

Abstract

Metalation of N-mesityl-P,P-diphenylphosphinic amide Ph2P(O)-NHMes (HL, I) with MgBu2 and Ae{N(SiMe3)2}2 (Ae = Ca, Sr, and Ba) yields alkaline-earth metal complexes with the compositions of [(thf)nAe(L·HL)2] [Ae/n = Mg/0 (II), Ca/2 (III)] as well as of [Sr2L3(L·HL)(HL)] (1), [Ba2L3(L·HL)(HL)] (2), [Ba3L6] (3), and [(thf)2Ba3L6] (4). In III, 1, 2, and 3, the alkaline-earth metal atoms are in severely distorted octahedral environments, and the structural distortions are partially caused by the small O-Ae-N bite angles of the chelating Ph2P(O)-NMes anions. The substructures (L·HL) contain N–H···N hydrogen bridges, stabilizing the arrangement of the ligands in complexes II, III, 1, and 2. In the trinuclear barium complex [Ba(μ-L)3Ba(μ-L)3Ba] (3), a rigid adjustment of the anionic L bases leads to a C3-symmetric molecule in the crystalline state with bridging oxygen atoms. Due to the small O–Ba–N bite angles of the chelating anions, vacant coordination sites are available at the outer barium centers. Coordination of thf bases in these gaps yields the complex [(thf)Ba(μ-L)3Ba(μ-L)3Ba(thf)] (4). However, THF is unable to deaggregate the trinuclear complexes into smaller barium-containing moieties. Increasing the radius of the alkaline-earth metals and increasing the nuclearity of these compounds lead to decreasing solubility in common organic solvents. NMR studies verify that the molecular structures of these alkaline-earth metal complexes are maintained in ethereal solvents and toluene.
Introduction
Phosphinates of group 2 metals (Ae) such as alkaline-earth metal diphenylphosphinates (L)nAe[OP(O)Ph2]2 are well known since many decades and have been studied by IR spectroscopy already in the 1970s, verifying charge delocalization within the O–P–O moiety.1 These compounds are only sparingly soluble in common organic solvents and form coordination polymers in the solid state; strong bases such as DMF and DMSO are required to crystallize strand structures with bridging diphenylphosphinato ligands.2,3 In the heteroleptic thf adduct of Ph2P(O)O–Ca–X (X = Br, I),2,4 the phosphinato anion shows a bridging coordination mode.4 In homologous dithiophosphinates, the Ph2P(S)S– ligands act as bidentate Lewis bases at one metal atom, allowing the isolation of highly soluble discrete molecular complexes.2
Substitution of one or both oxygen atoms in Ph2P(O)OR’ (hydrocarbyl diarylphosphinates) by amino groups leads to isoelectronic Ar2P(O)-NHR’ (hydrocarbylamino-P,P-diarylphosphane oxides or N-hydrocarbyl-P,P-diarylphosphinic amides)5 and R2P(=NR′)–NHR’ (N,N′-dihydrocarbyl-P,P-diarylphosphinimidic amides),6 as depicted in Scheme 1, which established substance classes with manifold applications in organic, medicinal, and coordination chemistry. Bulky N-bound trimethylsilyl groups ensure solubility in common organic solvents as has been demonstrated by Stalke and coworkers for the mononuclear alkaline-earth metal complexes [{Ph2P(NSiMe3)2}2Ae(thf)n] (Ae/n = Be/0, Mg/0, Ca/1, Sr/2, Ba/2) as well as [{Py2P(NSiMe3)2}2Ae(L)] (Ae/L = Sr/thf, Ba/4,4′-bpy).7 Alkaline-earth metal salts of P,P-di(hydrocarbyl)phosphinic amides are very scarce despite the fact that in early reports the adducts of dimethylamino-diphenylphosphane oxide at AeCl2 and Ae(ClO4)2 (Ae = Mg, Ca) suggest isomorphous pairs of complexes based on X-ray powder diagrams.8 The structural authentication of the solvent-separated ion pair [{(2,6-Me2C6H3–NH)Ph2P=O}2Ca(I)(thf)3]I verified a monodentate coordination mode via the oxygen atom [Ca–O 225.9(3) and 226.7(3), P–O 149.8(3) and 150.0(3), as well as P–N 163.9(3) and 164.0(3) pm].9 A comparable coordination mode has been elucidated for bis[1,3-bis(diphenylphosphinoylimino)isoindolinato]calcium(II) with similar structural parameters [Ca–O 225.8(2) and 226.5(2), P–O 148.4(2) and 148.9(2), and P–N 164.1(2) and 163.1(2) pm].10 Very recently, the reaction of N-mesityl-P,P-diphenylphosphinic amide (I) with MgBu2 and Ca[N(SiMe3)2]2 yielded the mononuclear complexes [(MesNH–Ph2PO)2(MesN–Ph2PO)2Ae(thf)n] [Ae/n = Mg/0 (II), Ca/2 (III)], containing equimolar amounts of deprotonated [(diphenylphosphoryl)-(mesityl)amide L, red] and unreacted (N-mesityl-P,P-diphenylphosphinic amide HL, black) substrate ligands regardless of the applied stoichiometry, as depicted in Scheme 2.11 The 1:1 ratio of protonated and deprotonated ligands in the molecular structures has been verified in solution as well as in the crystalline state, excluding the intermolecular exchange or dissociation reactions. These complexes are soluble in common organic solvents and stabilized by intramolecular N–H···N hydrogen bridges and bind only via the oxygen atoms to the alkaline-earth metals. Reduced electrostatic attraction leads to the elongation of the Ca–OHL bond [237.8(1) pm] compared to the Ca–OL distance of 228.7(1) pm.
Scheme 1. Substitution of O in Diarylphosphinates (Left) by NH Leads to Isoelectronic Substance Classes of Diarylphosphinic Amides (Middle) and Diarylphosphinimidic Amides (Right).

Scheme 2. Metalation of Ph2P(O)–NHMes (I) with MgBu2 and [(thf)2Ca{N(SiMe3)2}2] in THF at Room Temperature (Red Deprotonated and Black Electroneutral Ligand) Yielding the Mg (II) and Ca Complexes (III).

There are no reports on complexes with the heavier alkaline-earth metals strontium and barium of type [R2P(O)–NR’]2Ae(L)n, most probably due to poor solubility in common organic solvents and the enormous reactivity initiating ether degradation reactions.12 However, substitution of the hard oxygen base by a soft selenium atom allows the synthesis and isolation of selenophosphinic amides of type [{Ph2P(Se)N–R}2Ae(thf)2] (Ae = Ca, Sr, and Ba) with bidentate ligands in mononuclear complexes with distorted octahedrally coordinated alkaline-earth metal atoms.13 Lack of information on [R2P(O)–NR′]2Ae(L)n congeners initiated our investigation choosing N-mesityl-P,P-diphenylphosphinic amide (I) as an easily available substrate,14 allowing the synthesis of oligonuclear strontium and barium complexes containing the rather bulky (diphenylphosphoryl)-(mesityl)amide anions. Due to charge delocalization, these anions exhibit negatively polarized O and N atoms, as depicted in Scheme 3, making these ligands suitable for a bidentate binding mode.
Scheme 3. Mesomeric Forms of Deprotonated N-Mesityl-P,P-diphenylphosphinic Amide, Clarifying the Negatively Charged Nitrogen [Left, (Diphenylphosphoryl)-(mesityl)amide] and Oxygen Atoms (Right, N-Mesityl-P,P-diphenylphosphinimidate) as Well as the Positively Polarized P Atom.

Results and Discussion
Synthesis
Suitable metalation reagents of the heavy alkaline-earth metals are the well-known bis(trimethylsilyl)amides of calcium,15,16 strontium,15 and barium.15,17 The reaction of N-mesityl-P,P-diphenylphosphinic amide (HL) with [(thf)2Sr{N(SiMe3)2}2] in THF yields sparingly soluble, colorless, and dinuclear [Sr2L3(L·HL)(HL)] (1), as depicted in Scheme 4, contrary to the metalation of HL with the calcium congener [(thf)2Ca{N(SiMe3)2}2] that led to the formation of highly soluble mononuclear [(thf)2Ca(L·HL)2] (III).11
Scheme 4. Reaction of [(thf)2Ae{N(SiMe3)2}2] (Ae = Ca, Sr) and [(Me3Si)2N–Ba{μ-N(SiMe3)2}]2 with Ph2P(O)–NHMes (I) in THF at Room Temperature (Red Deprotonated, Black Electroneutral Ligand) Yielding Complexes III, 1, 2, 3, and 4.

For the deprotonation of HL, ether-free [(Me3Si)2N–Ba{μ-N(SiMe3)2}]2 was applied in toluene. Already after 1 h, no starting HL was monitored by NMR spectroscopy and the precipitate was isolated, washed with n-pentane, dried in vacuo, and recrystallized from toluene. This sparingly soluble trinuclear complex [Ba3L6] (3) only contained deprotonated L– ligands. The workup of the mother liquor allowed the isolation of a minor amount of [Ba2L3(L·HL)(HL)] (2), which is very similar to the homologous Sr congener. This finding suggested that the reaction of [(Me3Si)2N–Ba{μ-N(SiMe3)2}]2 with HL to [Ba3L6] (3) proceeded via intermediately formed [Ba2L3(L·HL)(HL)] (2). The use of insoluble Ba(NH2)2 for the metalation of HL proved to be disadvantageous. The storage of [Ba3L6] (3) in THF led to ether degradation but small amounts of rather pure [(thf)2Ba3L6] (4) were isolated, as depicted in Scheme 4. In agreement with earlier reports,12 slow THF cleavage occurred in this ethereal solvent with Sr complex 1, a significantly faster ether degradation was observed for barium complexes 3 and 4.
Molecular Structures
Molecular structure and the atom-labeling scheme of dinuclear [Sr2L3(L·HL)(HL)] (1) are depicted in Figure 1. The six ligands in the structure are marked with the letters A to F and show diverse coordination modes. The two ligands A and B appear as a bidentate unit (L–H·L)−, interconnected by a N1A–H1A···N1B hydrogen bridge, and bind via the oxygen bases O1A and O1B. This binding behavior has been observed earlier for the lighter congeners of magnesium (II) and calcium (III, see Scheme 2).11 The oxygen atoms of the anionic ligands C, D, and E occupy bridging positions between the strontium atoms, leading to a rather short non-bonding Sr1···Sr2 distance of 376.75(5) pm. The nitrogen base N1C coordinates to Sr1, whereas the nitrogen atoms of the two other bridging ligands D and E bind to Sr2. The coordination sphere of Sr2 is saturated by another monodentate ligand F of the type HL, binding via O1F. Selected bond lengths and angles are listed in Table 1.
Figure 1.

Molecular structure and atom-labeling scheme of dinuclear strontium complex 1. The ellipsoids represent a probability of 30%, H atoms are neglected for the sake of clarity except the N-bound hydrogen atoms. The ligands are distinguished by the letters A to F.
Table 1. Selected Bond Lengths (pm) and Bond Angles (Deg.) of Complex [Sr2L3(L·HL)(HL)] (1).
| ligand | A | B | C | D | E | F |
|---|---|---|---|---|---|---|
| Sr1–O1 | 244.3(2) | 247.5(3) | 278.2(2) | 255.7(2) | 251.7(2) | |
| Sr2–O1 | 239.8(2) | 262.2(2) | 265.6(2) | 243.8(2) | ||
| Sr1–N1 | 262.0(3) | |||||
| Sr2–N1 | 260.3(3) | 261.7(3) | ||||
| P1–O1 | 150.1(3) | 151.3(3) | 152.1(3) | 152.1(3) | 152.4(3) | 149.4(3) |
| P1–N1 | 162.5(3) | 158.9(3) | 158.0(3) | 159.1(3) | 158.2(3) | 164.9(3) |
| P1–C1 | 179.9(4) | 181.2(4) | 181.8(4) | 180.7(4) | 181.5(4) | 179.8(4) |
| P1–C7 | 180.6(4) | 183.4(4) | 181.8(4) | 181.9(4) | 179.9(4) | 180.4(4) |
| N1–C13 | 143.0(5) | 140.8(5) | 141.8(5) | 141.9(5) | 141.4(5) | 144.4(5) |
| N1–P1–O1 | 117.7(2) | 117.1(2) | 108.0(2) | 108.8(2) | 109.2(2) | 116.7(2) |
| P1–N1–C13 | 128.7(3) | 130.2(3) | 129.7(3) | 128.6(2) | 134.1(3) | 125.3(3) |
Starting Ph2P(O)–NHMes (I) forms N–H···O hydrogen bridges in the crystalline phase.14 Therefore, we assess the influence of coordination on the bonding parameters of ligated Ph2P(O)–NMes anions (B, C, D, and E) and electroneutral Ph2P(O)–NHMes ligands (A and F) in comparison to ligand F despite the fact that rather short Sr2–O1F bonds are formed. The formation of the N1A–H1A···N1B hydrogen bridge slightly widens the O1–P1–N1 and P1–N1–C13 bond angles for A and B. The deprotonated ligands C, D, and E in bridging modes between the Sr atoms show significantly deviating bonding parameters. The very narrow bite angles O1C–Sr1–N1C [55.25(8)°], O1D–Sr2–N1D [57.92(9)°], and O1E–Sr2–N1E [57.42(9)°] cause a severe reduction of the O1–P1–N1 bond angles of the respective ligands by approx. 8°. In addition, the P1–N1–C13 bond angles of these anions are widened. Regardless of the fact that A and F are protonated and B to E are anionic ligands, the P1–O1 bond lengths vary in a very small range of 150.4 ± 1°. The presence of an N-bound hydrogen atom leads to an elongation of the P1–N1 bonds (A and F). Ligands binding solely via the oxygen atoms to the Sr atoms show Sr–O distances around 245 pm. Much larger spreading of the Sr–O bond lengths is observed for the bridging oxygen atoms, especially for ligand C with intramolecular strain causing this asymmetric coordination behavior.
The molecular structure and atom-labeling scheme of isotypic [Ba2L3(L·HL)(HL)] (2) are depicted in the Supporting Information. The differentiation of the ligands in the coordination sphere of the barium atoms is identical to the Sr homologue, allowing a comparison of the structural parameters of these isostructural molecules. Selected structural data are summarized in Table 2. The similarities are based on very similar Allred–Rochow electronegativity values of 0.99 and 0.97 for strontium and barium, suggesting highly ionic complexes in both cases. The Ba–O and Ba–N distances are larger than in Sr derivative 4 according to the larger cation radius. The fact that anionic ligands L– occupy bridging positions and that electroneutral HL are bound terminally, the Ae–O bond lengths vary in the same order of magnitude of 259.0 ± 19.2 for 1 (Ae = Sr) and 274.0 ± 18.9 pm for 2 (Ae = Ba). The Sr–N bonds show very similar values around 262 pm, whereas the Ba1–N1C bond length is significantly larger than the Ba2–N1D/E values. The triply bridged barium ions enforce a rather small non-bonding Ba1···Ba2 contact of 406.06(2) pm.
Table 2. Selected Bond Lengths (pm) and Bond Angles (Deg.) of Complex [Ba2L3(L·HL)(HL)] (2).
| ligand | A | B | C | D | E | F |
|---|---|---|---|---|---|---|
| Ba1–O1 | 260.4(2) | 255.1(2) | 292.8(2) | 262.3(2) | 271.8(2) | |
| Ba2–O1 | 262.1(2) | 280.2(2) | 279.8(2) | 262.9(2) | ||
| Ba1–N1 | 290.5(2) | |||||
| Ba2–N1 | 271.3(3) | 277.0(2) | ||||
| P1–O1 | 150.3(2) | 151.2(2) | 152.2(2) | 151.1(2) | 151.8(2) | 149.2(2) |
| P1–N1 | 163.0(3) | 158.7(3) | 157.3(2) | 158.3(3) | 158.4(3) | 165.0(3) |
| P1–C1 | 180.7(3) | 181.1(3) | 182.9(3) | 181.8(3) | 182.0(3) | 179.8(4) |
| P1–C7 | 180.7(4) | 181.9(3) | 180.5(3) | 181.2(3) | 181.6(3) | 179.1(3) |
| N1–C13 | 142.9(4) | 140.6(4) | 141.5(4) | 140.6(4) | 140.2(4) | 143.1(5) |
| N1–P1–O1 | 119.28(14) | 119.54(14) | 110.27(12) | 108.37(13) | 108.72(12) | 119.31(14) |
| P1–N1–C13 | 129.7(2) | 129.2(2) | 130.0(2) | 133.2(2) | 132.3(2) | 129.0(2) |
The molecular structure and atom-labeling scheme of trinuclear barium complex 3 are depicted in Figure 2. The molecule lies on a crystallographic C3 axis with a linear arrangement of the Ba atoms. This trinuclear complex only contains anionic ligands with bridging oxygen atoms. The small bite angles cause severely distorted octahedral environments of the barium atoms. The different Ba2–O1 and Ba2–O2 distances strongly influence the steric repulsion between the ligands, leading to significantly different P1–N1–C1 and P2–N2–C22 bond angles of 134.3(4) and 123.0(3)° on the one hand as well as Ba1–N1–C1 and Ba3–N2–C22 values of 124.3(3) and 137.1(3)° on the other. The varying coordination mode also leads to different non-bonding Ba1···Ba2 and Ba2···Ba3 contacts of 396.17(6) and 411.59(6) pm. Due to the small bite of the chelating bidentate ligands, the Ba1–N1–P1 and Ba3–N2–P2 bond angles of 99.78(18) and 98.55(17)°, respectively, are very similar.
Figure 2.
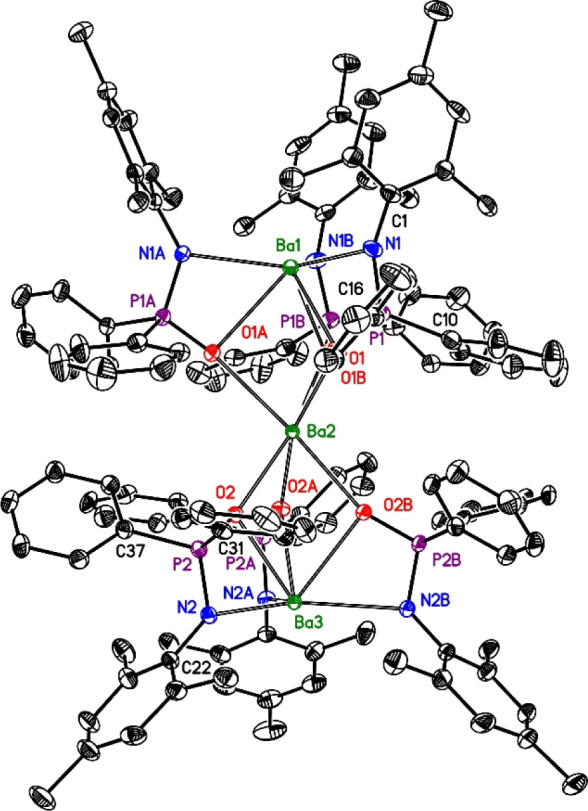
Molecular structure and atom-labeling scheme of trinuclear barium complex 3. The ellipsoids represent a probability of 30%, and H atoms are omitted for clarity reasons. Symmetry-equivalent ligands are marked with letters A and B. Selected bond lengths (pm): Ba1–O1 280.7(3), Ba2–O1 270.6(3), Ba2–O2 263.7(3), Ba3–O2 281.9(3), Ba1–N1 269.4(4), Ba3–N2 279.2(3), P1–O1 152.3(3), P1–N1 157.9(4), P1–C10 180.9(6), P1–C16 180.1(5), N1–C1 140.7(6), P2–O2 151.8(3), P2–N2 158.3(4), P2–C31 180.7(6), P2–C37 180.1(5), N2–C22 142.8(6); bond angles (deg): O1–P1–N1 108.9(2), P1–N1–C1 134.3(4), O2–P2–N2 108.5(2), P2–N2–C22 123.0(3), O1–Ba1–N1 54.57(11), O2–Ba3–N2 53.31(10).
The vacant coordination sites of the outer barium atoms Ba1 and Ba3 can be occupied by thf ligands, enhancing the coordination numbers of these metal atoms to seven as observed in complex 4. Due to the fact that the crystallographic C3 axis is maintained, the thf ligands are disordered around this axis. Surprisingly, this coordination of additional Lewis bases leads to a smaller unit cell, hinting toward a denser packing of the complexes. The calculated densities of 1.018 and 1.247 g cm–3 for compounds 3 and 4 reflect this finding. The molecular structure and atom-labeling scheme of molecule A of the trinuclear barium complex 4 are depicted in Figure 3, the very similar molecule B is shown in the Supporting Information. In molecule B, the N-bound mesityl substituent is disordered on two sites, limiting the discussion of the structural parameters. Nevertheless, the far-reaching relationship with complex 3 is obvious. The additional thf ligands induce steric repulsion and push the N-bound mesityl substituents to the outside of the complex, leading to very similar Ba1A–N1A–C21A and Ba3A–N2A–C51A bond angles of 130.2(7) and 132.0(7)°, respectively. Nevertheless, the presence of ligated thf bases verifies that the central complex of the type [Ba3L6] represents a strong structural motif and cannot be deaggregated by strong Lewis bases such as THF.
Figure 3.
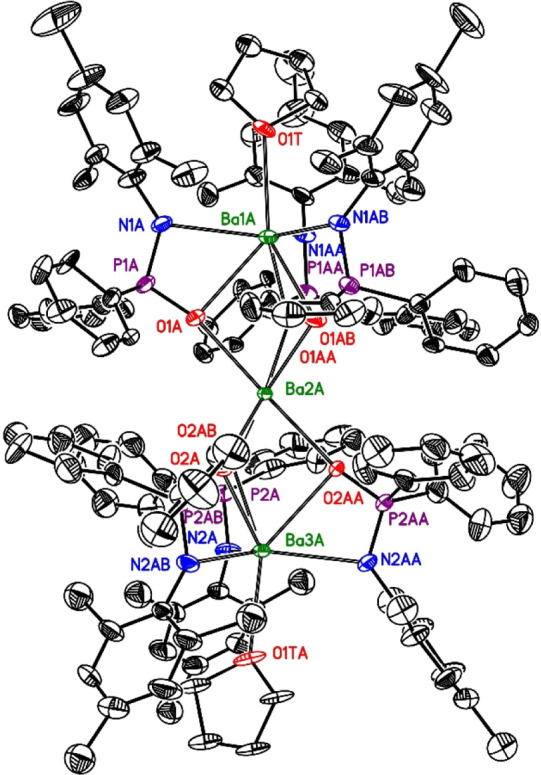
Molecular structure and atom-labeling scheme of molecule A of trinuclear barium complex 4. The ellipsoids represent a probability of 30%, and H atoms are omitted for clarity reasons. Symmetry-equivalent ligands are marked with additional letters A and B. Only one site of the disordered thf ligands is shown. Selected bond lengths (pm): P1A–O1A 151.6(7), P1A–N1A 157.1(9), P1A–C1A 178.3(12), P1A–C11A 177.3(12), N1A–C21A 139.9(10), P2A–O2A 151.0(7), P2A–N2A 156.1(10), P2A–C31A 175.2(8), P2A–C41A 183.6(7), N2A–C51A 140.9(14), Ba–O and Ba–N distances, see Table 2; bond angles (deg): O1A–P1A–N1A 108.1(4), P1A–N1A–C21A 127.2(7), O2A–P2A–N2A 108.5(4), P2A–N2A–C51A 127.8(8), O1A–Ba1A–N1A 54.1(2), O2A–Ba3A–N2A 53.5(2).
The asymmetric coordination of the Ph2P(O)–NMes anions that has been observed in thf-free 3 is significantly reduced. Indicative for the asymmetric binding mode in 3 are strikingly different structural pairs of Ba1–N1/Ba3–N2 bond lengths as well as Ba1–N1–C1/Ba3–N2–C22 and P1–N1–C1/P2–N2–C22 bond angles. The respective data of molecule A of the thf adduct 4 are leveled out for the structural parameters of the Ba2–O1/Ba2–O2 and Ba1A–N1A/Ba3A–N2A pairs as well as for the bond angle pairs Ba1A–N1A–C21A/Ba3A–N2A–C51A and P1A–N1A–C21A/P2A–N2A–C51A, making the symmetrification of complex 4 upon coordination of thf evident (Table 3). It is also obvious that there is no significant elongation of the Ba–O and Ba–N bonds due to the additional thf ligands at Ba1A/B and Ba3A/B despite the enhanced coordination numbers of seven. Furthermore, the Ba1···Ba2 and Ba2···Ba3 contacts become very similar upon the coordination of the thf molecules at Ba1 and Ba3. This finding suggests that the thf ligands induce only a small steric pressure and are occupying vacant coordination sites. The large coordination gaps also allow rather free motion and rotation of the thf ligands around the crystallographic C3 axis, leading to disordering of these ligated ether ligands and hence to a lower quality of the molecular structure of 4.
Table 3. Comparison of Ba–O and Ba–N Bond Lengths (pm) as Well as Selected Bond Angles (Deg) in Trinuclear Complexes 3 and 4.
| 3 | 4 | 4 | |
|---|---|---|---|
| complex | molecule A | molecule B | |
| Ba1–O1 | 280.7(3) | 276.9(8) | 283.0(7) |
| Ba2–O1 | 270.6(3) | 260.9(7) | 264.5(7) |
| Ba2–O2 | 263.7(3) | 262.3(7) | 264.3(9) |
| Ba3–O2 | 281.9(3) | 281.7(7) | 277.6(9) |
| Ba1–N1 | 269.4(4) | 272.5(8) | 273.3(8) |
| Ba3–N2 | 279.2(3) | 271.9(8) | 270.6(10) |
| Ba1–N1–CMes | 124.3(3) | 130.2(7) | a |
| Ba3–N2–CMes | 137.1(3) | 132.0(7) | 125.8(10) |
| P1–N1–CMes | 134.3(4) | 127.2(7) | a |
| P2–N2–CMes | 123.0(3) | 127.8(8) | 133.3(10) |
| Ba1···Ba2 | 396.17(6) | 396.17(13) | 398.24(16) |
| Ba2···Ba3 | 411.59(6) | 396.89(13) | 397.84(18) |
The mesityl group at N1B in molecule 4B is disordered on two sites with Ba1–N1–CMes and P1–N1–CMes bond-angle pairs of 120.5(13)/131.8(17)° and 138.3(13)/127.8(17)°, respectively.
IR Spectroscopy
The different structures of the complexes [Sr2L3(L·HL)(HL)] (1) and [Ba2L3(L·HL)(HL)] (2) on the one hand and the Ba congeners [Ba3L6] (3) and [(thf)2Ba3L6] (4) on the other are evident from the weak N–H stretching vibrations at 3251 and 3267 cm–1 for 1 and 2, respectively, whereas no bands are present in the appropriate IR region of the barium congeners 3 and 4. The very similar structures of 3 and 4 lead to comparable IR spectra of these complexes with strong bands at 1119, 1062, 990, 693, and 534 cm–1 for 3 and at 1121, 1064, 993, 693, and 535 cm–1 for thf adduct 4. The coordination of thf leads to additional intense vibrations in the IR spectrum of 4 at 1207 and 900 cm–1.
NMR Spectroscopy
The NMR spectra of the alkaline-earth metal complexes [Sr2L3(L·HL)(HL)] (1) and [Ba2L3(L·HL)(HL)] (2) show temperature-dependent dynamic behavior. At low temperatures, two NH groups are observed at δ = 11.76 (N–H···N hydrogen bridge) and 3.01 ppm (isolated N–H group) in the 1H NMR spectrum for the strontium congener 1, in agreement with the molecular structure in the crystalline state. At higher temperatures, both resonances broaden and finally disappear due to exchange processes being fast on the NMR time scale. The Gutowsky–Holm equation allows one to deduce the exchange energy barrier from the splitting at low temperatures (Δδ = 8.75 ppm, 3501 Hz), the spectrometer frequency (400.13 MHz), and the coalescence temperature (283 K), giving a value of 48 kJ mol–1. An exchange of these two N–H resonances requires the reorientation of the ligands in the vicinity of the strontium atoms. Nevertheless, the energy barrier is lower than observed for the proton exchange between the adjacent nitrogen bases in mononuclear calcium complex III (52.4 kJ mol–1).11 The NMR pattern of the aryl groups is quite complex at 233 K (probably also due to the hindered rotation of the Mes substituents around the N–Cipso bond) but becomes very clear at 323 K due to signal averaging in consequence of exchange processes being fast on the NMR time scale.
The 31P NMR spectrum of 1 at room temperature (293 K, 162 MHz) shows one broad resonance that splits into four major signals at 233 K with an intensity ratio of approx. 1:2:1:2 for the resonances at δ = 20.5, 14.4, 13.9, and 11.0 ppm (Figure 4). This fine structure reflects the solid-state structure containing four different ligand systems HL, (L–H···L)−, and two chemically different coordination modes of L– anions. A second minor isomer (approx. 10%) shows slightly low-field-shifted resonances at δ = 22.8, 16.5, and 11.7 ppm with an intensity ratio of 1:3:2. All these signals of both isomers merge to a broad singlet above 303 K. as depicted in Figure 4. A distinct assignment to different scrambling processes is impossible due to rather similar energy barriers for different exchange processes. For comparison, an energy barrier of 53 kJ mol–1 has been determined for the exchange of terminal and bridging bis(trimethylsilyl)amido ligands in dimeric [(Me3Si)2N–Sr{μ-N(SiMe3)2}]2 via the breaking of an endocyclic Sr–N bond, rotation around the remaining Sr–N bond, and reformation of a Sr–N bond.15a
Figure 4.

31P NMR spectra (162 MHz) of strontium complex [Sr2L3(L·HL)(HL)] (1) in [D8]toluene at 233, 243, 263, 283, 303, and 323 K (from bottom to top spectrum).
Crystals of [Ba2L3(L·HL)(HL)] (2) were dissolved in [D8]toluene and studied at different temperatures by 31P NMR spectroscopy to elucidate the dynamic behavior via proton exchange and ligand scrambling. The resonance broadens upon cooling and splits into several broad signals with a pattern of [Ba2L3(L·HL)(HL)] (2), as depicted in Figure 5. This spectrum at 233 K has a similar appearance as the spectrum of homologous [Sr2L3(L·HL)(HL)] (1) at 263 K (see Figure 4). However, an additional narrow singlet can be observed at a chemical shift that is characteristic for free Ph2P(O)–NHMes (I, HL). We interpret this finding in the sense of an operative equilibrium between [Ba2L3(L·HL)(HL)] (2) on the one side and [Ba3L6] (3) and HL (I) on the other, as depicted in Scheme 5. Free HL can bind to [Ba3L6] (3) leading initially to [{(HL)Ba}2BaL6] (in analogy to thf adduct 4) and finally to a breakdown of this complex and interconversion of the barium congeners 2 and 3.
Figure 5.

31P NMR spectra (162 MHz) of strontium complex [Ba2L3(L·HL)(HL)] (2) in [D8]toluene at 233, 253, 273, 297, 313, and 333 K (from bottom to top spectrum).
Scheme 5. Proposed Equilibrium between [Ba2L3(L·HL)(HL)] (2) as Well as [Ba3L6] (3) and HL (I) after the Dissolution of Complex 2 in [D8]Toluene.

Isolation of highly pure 3 proved to be quite challenging and repeated recrystallization of 3 from hot toluene was required. The dynamic processes probably hampered crystallization and isolation of highly pure 3. Furthermore, inadvertent traces of moisture during the handling and isolation of 3 could also lead to trace amounts of complex 2.
The barium complexes 3 and 4 only contain deprotonated anionic N-mesityl-P,P-diphenylphosphinic amide ligands, which lead to singlets in the NMR spectra at δ = 13.0 and 8.3 ppm, respectively.
The addition of THF again leads to dynamic and temperature-dependent 31P NMR spectra of barium complex 3 in [D8]THF solution. However, two singlets of low intensity at δ = 11.8 and 10.5 ppm remain unchanged and are most probably caused by ether degradation products, whereas the other resonances are involved in dynamic processes, suggesting different coordination modes of the N-mesityl-P,P-diphenylphosphinic amide anions at the barium atoms; deprotonation of THF yielded HL that underwent similar equilibria as discussed above (see Scheme 5). In summary, the N-mesityl-P,P-diphenylphosphinic amide ligands show a quite flexible coordination behavior at large and soft alkaline-earth metal cations, whereas more rigid complexes are formed with the harder congeners, magnesium and calcium, due to the formation of strong N–H···N hydrogen bridges between adjacent ligands.
Conclusions
This investigation focussed on the metalation of N-mesityl-P,P-diphenylphosphinic amide [Ph2P(O)–NH–Mes, HL, I] in the dependency of the metalation power of the alkaline-earth metal reagents [(thf)2Ae{N(SiMe3)2}2] (Ae = Ca, Sr) and [(Me3Si)2N–Ba{μ-N(SiMe3)2}]2 and on the coordination behavior of the N-mesityl-P,P-diphenylphosphinic amide anions (L–) with respect to the size of the metal ions. Under similar reaction conditions, the stoichiometry of the reactants varies regardless of the applied ratio of the metalation reagent to HL. Thus, four equivalents of HL are required for the formation of the calcium complex [Ca(L···HL)2] (III), three equivalents of HL per Sr and Ba atoms led to the formation of the dinuclear congeners [Sr2L3(L·HL)(HL)] (1) and [Ba2L3(L·HL)(HL)] (2), and quantitative metalation allowed the isolation of trinuclear [Ba3L6] (3). Increasing reactivity with increasing size and decreasing electronegativity of the alkaline-earth metals also led to growing tendency for THF degradation reactions. Thus, no THF decomposition was observed for 3 and very slow THF degradation was found for 1 in the NMR spectra within a few weeks. Contrary to these findings, barium complexes 2 and 3 had to be prepared in the absence of THF and the ether-free metalation reagent [(Me3Si)2N–Ba{μ-N(SiMe3)2}]2 had to be chosen. Nevertheless, the isolation of a barium complex from THF gave trinuclear [(thf)2Ba3L6] (4) because the strong THF base was unable to deaggregate 3 and hence complexes with smaller nuclearity were inaccessible by this route.
The molecular structures of the alkaline-earth metal complexes with N-mesityl-P,P-diphenylphosphinic amide ligands show several characteristic features. Electroneutral ligands HL and anionic (L–H···L)− ligands with N–H···N hydrogen bridges generally bind terminally via the oxygen donors. Free L– anions exhibit a μ-coordination mode with the oxygen bases between two metal atoms, whereas the nitrogen donors show backbonding to one of the adjacent metal centers turning these anions into chelating ligands.
In solution, diverse dynamic processes led to a scrambling of the N-mesityl-P,P-diphenylphosphinic amide ligands in [Sr2L3(L·HL)(HL)] (1) and [Ba2L3(L·HL)(HL)] (2) in [D8]toluene such as the dissociation and reformation of Ae–N bonds (Ae = Sr, Ba) as well as proton migration via N–H···N hydrogen bridges. At 233 K, two very similar isomers of [Sr2L3(L·HL)(HL)] (4) with an intensity ratio of approx. 9:1 were monitored in the [D8]toluene solution by 31P NMR spectroscopy. As expected, on the basis of the solid-state structure, four resonances were observed, namely, for HL, (L···HL)−, and two for the chemically different bridging L– ligands. Contrary to the strontium derivative 1, complex [Ba2L3(L·HL)(HL)] (2) showed an equilibrium with [Ba3L6] 3 via the loss of HL, thus impeding the isolation of pure 2 from toluene solutions. For pure complex 3, a singlet was observed in the 31P NMR spectrum. The addition of THF yielded [(thf)2Ba3L6] (4) and ether degradation occurred in this solution that significantly decreased the isolated yield of this thf adduct.
In summary, N-mesityl-P,P-diphenylphosphinic amide shows an unexpectedly rich coordination chemistry with alkaline-earth metal ions with competition between a chelating and a μ-bridging binding mode on the one hand and formation of N–H···N hydrogen bridges on the other, which can impede complete deprotonation and, hence, leads to anionic (L···HL)− ligands. The metalation force of Ae{N(SiMe3)2}2 clearly increases with the radius of the alkaline-earth metals in the row Ca < Sr < Ba.
Experimental Section
General
All manipulations were carried out under an inert nitrogen atmosphere using standard Schlenk techniques. The solvents were dried over KOH and subsequently distilled over sodium/benzophenone under a nitrogen atmosphere prior to use. Deuterated solvents were dried over sodium, degassed, and distilled in a nitrogen atmosphere. The yields given are not optimized. 1H, 13C{1H}, and 31P NMR spectra were recorded on Bruker AC 400 and AC 600 spectrometers. Chemical shifts are reported in parts per million relative to SiMe4 and aqueous phosphoric acid as external standards. The residual signals of the deuterated solvents [D8]THF, [D8]toluene, and [D6]benzene were used as internal standards for the interpretation of 1H NMR spectra. Due to poor solubility and various dynamic exchange processes, the 13C{1H} NMR spectra of these alkaline-earth metal compounds were not meaningful. The 1H and 31P NMR spectra are depicted in the Supporting Information. Substrates were purchased from Sigma-Aldrich, Merck, or Alfa Aesar and used without further purification. Substrate I [HL, δ(31P) = 16.9 ppm]11,14 and calcium complex III(11) [(thf)2Ae{N(SiMe3)2}2] of calcium15,16 and strontium,15 as well as [(Me3Si)2N–Ba{μ-N(SiMe3)2}]2,15,17 were prepared according to literature procedures.
Synthesis of [Sr2L3(L·HL)(HL)] (1)
In a Schlenk flask, 372 mg of (thf)2Sr[N(SiMe3)2]2 (0.67 mmol) was dissolved in 10 mL of THF and transferred to a separate Schlenk flask containing 452 mg of N-mesityl-P,P-diphenylphosphinic amide (1.35 mmol of I). The mixture was stirred for 2 days and then the volume was reduced to 2/3 of the original volume. The addition of 2 mL of n-hexane led to the precipitation of a crystalline powder, which was collected and dried in vacuum. Then, this solid was redissolved in 20 mL of Et2O, and the solution was filtered through a frit covered with diatomaceous earth. The filtrate was connected to a flask with anhydrous n-pentane for gas diffusion. After approx. 3 weeks, 180 mg of suitable single crystals of 1 for X-ray diffraction experiments were isolated (82.4 μmol, 36.6%). No trials were undertaken to extract the additional compound from the mother liquor. Elemental analysis (C126H128N6O6P6Sr2, 2183.40 g mol–1)—calc: C 69.31, H 5.91, and N 3.85; found: C 69.07, H 6.16, and N 3.85. IR (ATR, cm–1): 3252 w (NH), 3052 w, 2912 w, 1477 m, 1434 m, 1315 m, 1275 m, 1254 m, 1216 w, 1159 m, 1122 s, 1086 s, 1066 m, 991 m, 946 w, 853 m, 789 w, 714 vs, 693 vs, 664 m, 586 s, 539 vs, 518 s. 31P NMR (162 MHz, 233 K, [D8]toluene): δ 20.5, 14.4, 13.9, and 11.0 with an intensity ratio of 1:2:1:2 (major isomer, approx. 90%) as well as 22.8, 16.5, and 11.7 with an intensity ratio of 1:3:2 (minor component, approx. 10%). 1H NMR (400 MHz, 303 K, [D8]toluene): δ 7.43 (br, Ph, 4H), 6.8–6.5 (m, Ph, 6H), 6.29 (s, CHMes, 2H), 1.80 (br, p-MeMes, 3H), 1.70 (br, o-MeMes, 6H).
Synthesis of [Ba2L3(L·HL)(HL)] (2) and [Ba3L6] (3)
In a Schlenk flask, 2.26 g of [Ba{N(SiMe3)2}2]2 (2.46 mmol) was dissolved in 10 mL of toluene and added to 3.31 g of the solid N-mesityl-P,P-diphenylphosphinic amide (9.87 mmol of I) which immediately dissolved. After 2 minutes, a colorless solid precipitated and additional 10 mL of toluene was added to ease the stirring of this suspension. After 1 h, no N-mesityl-P,P-diphenylphosphinic amide was detected by NMR spectroscopy, and the solid was removed from the reaction mixture via filtration. The colorless solid was washed with n-pentane and dried in vacuum yielding a crude mixture of 2 and 3 (3.54 g, 89% with respect to the HL substrate I).
Part of this solid mixture (1.1 g, 4.55 mmol) was recrystallized from hot toluene. Upon slow cooling to ambient temperature, crystallization took place yielding 0.774 g of single-crystalline 3 (recovery, 70%), suitable for X-ray diffraction experiments. An additional batch of crystals was isolated after the condensation of n-pentane into the mother liquor leading to a crystal fraction of 2 and 3, thereby allowing solving the structure of [Ba2L3(L·HL)(HL)] (2); however, due to dynamic processes, it was impossible to isolate highly pure 2 from the toluene solution (see text and Scheme 5).
IR of [Ba2L3(L·HL)(HL)] (2, ATR, cm–1): 3267 w, 3051 w, 2911 w, 2849 w, 1473 m, 1434 m, 1319 m, 1276 m, 1262 m, 1216 w, 1156 m, 1119 s, 1083 s, 1064 m, 1028 w, 991 m, 851 m, 747 s, 712 vs, 694 vs, 585 m, 537 vs, 512 s, 463 m, 427 w.
Characterization of [Ba3L6] (3)
Elemental analyses of complex 3 showed low carbon values due to carbonate formation during the combustion of the samples. Elemental analysis (C126H126N6Ba3O6P6, 2418.16 g mol–1)—calc: C 62.58, H 5.25, and N 3.48; found: C 60.70, H 5.13, and N 3.23. IR (ATR, cm–1): 3051 w, 2911 w, 1471 m, 1434 m, 1386 w, 1308 m, 1263 m, 1161 w, 1119 s, 1081 s, 1062 s, 1029 w, 990 s, 946 m, 852 m, 750 m, 714 vs, 693 s, 665 m, 584 m, 534 vs, 512 vs, 466 m. 31P NMR (162 MHz, [D8]toluene): δ 13.0. 1H NMR (400 MHz, 313 K, [D8]toluene): δ 7.36 (m, Ph, 6H), 6.59 (m, Ph, 4H), 6.36 (br, CHMes, 2H), 1.86 (s, pMeMes, 3H), 1.34 (s, br, o-MeMes, 6H).
Synthesis of [(thf)2Ba3L6] (4)
N-Mesityl-P,P-diphenylphosphinic amide (3.6 g, 10.73 mmol) and Ba(NH2)2 (1.0 g, 5.90 mmol) were each weighed in separate Schlenk flasks and suspended in 15 mL of toluene. Both suspensions were combined and heated under reflux for 90 min. The hot reaction solution was filtered, and the pure ligand crystallized upon cooling to r.t. The precipitate was removed from the reaction mixture and the solution was stored at −20 °C. The low amount of precipitate was removed and the solution was evaporated to dryness. Then, the residue was suspended in 5 mL of toluene and filtered. The filter cake was washed with 5 mL of toluene and the combined toluene solutions were stored at −40 °C. The precipitate was collected and dried in vacuum giving 700 mg of a colorless powder (15.3%). Then, the powder was redissolved in 5 mL of THF, and slow diffusion of anhydrous n-pentane into the THF solution led to crystallization of the thf adduct 4. After several hours, 110 mg of crystalline 4 (2.4%) was isolated and proved to be suitable for X-ray diffraction experiments These crystals contained intercalated solvents. Further attempts to increase the yield were not successful because this complex underwent THF degradation processes that led to an increasing number of resonances in the 31P NMR spectrum. The elemental analysis gave no reliable results due to uncontrolled loss of intercalated solvent molecules and probably also ligated THF. IR (ATR, cm–1): 3051 w, 2913 w, 1475 m, 1435 s, 1391 m, 1307 w, 1270 w, 1208 m, 1186 s, 1145 w, 1121 s, 1105 s, 1082 m, 1064 s, 1028 m, 993 m, 957 w, 901 m, 847 m, 785 w, 745 m, 723 s, 693 vs, 648 m, 580 m, 571 m, 535 vs, 475 s, 429 m. 31P NMR (162 MHz, 293 K, [D8]THF): δ 8.3. 1H NMR (400 MHz, 293 K, [D8]THF): δ 8.00–6.76 (m, Ph, 10H), 6.49 (s, CHMes, 2H), 2.03 and 1.91 (o-MeMes, 6H), 1.61 and 1.60 (p-MeMes, 3H); the spectrum contained resonances of intercalated solvent.
X-ray Structure Determination
The intensity data for the compounds were collected on a Nonius KappaCCD diffractometer using graphite-monochromated Mo-Kα radiation. Data were corrected for Lorentz and polarization effects; absorption was taken into account on a semi-empirical basis using multiple-scans.18−20 The structures were solved by intrinsic phases (SHELXT) and refined by full-matrix least squares techniques against Fo2.21 The hydrogen atoms bonded to the amine groups N1A and N1F of compounds 1 and 2 were included at calculated positions with fixed thermal parameters. All other hydrogen atoms were included at calculated positions with fixed thermal parameters. All non-hydrogen atoms were refined anisotropically.21 The structure of 4 was refined as a merohedral inversion twin. The crystals of 1, 2, 3, and 4 contain large voids, filled with disordered solvent molecules. The sizes of the voids are 475, 1675, 8544, and 362 Å3/unit cell, respectively. Their contribution to the structure factors was secured by back-Fourier transformation using the SQUEEZE routine of the program PLATON22 resulting in 103, 445, 3729, and 201 electrons/unit cell, respectively. Crystallographic data as well as structure solution and refinement details are summarized in Table S1 (see the Supporting Information). The program packages XP23 and POV-Ray24 were used for structure representations.
Acknowledgments
We acknowledge the valuable support of the NMR service platform (www.nmr.uni-jena.de) of the Faculty of Chemistry and Earth Sciences of the Friedrich Schiller University Jena, Germany.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.1c03732.
Accession Codes
Crystallographic data (excluding structure factors) has been deposited with the Cambridge Crystallographic Data Centre as supplementary publication CCDC-2022917 for 1, CCDC-2089848 for 2, CCDC-2022918 for 3, and CCDC-2022919 for 4. Copies of the data can be obtained free of charge on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK [E-mail: deposit@ccdc.cam.ac.uk].
The authors declare no competing financial interest.
Supplementary Material
References
- a Katzin L. I.; Mason G. W.; Peppard D. F. Infrared studies of binding and structure in monobasic organophosphorus acids and their salts. Spectrochim. Acta, Part A 1978, 34, 51–56. 10.1016/0584-8539(78)80185-8. [DOI] [Google Scholar]; b For recent reviews see e.g.:Montchamp J.-L. Phosphinate chemistry in the 21st century: a viable alternative to the use of phosphorus trichloride in organophosphorus synthesis. Acc. Chem. Res. 2014, 47, 77–87. 10.1021/ar400071v. [DOI] [PubMed] [Google Scholar]; c Tieqiao C.; Han L.-B. Optically active H-phosphinates and their stereospecific transformations into optically active P-stereogenic organophosphoryl compounds. Synlett 2015, 26, 1153–1163. 10.1055/s-0034-1379996. [DOI] [Google Scholar]; d Goura J.; Chandrasekhar V. Molecular metal phosphonates. Chem. Rev. 2015, 115, 6854–6965. 10.1021/acs.chemrev.5b00107. [DOI] [PubMed] [Google Scholar]; e Montchamp J.-L. Challenges and solutions in phosphinate chemistry. Pure Appl. Chem. 2019, 91, 113–120. 10.1515/pac-2018-0922. [DOI] [Google Scholar]
- Al-Shboul T. M. A.; Volland G.; Görls H.; Krieck S.; Westerhausen M. Oxidation Products of Calcium and Strontium Bis(diphenylphosphanide). Inorg. Chem. 2012, 51, 7903–7912. 10.1021/ic300975s. [DOI] [PubMed] [Google Scholar]; a for a review see:Westerhausen M.; Krieck S.; Langer J.; Al-Shboul T. M. A.; Görls H. Phosphanides of calcium and their oxidation products. Coord. Chem. Rev. 2013, 257, 1049–1066. 10.1016/j.ccr.2012.06.018. [DOI] [Google Scholar]
- Rood J. A.; Huttenstine A. L.; Schmidt Z. A.; White M. R.; Oliver A. G. Linear alkaline earth metal phosphinate coordination polymers: synthesis and structural characterization. Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater. 2014, 70, 602–607. 10.1107/s2052520614004247. [DOI] [PubMed] [Google Scholar]
- Koch A.; Krieck S.; Görls H.; Westerhausen M. Reduction of bromo- and iodo-2,6-bis(diphenylphosphanylmethyl)benzene with magnesium and calcium. Inorganics 2016, 4, 39. 10.3390/inorganics4040039. [DOI] [Google Scholar]
- a Goud E. V.; Sivaramakrishna A.; Vijayakrishna K. Aminophosphine oxides: a platform for diversified functions. Top. Curr. Chem. 2017, 375, 10–51. 10.1007/s41061-016-0090-7. [DOI] [PubMed] [Google Scholar]; b Kumar C. A.; Panda T. K. Recent development of aminophosphine chalcogenides and boranes as ligands in s-block metal chemistry. Phosphorus, Sulfur Silicon Relat. Elem. 2017, 192, 1084–1101. 10.1080/10426507.2017.1322967. [DOI] [Google Scholar]
- a Alajarín M.; López-Leonardo C.; Llamas-Lorente P. The chemistry of phosphinous amides (aminophosphanes): old reagents with new applications. Top. Curr. Chem. 2005, 250, 77–106. 10.1007/b100982. [DOI] [Google Scholar]; b Fei Z.; Dyson P. J. The chemistry of phosphinoamides and related compounds. Coord. Chem. Rev. 2005, 249, 2056–2074. 10.1016/j.ccr.2005.03.014. [DOI] [Google Scholar]
- a Fleischer R.; Stalke D. Syntheses and structures of [(THF)nM{NSiMe3)2PPh2}2] complexes (M = Be, Mg, Ca, Sr, Ba; n = 0-2): deviation of alkaline earth metal cations from the plane of an anionic ligand. Inorg. Chem. 1997, 36, 2413–2419. 10.1021/ic9613117. [DOI] [PubMed] [Google Scholar]; b Wingerter S.; Pfeiffer M.; Murso A.; Lustig C.; Stey T.; Chandrasekhar V.; Stalke D. Phosphorus-based ambidentate chelating ligands: pyridyl-N- and imido-N-metal coordination in the Py2P(NSiMe3)2 anion. J. Am. Chem. Soc. 2001, 123, 1381–1388. 10.1021/ja0023736. [DOI] [Google Scholar]
- De Bolster M. W. G.; Groeneveld W. L. The coordination chemistry of phosphoryl compounds containing the −N(CH3)2 group, part VIII: complexes of bisphenyldimethylaminophosphine oxide. Z. Naturforsch., B: Anorg. Chem., Org. Chem. 1972, 27, 759–763. 10.1515/znb-1972-0706. [DOI] [Google Scholar]
- Naktode K.; Kottalanka R. K.; Jana S. K.; Panda T. K. Synthesis and crystal structures of sodium and calcium complexes with the ligand N-(2,6-dimethylphenyl)diphenylphosphinic amide. Z. Anorg. Allg. Chem. 2013, 639, 999–1003. 10.1002/zaac.201300005. [DOI] [Google Scholar]
- Li Z.; Shang D.; Guo J. Bis[1,3-bis(diphenylphosphinoylimino)isoindolinato-κ3O,N,O’]calcium(II). Acta Crystallogr., Sect. E: Struct. Rep. Online 2008, 64, m160. 10.1107/s1600536807065440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schönherr P. R. W.; Hempel N. A.; Görls H.; Krieck S.; Westerhausen M. Magnesiated and calciated N-mesityl diphenylphosphinic amides. Eur. J. Inorg. Chem. 2020, 2020, 1902–1905. 10.1002/ejic.202000164. [DOI] [Google Scholar]
- Morales-Juárez J.; Cea-Olivares R.; Moya-Cabrera M. M.; Jancik V.; García-Montalvo V.; Toscano R. A. Oxidative degradation of ethers promoted by strontium and barium tetraphenylimidodiphosphinates. Inorg. Chem. 2005, 44, 6924–6926. 10.1021/ic050724h. [DOI] [PubMed] [Google Scholar]
- a Kottalanka R. K.; Naktode K.; Anga S.; Nayek H. P.; Panda T. K. Heavier alkaline earth metal complexes with phosphinoselenoic amides: evidence of direct M-Se contact (M = Ca, Sr, Ba). Dalton Trans. 2013, 42, 4947–4956. 10.1039/c2dt32339g. [DOI] [PubMed] [Google Scholar]; b Naktode K.; Bhattacharjee J.; Das Gupta S.; Pada Nayek H.; Mallik B. S.; Panda T. K. Unprecedented calcium metal-macrocycle having phosphinoselenoic amide and diphenylphosphinate in the coordination sphere. Z. Anorg. Allg. Chem. 2014, 640, 994–999. 10.1002/zaac.201300492. [DOI] [Google Scholar]
- Bîrzoi R. M.; Lungu D.; Jones P. G.; Bartsch R.; du Mont W.-W. Sterically tuned P-phosphanylamino phosphaalkenes (Me3Si)2C=PN(R)PPh2 and (iPrMe2Si)2C=PN(R)PPh2. Z. Anorg. Allg. Chem. 2018, 644, 381–390. 10.1002/zaac.201700446. [DOI] [Google Scholar]
- a Westerhausen M. Synthesis and spectroscopic properties of the bis(trimethylsilyl)amides of the alkaline earth metals magnesium, calcium, strontium and barium. Inorg. Chem. 1991, 30, 96–101. 10.1021/ic00001a018. [DOI] [Google Scholar]; b Westerhausen M.; Hartmann M.; Makropoulos N.; Wieneke B.; Wieneke M.; Schwarz W.; Stalkec D. Synthese von Erdalkalimetallocenen aus Erdalkalimetall- bis[bis(trimethylsilyl)amid] und 6-Methyl-6-phenylfulven/Synthese von Erdalkalimetallocenen aus Erdalkalimetallbis[ bis(trimethylsilyl)amid] und 6-Methyl-6-phenylfulven Synthesis of Alkaline Earth Metallocenes from Alkaline Earth Metal Bis[bis(trimethylsilyl)amide] and 6 -Methyl-6 -phenylfulvene. Z. Naturforsch., B: J. Chem. Sci. 1998, 53, 117–125. 10.1515/znb-1998-0120. [DOI] [Google Scholar]; c For reviews see:Westerhausen M. Dynamic behavior and structures of alkaline earth metal bis[bis(trialkylsilyl)amides] and -phosphanides]. Trends Organomet. Chem. 1997, 2, 89–105. [Google Scholar]; d Westerhausen M. Synthesis, properties, and reactivity of alkaline earth metal bis[bis(trialkylsilyl)amides]. Coord. Chem. Rev. 1998, 176, 157–210. 10.1016/s0010-8545(98)00116-7. [DOI] [Google Scholar]
- Krieck S.; Schüler P.; Peschel J.; Westerhausen M. Straightforward one-pot syntheses of silylamides of magnesium and calcium via an in situ Grignard metalation method. Synthesis 2019, 51, 1115–1122. 10.1055/s-0037-1610407. [DOI] [Google Scholar]
- Vaartstra B. A.; Huffman J. C.; Streib W. E.; Caulton K. G. Syntheses and structures of a series of very low coordinate barium compounds: Ba[N(SiMe3)2]2(THF)2, {Ba[N(SiMe3)2]2(THF)}2, and {Ba[N(SiMe3)2]2}2. Inorg. Chem. 1991, 30, 121–125. 10.1021/ic00001a023. [DOI] [Google Scholar]
- Hooft R.COLLECT, Data Collection Software; Nonius B.V.: Netherlands, 1998.
- Otwinowski Z.; Minor W. [20] Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997, 276, 307–326. 10.1016/s0076-6879(97)76066-x. [DOI] [PubMed] [Google Scholar]
- SADABS 2.10; Bruker-AXS Inc.: Madison, WI, USA, 2002.; b SADABS 2016/2:Krause L.; Herbst-Irmer R.; Sheldrick G. M.; Stalke D. Comparison of silver and molybdenum microfocus X-ray sources for single-crystal structure determination. J. Appl. Crystallogr. 2015, 48, 3–10. 10.1107/s1600576714022985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldrick G. Crystal structure refinement with SHELXL. Acta Crystallogr., Sect. C: Struct. Chem. 2015, 71, 3–8. 10.1107/S2053229614024218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spek A. L. PLATON SQUEEZE: a tool for the calculation of the disordered solvent contribution to the calculated structure factors. Acta Crystallogr., Sect. C: Struct. Chem. 2015, 71, 9–18. 10.1107/s2053229614024929. [DOI] [PubMed] [Google Scholar]
- XP, Siemens Analytical X-ray Instruments Inc., Karlsruhe, Germany, 1990.
- POV-Ray Persistence of Vision Raytracer, Victoria, Australia, 2007.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.