

Abstract
Experimental evidence suggests that DNA-mediated redox signaling between high-potential [Fe4S4] proteins is relevant to DNA replication and repair processes, and protein-mediated charge transfer (CT) between [Fe4S4] clusters and nucleic acids is a fundamental component of the signaling and repair mechanisms. We analyzed the dominant CT pathways in the base excision repair glycosylase MutY using molecular dynamics (MD) simulations and hole hopping pathway analysis. We find that the adenine nucleobase of the mismatched A·oxoG DNA base pair facilitates [Fe4S4]-DNA CT prior to adenine excision by MutY. We also find that the R153L mutation in MutY (linked to colorectal adenomatous polyposis) influences the dominant [Fe4S4]-DNA CT pathways and appreciably decreases their effective CT rates.
Graphical Abstract
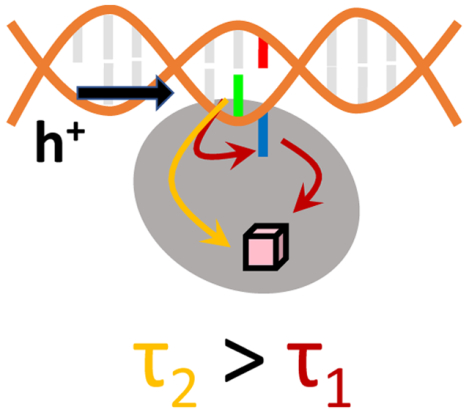
1. Introduction
CT processes are ubiquitous in cell biology, where they are used both to drive reactions1 and to convey signals.2 In protein-nucleic acid complexes, the protein medium can support charge transport between a redox cofactor and the nucleic acid duplex for purposes that might range from detection/repair of DNA defects2–5 to signaling between proteins for coordination of their activity.6–9 In these contexts, the π-stacking of DNA nucleobases allow the charge to propagate over distances up to the nanometer scale.10 CT also governs redox signaling that occurs in response to changes in reactive oxygen species (ROS) or reactive nitrogen species (RNS) levels in the cellular system.11–12 Redox signaling processes are paramount in the regulation of major cellular pathways. Structural changes in proteins and/or DNA can alter CT pathways, dysregulate cellular signaling, and thus result in the over proliferation of cells, contributing to human diseases like cancer.2, 13
DNA-mediated CT between high potential [Fe4S4]2+/3+ proteins is hypothesized as a means of coordinating DNA replication and repair.2–3 High-potential [Fe4S4] clusters can play a structural role, and experiments4, 6 suggest that these clusters are involved in the regulation of enzyme activity.14–15 Alternation between the reduced and oxidized states of a high-potential [Fe4S4] cofactor via DNA-mediated charge transport may synchronize the binding and unbinding events required for the function of [Fe4S4] proteins involved in DNA repair and replication. These events are believed to be governed to a great extent by electrostatic interactions in which a protein containing a [Fe4S4]3+ cluster is electrostatically more strongly attracted and binds more tightly to the negatively charged DNA backbone than a [Fe4S4]2+-containing protein.16 Anaerobic microscale thermophoresis indicates that the DNA-protein binding affinity increases by 550-fold in the presence of [Fe4S4]3+.16 This increased DNA binding also induces a shift of the [Fe4S4]2+/3+ redox couple to ca. +80 mV vs NHE.7, 17–18
Redox signaling between two [Fe4S4] proteins linked by a DNA duplex requires DNA-mediated charge transport, as well as protein-mediated charge transport between the iron-sulfur clusters and the duplex. Recent studies indicate the unidirectional nature of CT between [Fe4S4] clusters and nucleic acid duplexes,19 and the general feasibility of charge transport through a protein on μs-to-ms timescales by a multi-step hopping mechanism.19–20 Moreover, mutation effects on the preferential charge hopping routes through proteins were described.20 These findings motivate studies into the functional role of CT in protein-nucleic acid complexes, with the aim of providing insights into possible connections between mutation and health consequences.
Within the above context, base excision repair (BER) glycosylases are a class of [Fe4S4]2+/3+-containing proteins whose interface with DNA offers a rich medium for functional CT, including intra-DNA CT as well as direct and protein-mediated CT between the DNA and the [Fe4S4] cluster. Electrochemistry and atomic force microscopy experiments indicate that DNA-[Fe4S4] CT in these proteins is facilitated by the positioning of the enzyme in proximity to the base pair mismatch.4, 18, 21–22 One of the most studied BER enzymes is MutY,23–25 which excises an adenine residue (DA18 in Figure 1) from a mismatched A·oxoG base pair. This base mismatch arises in the presence of oxidative stress, when a guanine nucleobase is oxidized and a DNA polymerase inserts an adenine rather than a cytosine on the complementary strand. The excision of adenine by MutY produces an abasic site and a free adenine nucleobase. Repair polymerases and MutM eventually convert the abasic site and oxoG into the correct C·G base pair.
Figure 1.
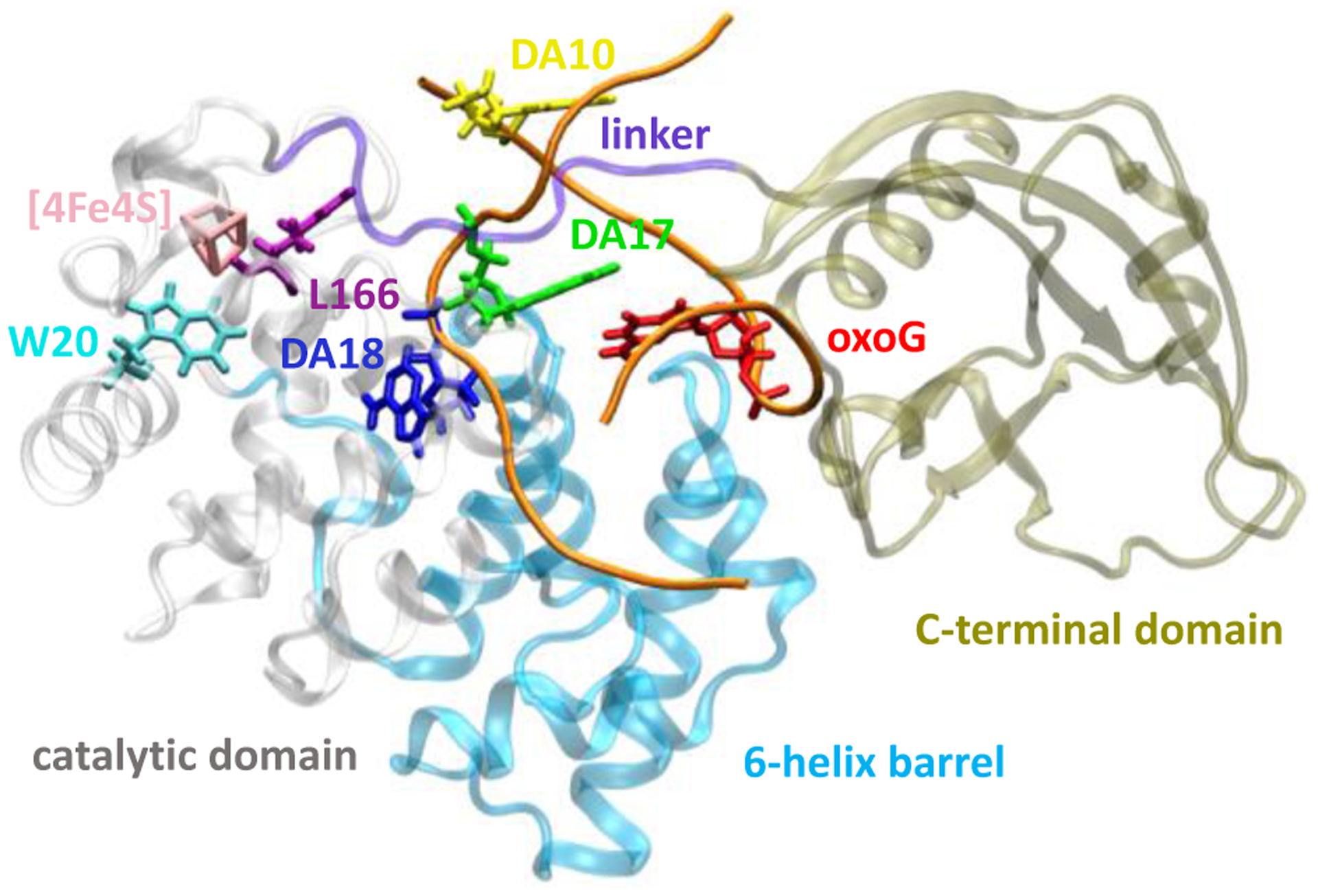
Structure of the MutY-DNA complex (PDB file 1RRQ23) containing the A·oxoG mismatched pair. The sugar-phosphate backbone of DNA is colored in orange.
In the MutY-DNA complex (PDB file 1RRQ; see Figure 1),23 the [Fe4S4] cluster is located in the catalytic domain, while the DNA is sandwiched between the catalytic and C-terminal domains. The adenine nucleobase of the A·oxoG pair (i.e., DA18, Figure 1) is extruded from the DNA double helix and extends into the catalytic domain. This extrusion is also observed in other DNA glycosylase complexes that repair single-base lesions.23 The nucleobase extrusion is attributed to the transition of the N-glycosidic bond of oxoG from a syn conformation to an anti conformation when the DNA binds to MutY (Figure 1).23 In the anti conformation, oxoG clashes sterically with DA18, thus promoting its extrusion. As DA18 fails to π-stack with other nucleobases and extends into the protein, DA18 could serve as a bridge site in the DNA-[Fe4S4] CT pathway, rather than as a terminal donor/acceptor site. Another adenine nucleobase (DA17) in the base pair next to A·oxoG (Figs. 1 and 2) seems to be a good nucleobase candidate for oxidizing the [Fe4S4]2+ cluster because of the DA17-cluster edge-to-edge distance of 17.7 Å. The next shortest distance between the cluster and other nucleobases in the double-stranded DNA is 20.0 Å (DA10-cluster).
Figure 2.
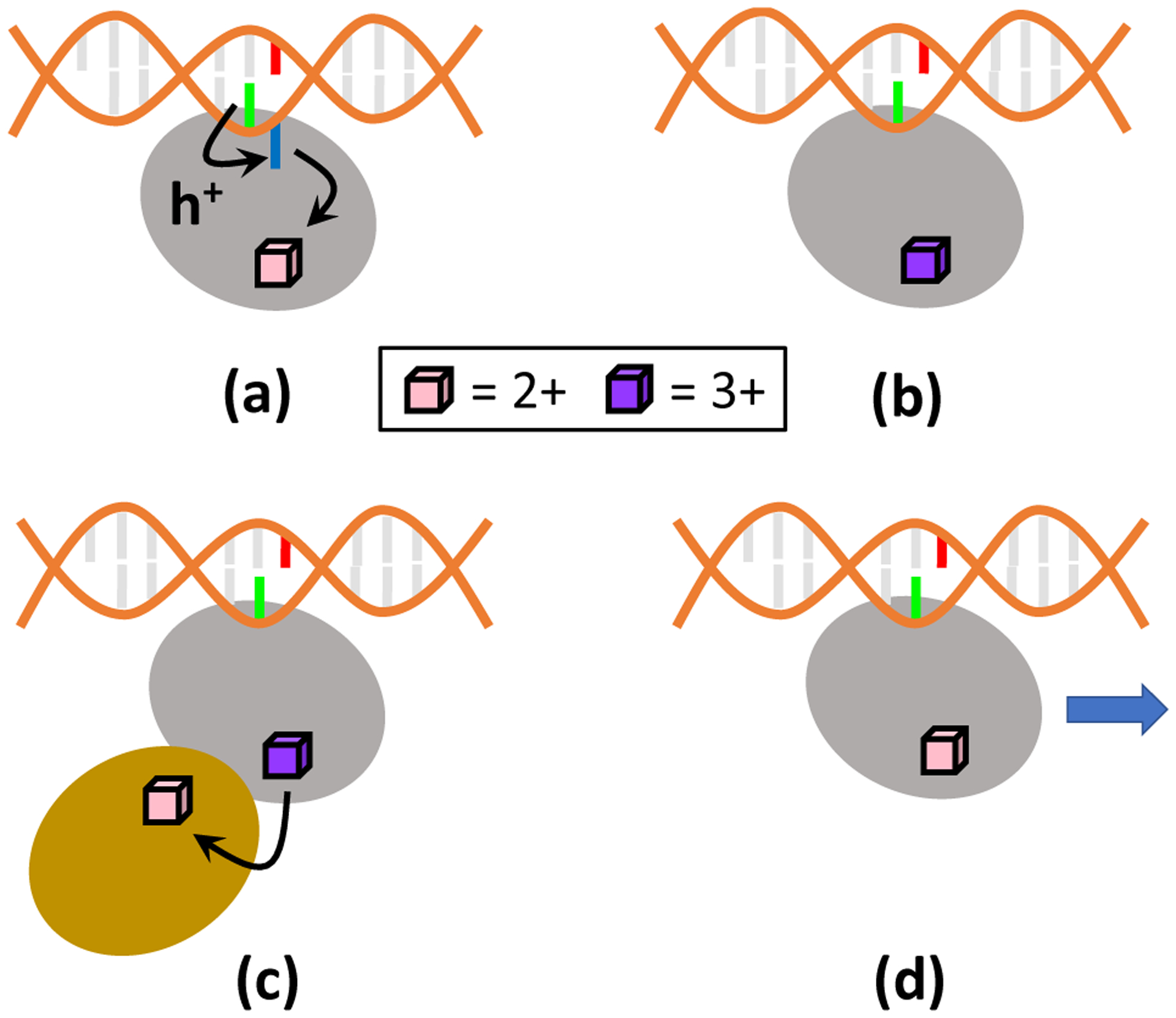
Proposed mechanism for the excision of mismatched DNA bases by MutY. The mechanism is substantially based on CT between the DNA and the [Fe4S4] cluster, and proceeds through the following steps: a) The oxidized nucleobase DA17 transfers the hole to [Fe4S4]2+ via a hopping pathway involving DA18, thus leading to [Fe4S4]3+. b) The DNA-bound MutY excises DA18, as part of the DNA repair process. c) CT between [Fe4S4]3+ in MutY and [Fe4S4]2+ in a partner repair protein may reduce [Fe4S4]3+ to [Fe4S4]2+. d) The absence of DA18 prevents the cluster from being easily re-oxidized by a nucleobase. The [Fe4S4]2+-containing MutY is thus able to slide along the DNA and search for further base mismatches. Color code: MutY (Grey), oxoG (red), DA17 (green), DA18 (blue), [Fe4S4] partner protein (gold).
While the feasibility of [Fe4S4]-DNA CT in the MutY complex was demonstrated experimentally,24–25 in this study we probe mechanistic characteristics of this CT process using MD simulations and an improved version of the EHPath code that identifies charge hopping pathways and assesses their corresponding transit times.19 We streamlined the prior hopping pathway search code19 to enable CT analysis of multiple MD trajectories using either a local computer or a computer cluster. The CT analysis described here assumes that signaling/repair by MutY is triggered by transport of a hole from an initially oxidized DNA molecule (i.e., a structure under oxidative stress) to the [Fe4S4] cluster. We found that the DA17 nucleobase is the most likely hole donor based on constraints of thermodynamics and distances that determine the CT kinetic timescales.26 It is thought that the change in the [Fe4S4] cluster charge (after oxidation) can oxidize a partner repair protein, which in turn allows the now-reduced MutY to slide along the DNA and identify other base mismatches.4, 24–25 Here, we propose a mechanistic interpretation of base excision repair by MutY-DNA based on the CT between the [Fe4S4] cluster and the DNA at the MutY-DNA interface. The proposed mechanism unifies the knowledge gained from the CT hopping pathway analysis with current understanding of the DNA repair process. Furthermore, we describe the influence of the R153L mutation in the MutY protein (which is associated with colorectal adenomatous polyposis27) on the pathways and transit times for CT between the [Fe4S4] cluster contained in the protein and the bound DNA.
2. Methods
2.1. System modeling.
The structure of the MutY-DNA complex was drawn from the PDB file 1RRQ.23 The missing residues (i.e., 230–233 and 288–291) were added using the MODELLER program28. Since the crystal structure contains several mutations (D144N, P164C, F347S, and K357E) that helped to stabilize the A·oxoG complex and amplify its expression, these mutated residues were replaced with the original residues using PyMOL, while our attention was focused on the effect of the R153L mutation.29 Moreover, while a study of Verdine et. al30 indicates that the D144N mutation may prevent full engagement of DA18 with MutY, we decided to use PDB 1RRQ as it contains unmodified DNA (versus the fluorinated DNA in PDB 3G0Q30). The resulting structure was used in the MD simulations.
2.2. MD simulations.
MD simulations (using NAMD 2.1131) of the wild-type (WT) MutY-DNA complex were performed using the A2+ and A3+ force fields (FFs) developed in ref. 20 for the [Fe4S4]2+ and [Fe4S4]3+ clusters. The [Fe4S4] cluster in MutY changes from its 2+ oxidation state to the 3+ state as it receives a hole from an oxidized nucleobase. As in other protein-DNA complexes,20 the timescale for CT between the nucleobase and [Fe4S4] is much slower than the accessible simulation timescale (vide infra). Ideally, the FFs used in the MD simulation should be updated as the charge hops from the hole donor in the DNA to the iron-sulfur cluster. As we recently observed,20 a more practical MD approach is to analyze the dynamics of the protein-DNA complex using both the A2+ and the A3+ FFs,20 while reactive FFs that allow for bond-breaking and formation were not considered for the purposes of this study that focuses on electron-hole transfer.32 The FF parameters for the oxoG moiety were drawn from ref.33. AMBER FFs ff14SB34 and ff99-bsc035–36 were used for the rest of the protein and DNA. We also mutated Arg153 to Leu and simulated the mutated MutY-DNA complex using the A2+ and A3+ FFs. The R153L mutation in the Bacillus stearothermophilus protein drawn from the 1RRQ23 PDB file corresponds to the R231L mutation that in the human homolog (hMYH) is associated with colorectal adenomatous polyposis.27
For each simulation, Na+ ions were added to neutralize the biomolecular system and TIP3P water was used to solvate the protein-DNA complex, extending 10 Å on each side of the unit cell. The resulting unit cell had a size of 72 × 79 × 101 Å3. We used the SHAKE algorithm to constrain the interatomic distances (H-O and H-H) in the water molecules.37 The electrostatic interaction energy was calculated every 2 MD time steps using the particle mesh Ewald summation method,38 with a grid spacing of 1 Å and a scaling factor of 0.833333 for 1–4 interactions. For non-bonded atomic pairs, the cutoff distance for the periodic calculation of their interaction energy was set to 14 Å, and the van der Waals interactions were truncated at 12 Å.
We conducted 8 × 104 steps of energy minimization, 150 ps of solvent equilibration at 298 K (namely, the crystallization temperature of the 1RRQ structure) using a Langevin thermostat with a damping coefficient of 1.0 ps−1. During this equilibration, the coordinates of the protein-nucleic acid complex were fixed. We next carried out another 125 ps of equilibration at 298 K, releasing all atoms in the protein-DNA complex. This equilibration was followed by another 1.5 ns of equilibration at constant temperature (298 K) and pressure (1 atm), using a Nosé-Hoover Langevin piston pressure control39–40 with a piston period of 100 fs, a damping coefficient of 2.0 ps−1, and a barostat damping time of 50 fs. Each MD production run lasted 60 ns, with a time step of 0.5 fs. The RMSDs along the MD trajectories are shown in Figure S1. Snapshots within the 10–60 ns time window were extracted every 0.5 ns to analyze the CT pathways between the [Fe4S4] cluster and DNA using EHPath_multirun.py (see github.com/etransfer/EHPath).
2.3. Kinetic modeling.
The CT rate constant k for each CT step was calculated using a non-adiabatic CT rate expression with Marcus’ high-temperature Franck-Condon factor,41
| (1) |
In eq 1, V is the electronic coupling between the initial and final electronic states, λ is the reorganization energy, ΔG° is the reaction free energy, and T is the temperature (in our study, T = 298 K). In the EHPath (and the updated EHPath_multirun.py) code,19 V is obtained from the charge donor-acceptor distance using a square-tunneling barrier model, λ is obtained using Marcus’ two-sphere model41–43 and ΔG° is approximated as the difference in the donor and acceptor redox potentials (see refs19, 42 for further details regarding the CT parameters).
Depending on the donor-acceptor distance in a given molecular conformation, the excess charge can either tunnel directly from the donor to the terminal acceptor site or it can follow a multi-step hopping pathway. Both charge transport processes are consistently described by birth-and-death kinetic models.44–46 As described previously, either the final charge acceptor behaves as an absorber42 or is in contact with a charge drain,8 the overall mean residence time τ of the charge in a path (the charge transit time) can be written in the compact form8 (a different derivation for the kinetic model with an absorber appears in ref.45)
| (2) |
This transit time is implemented in EHPath_multirun.py.19 N is the total number of hopping sites in the pathway and kn→n±1 is the rate constant for CT between nearest-neighbour redox sites n and n ± 1. n = 0 denotes the initial charge donor, n = 1 to N denotes the bridge sites, and site n = N + 1 is the terminal charge acceptor. The forward and reverse CT rate constants obtained with eq. 1 for each nearest-neighbor hop were used in eq 2. In our kinetic analysis, only the forward rate was considered for the CT step between site n = N and the terminal acceptor site. That is, the final CT step is irreversible. This irreversibility models rapid scavenging of the excess charge by redox agents in the cell42 or trapping of the charge on the terminal acceptor.20, 42–43
EHPath_multirun calculates τ for electron or hole transport pathways. With EHPath_multirun, one can either treat [Fe4S4]2+ as the electron donor and a nucleobase as the electron acceptor, or treat a nucleobase as the hole donor and [Fe4S4]2+ as the hole acceptor. These two options (with their different approximations in describing the individual CT steps and their different boundary conditions20) define kinetic models 1 and 2, respectively, in the following analysis. We investigated both electron and hole hopping pathways between [Fe4S4] and DNA using these two models (Section 3).
3. Results and discussion
3.1. Charge transport in the MutY-DNA complex.
With both choices of kinetic models and of FFs, we find that the dominant CT pathway between [Fe4S4] and DNA (namely, the fastest hole hopping route in most of the selected system’s snapshots) contains DA18 as a bridge site and DA17 as the hole donor in kinetic model 1 or as the electron acceptor in kinetic model 2 (see Table 1). Although DA17 could donate the hole to oxoG, since the reduction potential of oxoG is 0.74 V vs NHE,47 this hole can easily be “trapped” at oxoG, due to the higher reduction potentials of the adjacent purines. In the presence of an oxidative environment, it is very likely that another hole will arrive at DA17 through DNA hole transfer and proceed to other bridge sites such as DA18. In fact, all pathways (with the exception of 1% of the pathways for kinetic model 2 and A2+) contain DA18, and all hole (electron) hopping routes begin (end) at DA17. W20 is found to serve as a bridge hopping site in some of the CT pathways (Table 1), due to its proximity to [Fe4S4] and DA18 (Figure 1, Table S1 and S2), as well as its favorable energy landscape for CT. In fact, the oxidation potential of the Trp residue lies between the [Fe4S4] and adenine oxidation potentials.42 We note that a higher percentage of CT pathways involves W20 when the A2+ FF is used in the MD simulation (19.8%) than when A3+ is used (5.0%) (Table 1). Since the CT rate constant k in eq 1 drops exponentially with the donor-acceptor distance (due to the electronic coupling factor), and the reorganization energy grows with distance, this difference in the W20 contribution to the charge transport may be attributed to the larger W20-DA18 distance (15.8 Å, see Table S1) in the A3+ MD simulation compared to the A2+ simulation (15.0 Å). This increased distance produces a smaller W20-DA18 CT rate, so that other CT pathways that do not involve W20 are kinetically more favorable. Aside from this difference, the same qualitative picture emerges from the A2+ and A3+ simulations. In particular, the CT analysis of both MD simulations indicates that DA18 is an essential hopping site for charge transport between the DNA and the iron-sulfur cluster.
Table 1.
Strongest CT pathways computed for the WT MutY-DNA complex, using A2+ and A3+ to describe the iron-sulfur cluster in the MD simulation.
| FF | Kinetic model | CT pathways | % |
|---|---|---|---|
| A2+ | 1 | [Fe4S4]-DA18-DA17 | 80.2 |
| [Fe4S4]-W20-DA18-DA17 | 19.8 | ||
| 2 | DA17-DA18-[Fe4S4] | 79.2 | |
| DA17-DA18-W20-[Fe4S4] | 19.8 | ||
| DA17-[Fe4S4] | 1.0 | ||
| A3+ | 1 | [Fe4S4]-DA18-DA17 | 95.0 |
| [Fe4S4]-W20-DA18-DA17 | 5.0 | ||
| 2 | DA17-DA18-[Fe4S4] | 95.0 | |
| DA17-DA18-W20-[Fe4S4] | 5.0 |
The [Fe4S4]-to-DA17 mean transit time averaged over the MD snapshots, 〈τ〉, ranges from 0.7 to 1.7 ms (Table 2), which is within the millisecond (1–4 ms48) half-life of adenine radicals. Therefore, CT to the iron-sulfur cluster provides a viable route for the reduction of adenine.
Table 2.
Average mean residence times 〈τ〉 (in ms) computed for the WT MutY-DNA complex using the selected MD snapshots.
| FF | 〈τ〉kinetic model 1 | 〈τ〉kinetic model 2 |
|---|---|---|
| A2+ | 1.3 ± 1.6 | 1.7 ± 1.9 |
| A3+ | 0.7 ± 0.3 | 0.9 ±0.4 |
To further analyze the role of DA18 in the charge transport chain, we computed the CT pathways between [Fe4S4] and DA17 excluding DA18 as one of the possible bridge sites. Using both kinetic models 1 and 2, and both A2+ and A3+ FFs, we found that the average charge travel time 〈τ〉 increases by about an order of magnitude (〈τ〉 is in the range 11.2–13.6 ms; see Table 3) compared to the value that is obtained including DA17 in the hopping pathway analysis. The comparison of the charge transit times in Tables 2 and 3 with the adenine radical half-life (1–4 ms) indicates that the DA18 base to be excised serves as an essential bridge for sufficiently rapid oxidation of the [Fe4S4]2+ cluster by the DA17 radical cation, prior to DA17 reduction by other redox agents in the cell.
Table 3.
Average mean residence times 〈τ〉 (in ms) for the WT MutY-DNA complex excluding DA18 from the CT pathways.
| FF | 〈τ〉kinetic model 1 | 〈τ〉kinetic model 2 |
|---|---|---|
| A2+ | 11 ± 14 | 14 ± 16 |
| A3+ | 11 ± 10 | 11 ±10 |
The extrusion of DA18, upon DNA recognition by MutY, appears to optimally position DA18 both to serve as a key charge hopping site for DNA reduction/[Fe4S4] cluster oxidation on a millisecond timescale, and also to enable the base excision by the catalytic domain. The computed difference in 〈τ〉 with and without DA18 suggests a possible mechanism for the nucleobase excision preceding DNA repair. As MutY is near the mismatched A·oxoG pair, the [Fe4S4]2+ cluster of MutY is oxidized to the 3+ state by DA17, via DA18 (Figure 2a). The oxidation of the iron-sulfur cluster is expected to strengthen the MutY-DNA binding,7 thus favoring the excision of the mismatched DA18 nucleobase by MutY’s catalytic domain (Figure 2b). Then, the [Fe4S4]3+ cluster might be reduced to [Fe4S4]2+ by a partner protein (e.g., a nearby [Fe4S4]-containing repair polymerase that is then responsible for inserting the correct nucleobase at the excision site3, 49) (Figure 2c). The switch in [Fe4S4] redox state would weaken the MutY-DNA binding and thus allow MutY to slide along the DNA duplex and continue its search for other base-pair mismatches (Figure 2d). Furthermore, since the timescale for an initially oxidized nucleobase to transfer the hole to the [Fe4S4] cluster is larger without DA18, the excision of DA18 by MutY also discourages another hole on DNA from re-oxidizing the [Fe4S4] cluster (which would hinder the protein sliding) until the next DNA mismatch is encountered. In vitro experiments would be necessary to test the proposed mechanism, since it involves the coordination between MutY and its partner protein. In particular, future experiments should verify that the reduction of the [Fe4S4]3+ cluster of MutY does not occur before the excision of DA18.
3.2. Mutation effect on MutY-DNA charge transport.
The R153L mutation (associated with colorectal adenomatous polyposis27) influences the occurrences and transit times of the [Fe4S4]-DNA CT pathways. However, the computed mutation effects depend somewhat on the FF used for the [Fe4S4] cluster.
In the MD simulations of the mutated protein complex using the A2+ FF, the dominant CT route is either direct [Fe4S4]-DA17 tunneling (kinetic model 1) or DA17-DA18-W20-[Fe4S4] (kinetic model 2) (see Table 4). In contrast, [Fe4S4]-DA18-DA17 (kinetic model 1) and, conversely, DA17-DA18-[Fe4S4] (kinetic model 2) were the dominant CT pathways found for the WT protein-DNA complex using the same A2+ FF (and, indeed, also using the A3+ FF; see Table 1). These differences in CT pathways are consistent with the fact that the average [Fe4S4]-DA18 distance increases from 18.8 Å in the WT protein complex to 20.4 Å in the R153L protein complex (Tables S1 and S2), while the average [Fe4S4]-DA17 distance only experiences a modest increase from 22.0 Å to 22.2 Å upon mutation. The mutation also leads to the observation of direct CT between the cluster and DA10, which was not found in the WT complex. This observation is consistent with the decrease in the average [Fe4S4]-DA10 distance from 22.3 Å to 21.6 Å upon mutation (Table S2).
Table 4.
Distribution of CT pathways computed for the R153L MutY-DNA complex, using A2+ and A3+ to describe the iron-sulfur cluster in the MD simulation.
| FF | Kinetic model | CT pathways | % |
|---|---|---|---|
| A2+ | 1 | [Fe4S4]-DA17 | 77.2 |
| [Fe4S4]-DA18-DA17 | 14.9 | ||
| [Fe4S4]-DA10 | 6.9 | ||
| [Fe4S4]-W20-DA18-DA17 | 1.0 | ||
| 2 | DA17-DA18-W20-[Fe4S4] | 82.2 | |
| DA17-DA18-[Fe4S4] | 16.8 | ||
| DA17-[Fe4S4] | 1.0 | ||
| A3+ | 1 | [Fe4S4]-DA18-DA17 | 80.2 |
| [Fe4S4]-W20-DA18-DA17 | 19.8 | ||
| 2 | DA17-DA18-[Fe4S4] | 80.2 | |
| DA17-DA18-W20-[Fe4S4] | 19.8 |
The MD simulation of the mutated system with the A3+ FF leads, instead, to the same dominant CT pathways as in the WT protein complex (compare the bottom panels in Tables 1 and 4). However, the occurrence frequencies and transit times for these pathways change appreciably upon protein mutation. The mutation increases the average distance between the initial charge donor and final acceptor from 21.96 Å to 23.06 Å, with a consequent decrease in the electronic coupling. This change favors more frequent charge hopping through W20 compared to the WT system and, overall, decreases the CT speed (see Table 5). A decrease in the effective CT rate (1⁄τ) is consistently obtained from the MD simulations using the A2+ and A3+ FFs, despite the structural differences that emerge from the two MD simulations and related changes in the CT pathways. It is noteworthy that the structural differences arising in the two MD simulations are relatively small, but they significantly affect the CT routes, primarily because of the exponential dependence of the electronic couplings on the donor-acceptor distances. Since the [Fe4S4]-DA17 and [Fe4S4]-DA18 distances are significantly larger than the DA17-DA18 distance in both the WT and R153L protein complexes, we expect that the A2+ reduced-cluster FF describes the CT system more realistically than the A3+ FF. However, this expectation is mitigated by the transient charge localization on the W20 residue (which is midway between the [Fe4S4] cluster and the DNA) in the dominant CT pathways (see Section 2.2 and ref. 20).
Table 5.
Average mean residence times 〈τ〉 (ms) computed for the R153L MutY-DNA complex across the selected MD snapshots.
| FF | 〈τ〉kinetic model 1 | 〈τ〉kinetic model 2 |
|---|---|---|
| A2+ | 2.0 ± 0.8 | 3.0 ± 2.3 |
| A3+ | 1.9 ± 1.3 | 2.3 ± 1.4 |
Irrespective of the computational approach used, we find that the 〈τ〉 value for the WT protein-DNA system (Table 2) increases by approximately 1 ms upon R153L mutation (cf. Table 5). This increase in 〈τ〉 should be considered cautiously, because of the approximations in the simulations. Nonetheless, this increase may have important mechanistic implications if experimentally validated. To understand the implications, we note that the half-life of adenine radicals ranges from 1 to 4 ms in experiments.48 Thus, if the half-life of the DA17 cation in the local DNA environment is 1 ms for example, the predicted increase in 〈τ〉 upon protein mutation makes CT to the [Fe4S4] cluster non-competitive with other mechanisms for DA17 reduction. In these circumstances, mutation would significantly limit cluster oxidation, thus preventing tight protein-DNA binding, favoring the continuous sliding of R153L MutY along the DNA, and hampering the DA18 excision.
4. Conclusions
We studied functional CT at the interface between the [Fe4S4]-containing BER enzyme MutY and DNA using MD simulation and CT analysis with an enhanced version of our program19 to search for CT hopping pathways on numerous structures derived from MD simulations (EHPath_multirun.py).
After structural refinement and simulation of the protein-DNA complex from the PDB file 1RRQ,23 we found that hole transport occurs preferentially between the [Fe4S4] cluster and the DA17 nucleobase, and that the mismatched DA18 base acts as a critical hole hopping site prior to its excision. DA18 reduces, by an order of magnitude, the time required for the iron-sulfur cluster oxidation, which strengthens the protein-DNA binding and favors the DA18 excision by the catalytic domain.
Our analysis indicates that the R153L mutation in MutY slows the rate of [Fe4S4]2+ oxidation by reduction of DA17. Therefore, in the mutated system, the DA17 radical cation can be more readily reduced by other competing processes in the cell. We argue that the decrease in the [Fe4S4]2+ oxidation rate can be linked to a decreased ability of the R153L mutant to excise DA18, which may allow an interpretation of the relationship between the R153L mutation and colorectal adenomatous polyposis in terms of mutation effects on the functional charge transport at the protein-DNA interface.
Future experiments involving DNA-mediated electrochemical assays could examine possible changes in the DNA-[Fe4S4] charge transit time by exploring different DNA sequences bound to MutY. As the function of MutY is to excise adenine from its mispaired oxoG partner, MutY positions its [Fe4S4]-containing catalytic domain in proximity to the A·oxoG base pair for eventual excision. This invariably increases the likelihood for DNA-[Fe4S4] charge transfer, which may unlikely alter the charge transit time significantly.
Supplementary Material
ACKNOWLEDGMENT
We thank Tomasz Janowski for technical support with the computing cluster at Duke University. This research is supported by the National Institutes of Health (grant GM-48043) and the Blue Waters sustained-petascale computing project (R.D.T.), which is funded by the National Science Foundation (Awards OCI-0725070 and ACI-1238993) and the State of Illinois.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Supporting Information
The supporting information contains the RMSDs of the (wildtype and mutant) MutY-DNA complexes during the MD production run, and the center-to-center distances of the redox pairs obtained using the A3+ and A2+ FFs.
REFERENCES
- (1).Beratan DN; Liu CR; Migliore A; Polizzi NF; Skourtis SS; Zhang P; Zhang YQ Charge Transfer in Dynamical Biosystems, or the Treachery of (Static) Images. Acc. Chem. Res 2015, 48, 474–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Arnold AR; Grodick MA; Barton JK DNA Charge Transport: From Chemical Principles to the Cell. Cell Chem. Biol 2016, 23, 183–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Barton JK; Silva RMB; O’Brien E Redox Chemistry in the Genome: Emergence of the [4Fe4S] Cofactor in Repair and Replication. Annu. Rev. Biochem 2019, 88, 163–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Boal AK; Genereux JC; Sontz PA; Gralnick JA; Newman DK; Barton JK Redox Signaling Between DNA Repair Proteins for Efficient Lesion Detection. Proc. Natl. Acad. Sci. U.S.A 2009, 106, 15237–15242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Sato R; Kitoh-Nishioka H; Ando K; Yamato T Electron Transfer Pathways of Cyclobutane Pyrimidine Dimer Photolyase Revisited. J. Phys. Chem. B 2018, 122, 6912–6921. [DOI] [PubMed] [Google Scholar]
- (6).Grodick MA; Segal HM; Zwang TJ; Barton JK DNA-Mediated Signaling by Proteins with 4Fe–4S Clusters is Necessary for Genomic Integrity. J. Am. Chem. Soc 2014, 136, 6470–6478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).O’Brien E; Holt ME; Thompson MK; Salay LE; Ehlinger AC; Chazin WJ; Barton JK The [4Fe4S] Cluster of Human DNA Primase Functions as a Redox Switch Using DNA Charge Transport. Science 2017, 355, eaag1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Teo RD; Rousseau BJG; Smithwick ER; Di Felice R; Beratan DN; Migliore A Charge Transfer Between [4Fe4S] Proteins and DNA is Unidirectional. Implications for Biomolecular Signaling. Chem 2019, 5, 122–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Beratan DN Why are DNA and Protein Electron Transfer so Different? Annu. Rev. Phys. Chem 2019, 70, 71–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Slinker JD; Muren NB; Renfrew SE; Barton JK DNA Charge Transport Over 34 nm. Nat. Chem 2011, 3, 228–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Di Meo S; Reed TT; Venditti P; Victor VM Harmful and Beneficial Role of ROS. Oxidative Med. Cell. Longev 2016, 2016, 7909186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Schieber M; Chandel NS ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol 2014, 24, R453–R462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Yang WY; Zou LZ; Huang CH; Lei YL Redox Regulation of Cancer Metastasis: Molecular Signaling and Therapeutic Opportunities. Drug Dev. Res 2014, 75, 331–341. [DOI] [PubMed] [Google Scholar]
- (14).Fuss JO; Tsai C-L; Ishida JP; Tainer JA Emerging Critical Roles of Fe–S Clusters in DNA Replication and Repair. Biochim. Biophys. Acta-Mol. Cell Res 2015, 1853, 1253–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Johnson DC; Dean DR; Smith AD; Johnson MK Structure, Function, and Formation of Biological Iron-Sulfur Clusters. Annu. Rev. Biochem 2005, 74, 247–281. [DOI] [PubMed] [Google Scholar]
- (16).Tse ECM; Zwang TJ; Barton JK The Oxidation State of [4Fe4S] Clusters Modulates the DNA-Binding Affinity of DNA Repair Proteins. J. Am. Chem. Soc 2017, 139, 12784–12792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Bartels PL; Zhou A; Arnold AR; Nuñez NN; Crespilho FN; David SS; Barton JK Electrochemistry of the [4Fe4S] Cluster in Base Excision Repair Proteins: Tuning the Redox Potential with DNA. Langmuir 2017, 33, 2523–2530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Gorodetsky AA; Boal AK; Barton JK Direct Electrochemistry of Endonuclease III in the Presence and Absence of DNA. J. Am. Chem. Soc 2006, 128, 12082–12083. [DOI] [PubMed] [Google Scholar]
- (19).Teo RD; Wang R; Smithwick ER; Migliore A; Therien MJ; Beratan DN Mapping Hole Hopping Escape Routes in Proteins. Proc. Natl. Acad. Sci. U.S.A 2019, 116, 15811–15816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Teo RD; Migliore A; Beratan DN Mutation Effects on Charge Transport Through the p58c Iron-Sulfur Protein. Chem. Sci 2020, 11, 7076–7085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Boal AK; Yavin E; Lukianova OA; O’Shea VL; David SS; Barton JK DNA-Bound Redox Activity of DNA Repair Glycosylases Containing [4Fe-4S] Clusters. Biochemistry 2005, 44, 8397–8407. [DOI] [PubMed] [Google Scholar]
- (22).Sontz PA; Mui TP; Fuss JO; Tainer JA; Barton JK DNA Charge Transport as a First Step in Coordinating the Detection of Lesions by Repair Proteins. Proc. Natl. Acad. Sci. U.S.A 2012, 109, 1856–1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Fromme JC; Banerjee A; Huang SJ; Verdine GL Structural Basis for Removal of Adenine Mispaired with 8-Oxoguanine by MutY Adenine DNA Glycosylase. Nature 2004, 427, 652–656. [DOI] [PubMed] [Google Scholar]
- (24).Boon EM; Pope MA; Williams SD; David SS; Barton JK DNA-Mediated Charge Transport as a Probe of MutY/DNA Interaction. Biochemistry 2002, 41, 8464–8470. [DOI] [PubMed] [Google Scholar]
- (25).Boon EM; Livingston AL; Chmiel NH; David SS; Barton JK DNA-Mediated Charge Transport for DNA Repair. Proc. Natl. Acad. Sci. U.S.A 2003, 100, 12543–12547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Lewis FD; Young RM; Wasielewski MR Tracking Photoinduced Charge Separation in DNA: From Start to Finish. Acc. Chem. Res 2018, 51, 1746–1754. [DOI] [PubMed] [Google Scholar]
- (27).Bai H; Grist S; Gardner J; Suthers G; Wilson TM; Lu AL Functional Characterization of Human MutY Homolog (hMYH) Missense Mutation (R231L) That is Linked With hMYH-Associated Polyposis. Cancer Lett. 2007, 250, 74–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Šali A; Blundell TL Comparative Protein Modelling by Satisfaction of Spatial Restraints. J. Mol. Biol 1993, 234, 779–815. [DOI] [PubMed] [Google Scholar]
- (29).Schrödinger. The PyMOL Molecular Graphics System, Version 2.1; LLC.
- (30).Lee S; Verdine GL Atomic Substitution Reveals the Structural Basis for Substrate Adenine Recognition and Removal by Adenine DNA Glycosylase. Proc. Natl. Acad. Sci. U.S.A 2009, 106 (44), 18497–18502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Phillips JC; Braun R; Wang W; Gumbart J; Tajkhorshid E; Villa E; Chipot C; Skeel RD; Kalé L; Schulten K Scalable Molecular Dynamics with NAMD. J. Comput. Chem 2005, 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Senftle TP; Hong S; Islam MM; Kylasa SB; Zheng YX; Shin YK; Junkermeier C; Engel-Herbert R; Janik MJ; Aktulga HM, et al. The ReaxFF Reactive Force-Field: Development, Applications and Future Directions. Npj Comput. Mater 2016, 2. [Google Scholar]
- (33).Cheng X; Kelso C; Hornak V; de los Santos C; Grollman AP; Simmerling C Dynamic Behavior of DNA Base Pairs Containing 8-Oxoguanine. J. Am. Chem. Soc 2005, 127, 13906–13918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Maier JA; Martinez C; Kasavajhala K; Wickstrom L; Hauser KE; Simmerling C ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput 2015, 11, 3696–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Wang J; Cieplak P; Kollman PA How Well Does a Restrained Electrostatic Potential (RESP) Model Perform in Calculating Conformational Energies of Organic and Biological Molecules? J. Comput. Chem 2000, 21, 1049–1074. [Google Scholar]
- (36).Pérez A; Marchán I; Svozil D; Sponer J; Cheatham TE; Laughton CA; Orozco M Refinement of the AMBER Force Field for Nucleic Acids: Improving the Description of α/γ Conformers. Biophys. J 2007, 92, 3817–3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Ryckaert J-P; Ciccotti G; Berendsen HJC Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-Alkanes. J. Comput. Phys 1977, 23, 327–341. [Google Scholar]
- (38).Darden T; York D; Pedersen L Particle Mesh Ewald: An N·log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys 1993, 98, 10089–10092. [Google Scholar]
- (39).Martyna GJ; Tobias DJ; Klein ML Constant Pressure Molecular Dynamics Algorithms. J. Chem. Phys 1994, 101, 4177–4189. [Google Scholar]
- (40).Feller SE; Zhang YH; Pastor RW; Brooks BR Constant Pressure Molecular-Dynamics Simulation: the Langevin Piston Method. J. Chem. Phys 1995, 103, 4613–4621. [Google Scholar]
- (41).Marcus RA; Sutin N Electron Transfers in Chemistry and Biology. Biochim. Biophys. Acta 1985, 811, 265–322. [Google Scholar]
- (42).Teo RD; Smithwick ER; Migliore A 2′-Deoxy-2′-Fluoro-Arabinonucleic Acid: A Valid Alternative to DNA for Biotechnological Applications Using Charge Transport. Phys. Chem. Chem. Phys 2019, 21, 22869–22878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Teo RD; Terai K; Migliore A; Beratan DN Electron Transfer Characteristics of 2′-Deoxy-2′-Fluoro-Arabinonucleic Acid, a Nucleic Acid with Enhanced Chemical Stability. Phys. Chem. Chem. Phys 2018, 20, 26063–26067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Oppenheim I; Shuler KE; Weiss GH Stochastic Theory of Nonlinear Rate Processes with Multiple Stationary States. Physica A 1977, 88, 191–214. [Google Scholar]
- (45).Procaccia I; Mukamel S; Ross J On the Theory of Unimolecular Reactions: Application of Mean First Passage Time to Reaction Rates. J. Chem. Phys 1978, 68, 3244–3253. [Google Scholar]
- (46).Bar-Haim A; Klafter J On Mean Residence and First Passage Times in Finite One-Dimensional Systems. J. Chem. Phys 1998, 109, 5187–5193. [Google Scholar]
- (47).Steenken S; Jovanovic SV How Easily Oxidizable is DNA? One-Electron Reduction Potentials of Adenosine and Guanosine Radicals in Aqueous Solution. J. Am. Chem. Soc 1997, 119 (3), 617–618. [Google Scholar]
- (48).Banyasz A; Ketola T-M; Muñoz-Losa A; Rishi S; Adhikary A; Sevilla MD; Martinez-Fernandez L; Improta R; Markovitsi D UV-Induced Adenine Radicals Induced in DNA A-Tracts: Spectral and Dynamical Characterization. J. Phys. Chem. Lett 2016, 7, 3949–3953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Prindle MJ; Loeb LA DNA Polymerase Delta in DNA Replication and Genome Maintenance. Environ. Mol. Mutagen 2012, 53, 666–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.