

Structured abstract:
Genetically-encoded fluorescent sensors have been widely used to illuminate secretory vesicle dynamics and the vesicular lumen, including Zn2+ and pH, in living cells. However, vesicular sensors have a tendency to mislocalize and are susceptible to the acidic intraluminal pH. In this study, we performed a systematic comparison of five different vesicular proteins to target the fluorescent protein mCherry and a Zn2+ FRET sensor to secretory vesicles. We found that motifs derived from vesicular cargo proteins, including Chromogranin A (CgA), target vesicular puncta with greater efficacy than transmembrane proteins. To characterize vesicular Zn2+ levels, we developed CgA-Zn2+ FRET sensor fusions with existing sensors ZapCY1 and eCALWY-4, and characterized subcellular localization and the influence of pH on sensor performance. We simultaneously monitored Zn2+ and pH in individual secretory vesicles by leveraging the acceptor fluorescent protein as a pH sensor and found that pH influenced FRET measurements in situ. While unable to characterize vesicular Zn2+ levels at the single vesicle level, we were able to monitor Zn2+ dynamics in populations of vesicles and detected high vesicular Zn2+ in MIN6 cells compared to lower levels in the prostate cancer cell line LnCaP. The combination of CgA-ZapCY1 and CgA-eCALWY-4 allows for measurement of Zn2+ from pM to nM ranges.
Keywords: Secretory vesicle, genetically-encoded, fluorescent sensor, FRET, zinc, pH, targeting signal, Chromogranin A
Graphical Abstract
Proteins that are secreted from the cell constitute over one-third of the human proteome 1–2. Secretory proteins enter the secretory pathway through the ER and must traverse the Golgi network before being packaged into secretory vesicles. Vesicles are responsible for trafficking the myriad of secretory proteins to the plasma membrane for secretion 3. At the organismal level, protein secretion is required for central processes including synaptic plasticity 4 and glucose homeostasis 5. While all cell types constitutively secrete proteins, professional secretory cells, including pancreatic β-cells and prostate cells, have highly-developed secretion systems and contain a specialized type of vesicle called the granule. A wide range of human diseases are linked with impaired secretion from professional secretory cells 6, including diabetes which is characterized by deficient glucose-stimulated insulin secretion from pancreatic β-cells 7.
Secretory vesicles and granules must maintain an intraluminal ionic environment unlike any other cellular compartment 8–10 in order to carry out processing 11 and storage 12 of secretory proteins like insulin. One of the defining characteristics of vesicles is their acidic intraluminal pH, which is maintained near 5.5 13. High levels of Zn2+ are present in granules in pancreatic β-cells 14, among other secretory cells 15–18. In pancreatic β-cells, the Zn2+ transporter ZnT8 is responsible for concentrating Zn2+ inside granules 19, and reduced ZnT8 expression 20 and ZnT8 gene mutations 21 are both associated with an increased risk of developing diabetes. Zn2+ is required for secretory protein aggregation 22–25 and vesicle trafficking 26 and secretion 27–28. During exocytosis, Zn2+ is coreleased with vesicular cargo and can contribute to autocrine 29 and paracrine 30 signaling. It’s also been suggested that vesicular Zn2+ serves as a cellular Zn2+ reservoir that’s released into the cytosol 31.
Genetically-encoded fluorescent tools are widely used to study vesicle dynamics, including exocytosis 32–33 and endocytosis 34, intracellular vesicle trafficking 35–37, and to differentiate among vesicle pools 38–39. To express genetically-encoded fluorescent sensors within vesicles, targeting domains derived from exocytotic transmembrane proteins 40 and vesicular cargo proteins are attached to the sensor 41–43. However, these tools are sometimes present in multiple locations44–45 Furthermore, there is not a clear consensus as to which protein domain is the most effective at targeting. Genetically-encoded fluorescent sensors have also been used to characterize the intraluminal milieu of vesicles, including Ca2+ and pH levels 9, 46–49. Acidic pH can influence fluorescent sensor behavior 9–10, 50, making it difficult to quantitatively measure other vesicular ions. Dual fluorescent sensors of vesicular pH and Ca2+ or Cl- have overcome this limitation 9–10, but this concept has not yet been applied to fluorescent Zn2+ sensors.
In this study, we performed a systematic comparison of five targeting domains derived from vesicular proteins to express a fluorescent protein and fluorescence resonance energy transfer (FRET) sensor in secretory vesicles/granules. To identify a targeting motif that can be used across cell types, we performed our comparison in both normal and secretory cell lines. Using this information, we leveraged two existing genetically-encoded fluorescent sensors that can simultaneously detect Zn2+ and pH levels in vesicles/granules. These sensors (ZapCY1 and eCALWY-4) were chosen because they have the highest dynamic range of available FRET-based Zn2+ sensors and have previously been successfully targeted to organelles (mitochondria and ER). This study provides a roadmap for rigorous characterization of subcellular localization and the influence of pH on fluorescent sensor measurements in vitro and in situ. Using the dual Zn2+ and pH sensor, we detected Zn2+ dynamics and pH levels in single vesicles and measured relative Zn2+ levels in vesicle populations in two different secretory cell lines.
EXPERIMENTAL
Experimental details on materials, molecular cloning, cell culture and transfection, and the microscopes used in this study can be found in Supporting information.
Vesicular puncta detection and quantification.
Images of HeLa, MIN6 and LnCaP cells expressing mCherry fusions (VAMP2, Syp, CgA, NPY, INS), ZapCY1 fusions (VAMP2, CgA, NPY) and CgA-eCALWY-4 were collected using the wide field fluorescence microscope and spinning disk confocal microscope. The wide field images (Figure S-1, Figure S-2) were not used to quantify fluorescent puncta. For the spinning disk, the laser power was 100% for all channels (CFP, YFP, mCherry), and the exposure was 100 ms (YFP), 500 ms (CFP) or varied (mCherry).
Images were processed using NIS-Elements image analysis software v4.51. First, images were background subtracted by placing a rectangular region of interest (ROI) in a region without cells. Second, the Auto-Contrast function was used to account for differences in fluorescent signal between samples. Using the Spot Detection module, we determined the number of fluorescent puncta in each cell (CFP and YFP for CgA-ZapCY1, CFP only for CgA-eCALWY-4, mCherry for mCherry fusions) using the following parameters: bright/different sizes; output = circular area; typical diameter = 0.5 μm; contrast = 15,000; object symmetry = medium objects. We also determined the surface area (μm2) by drawing a ROI around each cell. For each cell, the number of fluorescent puncta was normalized to cell surface area. Cellular measurements (# fluorescent puncta/μm2) for 14–20 cells (mCherry in HeLa), 20–24 cells (mCherry in MIN6), 15–19 cells (CgA-ZapCY1 in HeLa), 22–31 cells (CgA-ZapCY1 in MIN6), 8 cells (CgA-eCALWY-4 in HeLa) and 7 cells (CgA-eCALWY-4 in MIN6) collected from three independent experiments were pooled together for the different comparisons performed in this study.
Colocalization between CgA-Zn2+ FRET sensor and fluorescent organelle markers.
Images of HeLa and MIN6 cells coexpressing CgA-ZapCY1 and mCherry organelle markers (Sec61, GalT, Rab5a, Rab7, LAMP1, VAMP2, NPY, INS) were collected using the spinning disk confocal microscope. The laser power was 100% for all channels (CFP, YFP, mCherry), and the exposure was 500 ms (CFP, YFP) or varied (mCherry). Images were processed using NIS-Elements image analysis software v4.51. First, images were background subtracted by placing a rectangular ROI in a region without cells. Second, an ROI was drawn around each cell and using the Colocalization module, we determined the Pearson correlation coefficient (PCC) between mCherry and CFP or YFP. Cellular measurements (PCC values) for 10–15 cells (HeLa) or 13–21 cells (MIN6) collected from three independent experiments were pooled together for the different comparisons performed in this study.
Zn2+ and pH calibrations in individual secretory vesicles.
Zn2+ and pH calibrations of individual secretory vesicles was performed in HeLa and MIN6 cells expressing CgA-ZapCY1 using the spinning disk confocal microscope. The laser power was 20% (CFP, FRET) or 5% (YFP). For all three channels, the exposure was 200ms and 2×2 binning was used. The FRET ratio (FRET channel/CFP channel) and YFP channel were monitored for Zn2+ calibrations and pH calibrations, respectively. Images were collected every 5 seconds over the course of each experiment. For each experiment (consisting of one cell), we recorded a 1–2-minute baseline and performed a Zn2+ calibration followed by a pH calibration. For the Zn2+ calibration, cells were placed in either phosphate-free HHBSS or phosphate-, calcium- and magnesium-free HHBSS. TPA (50 μM) or a combination of ZnCl2 (10 μM), pyrithione (5 μM) and saponin (0.001%) were added to the dish, respectively. TPA was because previous work has shown that the TPA acts faster and its binding/unbinding kinetics are less sensitive to pH than TPEN 51. The FRET ratio was monitored for ~5 minutes. For the pH calibration, the Zn2+ perturbations were removed and replaced with 20 mM NH4Cl (pH 8.5). We monitored the YFP signal for an additional ~5 minutes.
Zn2+ and pH calibrations of individual secretory vesicles were processed using Imaris image analysis software v9.5. Using the Object Detection and Tracking module, we tracked individual secretory vesicles from the baseline of each experiment through the Zn2+ and pH calibrations. The following object detection and tracking parameters were used: Algorithm menu: segment only a ROI, tracking spots over time; Source channel menu: CFP (MIN6 cells) or YFP (HeLa cells), background subtraction, estimated X-Y diameter = 0.5 μm; Classify spots menu: adjusted quality score to remove poor spots; Tracking menu: Brownian, max distance between frames = 1 μm, max gap size = 0; Classify tracks menu: adjusted track duration to keep longest tracks. In order to be stringent, some vesicles identified by the software were manually removed if we determined that a single track was linked to more than one vesicle. To determine if our tracking approach was robust, we calculated the number of vesicles successfully tracked (29 in HeLa and 24 in MIN6) as a percentage of the estimated starting number (127 in HeLa and 120 in MIN6). These percentages are reported in the Results section. We estimated the total starting number by multiplying the number of cells (5 in HeLa and 13 in MIN6) by the average number of fluorescent puncta/μm2 (~0.02 puncta/μm2 in HeLa and ~0.06 puncta/μm2 in MIN6) and average cell area (1231 μm2 in HeLa and 143.5 μm2 in MIN6).
For individual secretory vesicles, we plotted the FRET ratio (Zn2+ calibration traces in Figure 4B–E) and normalized YFP signal (pH calibration traces in Figure 5A&B) versus time. The resting FRET ratio for single vesicles was determined by calculating the average FRET ratio during the baseline measurements (Figure 4A). In the Results section, we report the robust coefficient of variation, or . The unnormalized YFP signal for single vesicles was determined by calculating the average raw YFP signal during the baseline measurements (Figure S-9A). The normalized YFP signal for individual secretory vesicles was determined by dividing the unnormalized YFP signal by the maximum YFP signal observed over the course of the entire experiment, which usually occurred after NH4Cl addition (Figure 5C). Finally, the normalized YFP signal was converted into an intraluminal pH using the standard curves in Figure S-8 (Figure 5D). By the end of our analysis pipeline, four values theoretically should have been reported for every secretory vesicle (resting FRET ratio, unnormalized YFP signal, normalized YFP signal and intraluminal pH). However, in some cases, the pH calibration could not be completed, so there are some vesicles with a resting FRET ratio/unnormalized YFP signal but without a normalized YFP signal/intraluminal pH. For other vesicles, the intraluminal pH could not be interpolated from the standard curves.
Figure 4: CgA-Zn2+ FRET sensor shows resting FRET ratio heterogeneity among vesicles and the response of single vesicles to Zn2+ perturbations.

HeLa and MIN6 cells expressing CgA-ZapCY1 were imaged using a spinning disk confocal microscope, and individual vesicles were identified and tracked over the course of each experiment. (A) Dot plot of the resting FRET ratio among secretory vesicles in HeLa cells (n=29 vesicles) and MIN6 cells (n=24 vesicles). One outlier was identified in the MIN6 group using the ROUT method (Q=1%) and removed from further analysis. Statistical analysis was performed using an unpaired t-test (ns = not significant). (B) Time-lapse of the FRET ratio of three secretory vesicles expressing CgA-ZapCY1 (HeLa cell) following the addition of 50 μM TPA at t = 0 min. (C) Time-lapse of the FRET ratio of a three secretory vesicles expressing CgA-ZapCY1 (HeLa cell) following the addition of 10 μM ZnCl2 + 5 μM pyrithione + 0.001% saponin (Zn2+/Pyr) at t = 0 min. (D) Time-lapse of the FRET ratio of three secretory vesicles expressing CgA-ZapCY1 (MIN6 cell) following the addition of 50 μM TPA at t = 0 min. (E) Time-lapse of the FRET ratio of three secretory vesicles expressing CgA-ZapCY1 (MIN6 cell) following the addition of 10 μM ZnCl2 + 5 μM pyrithione + 0.001% saponin at t = 0 min.
Figure 5: Quantification of pH levels in individual vesicles using CgA-Zn2+ FRET sensor reveals a positive correlation between the resting FRET ratio and pH levels.

HeLa and MIN6 cells expressing CgA-ZapCY1 were imaged using a spinning disk confocal microscope, and individual vesicles were identified and tracked over the course of each experiment. (A) Four representative traces of the normalized YFP signal (F/Fmax) in individual secretory vesicles expressing CgA-ZapCY1 in HeLa cells. To achieve the maximum YFP signal (Fmax), 20 mM NH4Cl (pH 8.5) was added at t = 0 min. (B) Four representative traces of YFP F/Fmax in individual secretory vesicles expressing CgA-ZapCY1 in MIN6 cells. To achieve the maximum YFP signal (Fmax), 20 mM NH4Cl (pH 8.5) was added at t = 0 min. For panels A and B, NH4Cl addition was time-adjusted such that it occurred at t = 0 minutes. (C) Dot plot of YFP F/Fmax prior to NH4Cl addition in individual secretory vesicles expressing CgA-ZapCY1 in HeLa cells (n=22 vesicles) and MIN6 cells (n=22 vesicles). Statistical analysis was performed using an unpaired t-test (*, P < 0.05). (D) Dot plot of resting pH levels in individual secretory vesicles expressing CgA-ZapCY1 in HeLa cells (n=22 vesicles) and MIN6 cells (n=18 vesicles). YFP F/Fmax values were converted to pH using the pH titration curves provided in Figure S-8. Statistical analysis was performed using an unpaired t-test (ns = not significant). (E) Scatter plot showing the linear relationship between resting FRET ratio and pH in HeLa cells (n=22 vesicles). Points were fit with linear regression, and the Pearson correlation coefficient (r) was used to determine whether a correlation exists (p < 0.05 is considered significant). (F) Scatter plot showing the linear relationship between resting FRET ratio and pH in MIN6 cells (n=18 vesicles).
Individual secretory vesicle measurements (resting FRET ratio, unnormalized YFP signal, normalized YFP signal, intraluminal pH) for 24 vesicles (HeLa cells) or 29 vesicles (MIN6 cells) collected from several independent experiments were pooled together for the different comparisons (Figure 4A: Resting FRET ratio in HeLa and MIN6, Figure S-9A: Unnormalized YFP signal in HeLa and MIN6, Figure 5C: Normalized YFP signal in HeLa and MIN6, Figure 5D: Intraluminal pH in HeLa and MIN6) and linear regression analysis (Figure 5E: Resting FRET ratio and pH in HeLa, Figure 5F: Resting FRET ratio and pH in MIN6, Figure S-9B: Resting FRET ratio and unnormalized YFP signal in HeLa, Figure S-9C: Resting FRET ratio and unnormalized YFP signal in MIN6) performed in this study. Based on FRET ratio traces like the ones shown in Figure 4B–E, we classified individual vesicles as non-responders (NR) or responders (R) to Zn2+ perturbations. In Figure S-10, we directly compared the intraluminal pH and unnormalized YFP signal between these two groups.
In situ pH titration of YFP signal and generating standard curves.
In situ pH titration of the YFP signal was performed in HeLa and MIN6 cells expressing the CgA-ZapCY1 using the spinning disk confocal microscope. The same imaging parameters used for Zn2+ and pH calibrations were used in this series of experiments. The CFP channel (reference) and YFP channel (measurement) were monitored during each pH titration. pH clamp buffers (pH 3.5–7.5) containing 10 μM monensin, 10 μM nigericin and 5 μM digitonin were prepared. For each experiment, we captured an image of a cell incubated in the first pH clamp buffer (pH 7.5). The pH 7.5 buffer was then removed and replaced with the pH 7 buffer. Following a 1-minute incubation to equilibrate the cell in the new buffer, we captured another image of the same cell (using the CFP channel to focus). The pH 7 buffer was removed and replaced with the pH 6.5 buffer and following another 1-minute incubation, we captured a third image of the cell. This process was repeated until the final image was collected in the pH 3.5 buffer. Only cells that remained viable throughout the entire pH titration were included in our analysis.
Images were processed using Imaris image analysis software v9.5. Using the Object Detection module, we identified individual secretory vesicles in each image. The following object detection (no tracking) parameters were used: Algorithm menu: segment only a ROI, tracking spots over time; Source channel menu: CFP, background subtraction, estimated X-Y diameter = 0.5 μm; Classify spots menu: adjusted quality score to remove poor spots. Individual secretory vesicles were identified in the CFP channel and measurements were collected in the YFP channel. For each experiment, we determined the average YFP signal of individual secretory vesicles at each pH and normalized all values to the maximum YFP signal, which we observed between pH 6.5–7.5. Individual secretory vesicle measurements (normalized YFP signal) for 26–89 vesicles (HeLa cells) or 105–138 vesicles (MIN6 cells) collected from four independent experiments were pooled together to generate the final plots, pH v. normalized YFP signal in HeLa (Figure S-8A) and MIN6 cells (Figure S-8B). To interpolate intraluminal pH values from normalized YFP signals (determined using pH calibration method above) for individual secretory vesicles, the curve was fit with a four-parameter logistic regression model. We calculated a pKa of 5.8 from both curves, which is close to the reported Citrine pKa of 5.7 52 and suggests that our pH titration method is valid.
Zn2+ calibrations of vesicle populations.
Zn2+ calibrations of vesicle populations were performed in MIN6 cells expressing CgA-ZapCY1 and CgA-eCALWY-4 and LnCaP cells expressing CgA-ZapCY1 using the wide field fluorescence microscope. Cells expressing CgA-ZapCY1 or CgA-eCALWY-4 were selected if they had a resting FRET ratio between 2–6 or above 0.7, respectively. For Zn2+ calibrations in vesicle populations, the FRET ratio (FRET channel/CFP channel) was monitored and images were collected every 10 seconds. For the Zn2+ calibration, cells were placed in either phosphate-free HHBSS or phosphate-, calcium- and magnesium-free HHBSS. TPA (150 μM) or a combination of ZnCl2 (10 μM), pyrithione (5 μM) and saponin (0.001%) were added to the dish, respectively. In some cases, a full Zn2+ calibration in which TPA and Zn2+ were both added to the same cell was performed. The FRET ratio was monitored for an additional 15–35 minutes.
Zn2+ calibrations of vesicle populations were processed using a custom MATLAB R2018A (Mathworks) pipeline for background subtraction, cell tracking, segmentation and reporting the average FRET and CFP signals used to calculate the FRET. The code is available here: https://biof-git.colorado.edu/palmer-lab/cellular-FRET. For all vesicle populations, we plotted the FRET ratio versus time in MIN6 cells expressing CgA-ZapCY1 (Figure 6A&B) or CgA-eCALWY-4 (Figure 6G, Figure S-13E) and LnCaP cells expressing CgA-ZapCY1 (Figure 6D&E). We determined the resting FRET ratio for all experiments by calculating the average FRET ratio during the baseline measurements. For experiments where TPA was added, we determined the minimum FRET ratio across the entire time-course. For experiments where Zn2+ was added, we determined the maximum FRET ratio across the entire time-lapse. Vesicle population measurements (resting FRET ratio, minimum FRET ratio, maximum FRET ratio) for 19 cells (MIN6 cells) or 17 cells (LnCaP cells) were pooled together for different comparisons (Figure 6C: MIN6 cells, Figure 6F: LnCaP cells). For Figure 6H, we calculated the difference FRET ratio (ΔR) between the resting FRET ratio and FRET ratio achieved following treatment with TPA or Zn2+. This value was divided by the resting FRET ratio to yield ΔR/R, and we set the baseline equal to one (dashed line) by adding one to all values.
Figure 6: Imaging vesicle populations with CgA-targeted Zn2+ FRET sensors reveals different vesicular Zn2+ environments in MIN6 cells and LnCaP cells.

(A) Time-lapse of FRET ratio in single MIN6 cells expressing CgA-ZapCY1 following the addition of 150 μM TPA (n=10 cells). (B) Time-lapse of FRET ratio in single MIN6 cells expressing CgA-ZapCY1 following the addition of 10 μM ZnCl2 + 5 μM pyrithione + 0.001% saponin (n=9 cells). (C) Dot plot displaying the FRET ratio of single MIN6 cells expressing CgA-ZapCY1 under three conditions: the resting FRET ratio prior to drug addition (resting), the minimum FRET ratio following TPA addition (TPA) and the maximum FRET ratio following Zn2+ addition (Zn2+). At least three independent experiments were performed per condition, and the average ± standard deviation is shown for n=19 cells (resting), n=10 cells (TPA) and n=9 cells (Zn2+). Statistical analysis was performed using a One-Way ANOVA test with post hoc Tukey (**, P < 0.01 compared with resting; #, P < 0.05 compared with TPA).(D) Time-lapse of FRET ratio in single LnCaP cells expressing CgA-ZapCY1 following the addition of 150 μM TPA (n=9 cells). (E) Time-lapse of FRET ratio in single LnCaP cells expressing CgA-ZapCY1 following the addition of 10 μM ZnCl2 + 5 μM pyrithione + 0.001% saponin (n=8 cells). (F) Dot plot displaying the FRET ratio of single LnCaP cells expressing CgA-ZapCY1 under resting, TPA and Zn2+ conditions. At least three independent experiments were performed per condition, and the average ± standard deviation is shown for n=17 cells (resting), n=9 cells (TPA) and n=8 cells (Zn2+). Statistical analysis was performed using a One-Way ANOVA test with post hoc Tukey (****, P < 0.0001 compared with resting; ####, P < 0.0001 compared with TPA). (G) Representative trace of full Zn2+ calibration for CgA-ZapCY1 (blue trace) and CgA-eCALWY-4 (red trace) performed in MIN6 cells. The FRET ratios on the left (blue) and right (red) correspond with CgA-ZapCY1 and CgA-eCALWY-4, respectively. (H) Dot plot of ΔR/R values (normalized to baseline of one) of CgA-ZapCY1 in MIN6 cells, CgA-ZapCY1 in LnCaP cells and CgA-eCALWY-4 in MIN6 cells. For each sensor/cell line combination, ΔR/R was compared between the TPA and Zn2+/Pyr treatments using an unpaired t-test (****, P < 0.0001).
Sensor purification and in vitro characterization.
ZapCY1 (pBAD) was expressed in TOP10 E. Coli and induced with 0.2% arabinose. Bacterial cell pellets were lysed with B-PER Bacterial Protein Extraction Reagent (Thermo Fisher Scientific). The Zn2+ sensor was purified on a Ni-NTA agarose column (Qiagen) and eluted into one of three buffers (pH 5.5, pH 6.5 or pH 7.5) containing 50 mM HEPES (pH 7.4) or 50 mM MES (pH 5.5 or pH 6.5), 10 mM NaCl and 10% glycerol. Immediately following elution, purified Zn2+ sensor was incubated with EDTA (1 mM). Zn2+ sensor concentration was determined at A516 using ε = 77,000 M−1cm−1 and diluted to a final concentration of 500 nM.
To determine the Kd’ of the Zn2+ FRET sensor at pH 5.5, pH 6.5 and pH 7.4, purified sensor was titrated with buffered Zn2+ solutions. Buffered Zn2+ solutions (EDTA, HEDTA, HEDTA/Sr2+, HEDTA/Ca2+, EGTA) were prepared as described previously 53. In brief, concentrations of free Zn2+ were determined using the chelator stability constants for Zn2+ and competing metals (Sr2+, Ca2+) and varying the ratio of Zn2+ to Zn2+/chelator or Zn2+/chelator/competing metal. The free Zn2+ concentrations for the buffered Zn2+ solutions at pH 5.5, pH 6.5 and pH 7.4 are shown in Table S-2. 100x buffered Zn2+ solutions (1 μL) were added to a black-walled 96-well plate. For pH 7.4 and pH 6.5, we used all five buffered Zn2+ solutions. For pH 5.5, we only used the HEDTA and EGTA buffered Zn2+ solutions. Prior to measurements, purified Zn2+ sensor was incubated with the reducing agent TCEP for 10 minutes. 99 μL protein was added on top of the buffered Zn2+ solutions to promote mixing. In vitro fluorescence measurements were performed using a SpectraMax iD3 Multi-Mode Microplate Reader (Molecular Devices) using the following parameters: 450 nm excitation and 460–660 nm emission with a 10 nm bandwidth. To determine the FRET ratio, the fluorescence emission at 480 nm (CFP) and 530 nm (FRET) were used. At each pH, we plotted the FRET ratio versus free Zn2 concentration and fit the curve with a model reported by Pomorski et al that can be used to determine the Kd’ from ratiometric fluorescence measurements 54:
where FRETbound is the maximum FRET signal (three data point average), FRETunbound is the minimum FRET signal (three data point average), CFPbound is the maximum CFP signal (three data point average), CFPunbound is the minimum CFP signal (three data point average) and [Zn2+] is the concentration of free Zn2+.
Image processing and data analysis.
All images shown were processed in Image J, including the following functions: background subtraction, scale bar addition, contrast adjustments, cropping and merging. All data was plotted in GraphPad Prism v.8.0.1. Statistical tests were also performed using this software. Outliers were identified using the ROUT method with Q = 1%. For comparisons, we used either an unpaired t-test for two groups or One-Way ANOVA test with post-hoc Tukey for greater than two groups (p < 0.05 was considered significant). For linear regression analysis, we used the Pearson correlation coefficient (p < 0.05 was considered significant). For curve fitting, the equations used in Figure S-7 and Figure S-8 are described above.
RESULTS
Systematic comparison of the vesicular targeting ability of secretory protein domains
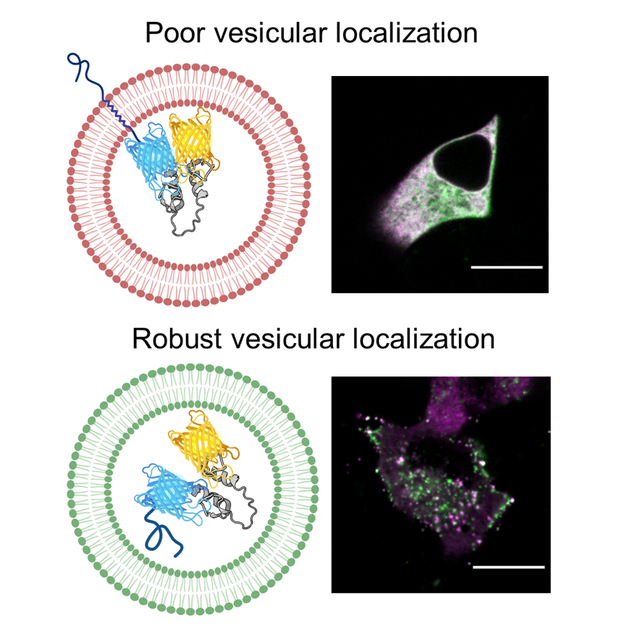
To develop a Zn2+ FRET sensor that can measure Zn2+ in secretory vesicles, we first required a robust vesicular targeting motif. Protein motifs derived from the transmembrane proteins, VAMP2 47, 55–57 and Synaptophysin (Syp) 34, 38, 48, and the soluble cargo proteins, Chromogranin A (CgA) 46, 49, Neuropeptide Y (NPY) 10, 32 and insulin (INS) 58, have been used to target fluorescent proteins and sensors to secretory vesicles and granules. However, their performance has never been directly compared. Here, we performed a systematic comparison of the ability of VAMP2, Syp, CgA and NPY to target the pH-stable fluorescent protein mCherry 32, 59 to vesicular puncta in live cells. We transfected HeLa and MIN6 cells with mCherry fused to VAMP2, Syp, CgA,NPY and INS and imaged cells using a spinning disk confocal microscope (Figure 1) and wide-field fluorescence microscope (Figure S-1). We found that VAMP2 accumulated in vesicular puncta and at the plasma membrane in both cell types (Figure 1A). In contrast, Syp was predominantly expressed in a region near the nucleus consistent with the Golgi 60. Bright vesicular puncta were abundant in HeLa and MIN6 cells expressing CgA, NPY and INS fusions. We determined the number of vesicular puncta per cell and found that as mCherry fusions, CgA, NPY and INS were present in a significantly greater number than either VAMP2 or Syp in HeLa cells (Figure 1B) and MIN6 cells (Figure 1C). Puncta were difficult to resolve in wide field images (Figure S-1), but accumulation of VAMP2 and Syp in the Golgi was evident. Furthermore, the background signal in MIN6 cells expressing INS-mCherry was noticeably higher than in cells expressing CgA or NPY.
Figure 1. Secretory targeting domains fused with a fluorescent protein accumulate in vesicular puncta in HeLa cells and MIN6 cells.

(A) Representative images of HeLa cells (top row) and MIN6 cells (bottom row) expressing VAMP2-mCherry, Syp-mCherry, CgA-mCherry,NPY-Cherry and INS-mCherry (MIN6 only) that were collected using a spinning disk confocal microscope. Scale bar = 10 μm. (B) Dot plot of mCherry puncta detected per HeLa cell, normalized to cell area (μm2). Three independent experiments were performed per construct, and the average ± standard deviation is shown for n=16 cells (VAMP2), n=14 cells (Syp), n=14 cells (CgA) and n=20 cells (NPY). One outlier was identified in the Syp group using the ROUT method (Q=1%) and was removed from further analysis. Statistical analysis was performed using a One-Way ANOVA test with post hoc Tukey (****, P < 0.0001, *, P < 0.05 compared with Syp; ##, P < 0.01, #, P < 0.05 compared with VAMP2). (C) Dot plot of mCherry puncta detected per cell, normalized to cell area (μm2). Three independent experiments were performed per construct, and the average ± standard deviation is shown for n=20 cells (VAMP2), n=22 cells (Syp), n=22 cells (CgA), n=24 cells (NPY) and n=22 cells (INS). Using the ROUT method (Q=1%), one outlier was identified in the Syp group and one outlier was identified in the NPY group, and both outliers were removed from further analysis. Statistical analysis was performed using a One-Way ANOVA test with post hoc Tukey (****, P < 0.0001 compared with VAMP2; ####, P < 0.0001 compared with Syp).
Based on the superior performance of CgA and NPY to target a single fluorescent protein to vesicular puncta in HeLa and MIN6 cells, we elected to test their performance as Zn2+ FRET sensor fusions 61. We included VAMP2 despite its poor performance as an mCherry fusion as it was used previously to target Zn2+ FRET sensor platforms to secretory vesicles 56–57. We initially selected ZapCY1 as the FRET sensor for this study because it has a high dynamic range and was previously shown to be the most robust sensor when targeted to the ER 53.ZapCY1 contains the pH-stable fluorescent protein ECFP 62 and pH-sensitive yellow fluorescent protein (YFP) Citrine 52. Since YFP is quenched in acidic compartments and CFP is not, we leveraged the YFP intensity of CgA-ZapCY1relative to CFP intensity, to assess sensor localization in the Golgi 63 and vesicles 64, as previously reported 65. HeLa and MIN6 cells were transfected with ZapCY1 fusions with VAMP2, CgA and NPY and images (CFP channel and YFP channel) were collected using a confocal microscope (Figure2A) and wide-field microscope (Figure S-2). We predicted that YFP would be quenched in acidic vesicles; therefore, we calculated the number of vesicular puncta per cell using the CFP channel (Figure 2B&C). When fused with ZapCY1, VAMP2 was not detected in vesicular puncta in either cell type (Figure 2B&C). In contrast, both CgA-ZapCY1 and NPY-ZapCY1 were expressed in vesicular puncta in HeLa and MIN6 cells, which were embedded among ER tubules in HeLa cells but localized near the plasma membrane or Golgi in MIN6 cells (Figure 2A). Using the wide field microscope, vesicular puncta could be seen at the Golgi, but not the plasma membrane, of MIN6 cells (Figure S-2). In both cell types, the vesicular puncta that we observed were dynamic (Supporting Video S1 and Supporting Video S2) and the YFP signal was quenched to a significant degree, suggesting that CgA-ZapCY1 and NPY-ZapCY1 were present in acidic vesicles. In summary, CgA and NPY, but not VAMP2 fusions, accumulated in vesicular puncta in HeLa and MIN6 cells (Figure 3B, Supporting Video S1).
Figure 2: Targeting domains derived from vesicular cargo proteins fused with ZapCY1 accumulate in vesicular puncta in HeLa cells and MIN6 cells.

(A) Representative images of HeLa cells (top row) and MIN6 cells (bottom row) expressing VAMP2-ZapCY1, CgA-ZapCY1 and NPY-ZapCY1 that were collected using a spinning disk microscope. The images shown are an overlay of the CFP channel (green) and YFP channel (magenta). Scale bar = 10 μm. (B) Dot plot of CFP puncta detected per HeLa cell, normalized to cell area (μm2). Three independent experiments were performed per construct, and the average ± standard deviation is shown for n=15 cells (VAMP2), n=19 cells (CgA) and n=16 cells (NPY). Two outliers were identified in the VAMP2 group using the ROUT method (Q=1%) and removed from further analysis. (C) Dot plot of CFP puncta detected per MIN6 cell, normalized to cell area (μm2). Three independent experiments were performed per construct, and the average ± standard deviation is shown for n=22 cells (VAMP2), n=31 cells (CgA) and n=23 cells (NPY). Four outliers were identified in the VAMP2 group using the ROUT method (Q=1%) and removed from further analysis. Statistical analysis for B and C was performed using a One-Way ANOVA test with post hoc Tukey (**** P < 0.0001, ** P < 0.01 compared with VAMP2).
Figure 3: CgA-Zn2+ FRET sensor is present in secretory vesicles/granules of HeLa cells and MIN6 cells.

(A&C) Representative images of HeLa cells (A) and MIN6 cells (C) coexpressing CgA-ZapCY1 with mCherry organelle markers: Sec61 and NPY (HeLa) and INS and NPY (MIN6). Spinning disk confocal images collected in the mCherry channel and CFP channel are shown in greyscale, and an overlay of both images is shown in color (mCherry = magenta, CFP = green). Scale bar = 10 μm. (B&D) Dot plot of PCC values between CFP and mCherry calculated in HeLa cells (B) and MIN6 cells (D). mCherry markers of compartments in the secretory pathway and endo-lysosomal pathway are shaded in yellow and purple, respectively. In panel B, three independent experiments were performed per mCherry marker, and the average ± standard deviation is shown for n=11 cells (Sec61), n=10 cells (GalT), n=14 cells (VAMP2), n=15 cells (NPY), n=13 cells (Rab5a), n=10 cells (Rab7) and n=11 cells (LAMP1). One outlier was identified in the GalT group using the ROUT method (Q=1%) and removed from further analysis. In panel D, three independent experiments were performed per mCherry marker, and the average ± standard deviation is shown for n=15 cells (Sec61), n=13 cells (GalT), n=16 cells (VAMP2), n=20 cells (INS), n=17 cells (NPY), n=14 cells (Rab5a), n=15 cells (Rab7) and n=17 cells (LAMP1). Using the ROUT method (Q=1%), one outlier was identified in the INS group and one outlier was identified in the Rab5a group, and both outliers were removed from further analysis. Statistical analysis was performed using a One-Way ANOVA test with post hoc Tukey (**, P < 0.01, *, P < 0.05 compared with Sec61; ####, P < 0.0001, ##, P < 0.01, #, P < 0.05 compared with GalT; ††††, P < 0.0001, ††, P < 0.01 compared with LAMP1).
In order to further characterize the types of vesicles targeted by VAMP2, CgA and NPY, when fused with the Zn2+ FRET sensor, we calculated the number of vesicular puncta using the YFP channel, rather than the CFP channel. Surprisingly, we detected vesicular puncta in MIN6 cells expressing CgA-, but not NPY-ZapCY1 fusions (Figure S-3B). This suggests that the CgA-ZapCY1 may be present in secretory vesicles with diverse pH environments, including those with a less acidic lumen. Measuring Zn2+ levels in different types of vesicles 13, 66 was a critical part of our investigation; therefore, we elected to move forward with the CgA-ZapCY1 in further experiments.
Characterizing the subcellular localization of the CgA-ZapCY1
Many types of acidic vesicles are present in eukaryotic cells, including secretory vesicles, endosomes and lysosomes 64, 67. To validate that CgA-ZapCY1 was expressed in secretory vesicles, we examined colocalization between our sensor and fluorescent organelle markers using the Pearson correlation coefficient (PCC). We coexpressed the CgA-ZapCY1 and fluorescent markers of secretory vesicles/granules (VAMP2, INS, NPY), early and late endosomes (Rab5a, Rab7), and lysosomes (LAMP1) in HeLa and MIN6 cells and imaged live cells using a confocal microscope. Since we suspected that the sensor localized to the ER and Golgi, especially in HeLa cells, we also included fluorescent markers of the ER (Sec61) and Golgi (GalT). YFP is quenched in acidic cellular compartments 52, 65, and indeed we observed a decrease in PCC values between the YFP signal and acidic organelle markers (Figure S-4). Therefore, we assessed colocalization using the CFP signal. As shown in Figure 3A&B, in HeLa cells, CgA-ZapCY1 showed the highest colocalization with Sec61- and Rab7-mCherry, suggesting localization to both the ER and late endosomes, although there was some colocalization with the Golgi and multiple vesicular compartments (Figure S-5), suggesting the sensor was also present in secretory vesicles. In MIN6 cells, there was strong correlation between vesicles with CgA-ZapCY1 and NPY-mCherry or INS-mCherry signal (Figure 3C&D, Figure S-6). While there was little evidence of sensor localization to the ER and Golgi, CgA-ZapCY1 did colocalize to some degree with endosomal markers (Figure 3D). Taken together, CgA-ZapCY1 accumulates in secretory vesicles, especially in MIN6 cells.
CgA-ZapCY1 can detect intraluminal Zn2+ changes and pH in individual vesicles
Having identified CgA as a robust secretory targeting domain, our primary goal was to determine if the CgA-ZapCY1 sensor could measure Zn2+ in individual vesicles. The resting FRET ratio (ratio of YFP signal to CFP signal upon CFP excitation) can be converted into a Zn2+ concentration with knowledge of the binding affinity and by performing a Zn2+ calibration to determine the minimum and maximum FRET ratios 61, 68–69. We focused on single vesicles because we were interested in studying vesicle heterogeneity. We successfully tracked 29 vesicles in five HeLa cells and 24 vesicles in thirteen MIN6 cells, which represent 23% and 20% of our starting population, respectively. We found that the average resting FRET ratio among secretory vesicles in HeLa and MIN6 cells was not significantly different (Figure 4A). However, the range of resting FRET ratios was greater in MIN6 cells (ranging from 0.7 to 2.6) than HeLa cells (ranging from 1.3 to 2.1). Furthermore, the robust coefficient of variation (RCV), which can also be used to describe variation in a population, was greater in MIN6 cells (53.9%) compared with HeLa cells (13.6%). We predicted that the resting FRET ratio heterogeneity in MIN6 cells might be attributed to differences in vesicular Zn2+ levels. However, since vesicles were difficult to track over long periods, we could not perform an entire Zn2+ calibration. Therefore, we performed half-calibrations, where the Zn2+ chelator TPA was added to one set of cells and Zn2+ along with the ionophore pyrithione was added to another set of cells. In response to TPA, 41% vesicles in HeLa cells and 47% vesicles in MIN6 cells showed a FRET ratio decrease. Furthermore, 42% vesicles in HeLa cells and 29% vesicles in MIN6 cells showed a FRET ratio increase in response to Zn2+ and pyrithione. Representative traces of the TPA and Zn2+ response of individual secretory vesicles in HeLa cells (Figure 4B&C) and MIN6 cells (Figure 4D&E) are shown. More than half of secretory vesicles did not respond to Zn2+ perturbations, and among those that did respond, changes in FRET ratio were difficult to interpret (drifting baseline, poor signal-to-noise). Therefore, we were unable to determine the minimum and maximum FRET ratios and ultimately could not determine Zn2+ levels in individual vesicles.
It’s possible that the resting FRET ratio heterogeneity observed in MIN6 cells could also be due to differences in secretory vesicle pH. We’ve previously shown that alkaline pH environments alter the Zn2+ -binding affinity of the Zn2+ FRET sensor used here 69. To test if acidic pH alters the Zn2+ binding-affinity, we purified ZapCY1 and performed in vitro Zn2+ titrations at pH 5.5 and pH 6.5, which are the approximate pH of immature 66 and mature 13 secretory granules, respectively. We also performed this set of experiments at pH 7.4. We found that the apparent dissociation constant was 115 pM at pH 6.5 compared with 4.3 pM at pH 7.4 (Figure S-7). Furthermore, at pH 5.5, the Zn2+ FRET sensor did not show a FRET ratio increase, even at nanomolar Zn2+ concentrations. Thus, we might not expect to see a Zn2+ response if our sensor is expressed in secretory vesicles with pH 5.5 or lower. From our in vitro characterization, we also found that the FRET ratio range varied at pH 5.5 (~0.8), pH 6.5 (ranging from 1.3 to 1.9) and pH 7.4 (ranging from 1.5–2.3), suggesting that differences in pH may account for differences in resting FRET ratio.
To test if vesicular pH is a determinant of the resting FRET ratio in situ, we determined the pH of individual vesicles directly alongside our FRET ratio/Zn2+ response measurements. To calculate the pH of single vesicles, we used an approach developed by Dickson et al 9 that leverages the FRET acceptor YFP as a pH sensor. The pKa of the YFP variant Citrine is 5.7 and hence close to the pH of secretory vesicles 10, 13, 55 and its signal is independent of FRET. Here, we monitored the YFP signal of single vesicles in HeLa cells (Figure 5A) and MIN6 cells (Figure 5B). Following each Zn2+ calibration, we performed a pH calibration by adding NH4Cl in order to dequench YFP and achieve a maximum YFP signal (Fmax). Figure 5C shows the normalized YFP signal (F/Fmax) of secretory vesicles in HeLa cells and MIN6 cells. We found that the resting YFP signal (F/Fmax) prior to NH4Cl addition was significantly less in MIN6 cells compared with HeLa cells (Figure 5C). Using standard curves generated in both cell lines (Figure S-8), we converted the YFP signal (F/Fmax) into an intraluminal pH for each vesicle. We found that the intraluminal pH level among vesicles in MIN6 cells (intraluminal pH = 6.2) and HeLa cells (intraluminal pH = 6.5) was quite similar (Figure 5D). Furthermore, the intraluminal pH ranged from 5.7 to 7.2 in HeLa cells and ranged from 5.1 to 8.0 in MIN6 cells, supporting our prediction that CgA-ZapCY1 is expressed in vesicles with diverse pH environments. Finally, to determine if the intraluminal pH correlated with the FRET ratio, we plotted both variables and evaluated their linear relationship using the Pearson correlation coefficient (r). An r-value of zero suggests that there is no relationship, and r-values greater or lesser than zero indicate a positive or negative linear relationship, respectively. Indeed, there was a positive correlation between resting FRET ratio and pH in HeLa cells (Figure 5E) and MIN6 cells (Figure 5F).
We also evaluated the correlation between the resting FRET ratio and raw YFP signal (not pH-calibrated). The latter can provide information about FRET sensor expression levels 61, in addition to pH. We found that the raw YFP signal was significantly greater in MIN6 cells than HeLa cells (Figure S-9A), and there was a strong positive correlation between the resting FRET ratio and raw YFP signal in HeLa cells (Figure S-9B) and MIN6 cells (Figure S-9C). Taken together, the results in Figure 5 and Figure S-9 demonstrate that the intraluminal pH level and vesicular sensor expression are drivers of the resting FRET ratio in individual vesicles when data were collected on a spinning disc confocal. It is possible that one or both variables may be responsible for the observation that more than half of vesicles did not respond to Zn2+ perturbations. Although we did not see significant differences in intraluminal pH or raw YFP signal between non-responding and responding vesicles (Figure S-10).
CgA-targeted Zn2+ FRET sensors can provide Zn2+ measurements in vesicle populations
We encountered many challenges in our attempt to measure Zn2+ levels in individual secretory vesicles. Some were related to the equipment (e.g. laser photobleaching and poor signal-to-noise on confocal microscope) and others to the biological system (e.g. vesicle crowding and dynamics in MIN6 cells). As an alternative approach, we evaluated the response of secretory vesicle populations to Zn2+ perturbations using a wide-field fluorescence microscope. While we were unable to resolve single vesicles in cells expressing CgA-ZapCY1using a wide-field microscope (Figure S-2), colocalization analysis showed that our sensor primarily localized to secretory granules in MIN6 cells (Figure 3). Therefore, we subjected MIN6 cells expressing CgA-ZapCY1 to Zn2+ perturbations and measured vesicle populations in a region of interest. In contrast with the variable response that we observed in single vesicles, TPA resulted in a consistent and robust FRET ratio decrease (Figure 6A, Figure S-11A&B). On the other hand, addition of Zn2+ and pyrithione had little effect on the FRET ratio (Figure 6B). As shown in Figure 6C, the resting FRET ratio was comparable to the maximum FRET ratio of the saturated sensor upon addition of Zn2+ and pyrithione, suggesting that the CgA-ZapCY1 is saturated under resting conditions in MIN6 cells and that vesicular Zn2+ levels at the population level are high. This finding is in agreement with previous reports demonstrating high Zn2+ levels in pancreatic β-cell secretory granules 14, 23.
Prostate cells, like β-cells, are a secretory cell type with granules that contain large amounts of Zn2+ 15. While the prostate gland is a Zn2+-rich tissue 70–71, prostate cancer cells exhibit less cellular Zn2+ 72–73. Whether a reduction in total cellular zinc causes depletion of vesicular Zn2+ in these cells is unclear. In this study, we sought out to determine the relative level of vesicular Zn2+ in the prostate cancer cell line, LnCaP 74. In LnCaP cells vesicular puncta were abundant in cells expressing CgA-ZapCY1 (Figure S-12A, Supporting Video S3), with significantly more vesicular puncta detected in LnCaP cells compared with MIN6 cells (Figure S-12B). In contrast with MIN6 cells, we found that most vesicle populations did not respond to TPA (Figure 6D). However, LnCaP cells showed a consistent rise in FRET ratio following addition of Zn2+ and pyrithione (Figure 6E, Figure S-11C&D). Figure 6F shows that there was no difference between the resting and minimum FRET ratios, but both were less than the maximum FRET ratio. This suggests that the CgA-ZapCY1 is deprived of Zn2+ under resting conditions in LnCaP cells and that vesicular Zn2+ levels are, in fact, lower than MIN6 cells.
ZapCY1 was originally chosen as the FRET-based Zn2+ sensor because it had the highest dynamic range and least variability when targeted to the secretory pathway (specifically the ER) compared to two other FRET-based sensor platforms 53. A high dynamic range is an important sensor characteristic for accurate evaluation of Zn2+ levels as low dynamic range sensors have been shown to overestimate Zn2+ concentrations 69. However, an equally important factor to consider is the Kd of the sensor because a sensor is most accurate when the Kd value is close to the physiological Zn2+ concentration. Indeed, the ZapCY1 sensor was saturated at rest in MIN6 cells due to the relatively high affinity (Kd ~ 115 pM at pH 6.5), demonstrating that it was ill-suited for quantifying Zn2+ in these cells. Therefore, we created a CgA fusion with eCALWY4, another high dynamic range FRET-based Zn2+ sensor that has a lower affinity (Kd ~630 pM at 7.1) 75. CgA-eCALWY-4 localized to vesicles in both HeLa cells (Figure S-13A&B) and MIN6 cells (Figure S-13C&D, further demonstrating the robustness of this targeting motif. In MIN6 cells CgA-eCALWY-4 responded to both TPA and Zn2+/pyrithione (Figure 6G & Figure S-13), demonstrating that it was 26% saturated under resting conditions. Figure 6H shows the ΔR/R0 for calibration conditions showing that CgA-ZapCY1 is saturated at rest (Zn2+/Pyr is comparable to the normalized resting ratio of 1, while CgA-eCALWY-4 is partially saturated, responding to both TPA and Zn2+/Pyr).
DISCUSSION
Development and characterization of a vesicular fluorescent Zn2+ and pH sensor
Secretory vesicles and granules package and traffic thousands of secretory proteins to the plasma membrane for secretion 2. Once released, secretory proteins can participate in physiological processes including hormone signaling 58 and construction of the extracellular matrix 76. Impaired protein secretion, especially from professional secretory cells, is associated with human diseases, such as pancreatic β-cell dysfunction in diabetes 6. To promote protein storage, secretory vesicles/granules maintain high intraluminal Zn2+ 14–18 and an acidic pH 13, 55. Dysregulation of Zn2+ and pH homeostasis in the vesicular lumen can have pathophysiological consequences 19, 77. To understand the molecular mechanisms behind vesicular trafficking/secretion 34, 36–37, 55 and to characterize the vesicular environment 9–10, 57, researchers have turned to genetically-encoded fluorescent tools. To target fluorescent sensors to secretory vesicles/granules, a common strategy is to attach a targeting domain derived from a vesicular protein. However, it’s unclear which protein domain can target secretory vesicles with the greatest efficacy. To make matters worse, the acidic intraluminal pH of vesicles can alter fluorescent protein brightness 50 and fluorescent sensor performance 41, making it difficult to perform quantitative measurements without taking pH into account. The identification of a robust vesicular targeting motif, along with a detailed roadmap for characterizing the localization and pH-dependent properties of vesicular sensors, would be useful to guide the development of new sensors of vesicular dynamics and intraluminal environment.
In this study, we performed a systematic comparison of the vesicular targeting ability of two transmembrane proteins (VAMP2 and Syp) and three cargo proteins (CgA, NPY and INS). Overall, we found that targeting motifs derived from vesicular cargo exhibit better vesicular localization compared with transmembrane proteins. We leveraged this information to target existing FRET-based Zn2+ sensors (ZapCY1 and eCALWY-4) which can serve as genetically-encoded dual Zn2+/pH sensors in secretory vesicles/granules in both normal and secretory cells. Given the different Zn2+ binding affinities, CgA-ZapCY1 and CgA-eCALWY-4 are capable of detecting Zn2+ in vesicles/secretory granules over a wide concentration range.
Vesicular cargo proteins are robust secretory vesicle targeting signals
To our knowledge, this study represents the first side-by-side comparison of vesicular targeting motifs in the context of a fluorescent protein or FRET sensor. Overall, we found that the secretory proteins CgA, NPY and INS, but not VAMP2 or Syp, accumulated in vesicular puncta when attached to mCherry (Figure 1). This suggests that soluble cargo proteins, but not transmembrane proteins, should generally be used to construct vesicular sensors. This result was consistent with the VAMP2-, CgA- and NPY-ZapCY1 fusions (Figure 2). Given that VAMP2 is one of the most widely used targeting motifs, the lackluster ability of VAMP2 to accumulate in vesicles was surprising, 33, 39, 47, 55, 57. Previous reports suggest that both CgA 46, 49 and NPY 10, 32 are robust targeting signals for vesicular sensors. Consistent with this, CgA and NPY targeted vesicular structures with similar efficacy in mCherry and ZapCY1 fusions, regardless of the cell type. INS-mCherry, which was constructed by inserting mCherry in between the A- and B-chains of proinsulin 78, was on par with CgA and NPY. Based on vesicular puncta detection in the YFP channel (Figure S-3), we predicted that the NPY Zn2+ FRET sensor fusion was selectively-expressed in acidic vesicles 13, while the CgA-Zn2+ FRET sensor fusion was expressed in a more heterogenous population with diverse intraluminal environments 9, 66. Furthermore, we observed that vesicles expressing CgA-ZapCY1 were found in one of two places in MIN6 cells: the Golgi or plasma membrane (Figure 3). It’s possible that these pools correspond with the reserve and readily-releasable pools of insulin-containing granules in pancreatic β-cells 58.
Rigorous evaluation of subcellular localization and pH-dependence of vesicular sensors
Fluorescent sensors targeted to vesicles must traverse the entire secretory pathway and hence it’s imperative to rigorously define subcellular localization. In this study, we evaluated the colocalization (Pearson correlation coefficient) between CgA-ZapCY1 and fluorescent organelle markers in live cells. Indeed, we found that our sensor localized to the ER and Golgi in HeLa cells (Figure 3). We previously showed that the Zn2+ FRET sensor used in this study forms higher-order oligomers in the ER 53, which in our case might prevent progression to secretory vesicles. In MIN6 cells, the CgA-Zn2+ FRET sensor colocalized with secretory granule markers but not the ER or Golgi. Thus, the secretory pathway of MIN6 cells might be better equipped to handle vesicular sensor expression. Another general concern with vesicular sensor targeting is that the sensor might be recycled into endosomal compartments upon vesicle fusion with the plasma membrane. Indeed, we found that the CgA-Zn2+ FRET sensor sometimes colocalized with a late endosome marker in HeLa and MIN6 cells. The Ca2+ FRET sensor D1-SG also exhibited significant colocalization with an endosomal marker 9, suggesting that endosome mislocalization is a common problem.
The intraluminal pH of secretory vesicles ranges from 5.2–5.8 9–10, 13, 55. Since pH strongly influences fluorescent sensor measurements 41, the effect of acidic pH on vesicular sensor performance should be carefully considered. For example, granular Ca2+ and Cl- levels were quantitatively measured using the genetically-encoded Ca2+ sensor D1-SG 9 and Cl- sensor NPY-ClopHensor 10. This was accomplished by rigorously characterizing the effect of pH on in vitro binding-affinity and performing in situ pH calibrations alongside Ca2+ or Cl- measurements. In this study, we leveraged a similar approach with a fluorescent Zn2+ sensor. However, we were unable to perform reliable and consistent Zn2+ calibrations at the single vesicle level. It was difficult to track single vesicles during consecutive Zn2+ and pH calibrations due to vesicle crowding and the tendency of vesicles to move in and out of the confocal plane. Therefore, we were only able to track ~20% of the vesicles that we began with. Among these vesicles, we found that only 29–47% exhibited a FRET ratio change in response to Zn2+ perturbations, which we attribute to the poor signal-to-noise on the confocal microscope. Ultimately, we could make no conclusions about free Zn2+ levels in individual vesicles. From our in vitro characterization, we learned that the Zn2+ binding-affinity and FRET ratio range were diminished at pH 5.5 and pH 6.5 (Figure S-7), which correspond with the pH of immature 66 and mature 13 secretory granules, respectively. From our in situ characterization, we found that the resting FRET ratio in individual vesicles was strongly driven by differences in intraluminal pH rather than Zn2+ levels. Thus, we offer a word of caution to not overinterpret FRET ratio measurements in the absence of sensor calibrations.
Estimating granular Zn2+ levels in pancreatic β-cells
Granules in secretory cells, including pancreatic β-cells, contain high levels of Zn2+ 14–18. The genetically-encoded Zn2+ sensors ZnGreen1 79 and ZIBG2 80 were targeted to the outer leaflet of the plasma membrane and detected Zn2+ release from pancreatic β-cells in response to glucose stimulation. Given their low affinity for Zn2+, this suggests that vesicular Zn2+ levels are quite high. Merkx and colleagues created vesicular sensors VAMP2-eCALWY-1 and VAMP2-eZinCh 56. While these sensors did not respond to Zn2+ perturbations, perhaps because the perturbations did not sufficiently change Zn2+ levels within vesicles, eZinCh did respond to an increase in pH. In a subsequent study, VAMP2-eZinCh-2 responded to Zn2+ perturbations, albeit with a low dynamic range, and was used to estimate that granular Zn2+ levels were ~120 nM in INS-1 cells 57. Using the Zn2+ FRET sensor NPY-ZnT72R, another group reported that granular Zn2+ levels were ~41 μM in primary mouse pancreatic β-cells 81.
In our study, we went a step further to define the intraluminal pH in single vesicles in MIN6 cells (Figure 5). However, we were unable to define Zn2+ levels in single vesicles as we had hoped. At the population level, CgA-ZapCY1 exhibited a large dynamic range (DR) of 2.03 but was saturated under resting conditions in MIN6 cells (Figure 6). To further enable measurement of Zn2+ in vesicles, we used the CgA targeting motif to target another high dynamic range, but lower affinity FRET-based Zn2+ sensor (eCALWY4) to vesicles. Indeed, CgA-eCALWY-4 showed robust localization to vesicles and response to in situ calibration (DR ~ 1.65). This lower affinity sensor (Kd 630 pM at pH 7.1) was only partially saturated (26%) at rest in MIN6 cells. With knowledge of the average vesicular pH (pH 6.2) in MIN6 cells and our in vitro data showing that the Zn2+ FRET sensor was saturated at concentrations greater than 1 nM at pH 6.5, using CgA-ZapCY1 we estimate granular Zn2+ levels in MIN6 cells to be greater than 1 nM. The Kd of eCALWY-4 at pH 5.5–6.5 is unknown. However, we predict the Kd value would be ~ 5–10-fold lower than eCALWY-6 which is 500 nM at pH 6.0 75. Combined with the data that CgA-eCALWY4 is 26% saturated, we estimate the labile Zn2+ concentration in vesicles to be 20–40 nM. This is close to the result of Hessels et al 57 and three orders of magnitude lower than those reported in Xian et al 81. In summary, the two vesicular sensors show that labile Zn2+ levels are higher in granules compared with other organelles, including the ER, Golgi and mitochondria 56–57, 61, 69.
Outlook
The findings in this study can be used to inform fluorescent sensor design and application in the context of secretory vesicles and streamline the development and characterization vesicular fluorescent sensors. Our rigorous comparison of the five most widely used targeting motifs revealed that CgA and NPY are the most robust in normal and secretory cells and therefore should be used more broadly to develop vesicular fluorescent sensors. Furthermore, we provide a roadmap for vesicular sensor characterization, including rigorously defining subcellular localization and the influence of pH on sensor measurements. Combined, CgA-ZapCY1 and CgA-eCALWY-4 can be used to measure differences in vesicular Zn2+ levels in a range of cells. In the future these tools can be used to define how vesicular Zn2+ is regulated, whether vesicular Zn2+ is dynamic and if vesicular Zn2+ levels are altered in diseases such as diabetes or cancers derived from secretion specialized cells.
Supplementary Material
Acknowledgements
This research was supported by an NIH Director’s Pioneer Award (DP1-GM114863 to AEP), NSF GRF (1650115 to KJA), NIH Molecular Biophysics Training Grant (T32 GM-065103 to KJA), and undergraduate fellowships from the Biological Sciences Initiative and UROP to JT. We would like to acknowledge the BioFrontiers Institute Advanced Light Microscopy Core, where fluorescence imaging and analysis were performed. We performed spinning disc confocal microscopy on a Nikon Ti-E microscope supported by the BioFrontiers Institute and the Howard Hughes Medical Institute. The Analysis Workstation and the software package Imaris were supported by NIH 1S10RR026680-01A1. We would also like to acknowledge the Biochemistry Cell Culture Core Facility at the University of Colorado Boulder for supporting this work. Finally, we would like to thank Dr. Jian Wei Tay for developing the MATLAB pipeline used in this study.
Footnotes
Supporting Information Available: The following files are available free of charge: Supporting Information. This file contains supporting methods and figures.
REFERENCES
- 1.Uhlen M; Fagerberg L; Hallstrom BM; Lindskog C; Oksvold P; Mardinoglu A; Sivertsson A; Kampf C; Sjostedt E; Asplund A; Olsson I; Edlund K; Lundberg E; Navani S; Szigyarto CA; Odeberg J; Djureinovic D; Takanen JO; Hober S; Alm T; Edqvist PH; Berling H; Tegel H; Mulder J; Rockberg J; Nilsson P; Schwenk JM; Hamsten M; von Feilitzen K; Forsberg M; Persson L; Johansson F; Zwahlen M; von Heijne G; Nielsen J; Ponten F, Proteomics. Tissue-based map of the human proteome. Science 2015, 347 (6220), 1260419. doi: 10.1126/science.1260419 [DOI] [PubMed] [Google Scholar]
- 2.Uhlen M; Karlsson MJ; Hober A; Svensson AS; Scheffel J; Kotol D; Zhong W; Tebani A; Strandberg L; Edfors F; Sjostedt E; Mulder J; Mardinoglu A; Berling A; Ekblad S; Dannemeyer M; Kanje S; Rockberg J; Lundqvist M; Malm M; Volk AL; Nilsson P; Manberg A; Dodig-Crnkovic T; Pin E; Zwahlen M; Oksvold P; von Feilitzen K; Haussler RS; Hong MG; Lindskog C; Ponten F; Katona B; Vuu J; Lindstrom E; Nielsen J; Robinson J; Ayoglu B; Mahdessian D; Sullivan D; Thul P; Danielsson F; Stadler C; Lundberg E; Bergstrom G; Gummesson A; Voldborg BG; Tegel H; Hober S; Forsstrom B; Schwenk JM; Fagerberg L; Sivertsson A, The human secretome. Sci Signal 2019, 12 (609). doi: 10.1126/scisignal.aaz0274 [DOI] [PubMed] [Google Scholar]
- 3.Burgoyne RD; Morgan A, Secretory granule exocytosis. Physiol Rev 2003, 83 (2), 581–632. doi: 10.1152/physrev.00031.2002 [DOI] [PubMed] [Google Scholar]
- 4.Alabi AA; Tsien RW, Synaptic vesicle pools and dynamics. Cold Spring Harb Perspect Biol 2012, 4 (8), a013680. doi: 10.1101/cshperspect.a013680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rorsman P; Renstrom E, Insulin granule dynamics in pancreatic beta cells. Diabetologia 2003, 46 (8), 1029–45. doi: 10.1007/s00125-003-1153-1 [DOI] [PubMed] [Google Scholar]
- 6.Lin WJ; Salton SR, The regulated secretory pathway and human disease: insights from gene variants and single nucleotide polymorphisms. Front Endocrinol (Lausanne) 2013, 4, 96. doi: 10.3389/fendo.2013.00096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davis SN; Piatti PM; Monti L; Brown MD; Branch W; Hales CN; Alberti KG, Proinsulin and insulin concentrations following intravenous glucose challenges in normal, obese, and non-insulin-dependent diabetic subjects. Metabolism 1993, 42 (1), 30–5. doi: 10.1016/0026-0495(93)90168-n [DOI] [PubMed] [Google Scholar]
- 8.Estevez-Herrera J; Dominguez N; Pardo MR; Gonzalez-Santana A; Westhead EW; Borges R; Machado JD, ATP: The crucial component of secretory vesicles. Proc Natl Acad Sci U S A 2016, 113 (28), E4098–106. doi: 10.1073/pnas.1600690113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dickson EJ; Duman JG; Moody MW; Chen L; Hille B, Orai-STIM-mediated Ca2+ release from secretory granules revealed by a targeted Ca2+ and pH probe. Proc Natl Acad Sci U S A 2012, 109 (51), E3539–48. doi: 10.1073/pnas.1218247109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arosio D; Ricci F; Marchetti L; Gualdani R; Albertazzi L; Beltram F, Simultaneous intracellular chloride and pH measurements using a GFP-based sensor. Nat Methods 2010, 7 (7), 516–8. doi: 10.1038/nmeth.1471 [DOI] [PubMed] [Google Scholar]
- 11.Williamson DM; Elferich J; Shinde U, Mechanism of Fine-tuning pH Sensors in Proprotein Convertases: IDENTIFICATION OF A pH-SENSING HISTIDINE PAIR IN THE PROPEPTIDE OF PROPROTEIN CONVERTASE 1/3. J Biol Chem 2015, 290 (38), 23214–25. doi: 10.1074/jbc.M115.665430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yoo SH, pH- and Ca(2+)-dependent aggregation property of secretory vesicle matrix proteins and the potential role of chromogranins A and B in secretory vesicle biogenesis. J Biol Chem 1996, 271 (3), 1558–65. [PubMed] [Google Scholar]
- 13.Wu MM; Grabe M; Adams S; Tsien RY; Moore HP; Machen TE, Mechanisms of pH regulation in the regulated secretory pathway. J Biol Chem 2001, 276 (35), 33027–35. doi: 10.1074/jbc.M103917200 [DOI] [PubMed] [Google Scholar]
- 14.Kristiansen LH; Rungby J; Sondergaard LG; Stoltenberg M; Danscher G, Autometallography allows ultrastructural monitoring of zinc in the endocrine pancreas. Histochem Cell Biol 2001, 115 (2), 125–9. doi: 10.1007/s004180000241 [DOI] [PubMed] [Google Scholar]
- 15.Chandler JA; Timms BG; Morton MS, Subcellular distribution of zinc in rat prostate studied by x-ray microanalysis: I. Normal prostate. Histochem J 1977, 9 (1), 103–20. doi: 10.1007/BF01007013 [DOI] [PubMed] [Google Scholar]
- 16.Cole TB; Wenzel HJ; Kafer KE; Schwartzkroin PA; Palmiter RD, Elimination of zinc from synaptic vesicles in the intact mouse brain by disruption of the ZnT3 gene. Proc Natl Acad Sci U S A 1999, 96 (4), 1716–21. doi: 10.1073/pnas.96.4.1716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ho LH; Ruffin RE; Murgia C; Li L; Krilis SA; Zalewski PD, Labile zinc and zinc transporter ZnT4 in mast cell granules: role in regulation of caspase activation and NF-kappaB translocation. J Immunol 2004, 172 (12), 7750–60. doi: 10.4049/jimmunol.172.12.7750 [DOI] [PubMed] [Google Scholar]
- 18.Podany AB; Wright J; Lamendella R; Soybel DI; Kelleher SL, ZnT2-Mediated Zinc Import Into Paneth Cell Granules Is Necessary for Coordinated Secretion and Paneth Cell Function in Mice. Cell Mol Gastroenterol Hepatol 2016, 2 (3), 369–383. doi: 10.1016/j.jcmgh.2015.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davidson HW; Wenzlau JM; O’Brien RM, Zinc transporter 8 (ZnT8) and beta cell function. Trends Endocrinol Metab 2014, 25 (8), 415–24. doi: 10.1016/j.tem.2014.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dwivedi OP; Lehtovirta M; Hastoy B; Chandra V; Krentz NAJ; Kleiner S; Jain D; Richard AM; Abaitua F; Beer NL; Grotz A; Prasad RB; Hansson O; Ahlqvist E; Krus U; Artner I; Suoranta A; Gomez D; Baras A; Champon B; Payne AJ; Moralli D; Thomsen SK; Kramer P; Spiliotis I; Ramracheya R; Chabosseau P; Theodoulou A; Cheung R; van de Bunt M; Flannick J; Trombetta M; Bonora E; Wolheim CB; Sarelin L; Bonadonna RC; Rorsman P; Davies B; Brosnan J; McCarthy MI; Otonkoski T; Lagerstedt JO; Rutter GA; Gromada J; Gloyn AL; Tuomi T; Groop L, Loss of ZnT8 function protects against diabetes by enhanced insulin secretion. Nat Genet 2019, 51 (11), 1596–1606. doi: 10.1038/s41588-019-0513-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sladek R; Rocheleau G; Rung J; Dina C; Shen L; Serre D; Boutin P; Vincent D; Belisle A; Hadjadj S; Balkau B; Heude B; Charpentier G; Hudson TJ; Montpetit A; Pshezhetsky AV; Prentki M; Posner BI; Balding DJ; Meyre D; Polychronakos C; Froguel P, A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature 2007, 445 (7130), 881–5. doi: 10.1038/nature05616 [DOI] [PubMed] [Google Scholar]
- 22.Xu Y; Yan Y; Seeman D; Sun L; Dubin PL, Multimerization and aggregation of native-state insulin: effect of zinc. Langmuir 2012, 28 (1), 579–86. doi: 10.1021/la202902a [DOI] [PubMed] [Google Scholar]
- 23.Nicolson TJ; Bellomo EA; Wijesekara N; Loder MK; Baldwin JM; Gyulkhandanyan AV; Koshkin V; Tarasov AI; Carzaniga R; Kronenberger K; Taneja TK; da Silva Xavier G; Libert S; Froguel P; Scharfmann R; Stetsyuk V; Ravassard P; Parker H; Gribble FM; Reimann F; Sladek R; Hughes SJ; Johnson PR; Masseboeuf M; Burcelin R; Baldwin SA; Liu M; Lara-Lemus R; Arvan P; Schuit FC; Wheeler MB; Chimienti F; Rutter GA, Insulin storage and glucose homeostasis in mice null for the granule zinc transporter ZnT8 and studies of the type 2 diabetes-associated variants. Diabetes 2009, 58 (9), 2070–83. doi: 10.2337/db09-0551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jain RK; Chang WT; Geetha C; Joyce PB; Gorr SU, In vitro aggregation of the regulated secretory protein chromogranin A. Biochem J 2002, 368 (Pt 2), 605–10. doi: 10.1042/BJ20021195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jacob RS; Das S; Ghosh S; Anoop A; Jha NN; Khan T; Singru P; Kumar A; Maji SK, Amyloid formation of growth hormone in presence of zinc: Relevance to its storage in secretory granules. Sci Rep 2016, 6, 23370. doi: 10.1038/srep23370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee S; Rivera OC; Kelleher SL, Zinc transporter 2 interacts with vacuolar ATPase and is required for polarization, vesicle acidification, and secretion in mammary epithelial cells. J Biol Chem 2017, 292 (52), 21598–21613. doi: 10.1074/jbc.M117.794461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lavoie N; Jeyaraju DV; Peralta MR 3rd; Seress L; Pellegrini L; Toth K, Vesicular zinc regulates the Ca2+ sensitivity of a subpopulation of presynaptic vesicles at hippocampal mossy fiber terminals. J Neurosci 2011, 31 (50), 18251–65. doi: 10.1523/JNEUROSCI.4164-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Petkovic V; Miletta MC; Eble A; Iliev DI; Binder G; Fluck CE; Mullis PE, Effect of zinc binding residues in growth hormone (GH) and altered intracellular zinc content on regulated GH secretion. Endocrinology 2013, 154 (11), 4215–25. doi: 10.1210/en.2013-1089 [DOI] [PubMed] [Google Scholar]
- 29.Toth K, Zinc in neurotransmission. Annu Rev Nutr 2011, 31, 139–53. doi: 10.1146/annurev-nutr-072610-145218 [DOI] [PubMed] [Google Scholar]
- 30.Egefjord L; Petersen AB; Rungby J, Zinc, alpha cells and glucagon secretion. Curr Diabetes Rev 2010, 6 (1), 52–7. doi: 10.2174/157339910790442655 [DOI] [PubMed] [Google Scholar]
- 31.Jeong J; Walker JM; Wang F; Park JG; Palmer AE; Giunta C; Rohrbach M; Steinmann B; Eide DJ, Promotion of vesicular zinc efflux by ZIP13 and its implications for spondylocheiro dysplastic Ehlers-Danlos syndrome. Proc Natl Acad Sci U S A 2012, 109 (51), E3530–8. doi: 10.1073/pnas.1211775110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gandasi NR; Vesto K; Helou M; Yin P; Saras J; Barg S, Survey of Red Fluorescence Proteins as Markers for Secretory Granule Exocytosis. PLoS One 2015, 10 (6), e0127801. doi: 10.1371/journal.pone.0127801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sinha R; Ahmed S; Jahn R; Klingauf J, Two synaptobrevin molecules are sufficient for vesicle fusion in central nervous system synapses. Proc Natl Acad Sci U S A 2011, 108 (34), 14318–23. doi: 10.1073/pnas.1101818108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Granseth B; Odermatt B; Royle SJ; Lagnado L, Clathrin-mediated endocytosis is the dominant mechanism of vesicle retrieval at hippocampal synapses. Neuron 2006, 51 (6), 773–86. doi: 10.1016/j.neuron.2006.08.029 [DOI] [PubMed] [Google Scholar]
- 35.Hao M; Li X; Rizzo MA; Rocheleau JV; Dawant BM; Piston DW, Regulation of two insulin granule populations within the reserve pool by distinct calcium sources. J Cell Sci 2005, 118 (Pt 24), 5873–84. doi: 10.1242/jcs.02684 [DOI] [PubMed] [Google Scholar]
- 36.Ohara-Imaizumi M; Nakamichi Y; Tanaka T; Katsuta H; Ishida H; Nagamatsu S, Monitoring of exocytosis and endocytosis of insulin secretory granules in the pancreatic beta-cell line MIN6 using pH-sensitive green fluorescent protein (pHluorin) and confocal laser microscopy. Biochem J 2002, 363 (Pt 1), 73–80. doi: 10.1042/0264-6021:3630073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rajan S; Eames SC; Park SY; Labno C; Bell GI; Prince VE; Philipson LH, In vitro processing and secretion of mutant insulin proteins that cause permanent neonatal diabetes. Am J Physiol Endocrinol Metab 2010, 298 (3), E403–10. doi: 10.1152/ajpendo.00592.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rose T; Schoenenberger P; Jezek K; Oertner TG, Developmental refinement of vesicle cycling at Schaffer collateral synapses. Neuron 2013, 77 (6), 1109–21. doi: 10.1016/j.neuron.2013.01.021 [DOI] [PubMed] [Google Scholar]
- 39.Sankaranarayanan S; De Angelis D; Rothman JE; Ryan TA, The use of pHluorins for optical measurements of presynaptic activity. Biophys J 2000, 79 (4), 2199–208. doi: 10.1016/S0006-3495(00)76468-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Takamori S; Holt M; Stenius K; Lemke EA; Gronborg M; Riedel D; Urlaub H; Schenck S; Brugger B; Ringler P; Muller SA; Rammner B; Grater F; Hub JS; De Groot BL; Mieskes G; Moriyama Y; Klingauf J; Grubmuller H; Heuser J; Wieland F; Jahn R, Molecular anatomy of a trafficking organelle. Cell 2006, 127 (4), 831–46. doi: 10.1016/j.cell.2006.10.030 [DOI] [PubMed] [Google Scholar]
- 41.Shinoda H; Shannon M; Nagai T, Fluorescent Proteins for Investigating Biological Events in Acidic Environments. Int J Mol Sci 2018, 19 (6). doi: 10.3390/ijms19061548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bencina M, Illumination of the spatial order of intracellular pH by genetically encoded pH-sensitive sensors. Sensors (Basel) 2013, 13 (12), 16736–58. doi: 10.3390/s131216736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Suzuki J; Kanemaru K; Iino M, Genetically Encoded Fluorescent Indicators for Organellar Calcium Imaging. Biophys J 2016, 111 (6), 1119–1131. doi: 10.1016/j.bpj.2016.04.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Watkins S; Geng X; Li L; Papworth G; Robbins PD; Drain P, Imaging secretory vesicles by fluorescent protein insertion in propeptide rather than mature secreted peptide. Traffic 2002, 3 (7), 461–71. doi: 10.1034/j.1600-0854.2002.30703.x [DOI] [PubMed] [Google Scholar]
- 45.Park JJ; Gondre-Lewis MC; Eiden LE; Loh YP, A distinct trans-Golgi network subcompartment for sorting of synaptic and granule proteins in neurons and neuroendocrine cells. J Cell Sci 2011, 124 (Pt 5), 735–44. doi: 10.1242/jcs.076372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mahapatra NR; Mahata M; Hazra PP; McDonough PM; O’Connor DT; Mahata SK, A dynamic pool of calcium in catecholamine storage vesicles. Exploration in living cells by a novel vesicle-targeted chromogranin A-aequorin chimeric photoprotein. J Biol Chem 2004, 279 (49), 51107–21. doi: 10.1074/jbc.M408742200 [DOI] [PubMed] [Google Scholar]
- 47.Mitchell KJ; Pinton P; Varadi A; Tacchetti C; Ainscow EK; Pozzan T; Rizzuto R; Rutter GA, Dense core secretory vesicles revealed as a dynamic Ca(2+) store in neuroendocrine cells with a vesicle-associated membrane protein aequorin chimaera. J Cell Biol 2001, 155 (1), 41–51. doi: 10.1083/jcb.200103145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Egashira Y; Takase M; Watanabe S; Ishida J; Fukamizu A; Kaneko R; Yanagawa Y; Takamori S, Unique pH dynamics in GABAergic synaptic vesicles illuminates the mechanism and kinetics of GABA loading. Proc Natl Acad Sci U S A 2016, 113 (38), 10702–7. doi: 10.1073/pnas.1604527113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Poea-Guyon S; Pasquier H; Merola F; Morel N; Erard M, The enhanced cyan fluorescent protein: a sensitive pH sensor for fluorescence lifetime imaging. Anal Bioanal Chem 2013, 405 (12), 3983–7. doi: 10.1007/s00216-013-6860-y [DOI] [PubMed] [Google Scholar]
- 50.Cranfill PJ; Sell BR; Baird MA; Allen JR; Lavagnino Z; de Gruiter HM; Kremers GJ; Davidson MW; Ustione A; Piston DW, Quantitative assessment of fluorescent proteins. Nat Methods 2016, 13 (7), 557–62. doi: 10.1038/nmeth.3891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang Z; Zhang XA; Bosch M; Smith SJ; Lippard SJ, Tris(2-pyridylmethyl)amine (TPA) as a membrane-permeable chelator for interception of biological mobile zinc. Metallomics 2013, 5 (6), 648–55. doi: 10.1039/c3mt00103b [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Griesbeck O; Baird GS; Campbell RE; Zacharias DA; Tsien RY, Reducing the environmental sensitivity of yellow fluorescent protein. Mechanism and applications. J Biol Chem 2001, 276 (31), 29188–94. doi: 10.1074/jbc.M102815200 [DOI] [PubMed] [Google Scholar]
- 53.Carter KP; Carpenter MC; Fiedler B; Jimenez R; Palmer AE, Critical Comparison of FRET-Sensor Functionality in the Cytosol and Endoplasmic Reticulum and Implications for Quantification of Ions. Anal Chem 2017, 89 (17), 9601–9608. doi: 10.1021/acs.analchem.7b02933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pomorski A; Kochanczyk T; Miloch A; Krezel A, Method for accurate determination of dissociation constants of optical ratiometric systems: chemical probes, genetically encoded sensors, and interacting molecules. Anal Chem 2013, 85 (23), 11479–86. doi: 10.1021/ac402637h [DOI] [PubMed] [Google Scholar]
- 55.Miesenbock G; De Angelis DA; Rothman JE, Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature 1998, 394 (6689), 192–5. doi: 10.1038/28190 [DOI] [PubMed] [Google Scholar]
- 56.Chabosseau P; Tuncay E; Meur G; Bellomo EA; Hessels A; Hughes S; Johnson PR; Bugliani M; Marchetti P; Turan B; Lyon AR; Merkx M; Rutter GA, Mitochondrial and ER-targeted eCALWY probes reveal high levels of free Zn2+. ACS Chem Biol 2014, 9 (9), 2111–20. doi: 10.1021/cb5004064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hessels AM; Chabosseau P; Bakker MH; Engelen W; Rutter GA; Taylor KM; Merkx M, eZinCh-2: A Versatile, Genetically Encoded FRET Sensor for Cytosolic and Intraorganelle Zn(2+) Imaging. ACS Chem Biol 2015, 10 (9), 2126–34. doi: 10.1021/acschembio.5b00211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hou JC; Min L; Pessin JE, Insulin granule biogenesis, trafficking and exocytosis. Vitam Horm 2009, 80, 473–506. doi: 10.1016/S0083-6729(08)00616-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shaner NC; Campbell RE; Steinbach PA; Giepmans BN; Palmer AE; Tsien RY, Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol 2004, 22 (12), 1567–72. doi: 10.1038/nbt1037 [DOI] [PubMed] [Google Scholar]
- 60.Tie HC; Mahajan D; Chen B; Cheng L; VanDongen AM; Lu L, A novel imaging method for quantitative Golgi localization reveals differential intra-Golgi trafficking of secretory cargoes. Mol Biol Cell 2016, 27 (5), 848–61. doi: 10.1091/mbc.E15-09-0664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Qin Y; Dittmer PJ; Park JG; Jansen KB; Palmer AE, Measuring steady-state and dynamic endoplasmic reticulum and Golgi Zn2+ with genetically encoded sensors. Proc Natl Acad Sci U S A 2011, 108 (18), 7351–6. doi: 10.1073/pnas.1015686108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Heim R; Tsien RY, Engineering green fluorescent protein for improved brightness, longer wavelengths and fluorescence resonance energy transfer. Curr Biol 1996, 6 (2), 178–82. doi: 10.1016/s0960-9822(02)00450-5 [DOI] [PubMed] [Google Scholar]
- 63.Llopis J; McCaffery JM; Miyawaki A; Farquhar MG; Tsien RY, Measurement of cytosolic, mitochondrial, and Golgi pH in single living cells with green fluorescent proteins. Proc Natl Acad Sci U S A 1998, 95 (12), 6803–8. doi: 10.1073/pnas.95.12.6803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Paroutis P; Touret N; Grinstein S, The pH of the secretory pathway: measurement, determinants, and regulation. Physiology (Bethesda) 2004, 19, 207–15. doi: 10.1152/physiol.00005.2004 [DOI] [PubMed] [Google Scholar]
- 65.Burgstaller S; Bischof H; Gensch T; Stryeck S; Gottschalk B; Ramadani-Muja J; Eroglu E; Rost R; Balfanz S; Baumann A; Waldeck-Weiermair M; Hay JC; Madl T; Graier WF; Malli R, pH-Lemon, a Fluorescent Protein-Based pH Reporter for Acidic Compartments. ACS Sens 2019, 4 (4), 883–891. doi: 10.1021/acssensors.8b01599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Urbe S; Dittie AS; Tooze SA, pH-dependent processing of secretogranin II by the endopeptidase PC2 in isolated immature secretory granules. Biochem J 1997, 321 ( Pt 1), 65–74. doi: 10.1042/bj3210065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hu YB; Dammer EB; Ren RJ; Wang G, The endosomal-lysosomal system: from acidification and cargo sorting to neurodegeneration. Transl Neurodegener 2015, 4, 18. doi: 10.1186/s40035-015-0041-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Park JG; Palmer AE, Quantitative measurement of Ca2+ and Zn2+ in mammalian cells using genetically encoded fluorescent biosensors. Methods Mol Biol 2014, 1071, 29–47. doi: 10.1007/978-1-62703-622-1_3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Park JG; Qin Y; Galati DF; Palmer AE, New sensors for quantitative measurement of mitochondrial Zn(2+). ACS Chem Biol 2012, 7 (10), 1636–40. doi: 10.1021/cb300171p [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Franklin RB; Costello LC, Zinc as an anti-tumor agent in prostate cancer and in other cancers. Arch Biochem Biophys 2007, 463 (2), 211–7. doi: 10.1016/j.abb.2007.02.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gyorkey F; Min KW; Huff JA; Gyorkey P, Zinc and magnesium in human prostate gland: normal, hyperplastic, and neoplastic. Cancer Res 1967, 27 (8), 1348–53. [PubMed] [Google Scholar]
- 72.Iguchi K; Otsuka T; Usui S; Ishii K; Onishi T; Sugimura Y; Hirano K, Zinc and metallothionein levels and expression of zinc transporters in androgen-independent subline of LNCaP cells. J Androl 2004, 25 (1), 154–61. doi: 10.1002/j.1939-4640.2004.tb02771.x [DOI] [PubMed] [Google Scholar]
- 73.Zaichick V; Sviridova TV; Zaichick SV, Zinc in the human prostate gland: normal, hyperplastic and cancerous. Int Urol Nephrol 1997, 29 (5), 565–74. doi: 10.1007/BF02552202 [DOI] [PubMed] [Google Scholar]
- 74.Horoszewicz JS; Leong SS; Kawinski E; Karr JP; Rosenthal H; Chu TM; Mirand EA; Murphy GP, LNCaP model of human prostatic carcinoma. Cancer Res 1983, 43 (4), 1809–18. [PubMed] [Google Scholar]
- 75.Vinkenborg JL; Nicolson TJ; Bellomo EA; Koay MS; Rutter GA; Merkx M, Genetically encoded FRET sensors to monitor intracellular Zn2+ homeostasis. Nat Methods 2009, 6 (10), 737–40. doi: 10.1038/nmeth.1368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Unlu G; Levic DS; Melville DB; Knapik EW, Trafficking mechanisms of extracellular matrix macromolecules: insights from vertebrate development and human diseases. Int J Biochem Cell Biol 2014, 47, 57–67. doi: 10.1016/j.biocel.2013.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pamarthy S; Kulshrestha A; Katara GK; Beaman KD, The curious case of vacuolar ATPase: regulation of signaling pathways. Mol Cancer 2018, 17 (1), 41. doi: 10.1186/s12943-018-0811-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schifferer M; Yushchenko DA; Stein F; Bolbat A; Schultz C, A Ratiometric Sensor for Imaging Insulin Secretion in Single beta Cells. Cell Chem Biol 2017, 24 (4), 525–531 e4. doi: 10.1016/j.chembiol.2017.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chen Z; Ai HW, Single Fluorescent Protein-Based Indicators for Zinc Ion (Zn(2+)). Anal Chem 2016, 88 (18), 9029–36. doi: 10.1021/acs.analchem.6b01653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chen M; Zhang S; Xing Y; Li X; He Y; Wang Y; Oberholzer J; Ai HW, Genetically Encoded, Photostable Indicators to Image Dynamic Zn(2+) Secretion of Pancreatic Islets. Anal Chem 2019, 91 (19), 12212–12219. doi: 10.1021/acs.analchem.9b01802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Xian Y; Zhou M; Han S; Yang R; Wang Y, A FRET biosensor reveals free zinc deficiency in diabetic beta-cell vesicles. Chinese Chemical Letters 2020, 31 (2), 468–472. doi: 10.1016/j.cclet.2019.08.012 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.