

Abstract
Non-small cell lung cancer (NSCLC) has a poor prognosis with a 5 year survival rate of only ~ 10%. Important driver mutations underlying NSCLC affect the epidermal growth factor receptor (EGFR) causing the constitutive activation of its tyrosine kinase domain. There are efficient EGFR tyrosine kinase inhibitors (TKIs), but patients develop inevitably a resistance against these drugs. On the other hand, KCa3.1 channels contribute to NSCLC progression so that elevated KCa3.1 expression is a strong predictor of poor NSCLC patient prognosis. The present study tests whether blocking KCa3.1 channels increases the sensitivity of NSCLC cells towards the EGFR TKI erlotinib and overcomes drug resistance. mRNA expression of KCa3.1 channels in erlotinib-sensitive and -resistant NSCLC cells was analysed in datasets from Gene expression omnibus (GEO) and ArrayExpress. We assessed proliferation and migration of NSCLC cells. These (live cell-imaging) experiments were complemented by patch clamp experiments and Western blot analyses. We identified three out of four datasets comparing erlotinib-sensitive and -resistant NSCLC cells which revealed an altered expression of KCa3.1 mRNA in erlotinib-resistant NSCLC cells. Therefore, we evaluated the combined effect of erlotinib and the KCa3.1 channel inhibition with sencapoc. Erlotinib elicits a dose-dependent inhibition of migration and proliferation of NSCLC cells. The simultaneous application of the KCa3.1 channel blocker senicapoc increases the sensitivity towards a low dose of erlotinib (300 nmol/L) which by itself has no effect on migration and proliferation. Partial erlotinib resistance can be overcome by KCa3.1 channel blockade. The sensitivity towards erlotinib as well as the potentiating effect of KCa3.1 blockade is further increased by mimicking hypoxia. Our results suggest that KCa3.1 channel blockade may constitute a therapeutic concept for treating NSCLC and overcome EGFR TKI resistance. We propose that this is due to complementary mechanisms of action of both blockers.
Subject terms: Physiology, Cancer, Cancer therapy, Lung cancer
Introduction
Non-small cell lung cancer (NSCLC) accounts for the majority of all lung cancers1. The prognosis of lung cancer patients is still rather poor. Even after complete resection of early stage NSCLC ~ 50% of the patients will die from recurring tumors, and the mean 5 year survival rate of European lung cancer patients is only 13%2. It has become clear, that an efficient therapy needs to take into account not only the pathological classification1 but also the presence of genetic alterations3, 4. In this context, mutations of the epidermal growth factor receptor (EGFR) are of great interest. They are found in 15–50% of the non-squamous tumors. Most mutations occur in the tyrosine kinase domain of the EGFR causing its constitutive activation5. The knowledge of the presence or absence of EGFR mutations has great therapeutic implications since it determines the sensitivity towards blockers of the EGFR tyrosine kinase domain like erlotinib. Unfortunately, almost all patients initially responding to erlotinib and related compounds eventually develop acquired resistance towards these drugs6, 7. Thus, there is still an unmet need for improved therapeutic concepts for the treatment of lung cancer.
We have recently shown that KCa3.1 channels play an important pathophysiological role in processes underlying NSCLC progression. They are upregulated in NSCLC biopsies, and the increased expression as well as the hypomethylation of its promoter are strong predictors for poor patient prognosis8 (reviewed in9). Increased KCa3.1 channel expression promotes aggressiveness of NSCLC cells by controlling their proliferation and migration. Mechanistically, this is due to the indirect impact of KCa3.1 channels on the intracellular Ca2+ concentration. Consequently, a blocker of KCa3.1 channels, senicapoc, strongly slows down tumor progression when NSCLC cells are transplanted into mice8. Moreover, KCa3.1 channels regulate the expression of the intercellular adhesion molecule (ICAM)1 and adhesion of NSCLC cells to endothelial cells which is an important step of the metastatic cascade10. While these findings clearly point to a role of KCa3.1 channels in NSCLC progression, they do not address the clinical problem of resistance against tyrosine kinase inhibitors (TKI).
However, there is increasing evidence that ion channels are important players in (acquired) chemo- and/or radioresistance of cancer cells11–14. In ovarian cancer inhibition or silencing of KCa1.1 channels leads to increased cisplatin resistance15. In melanoma cells the KCa3.1 inhibitor TRAM-34 overcomes the resistance towards the BRAF inhibitor vemurafenib16. In NSCLC a link between ion channels and chemoresistance has been drawn for a voltage-gated K+ channel (KV1.1;17).
Here we show that KCa3.1 channel expression differs between erlotinib-sensitive and -resistant NSCLC cells. We present evidence that KCa3.1 and KCa1.1 channel inhibition with senicapoc can exert additive inhibitory effects with the TKI erlotinib. It may even partially overcome the resistance against erlotinib.
Methods
Bioinformatic analysis
We used the Gene Expression Omnibus database (GEO; http://www.ncbi.nlm.nih.gov/geo), and ArrayExpress transcriptomic repository (https://www.ebi.ac.uk/arrayexpress/) to compare drug sensitive and resistant cell lines18, 19. We searched for microarray datasets allowing the comparison of human erlotinib-sensitive NSCLC cell lines with human erlotinib-resistant NSCLC cell lines. The raw data of four datasets (GSE31625, GSE38310, GSE38404 and GSE80344) were processed using the Bioconductor package in R (Gentleman et al. 2004). They were background corrected and then normalized with a log2 conversion. A matrix was created for each dataset after the processing optimization, in order to compare expression data from erlotinib-sensitive and erlotinib-resistant NSCLC cell lines. The analysis of the matrix was carried out with a linear model (limma software in R), in order to identify the differentially expressed genes in the two groups20. An impartial cut-off of 0.05 was imposed on the section for identifying the differentially expressed genes between the two groups.
Cell culture
A549 lung adenocarcinoma cells were cultured at 37 °C in a 5% CO2 atmosphere in Dulbecco’s modified Eagle’s medium (Invitrogen, Carlsbad, CA) supplemented with 10% fetal calf serum. The generation of the highly aggressive A549 cell line, designated as A549-3R, was described previously21. H1975 cells were cultured in RPMI1640 medium supplemented with L-glutamine and 10% FCS. Hypoxia was produced by incubating the A549 cells in 5% CO2 and 0.2% O2. To obtain A549-3R cells that are partially resistant against erlotinib they were cultured in the presence of 10 µmol/L erlotinib (Chemos GmbH, Germany) and 10 ng/mL EGF (Sigma Aldrich) for nine weeks. Thereby, we wanted to mimick the situation encoutered in patients whose serum levels were determined to be ~ 2 ng/ml22. After thawing cryoconserved erlotinib-resistant A549-3R cells they were cultured for two weeks in the presence of erlotinib and EGF prior to use for experiments.
Proliferation
50.000 cells each were seeded in 12-well plates in triplicate (Falcon Multiwell 12 Well Tissue Culture Plate, Becton Dickinson, NJ, USA). After 24 h growth in standard cell culture medium, cells were starved in serum-free medium for another 24 h. Thereafter, experimental media supplemented with 10% FCS, 100 ng/mL EGF and drugs (erlotinib, senicapoc, DMSO, dimethyl-oxaloyl-glycine (DMOG; 1 mmol/l) as indicated) were added. Experimental media were changed every other 24 h. Cells were counted in a Neubauer chamber after serum starvation (t = 0) and following trypsinization after 24, 48 and 72 h, respectively.
Cell migration
Cell migration experiments were performed as described previously8. Briefly, A549-3R cells were seeded on matrices of the following composition: RPMI (10.4 g/L), HEPES (10 mmol/L), laminin (20 µg/mL; Sigma Aldrich), fibronectin (40 µg/µL; Becton Dickinson, USA), collagen IV (5.4 µg/µL; Becton Dickinson), collagen III (12 µg/µL; Becton Dickinson), collagen I (800 µg/mL; Biochrom, Germany), H2O ad 1000 µL, titrated to pH 7.4 with 1 N NaOH. We used ~ 200 µL of the matrix per 12.5 cm2 culture flask. The next day A549-3R cells were seeded onto the matrix. They adhered to the matrix for 4 h prior to the start of the experiment. The culture medium used for the migration experiments was supplemented with 100 ng/mL EGF and different concentrations of senicapoc and/or erlotinib. The solvent, DMSO, was added in equal amounts (1:1000) for control experiments. Images were acquired in 10 min intervals for a total duration of 10 h and migration was quantified from the movement of the cell center as described previously23. Migration speed is calculated as a three point difference quotient for each time interval, translocation refers to the net movement during the course of the experiment, i. e. the distance between the start and end position of the cell. The directionality is determined by dividing the translocation by the total path length covered by the cell during the course of the experiment. Thus, directionality can be seen as a measure of the tortuosity of the cell path.
Single-cell electrophysiology
Whole-cell patch clamp recordings were performed at room temperature using borosilicate glass pipettes (GC150TF-10, Clark Electromedical Instruments, Pangbourne, UK) connected to an EPC-10 amplifier (HEKA Electronics, Lambrecht, Germany) as described previously24, 25. The typical electrode resistance was 4–5 MΩ, while series resistance was in the range of 8–15 MΩ. Series resistance compensation of > 30% was used. Voltage clamp experiments on cultured cells were controlled by Patch Master software (HEKA Electronics, Lambrecht, Germany). Current density was calculated by dividing the current amplitude determined at the end of the depolarizing voltage ramp to + 60 mV by the membrane capacitance obtained from slow capacitance compensation. The following recording solutions were used: (a) extracellular solution (in millimoles per liter): NaCl, 140; KCl, 5; HEPES, 10; MgCl2, 1; CaCl2, 1; pH 7.4 with NaOH. (b) Intracellular solution (in millimoles per liter): KCl, 140; HEPES, 10; EGTA, 1.3; CaCl2, 1.217; MgCl2,1; pH 7.4 with KOH. The calculated free Ca2+ concentration of the internal solution was set to 1 μmol/l in order to obtain full activation of the KCa channels during the patch clamp experiments. A liquid junction potential of − 3.2 mV was not taken into account. Drugs were dissolved in DMSO and added to the standard extracellular solution and applied for a time period of 10 min. A multi-barrel application pipette with a tip diameter of about 100 μm was used for drug application close to the recorded cell. Recordings were analyzed using Fit Master and Origin 7.5 software. First the total transmembrane current flow (sum of inward and outward currents mediated by all open channels) in response to the voltage clamp protocol was measured as control. In order to increase the current proportion mediated by members of the family of calcium-activated K+ channels, the activator 1-EBIO (50 µmol/L) was added to the bath solution. Next, the KCa1.1 channel blocker paxilline (10 µmol/L) was applied to the bath solution containing 1-EBIO. Finally, the KCa3.1 channel blocker senicapoc (1 µmol/L) was added to the cocktail. KCa1.1- and KCa3.1-mediated currents are represented by the paxilline-sensitive and senicapoc-sensitive current components, respectively.
Western blotting
We employed standard techniques for Western blotting described previously8 and used the following antibodies: anti-KCa3.1 (1:500; Sigma), anti-EGFR (1:250; Santa Cruz Inc., USA), anti-HIF-1α (1:1000; Becton Dickinson), anti-phospho-p44/42 (pERK1/2; 1:1,000; Cell Signaling Technology Inc., USA; cat. # 9106), anti-actin (1:10,000, Sigma Aldrich). When PVDF membranes were probed successively with multiple antibodies, they were stripped by incubation in 200 mmol/L NaOH for 5 min and subsequent washing.
Statistics
Data are presented as mean ± SEM unless otherwise indicated. N reflects the number of independent experiments, n represents the number of individual cells. Extreme values (3 interquartile ranges above the third or below the first quartile) were removed and data were tested for normality. Significance (p < 0.05) was assessed by One-way-ANOVA or Kruskal–Wallis test followed by Bonferroni or Fisher post hoc test. Migration experiments were also evaluated with mixed-measures ANOVA and automatic post hoc test.
Results
Altered expression of KCa3.1 channels in erlotinib-resistant NSCLC cells
We searched the GEO and ArrayExpress databases for mRNA expression obtained from microarrays of human erlotinib-sensitive and erlotinib-resistant cell lines and identified four datasets of interest: GSE31625, GSE38310, GSE38404 and GSE80344. They contained data from 18, 6, 2 and 4 NSCLC erlotinib-sensitive cell lines as well as 28, 12, 6, and 12 NSCLC erlotinib-resistant cell lines, respectively. On average, these datasets displayed ~ 4.100 genes that were differentially expressed in the two groups when applying a threshhold of 0.05 for the adjusted p-value. Using the online software Venny 2.0 (http://bioinfogp.cnb.csic.es/tools/venny/index.html) we found 23 differentially expressed genes that are common to all four datasets, and 504 are common at least to 3 of the 4 datasets. We identified three ion channel genes that are expressed at a reduced level in erlotinib-resistant NSCLC cell lines. KCNN4 (encoding the KCa3.1 channel) and TRPM4 (encoding TRPM4 channels) are listed in GSE31625, GSE38310 and GSE38404 with logFC values (number of times a gene is over/under-expressed) of − 2.82, 0.41 and − 0.86 (KCNN4) as well as − 2.16, − 0.40 and − 0.75 (TRPM4), respectively. KCNS3 which encodes the KV9.3 channel is detected in GSE31625, GSE80344 and GSE38404, with logFC values of − 1.21, − 1.81 and − 0.75, respectively. These results are summarized in Fig. 1A. The expression of other KCa channels such as KCa1.1 does not differ between erlotinib-sensitive and -resistant cell lines in these data sets.
Figure 1.
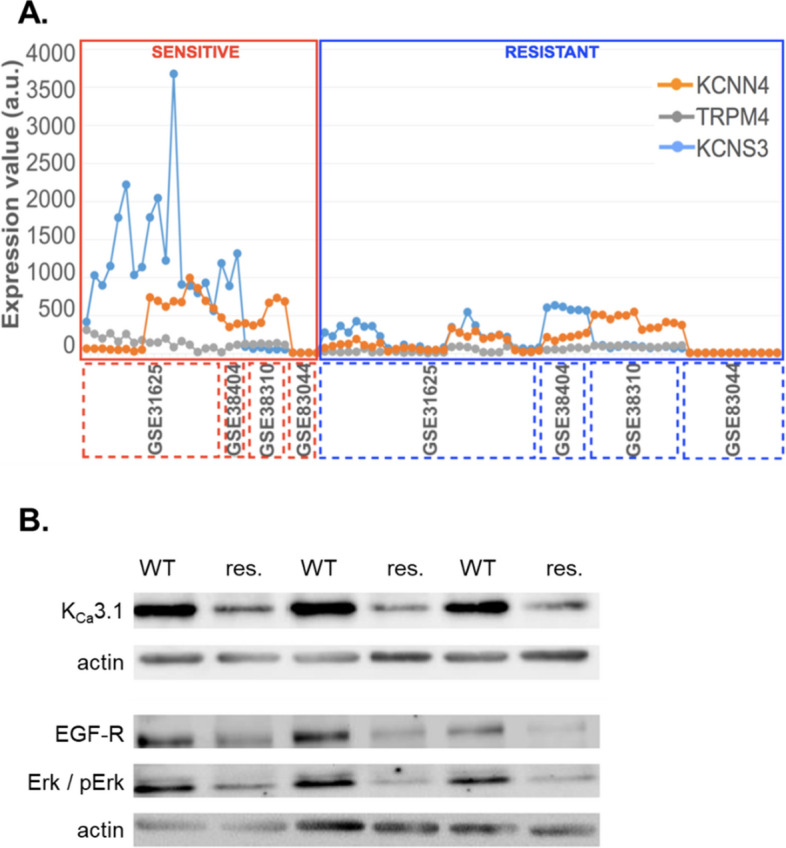
(A) Gene expression values (estimate of the number of RNA transcripts) of KCNN4, TRPM4 and KCNS3 for each cell line represented by the GSE31625, GSE38310, GSE80344 and GSE38404 microarray datasets. The gene expression values have been normalized by removing non-biological variation, and generating the final probe set expression values. GSE31625 gene expression was evaluated with MAS5 normalization, GSE38310, GSE80344 and GSE38404 were evaluated with RMA normalization. The x axis represents all the samples from the four datasets. (B) Western blot analysis of KCa3.1 channels, EGFR and phospho-p44/42 (pERK1/2) in wildtype A549-3R (WT) and partially erlotinib resistant A549-3R (res.) cells. Western blots were repeated three times (N = 3).
In the present study we followed up on KCa3.1 channels and their potential functional role in erlotinib-ressitance of NSCLC cells using the A549-3R cell model that we had characterized previously8. To learn more about the role of EGFR signaling and KCa3.1 in NSCLC cells, we generated partially erlotinib-resistant A549-3R cells by culturing them for ~ 9 weeks in the presence of 10 µmol/L erlotinib and 100 ng/ml EGF. Western bot analysis recapitulates our in silico analyses and reveals that KCa3.1 expression is reduced in these cells (N = 3). Moreover, EGFR expression and ERK phosphorylation are also diminished (see Figs. 1B and 1Ba-suppl). For comparison, we also tested H1975 cells that are erlotinib-resistant because of an activating mutation of the EGFR which is then constitutively active. They are also represented in arrays mentioned above and have a lower mRNA and protein expression of KCa3.1 channels than erlotinib-sensitive NSCLC cells (data not shown). KCa3.1 expression is lower in H1975 cells than in wt A549-3R cells (N = 3; Fig. 1Bb-suppl.).
Functional KCa channels in wildtype and partially erlotinib-resistant A549-3R cells
We next tested whether the altered KCa3.1 expression on mRNA and protein level also affects channel activity. We therefore performed patch clamp experiments with wildtype and partially erlotinib-resistant A549-3R cells. First we compared KCa current densities when cells were kept under standard cell culture conditions (ambient pO2). We had characterized KCa3.1-mediated current in A549-3R cells already previously8. We slightly modified the protocol and also probed for KCa1.1 channels (paxillin-sensitive current component) because they are known to be activated by 1-EBIO, too26. Moreover, we used senicapoc to inhibit KCa3.1-mediated currents. Figure 2 provides a summary of the patch clamp experiments. Current density (@ + 60 mV) in A549-3R WT cells rises ~ fourfold from 12.8 ± 1.4 pA/pF to 44.1 ± 3.1 pA/pF (n = 9) following the application of 1-EBIO (50 µmol/L). The 1-EBIO-induced increase of the current density is largely due to the activation of paxilline-sensitive KCa1.1 channels. Under normoxic conditions the paxilline-sensitive current density amounts to 21.2 ± 2.9 pA/pF, while the senicapoc-sensitive current component is only 12.0 ± 1.6 pA/pF.
Figure 2.
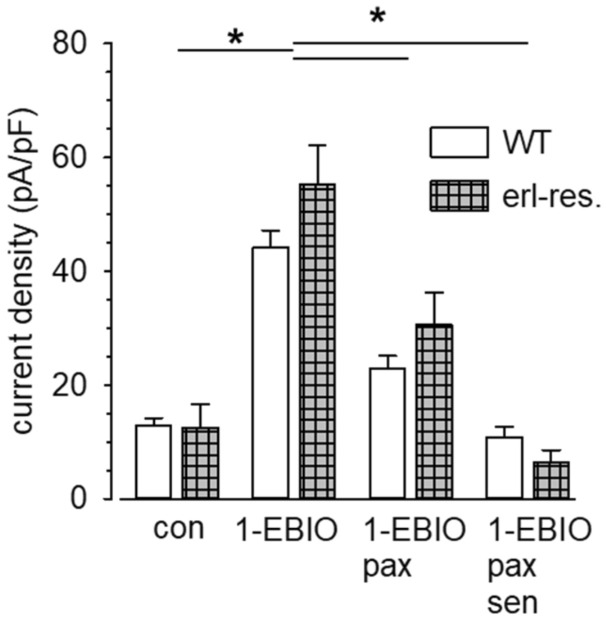
Summary of patch clamp experiments. We compared the mean current densities @ + 60 mV of erlotinib-sensitive WT A549-3R and erlotinib-resistant A549-3R cells. The contribution of KCa3.1 and KCa1.1 channels to the outward current induced by the application of 1-EBIO (50 µmol/L) is disclosed by adding the KCa1.1 blocker paxilline (pax; 10 µmol/l) or the KCa3.1 blocker senicapoc (sen; 1 µmol/L). A549-3R WT: N = 9, erlotinib-resistant cells: N = 10. * indicates p < 0.05.
Partially erlotinib-resistant cells respond in a similar fashion (see Fig. 2). There is a trend towards higher KCa3.1 current density in partially erlotinib-resistant than in A549-3R WT cells. In these cells it mediates almost as much current as KCa1.1 channel activity. However, this does not reach statistical significance when compared with A549-3R WT cells. Thus, KCa3.1 channel activity behaves differently than the channel expression on mRNA and protein level. We next tested the impact of these channels on the sensitivity of A549-3R cells towards erlotinib, an EGFR TKI.
Erlotinib inhibits migration and proliferation of A549-3R cells
In a first set of functional experiments we quantified the inhibitory effect of erlotinib, an inhibitor of the EGFR tyrosine kinase, on migration of A549-3R cells. Erlotinib was applied in a concentration range of 10 nmol/l to 3 µmol/L. Control experiments in the presence of DMSO (1:1000) were always performed in parallel. Under control conditions migration speed is 0.40 ± 0.02 µm/min and translocation amounts to 77.9 ± 4.6 µm (n = 168 cells from N = 14 experiments). Figure 3A depicts the trajectories normalized to a common starting point for cells migrating under control conditions and in the presence of 1 µmol/L erlotinib. Erlotinib leads to a marked inhibition of migration. The dose-response curve shown in Fig. 3B indicates that the IC50 value is in the order of ~ 500 nmol/L. Similarly, proliferation of A549-3R cells is inhibited by erlotinib (Fig. 3C). Consistent with published results27 the estimated IC50 value is in the order of 5 µmol/L. Thus, erlotinib inhibits migration more efficiently than it prevents an increase of the cell number. Moreover, these concentrations correspond well to clinically achievable plasma concentrations in patients28.
Figure 3.
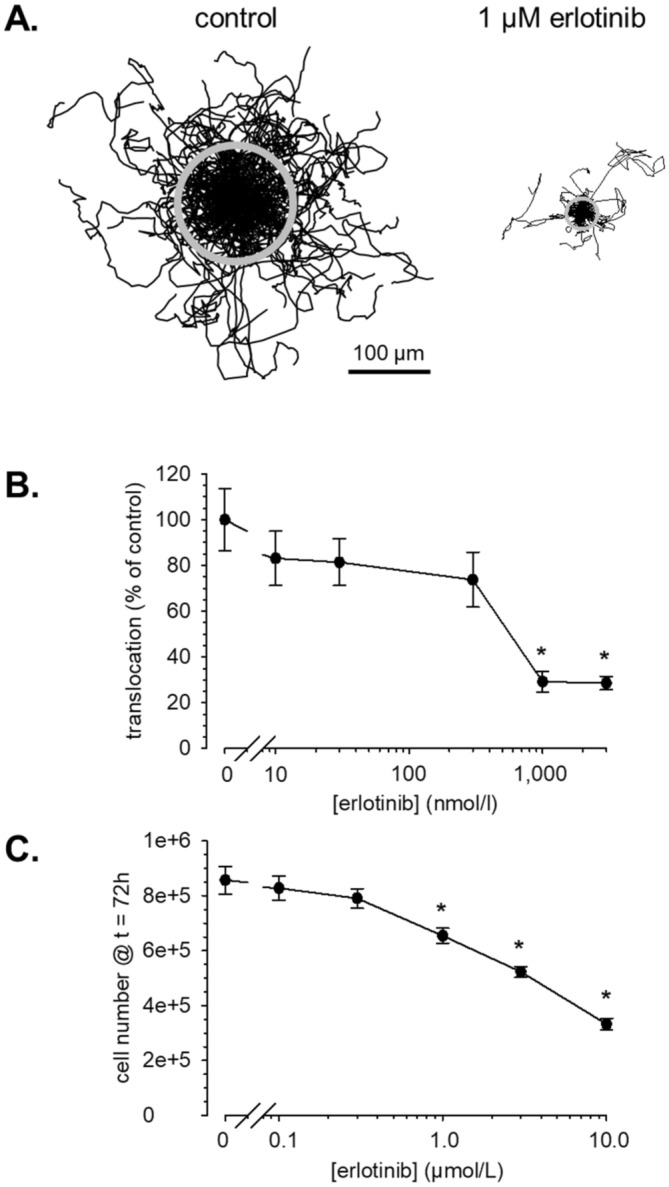
Erlotinib induces a dose-dependent inhibition of migration and proliferation of A549-3R cells. (A) Trajectories of migrating A549-3R cells. Each line corresponds to the path of an individual cell. Trajectories are normalized to a common starting point. The radius of the grey circle represents the translocation, i. e. the mean distance between start and end point of the cells. N/n = 14/168 for control and N/n = 5/66 for 1 µmol/L erlotinib. (B) Summary of migration experiments. Erlotinib reduces translocation dose-dependently. Translocation is normalized to the respective control that were always run in parallel. (C) Proliferation of A549-3R cells is also inhibited dose-dependently by erlotinib (N = 4). The cell number @ t = 72 h after plating is normalized to the respective control values. * indicates p < 0.05.
Based on these results, the experiments combining erlotinib with the KCa3.1 blocker senicapoc or the KCa1.1 blocker paxilline will be performed either with 300 nmol/L, 3 µmol/L or 10 µmol/L erlotinib. These concentrations are not yet effective in inhibiting migration and proliferation (300 nmol/L), or they produce the maximal effects (3 µmol/L and 10 µmol/L).
Combining erlotinib with senicapoc or paxilline elicits additive effects on migration and proliferation
We combined 300 nmol/L erlotinib with 3 µmol/L or 30 µmol/L senicapoc in migration experiments. The results of these experiments are summarized in Fig. 4. The combination of both drugs reduces the translocation (Fig. 4A), while either drug alone has no significant impact on migration. The inhibition of migration by the drug combination can be accounted for by a reduction of the directionality of migration (Fig. 4B). When combining a tenfold higher concentration of erlotinib (3 µmol/L) with senicapoc, the KCa3.1 channel blocker does not augment the inhibition of migration that is already elicited by erlotinib alone. The respective mean values of the translocation are (N = 3 experiments each): DMSO: 101 ± 12 µm (n = 30); 3 µmol/L erlotinib: 45 ± 8 µm (n = 30); 3 µmol/L erlotinib plus 3 µmol/L senicapoc: 45 ± 7 µm (n = 30); 3 µmol/L erlotinib plus 30 µmol/L senicapoc: 52 ± 8 µm (n = 29).
Figure 4.
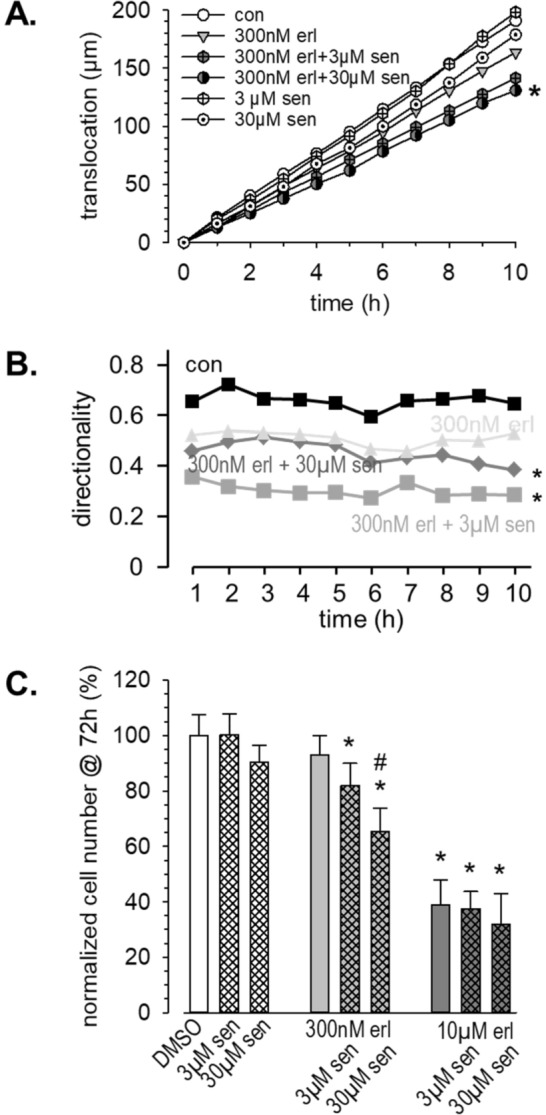
Combining erlotinib with the KCa3.1 inhibitor senicapoc elicits additive effects on migration and proliferation. (A) Summary of migration experiments. We plotted the cumulative translocation (binned in 1 h intervals) during the course of the experiments. For clarity we omitted the error bars. (N/n ≥ 3/30). (B) Combining erlotinib and senicapoc leads to a reduction of the directionality of migrating cells. (C) Summary of proliferation experiments (N = 4). * indicates p < 0.05 with respect to the DMSO control; # indicates p < 0.05 with respect to 300 nmol/L erlotinib alone.
We observed a similar pattern in proliferation experiments (Fig. 4C). Combining low dose erlotinib (300 nmol/L) with the KCa3.1 channel blocker senicapoc inhibits proliferation. It is reduced by 18% and 35% in the combined presence of 300 nmol/L erlotinib and 3 or 30 µmol/L senicapoc, respectively. Moreover, the cell number is significantly lower in the combined presence of 300 nmol/L erlotinib and 30 µmol/L senicapoc than in the presence of erlotinib alone. We also tested the combination of senicapoc (3 and 30 µmol/L) with 10 µmol/L erlotinib. Erlotinib alone and in combination with senicapoc reduces the cell number by 61%, 62% and 68%, respectively. Thus, KCa3.1 channel inhibition has only a minor additional effect in the presence of a high dose of erlotinib.
We also tested the effect of inhibiting KCa1.1 channels in A549-3R cells (Fig. 5). Figure 5A displays the trajectories of A549-3R cells migrating under control conditions and in the presence of 10 µmol/l paxilline. It is evident that KCa1.1 channel inhibition by itself does not affect migration. Translocation of control and paxilline-treated cells amounts to 72.7 ± 7.3 µm (n = 59; Fig. 5A left panel and Fig. 5B) and 69.8 ± 6.4 µm (n = 60; Fig. 5A, right panel and Fig. 5B), respectively. However, paxilline and erlotinib elicit strongly potentiated inhibitory effects when they are combined. The additional application of the low dose of erlotinib (300 nmol/l) to 10 µmol/l paxilline reduces the translocation by ~ 75% to 16.8 ± 3.9 µm. Combining 10 µmol/l paxilline with 10 µmol/l erlotinib causes a reduction of the translocation by ~ 85% to 9.6 ± 1.5 µm (n = 30 each; Fig. 5B). We plotted the cumulative translocation (covered during 1 h intervals) as a function of time in Fig. 5C. It becomes evident that the inhibition of migration is effective during the entire course of the experiment. In addition to the speed of migration, the combination of erlotinib and paxilline also reduces the directionality by ~ 25% (Fig. 5D).
Figure 5.
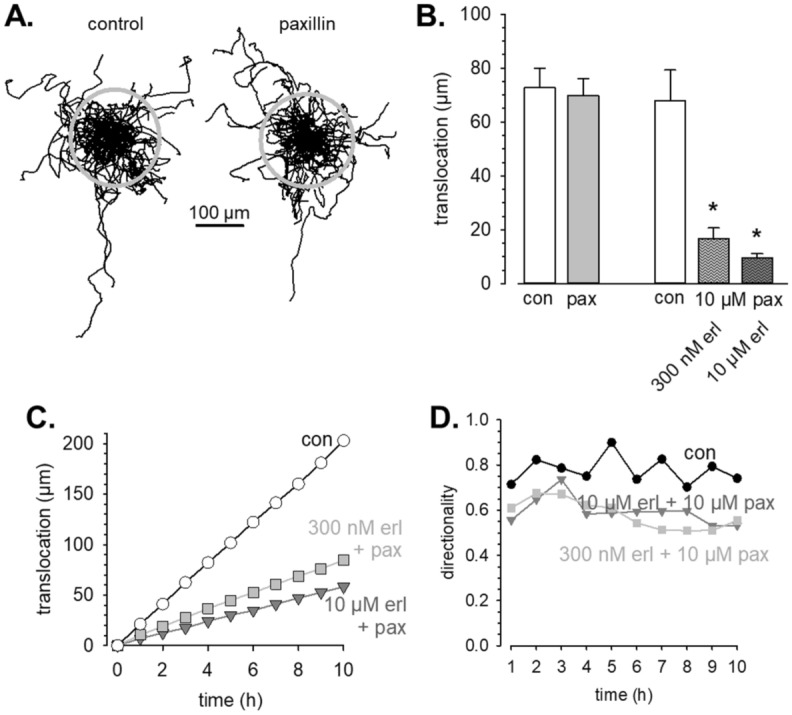
KCa1.1 channel blockade potentiates the inhibitory effect of erlotinib on migration of A549-3R cells. (A) Trajectories of individual A549-3R cells migrating under control conditions (left panel; n = 59) or in the presence of 10 µmol/L paxilline (right panel; n = 60). Cell paths are normalized to common starting points and the grey circles represent the mean translocations covered during the course of the experiment. (B) Summary of the migration experiments in which we probed the contribution of KCa1.1 channels with paxilline (pax; 10 µmol/L) under control conditions (con; left part of the panel) and in combination with erlotinib (erl; right part of the panel; N/n = 3/30 for each condition). (C) Cumulative translocation binned in 1 h intervals of A549-3R cells migrating under control conditions and in the presence of paxilline and erlotinib. (D) The directionality of A549-3R cells is reduced in the presence of paxilline and erlotinib.
Partial erlotinib resistance can be overcome by KCa3.1 or KCa1.1 channel blockade
As expected, proliferation with erotinib-resistant cells experiments revealed a ~ tenfold lower sensitivity towards EGFR tyrosine kinase inhibition with erlotinib (Fig. 6A). 10 µmol/L erlotinib only cause a reduction of the cell number @ t = 72 h by 27%. For comparison, 1 µmol/L erlotinib inhibits proliferation by 24% in control cells. We then combined erlotinib with the KCa3.1 channel blocker senicapoc. When using 300 nmol/L erlotinib which by itself is ineffective (− 6%) together with 3 µmol/L or 30 µmol/L senicapoc, proliferation is reduced by 15% or 35%, respectively. When using 10 µmol/L erlotinib together with 3 µmol/L or 30 µmol/L senicapoc, proliferation is reduced by up to ~ 70% (Fig. 6B). Thus, in partially erlotinib-resistant cells KCa3.1 channel blockade elicits a more pronounced effect than in control cells and thereby partially overcomes erlotinib resistance.
Figure 6.

KCa3.1 and KCa1.1 inhibition can overcome partial erlotinib resistance. (A) Dose–response curves of proliferation for erlotinib. For comparison, we replotted data from Fig. 1A for wildtype cells (grey symbols). (B) Proliferation of partially erlotinib resistant A549-3R cells is efficiently inhibited by the KCa3.1 channel blocker senicapoc. (C, D) Summary of migration experiments with partially erlotinib-resistant A549-3R cells (N/n = 3/30). Blocking KCa1.1 channels with paxillin causes a marked inhibition of migration.
The KCa1.1 channel blocker paxilline (10 µmol/l) produces comparable results in migration experiments with partially erlotinib-resistant A549-3R cells. It is notable that the translocation of these cells is strongly reduced when compared with the parental WT cells. They cover only 27.2 ± 4.0 µm during the course of the experiments compared to 67.8 ± 11.5 µm. In the combined presence of paxilline and erlotinib the translocation of the partially erlotinib-resistant cells is reduced by further ~ 50% (N/n = 3/30 each; Fig. 6C,D).
Hypoxia augments the contribution of KCa3.1 channels to migration and proliferation
Finally, we assessed the impact of KCa channel activity on NSCLC cells when they were kept under hypoxic conditions. We included the analysis of hypoxia because it has been known for many years that the NSCLC tumor stroma is hypoxic29. Moreover, it has been shown that KCa3.1 channels are upregulated in melanoma cells when they are cultured under hypoxic conditions30.
We pretreated A549-3R cells with 1 mmol/L DMOG or kept them under hypoxic conditions (0.2% O2) for up to 72 h. Six hours of hypoxia lead to a massively elevated expression of HIF-1α in A549-3R (Fig. 7A). Thereafter, HIF-1α decreases but still remains on a higher level than in control cells. The EGFR expression is also strongly increased and remains steadily on a higher level. In contrast, KCa3.1 expression itself is not altered by hypoxia which is in contrast to results obtained from melanoma cells30. Similarly, KCa1.1 channels were shown to be activated in glioblastoma cells in response to hypoxia31. In order to prevent a transient reoxygenation upon addition of inhibitors during migration and proliferation experiments, we induced a chemical hypoxia during the functional assays. Although the numerical values of the observed changes of current densities are bigger in hypoxia-adapted cells, they do not reach statistical significance when compared with the values obtained under normoxia. Under hypoxic condition current density rises almost sevenfold, from 9.8 ± 2.2 pA/pF to 67.7 ± 7.7 pA/pF (n = 12). The KCa1.1-mediated K+ current density doubles and rises to 38.6 ± 9.8 pA/pF. KCa3.1-mediated current increases by 64% and reaches 18.7 ± 6.8 pA/pF (see Fig. 7B). Thus, our patch clamp experiments confirm the Western blot results in that KCa3.1 channel activity, blocked by senicapoc (1 µmol/L), is not massively increased by hypoxia. WT cells (erlotinib-sensitive) and erlotinib and erlotinib-resistant cells behave alike in this respect.
Figure 7.

Hypoxia sensitizes A549-3R cells for EGFR and KCa3.1 channel inhibition. (A) Western blot analysis reveals increased expression of HIF-1α and EGFR while that of KCa3.1 channels is not altered. The blots are representative for N = 3 experiments. (B) Current densities of erlotinib-sensitive WT A549-3R (N = 12) and erlotinib-resistant A549-3R cells (N = 8). C. Comparison of the cumulative translocation under normoxia and under hypoxia. In the presence of chemical hypoxia both low dose erlotinib (300 nmol/L) and KCa3.1 inhibition reduce migration more effectively than under normoxic conditions (N/n = 3/30). (D) Summary of proliferation experiments in the presence of chemical hypoxia. Note that the proliferation rate is only ~ 1/3 of that in normoxic conditions. *Indicates p < 0.05 with respect to the DMSO control. (E) Hypoxia does not increase the sensitivity of A549-3R cells towards inhibition of KCa1.1 channels. For comparison, we replotted the data for normoxia from Fig. 5B.
It is notable that DMOG-pretreated A549-3R cells migrate considerably faster than control cells. Speed of migration and translocation amount to 0.51 ± 0.05 µm/min and 102 ± 15 µm, respectively. Furthermore, cells are sensitized towards erlotinib (300 nmol/L) in the presence of chemical hypoxia: Migration is reduced more efficiently than under normoxic conditions (0.31 ± 0.03 µm/min and 56 ± 9 µm corresponding to a reduction of ~ 40% ). The same holds true for the additional effect of KCa3.1 channel blockade. The respective values are 0.25 ± 0.03 µm/min and 50 ± 11 µm for 300 nmol/L erlotinib plus 3 µmol/L senicapoc and 0.21 ± 0.03 µm/min and 47 ± 14 µm for 300 nmol/L erlotinib plus 30 µmol/L senicapoc which correspond to reductions by ~ 50% and ~ 60%, respectively (N/n = 3/30; p < 0.05). This effect of the KCa3.1 channel blocker senicapoc is additive when cells are treated with 3 µmol/L erlotinib (data not shown). The increased efficacy of the combination of low concentrations of erlotinib and senicapoc to inhbit migration of A549-3R cells under hypoxic conditions becomes particularly evident when plotting the cumulative translocation (see Fig. 7C).
Proliferation of A549-3R cells is much slower in the presence of chemical hypoxia (Fig. 7D). Yet, proliferation is further reduced by erlotinib and by its combination with the KCa3.1 channel blocker senicapoc. Notably, the lowest (combined) concentrations of erlotinib and senicapoc already elicit significant effects and reduce the cell number by 23% (300 nmol/l erlotinib) or 34% (300 nmol/l erlotinib plus 3 µmol/l senicapoc; Fig. 7D; N = 4). Taken together, the combined application of EGFR tyrosine kinase and KCa3.1 channel blockade leads to an effective inhibition of migration in the presence of chemical hypoxia.
In contrast, chemical hypoxia does not increase the sensitivity of A549 cells to KCa1.1 channel blockade. Paxilline (10 µmol/l) has no effect on the migration of A549-3R cells pretreated with DMOG (Fig. 7E). For cells kept under hypoxia, the respective values are 95.2 ± 9.2 µm (n = 55) and 97.6 ± 7.8 µm (n = 53). This underlines the distinct roles played by KCa3.1 and KCa1.1 channels in regulating NSCLC cell behavior.
Discussion
Our study addressed the question whether the therapeutic efficacy of the EGFR tyrosine kinase inhibitor erlotinib can be enhanced and therapy resistance be overcome by combining it with KCa channel blockers. Since TKI resistance is a particular problem in NSCLC we performed our experiments with the aggressive lung adenocarcinoma cell line A549-3R. Our key results are the following: (i) Depending on the concentration of erlotinib, the simultaneous application of the KCa3.1 inhibitor senicapoc or the KCa1.1 blocker paxilline elicit additive effects on the reduction of proliferation and cell migration. (ii) Hypoxia and partial erlotinib resistance increase the susceptibility to KCa3.1 channel inhibition while this appears not to be the case for KCa1.1 blockade. Thus, KCa channel inhibition has the potential to strengthen the therapeutic efficacy of drugs such as erlotinib and to potentially overcome EGFR TKI resistance.
At first sight, the senicapoc concentrations used in the migration and proliferation experiments appear to be exceedingly high. The nominal concentrations are much higher than those used in patch clamp experiments. However, it has to be kept in mind that KCa3.1 channel inhibitors having a similar structure as the lead compound clotrimazole, are strongly bound by plasma proteins so that the free concentration is up to 50 times lower32. In this submicromolar concentration range senicapoc is a highly specific inhibitor of KCa3.133. Moreover, it is conceivable that the observed effects may at least be in part be mediated by mitochondrial KCa3.1 channels (see discussion below). Then the blockers have to cross the plasma membrane and the outer mitochondrial membrane to reach the KCa3.1 channel protein in the inner mitochondrial membrane. Obviously, effective drug concentrations are not easy to predict at this location.
The additive effects of EGFR TKIs and KCa3.1 and KCa1.1 channel blockers can be explained by considering the differential mechanisms by which both membrane proteins regulate cell proliferation and migration. EGFR activates several signaling pathways such as RAS/RAF/MAPK, PI3K/AKT and JAK/STAT thereby leading to progression of the cell cycle, proliferation and an inhibition of apoptosis34. Inhibition of these signaling cascades causes a cell cycle arrest in G1/G035. Moreover, there is evidence that erlotinib promotes apoptosis by elevating the intracellular ROS production36. KCa3.1 inhibition has complementary and synergistic effects. It arrests the cell cycle at the G1/S phase transition37, 38. KCa3.1 and KCa1.1 channels are also expressed in the inner mitochondrial membrane of cancer cells39, 40. Blocking the channels is expected to elicit a hyperpolarization of the mitochondrial membrane potential which could, like erlotinib, also lead to increased ROS production41, 42. In melanoma cells the combination of the BRAF inhibitor vemurafenib with the KCa3.1 blocker TRAM-34 resulted in the release of proapoptotic, mitochondrial factors16. As far as the interpretation of our own results is concerned the reduced cell number @ t = 72 h can of course be a consequence of both effects: induction of apoptosis or inhibition of the cell cycle. In melanoma cells the authors noted a depolarization of the mitochondrial membrane potential instead of the anticipated hyperpolarization. In our view this is likely a secondary effect. The combined application of vemurafenib and TRAM-34 could have caused a ROS-induced secondary opening of the permeability transition pore in the inner mitochondrial membrane and thereby caused the mitochondrial depolarization41. Finally, KCa3.1 channels can also indirectly affect EGFR-dependent proliferation by providing the electrochemical driving force for TRPC1-mediated Ca2+ entry43.
Cell migration critically depends on cytoskeletal dynamics44 as well as on tightly regulated ion and water fluxes45. EGFR activation promotes migration in multiple ways (reviewed in46). For example, in oesophageal cancer erlotinib inhibits cell migration by impairing Rho GTPase or focal adhesion kinase (FAK) activity47. KCa1.1 channels are known to interact with FAK48. By controlling (local) volume homeostasis KCa3.1 and KCa1.1 channels indirectly also affect actin dynamics in migrating cells49, 50.
Because of their synergistic and complementary effects it is a meaningful therapeutic strategy to combine EGFR inhibitors such as erlotinib or related compounds and KCa channel blockers such as senicapoc or paxilline. If the results of our in vitro experiments are applicable in an in vivo setting, one of the advantages will be a dose reduction of the drugs. Our results show that the drug combination inhibits migration and proliferation already at low concentrations that are ineffective when given as “monotherapy”. Dose reduction of erlotinib is a relevant issue since there is ample evidence that the frequency and severity of its side effects correlate with the plasma concentration51. It is not yet known whether this would be critical for senicapoc as well. So far, only a few hundred patients with sickle cell anemia have been treated with senicapoc. No major adverse effects had been observed in this patient cohort52.
Supplementary Information
Acknowledgements
F.G. was member of the MedK of the Medical Faculty of the Westfälische Wilhelms-Universität Münster. A.S. was supported by Cells-in-Motion Cluster of Excellence (EXC 1003–CiM), University of Münster, Germany. L.M.T., P.H., T.B, S.G.M. and A.S. were supported by DFG (GRK 2515/1, Chembion).
Author contributions
F.G., P.H., E.B., L.M.T., S.S. and E.N. conducted experiments; F.G., P.H., E.B., L.M.T., S.S., E.N., T.B., S.G.M. and A.S. analysed data; F.G., L.M.T., T.B., S.G.M. and A.S. wrote the manuscript; A.S. conceived and designed research.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-021-97406-0.
References
- 1.Zugazagoitia J, Enguita AB, Nunez JA, Iglesias L, Ponce S. The new IASLC/ATS/ERS lung adenocarcinoma classification from a clinical perspective: Current concepts and future prospects. J. Thorac. Dis. 2014;6:S526–536. doi: 10.3978/j.issn.2072-1439.2014.01.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De Angelis R, et al. Cancer survival in Europe 1999–2007 by country and age: Results of EUROCARE–5-a population-based study. Lancet Oncol. 2014;15:23–34. doi: 10.1016/S1470-2045(13)70546-1. [DOI] [PubMed] [Google Scholar]
- 3.Langer CJ, Besse B, Gualberto A, Brambilla E, Soria JC. The evolving role of histology in the management of advanced non-small-cell lung cancer. J. Clin. Oncol. 2010;28:5311–5320. doi: 10.1200/JCO.2010.28.8126. [DOI] [PubMed] [Google Scholar]
- 4.Besse B, et al. 2nd ESMO consensus conference on lung cancer: Non-small-cell lung cancer first-line/second and further lines of treatment in advanced disease. Ann. Oncol. 2014;25:1475–1484. doi: 10.1093/annonc/mdu123. [DOI] [PubMed] [Google Scholar]
- 5.Antonicelli A, et al. EGFR-targeted therapy for non-small cell lung cancer: Focus on EGFR oncogenic mutation. Int. J. Med. Sci. 2013;10:320–330. doi: 10.7150/ijms.4609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Diaz-Serrano A, Gella P, Jimenez E, Zugazagoitia J, Paz-Ares Rodriguez L. Targeting EGFR in lung cancer: Current standards and developments. Drugs. 2018;78:893–911. doi: 10.1007/s40265-018-0916-4. [DOI] [PubMed] [Google Scholar]
- 7.Lin Y, Wang X, Jin H. EGFR-TKI resistance in NSCLC patients: Mechanisms and strategies. Am. J. Cancer Res. 2014;4:411–435. [PMC free article] [PubMed] [Google Scholar]
- 8.Bulk E, et al. Epigenetic dysregulation of KCa 3.1 channels induces poor prognosis in lung cancer. Int. J. Cancer. 2015;137:1306–1317. doi: 10.1002/ijc.29490. [DOI] [PubMed] [Google Scholar]
- 9.Bulk E, Todesca LM, Schwab A. Ion channels in lung cancer. Rev. Physiol. Biochem. Pharmacol. 2020 doi: 10.1007/112_2020_29. [DOI] [PubMed] [Google Scholar]
- 10.Bulk E, et al. KCa3.1 channel inhibition leads to an ICAM-1 dependent increase of cell-cell adhesion between A549 lung cancer and HMEC-1 endothelial cells. Oncotarget. 2017;8:112268–112282. doi: 10.18632/oncotarget.22735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hoffmann EK, Lambert IH. Ion channels and transporters in the development of drug resistance in cancer cells. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2014;369:20130109. doi: 10.1098/rstb.2013.0109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huber SM, et al. Role of ion channels in ionizing radiation-induced cell death. Biochem. Biophys. Acta. 1848;2657–2664:2015. doi: 10.1016/j.bbamem.2014.11.004. [DOI] [PubMed] [Google Scholar]
- 13.He DX, Ma X. Transient receptor potential channel C5 in cancer chemoresistance. Acta Pharmacol. Sin. 2016;37:19–24. doi: 10.1038/aps.2015.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kischel P, et al. Ion channels: New actors playing in chemotherapeutic resistance. Cancers (Basel) 2019 doi: 10.3390/cancers11030376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Samuel P, et al. Over-expression of miR-31 or loss of KCNMA1 leads to increased cisplatin resistance in ovarian cancer cells. Tumour Biol. 2016;37:2565–2573. doi: 10.1007/s13277-015-4081-z. [DOI] [PubMed] [Google Scholar]
- 16.Bauer D, Werth F, Nguyen HA, Kiecker F, Eberle J. Critical role of reactive oxygen species (ROS) for synergistic enhancement of apoptosis by vemurafenib and the potassium channel inhibitor TRAM-34 in melanoma cells. Cell Death Dis. 2017;8:e2594. doi: 10.1038/cddis.2017.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jeon WI, Ryu PD, Lee SY. Effects of voltage-gated K+ channel blockers in gefitinib-resistant H460 non-small cell lung cancer cells. Anticancer Res. 2012;32:5279–5284. [PubMed] [Google Scholar]
- 18.Athar A, et al. ArrayExpress update—From bulk to single-cell expression data. Nucleic Acids Res. 2019;47:D711–D715. doi: 10.1093/nar/gky964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barrett T, et al. NCBI GEO: Archive for functional genomics data sets–update. Nucleic Acids Res. 2013;41:D991–995. doi: 10.1093/nar/gks1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Phipson B, Lee S, Majewski IJ, Alexander WS, Smyth GK. Robust hyperparameter estimation protects against hypervariable genes and improves power to detect differential expression. Ann. Appl. Stat. 2016;10:946–963. doi: 10.1214/16-AOAS920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hascher A, et al. DNA methyltransferase inhibition reverses epigenetically embedded phenotypes in lung cancer preferentially affecting polycomb target genes. Clin. Cancer Res. 2014;20:814–826. doi: 10.1158/1078-0432.CCR-13-1483. [DOI] [PubMed] [Google Scholar]
- 22.Garcia B, et al. Effective inhibition of the epidermal growth factor/epidermal growth factor receptor binding by anti-epidermal growth factor antibodies is related to better survival in advanced non-small-cell lung cancer patients treated with the epidermal growth factor cancer vaccine. Clin. Cancer Res. 2008;14:840–846. doi: 10.1158/1078-0432.CCR-07-1050. [DOI] [PubMed] [Google Scholar]
- 23.Schwab A, et al. Subcellular distribution of calcium-sensitive potassium channels (IK1) in migrating cells. J. Cell. Physiol. 2006;206:86–94. doi: 10.1002/jcp.20434. [DOI] [PubMed] [Google Scholar]
- 24.Storck H, et al. Ion channels in control of pancreatic stellate cell migration. Oncotarget. 2017;8:769–784. doi: 10.18632/oncotarget.13647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brömmel K, et al. Co-staining of KCa3.1 channels in NSCLC cells with a small-molecule fluorescent probe and antibody-based indirect immunofluorescence. ChemMedChem. 2020;15:2462–2469. doi: 10.1002/cmdc.202000652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu YC, Lo YK, Wu SN. Stimulatory effects of chlorzoxazone, a centrally acting muscle relaxant, on large conductance calcium-activated potassium channels in pituitary GH3 cells. Brain Res. 2003;959:86–97. doi: 10.1016/S0006-8993(02)03730-7. [DOI] [PubMed] [Google Scholar]
- 27.Jin Y, et al. Effect of an epidermal growth factor receptor tyrosine kinase inhibitor on actin remodeling in an in vitro bladder cancer carcinogenesis model. Mol. Cancer Ther. 2006;5:1754–1763. doi: 10.1158/1535-7163.MCT-06-0043. [DOI] [PubMed] [Google Scholar]
- 28.Petty WJ, et al. Epidermal growth factor receptor tyrosine kinase inhibition represses cyclin D1 in aerodigestive tract cancers. Clin. Cancer Res. 2004;10:7547–7554. doi: 10.1158/1078-0432.CCR-04-1169. [DOI] [PubMed] [Google Scholar]
- 29.Rasey JS, et al. Quantifying regional hypoxia in human tumors with positron emission tomography of [18F]fluoromisonidazole: A pretherapy study of 37 patients. Int. J. Radiat. Oncol. Biol. Phys. 1996;36:417–428. doi: 10.1016/S0360-3016(96)00325-2. [DOI] [PubMed] [Google Scholar]
- 30.Tajima N, et al. Ca2+-activated K+ channels in human melanoma cells are up-regulated by hypoxia involving hypoxia-inducible factor-1alpha and the von Hippel-Lindau protein. J. Physiol. 2006;571:349–359. doi: 10.1113/jphysiol.2005.096818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rosa P, et al. BK channels blockage inhibits hypoxia-induced migration and chemoresistance to cisplatin in human glioblastoma cells. J. Cell. Physiol. 2018;233:6866–6877. doi: 10.1002/jcp.26448. [DOI] [PubMed] [Google Scholar]
- 32.Wienen F, et al. Determination of clotrimazole in mice plasma by capillary electrophoresis. J. Pharm. Biomed. Anal. 2003;30:1879–1887. doi: 10.1016/s0731-7085(02)00531-9. [DOI] [PubMed] [Google Scholar]
- 33.Brown BM, Pressley B, Wulff H. KCa3.1 channel modulators as potential therapeutic compounds for glioblastoma. Curr. Neuropharmacol. 2018;16:618–626. doi: 10.2174/1570159X15666170630164226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang L, Fu L. Mechanisms of resistance to EGFR tyrosine kinase inhibitors. Acta Pharm. Sin. B. 2015;5:390–401. doi: 10.1016/j.apsb.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huether A, Hopfner M, Sutter AP, Schuppan D, Scherubl H. Erlotinib induces cell cycle arrest and apoptosis in hepatocellular cancer cells and enhances chemosensitivity towards cytostatics. J. Hepatol. 2005;43:661–669. doi: 10.1016/j.jhep.2005.02.040. [DOI] [PubMed] [Google Scholar]
- 36.Shan F, Shao Z, Jiang S, Cheng Z. Erlotinib induces the human non-small-cell lung cancer cells apoptosis via activating ROS-dependent JNK pathways. Cancer Med. 2016;5:3166–3175. doi: 10.1002/cam4.881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ouadid-Ahidouch H, et al. Functional and molecular identification of intermediate-conductance Ca2+-activated K+ channels in breast cancer cells: Association with cell cycle progression. Am. J. Physiol. Cell Physiol. 2004;287:C125–134. doi: 10.1152/ajpcell.00488.2003. [DOI] [PubMed] [Google Scholar]
- 38.Du Y, et al. The potassium channel KCa3.1 promotes cell proliferation by activating SKP2 and metastasis through the EMT pathway in hepatocellular carcinoma. Int. J. Cancer. 2019;145:503–516. doi: 10.1002/ijc.32121. [DOI] [PubMed] [Google Scholar]
- 39.De Marchi U, et al. Intermediate conductance Ca2+-activated potassium channel (KCa3.1) in the inner mitochondrial membrane of human colon cancer cells. Cell Calcium. 2009;45:509–516. doi: 10.1016/j.ceca.2009.03.014. [DOI] [PubMed] [Google Scholar]
- 40.Toro L, et al. MaxiK channel and cell signalling. Pflugers Arch. 2014;466:875–886. doi: 10.1007/s00424-013-1359-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Szabo I, Zoratti M. Mitochondrial channels: Ion fluxes and more. Physiol. Rev. 2014;94:519–608. doi: 10.1152/physrev.00021.2013. [DOI] [PubMed] [Google Scholar]
- 42.Leanza L, et al. Pharmacological modulation of mitochondrial ion channels. Br. J. Pharmacol. 2018 doi: 10.1111/bph.14544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tajeddine N, Gailly P. TRPC1 protein channel is major regulator of epidermal growth factor receptor signaling. J. Biol. Chem. 2012;287:16146–16157. doi: 10.1074/jbc.M112.340034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fife CM, McCarroll JA, Kavallaris M. Movers and shakers: Cell cytoskeleton in cancer metastasis. Br. J. Pharmacol. 2014;171:5507–5523. doi: 10.1111/bph.12704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schwab A, Fabian A, Hanley PJ, Stock C. Role of ion channels and transporters in cell migration. Physiol. Rev. 2012;92:1865–1913. doi: 10.1152/physrev.00018.2011. [DOI] [PubMed] [Google Scholar]
- 46.Appert-Collin A, Hubert P, Crémel G, Bennasroune A. Role of ErbB receptors in cancer cell migration and invasion. Front Pharmacol. 2015;6:283. doi: 10.3389/fphar.2015.00283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fichter CD, et al. ErbB targeting inhibitors repress cell migration of esophageal squamous cell carcinoma and adenocarcinoma cells by distinct signaling pathways. J. Mol. Med. (Berl.) 2014;92:1209–1223. doi: 10.1007/s00109-014-1187-5. [DOI] [PubMed] [Google Scholar]
- 48.Rezzonico R, et al. Focal adhesion kinase pp125FAK interacts with the large conductance calcium-activated hSlo potassium channel in human osteoblasts: Potential role in mechanotransduction. J. Bone Miner. Res. 2003;18:1863–1871. doi: 10.1359/jbmr.2003.18.10.1863. [DOI] [PubMed] [Google Scholar]
- 49.Schwab A, Schuricht B, Seeger P, Reinhardt J, Dartsch PC. Migration of transformed renal epithelial cells is regulated by K+ channel modulation of actin cytoskeleton and cell volume. Pflugers Arch. 1999;438:330–337. doi: 10.1007/s004240050917. [DOI] [PubMed] [Google Scholar]
- 50.Thompson EG, Sontheimer H. A role for ion channels in perivascular glioma invasion. Eur. Biophys. J. 2016;45:635–648. doi: 10.1007/s00249-016-1154-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bigot F, et al. Erlotinib pharmacokinetics: A critical parameter influencing acute toxicity in elderly patients over 75 years-old. Investig. New Drugs. 2017;35:242–246. doi: 10.1007/s10637-016-0400-5. [DOI] [PubMed] [Google Scholar]
- 52.Ataga KI, et al. Improvements in haemolysis and indicators of erythrocyte survival do not correlate with acute vaso-occlusive crises in patients with sickle cell disease: A phase III randomized, placebo-controlled, double-blind study of the Gardos channel blocker senicapoc (ICA-17043) Br. J. Haematol. 2011;153:92–104. doi: 10.1111/j.1365-2141.2010.08520.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.