutants tagged with 3XFLAG were co-transfected with either TRIM8 or a vector control in 293T cells. After 48 h, cells were lysed and immunoblotted with the indicated antibodies.
(C) 293T cells were co-transfected with TRIM8 and EWS/FLI, N-EWS, C-FLI, or a vector control for 48 h, treated with 500 nM carfilzomib for 6 h, lysed, immunoprecipitated, and immunoblotted with the indicated antibodies.
(D) 293T cells transfected with the indicated constructs for 48 h, treated with 500 nM carfilzomib for 6 h, lysed, immunoprecipitated, and immunoblotted with the indicated antibodies.
(E) Schematic of TRIM8 structural domains and mutants.
(F and G) 293T cells transfected with the indicated constructs for 48 h, treated with 500 nM carfilzomib for 6 h, lysed with either lysis buffer (F) or ubiquitination lysis buffer (G), immunoprecipitated, and immunoblotted with the indicated antibodies. Data representative of three independent experiments.
(H) Schematic depicting fusion oncoprotein-specific regulators as potential therapeutic targets. EWS/FLI-specific regulation of TRIM8 is shown as an example.
The low complexity domain of EWS/FLI has been reported to have the propensity for phase separation via its multivalent interactions, which is critical for its pioneer transcription factor activity (Boulay et al., 2017). To test whether this interaction behavior of EWS/FLI confers specificity for TRIM8 regulation, we generated mutants that contain minimal low complexity domains in EWS fused to C-terminal FLI (Figure S7F). Similar to ΔC1 and ΔC5 mutants, we observed that the phase transition-like property of EWS/FLI is insufficient to promote efficient TRIM8-mediated ubiquitination and degradation (Figures S7G–S7I).
Furthermore, we generated structural domain deletion mutants of TRIM8 to identify the functional domain(s) required to regulate EWS/FLI protein levels (Figure7E). We observed that deletions of the RING, B-box 1, B-box 2, and coiled-coil domains largely did not affect binding to EWS/FLI but completely abolished TRIM8-mediated ubiquitination of EWS/FLI (Figures 7F and 7G). Interestingly, we observed that deletion of RFP-like domain of TRIM8 abrogated its ability to bind and ubiquitinate EWS/FLI (Figures 7F and 7G). In sum, our data suggest that (1) EWS/FLI acts as a neomorphic substrate for TRIM8, and (2) binding to TRIM8 alone is not sufficient for ubiquitination and degradation. The EWS/FLI-TRIM8 complex likely adopts a specific structural conformation or a cellular context that allows for efficient ubiquitination and degradation (Figure 7H).
K334 is critical for TRIM8-mediated degradation of EWS/FLI
We next asked whether we could identify potential lysines on EWS/FLI that are ubiquitinated by TRIM8. EWS/FLI has a total of 14 lysines, and a previous study has shown that K380 on the ETS domain is critical for EWS/FLI protein stability (Gierisch et al., 2016). Of note, K380 in the previous study corresponds to K334 in WT FLI amino acid sequence, which we will refer to for lysine positions. K144 is the only lysine on the N-terminal EWS of all EWS/ETS fusions in Ewing sarcoma. K144R mutant partially rescued, whereas K334R fully rescued, TRIM8-mediated degradation (Figures 8A and 8B). Moreover, we noted the presence of two lysines, K217 and K240, in the C1 domain of EWS/FLI that, when deleted, rescued TRIM8-mediated degradation. However, mutating these lysines alone or in combination did not phenocopy the C1 deletion mutant (Figure 8C). Furthermore, we noted that K334 is in the C3 domain of EWS/FLI that, when deleted, is still degraded by TRIM8. This raised the possibility that TRIM8 ubiquitinates an alternative lysine on the EWS/FLIΔC3 mutant for degradation. To test this hypothesis, we generated EWS/FLIΔC3-K144R mutant given our data that the EWS/FLI-K144R mutant showed partial but significant rescue of TRIM8-mediated degradation. We observed that the K144R mutation led to increased protein stability of EWS/FLIΔC3 and that it rescued TRIM8-mediated degradation (Figure S8). Taken together, these results suggest that TRIM8 can likely ubiquitinate multiple lysines for degradation in a context-dependent manner and that K144 and K334 are critical for TRIM8-mediated degradation of full-length EWS/FLI.
Figure 8.

K334 is critical for TRIM8-mediated degradation
(A) Schematic of lysine mutants of EWS/FLI. Lysine positions correspond to the positions found in WT EWS and FLI1 proteins.
(B and C) Indicated EWS/FLI lysine mutants were co-transfected with either TRIM8 or a vector control in 293T cells. After 48 h, cells were lysed and immunoblotted with the indicated antibodies. Data representative of three independent experiments.
Discussion
Massively parallel sequencing of cancer genomes led to the discovery of readily druggable therapeutic targets in some malignancies. Pediatric cancers, however, often have quiet genomes with few recurrent mutations that can be exploited for therapeutic intervention. For example, loss-of-function mutations in the gene STAG2, which has been shown to play a critical role in cis-chromatin interactions and metastasis, were identified as one of the only recurrent mutations (∼15%) in Ewing sarcoma tumors (Adane et al., 2021; Brohl et al., 2014; Crompton et al., 2014; Surdez et al., 2021; Tirode et al., 2014). Our work suggests an alternative therapeutic strategy for Ewing sarcoma by targeting TRIM8, an E3 ligase for EWS/FLI. Importantly, TRIM8 knockout mice are viable with only a minor immune-related phenotype (Ye et al., 2017). This knockout mouse data, the lack of essentiality of TRIM8 in all non-Ewing sarcoma cancer cell lines screened in the Dependency Map, and the selectivity of TRIM8 for EWS/ETS fusions, but not the WT components, suggests a high therapeutic window for TRIM8 inhibitors. Using genetic approaches, we were unable to select for clones that sustained TRIM8 knockout in vitro or in vivo, consistent with the essentiality of TRIM8 in Ewing sarcoma cells. Nevertheless, further preclinical studies using TRIM8 chemical inhibitors, when such molecules are developed, will be needed to better understand the therapeutic potential and the mechanisms of resistance in targeting TRIM8 in Ewing sarcoma. TRIM8 previously has been described as a regulator of various immune signaling pathways, including TNF-α and IFN-γ (Li et al., 2011; Maarifi et al., 2019). Although these signaling pathways are not regulated by TRIM8 in Ewing sarcoma cells, therapeutic targeting of TRIM8 might elicit modulation of immune responses in patients, highlighting the importance of establishing an immunocompetent mouse model for Ewing sarcoma to enable assessment of immunomodulation with targeted therapies.
Oncogenes activate molecular pathways that promote cellular transformation leading to tumorigenesis. The activation of oncogenes in untransformed cells, however, induces cellular stress that imparts a barrier to transformation. Accordingly, tumors possess driver oncogenic events coupled with multiple cooperating oncogenes or the inactivation of tumor suppressors. For example, expression of oncogenic Ras in primary human cells causes oncogene-induced senescence that is dependent on p53 signaling (Serrano et al., 1997). However, the cellular consequences of oncogene overdose in transformed cells remain elusive. Here, we demonstrate that suppression of TRIM8 disrupts the oncoprotein rheostat, leading to excessive EWS/FLI that cannot be tolerated by Ewing sarcoma cells. Consistent with our findings, a recent single-cell sequencing study of Ewing sarcoma PDX models showed that single cells with either low or high EWS/FLI gene expression programs were non-cycling cells, whereas cells with an “optimal” EWS/FLI program were actively proliferating (Aynaud et al., 2020).
We observed that TRIM8 repression in Ewing sarcoma cells led to increased DNA damage, which rendered cells sensitive to DNA damage-related inhibitors, such as olaparib. Moreover, there is a correlation between TRIM8 dependency and sensitivity to PARP inhibitors with strong PARP trapping potency. However, the exact mechanisms by which further upregulation of EWS/FLI by TRIM8 inhibition induces DNA damage and causes apoptosis remain unknown. EWS/FLI has been reported to increase basal transcription in Ewing sarcoma cells resulting in R loop formation (Gorthi et al., 2018). Unresolved R loops can also lead to stalled/collapsed replication forks, DNA damage, and replicative stress in non-transformed primary cells (Su et al., 2021). It is possible that increased R loops and replicative stress are, in part, responsible for the DNA damage phenotype observed with further upregulated EWS/FLI expression by TRIM8 inhibition, the subject of future investigations.
E3 ligases are notoriously difficult to target; however, it has been proven feasible. For example, the E3 ligase MDM2 has been successfully targeted, with multiple MDM2 inhibitors in clinical trials, including a stapled peptide that targets MDM2 and MDM4, and small molecules that inhibit the protein-protein interaction between MDM2 and p53 (Tisato et al., 2017; Wachter et al., 2017). In addition, recent studies have successfully identified novel binders for E3 ligases, including TRIM24, DCAF16, RNF4, RNF114, KEAP1, and CIAP1/XIAP, suggesting that this class is broadly targetable (Gechijian et al., 2018; Spradlin et al., 2019; Sun et al., 2014; Ward et al., 2019; Winkel et al., 2015; Zhang et al., 2019). Moreover, recent advances in medicinal chemistry, including proteolysis targeting chimeras (PROTACs), provide additional approaches to degrade challenging targets. Biochemical characterization has revealed EWS/FLI to be a highly disordered protein, which makes it difficult to determine the conformational structure at atomic resolution (Ng et al., 2007; Uren et al., 2004). Indeed, no crystal structure of the native EWS/FLI has been solved to date. It is possible that TRIM8 binding can help stabilize the EWS/FLI structure and that co-crystallization of the EWS/FLI-TRIM8 complex may be feasible. Importantly, future investigations utilizing an in vitro reconstitution strategy to express full-length and small fragments of EWS/FLI and TRIM8 will be critical for a more in-depth characterization of their interaction and the development of small molecules that can specifically inhibit the EWS/FLI-TRIM8 interaction. Moreover, alternative strategies can be exploited, including chemical screens to identify “molecular glues” that can bring TRIM8 in close proximity to EWS/FLI for degradation, allosteric agonists that increase TRIM8 enzymatic activity to induce EWS/FLI degradation, and covalent fragment binders to serve as a starting point for drug development. All told, these examples provide optimism for the discovery of TRIM8 binders, which might be further developed into TRIM8 degraders.
Our finding of the “Goldilocks principle”—that too little or too much oncoprotein is lethal and “just-the-right” level is required for cancer cell survival—is likely important in other fusion-TF-driven cancers. Hence, the identification and investigation of mechanisms for fusion oncoprotein-specific regulators, such as E3 ligases, warrants further exploration and may offer new inroads into therapeutic intervention.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| GAPDH | Cell signaling | Cat#2118; RRID:AB_561053 |
| β-actin | Cell signaling | Cat#58169; RRID:AB_2750839 |
| HA | Cell signaling | Cat#3724; RRID:AB_1549585 |
| HA | Cell signaling | Cat#2367; RRID:AB_10691311 |
| V5 | Cell signaling | Cat#13202; RRID:AB_2687461 |
| Cleaved-PARP | Cell signaling | Cat#5625; RRID:AB_10699459 |
| GFP | Cell signaling | Cat#2555; RRID:AB_10692764 |
| FLAG | Cell signaling | Cat#14793; RRID:AB_2572291 |
| ERG | Cell signaling | Cat#97249; RRID:AB_2721841 |
| FLI | Abcam | Cat#15289; RRID:AB_301825 |
| NKX2-2 | Abcam | Cat#187375; RRID:AB_2889166 |
| FLAG-M2 | Sigma | Cat#F3165; RRID:AB_259529 |
| TRIM8 | Santa Cruz Biotechnology | Cat#sc-398878; RRID:AB_2889167 |
| EWS | Santa Cruz Biotechnology | Cat#sc-28327; RRID:AB_675526 |
| FLAG | Novus Biologicals | Cat#NBP1-06712; AB_1625981 |
| Ubiquitin | LifeSensors | Cat#VU191; RRID:AB_2716558 |
| Bacterial and virus strains | ||
| One Shot™ Stbl3 E.coli | ThermoFisher Scientific | Cat#C737303 |
| Biological samples | ||
| EW_PDX001 | Garcia-Dominguez et al.2018 (PMID: 30140378) | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Lipofectamine RNAiMAX | ThermoFisher Scientific | Cat#13778030 |
| dTAGV-1 | Nabet et al., 2020 (PMID: 32948771) | N/A |
| Dynabeads™ Protein G | Invitrogen | Cat#10004D |
| Dynabeads™ Protein A | Invitrogen | Cat#10001D |
| BD Pharmingen™ Transcription-Factor Buffer Set | FisherScientific | Cat#BDB562574 |
| FuGENE® HD Transfection Reagent | Promega | Cat#E2311 |
| UM402M: TUBE 2 (Magnetic Beads) | LifeSensors | Cat#UM402M |
| Janelia Fluor® 646 HaloTag® Ligands | Promega | Cat#GA1120 |
| Duolink® In Situ PLA® Probe Anti-Mouse MINUS Affinity purified Donkey anti-Mouse IgG (H+L) | Sigma | Cat#DUO92004 |
| Duolink® In Situ PLA® Probe Anti-Rabbit PLUS Affinity purified Donkey anti-Rabbit IgG (H+L) | Sigma | Cat#DUO92002 |
| Critical commercial assays | ||
| CellTiter-Glo® 2.0 Cell Viability Assay | Promega | Cat#G9241 |
| Duolink™ flowPLA Detection Kit - Green | Sigma | Cat#DUO94002 |
| Click-iT™ EdU Pacific Blue™ Flow Cytometry Assay Kit | ThermoFisher Scientific | Cat#C10418 |
| Deposited data | ||
| Raw and analyzed data | This study | GEO: GSE150244 |
| Flow cytometry-based CRISPR screen | This study | Table S1 |
| Proteomics experiment | This study | Table S2 |
| DepMap 20Q1 genome-scale CRISPR-Cas9 screening and cell line characterization data | Broad Institute DepMap |
https://depmap.org; https://figshare.com/articles/DepMap_20Q1_Public/11791698/3 |
| Experimental models: cell lines | ||
| EWS502 N-dTAG E/F | Nabet et al., 2020(PMID: 32948771) | N/A |
| TC32 N-dTAG TRIM8 | This study | N/A |
| TC32 C-dTAG TRIM8 | This study | N/A |
| TC71 N-dTAG TRIM8 | This study | N/A |
| TC71 C-dTAG TRIM8 | This study | N/A |
| 293T EWS/FLI-GFP-IRES-mCherry-1 | This study | N/A |
| Experimental models: organisms/strains | ||
| NU/NU Nude Mouse - Crl:NU-Foxn1nu | Charles River | Cat#088; RRID:IMSR_CRL:088 |
| NGS Mouse - NOD-scid IL2Rgammanull | The Jackson Laboratory | Cat#005557; RRID:IMSR_JAX:005557 |
| Oligonucleotides | ||
| sgNT: 5’ GTAGCGAACGTGTCCGGCGT3’ | This study | N/A |
| sgTRIM8-1: 5’ CTACCGCCTCTACCACTGCG 3’ | This study | N/A |
| sgTRIM8-2: 5’ GCACGTGGAGAAGCCGCCGG 3’ | This study | N/A |
| sgTRIM8-3: 5’ GCGCAGGCAGACCTTCTGCG 3’ | This study | N/A |
| ON-TARGETplus Non-targeting Control siRNA | Dharmacon | Cat#001810 |
| SMARTpool TRIM8 siRNA | Dharmacon | Cat#81603 |
| Recombinant DNA | ||
| Artichoke-EWS/FLI (EWS/FLI-GFP-IRES-mCherry) | This study | N/A |
| pLEX305-N-dTAG-EWS/FLI | Nabet et al., 2020 (PMID: 32948771) | N/A |
| pLEX305-N-dTAG-TRIM8 | This study | N/A |
| pLEX305-C-dTAG-TRIM8 | This study | N/A |
| pLX307-TRIM8-V5 | This study | N/A |
| pLX307-TRIM8ΔRING-V5 | This study | N/A |
| pLX307-TRIM8ΔB-box1-V5 | This study | N/A |
| pLX307-TRIM8ΔB-box2-V5 | This study | N/A |
| pLX307-TRIM8ΔCC-V5 | This study | N/A |
| pLX307-TRIM8ΔRFP-like-V5 | This study | N/A |
| pLX307-TRIM8-EGFP | This study | N/A |
| pLX307-EWS/FLI-3XFLAG | This study | N/A |
| pLX307-N-EWS-3XFLAG | This study | N/A |
| pLX307-C-FLI-3XFLAG | This study | N/A |
| pLX307-EWS/ERG-3XFLAG | This study | N/A |
| pLX307-ERG-3XFLAG | This study | N/A |
| pLX307-N1-FLI-3XFLAG | This study | N/A |
| pLX307-N2-FLI-3XFLAG | This study | N/A |
| pLX307-EWS/FLIΔC1-3XFLAG | This study | N/A |
| pLX307-EWS/FLIΔC2-3XFLAG | This study | N/A |
| pLX307-EWS/FLIΔC3-3XFLAG | This study | N/A |
| pLX307-EWS/FLIΔC4-3XFLAG | This study | N/A |
| pLX307-EWS/FLIΔC5-3XFLAG | This study | N/A |
| pLX307-EWS-C1-3XFLAG | This study | N/A |
| pLX307-EWS-C5-3XFLAG | This study | N/A |
| pLX307-EWS/FLI-K144R-3XFLAG | This study | N/A |
| pLX307-EWS/FLI-K334R-3XFLAG | This study | N/A |
| pLX307-EWS/FLI-K217R-3XFLAG | This study | N/A |
| pLX307-EWS/FLI-K240R-3XFLAG | This study | N/A |
| pLX307-EWS/FLI-K217/240R-3XFLAG | This study | N/A |
| pLX307-EWS/FLIΔC3-K144R-3XFLAG | This study | N/A |
| pLX307-HA-Ubiquitin | This study | N/A |
| pLIV-V5-EWS-FLI1 | Boulay et al., 2017 (PMID: 28844694) | N/A |
| pINDUCER20-EWS/FLI-HA | This study | N/A |
| Software and algorithms | ||
| limma v3.42.2 R package | Ritchie et al., 2015 (PMID: 25605792) | https://bioconductor.org/packages/release/bioc/html/limma.html |
| FASTQC v0.11.9 software | N/A | www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| multiqc v1.6 tool | Ewels et al., 2016 (PMID: 27312411) | https://multiqc.info |
| STAR v2.7.2b | Dobin et al., 2013 (PMID: 23104886) | https://github.com/alexdobin/STAR |
| ercc dashboard R package available from Bioconductor v3.10 | Munro et al., 2014 (PMID: 25254650) | https://bioconductor.org/packages/release/bioc/html/erccdashboard.html |
| SARTools v1.7.3 | Varet et al., 2016 (PMID: 27280887) | https://github.com/PF2-pasteur-fr/SARTools |
| featureCounts v1.6.3 method implemented in the Subread v2.0.0 R package | Liao et al., 2014 (PMID: 24227677) | http://subread.sourceforge.net |
| DESeq2 v1.24.0 | Love et al., 2014 (PMID: 25516281) | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| enricher function in the clusterProfiler v 3.14.3 R package | Yu et al., 2012 (PMID: 22455463) | https://www.bioconductor.org/packages/release/bioc/html/clusterProfiler.html |
| Proteome Discoverer 2.2 | ThermoFisher | RRID:SCR_014477 |
| Other | ||
| Code used to analyze data and generate figures | This study |
https://github.com/ndharia-broad/TRIM8_Ewing DOI: https://doi.org/10.6084/m9.figshare.14888382.v1 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Kimberly Stegmaier (Kimberly_stegmaier@dfci.harvard.edu).
Materials availability
Plasmids and cell lines generated in this study are available upon request.
Data and code availability
The raw data for flow cytometry-based CRISPR screen is available in Table S1. The raw data for proteomics experiment is available in Table S2. The RNA-seq data for this study is available for download from the Gene Expression Omnibus (GEO) repository (GSE150244).
The genomic data used for CRISPR-Cas9 dependency screening were taken from the DepMap 20Q1 release. CRISPR dependency data from Project Achilles can be downloaded from the Figshare repository (https://figshare.com/articles/DepMap_20Q1_Public/11791698/3). The custom code used to analyze the data is uploaded to Figshare repository (https://doi.org/10.6084/m9.figshare.14888382.v1) and are available at https://github.com/ndharia-broad/TRIM8_Ewing.
Experimental model and subject details
Cell lines
A673, SK-PN-DW, and HEK293T cells were grown in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% Fetal Bovine Serum (FBS) and 1% Penicillin-Streptomycin-Glutamine (PSQ). RDES and SK-N-EP1 were grown in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 15% Fetal Bovine Serum (FBS) and 1% Penicillin-Streptomycin-Glutamine (PSQ). TC32, TC71, MOLM14, and U937 cells were grown in Roswell Park Memorial Institute (RPMI) media supplemented with 10% FBS, and 1% PSQ. EWS502 cells were grown in RPMI supplemented with 15% FBS, and 1% PSQ. All cell lines have been STR-profiled at the Dana-Farber Cancer Institute. For antibiotic selection, puromycin (1 μg/ml), blasticidin (5 μg/ml), and G418 (500 μg/ml) were used. A patient-derived Ewing sarcoma xenograft (HSJD-ES-PDX-001) (Garcia-Dominguez et al., 2018) was generously provided by Dr. Jaume Mora (Sant Joan de Déu Hospital, Barcelona). To generate minimally passaged cell line (ES-PDX-001), PDX tumors were harvested, minced, filtered, and cultured. Immunoblot was performed to confirm EWS/FLI expression.
Generation of the EWS/FLI-GFP reporter cell line
To generate the EWS/FLI-GFP reporter line, 293T cells were infected with lentivirus encoding EWS/FLI-GFP-IRES-mCherry (Artichoke-EWS/FLI) and selected with puromycin. After several passages, cells with low GFP were sorted and single cell clones were generated. EWS/FLI-GFP expression and nuclear localization were confirmed by immunoblot and imaging. Artichoke-EWS/FLI was cloned using gateway cloning (Invitrogen).
Generation of dTAG cell lines
To generate the TRIM8 dTAG cell lines, TC32 and TC71 cells were co-infected with FKBP12F36V-2XHA tagged TRIM8 and sgRNAs targeting TRIM8 to replace endogenous TRIM8 with FKBP12F36V-2XHA tagged TRIM8. Cells were then selected with both puromycin (FKBP12F36V-2XHA tagged TRIM8) and blasticidin (sgRNA). PAM sequences in the FKBP12F36V-2XHA tagged TRIM8 construct were mutated by Gibson assembly (New England Biolabs) using the manufacturer’s instructions to prevent cutting from infected sgRNAs. pLEX_305-TRIM8-dTAG were generated by cloning TRIM8 into pLEX_305-N-dTAG and pLEX_305-C-dTAG using gateway recombination cloning technology (Invitrogen) as previously described (Nabet et al., 2020). dTAGV-1 was synthesized as previously described (Nabet et al., 2020).
In vivo xenograft studies
For xenograft assays, eight-week old female nude mice (RRID:IMSR_CRL:088) were used for TC32, TC138 and ES-PDX-001 cells and eight-week old female NSG mice (RRID:IMSR_JAX:005557) were used for TC71 cells. Mice were subcutaneously implanted with cells infected with the indicated constructs. Infected cells were selected with puromycin for at least 4 days before implantation into mice. The tumor growth was measured at least weekly by the caliper method. Animals were sacrificed when tumors reached 2 cm in any one dimension or reached humane endpoints such as ulcerations. All animal protocols were approved by the Dana-Farber Cancer Institute Animal Care and Use Committee. Nude mice were maintained according to institutional guidelines.
Method details
CRISPR-Cas9 knockout, lentiviral infection, and RNA interference
For CRISPR-Cas9 knockout, sgRNAs targeting specific genes were cloned into lentiCRISPR v2 plasmid. The sgRNA sequences are as follows: non-targeting (GTAGCGAACGTGTCCGGCGT), TRIM8-1 (CTACCGCCTCTACCACTGCG), TRIM8-2 (GCACGTGGAGAAGCCGCCGG), and TRIM8-3 (GCGCAGGCAGACCTTCTGCG). Cells were infected with lentivirus using polybrene (8 μg/mL). Lentiviral particles were produced in HEK293T cells. For RNA interference, siControl (Dharmacon, 001810) and siTRIM8 SMARTpool (Dharmacon, 81603) were transfected with Lipofectamine RNAiMAX reagent using the manufacturer’s instructions.
Cell viability and proliferation assays
Cell viability/growth was assessed by serial cell counting using trypan blue and the CellTiter-Glo luminescent assay kit (Promega) according to the manufacturer’s instructions. Luminescence was read on a Fluostar Omega Reader (BMG Labtech). Cell proliferation was assessed using Click-iT EdU kit (Thermo Fisher) according to the manufacturer’s instructions. Annexin V staining was performed with an APC Annexin V Apoptosis Detection Kit with 7-AAD (640930, BioLegend), according to the manufacturer's instructions.
Immunoblot and immunoprecipitation
For immunoblotting, cells were scraped and lysed with Cell Lysis Buffer (Cell Signaling Technology) supplemented with cOmplete, EDTA-free Protease Inhibitor Cocktail (Roche). Proteins were separated by SDS-PAGE and transferred to PVDF membranes, which were blocked with 5% milk in TBS-T and incubated with primary antibodies overnight. Membranes were washed 5 times with TBS-T for 5 min and incubated with the appropriate horseradish peroxidase-conjugated secondary antibodies (anti-mouse, 45000680 and anti-rabbit, 45000682 from Thermo Fischer Scientific). Primary antibodies used include: GAPDH (2118), β-actin (58,169), HA (3724), V5 (13,202), cleaved-PARP (5625), GFP (2555), FLAG (14,793) and ERG (97,249) from Cell Signaling; FLI (15,289) and NKX2-2 (187375) from Abcam; FLAG-M2 (F3165) from Sigma Aldrich; TRIM8 (398878) and EWS (28,327) from Santa Cruz; and Ubiquitin from LifeSensors (VU191). For immunoprecipitation, Protein A or G Dynabeads (Thermo Fisher) were pre-washed twice with TBS-T and blocked with 1% milk in TBS-T for 1 hr at room temperature. The beads were then washed twice and incubated with appropriate antibodies for at least 3 hr in 4⁰ C. The beads were washed twice and incubated with protein lysates overnight rotating in 4⁰ C. The beads were washed 3 times 15 min each with IP lysis buffer (Thermo Fisher) and proteins were eluted by adding 2X loading buffer and boiling for 5 min.
Proximity ligation assay (PLA)
For the PLA experiment, TRIM8 dTAG cells were first fixed and permeabilized using BD PharmingenTM Transcription-Factor Buffer Set (Thermo Fisher Scientific, 562574) according to the manufacturer's protocol. Fixed cells were used to perform PLA using the DuolinkTM flowPLA Detection Kit (Sigma Aldrich, DUO94002) according to the manufacturer's protocol. The amplification reaction was performed overnight to increase the signal due to low expression of TRIM8.
Ubiquitination assay
For 293T over-expression experiments, 293T cells were transfected with indicated plasmids for 48 hr using FuGENE 6 transfection reagent (Promega, E2691). Cells were scraped, pelleted, washed with PBS, and lysed by adding 200 μL of SDS ubiquitination lysis buffer (2% SDS, 20 mM N-Ethylmaleimide, 25 mM Tris-HCl pH 7.4, 150 mM NaCl, 1% NP-40, 1 mM EDTA, 5% glycerol), supplemented with protease and phosphatase inhibitors and boiled for 10 min. After sonication, the lysates were diluted 1 to 10 using ubiquitination lysis buffer (20 mM N-Ethylmaleimide, 25 mM Tris-HCl pH 7.4, 150 mM NaCl, 1% NP-40, 1 mM EDTA, 5% glycerol) and rotated for 30 min at 4⁰ C before proceeding with immunoprecipitation. For the TUBE enrichment experiments, TC71 cells were infected with either non-targeting or TRIM8-targeting sgRNA for 5 days, harvested, and lysed. Lysates were used to immunoprecipitate all polyubiquitinated proteins using TUBE2 beads (LifeSensors, UM402M) according to manufacturer's protocol and were immunoblotted.
Flow cytometry
All flow cytometry analytical experiments were conducted in the Hematologic Neoplasia Flow Cytometry core at the Dana-Farber Cancer Institute using a Fortessa X-20 (BD Biosciences) analyzer. An Aria II SORP (BD Biosciences) sorter was used for all Fluorescence-activated cell sorting (FACS) experiments.
Flow cytometry-based CRISPR-Cas9 screen
293T-Cas9 cells expressing EWS/FLI-GFP were infected with the genome-wide CRISPR Avana4 library at a 0.8 multiplicity of infection (MOI) in two biological replicates. Fifty million cells were infected per replicate and the guide representation was 500 cells per guide. Cells were passaged for 7 days, and 100 million cells per replicate were sorted into GFP high and GFP low populations using FACS. One hundred million pre-sort cells were collected for reference. 293T EWS/FLI-GFP cells infected with non-targeting and FLI1-targeting sgRNAs were used to guide gates for sorting.
PCR, deconvolution and analysis of genome-scale CRISPR screens
For all screens, genomic DNA (gDNA) was isolated using Maxi (Pre-sort samples) or Mini (sorted samples) kits according to the manufacturer's protocol (Qiagen). PCR and sequencing were performed as previously described (Doench et al., 2016; Piccioni et al., 2018). Samples were sequenced on a HiSeq2000 (Illumina) and the read counts normalized to reads per million and then log2 transformed. The log2 fold-change of each sgRNA was determined relative to the plasmid DNA (pDNA) for each biological replicate. For every gene, we calculated the average log-fold change between the 4 guides and the p values using a hypergeometric distribution without replacement based on the rank order of the log-fold-change of the perturbations.
CRISPR-Cas9 dependency screen analysis
The genome-scale CRISPR-Cas9 screens were performed using the Broad Institute's GeCKOv2 and Avana libraries (Doench et al., 2016; Sanjana et al., 2014). Forty-three cancer cell lines (including 11 Ewing sarcoma) were screened with the GeCKOv2 library, containing approximately 123,411 guides and an average of 6 guides per gene, while 739 cancer cell lines (including 14 Ewing sarcoma) were screened with the Avana library, containing 73,372 guides and an average of 4 guides per gene (Doench et al., 2016; Sanjana et al., 2014). The screens were conducted in a pooled experiment as previously described (Aguirre et al., 2016; Meyers et al., 2017). For both CRISPR-Cas9 datasets, genetic dependencies that are enriched in Ewing sarcoma cell lines were identified using linear-model analysis from the limma v3.42.2 R package (Ritchie et al., 2015) by performing a two-tailed t test for the difference in distribution of gene dependency scores in Ewing sarcoma compared to all other cell lines screened. Statistical significance was calculated as a q-value derived from the p value corrected for multiple hypothesis testing using the Benjamini & Hochberg method (https://www.jstor.org/stable/2346101). The context-specific enrichment for all dependencies in the Avana 20Q1 Public data were downloaded from depmap.org. A PubMed search was performed on April 23, 2020 using the context name and gene name for genetic dependencies with a p value <10–60 using the rentrez v1.2.2 R package.
RNA sequencing and gene set enrichment analysis
RNA was extracted using Qiagen RNeasy Plus Mini Kit (Cat. 74,134) according to manufacturer's instructions. ERCC RNA spike-in mix (ThermoFisher, Cat. 4456740) was added to the samples according to manufacturer's instructions. RNA sequencing library preparation and paired-end sequencing were performed by Novogene Corporation Inc. Quality control tests for the 150 bp paired-end human reads were performed using the FASTQC v0.11.9 software (www.bioinformatics.babraham.ac.uk/projects/fastqc/) and summarized with the multiqc v1.6 tool (Ewels et al., 2016). The human reads were mapped to the GRCh37/hg19 human genome using STAR v2.7.2b (Dobin et al., 2013) with standard parameters. Technical performance for differential expression was assessed using External RNA Controls Consortium (ERCC) spike-in ratio mixtures kit 1. The spike-in ERCC92 control reads were mapped to ERCC92.fa genome data using the same protocol employed for the human reads. The performance metrics were computed by using the ercc dashboard R package available from Bioconductor v3.10 (Munro et al., 2014). Quality control for the mapped reads and for replicate reproducibility were performed using SARTools v1.7.3 (Varet et al., 2016). Gene level reads were summarized by counting the reads that overlapped the gencode v19 annotated gene exons, by using the featureCounts v1.6.3 method implemented in the Subread v2.0.0 R package (Liao et al., 2014). Gene-level expression was estimated as log2(1 + transcripts per million (TPM)) (Wagner et al., 2012). Gene counts were then used to quantify differentially expressed genes between the experimental and control conditions using DESeq2 v1.24.0 based on the shrunken log2 fold changes and the approximate posterior estimation for GLM coefficients (apeglm v1.6) method for effect size (Love et al., 2014). Statistically significantly differentially expressed genes were identified as with an adjusted p value of <0.05 from DESeq2. Gene set enrichment analysis (GSEA) was performed on these lists of genes using the enricher function in the clusterProfiler v 3.14.3 R package (Yu et al., 2012) and mSigDB C2 curated gene set collection v7.0 (Subramanian et al., 2005). The RNA-seq data for this study is available for download from the Gene Expression Omnibus (GEO) repository (GSE150244).
TMT LC-MS3 mass spectrometry and data analysis
TC71 cells expressing FKBP12F36V-TRIM8 were treated with DMSO or 1 μM of dTAGV-1 for 6 hr in biological triplicates, or 1 μM pomalidomide for 6 hr and cells harvested by centrifugation. Lysis was performed by addition of Urea Lysis buffer (8 M Urea, 50 mM NaCl, 50 mM 4-(phydroxyethyl)-1-piperazineethanesulfonic acid (EPPS) pH 8.5, Protease and Phosphatase inhibitors from Roche) to the cell pellets followed by manual homogenization with 20 passes through a 21-gauge (1.25 in. long) needle. TMT LC-MS3 mass spectrometry sample preparation and analysis were performed as previously described (Donovan et al., 2018). Proteome Discoverer 2.2 (Thermo Fisher) was used for.RAW file processing, controlling peptide and protein level false discovery rates, assembling proteins from peptides, and protein quantification from peptides. MS/MS spectra were searched against a Uniprot human database (September 2016) containing C-dTAG TRIM8 in place of TRIM8 and EWS/FLI in place of FLI1 with both the forward and reverse sequences. Database search criteria were as follows: tryptic with two missed cleavages, a precursor mass tolerance of 20 ppm, fragment ion mass tolerance of 0.6 Da, static alkylation of cysteine (57.02146 Da), static TMT labeling of lysine residues and N-termini of peptides (229.16293 Da), and variable oxidation of methionine (15.99491 Da). TMT reporter ion intensities were measured using a 0.003 Da window around the theoretical m/z for each reporter ion in the MS3 scan. Peptide spectral matches with poor quality MS3 spectra were excluded from quantitation (summed signal-to-noise across 11 channels <200 and precursor isolation specificity <0.5). Reporter ion intensities were normalized and scaled using in-house scripts in the R framework (R Core Team, 2014). Statistical analysis was carried out using the limma package within the R framework (Ritchie et al., 2015).
Airyscan confocal super-resolution imaging and analyses
To prepare cell samples for simultaneous imaging of endogenous EWS/FLI and transiently expressed TRIM8-EGFP, we plated the knock-in A673 cells on 18 mm circular No. 1 cover glasses (VWR VistaVision, 16,004-300) and transfected the cells with a plasmid encoding TRIM8-EGFP using Lipofectamine 3000 (ThermoFisher, L3000001). 24 hrs after transfection, we labeled cells with 200 nM HaloTag ligand JF646 for 15 min and washed twice (for each wash: remove medium, rinse twice with PBS, incubate in fresh medium for 30 min). We then fixed cells with 4% paraformaldehyde, stained the cells with 8 μM Hoechst solution (ThermoFisher, 62,249), and mounted the cover glass on a microscope slide (Fisher Scientific, 12-550-15) with VECTASHIELD mounting medium (Vector Laboratories, H-1000) between the glass surfaces.
Fluorescence imaging of knock-in A673 cells was performed on an inverted laser scanning confocal microscope with Airyscan super-resolution capability (Zeiss, LSM 900 with Airyscan 2). We acquired z stacks of fixed cell samples with a slice interval of 0.2 μm on the microscope in the Airyscan super-resolution (SR) mode using a 40x oil objective (Zeiss Plan-Apochromat 40x/1.3 Oil DIC). 405 nm, 488 nm, and 633 nm lasers were used to excite the fluorescence of Hoechst-labeled nuclei, TRIM8-EGFP, and JF646-labeled EWS/FLI-Halo in the cells, respectively. Before acquiring any fluorescence image, we carefully set the laser intensity and microscope detectors to make sure that no pixel in the image was saturated. We used proper emission filters for three-color imaging and ensured no bleed-through between the three channels by imaging cell samples that contain only one of the three fluorophores (Hoechst, EGFP, JF646) under the three-color imaging settings.
To evaluate enrichment of TRIM8-EGFP at JF646-labeled EWS/FLI-Halo hubs, we performed maximum z projection for all the slices in the JF646-channel z stack that covers the nuclear fluorescence and manually located the center pixels of individual EWS/FLI hubs in the maximum JF646 image. Next, for each EWS/FLI hub, we searched in the JF646 stack for the slice where a square region (5 x 5 pixels) centering the hub center pixel has the highest total fluorescence intensity among all the slices in the stack. Then we cropped a square region (2.32 μm × 2.32 μm) centering the EWS/FLI hub center pixel from the above selected slice in the JF646 stack as well as from the slice with the same z position in the simultaneously acquired EGFP-channel z stack. We processed 6872 EWS/FLI hubs in 15 cells with various TRIM8-EGFP expression levels following the above procedure and averaged the cropped square images for both channels. To acquire the average fluorescence image of random nuclear locations, we opened the slice with a central z position in both JF646 and EGFP stacks of each cell, randomly selected N pixels within the cell nucleus in the JF646 image (N equals to 4 times the number of EWS/FLI hubs analyzed in the cell), cropped a square region (2.32 μm × 2.32 μm) centering each pixel from the slice in both channels, and averaged the cropped square images, which provides the baseline nuclear fluorescence image for both channels. We subtracted the baseline image from the average image centering EWS/FLI hubs in each channel to acquire the background-free two-color image in Figure 5E and plotted the radial profiles of EGFP and JF646 intensity surrounding the center pixel in the background-free image to acquire Figure 5F. The HaloTag ligand JF646 was a gift from Luke Lavis to the Tjian-Darzacq Lab.
Quantification and statistical analysis
Microsoft excel, GSEA v2.1.0, GraphPad PRISM 7, R, and Proteome Discoverer 2.2 software packages were used to perform the statistical analyses. Statistical tests used are specified in the figure legends. Errors bars represent standard deviation, unless otherwise stated. The threshold for statistical significance is p < 0.05, unless otherwise specified in the figure legends.
Data and software availability
RNA-sequencing data generated in the manuscript is deposited in GEO: GSE63473. All codes used for the manuscript are deposited in github: https://github.com/ndharia-broad/TRIM8_Ewing.
Acknowledgments
K.S. was supported by the National Cancer Institute R35 CA210030, R01 CA204915, and U54 CA231637, a St. Baldrick's Foundation Robert J. Arceci Innovation Award, and the Brian MacIsaac Sarcoma Foundation. B.K.A.S. was supported by Department of Defense PRCRP Horizon Award (CA181249), N.V.D. by the Julia's Legacy of Hope St. Baldrick's Foundation Fellowship, X.D. by U54 CA231641-01, and B.N. by the American Cancer Society Postdoctoral Fellowship PF-17-010-01-CDD. B.N. and N.S.G. were supported by the Katherine L. and Steven C. Pinard Research Fund and the Hale Center for Pancreatic Cancer. We thank Robert Tjian for scientific input on the super-resolution imaging experiments.
Author contributions
B.K.A.S. conceived the study, designed, and performed the experiments, analyzed the data, interpreted the results and wrote the manuscript. N.V.D. analyzed the CRISPR dependency screens, flow cytometry-based CRISPR screen, and RNA-seq data and interpreted the results. S.L. assisted with designing and performing the immunoprecipitation and the ubiquitination experiments, analyzed the data, and interpreted the results. K.A.D. performed mass spectrometry, analyzed the data, and interpreted the results. S.C. performed Airyscan confocal super-resolution imaging, analyzed the data, and interpreted the results. A.R. and A.C. performed in vivo studies, analyzed the data, and interpreted the results. L.R., G.A., B.A., and A.H. provided technical assistance, analyzed the data, and interpreted the results. B.N., F.M.F., and N.S.G. synthesized and provided the dTAGV-1 molecule. B.S. and E.J.W assisted with TRIM8 CRISPR knockout and EWS/FLI overexpression experiments, analyzed the data, and interpreted the results. J.S. provided technical assistance and intellectual input. F.P. assisted with designing the flow cytometry-based CRISPR screen, analyzed the data, and interpreted the results. E.S.F. analyzed the mass spectrometry data, interpreted the results, and supervised the study. K.S. conceived the study, designed the experiments, interpreted the results, and supervised and funded the study. All authors read, edited, and approved the final manuscript.
Declaration of interests
K.S. has funding from Novartis Institutes for BioMedical Research, consults for and has stock options in Auron Therapeutics and served as an advisor for Kronos Bio and AstraZeneca all on topics unrelated to this manuscript. E.S.F. is a founder, scientific advisory board (SAB) member, and equity holder of Civetta Therapeutics, Jengu Therapeutics (board member), and Neomorph Inc., an equity holder in C4 Therapeutics, and is a consultant to Novartis, Sanofi, EcoR1 capital, Astellas, and Deerfield. The Fischer lab receives or has received research funding from Novartis, Ajax, Deerfield, and Astellas on topics unrelated to this manuscript. B.N. is an inventor on patent applications related to the dTAG system (WO/2017/024318, WO/2017/024319, WO/2018/148440, and WO/2018/148443). F.M.F., B.N., and N.S.G. are inventors on a patent related to the dTAG system and molecules described in this manuscript (WO/2020/146250). F.M.F. is a consultant to RA Capital and Santi Therapeutics. N.S.G. is a founder, SAB member and equity holder in Syros, C4, Allorion, Jengu, B2S, Inception, EoCys, Larkspur (board member), and Soltego (board member). The Gray lab receives or has received research funding from Novartis, Takeda, Astellas, Taiho, Jansen, Kinogen, Arbella, Deerfield, and Sanofi on topics unrelated to this manuscript. N.V.D. is a current employee of Genentech, Inc., a member of the Roche Group. K.A.D. is a consultant to Kronos Bio.
Inclusion and diversity
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science. One or more of the authors of this paper self-identifies as a member of the LGBTQ+ community. One or more of the authors of this paper received support from a program designed to increase minority representation in science.
Published: July 29, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ccell.2021.07.003.
Supplemental information
References
- Adane B., Alexe G., Seong B.K.A., Lu D., Hwang E.E., Hnisz D., Lareau C.A., Ross L., Lin S., Dela Cruz F.S. STAG2 loss rewires oncogenic and developmental programs to promote metastasis in Ewing sarcoma. Cancer Cell. 2021;39:827–844 e810. doi: 10.1016/j.ccell.2021.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguirre A.J., Meyers R.M., Weir B.A., Vazquez F., Zhang C.Z., Ben-David U., Cook A., Ha G., Harrington W.F., Doshi M.B. Genomic copy number dictates a gene-independent cell response to CRISPR/Cas9 targeting. Cancer Discov. 2016;6:914–929. doi: 10.1158/2159-8290.CD-16-0154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aynaud M.M., Mirabeau O., Gruel N., Grossetete S., Boeva V., Durand S., Surdez D., Saulnier O., Zaidi S., Gribkova S. Transcriptional programs define intratumoral heterogeneity of Ewing sarcoma at single-cell resolution. Cell Rep. 2020;30:1767–1779 e1766. doi: 10.1016/j.celrep.2020.01.049. [DOI] [PubMed] [Google Scholar]
- Behan F.M., Iorio F., Picco G., Goncalves E., Beaver C.M., Migliardi G., Santos R., Rao Y., Sassi F., Pinnelli M. Prioritization of cancer therapeutic targets using CRISPR-Cas9 screens. Nature. 2019 doi: 10.1038/s41586-019-1103-9. [DOI] [PubMed] [Google Scholar]
- Boulay G., Sandoval G.J., Riggi N., Iyer S., Buisson R., Naigles B., Awad M.E., Rengarajan S., Volorio A., McBride M.J. Cancer-specific retargeting of BAF complexes by a prion-like domain. Cell. 2017;171:163–178 e119. doi: 10.1016/j.cell.2017.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner J.C., Feng F.Y., Han S., Patel S., Goyal S.V., Bou-Maroun L.M., Liu M., Lonigro R., Prensner J.R., Tomlins S.A., Chinnaiyan A.M. PARP-1 inhibition as a targeted strategy to treat Ewing's sarcoma. Cancer Res. 2012;72:1608–1613. doi: 10.1158/0008-5472.CAN-11-3648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brohl A.S., Solomon D.A., Chang W., Wang J., Song Y., Sindiri S., Patidar R., Hurd L., Chen L., Shern J.F. The genomic landscape of the Ewing sarcoma family of tumors reveals recurrent STAG2 mutation. PLoS Genet. 2014;10:e1004475. doi: 10.1371/journal.pgen.1004475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong S., Dugast-Darzacq C., Liu Z., Dong P., Dailey G.M., Cattoglio C., Heckert A., Banala S., Lavis L., Darzacq X., Tjian R. Imaging dynamic and selective low-complexity domain interactions that control gene transcription. Science. 2018;361 doi: 10.1126/science.aar2555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocco E., Scaltriti M., Drilon A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat. Rev. Clin. Oncol. 2018;15:731–747. doi: 10.1038/s41571-018-0113-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crompton B.D., Stewart C., Taylor-Weiner A., Alexe G., Kurek K.C., Calicchio M.L., Kiezun A., Carter S.L., Shukla S.A., Mehta S.S. The genomic landscape of pediatric Ewing sarcoma. Cancer Discov. 2014;4:1326–1341. doi: 10.1158/2159-8290.CD-13-1037. [DOI] [PubMed] [Google Scholar]
- Delattre O., Zucman J., Plougastel B., Desmaze C., Melot T., Peter M., Kovar H., Joubert I., de Jong P., Rouleau G. Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature. 1992;359:162–165. doi: 10.1038/359162a0. [DOI] [PubMed] [Google Scholar]
- Deshaies R.J. Proteotoxic crisis, the ubiquitin-proteasome system, and cancer therapy. BMC Biol. 2014;12:94. doi: 10.1186/s12915-014-0094-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dharia N.V., Kugener G., Guenther L.M., Malone C.F., Durbin A.D., Hong A.L., Howard T.P., Bandopadhayay P., Wechsler C.S., Fung I. A first-generation pediatric cancer dependency map. Nat. Genet. 2021;53:529–538. doi: 10.1038/s41588-021-00819-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench J.G., Fusi N., Sullender M., Hegde M., Vaimberg E.W., Donovan K.F., Smith I., Tothova Z., Wilen C., Orchard R. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016;34:184–191. doi: 10.1038/nbt.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donovan K.A., An J., Nowak R.P., Yuan J.C., Fink E.C., Berry B.C., Ebert B.L., Fischer E.S. Thalidomide promotes degradation of SALL4, a transcription factor implicated in Duane Radial Ray syndrome. Elife. 2018;7 doi: 10.7554/eLife.38430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewels P., Magnusson M., Lundin S., Kaller M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics. 2016;32:3047–3048. doi: 10.1093/bioinformatics/btw354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Dominguez D.J., Hontecillas-Prieto L., Rodriguez-Nunez P., Pascual-Pasto G., Vila-Ubach M., Garcia-Mejias R., Robles M.J., Tirado O.M., Mora J., Carcaboso A.M., de Alava E. The combination of epigenetic drugs SAHA and HCI-2509 synergistically inhibits EWS-FLI1 and tumor growth in Ewing sarcoma. Oncotarget. 2018;9:31397–31410. doi: 10.18632/oncotarget.25829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gechijian L.N., Buckley D.L., Lawlor M.A., Reyes J.M., Paulk J., Ott C.J., Winter G.E., Erb M.A., Scott T.G., Xu M. Functional TRIM24 degrader via conjugation of ineffectual bromodomain and VHL ligands. Nat. Chem. Biol. 2018;14:405–412. doi: 10.1038/s41589-018-0010-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gierisch M.E., Pfistner F., Lopez-Garcia L.A., Harder L., Schafer B.W., Niggli F.K. Proteasomal degradation of the EWS-FLI1 fusion protein is regulated by a single lysine residue. J. Biol. Chem. 2016;291:26922–26933. doi: 10.1074/jbc.M116.752063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorthi A., Romero J.C., Loranc E., Cao L., Lawrence L.A., Goodale E., Iniguez A.B., Bernard X., Masamsetti V.P., Roston S. EWS-FLI1 increases transcription to cause R-loops and block BRCA1 repair in Ewing sarcoma. Nature. 2018;555:387–391. doi: 10.1038/nature25748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hjerpe R., Aillet F., Lopitz-Otsoa F., Lang V., England P., Rodriguez M.S. Efficient protection and isolation of ubiquitylated proteins using tandem ubiquitin-binding entities. EMBO Rep. 2009;10:1250–1258. doi: 10.1038/embor.2009.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iniguez A.B., Stolte B., Wang E.J., Conway A.S., Alexe G., Dharia N.V., Kwiatkowski N., Zhang T., Abraham B.J., Mora J. EWS/FLI confers tumor cell synthetic lethality to CDK12 inhibition in Ewing sarcoma. Cancer Cell. 2018;33:202–216 e206. doi: 10.1016/j.ccell.2017.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantarjian H., Sawyers C., Hochhaus A., Guilhot F., Schiffer C., Gambacorti-Passerini C., Niederwieser D., Resta D., Capdeville R., Zoellner U. Hematologic and cytogenetic responses to imatinib mesylate in chronic myelogenous leukemia. N. Engl. J. Med. 2002;346:645–652. doi: 10.1056/NEJMoa011573. [DOI] [PubMed] [Google Scholar]
- Kronke J., Udeshi N.D., Narla A., Grauman P., Hurst S.N., McConkey M., Svinkina T., Heckl D., Comer E., Li X. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science. 2014;343:301–305. doi: 10.1126/science.1244851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q., Yan J., Mao A.P., Li C., Ran Y., Shu H.B., Wang Y.Y. Tripartite motif 8 (TRIM8) modulates TNFalpha- and IL-1beta-triggered NF-kappaB activation by targeting TAK1 for K63-linked polyubiquitination. Proc. Natl. Acad. Sci. U S A. 2011;108:19341–19346. doi: 10.1073/pnas.1110946108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin A.W., Barradas M., Stone J.C., van Aelst L., Serrano M., Lowe S.W. Premature senescence involving p53 and p16 is activated in response to constitutive MEK/MAPK mitogenic signaling. Genes Dev. 1998;12:3008–3019. doi: 10.1101/gad.12.19.3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y., Smyth G.K., Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- Lo-Coco F., Avvisati G., Vignetti M., Thiede C., Orlando S.M., Iacobelli S., Ferrara F., Fazi P., Cicconi L., Di Bona E. Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N. Engl. J. Med. 2013;369:111–121. doi: 10.1056/NEJMoa1300874. [DOI] [PubMed] [Google Scholar]
- Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu G., Middleton R.E., Sun H., Naniong M., Ott C.J., Mitsiades C.S., Wong K.K., Bradner J.E., Kaelin W.G., Jr. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science. 2014;343:305–309. doi: 10.1126/science.1244917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maarifi G., Smith N., Maillet S., Moncorge O., Chamontin C., Edouard J., Sohm F., Blanchet F.P., Herbeuval J.P., Lutfalla G. TRIM8 is required for virus-induced IFN response in human plasmacytoid dendritic cells. Sci. Adv. 2019;5:eaax3511. doi: 10.1126/sciadv.aax3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon S.B. MYC and the control of apoptosis. Cold Spring Harb. Perspect. Med. 2014;4:a014407. doi: 10.1101/cshperspect.a014407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers R.M., Bryan J.G., McFarland J.M., Weir B.A., Sizemore A.E., Xu H., Dharia N.V., Montgomery P.G., Cowley G.S., Pantel S. Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat Genet. 2017;49:1779–1784. doi: 10.1038/ng.3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitelman F., Johansson B., Mertens F. The impact of translocations and gene fusions on cancer causation. Nat. Rev. Cancer. 2007;7:233–245. doi: 10.1038/nrc2091. [DOI] [PubMed] [Google Scholar]
- Munro S.A., Lund S.P., Pine P.S., Binder H., Clevert D.A., Conesa A., Dopazo J., Fasold M., Hochreiter S., Hong H. Assessing technical performance in differential gene expression experiments with external spike-in RNA control ratio mixtures. Nat Commun. 2014;5:5125. doi: 10.1038/ncomms6125. [DOI] [PubMed] [Google Scholar]
- Nabet B., Ferguson F.M., Seong B.K.A., Kuljanin M., Leggett A.L., Mohardt M.L., Robichaud A., Conway A.S., Buckley D.L., Mancias J.D. Rapid and direct control of target protein levels with VHL-recruiting dTAG molecules. Nat. Commun. 2020;11:4687. doi: 10.1038/s41467-020-18377-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabet B., Roberts J.M., Buckley D.L., Paulk J., Dastjerdi S., Yang A., Leggett A.L., Erb M.A., Lawlor M.A., Souza A. The dTAG system for immediate and target-specific protein degradation. Nat. Chem. Biol. 2018;14:431–441. doi: 10.1038/s41589-018-0021-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng K.P., Potikyan G., Savene R.O., Denny C.T., Uversky V.N., Lee K.A. Multiple aromatic side chains within a disordered structure are critical for transcription and transforming activity of EWS family oncoproteins. Proc. Natl. Acad. Sci. U S A. 2007;104:479–484. doi: 10.1073/pnas.0607007104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okumura F., Matsunaga Y., Katayama Y., Nakayama K.I., Hatakeyama S. TRIM8 modulates STAT3 activity through negative regulation of PIAS3. J. Cell Sci. 2010;123:2238–2245. doi: 10.1242/jcs.068981. [DOI] [PubMed] [Google Scholar]
- Piccioni F., Younger S.T., Root D.E. Pooled lentiviral-delivery genetic screens. Curr Protoc Mol Biol. 2018;121 doi: 10.1002/cpmb.52. 32 31 31-32 31 21. [DOI] [PubMed] [Google Scholar]
- R Core Team R: A language and environment for statistical computing. 2014. https://www.R-project.org/
- Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana N.E., Shalem O., Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods. 2014;11:783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuijers J., Manteiga J.C., Weintraub A.S., Day D.S., Zamudio A.V., Hnisz D., Lee T.I., Young R.A. Transcriptional dysregulation of MYC reveals common enhancer-docking mechanism. Cell Rep. 2018;23:349–360. doi: 10.1016/j.celrep.2018.03.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano M., Lin A.W., McCurrach M.E., Beach D., Lowe S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- Shangary S., Wang S. Small-molecule inhibitors of the MDM2-p53 protein-protein interaction to reactivate p53 function: a novel approach for cancer therapy. Annu. Rev. Pharmacol. Toxicol. 2009;49:223–241. doi: 10.1146/annurev.pharmtox.48.113006.094723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw A.T., Kim D.W., Mehra R., Tan D.S., Felip E., Chow L.Q., Camidge D.R., Vansteenkiste J., Sharma S., De Pas T. Ceritinib in ALK-rearranged non-small-cell lung cancer. N. Engl. J. Med. 2014;370:1189–1197. doi: 10.1056/NEJMoa1311107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spradlin J.N., Hu X., Ward C.C., Brittain S.M., Jones M.D., Ou L., To M., Proudfoot A., Ornelas E., Woldegiorgis M. Harnessing the anti-cancer natural product nimbolide for targeted protein degradation. Nat. Chem. Biol. 2019;15:747–755. doi: 10.1038/s41589-019-0304-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su X.A., Ma D., Parsons J.V., Replogle J.M., Amatruda J.F., Whittaker C.A., Stegmaier K., Amon A. RAD21 is a driver of chromosome 8 gain in Ewing sarcoma to mitigate replication stress. Genes Dev. 2021;35:556–572. doi: 10.1101/gad.345454.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H., Lu J., Liu L., Yang C.Y., Wang S. Potent and selective small-molecule inhibitors of cIAP1/2 proteins reveal that the binding of Smac mimetics to XIAP BIR3 is not required for their effective induction of cell death in tumor cells. ACS Chem. Biol. 2014;9:994–1002. doi: 10.1021/cb400889a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surdez D., Zaidi S., Grossetete S., Laud-Duval K., Ferre A.S., Mous L., Vourc'h T., Tirode F., Pierron G., Raynal V. STAG2 mutations alter CTCF-anchored loop extrusion, reduce cis-regulatory interactions and EWSR1-FLI1 activity in Ewing sarcoma. Cancer Cell. 2021;39:810–826 e819. doi: 10.1016/j.ccell.2021.04.001. [DOI] [PubMed] [Google Scholar]
- Tirode F., Surdez D., Ma X., Parker M., Le Deley M.C., Bahrami A., Zhang Z., Lapouble E., Grossetete-Lalami S., Rusch M. Genomic landscape of Ewing sarcoma defines an aggressive subtype with co-association of STAG2 and TP53 mutations. Cancer Discov. 2014;4:1342–1353. doi: 10.1158/2159-8290.CD-14-0622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tisato V., Voltan R., Gonelli A., Secchiero P., Zauli G. MDM2/X inhibitors under clinical evaluation: perspectives for the management of hematological malignancies and pediatric cancer. J. Hematol. Oncol. 2017;10:133. doi: 10.1186/s13045-017-0500-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsherniak A., Vazquez F., Montgomery P.G., Weir B.A., Kryukov G., Cowley G.S., Gill S., Harrington W.F., Pantel S., Krill-Burger J.M. Defining a cancer dependency map. Cell. 2017;170:564–576 e516. doi: 10.1016/j.cell.2017.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uren A., Tcherkasskaya O., Toretsky J.A. Recombinant EWS-FLI1 oncoprotein activates transcription. Biochemistry. 2004;43:13579–13589. doi: 10.1021/bi048776q. [DOI] [PubMed] [Google Scholar]
- Varet H., Brillet-Gueguen L., Coppee J.Y., Dillies M.A. SARTools: A DESeq2- and EdgeR-based R pipeline for comprehensive differential analysis of RNA-Seq data. PLoS One. 2016;11:e0157022. doi: 10.1371/journal.pone.0157022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wachter F., Morgan A.M., Godes M., Mourtada R., Bird G.H., Walensky L.D. Mechanistic validation of a clinical lead stapled peptide that reactivates p53 by dual HDM2 and HDMX targeting. Oncogene. 2017;36:2184–2190. doi: 10.1038/onc.2016.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner G.P., Kin K., Lynch V.J. Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples. Theory Biosci. 2012;131:281–285. doi: 10.1007/s12064-012-0162-3. [DOI] [PubMed] [Google Scholar]
- Ward C.C., Kleinman J.I., Brittain S.M., Lee P.S., Chung C.Y.S., Kim K., Petri Y., Thomas J.R., Tallarico J.A., McKenna J.M. Covalent ligand screening uncovers a RNF4 E3 ligase recruiter for targeted protein degradation applications. ACS Chem. Biol. 2019;14:2430–2440. doi: 10.1021/acschembio.8b01083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willumsen B.M., Christensen A., Hubbert N.L., Papageorge A.G., Lowy D.R. The p21 ras C-terminus is required for transformation and membrane association. Nature. 1984;310:583–586. doi: 10.1038/310583a0. [DOI] [PubMed] [Google Scholar]
- Winkel A.F., Engel C.K., Margerie D., Kannt A., Szillat H., Glombik H., Kallus C., Ruf S., Gussregen S., Riedel J. Characterization of RA839, a noncovalent small molecule binder to Keap1 and selective activator of Nrf2 signaling. J. Biol. Chem. 2015;290:28446–28455. doi: 10.1074/jbc.M115.678136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye W., Hu M.M., Lei C.Q., Zhou Q., Lin H., Sun M.S., Shu H.B. TRIM8 negatively regulates TLR3/4-mediated innate immune response by blocking TRIF-TBK1 interaction. J. Immunol. 2017;199:1856–1864. doi: 10.4049/jimmunol.1601647. [DOI] [PubMed] [Google Scholar]
- Yu G., Wang L.G., Han Y., He Q.Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Crowley V.M., Wucherpfennig T.G., Dix M.M., Cravatt B.F. Electrophilic PROTACs that degrade nuclear proteins by engaging DCAF16. Nat. Chem. Biol. 2019;15:737–746. doi: 10.1038/s41589-019-0279-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J., Lallemand-Breitenbach V., de The H. Pathways of retinoic acid- or arsenic trioxide-induced PML/RARalpha catabolism, role of oncogene degradation in disease remission. Oncogene. 2001;20:7257–7265. doi: 10.1038/sj.onc.1204852. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw data for flow cytometry-based CRISPR screen is available in Table S1. The raw data for proteomics experiment is available in Table S2. The RNA-seq data for this study is available for download from the Gene Expression Omnibus (GEO) repository (GSE150244).
The genomic data used for CRISPR-Cas9 dependency screening were taken from the DepMap 20Q1 release. CRISPR dependency data from Project Achilles can be downloaded from the Figshare repository (https://figshare.com/articles/DepMap_20Q1_Public/11791698/3). The custom code used to analyze the data is uploaded to Figshare repository (https://doi.org/10.6084/m9.figshare.14888382.v1) and are available at https://github.com/ndharia-broad/TRIM8_Ewing.
RNA-sequencing data generated in the manuscript is deposited in GEO: GSE63473. All codes used for the manuscript are deposited in github: https://github.com/ndharia-broad/TRIM8_Ewing.