

Abstract
Background
Rapid deployment of technologies capable of high-throughput and high-resolution screening is imperative for timely response to viral outbreaks. Risk mitigation in the form of leveraging multiple advanced technologies further increases the likelihood of identifying efficacious treatments in aggressive timelines.
Methods
In this study, we describe two parallel, yet distinct, in vivo approaches for accelerated discovery of antibodies targeting the severe acute respiratory syndrome coronavirus-2 spike protein. Working with human transgenic Alloy-GK mice, we detail a single B-cell discovery workflow to directly interrogate antibodies secreted from plasma cells for binding specificity and ACE2 receptor blocking activity. Additionally, we describe a concurrent accelerated hybridoma-based workflow utilizing a DiversimAb™ mouse model for increased diversity.
Results
The panel of antibodies isolated from both workflows revealed binding to distinct epitopes with both blocking and non-blocking profiles. Sequence analysis of the resulting lead candidates uncovered additional diversity with the opportunity for straightforward engineering and affinity maturation.
Conclusions
By combining in vivo models with advanced integration of screening and selection platforms, lead antibody candidates can be sequenced and fully characterized within one to three months.
Keywords: beacon, single B cell, carterra, epitope binning, neutralizing antibodies, therapeutic, human antibody, COVID
Statement of Significance
Rapid antibody discovery methods for functional lead candidates using in vivo techniques and high-throughput single cell platforms.
INTRODUCTION
The pandemic caused by severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2), or coronavirus disease 2019 (COVID-19), has received unprecedented attention from the scientific community in an effort to rapidly develop efficacious treatments and vaccines. Within weeks of the emergence of viral pneumonia outbreaks in Wuhan, China, deep sequencing had identified the cause [1], and the resulting mobilization of widespread therapeutic and prophylactic discovery efforts ensued. The response to the COVID-19 pandemic mirrored that of other recent viral outbreaks, including, but not limited to, H1N1 influenza in 2009 [2], Ebola Virus in 2014 [3, 4] and Zika Virus in 2015 [5]. Lessons learned from these public health threats helped guide the strategy for the accelerated response to COVID-19. In particular, the understanding that neutralizing antibody function is fundamental to combating disease progression [6] helped streamline early antibody-based drug therapy discovery strategies.
Beyond direct therapeutic use, antibodies can help inform vaccine design to enable next-generation vaccine development with a focus on relevant viral epitopes [7]. In general, the most valuable and broadly applicable antiviral antibodies are those that exhibit cross-reactivity to related viruses and are unaffected by escape mutant evolutionary pressures [8]. These antibodies, which can function either alone or in combination with oligoclonal mixtures of non-competing antibodies [9], harbor basic properties like receptor blocking activity and high affinity. When taken in aggregate, these criteria are quite stringent and therefore necessitate efficient, high-resolution screening strategies to identify valuable lead candidates.
Development of viable therapeutic antibody candidates typically follows one of two principal methodologies: in vitro or in vivo discovery [10]. Both have served the industry well for decades; however, in recent years the traditionally lengthy timeline required to move from target identification through lead candidate discovery has been challenged [11]. The ability to rapidly perform the discovery tasks within this scope requires novel technologies, strategies and efficiencies to maintain the required throughput and depth of screening [12]. For example, traditional in vitro naïve library panning or in vivo hybridoma generation can take 6 months or longer to move from target ID to lead candidate selection. The present study focuses on optimizing the in vivo discovery timeline by introducing compressed workflows for immunization, primary cell screening and antibody characterization while maintaining or improving screening depth to elucidate desired properties faster.
This report highlights several different techniques and antibody discovery workflows leveraged in the discovery and characterization of antibody panels targeting the spike protein (S) of SARS-CoV-2 and showcases screening results for a subset of representative candidates. Across the different workflows (Fig. 1), two separate mouse strains were immunized with the S1 subunit (which contains the receptor binding domain): a humanized strain to facilitate the discovery of fully human antibodies (Alloy-GK mice), and an engineered mouse strain designed to elicit greater epitopic diversity and overall immune response (Abveris DiversimAb™ mice). Furthermore, two distinct upstream discovery methods were applied: a hybridoma discovery platform optimized for high-content screening and efficiency (Abveris Hybridoma Workflow) and a high-throughput state-of-the-art single B cell screening platform (Abveris Single B Cell Workflow enabled by the Berkeley Lights Beacon®). Final characterization and candidate analyses were performed on the Carterra LSA™.
Figure 1.
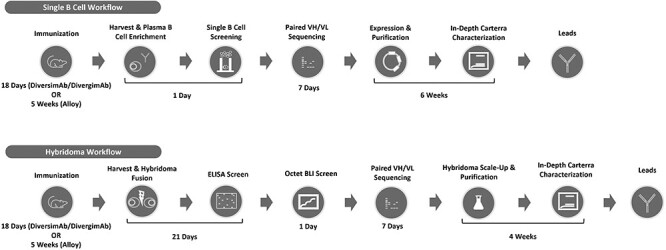
Outline of the workflow for both single B cell and hybridoma discovery platforms. The time frame required for each stage of the workflow is indicated.
METHODS AND RESULTS
Immunization of DiversimAb and Alloy-GK mice was completed in 16 and 35 days, respectively; both protocols resulted in an appreciable immune response as indicated by detectible serum titer to the S1 protein at serum dilution factors of at least 1:70,000 or higher (Supplementary Fig. 1). Following a high-efficiency electrofusion to generate hybridoma lines [27], resulting colonies were initially screened by enzyme-linked immunosorbent assay (ELISA) to identify S-protein-reactive clones. Preliminary clones of interest were subsequently screened via high-throughput biolayer interferometry (BLI) kinetic screening on the ForteBio Octet® system to select candidates for scale up and antibody purification from hybridoma cultures. Simultaneously, the sequencing of immunoglobulin genes was performed following a high-throughput hybridoma sequencing procedure and sequence analysis was performed using Geneious Biologics. Screening results from a subset of representative candidates are shown in Table 1.
Table 1.
Binding characteristics of anti-S protein candidates, including ELISA binding, single point kinetic measurements to the trimeric S protein, affinity characterization to the monomeric S1 protein and effect on ACE2 binding to S1 protein (S1:ACE2 interaction blocking activity)
| Indirect ELISA (OD 450 nm) | BLI Kinetics (ForteBio); Trimeric S Protein (100 nM) | SPR Kinetics (Carterra); Monomeric S1 Protein (500 nM, 125 nM, 31.25 nM, 7.81 nM, 1.95 nM, 0.49 nM) | S1:ACE2 Interaction (Carterra) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ID | Mouse strain | S Protein (+) | Irrelevant AVI-His Tagged Protein (−) | ka (M-1 s-1) | kd (s-1)* | KD (nM)** | R2 | ka (M-1 s-1) | kd (s-1) | KD (nM) | Rmax | Res SD | % Change ACE2 binding*** |
| 15G11 | DivergimAb | 0.879 | 0.041 | 1.05E+05 | 2.23E-03 | 21.3 | 0.9582 | 3.23E+04 | 1.12E-03 | 38.50 | 154 | 7.725 | 51.95 |
| 16F2 | DivergimAb | 1 | 0.04 | 8.85E+04 | <1E-05 | <1 | 0.9924 | 7.13E+04 | 8.50E-04 | 12.58 | 326 | 8 | -7.73 |
| 18F4 | DivergimAb | 1.05 | 0.042 | 7.03E+04 | <1E-05 | <1 | 0.9467 | 6.95E+04 | 4.65E-04 | 7.48 | 299 | 7.025 | -3.44 |
| 1E5 | DiversimAb | 1.216 | 0.041 | 1.10E+05 | <1E-05 | <1 | 0.9522 | 1.30E+05 | 5.78E-04 | 5.73 | 246 | 11.725 | 16.55 |
| 1G3 | DiversimAb | 0.818 | 0.041 | 6.32E+04 | <1E-05 | <1 | 0.958 | 3.85E+04 | 6.55E-04 | 18.00 | 250 | 10.45 | 3.77 |
| 21C3 | DivergimAb | 0.81 | 0.043 | 7.18E+04 | <1E-05 | <1 | 0.9872 | 3.68E+04 | 6.63E-04 | 19.50 | 269 | 8.425 | -1.27 |
| 22D9 | DivergimAb | 0.8 | 0.042 | 7.63E+04 | <1E-05 | <1 | 0.9866 | 6.30E+04 | 6.08E-04 | 11.83 | 295 | 11.6 | -0.43 |
| 23D11 | DivergimAb | 1.241 | 0.042 | 9.03E+04 | 1.13E-04 | 1.25 | 0.9903 | 1.20E+05 | 1.09E-03 | 9.73 | 234 | 8.325 | 3.34 |
| 26E2 | DivergimAb | 1.113 | 0.044 | 8.23E+04 | 1.66E-03 | 20.2 | 0.9871 | 8.08E+04 | 1.11E-03 | 14.25 | 290 | 13.25 | 44.66 |
| 29F7 | DivergimAb | 0.714 | 0.041 | 1.12E+05 | 1.63E-04 | 1.46 | 0.9267 | 5.13E+04 | 3.55E-04 | 7.88 | 335 | 9.725 | 52.71 |
| 3B3 | DiversimAb | 1.100 | 0.041 | 8.39E+04 | 1.19E-03 | 14.2 | 0.9852 | 6.10E+04 | 5.40E-04 | 10.10 | 250 | 11.75 | 35.91 |
| 3F2 | DiversimAb | 1.261 | 0.045 | 9.26E+04 | <1E-05 | <1 | 0.9539 | 1.60E+05 | 7.60E-04 | 4.95 | 181 | 6.85 | 48.47 |
| D59047-11955 | DivergimAb | 0.879 | 0.042 | 7.40E+04 | <1E-05 | <1 | 0.9962 | 4.13E+04 | 7.03E-04 | 18.00 | 176 | 5.8 | -7.98 |
| D70678-12637-S1 | Alloy | 1.059 | 0.043 | 7.94E+03 | <1E-05 | <1 | 0.9289 | 3.48E+04 | 3.83E-04 | 11.13 | 196 | 3.875 | 67.80 |
| D70678-12799-S1 | Alloy | 1.489 | 0.043 | 6.83E+04 | 2.81E-04 | 4.12 | 0.8743 | 1.56E+04 | 2.43E-03 | 331.75 | 166 | 4.575 | 89.15 |
| D70678-13531-S1 | Alloy | 1.627 | 0.043 | 3.52E+04 | <1E-05 | <1 | 0.9954 | 5.93E+04 | 1.32E-04 | 2.38 | 149 | 3.375 | 55.47 |
| D70678-13576-S1 | Alloy | 1.134 | 0.043 | 3.04E+04 | 2.85E-04 | 9.38 | 0.9705 | 5.73E+04 | 1.60E-04 | 2.88 | 80 | 3.075 | 71.73 |
| D70678-14004-S2 | Alloy | 1.408 | 0.043 | 1.21E+04 | <1E-05 | <1 | 0.9456 | 1.98E+04 | 9.13E-04 | 47.75 | 95 | 2.9 | 40.60 |
| D70678-14027-S2 | Alloy | 1.510 | 0.043 | 2.63E+04 | 2.85E-04 | 10.8 | 0.9984 | 5.15E+04 | 3.85E-04 | 7.60 | 238 | 5.35 | 34.02 |
| D70678-2155-S1 | Alloy | 1.65 | 0.051 | 1.15E+04 | <1E-05 | <1 | 0.9966 | 4.55E+04 | 2.05E-04 | 4.63 | 225 | 4.975 | 35.94 |
| D70678-2743-S1 | Alloy | 1.496 | 0.044 | 5.33E+04 | <1E-05 | <1 | 0.9785 | 1.57E+04 | 2.00E-04 | 15.35 | 47 | 2.25 | 38.16 |
| D70678-5521-S2 | Alloy | 1.658 | 0.044 | 2.90E+04 | <1E-05 | <1 | 0.991 | 5.45E+04 | 1.60E-04 | 2.98 | 193 | 4.35 | 44.93 |
*Values < 1E-05 (s-1) are beyond the limit of detection in this experiment and are estimated.
**Values < 1 nM are beyond the limit of detection in this experiment and are estimated.
***Calculated as % increase or decrease in signal upon exposure of Antibody:S1 complex to 100 nM ACE2 (as compared to Antibody:S1 signal).
Concurrently, a single B cell discovery approach was employed whereby plasma cells were enriched from primary tissues prior to loading onto OptoSelect™ chips on the Berkeley Lights Beacon. Following single cell deposition into NanoPens, assay mixtures containing capture beads and fluorescently labeled target proteins were imported into the channels above the NanoPens. Throughout the course of the assay, antibody secreted from the plasma B cells diffused from the NanoPen chambers into the channel above. Upon bead binding, fluorescence from either directly labeled protein(s) or secondary detection antibodies was concentrated on the surface of the bead, resulting in the time-dependent development of fluorescent halos in the channels above the pens containing antigen-specific plasma cells (Fig. 2). Plasma cells exhibiting on-Beacon binding profiles of interest were exported for immunoglobulin sequence capture and analysis with Geneious Biologics. The resulting naturally paired heavy and light chains were cloned into expression plasmids and recombinantly expressed. Purified antibodies were screened similarly to the strategy used for hybridoma candidates via ELISA and BLI with a subset of representative candidates displayed in Table 1.
Figure 2.
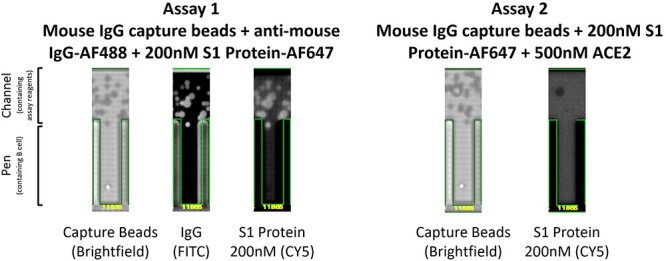
Example Beacon screening data for candidate D59047-11955. Anti-mouse IgG capture beads (brightfield image) were imported into channels above the pens. Antibody secretion from a single B cell contained within a pen bound to capture beads at the mouth of the pen. In assay 1, antibody secretion was assessed by detection of total IgG in the FITC detection channel via binding of an anti-mouse IgG AF488 conjugated secondary and simultaneously the specificity for S1 protein was determined in the CY5 detection channel with AF647 conjugated S1 protein at 200 nM. In assay 2, binding competition between secreted antibody from the B cell and ACE2 receptor was assessed by precomplexing AF647 conjugated S1 protein with a molar excess of recombinant ACE2. A lack of antibody binding to S1 protein under these conditions demonstrated binding to a similar epitope as ACE2, thus indicative of a potential blocking candidate.
High-throughput and high-content screening on lead candidates was performed on the Carterra LSA to elucidate kinetic profiles to the monovalent S1 protein (Table 1, Fig. 3). Full kinetic profiles were assessed in triplicate under regenerative and non-regenerative conditions with both purified antibodies and crude supernatant samples. Target S1 protein was used as an analyte in an ascending concentration series ranging from 0.49 to 500 nM with 4-fold dilutions (Fig. 3). All conditions (regeneration vs. non-regeneration and purified vs. crude antibody samples) yielded similar affinity values. The average resulting values from all assays is reported in Table 1.
Figure 3.
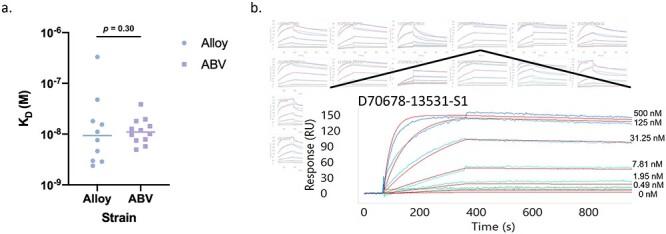
Carterra affinity analysis and example sensogram. With antibody captured on the chip surface, various concentrations of the target S1 protein were assessed for association and dissociation rates to calculate (a) final KD values. (b) Array view of sensograms for each clone in the background with an example sensogram highlighted in the foreground. Each colored line indicates a distinct analyte concentration with red lines representing the curve fit analysis for rate constant calculations.
Additionally, candidates were assayed on the Carterra LSA for the ability to block the S1:ACE2 binding interaction (Table 1, Fig. 4). ACE2 receptor blocking activity was interrogated by forming an antibody-S1 protein complex on the chip surface in a sequential format. Following complex formation, ACE2 was introduced as an analyte at 100 nM (Fig. 4) and the percent increase in RU value as a result of ACE2 binding was quantified using the RU signal from antibody-S1 complex formation as the baseline. The average of triplicate measurements is reported in Table 1. Non-blocking candidates resulted in a 50.8% ± 15.3% increase in signal from ACE2 binding, while blocking candidates completely prevented ACE2 binding (−1.96% ± 4.40% change in signal). One candidate, 1E5, demonstrated intermediate blocking characteristics with a 16.6% increase in signal upon ACE2 binding, perhaps indicating a weak or partial blocking profile.
Figure 4.
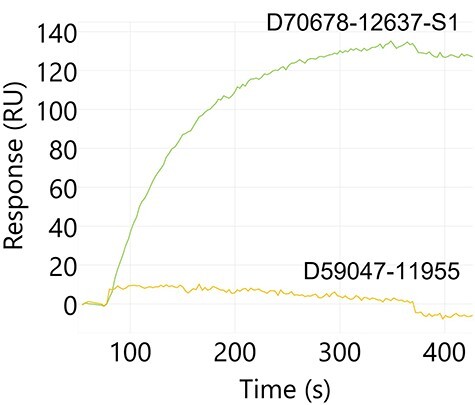
Example Carterra sensogram for antibody blocking of the S1:ACE2 interaction. Antibody:S1 protein complex was captured on the chip surface and ACE2 protein binding was assessed. A non-blocking candidate, D70678-12637-S1, complexed with S1 protein does not inhibit interaction with ACE2 (green). Conversely, a blocking candidate, D59047-11955, prevents ACE2 binding when S1 is complexed with the antibody (yellow).
Lastly, candidate antibodies were assayed in a classical binning competition format (Fig. 5) to identify the relative binding epitopes on the S1 protein. Epitope binning was performed in a sequential format and binding of each antibody combination was assessed simultaneously. Non-competitive binding pairs and competitive binding pairs are highlighted by green or red squares, respectively, in Fig. 5a. The data are also presented as a community network plot in Fig. 5b to better visualize the relative binding epitopes among the candidates assessed.
Figure 5.
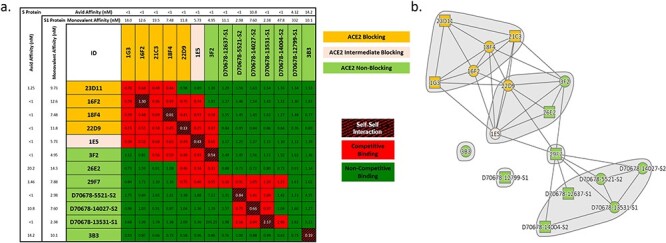
Select antibodies were characterized for competitive binding to the S1 protein to elucidate epitopic coverage. (a) All antibodies capable of binding S1 protein and preventing the S1:ACE2 interaction (ID highlighted in yellow) focused on a similar epitope (competitive binding indicated by red squares in grid). Antibodies that did not function as ACE2 blocking candidates (ID highlighted in light green) were distributed across two distinct core epitopes, with some antibodies binding at the interface of these epitopes, and an additional two clones appeared to bind distinct epitopes. Interestingly, an intermediate S1:ACE2 blocking candidate (1E5; ID highlighted in beige) bound at the interface between the ACE2 blocking epitope and a separate non-blocking epitope, thus supporting the partial blocking characteristic. No direct correlation was observed between affinity and binding epitope when assessed in either the monovalent binding format to the monomeric S1 protein or avid binding to the trimeric S protein. (b) A community network plot illustrates the bin clustering and distinct binding regions for each group of candidates.
Sequence information for each candidate is presented in Table 2, including full-paired heavy and light chain variable regions along with V-region and amino acid mutation rate analysis. Table 3 highlights common in silico liability assessments for each candidate based on published motifs [13, 14] for antibodies.
Table 2.
Heavy and light chain family and full sequence information for select characterized candidate antibodies
| ID | Mouse strain | Heavy V Gene | Heavy V Gene Identity % | VDJ Region |
|---|---|---|---|---|
| 15G11 | DivergimAb | IGHV1S137 | 98.64 | QVQLQQSGAELVRPGVSVKISCKGSGYTFTDYAMHWVKQSHAKSLEWIGVISTYYGDASYN QKFKGKATMTVDKSSSTAYMELARLTSEDSAIYYCARWANWGYYYAMDYWGQG TSVTVSS |
| 18F4 | DivergimAb | IGHV2-9-1 | 100 | QVQLKESGPGLVAPSQSLSITCTVSGFSLTSYAISWVRQPPGKGLEWLGVIWTGGGTNYN SALKSRLSISKDNSKSQVFLKMNSLQTDDTARYYCARKDYYGSSRNAMDYWGQGTSVT VSS |
| 1E5 | DiversimAb | IGHV1-55 | 100 | QVQLQQPGAELVKPGASVKMSCKASGYTFTSYWITWVKQRPGQGLEWIGDIYPGSGST NYNEKFKSKATLTVDTSSSTAYMQLSSLTSEDSAVYYCARSTVVATDAMDYWGQGTSVT VSS |
| 1G3 | DiversimAb | IGHV2-9-1 | 100 | QVQLKESGPGLVAPSQSLSITCTVSGFSLTSYAISWVRQPPGKGLEWLGVIWTGGGTNYNSALKS RLSISKDNSKSQVFLKMNSLQTDDTARYYCARFHYYGSSYGYFDYWGQGTTLTVSS |
| 21C3 | DivergimAb | IGHV2-9-1 | 100 | QVQLKESGPGLVAPSQSLSITCTVSGFSLTSYAISWVRQPPGKGLEWLGVIWTGGGTNYNSALK RLSISKDNSKSQVFLKMNSLQTDDTARYYCARIYYYGSSYFDYWGQGTTLTVSS |
| 22D9 | DivergimAb | IGHV1-54 | 100 | QVQLQQSGAELVRPGTSVKVSCKASGYAFTNYLIEWVKQRPGQGLEWIGVINPGSGGTNYN EKFKGKATLTADKSSSTAYMQLSSLTSEDSAVYFCARRHYYYGVDYWGQGTTLTVSS |
| 23D11 | DivergimAb | IGHV1S113 | 97.22 | EVQLQQSGPELVKPGASVKISCKTSGYTFTEYTMYWVKQSHGKSLEWIGGVNPNNGDTSYSQ KFKGKATLTVDKSSSTAYMELRSLTSEDSAVYYCARDGYDLYYGMDYWGQGTSVTVSS |
| 26E2 | DivergimAb | IGHV14-4 | 100 | EVQLQQSGAELVRPGASVKLSCTASGFNIKDDYMHWVKQRPEQGLEWIGWIDPENGDTEYA SKFQGKATITADTSSNTAYLQLSSLTSEDTAVYYCTKGYYGSSYDYFDYWGQGTTLTVSS |
| 29F7 | DivergimAb | IGHV1-82 | 100 | QVQLQQSGPELVKPGASVKISCKASGYAFSSSWMNWVKQRPGKGLEWIGRIYPGDGDTNYN GKFKGKATLTADKSSSTAYMQLSSLTSEDSAVYFCARDDYDEGDYWGQGTTLTVSS |
| 3B3 | DiversimAb | IGHV5-9-1 | 100 | DVKLVESGEGLVKPGGSLKLSCAASGFTFSSYAMSWVRQTPEKRLEWVAYISSGGDYIYYAD TVKGRFTISRDNARNTLYLQMSSLKSEDTAMYYCTRVARLYDGYFDYWGQGTTLTVSS |
| 3F2 | DiversimAb | IGHV14-4 | 97.95 | EVQLQQSGAELVRPGASVKLSCTASGFNIKDDYMHWVKQRPEQGLEWIGWIDPENGDTEYAS KFQGKATITVDISSNTAYLQLNSLTSEDTAVYYCSTLIYYYGSSNDYWGQGTTLTVSS |
| D59047-11955 | DivergimAb | IGHV2-9-1 | 100 | QVQLKESGPGLVAPSQSLSITCTVSGFSLTSYAISWVRQPPGKGLEWLGVIWTGGGTNYNSAL KSRLSISKDNSKSQVFLKMNSLQTDDTARYYCARIYYYGSSYFDYWGQGTTLTVSS |
| D70678-12637-S1 | Alloy | IGHV3-21 | 96.94 | EVQLVESGGGLVKPGGSLRLSCATSGFTFSHYSMNWVRQAPGKGLEWVSSISSSSSNIYYADS VKGRFTVSRDNAKNSLYLQMNSLRAEDTAVYYCARRGSSWSFDYWGQGTLVTVSS |
| D70678-12799-S1 | Alloy | IGHV1-2 | 97.95 | QVQLVQSGAEVKKPGASVKVSCKASGYTFTDYYMHWVRQAPGQGLEWMGWINPNSGGTN YAQKFQGRVTMTRDTSISTAYMELSRLRSDDTALYYCARLDYWSQGTLVTVSS |
| D70678-13531-S1 | Alloy | IGHV4-59 | 94.9 | QVQLQESGPGLVKPSETLSLTCTVSGGSISSYYWTWIRQSPGKGLEWIGYIYYSGSTYTNPSLKS RVTFSVDTSENQFSLKLNSVTAADTAIYFCARDNMDVWGKGTTVTVSS |
| D70678-13576-S1 | Alloy | IGHV4-59 | 94.58 | QVQLQESGPGLVKPSETLSLTCTVSGGSISNYYWTWIRQPPGKGLEWIGYIYYSGSTYTNPSLK SRVTISVDTSENQFSLKLNSVTAADTAIYYCARDNMDVWGKGTTVTVSS |
| D70678-14004-S2 | Alloy | IGHV1-18 | 97.64 | QVQLVQSGAEVKKPGASVKVSCKASGYTFSSYGIIWVRQAPGQGLEWMGWISAYNGNTNYA QKLQGRVTMTTDTSTSTAYMELRTLRSDDTAVYYCAREITLNWNYAGWFDPWGQGTLVTVSS |
| D70678-14027-S2 | Alloy | IGHV3-15 | 97.33 | EVQLVESGGGLVKPGGSLRLSCAASGFTFSYAWMTWVRQAPGKGLEWVGRIKTKSDGGTT DYASPVKGKFTISRDDSKNTLYLQMNSLQTEDTAVYYCTTHSSPDYWGQGTLVTVSS |
| D70678-2155-S1 | Alloy | IGHV3-15 | 94.06 | QVQLVESGGDLVKPGGSLRLSCAASGFTFSNAWMTWVRQAPGKELEWVGRIKTKSDGGTIE YGVSVKGRFTISRDDSKNTLFLQMNSLTTEDTAVYYCTTHSSPDYWGQGTLVTVSS |
| D70678-2743-S1 | Alloy | IGHV4-59 | 95.25 | QVQLQESGPGLVKPSETLSLTCTVSGGSISSYYWTWIRQPPGKGLEWIGYIYSSGSTYTNPSLK SRVTISVDTSENQFSLKLNSVTAADTAVYYCARDNMDVWGKGTTVTVSS |
| D70678-5521-S2 | Alloy | IGHV3-15 | 97 | EVQLVESGGGLVKPGGSLRLSCAASGFTFNYAWMTWVRQAPGKGLDWVGRIKTKTDSGTTD YAAPVKGRFTISRDDSKNTLYLQMNNLKTEDTAVYYCTTHSTPDYWGQGTLVTVSS |
| ID | Mouse strain | Light V Gene | Light V Gene Identity % | VJ Region |
| 15G11 | DivergimAb | IGLV1 | 99.66 | QAVVTQESALTTSPGETVTLTCRSSTGAVTTSNYANWVQEKPDHLFTGLIGGTNNRAPGVPAR FSGSLIGDKAALTITGAQTEDEAIYFCALWYSNHWVFGGGTKLTVL |
| 18F4 | DivergimAb | IGLV1 | 100 | QAVVTQESALTTSPGETVTLTCRSSTGAVTTSNYANWVQEKPDHLFTGLIGGTNNRAPGVPAR FSGSLIGDKAALTITGAQTEDEAIYFCALWYSNHWVFGGGTKLTVL |
| 1E5 | DiversimAb | IGKV2-109 | 100 | DIVMTQAAFSNPVTLGTSASISCRSSKSLLHSNGITYLYWYLQKPGQSPQLLIYQMSNLASGV PDRFSSSGSGTDFTLRISRVEAEDVGVYYCAQNLELPWTFGGGTKLEIK |
| 1G3 | DiversimAb | IGLV1 | 99.32 | QAVVTQESALTTSPGETVTLTCRSSTGAVTTSNYANWVQEKPDHLFTGLIGGTNNRAPGVPA RFSGSLIGDKAALTITGAQTEDEAIYFCALWYSNHWVFGGGTKLTVL |
| 21C3 | DivergimAb | IGLV1 | 99.66 | QAVVTQESALTTSPGETVTLTCRSSTGAVTTSNYANWVQEKPDHLFTGLIGGTNNRAPGVPA RFSGSLIGDKAALTITGAQTEDEAIYFCALWYSNQFIFGSGTKVTVL |
| 22D9 | DivergimAb | IGKV14-111 | 100 | DIKMTQSPSSMYASLGERVTITCKASQDINSYLSWFQQKPGKSPKTLIYRANRLVDGVPSR FSGSGSGQDYSLTISSLEYEDMGIYYCLQYDELYTFGGGTKLEIK |
| 23D11 | DivergimAb | IGKV6-15 | 99.3 | DIVMTQSQKFMSTSVGDRVSVTCKASQNVGTNVAWYQQKPGQSPKALIYSASYRYSGVPDR FTGSGSGTDFTLTISNVQSVDLAEYFCHQYNSYPWTFGGGTKLEIR |
| 26E2 | DivergimAb | IGKV19-93 | 99.65 | DIQMTQSPSSLSASLGGKVTITCKASQDINKYIAWYQHKPGKGPRLLIHYTSTLQPGIPSRFS GSGSGRDYSFSISNLEPEDIATYYCLQYDNLWTFGGGTKLEIK |
| 29F7 | DivergimAb | IGKV6-15 | 100 | DIVMTQSQKFMSTSVGDRVSVTCKASQNVGTNVAWYQQKPGQSPKALIYSASYRYSGVPDR FTGSGSGTDFTLTISNVQSEDLAEYFCQQYNSYPLYTFGGGTKLEIK |
| 3B3 | DiversimAb | IGKV3-10 | 100 | NIVLTQSPASLAVSLGQRATISCRASESVDSYGNSFMHWYQQKPGQPPKLLIYLASNLESGVP ARFSGSGSRTDFTLTIDPVEADDAATYYCQQNNEDPYTFGGGTKLEIK |
| 3F2 | DiversimAb | IGLV1 | 100 | QAVVTQESALTTSPGETVTLTCRSSTGAVTTSNYANWVQEKPDHLFTGLIGGTNNRAPGVPA RFSGSLIGDKAALTITGAQTEDEAIYFCALWYSNHWVFGGGTKLTVL |
| D59047-11955 | DivergimAb | IGLV1 | 99.66 | QAVVTQESALTTSPGETVTLTCRSSTGAVTTSNYANWVQEKPDHLFTGLIGGTNNRAPGVPA RFSGSLIGDKAALTITGAQTEDEAIYFCALWYSNQFIFGSGTKVTVL |
| D70678-12637-S1 | Alloy | IGKV3-20 | 97.19 | EIVLTQSPATLSLSPGERAILSCRASQSISSTYLAWNQQKPGQAPRLLIYGASSRATGIPDRFSG SGSGTDFTLTISRLEPEDFAVYYCQQYGSSWTFGQGTKVEIK |
| D70678-12799-S1 | Alloy | IGKV2-24 | 94.67 | DIVMTQTPLSLPVTLGQPASISCRSSQSLVHSDGNTYLSWLQQRPGQPPRLLIYKISNRFSGVP DRFSGSGAGTDFTLKISRVEAEDVGVYYCMQETQFTWTFGQGTKVEIK |
| D70678-13531-S1 | Alloy | IGKV2-30 | 97.99 | DVVMTQTPLSLPVTLGQPASISCRCSQSLVYSDGNTYLNWFQQRPGQSPRRLIYKVSIRDSGVP DRFSGSGSGTDFTLKISRVEAEDVGIYYCMQGTHRPITFGRGTRLEIK |
| D70678-13576-S1 | Alloy | IGKV4-1 | 94.7 | DIVMTQSPDSLAVSLGERATINCKSSQSVLYSSNNKNYLAWYQQKPGQPPKLLIYWASTRESGV PDRFSGSGSGTDFTLTISSLQAEDVAVYYCQQYYSTPYTFGQGTKLEIK |
| D70678-14004-S2 | Alloy | IGKV3-15 | 96.52 | EIVMTQSPATLSVSPGERATLSCRASQSIRNNLAWYQQKPGQAPRLLIYGAISRATGVPARFSG SGSGTEFTLTISSLQSEDFAVYHCQQYNNWLPYTFGQGTKLEIK |
| D70678-14027-S2 | Alloy | IGKV1-33;IGKV1D-33 | 99.3 | DIQMTQSPSSLSASVGDRVTITCQASQDIRNYLNWYHQKPGKAPKLLIYDASNLETGVPSRFS GSGSGTDFTFTISSLQPEDIATYYCQQYDNLPYTFGQGTKLEIK |
| D70678-2155-S1 | Alloy | IGKV1-33;IGKV1D-33 | 96.86 | DIQMTQSPSSLSASVGDRVTITCQASQDIRNYLNWYQQKPGKAPKLLIYDASTLETGVPSRFS GSGSGTDFTFTISSLQPEDIATYYCHQYGNLPLSFGGGTKVEIK |
| D70678-2743-S1 | Alloy | IGKV2-30 | 95.65 | DVVMTQTPLSLPVTLGQPASISCRSSQSLVYSDGNTYLNWFQQRPGQSPRRLIYKVSNRDSG VPDRFSGSGSGTDFTLKISRVEAEDVGIYYCMQGTHWPIIFGRGTRLEIK |
| D70678-5521-S2 | Alloy | IGKV1-33;IGKV1D-33 | 98.24 | DIQMTQSPSSLSASVGDRVTITCQASQDINNHLNWYQQKPGKAPNLLIYDASNLETGVPSR FSGSGSGTDFTFTISSLQPEDFATYYCQQYDNLPYTFGQGTKLEIK |
Table 3.
In silico sequence analysis of candidate antibodies for common liability motifs
| ID | Mouse strain | Liability (High) | Liability (Medium) | Liability (Low) |
|---|---|---|---|---|
| 15G11 | DivergimAb | DS (Isomerization, Heavy FR3); M (Oxidation, Heavy CDR3) | TS (Cleavage, Heavy FR3, Heavy FR4); TS (Cleavage, Light CDR1, Light FR1); NH (Deamidation, Light CDR3) | SN (Deamidation, Light CDR1, Light CDR3); TN (Deamidation, Light CDR2) |
| 18F4 | DivergimAb | DP (Cleavage, IGHG2B); NA (Deamidation, Heavy CDR3); NS (Deamidation, 3 * Heavy FR3); M (Oxidation, Heavy CDR3) | TS (Cleavage, Heavy CDR1, Heavy FR4); TS (Cleavage, Light CDR1, Light FR1); NH (Deamidation, Light CDR3) | TN (Deamidation, IGHV2-9-1); SN (Deamidation, Light CDR1, Light CDR3); TN (Deamidation, Light CDR2) |
| 1E5 | DiversimAb | DS (Isomerization, Heavy FR3); M (Oxidation, Heavy CDR3); NG (Deamidation, Light CDR1); M (Oxidation, Light CDR2) | TS (Cleavage, Heavy CDR1, 2 * Heavy FR3, Heavy FR4); TS (Cleavage, Light FR1); NP (Hydrolysis, Light FR1) | TN (Deamidation, IGHV1-55); SN (Deamidation, IGKV2-109, Light CDR1, Light FR1) |
| 1G3 | DiversimAb | NS (Deamidation, 3 * Heavy FR3, IGHG1) | TS (Cleavage, Heavy CDR1); TS (Cleavage, Light CDR1, Light FR1); NH (Deamidation, Light CDR3) | TN (Deamidation, IGHG1, IGHV2-9-1); SN (Deamidation, Light CDR1, Light CDR3); TN (Deamidation, Light CDR2) |
| 21C3 | DivergimAb | NS (Deamidation, 3 * Heavy FR3) | TS (Cleavage, Heavy CDR1); TS (Cleavage, Light CDR1, Light FR1) | TN (Deamidation, IGHV2-9-1); SN (Deamidation, Light CDR1, Light CDR3); TN (Deamidation, Light CDR2) |
| 22D9 | DivergimAb | DS (Isomerization, Heavy FR3); NS (Deamidation, Light CDR1); DG (Isomerization, Light FR3) | TS (Cleavage, Heavy FR1, Heavy FR3); NH (Deamidation, IGHG1); NP (Hydrolysis, Heavy CDR2) | TN (Deamidation, Heavy CDR1, IGHV1-54) |
| 23D11 | DivergimAb | NG (Deamidation, Heavy CDR2); DG (Isomerization, Heavy CDR3); DS (Isomerization, Heavy FR3); M (Oxidation, Heavy CDR3); NS (Deamidation, Light CDR3) | TS (Cleavage, Heavy FR1, Heavy FR3, Heavy FR4, IGHV1S113); NP (Hydrolysis, Heavy CDR2); TS (Cleavage, Light FR1) | SN (Deamidation, Light FR3); TN (Deamidation, Light CDR1) |
| 26E2 | DivergimAb | DP (Cleavage, Heavy CDR2); NG (Deamidation, Heavy CDR2); DS (Isomerization, IGHG2A;IGHG2C) | TS (Cleavage, 2 * Heavy FR3, IGHG2A;IGHG2C) | SN (Deamidation, Heavy FR3); SN (Deamidation, Light FR3) |
| 29F7 | DivergimAb | NG (Deamidation, Heavy FR3); DG (Isomerization, Heavy CDR2); DS (Isomerization, Heavy FR3, IGHG2C); NS (Deamidation, Light CDR3) | TS (Cleavage, Heavy FR3); TS (Cleavage, Light FR1) | TN (Deamidation, IGHV1-82); SN (Deamidation, Light FR3); TN (Deamidation, Light CDR1) |
| 3B3 | DiversimAb | NA (Deamidation, Heavy FR3); DG (Isomerization, Heavy CDR3); DP (Cleavage, Light CDR3, Light FR3); NS (Deamidation, Light CDR1); DS (Isomerization, Light CDR1) | SN (Deamidation, IGKV3-10) | |
| 3F2 | DiversimAb | DP (Cleavage, Heavy CDR2); NG (Deamidation, Heavy CDR2); NS (Deamidation, Heavy FR3); | TS (Cleavage, Heavy FR3); TS (Cleavage, Light CDR1, Light FR1); NH (Deamidation, Light CDR3) | SN (Deamidation, Heavy CDR3, Heavy FR3); SN (Deamidation, Light CDR1, Light CDR3); TN (Deamidation, Light CDR2) |
| D59047-11955 | DivergimAb | NS (Deamidation, 3 * Heavy FR3) | TS (Cleavage, Heavy CDR1); TS (Cleavage, Light CDR1, Light FR1) | TN (Deamidation, IGHV2-9-1); SN (Deamidation, Light CDR1, Light CDR3); TN (Deamidation, Light CDR2) |
| D70678-12637-S1 | Alloy | NA (Deamidation, Heavy FR3); NS (Deamidation, 2 * Heavy FR3); DS (Isomerization, Heavy FR3) | TS (Cleavage, Heavy FR1) | KN (Deamidation, Heavy FR3); SN (Deamidation, Heavy CDR2) |
| D70678-12799-S1 | Alloy | NS (Deamidation, Heavy CDR2); DG (Isomerization, Light CDR1); M (Oxidation, Light CDR3) | TS (Cleavage, Heavy FR3); NP (Hydrolysis, Heavy CDR2) | TN (Deamidation, IGHV1-2); SN (Deamidation, IGKV2-24) |
| D70678-13531-S1 | Alloy | NS (Deamidation, Heavy FR3); M (Oxidation, Heavy CDR3); 2 C (Extra Cysteine, Light FR1); DG (Isomerization, Light CDR1); DS (Isomerization, Light FR3); M (Oxidation, Light CDR3) | TS (Cleavage, Heavy FR3); NP (Hydrolysis, Heavy FR3) | TN (Deamidation, Heavy FR3) |
| D70678-13576-S1 | Alloy | NS (Deamidation, Heavy FR3); M (Oxidation, Heavy CDR3); 2 C (Extra Cysteine, Light FR1); DG (Isomerization, Light CDR1); DS (Isomerization, Light FR3); M (Oxidation, Light CDR3); DS (Isomerization, Light FR1) | TS (Cleavage, Heavy FR3); NP (Hydrolysis, Heavy FR3) | SN (Deamidation, Heavy CDR1); TN (Deamidation, Heavy FR3); KN (Deamidation, Light CDR1); SN (Deamidation, Light CDR1) |
| D70678-14004-S2 | Alloy | DP (Cleavage, Heavy CDR3); NG (Deamidation, Heavy CDR2); | TS (Cleavage, 2 * Heavy FR3) | TN (Deamidation, IGHV1-18) |
| D70678-14027-S2 | Alloy | NS (Deamidation, Heavy FR3); DG (Isomerization, Heavy CDR2); DS (Isomerization, Heavy FR3); | KN (Deamidation, Heavy FR3); SN (Deamidation, IGKV1-33;IGKV1D-33) | |
| D70678-2155-S1 | Alloy | NA (Deamidation, Heavy CDR1); NS (Deamidation, Heavy FR3); DG (Isomerization, Heavy CDR2); DS (Isomerization, Heavy FR3); | KN (Deamidation, Heavy FR3); SN (Deamidation, Heavy CDR1) | |
| D70678-2743-S1 | Alloy | NS (Deamidation, Heavy FR3); M (Oxidation, Heavy CDR3); DG (Isomerization, Light CDR1); DS (Isomerization, Light FR3); M (Oxidation, Light CDR3) | TS (Cleavage, Heavy FR3); NP (Hydrolysis, Heavy FR3) | TN (Deamidation, Heavy FR3); SN (Deamidation, IGKV2-30) |
| D70678-5521-S2 | Alloy | DS (Isomerization, Heavy CDR2, Heavy FR3) | NH (Deamidation, Light CDR1) | KN (Deamidation, Heavy FR3); SN (Deamidation, IGKV1-33;IGKV1D-33) |
DISCUSSION
The COVID-19 pandemic highlighted the importance of rapid discovery of antiviral drugs, both prophylactic and therapeutic, as critical to containing the spread of the virus [15]. In response to the COVID-19 outbreak, the scientific community collectively answered the public health call to action with extraordinary momentum, leveraging both new and traditional technologies with a strong emphasis on speed [16]. Continuing to build on COVID-19 research critical to public health while highlighting various methods that can be deployed for antibody discovery, this report outlines the use of a number of distinct workflows that enabled the accelerated identification of antiviral antibodies, some with promising therapeutic potential. The sequence information and corresponding characterization data for a panel of 21 antibody candidates are reported for unrestricted use.
We highlight the use of both DiversimAb and Alloy-GK mice in accelerated hybridoma-based and single B cell screening platforms, respectively. To enable rapid generation of monoclonal antibodies out of the DiversimAb platform, mice were immunized on a 18-day accelerated schedule followed by hybridoma generation. Concurrently, Alloy mice were immunized on a 5-week schedule and subsequently screened on the Beacon in a single day followed by a sequence recovery and analysis process spanning less than 1 week. Despite the divergent immunization and screening workflows, both campaigns yielded sequences in similar timeframes to enable simultaneous downstream characterization requiring 3 total days. In sum, both campaigns required fewer than 3 months to go from immunization start to fully characterized, purified antibody. Of course, alternative workflows are possible to further tighten the timeline for future campaigns. For example, characterization data were acquired using both purified or crude antibody samples to validate either source on the Carterra LSA™ for kinetic, neutralization and binning data. With equivalent results, the time and resources required for purification can be incorporated further downstream to expedite the early discovery timeline. In addition, Alloy mice can be immunized on a more accelerated timeline.
The resulting data set presented here is comprised of a representative subset of a diverse panel of candidate antibodies spanning multiple epitopes with high affinity and both receptor blocking and non-blocking activity. Interestingly, all candidate antibodies identified from the Alloy mice fell within a similar non-blocking bin, likely indicative of epitope immunodominance [17], which has been reported as a common result from COVID-19 infection [18, 19]. However, the extended immunization strategy employed for the Alloy mice resulted in high-affinity antibodies, including the top four highest affinity candidates presented here. The balance between diversity and affinity is a challenge for in vivo immunization models: with continued boosting, clonal expansion of the highest affinity germline B cells can often result in a limited overall diversity [20]. Alternative immunization workflows involving rapid schedules and/or immunogen manipulation are effective risk mitigation strategies to circumvent these challenges [21]. In this study, the DiversimAb mice were immunized following an accelerated strategy to maximize epitopic diversity, and although the overall average affinity of these candidates was lower as a result, a subset of high affinity blocking clones was discovered. Recent studies on SARS-CoV-2 variants underscore the importance of diverse epitopic coverage, highlighting a major benefit in working with a hyperimmune mouse strain for antibody discovery [22]. It is also important to note that recent studies have identified non-blocking neutralizing epiotpes [23], indicating further testing of non-blocking Alloy candidates identified here could reveal efficacious potential in viral neutralizing experiments.
A review of the lead candidate sequences further underscores the importance of risk mitigation in antibody discovery through the use of multiple strains of mice for added diversity. It is well documented that genetic backgrounds heavily influence the B cell repertoire diversity [24, 25]. Therefore, in cases where rapid discovery is imperative, starting with multiple strains can improve the diversity in gene usage for V(D)J recombination. In the case of the Abveris DiversimAb mice, two separate background strains were leveraged (DiversimAb vs. DivergimAb) to further increase the output sequence diversity. Interestingly, many of the candidates from these mice contained near germline V-regions—likely a result of the accelerated immunization approach employed for rapid discovery. Regardless, binding affinity is still maintained at the nanomolar level, which could be due to the unique and diverse CDR3 regions. The presence of germline sequences from DiversimAb mice using this immunization approach provides an opportunity for streamlined lead optimization via humanization and affinity maturation without the need to assess numerous V region backmutation permutations.
Traditional methods of in vivo antibody drug discovery suffer from timeline disadvantages associated with immunization, humanization and downstream lead optimization (if required) [26]. However, with the recent development of innovative new technologies to accelerate the upstream drug discovery process highlighted in this report, in vivo antibody discovery is now a viable option for accelerated response to novel viral threats. Humanized mouse strains can be leveraged synergistically with genetically engineered mice designed to increase epitopic diversity (DiversimAb) to provide lead and backup antibody drug candidates of desired therapeutic efficacy. When combined with efficient high-throughput downstream antibody capture and characterization tools, it becomes possible to go from immunization to sequence in as few as 29 days.
DATA AVAILABILITY
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary materials.
AUTHOR CONTRIBUTIONS
T.E.M. and C.A.S. conceived of and designed experiments, performed data analysis and authored the manuscript. R.A., J.B., A.S.B., N.K.D., N.T.D., A.M.D., C.E., J.G., B.G., K.S.P., K.R. and J.S. designed and executed experiments, performed data analysis and reporting, and participated in manuscript review.
CONFLICT OF INTEREST STATEMENT
T.E.M., R.A., J.B., A.S.B., N.K.D., A.M.D., C.E., J.G., B.G., K.S.P., K.R., J.S. and C.A.S. are employees of Abveris Inc.; N.T.D. is an employee of Carterra.
Supplementary Material
ACKNOWLEDGMENTS
We would like to thank Alloy Therapeutics for supplying the GK mice, Carterra for assistance and use of the LSA and Gary Ng of Abveris for preparation of the manuscript.
References
- 1.Huang, C, Wang, Y, Li, X et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020; 395: 497–506. 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fineberg, HV. Pandemic preparedness and response--lessons from the H1N1 influenza of 2009. N Engl J Med 2014; 370: 1335–42. 10.1056/NEJMra1208802. [DOI] [PubMed] [Google Scholar]
- 3.Furuyama, W, Marzi, A, Nanbo, A et al. Discovery of an antibody for pan-ebolavirus therapy. Sci Rep 2016; 6: 20514. 10.1038/srep20514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bornholdt, ZA, Turner, HL, Murin, CD et al. Isolation of potent neutralizing antibodies from a survivor of the 2014 Ebola virus outbreak. Science 2016; 351: 1078–83. 10.1126/science.aad5788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lowe, R, Barcellos, C, Brasil, P et al. The Zika virus epidemic in Brazil: from discovery to future implications. Int J Environ Res Public Health 2018; 15: 96. 10.3390/ijerph15010096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Klasse, PJ. Neutralization of virus infectivity by antibodies: old problems in new perspectives. Adv Biol 2014; 2014: 157895. 10.1155/2014/157895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Correia, BE, Bates, JT, Loomis, RJ et al. Proof of principle for epitope-focused vaccine design. Nature 2014; 507: 201–6. 10.1038/nature12966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ali, MG, Zhang, Z, Gao, Q et al. Recent advances in therapeutic applications of neutralizing antibodies for virus infections: an overview. Immunol Res 2020; 68: 325–39. 10.1007/s12026-020-09159-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.VanBlargan, LA, Goo, L, Pierson, TC. Deconstructing the antiviral neutralizing-antibody response: implications for vaccine development and immunity. Microbiol Mol Biol Rev 2016; 80: 989–1010. 10.1128/MMBR.00024-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abdiche, YN, Harriman, R, Deng, X et al. Assessing kinetic and epitopic diversity across orthogonal monoclonal antibody generation platforms. MAbs 2016; 8: 264–77. 10.1080/19420862.2015.1118596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kelley, B. Developing therapeutic monoclonal antibodies at pandemic pace. Nat Biotechnol 2020; 38: 540–5. 10.1038/s41587-020-0512-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Parola, C, Neumeier, D, Reddy, ST. Integrating high-throughput screening and sequencing for monoclonal antibody discovery and engineering. Immunology 2018; 153: 31–41. 10.1111/imm.12838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lu, X, Nobrega, RP, Lynaugh, H et al. Deamidation and isomerization liability analysis of 131 clinical-stage antibodies. MAbs 2019; 11: 45–57. 10.1080/19420862.2018.1548233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu, A, Kim, HS, Estee, S et al. Susceptibility of antibody CDR residues to chemical modifications can be revealed prior to antibody humanization and aid in the lead selection process. Mol Pharm 2018; 15: 4529–37. 10.1021/acs.molpharmaceut.8b00536. [DOI] [PubMed] [Google Scholar]
- 15.Zhu, Y, Li, J, Pang, Z. Recent insights for the emerging COVID-19: drug discovery, therapeutic options and vaccine development. Asian J Pharm Sci 2021 Jan; 16: 4–23. 10.1016/j.ajps.2020.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sempowski, GD, Saunders, KO, Acharya, P et al. Pandemic preparedness: developing vaccines and therapeutic antibodies for COVID-19. Cell 2020; 181: 1458–63. 10.1016/j.cell.2020.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Angeletti, D, Yewdell, JW. Understanding and manipulating viral immunity: antibody immunodominance enters Center stage. Trends Immunol 2018; 39: 549–61. 10.1016/j.it.2018.04.008. [DOI] [PubMed] [Google Scholar]
- 18.Andreano, E, Rappuoli, R. Immunodominant antibody germlines in COVID-19. J Exp Med 2021; 218: e20210281. 10.1084/jem.20210281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Farrera-Soler, L, Daguer, JP, Barluenga, S et al. Identification of immunodominant linear epitopes from SARS-CoV-2 patient plasma. PLoS One 2020; 15: e0238089. 10.1371/journal.pone.0238089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mesin, L, Schiepers, A, Ersching, J et al. Restricted Clonality and limited germinal Center Reentry characterize memory B cell reactivation by boosting. Cell 2020; 180: 92–106.e11. 10.1016/j.cell.2019.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Asensio, MA, Lim, YW, Wayham, N et al. Antibody repertoire analysis of mouse immunization protocols using microfluidics and molecular genomics. MAbs 2019; 11: 870–83. 10.1080/19420862.2019.1583995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kwon, D. This 'super antibody' for COVID fights off multiple coronaviruses. Nature 2021. 10.1038/d41586-021-01917-9. [DOI] [PubMed] [Google Scholar]
- 23.Yu, F, Xiang, R, Deng, X et al. Receptor-binding domain-specific human neutralizing monoclonal antibodies against SARS-CoV and SARS-CoV-2. Signal Transduct Target Ther 2020; 5: 212. 10.1038/s41392-020-00318-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chaaya, N, Shahsavarian, MA, Maffucci, I et al. Genetic background and immunological status influence B cell repertoire diversity in mice. Sci Rep 2019; 9: 14261. 10.1038/s41598-019-50714-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Greiff, V, Menzel, U, Miho, E et al. Systems analysis reveals high genetic and antigen-driven predetermination of antibody repertoires throughout B cell development. Cell Rep 2017; 19: 1467–78. 10.1016/j.celrep.2017.04.054. [DOI] [PubMed] [Google Scholar]
- 26.Lu, RM, Hwang, YC, Liu, IJ et al. Development of therapeutic antibodies for the treatment of diseases. J Biomed Sci 2020; 27: 1. 10.1186/s12929-019-0592-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hui, SW, Stenger, DA. Electrofusion of cells: hybridoma production by electrofusion and polyethylene glycol. Methods Enzymol 1993; 220: 212–27. 10.1016/0076-6879(93)20084-g. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary materials.