

Graphical abstract
Keywords: viral infection, cell death, inflammasome, inflammation, pyroptosis, apoptosis, necroptosis, PANoptosis, PANoptosome
Abstract
In the past decade, emerging viral outbreaks like SARS-CoV-2, Zika and Ebola have presented major challenges to the global health system. Viruses are unique pathogens in that they fully rely on the host cell to complete their lifecycle and potentiate disease. Therefore, programmed cell death (PCD), a key component of the host innate immune response, is an effective strategy for the host cell to curb viral spread. The most well-established PCD pathways, pyroptosis, apoptosis and necroptosis, can be activated in response to viruses. Recently, extensive crosstalk between PCD pathways has been identified, and there is evidence that molecules from all three PCD pathways can be activated during virus infection. These findings have led to the emergence of the concept of PANoptosis, defined as an inflammatory PCD pathway regulated by the PANoptosome complex with key features of pyroptosis, apoptosis, and/or necroptosis that cannot be accounted for by any of these three PCD pathways alone. While PCD is important to eliminate infected cells, many viruses are equipped to hijack host PCD pathways to benefit their own propagation and subvert host defense, and PCD can also lead to the production of inflammatory cytokines and inflammation. Therefore, PANoptosis induced by viral infection contributes to either host defense or viral pathogenesis in context-specific ways. In this review, we will discuss the multi-faceted roles of PCD pathways in controlling viral infections.
Introduction
Viral infections pose a significant global health threat, as seen by the ongoing coronavirus disease 2019 (COVID-19) pandemic, which is caused by the virus severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2). Beyond SARS-CoV-2, several other viruses have also caused recent outbreaks, including influenza, Ebola, and Zika, and many more continue to circulate and cause morbidity and mortality around the globe. In mammals, innate immunity stands as the first line of defense during viral infections. Upon infection, viral pathogen-associated molecular patterns (PAMPs) are sensed by host pattern recognition receptors (PRRs), including the membrane-bound Toll-like receptors (TLRs) and C-type lectin receptors (CLRs) and the cytosolic nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), retinoic acid-inducible gene-I (RIG-I)-like receptors (RLRs), and absent in melanoma 2 (AIM2)-like receptors (ALRs). PRR detection of viral PAMPs triggers the activation of the innate immune signaling pathways, such as transcription factor NF-κB and mitogen-activated protein (MAP) kinase signaling to produce inflammatory cytokines and interferons, priming the immune response.1 Activation of PRRs can often lead to diverse forms of cell death.2 At the cellular level, elimination of infected host cells through programmed cell death (PCD) pathways is crucial to stop viral spread, as viruses are intracellular pathogens that are entirely dependent on host cells for their propagation. On the other hand, at the organismal level, cell death can contribute to disease pathogenesis during viral infections; inflammatory products such as damage-associated molecular patterns (DAMPs), alarmins, additional PAMPs and inflammatory cytokines are released from the dying cells and can trigger inflammatory cytokine storms, organ damage, and lethality.3, 4, 5 Therefore, the activation of cell death must be carefully balanced for optimal host defense.
The most well-defined PCD pathways include pyroptosis, apoptosis and necroptosis, and each is activated in many viral infections. Furthermore, viral infections also elicit activation of multiple cell death pathways, as seen with influenza virus (IAV) infections6, 7, 8, 9 and herpes simplex virus (HSV).10, 11, 12, 130 The extensive crosstalk between the PCD pathways is increasingly recognized,6, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 130 providing evidence for the activation of PANoptosis, an inflammatory PCD pathway regulated by the multi-protein PANoptosome complex with key features of pyroptosis, apoptosis and/or necroptosis.6, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 63, 130 Studies of this crosstalk have identified the PANoptosome, which provides a molecular scaffold for contemporaneous engagement of key molecules from pyroptosis, apoptosis, and/or necroptosis.20, 21, 22, 27, 63, 130, 131 Additionally, work on the PCD pathways has opened discussions about the possibility that multiple cell death pathways are activated in infected cells as a stepwise backup-mechanism or as a simultaneous mechanistic death process that rapidly eliminates infected cells to limit viral spread and avoid detrimental host pathogenesis.
In this review, we will discuss the physiological importance of the interconnections between pyroptosis, apoptosis and necroptosis and how PANoptosis broadens our ability to understand the process of cell death induced during viral infections. Improved understanding of these processes is critical for developing new strategies to counteract deadly viral infections and respond to emerging and future viral pathogens.
Cell death pathways and viral infection
Pyroptosis and viral infection
Pyroptosis is a form of lytic, inflammatory cell death mediated by the inflammatory caspases, including caspase-1 and caspase-4/5 (caspase-11 in mouse), that results in membrane rupture to release PAMPs and DAMPs.31, 32 Several PRRs can be activated to form caspase-1-activating scaffolds known as inflammasomes, with the most well-characterized being NLR family pyrin domain (PYD)-containing 1 (NLRP1),33 NLRP3,34, 35, 36 NLR family caspase activation and recruitment domain (CARD)-containing 4 (NLRC4),37, 38 AIM239, 40 and Pyrin.41 Inflammasome formation is generally initiated by homotypic interactions between the PYD or CARD of the PRR sensor and the adaptor protein apoptosis associated speck-like protein containing a CARD (ASC). Then ASC can recruit caspase-1 through CARD-CARD interactions, facilitating caspase-1 activation. In the case of some PRRs, direct interactions between the sensor and caspase-1 can occur without the need for ASC.132, 133 The active form of caspase-1 subsequently cleaves pro–interleukin-1β (IL-1β) and pro–IL-18 to form the mature proinflammatory cytokines IL-1β and IL-18. Active caspase-1 and caspase-4/5 (caspase-11 in mice) also mediate the cleavage of gasdermin D (GSDMD) into its N-terminal and C-terminal fragments.42, 43 The GSDMD N-terminal fragment drives pyroptosis by forming transmembrane pores allowing potassium ion efflux and water influx, disrupting membrane potential and releasing DAMPs and the inflammatory cytokines IL-1β and IL-18 to the extracellular space, driving inflammation (Figure 1 ).43, 44, 45, 46, 47
Figure 1.

Molecular mechanisms of pyroptosis activation during viral infection. Inflammasome activation during viral infections occurs in response to PRRs sensing their specific PAMPs. Examples include direct sensing by AIM2 (dsDNA), CARD8 (HIV protease), and NLRP1 (long dsRNA); or indirect sensing by NLRP3 (ionic fluxioni, ATP or ROS). Inflammasomes are traditionally composed of a PRR sensor, the adaptor protein ASC, and caspase-1, where activated caspase-1 is released and subsequently proteolytically cleaves pro-IL-1β and pro-IL-18 into bioactive inflammatory cytokines. Simultaneously, active caspase-1 cleaves GSDMD to free the pore-forming N-terminal fragment (N-GSDMD) to assemble membrane pores and initiate the releases of active IL-1β and IL-18 and induce pyroptotic cell death. The Sendai and Nipah viral protein V can directly bind to NLRP3 to inhibit NLRP3 self-oligomerization and prevent the assembly of NLRP3 and ASC to activate the inflammasome. Additionally, the EV71 protease 3C can cleave either NLRP3 or GSDMD to inhibit pyroptosis. Created with BioRender.com.
Pyroptotic cell death has been reported in various viral infections. In HIV infection, CD4+ T cell depletion is driven by pyroptosis, through caspase-1 activation.48 Indeed, inhibition of the apoptotic caspase-3 does not protect CD4+ T cells from death in HIV-infected human lymphoid aggregate culture, while blocking caspase-1 using the caspase-1 inhibitor VX-765 significantly inhibits caspase-1 cleavage, IL-1β release, and CD4+ T cell death.48 Recently, caspase recruitment domain-containing protein 8 (CARD8) was reported to act as a sensor of the HIV protease to activate an ASC-independent inflammasome in HIV-infected CD4+ T cells and macrophages; deletion of CARD8 or caspase-1 blocks HIV infection-induced inflammasome activation and pyroptotic cell death in CD4+ T and THP-1 cells.49
In human keratinocytes and epithelial cells, NLRP1 acts as a dsRNA sensor during Semliki Forest virus (SFV) infection or synthetic dsRNA analog poly(I:C) transfection.50 NLRP1 directly interacts with dsRNA through its leucine-rich repeat domain to induce ATP hydrolysis and a conformational change that triggers NLRP1 oligomerization and subsequent inflammasome activation and pyroptosis.50 The sensing of long dsRNA by NLRP1 is specific for human but not murine NLRP1, given the low level of sequence conservation between human and rodent NLRP1.51
In dengue virus infection, inflammasome activation and pyroptosis have been reported in multiple cell types including macrophages, dendritic cells, platelets and neutrophils.52 C-type lectin domain family member A (CLEC5A) acts as a PRR to mediate dengue virus-induced NLRP3 inflammasome activation.53 The expression of CLEC5A and NLRP3 is higher in activated macrophages compared to resting macrophages. Upon dengue infection, NLRP3 expression is upregulated, while expression of other NLR sensors such as NLRC4 and NLRP1 is unchanged. In addition, knockdown of NLRP3 using siRNA reduces dengue infection-induced cleavage of caspase-1 and release of IL-1β and IL-18.53
Zkia virus, another member of the Flaviviridae family, also induces pyroptotic death. This occurs in neural progenitor cells, as evidenced by the cleavage of caspase-1 and GSDMD and the increased release of IL-1β and IL-18.54 Similar to the upregulation of NLRP3 expression observed during dengue virus infection, neural progenitor cells upregulate expression of NLRP3 but not NLRC4, AIM2, or Pyrin during Zika infection, suggesting that Zika infection-induced inflammasome activation is likely to be NLRP3-dependent.54 Furthermore, in a Zika-infected mouse model, genetic deletion of caspase-1 or inhibition with VX-765 significantly reduces brain atrophy-induced microcephaly, while inhibition of caspase-3 does not,54 further highlighting the role of inflammasome activation in the infection.
Activation of the inflammasome has also been reported in coronavirus infections16, 55, 56 including infections with the newly emerging SARS-CoV-2.57, 58 In primary human monocytes, SARS-CoV-2 infection induces the formation of an NLRP3 puncta, and NLRP3-positive cells can be found in the lung tissue of patients who have succumbed to COVID-19.57 Furthermore, inflammasome-associated inflammatory products in the plasma from patients with COVID-19, such as active caspase-1, IL-18,57 LDH, and GSDMD,58 are positively correlated with disease severity.57, 58
While there are many examples of inflammasome and pyroptosis activation during viral infections, viruses can also directly interfere with pyroptotic cell death by regulating inflammasome activation or targeting the pyroptotic executer GSDMD. In Sendai virus infection, Sendai virus V protein can directly bind to NLRP3 and inhibit NLRP3 self-oligomerization and NLRP3-ASC speck assembly, leading to a reduction in IL-1β release.59 The V proteins of Nipah virus and human parainfluenza virus type 2 can also interact with NLRP3 to inhibit inflammasome activation, suggesting that the ability to interfere with NLRP3 inflammasome activation and pyroptosis is conserved among V protein-containing viruses (Figure 1).59 In enterovirus 71 (EV71) infection, mouse strains that are deficient in NLRP3 inflammasome activation, such as Nlrp3-/-, Asc-/-, and Casp1-/- mice, develop more severe viral disease compared with wild-type mice, suggesting that NLRP3 inflammasome activation is important for host defense against EV71 infection.60 To overcome this host defense strategy, the EV71 protease 3C interacts with and cleaves NLRP3 to inhibit NLRP3 inflammasome activity to potentiate viral disease.61 The EV71 protease 3C also cleaves GSDMD to form an N-terminal fragment that is unable to trigger pyroptosis,60 suggesting that the EV71 protease 3C inactivates GSDMD and prevents pyroptosis (Figure 1). These and many other virulence and immune evasion strategies allow viruses to subvert host PCD-induced inflammation and coordination of a successful immune response, thereby maximizing the chances of virus survival at the expense of the host.
Apoptosis and viral infection
Apoptosis has historically been characterized as “immunologically silent” PCD; however, more recent evidence suggests that it can also be inflammatory, with key apoptotic regulators activating inflammatory mechanisms.18, 24, 26, 62, 63, 64, 65 Cells undergoing apoptosis exhibit morphological changes including nuclear and chromatin condensations, DNA fragmentation, and membrane blebbing to form apoptotic bodies which are cleared upon engulfment by phagocytes.66 Apoptosis can be initiated through extrinsic or intrinsic pathways. The extrinsic pathway is initiated by signals from outside the cell through the binding of death ligands such as Fas ligand (FasL), tumor necrosis factor (TNF) and TNF-related apoptosis-inducing ligand (TRAIL) to their respective receptors. Receptor engagement triggers the formation of the death-inducing signaling complex (DISC), which is comprised of Fas-associated death domain (FADD) and pro–caspase-8, through homotypic interactions, leading to caspase-8 activation.67 Activation of caspase-8 is tightly regulated by the anti-apoptotic cellular FADD-like IL-1β-converting enzyme inhibitory protein (cFLIP), which binds to FADD and/or caspase-8 to form an apoptosis inhibitory complex.68 Activated caspase-8 can directly proteolytically activate effectors and the executioners caspases-7 and -3 to induce apoptosis.69 Alternatively, it can indirectly induce intrinsic apoptosis by cleaving the BH3-only protein BID to its active form tBID, and driving the BAK/BAX-mediated mitochondrial outer membrane permeabilization (MOMP)-induced apoptosis (Figure 2 ).70, 71
Figure 2.

Molecular mechanisms of apoptosis activation during viral infection. Extrinsic apoptosis is activated through engagement of death receptors by their cognate ligands. One example is the activation of FAS by FASL expressed on cytotoxic T lymphocytes (CTLs) or natural killer (NK) cells. The adaptor protein FADD together with pro–caspase-8 are then recruited to the death receptor’s cytosolic domain, inducing caspase-8 activation. Active caspase-8 directly induces proteolytic cleavage of the executioner caspases, caspase-3 and -7, triggering extrinsic apoptosis. Active caspase-8 also can cleave the BH3-only protein BID to form tBID, inducing intrinsic apoptosis. Intrinsic apoptosis is triggered by internal cellular stress such as DNA damage or ER stress, inducing the activation of pro-apoptotic BH3-only proteins including NOXA, PUMA, BAD and BIM. This activation triggers BAX and BAK activation to induce mitochondrial outer membrane permeabilization (MOMP). MOMP leads to the release of apoptogenic factors including cytochrome C, which binds to apoptotic peptidase activating factor 1 (APAF1) and induces APAF1 oligomerization to form the apoptosome. Apoptosome formation activates the initiator caspase-9, which then cleaves executioner caspases, caspase-3 and -7 to drive intrinsic apoptosis. Intrinsic apoptosis can also be activated through active caspase-8 or in response to the release of protease granzyme B through the perforin pore initiated by CTLs and NK cells, mediating the cleavage of BID to its active form tBID, triggering BAX-BAK to induce MOMP and apoptosis. Furthermore, recognition of viral nucleic acids through TLRs in the endosome or cytosolic PRRs, such as cGAS-STING (vDNA), RIG-I (triphosphates-vRNA), and MDA5 (long vdsRNA), can trigger pro-apoptotic and inflammatory transcription factor NF-κB and type I IFN production, which can induce pro-apoptotic gene expression and subsequent apoptosis. Created with BioRender.com.
Unlike extrinsic apoptosis, which requires the presence of external ligands, intrinsic apoptosis is activated by nutrient or growth factor withdrawal, DNA damage or other internal cellular stresses. These events trigger the expression of pro-apoptotic members of the BCL-2 family including BIM, NOXA, PUMA and BAD, which bind and neutralize the pro-survival BCL-2 family BH3-only proteins such as BCL-2, BCL-XL, MCL-1 and BCL-2-related protein A1 (BCL-2A1), freeing the BAK/BAX complex to induce MOMP and release cytochrome C.72 The binding of cytochrome C to the apoptotic peptidase activation factor 1 (APAF1) in the cytoplasm prompts apoptosome formation, leading to the activation of initiator caspase-9 to induce activation of caspases-3 and -7 to drive apoptosis (Figure 2).73 To keep the system in balance, the inhibitors of apoptosis proteins (IAPs), such as X-linked inhibitor of apoptosis (XIAP), can promote proteasomal degradation of caspases to inhibit apoptosis.74 These IAPs can be blocked by MOMP-induced second mitochondrial activator of caspases (SMAC/DIABLO) and HTRA serine peptidase 2 (HTR2) to release the brake on cell death and facilitate apoptosis.75
Apoptosis is the most studied PCD in viral infections. Pro-apoptotic signaling is associated with nearly all stages of viral infection. At the viral entry stage, the binding of avian leukosis virus (ALV) envelope protein to the cytopathic ALSV receptor (CAR1) or the binding of bovine herpesvirus (BHV-1) glycoprotein gD to the herpesvirus entry mediator (HVEM) is sufficient to induce apoptosis.76, 77 Given that CAR1 and HVEM are death receptors themselves, extrinsic apoptosis likely plays important roles in the cell death induced by these viral attachments, though detailed mechanisms remain to be characterized.
After the virus has enters cell, sensing of the viral genomes by host PRRs to initiate apoptotic cell death is an extensively studied component of the innate immune response to viral infection. Viral genomes can be sensed in many forms, including viral DNA (vDNA), cDNA synthesized from viral RNA (v-cDNA), and viral single-stranded or double-stranded RNA (vRNA). Endosomal vDNA is recognized by TLR9 at the endosome, while cytosolic vDNA is sensed by cyclic GMP-AMP (cGAMP) synthase (cGAS) and the interferon (IFN)-inducible proteins AIM2, RNA Pol III, and IFN-γ–inducible protein 16 (IFI16). In the case of vRNA, TLR3 (dsRNA) and TLR7 and TLR8 (ssRNA) recognize the vRNA at the endosome, while cytosolic vRNA is sensed by RIG-I, melanoma differentiation-associated protein 5 (MDA5), probable ATP-dependent RNA helicase (DHX58) and other sensors.78
Recognition of viral genomes by host PRRs can directly result in apoptosis without transcription factor-induced gene expression. Studies in Sendai and dengue infections show that activation of RIG-I and MDA5 lead to the recruitment of mitochondrial antiviral signaling protein (MAVS), which disrupts mitochondrial membrane potential to induce caspase activation independent of NF-κB or IFN regulatory factor 3 (IRF3) transcriptional target genes (Figure 2).79, 80 In addition, IRF3 mutants that lack transcription factor activity can interact with BAX to induce translocation of IRF3-BAX to the mitochondria, triggering intrinsic apoptosis in Sendai and human T cell leukemia virus (HTLV) infections.81, 82 In the context of IRF3-mediated, transcription-dependent apoptosis, IRF3 drives the expression of the death ligand TRAIL83 and BH3-only protein NOXA84 in Sendai and reovirus infections, respectively, to trigger apoptosis.
The sensing of viral genome by PRRs also triggers a cascade of adaptor protein interactions. For example, RIG-I sensing of short 5′-triphosphate (ppp) viral dsRNA leads to an ATP-dependent conformational change and the formation of a filamentous tetramer of RIG-I through CARD-CARD homotypic interactions.85 The K63-ubiqitin chain linked to the RIG-I tetramer N-terminal CARDs forms oligomers with the CARD in the adaptor protein MAVS,86 recruiting downstream adaptors such as TNFR-associated factor 6 (TRAF6) or TNFR-associated factor 3 (TRAF3)87, 88 to activate NF-κB, IRF3, and IFN regulatory factor 7 (IRF7) and induce inflammatory cytokine and chemokine production and type I and III IFN responses. This signaling process positively regulates apoptosis (Figure 2). Indeed, IFN-α/β signaling through the IFN-α/β receptor (IFNAR) activates the Janus kinase/signal transducer and activator of transcription (JAK/STAT) signaling pathway to upregulate IFN-stimulated genes (ISGs) and promote apoptosis.89 For example, the activity of the ISG protein kinase R (PKR) is stimulated after sensing dsRNA and subsequently phosphorylates translation initiation factor eIF2a, blocking cellular translation and facilitating apoptosis.90 PKR also can induce apoptosis independently with its ability to inhibit translation through the FADD-caspase-8/-3 axis.91 Another ISG, 2′, 5′-oligoadenylate synthetase (OAS), detects dsRNA and activates the ISG RNase L to degrade host and viral RNA, resulting in apoptosis.92 IFN signaling can also induce p53 activation, which drives apoptosis through p53 transcriptional activity inducing pro-apoptotic BH3-only proteins PUMA and NOXA.93
Apoptotic cell death also plays an important role in cytotoxic T lymphocyte (CTL) and natural killer (NK) cell killing of viral infected cells to control virus infection. CTLs and NK cells produce and store perforin, a membrane pore-forming protein, and granzymes, serine proteases, in cytotoxic granules. Upon detection of virally infected cells, CTLs or NK cells target the infected cells and release cytotoxic granules, liberating granzymes into the cytoplasm of targeted cells and facilitating perforin oligomerization and the formation of a membrane attack complex to initiate intrinsic apoptosis (Figure 2).94 CTLs can also induce extrinsic apoptosis in virally infected cells through the engagement of FAS ligand on CTLs and FAS receptor on targeted cells.95
To enable immune evasion and viral survival strategies, many viruses encode virulence proteins that inhibit apoptotic death, such as caspase inhibitors. Examples include the murine cytomegalovirus (MCMV) vICA protein, which inhibits caspase-8,96 and the cowpox virus CrmA protein, which inhibits caspase-8 and granzyme B (Figure 2).97, 98 In addition to caspase inhibitors, viruses can also encode BCL-2 homologs to block BAX- and BAK-mediated MOMP-dependent apoptosis, such as the Adenovirus E1B19K99 and the Epstein-Barr virus BHRF1 and BALF1.100, 101 Despite these attempts by the virus to evade cell death, viral blockade of apoptosis commonly leads to the activation of alternative PCD pathways by host cells, highlighting the interconnection of PCD pathways, which will be discussed in detail later in this review.
Necroptosis and viral infection
Necroptosis is an additional PCD pathway that was often considered as a fail-safe to allow cell death to proceed in conditions where apoptosis was inhibited. Therefore, the signaling pathways involved in apoptosis and necroptosis have long been known to be intricately linked. Activation of the death receptor TNFR by the TNF ligand results in the formation of a ubiquitin scaffold complex (Complex I) composed of TRAF2 and TRADD adaptors, IAPs and LUBAC ubiquitin ligases, and receptor interacting serine/threonine kinase 1 (RIPK1), recruiting TAB1/2/3-TAK1 and NEMO-IKK1/2 complexes. Complex I activates NF-κB survival and inflammatory signals. Deactivation of this survival signaling complex by deubiquitinases like A20 and/or CYLD triggers the formation of different cell death complexes such as TRADD-FADD-caspase-8 or TRADD-RIPK1-FADD-caspase-8 to drive apoptosis.102 During infection or in response to pharmacological inhibition of caspase-8,103, 104 RIPK1 can interact with RIPK3 through their RIP homotypic interaction motif (RHIM)105 and form a cytosolic complex known as the necrosome. Once formed, the necrosome initiates RIPK3-mediated phosphorylation of the pseudo-kinase mixed lineage kinase domain-like (MLKL), changing its conformation (Figure 3 ). Phosphorylated MLKL is oligomerized to become a multimeric complex that disrupts the plasma membrane and leads to necroptotic cell death.106 The formation of the RIPK1-RIPK3 necrosome complex, in most physiological conditions, is inhibited by the cFLIP/caspase-8 heterodimeric complex, which is recruited to the RIPK1-RIPK3 complex through the adaptor FADD and can cleave both RIPK1 and RIPK3 to prevent necroptosis and promote apoptosis (Figure 3).103, 104, 107 Thus, inhibition of caspase-8, as occurs in response to viral effectors such as the MCMV vICA protein,96 is a key event triggering necroptosis. Alternatively, necroptosis can also be activated by the sensor Z-DNA-binding protein 1 (ZBP1), an ISG that contains RHIM domains to recruit and activate RIPK3-induced MLKL phosphorylation resulting in cell death.6, 10, 108, 109 ZBP1-mediated cell death has been reported in various viral infections including cytomegalovirus (CMV),108 HSV,10 vaccinia virus (VACV),109 West Nile virus (WNV),110 Zika virus,111 and IAV.6 In addition to its role in necroptosis, ZBP1 is also critically involved across PCD pathways,6 which will be discussed in depth in the next section.
Figure 3.

Molecular mechanisms of necroptosis activation during viral infection. Recognition of TNF by the TNFR1 receptor induces the formation of complex I, which includes the TNF receptor type 1 associated death domain (TRADD) adaptor protein, RIPK1 kinase, and E3 ubiquitin ligases IAPs and linear ubiquitin chain assembly complex (LUBAC). This formation creates a dense network of diverse ubiquitin chains that allow the recruitment of several kinase complexes, including TABs/TAK1 and NEMO/IKKs, triggering MAPK and NF-κB signaling pathways and allowing the expression of pro-survival and proinflammatory genes. The dissociation of complex I is mediated by the deubiquitinases A20 and CYLD, which results in the formation of several death-inducing complexes. TRADD recruits FADD-caspase-8 to form complex IIa, inducing caspase-8-mediated apoptosis. RIPK1 kinase can be recruited to complex IIa to form complex IIb and induce apoptosis. Inhibition of caspase-8 activity mediated by viral proteins such as vICAs or CrmA triggers the recruitment of RIPK3 kinase to complex IIb, inducing MLKL phosphorylation and necroptosis. The inhibition of caspase-8 by vICAs or CrmA at the TLR3-mediated complex TRIF-RIPK1-FADD-caspase-8 also results in the recruitment of RIPK3 to mediate MLKL phosphorylation and induce necroptosis. Alternatively, IAV Z-RNA is sensed by ZBP1, mediating RHIM-RHIM homotypic interactions between ZBP1 and RIPK3 to induce MLKL phosphorylation and mediate necroptosis. The vaccinia virus E3L protein can compete with ZBP1 for Z-nucleic acid binding, preventing necroptosis, while the MCMV viral protein M45 interferes with the ZBP1-RIPK3 interaction to inhibit necroptosis. Created with BioRender.com.
As is the case with other PCD pathways, many viruses are equipped with viral proteins to inhibit cellular necroptosis. VACV viral protein E3L has an N-terminal Zα domain similar to that found in ZBP1 that competes with ZBP1 to inhibit the ZBP1-RIPK3 axis and block necroptosis.112 Additionally, the MCMV viral protein M45 contains a RHIM domain, which can interfere with the RHIM-RHIM interaction between ZBP1 and RIPK3, leading to necroptosis inhibition (Figure 3).113 Recent evidence also suggests that some viruses can lead to elimination of RIPK3, as the viral inducer of RIPK3 degradation (vIRD) from cowpox virus and other orthopoxviruses induces the proteasomal degradation of RIPK3 to inhibit necroptosis.114
Interconnections between PCD during viral infections: PANoptosis
Clearance of infected cells is the ultimate goal to enable the host to survive pathogen infection. To counteract this strategy, intracellular pathogens such as viruses are equipped to hijack host cell death pathways to support their own propagation. Virus-mediated inhibition of one PCD pathway could evolutionarily favor the development of mechanisms that would potentiate other cell death executioners and effectors through a shared signaling scaffold under these conditions, underscoring the importance of understanding the interconnections between PCD pathways. As discussed above, apoptosis and necroptosis are both regulated by caspase-8 activity. In a physiological context, canonical signals from death receptors can trigger extrinsic apoptosis through the activation of caspase-8, which in turn also cleaves RIPK1 or/and RIPK3 to prevent necroptosis (Figure 3). The crosstalk between PCD pathways mediated through caspase-8 has been further shown through its connection with inflammasome activation and pyroptosis. Caspase-8 mediates the priming and activation of the canonical and noncanonical NLRP3 inflammasomes.26 Furthermore, caspase-8 can associate with the inflammasome adaptor ASC,115 and induce cleavage of GSDMD to trigger pyroptosis.29, 116, 28 The ASC-dependent activation of caspase-8 also can trigger apoptosis in the absence of caspase-1.117 Molecular connections between these PCD pathways that go beyond caspase-8 have also been found. The apoptotic caspase-7 can be cleaved by caspase-1 downstream of inflammasome activation,25 and the NLRP3 and NLRC4 inflammasomes can result in the cleavage of the apoptotic marker PARP1.19 Additionally, inflammasome-induced caspase-1 activation can trigger apoptosis in the absence of GSDMD through the BID, caspase-9, and caspase-3 axis.118 Activated caspase-3 can also cleave GSDME, a pore-forming protein belonging to the gasdermin protein family, triggering pyroptotic cell death.65, 118
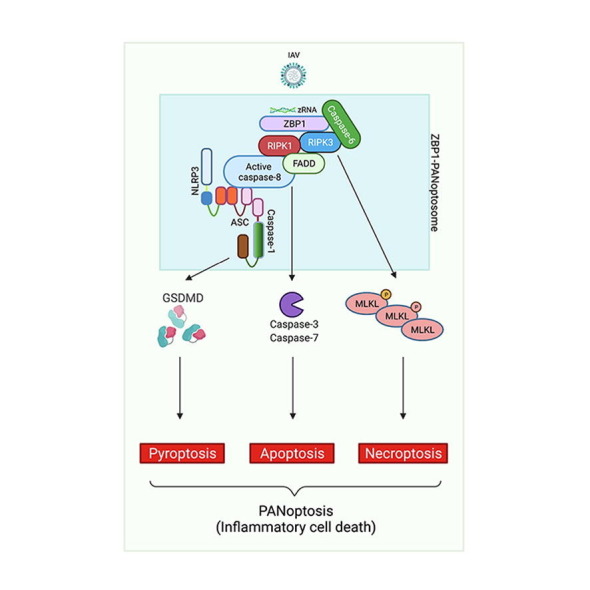
In the context of viral infection, evidence for crosstalk between PCD pathways has been the most well characterized with IAV infection. During IAV infection, ZBP1 acts as the upstream sensor to induce cell death with biochemical characteristics of pyroptosis, apoptosis and necroptosis.6 Deletion of ZBP1 inhibits activation of all these PCD pathways, including NLRP3 inflammasome-induced pyroptosis, RIPK1-RIPK3 necrosome-induced necroptosis, and caspase-8-driven apoptosis. However, individual deletion of any of the PCD pathways in isolation does not protect against cell death; IAV infection-induced ZBP1-dependent cell death is only prevented in cells lacking both RIPK3 and caspase-8 (Figure 4 ).6, 8 These findings suggest that ZBP1 acts as a master regulator of a unique inflammatory PCD pathway that contemporaneously engages key molecules from pyroptosis, apoptosis and/or necroptosis, establishing that PANoptosis occurs during viral infection (Figure 4). More recent studies have built upon this initial evidence to describe an important role for caspase-6 in promoting the interaction between ZBP1 and RIPK3 and to characterize the formation of a multi-protein complex that contains ZBP1, RIPK3, RIPK1, caspase-8, ASC and NLRP3, defined as the ZBP1-PANoptosome, to regulate PANoptosis (Figure 4).9, 27, 119, 120
Figure 4.

Molecular mechanism of PANoptosis activation during IAV infection. The sensing of IAV Z-RNA by ZBP1 allows the activation of proteins involved in pyroptosis, apoptosis, and necroptosis to form the ZBP1-PANoptosome and mediate PANoptosis. Caspase-6 promotes the interaction between ZBP1 and RIPK3 in the ZBP1-PANoptosome. RIPK3 interacts with RIPK1, which can associate with FADD and caspase-8. Caspase-8 can also be associated with ASC, a core component of the inflammasome, recruiting NLRP3 and caspase-1. The executioners GSDMD, caspase-3 and -7, and MLKL are all activated in response to PANoptosome formation to drive PANoptosis. Created with BioRender.com.
In murine hepatitis virus (MHV) infection, a model to study coronavirus infections in mice, biochemical features of pyroptosis, apoptosis and necroptosis are activated, highlighting a role of PANoptosis in this infection as well. Blocking the activation of pyroptotic effectors through deletion of NLRP3, caspase-1 and -11, or GSDMD leads to increased cell death characterized by increased activation of caspase-8, caspase-3, caspase-7, and MLKL, while combined deletion of caspase-8/RIPK3 inhibits MHV infection-induced cell death.16 In vesicular stomatitis virus (VSV) infection, PANoptosis is also observed. Blocking a single PCD, such as deletion of caspase-1/11, GSDMD, GSDMD/MLKL, or RIPK3, cannot prevent cell death during VSV infection. As is the case during MHV infection, VSV-induced cell death is significantly reduced in the combined absence of caspase-8 and RIPK3.27
In addition to NLRP3-containing PANoptosomes such as the ZBP1-PANoptosome, the AIM2-PANoptosome has also been identified and characterized during viral infection. During HSV1 infection, AIM2 regulates pyrin and ZBP1 expression and forms a PANoptosome complex along with these two sensors and ASC, caspase-1, caspase-8, RIPK3, RIPK1 and FADD to drive PANoptosis.130 This process is critical for host defense, as mice deficient in AIM2 are significantly more susceptible to lethality during HSV1 infection than WT mice are 130 Several viruses carry viral proteins that influence the biochemical cell death outcome between the different arms of PANoptosis. The HSV1 and HSV2 viral protein ICP6 contains a RHIM domain, which can bind to both RIPK1 and RIPK3 in mouse cells, triggering necrosome assembly and necroptotic cell death.122 By contrast, in human cells, ICP6 binds to both RIPK1 and RIPK3 and disrupts their interaction to block necrosome formation.11 Furthermore, human cells expressing ICP6 without the C-terminal, which is responsible for caspase-8 binding, undergo apoptotic death, while cells expressing ICP6 lacking the N-terminal, containing the RHIM domain, undergo necroptotic death.11
The CMV viral protein pUL36, encoded by the UL36 gene, interacts with pro–caspase-8 and inhibits caspase-8 activation, blocking apoptosis. CMV pUL36 also blocks necroptosis by inducing MLKL degradation.123 Other studies have found that CMV infection can induce both caspase-1 and caspase-11 activation and cleavage of GSDMD, causing pyroptosis-driven inflammation in an induced retinitis model in immunosuppressed mice.124, 125
Beyond the ability of viruses to directly activate PANoptosis and modulate the biochemical processes of cell death, virus-induced signaling and cytokine release also drive PANoptosis. One key example of this occurs during SARS-CoV-2 infection. A prominent feature of severe COVID-19 pathology is cytokine storm, which can cause severe inflammation and lethality in patients.3 Two of the key cytokines produced during cytokine storms are TNF-α and IFN-γ. Administration of TNF-α and IFN-γ in mice can mimic the clinical symptoms of COVID-19,4 highlighting their importance in disease pathogenesis. Together, these two cytokines can drive PANoptosis and tissue and organ damage.4 These findings led to the definition of cytokine storm as a life-threatening condition caused by excessive production of cytokines mediated by inflammatory cell death, PANoptosis.5 Treatment of mice with neutralizing antibodies against TNF-α and IFN-γ provides protection during SARS-CoV-2 infection,4 suggesting that these two cytokines and their induction of PANoptosis are a driving force in COVID-19 pathology.
Taken together, the emerging evidence of the interconnections between PCD pathways through PANoptosis opens a new window into the complexity of PCD during pathogenic viral infections. The existence of a central PANoptosome signaling scaffold allows for functionally redundant cell death effectors to simultaneously engage,9, 27, 119, 120, 130 which may reduce the chance for viral inhibition of the cell death execution. Further studies of PCD during viral infection should carefully examine the molecular mechanisms of PANoptosis and the impact of different viral proteins on the biochemical features of cell death to gain new insight into avenues of therapeutic development to regulate cell death and provide optimal host immune responses against viral infections.
Summary and future perspectives
Viruses are intracellular pathogens that require host cell machinery to advance their lifecycle. Due to this unique lifestyle, the survival of infected cells is necessary for viral spread. Thus, successful activation of any PCD pathway de-rails virus survival by limiting its replicative niche and exposing the virus to the immune system. PCD is an effective host defense strategy, but hyperactivation of the antiviral response and inflammatory PCD can lead to systemic inflammation and pathology. Therefore, the host must carefully balance PCD activation to prevent excess inflammation while clearing the infection and blocking viral disease potentiation. Interconnections among PCD pathways have defined PANoptosis as an inflammatory PCD involving key molecules from pyroptosis, apoptosis and/or necroptosis. PANoptosis has now been implicated in viral infections as well as in bacterial and fungal infections, cancer, and autoinflammatory diseases.2, 5, 6, 13, 14, 15, 16, 17, 18, 20, 21, 22, 23, 24, 26, 27, 126, 127, 128, 129, 130, 131 These discoveries have transformed our understanding of PCD pathways and identified the formation of a signaling complex containing initiators and effectors of pyroptosis, apoptosis and necroptosis, such as ASC/caspase-1, caspase-8 and RIPK1-RIPK3, the PANoptosome. This formation is likely to be context- and cell type-specific, with different sensors involved depending on the stimuli. Viruses have an extraordinary ability to block PCD, which could emerge troublesome for the immune system to contain viral diseases, and PANoptosis may serve as a mechanistic strategy to overcome the inhibition of PCD. Future studies on how the host immune system detects virally infected cells to activate or inhibit PANoptosis will help inform the development of therapeutic agents to selectively inhibit or activate PANoptosis for the benefit of the host immune system to contain and clear infected cells and limit systemic inflammation.
Acknowledgments
We apologize to our colleagues in the field whose work could not be cited due to space limitations. We thank all members of the Kanneganti laboratory for their comments and suggestions and R. Tweedell for scientific editing and writing support. Work from our laboratory is supported by the US National Institutes of Health (AI101935, AI124346, AI160179, AR056296 and CA253095 to T.-D.K.) and the American Lebanese Syrian Associated Charities (to T.-D.K.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Edited by Kate Fitzgerald
References
- 1.Carty M., Guy C., Bowie A.G. Detection of viral infections by innate immunity. Biochem. Pharmacol. 2020:114316. doi: 10.1016/j.bcp.2020.114316. [DOI] [PubMed] [Google Scholar]
- 2.Place D.E., Lee S., Kanneganti T.D. PANoptosis in microbial infection. Curr. Opin. Microbiol. 2021;59:42–49. doi: 10.1016/j.mib.2020.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fajgenbaum D.C., June C.H. Cytokine Storm. N. Engl. J. Med. 2020;383:2255–2273. doi: 10.1056/NEJMra2026131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Karki R., Sharma B.R., Tuladhar S., Williams E.P., Zalduondo L., Samir P., et al. Synergism of TNF-alpha and IFN-gamma Triggers Inflammatory Cell Death, Tissue Damage, and Mortality in SARS-CoV-2 Infection and Cytokine Shock Syndromes. Cell. 2021;184 doi: 10.1016/j.cell.2020.11.025. 149–68 e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Karki R.K. The ‘cytokine storm’: molecular mechanisms and therapeutic prospects. Trends Immunol. 2021;42(8):681–705. doi: 10.1016/j.it.2021.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kuriakose T., Man S.M., Malireddi R.K., Karki R., Kesavardhana S., Place D.E., et al. ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci. Immunol. 2016;1:aag2045. doi: 10.1126/sciimmunol.aag2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nogusa S., Thapa R.J., Dillon C.P., Liedmann S., Oguin T.H., 3rd, Ingram J.P., et al. RIPK3 Activates Parallel Pathways of MLKL-Driven Necroptosis and FADD-Mediated Apoptosis to Protect against Influenza A Virus. Cell Host Microbe. 2016;20:13–24. doi: 10.1016/j.chom.2016.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kesavardhana S., Kuriakose T., Guy C.S., Samir P., Malireddi R.K.S., Mishra A., et al. ZBP1/DAI ubiquitination and sensing of influenza vRNPs activate programmed cell death. J. Exp. Med. 2017;214:2217–2229. doi: 10.1084/jem.20170550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zheng M.K., Vogel R., Kanneganti P. TD Caspase-6 is a key regulator of innate immunity, inflammasome activation and host defense. Cell. 2020;181 doi: 10.1016/j.cell.2020.03.040. 674–87.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pham T.H., Kwon K.M., Kim Y.E., Kim K.K., Ahn J.H. DNA sensing-independent inhibition of herpes simplex virus 1 replication by DAI/ZBP1. J. Virol. 2013;87:3076–3086. doi: 10.1128/JVI.02860-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guo H., Omoto S., Harris Philip A., Finger Joshua N., Bertin J., Gough Peter J., et al. Herpes Simplex Virus Suppresses Necroptosis in Human Cells. Cell Host Microbe. 2015;17:243–251. doi: 10.1016/j.chom.2015.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hayes C.K., Wilcox D.R., Yang Y., Coleman G.K., Brown M.A., Longnecker R. ASC-dependent inflammasomes contribute to immunopathology and mortality in herpes simplex encephalitis. PLoS Pathog. 2021;17 doi: 10.1371/journal.ppat.1009285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kesavardhana S., Malireddi R.K.S., Burton A.R., Porter S.N., Vogel P., Pruett-Miller S.M., et al. The Zα2 domain of ZBP1 is a molecular switch regulating influenza-induced PANoptosis and perinatal lethality during development. J. Biol. Chem. 2020;295:8325–8330. doi: 10.1074/jbc.RA120.013752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Banoth B., Tuladhar S., Karki R., Sharma B.R., Briard B., Kesavardhana S., et al. ZBP1 promotes fungi-induced inflammasome activation and pyroptosis, apoptosis, and necroptosis (PANoptosis) J. Biol. Chem. 2020;295(52):18276–18283. doi: 10.1074/jbc.RA120.015924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Karki R., Sharma B.R., Lee E., Banoth B., Malireddi R.K.S., Samir P., et al. Interferon regulatory factor 1 regulates PANoptosis to prevent colorectal cancer. JCI Insight. 2020;5 doi: 10.1172/jci.insight.136720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zheng M., Williams E.P., Malireddi R.K.S., Karki R., Banoth B., Burton A., et al. Impaired NLRP3 inflammasome activation/pyroptosis leads to robust inflammatory cell death via caspase-8/RIPK3 during coronavirus infection. J. Biol. Chem. 2020;295(41):14040–14052. doi: 10.1074/jbc.RA120.015036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gurung P., Burton A., Kanneganti T.-D. NLRP3 inflammasome plays a redundant role with caspase 8 to promote IL-1β–mediated osteomyelitis. Proc. Natl. Acad. Sci. U. S. A. 2016;113:4452–4457. doi: 10.1073/pnas.1601636113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lukens J.R., Gurung P., Vogel P., Johnson G.R., Carter R.A., McGoldrick D.J., et al. Dietary modulation of the microbiome affects autoinflammatory disease. Nature. 2014;516:246–249. doi: 10.1038/nature13788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Malireddi R.K., Ippagunta S., Lamkanfi M., Kanneganti T.D. Cutting edge: proteolytic inactivation of poly(ADP-ribose) polymerase 1 by the Nlrp3 and Nlrc4 inflammasomes. J. Immunol. 2010;185:3127–3130. doi: 10.4049/jimmunol.1001512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Malireddi R.K.S., Gurung P., Kesavardhana S., Samir P., Burton A., Mummareddy H., et al. Innate immune priming in the absence of TAK1 drives RIPK1 kinase activity–independent pyroptosis, apoptosis, necroptosis, and inflammatory disease. J. Exp. Med. 2020;217 doi: 10.1084/jem.20191644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Malireddi R.K.S., Kesavardhana S., Karki R., Kancharana B., Burton A.R., Kanneganti T.D. RIPK1 Distinctly Regulates Yersinia-Induced Inflammatory Cell Death, PANoptosis. Immunohorizons. 2020;4:789–796. doi: 10.4049/immunohorizons.2000097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zheng M., Karki R., Vogel P., Kanneganti T.D. Caspase-6 Is a Key Regulator of Innate Immunity, Inflammasome Activation, and Host Defense. Cell. 2020;181 doi: 10.1016/j.cell.2020.03.040. 674–87.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karki R., Sharma B.R., Tuladhar S., Williams E.P., Zalduondo L., Samir P., et al. Synergism of TNF-α and IFN-γ Triggers Inflammatory Cell Death, Tissue Damage, and Mortality in SARS-CoV-2 Infection and Cytokine Shock Syndromes. Cell. 2021;184 doi: 10.1016/j.cell.2020.11.025. 149–68.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Malireddi R.K.S., Gurung P., Mavuluri J., Dasari T.K., Klco J.M., Chi H., et al. TAK1 restricts spontaneous NLRP3 activation and cell death to control myeloid proliferation. J. Exp. Med. 2018;215:1023–1034. doi: 10.1084/jem.20171922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lamkanfi M., Kanneganti T.D., Van Damme P., Vanden Berghe T., Vanoverberghe I., Vandekerckhove J., et al. Targeted peptidecentric proteomics reveals caspase-7 as a substrate of the caspase-1 inflammasomes. Mol. Cell. Proteomics. 2008;7:2350–2363. doi: 10.1074/mcp.M800132-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gurung P., Anand P.K., Malireddi R.K., Vande Walle L., Van Opdenbosch N., Dillon C.P., et al. FADD and caspase-8 mediate priming and activation of the canonical and noncanonical Nlrp3 inflammasomes. J. Immunol. 2014;192:1835–1846. doi: 10.4049/jimmunol.1302839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Christgen S., Zheng M., Kesavardhana S., Karki R., Malireddi R.K.S., Banoth B., et al. Identification of the PANoptosome: A Molecular Platform Triggering Pyroptosis, Apoptosis, and Necroptosis (PANoptosis) Front. Cell. Infect. Microbiol. 2020;10:237. doi: 10.3389/fcimb.2020.00237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fritsch M., Günther S.D., Schwarzer R., Albert M.C., Schorn F., Werthenbach J.P., et al. Caspase-8 is the molecular switch for apoptosis, necroptosis and pyroptosis. Nature. 2019;575:683–687. doi: 10.1038/s41586-019-1770-6. [DOI] [PubMed] [Google Scholar]
- 29.Newton K., Wickliffe K.E., Maltzman A., Dugger D.L., Reja R., Zhang Y., et al. Activity of caspase-8 determines plasticity between cell death pathways. Nature. 2019;575:679–682. doi: 10.1038/s41586-019-1752-8. [DOI] [PubMed] [Google Scholar]
- 30.Schwarzer R., Jiao H., Wachsmuth L., Tresch A., Pasparakis M. FADD and Caspase-8 Regulate Gut Homeostasis and Inflammation by Controlling MLKL- and GSDMD-Mediated Death of Intestinal Epithelial Cells. Immunity. 2020;52 doi: 10.1016/j.immuni.2020.04.002. 978–93.e6. [DOI] [PubMed] [Google Scholar]
- 31.Kuriakose T., Kanneganti T.D. Pyroptosis in Antiviral Immunity. Curr. Top. Microbiol. Immunol. 2019 doi: 10.1007/82_2019_189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Christgen S., Place D.E., Kanneganti T.D. Toward targeting inflammasomes: insights into their regulation and activation. Cell Res. 2020;30:315–327. doi: 10.1038/s41422-020-0295-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martinon F., Burns K., Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell. 2002;10:417–426. doi: 10.1016/s1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- 34.Kanneganti T.D., Ozoren N., Body-Malapel M., Amer A., Park J.H., Franchi L., et al. Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature. 2006;440:233–236. doi: 10.1038/nature04517. [DOI] [PubMed] [Google Scholar]
- 35.Mariathasan S., Weiss D.S., Newton K., McBride J., O'Rourke K., Roose-Girma M., et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440:228–232. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- 36.Martinon F., Petrilli V., Mayor A., Tardivel A., Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–241. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- 37.Franchi L., Amer A., Body-Malapel M., Kanneganti T.D., Ozoren N., Jagirdar R., et al. Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1beta in salmonella-infected macrophages. Nat. Immunol. 2006;7:576–582. doi: 10.1038/ni1346. [DOI] [PubMed] [Google Scholar]
- 38.Miao E.A., Alpuche-Aranda C.M., Dors M., Clark A.E., Bader M.W., Miller S.I., et al. Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1beta via Ipaf. Nat. Immunol. 2006;7:569–575. doi: 10.1038/ni1344. [DOI] [PubMed] [Google Scholar]
- 39.Fernandes-Alnemri T., Yu J.-W., Datta P., Wu J., Alnemri E.S. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature. 2009;458:509–513. doi: 10.1038/nature07710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hornung V., Ablasser A., Charrel-Dennis M., Bauernfeind F., Horvath G., Caffrey D.R., et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. 2009;458:514–518. doi: 10.1038/nature07725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xu H., Yang J., Gao W., Li L., Li P., Zhang L., et al. Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nature. 2014;513:237–241. doi: 10.1038/nature13449. [DOI] [PubMed] [Google Scholar]
- 42.Kayagaki N., Stowe I.B., Lee B.L., O’Rourke K., Anderson K., Warming S., et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 2015;526:666–671. doi: 10.1038/nature15541. [DOI] [PubMed] [Google Scholar]
- 43.Shi J., Zhao Y., Wang K., Shi X., Wang Y., Huang H., et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660–665. doi: 10.1038/nature15514. [DOI] [PubMed] [Google Scholar]
- 44.He W-t, Wan H., Hu L., Chen P., Wang X., Huang Z., et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 2015;25:1285–1298. doi: 10.1038/cr.2015.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ding J., Wang K., Liu W., She Y., Sun Q., Shi J., et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. 2016;535:111–116. doi: 10.1038/nature18590. [DOI] [PubMed] [Google Scholar]
- 46.Sborgi L., Ruhl S., Mulvihill E., Pipercevic J., Heilig R., Stahlberg H., et al. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J. 2016;35:1766–1778. doi: 10.15252/embj.201694696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu X., Zhang Z., Ruan J., Pan Y., Magupalli V.G., Wu H., et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 2016;535:153–158. doi: 10.1038/nature18629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Doitsh G., Galloway N.L.K., Geng X., Yang Z., Monroe K.M., Zepeda O., et al. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature. 2014;505:509–514. doi: 10.1038/nature12940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang Q., Gao H., Clark K.M., Mugisha C.S., Davis K., Tang J.P., et al. CARD8 is an inflammasome sensor for HIV-1 protease activity. Science. 2021;371:eabe1707. doi: 10.1126/science.abe1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bauernfried S., Scherr M.J., Pichlmair A., Duderstadt K.E., Hornung V. Human NLRP1 is a sensor for double-stranded RNA. Science. 2021;371:eabd0811. doi: 10.1126/science.abd0811. [DOI] [PubMed] [Google Scholar]
- 51.Chavarría-Smith J., Mitchell P.S., Ho A.M., Daugherty M.D., Vance R.E. Functional and evolutionary analyses identify proteolysis as a general mechanism for NLRP1 inflammasome activation. PLoS Pathog. 2016;12 doi: 10.1371/journal.ppat.1006052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shrivastava G., Valenzuela Leon P.C., Calvo E. Inflammasome Fuels Dengue Severity. Front. Cell. Infect. Microbiol. 2020;10:489. doi: 10.3389/fcimb.2020.00489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wu M.F., Chen S.T., Yang A.H., Lin W.W., Lin Y.L., Chen N.J., et al. CLEC5A is critical for dengue virus-induced inflammasome activation in human macrophages. Blood. 2013;121:95–106. doi: 10.1182/blood-2012-05-430090. [DOI] [PubMed] [Google Scholar]
- 54.He Z., An S., Chen J., Zhang S., Tan C., Yu J., et al. Neural progenitor cell pyroptosis contributes to Zika virus-induced brain atrophy and represents a therapeutic target. Proc. Natl. Acad. Sci. 2020;117:23869–23878. doi: 10.1073/pnas.2007773117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shi C.-S., Nabar N.R., Huang N.-N., Kehrl J.H. SARS-Coronavirus Open Reading Frame-8b triggers intracellular stress pathways and activates NLRP3 inflammasomes. Cell Death Discovery. 2019;5:101. doi: 10.1038/s41420-019-0181-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee S., Channappanavar R., Kanneganti T.D. Coronaviruses: Innate Immunity, Inflammasome Activation, Inflammatory Cell Death, and Cytokines. Trends Immunol. 2020;41:1083–1099. doi: 10.1016/j.it.2020.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rodrigues T.S., de Sá K.S.G., Ishimoto A.Y., Becerra A., Oliveira S., Almeida L., et al. Inflammasomes are activated in response to SARS-CoV-2 infection and are associated with COVID-19 severity in patients. J. Exp. Med. 2020;218 doi: 10.1084/jem.20201707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Junqueira C., Crespo Â., Ranjbar S., Ingber J., Parry B., Ravid S., et al. SARS-CoV-2 infects blood monocytes to activate NLRP3 and AIM2 inflammasomes, pyroptosis and cytokine release. medRxiv. 2021 [Google Scholar]
- 59.Komatsu T., Tanaka Y., Kitagawa Y., Koide N., Naiki Y., Morita N., et al. Sendai Virus V Protein Inhibits the Secretion of Interleukin-1β by Preventing NLRP3 Inflammasome Assembly. J. Virol. 2018;92 doi: 10.1128/JVI.00842-18. e00842–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lei X., Zhang Z., Xiao X., Qi J., He B., Wang J., et al. Enterovirus 71 Inhibits Pyroptosis through Cleavage of Gasdermin D. J. Virol. 2017;91:e01069–e01117. doi: 10.1128/JVI.01069-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang H., Lei X., Xiao X., Yang C., Lu W., Huang Z., et al. Reciprocal Regulation between Enterovirus 71 and the NLRP3 Inflammasome. Cell Rep. 2015;12:42–48. doi: 10.1016/j.celrep.2015.05.047. [DOI] [PubMed] [Google Scholar]
- 62.de Vasconcelos N.M., Van Opdenbosch N., Van Gorp H., Martin-Perez R., Zecchin A., Vandenabeele P., et al. An Apoptotic Caspase Network Safeguards Cell Death Induction in Pyroptotic Macrophages. Cell Rep. 2020;32 doi: 10.1016/j.celrep.2020.107959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Malireddi R.K.S., Gurung P., Kesavardhana S., Samir P., Burton A., Mummareddy H., et al. Innate immune priming in the absence of TAK1 drives RIPK1 kinase activity-independent pyroptosis, apoptosis, necroptosis, and inflammatory disease. J. Exp. Med. 2020;217 doi: 10.1084/jem.20191644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sarhan J., Liu B.C., Muendlein H.I., Li P., Nilson R., Tang A.Y., et al. Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection. Proc. Natl. Acad. Sci. U. S. A. 2018;115:E10888–E10897. doi: 10.1073/pnas.1809548115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang Y., Gao W., Shi X., Ding J., Liu W., He H., et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature. 2017;547:99–103. doi: 10.1038/nature22393. [DOI] [PubMed] [Google Scholar]
- 66.Galluzzi L., Vitale I., Aaronson S.A., Abrams J.M., Adam D., Agostinis P., et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25:486–541. doi: 10.1038/s41418-017-0012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dickens L.S., Boyd R.S., Jukes-Jones R., Hughes M.A., Robinson G.L., Fairall L., et al. A death effector domain chain DISC model reveals a crucial role for caspase-8 chain assembly in mediating apoptotic cell death. Mol. Cell. 2012;47:291–305. doi: 10.1016/j.molcel.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Feoktistova M., Geserick P., Kellert B., Dimitrova Diana P., Langlais C., Hupe M., et al. cIAPs Block Ripoptosome Formation, a RIP1/Caspase-8 Containing Intracellular Cell Death Complex Differentially Regulated by cFLIP Isoforms. Mol. Cell. 2011;43:449–463. doi: 10.1016/j.molcel.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Julien O., Wells J.A. Caspases and their substrates. Cell Death Differ. 2017;24:1380–1389. doi: 10.1038/cdd.2017.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Roucou X., Montessuit S., Antonsson B., Martinou J.C. Bax oligomerization in mitochondrial membranes requires tBid (caspase-8-cleaved Bid) and a mitochondrial protein. Biochem. J. 2002;368:915–921. doi: 10.1042/BJ20020972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chipuk J.E., Bouchier-Hayes L., Green D.R. Mitochondrial outer membrane permeabilization during apoptosis: the innocent bystander scenario. Cell Death Differ. 2006;13:1396–1402. doi: 10.1038/sj.cdd.4401963. [DOI] [PubMed] [Google Scholar]
- 72.Elmore S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007;35:495–516. doi: 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li P., Nijhawan D., Budihardjo I., Srinivasula S.M., Ahmad M., Alnemri E.S., et al. Cytochrome c and dATP-Dependent Formation of Apaf-1/Caspase-9 Complex Initiates an Apoptotic Protease Cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- 74.Holcik M., Gibson H., Korneluk R.G. XIAP: apoptotic brake and promising therapeutic target. Apoptosis. 2001;6:253–261. doi: 10.1023/a:1011379307472. [DOI] [PubMed] [Google Scholar]
- 75.Adrain C., Creagh E.M., Martin S.J. Apoptosis-associated release of Smac/DIABLO from mitochondria requires active caspases and is blocked by Bcl-2. EMBO J. 2001;20:6627–6636. doi: 10.1093/emboj/20.23.6627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Brojatsch J., Naughton J., Rolls M.M., Zingler K., Young J.A. CAR1, a TNFR-related protein, is a cellular receptor for cytopathic avian leukosis-sarcoma viruses and mediates apoptosis. Cell. 1996;87:845–855. doi: 10.1016/s0092-8674(00)81992-3. [DOI] [PubMed] [Google Scholar]
- 77.Hanon E., Meyer G., Vanderplasschen A., Dessy-Doize C., Thiry E., Pastoret P.P. Attachment but not penetration of bovine herpesvirus 1 is necessary to induce apoptosis in target cells. J. Virol. 1998;72:7638–7641. doi: 10.1128/jvi.72.9.7638-7641.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Briard B., Place D.E., Kanneganti T.D. DNA Sensing in the Innate Immune Response. Physiology (Bethesda, Md) 2020;35:112–124. doi: 10.1152/physiol.00022.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lei Y., Moore C.B., Liesman R.M., O'Connor B.P., Bergstralh D.T., Chen Z.J., et al. MAVS-mediated apoptosis and its inhibition by viral proteins. PLoS ONE. 2009;4 doi: 10.1371/journal.pone.0005466. e5466-e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yu C.-Y., Chiang R.-L., Chang T.-H., Liao C.-L., Lin Y.-L. The Interferon Stimulator Mitochondrial Antiviral Signaling Protein Facilitates Cell Death by Disrupting the Mitochondrial Membrane Potential and by Activating Caspases. J. Virol. 2010;84:2421–2431. doi: 10.1128/JVI.02174-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chattopadhyay S., Marques J.T., Yamashita M., Peters K.L., Smith K., Desai A., et al. Viral apoptosis is induced by IRF-3-mediated activation of Bax. EMBO J. 2010;29:1762–1773. doi: 10.1038/emboj.2010.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sze A., Belgnaoui S.M., Olagnier D., Lin R., Hiscott J., van Grevenynghe J. Host restriction factor SAMHD1 limits human T cell leukemia virus type 1 infection of monocytes via STING-mediated apoptosis. Cell Host Microbe. 2013;14:422–434. doi: 10.1016/j.chom.2013.09.009. [DOI] [PubMed] [Google Scholar]
- 83.Kirshner J.R., Karpova A.Y., Kops M., Howley P.M. Identification of TRAIL as an interferon regulatory factor 3 transcriptional target. J. Virol. 2005;79:9320–9324. doi: 10.1128/JVI.79.14.9320-9324.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Knowlton J.J., Dermody T.S., Holm G.H. Apoptosis induced by mammalian reovirus is beta interferon (IFN) independent and enhanced by IFN regulatory factor 3- and NF-κB-dependent expression of Noxa. J. Virol. 2012;86:1650–1660. doi: 10.1128/JVI.05924-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Myong S., Cui S., Cornish P.V., Kirchhofer A., Gack M.U., Jung J.U., et al. Cytosolic viral sensor RIG-I is a 5'-Triphosphate–dependent translocase on double-stranded RNA. Science. 2009;323:1070–1074. doi: 10.1126/science.1168352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Peisley A., Wu B., Xu H., Chen Z.J., Hur S. Structural basis for ubiquitin-mediated antiviral signal activation by RIG-I. Nature. 2014;509:110–114. doi: 10.1038/nature13140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Paz S., Vilasco M., Werden S.J., Arguello M., Joseph-Pillai D., Zhao T., et al. A functional C-terminal TRAF3-binding site in MAVS participates in positive and negative regulation of the IFN antiviral response. Cell Res. 2011;21:895–910. doi: 10.1038/cr.2011.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Liu S., Chen J., Cai X., Wu J., Chen X., Wu Y.T., et al. MAVS recruits multiple ubiquitin E3 ligases to activate antiviral signaling cascades. Elife. 2013;2 doi: 10.7554/eLife.00785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ivashkiv L.B., Donlin L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014;14:36–49. doi: 10.1038/nri3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kibler K.V., Shors T., Perkins K.B., Zeman C.C., Banaszak M.P., Biesterfeldt J., et al. Double-stranded RNA is a trigger for apoptosis in vaccinia virus-infected cells. J. Virol. 1997;71:1992–2003. doi: 10.1128/jvi.71.3.1992-2003.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Balachandran S., Kim C.N., Yeh W.C., Mak T.W., Bhalla K., Barber G.N. Activation of the dsRNA-dependent protein kinase, PKR, induces apoptosis through FADD-mediated death signaling. EMBO J. 1998;17:6888–6902. doi: 10.1093/emboj/17.23.6888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Castelli J.C., Hassel B.A., Wood K.A., Li X.L., Amemiya K., Dalakas M.C., et al. A study of the interferon antiviral mechanism: apoptosis activation by the 2–5A system. J. Exp. Med. 1997;186:967–972. doi: 10.1084/jem.186.6.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Villunger A., Michalak E.M., Coultas L., Müllauer F., Böck G., Ausserlechner M.J., et al. p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science. 2003;302:1036–1038. doi: 10.1126/science.1090072. [DOI] [PubMed] [Google Scholar]
- 94.Trapani J.A., Smyth M.J. Functional significance of the perforin/granzyme cell death pathway. Nat. Rev. Immunol. 2002;2:735–747. doi: 10.1038/nri911. [DOI] [PubMed] [Google Scholar]
- 95.Lowin B., Hahne M., Mattmann C., Tschopp J. Cytolytic T-cell cytotoxicity is mediated through perforin and Fas lytic pathways. Nature. 1994;370:650–652. doi: 10.1038/370650a0. [DOI] [PubMed] [Google Scholar]
- 96.Skaletskaya A., Bartle L.M., Chittenden T., McCormick A.L., Mocarski E.S., Goldmacher V.S. A cytomegalovirus-encoded inhibitor of apoptosis that suppresses caspase-8 activation. Proc. Natl. Acad. Sci. 2001;98:7829–7834. doi: 10.1073/pnas.141108798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Quan L.T., Caputo A., Bleackley R.C., Pickup D.J., Salvesen G.S. Granzyme B is inhibited by the cowpox virus serpin cytokine response modifier A. J. Biol. Chem. 1995;270:10377–10379. doi: 10.1074/jbc.270.18.10377. [DOI] [PubMed] [Google Scholar]
- 98.Zhou Q., Snipas S., Orth K., Muzio M., Dixit V.M., Salvesen G.S. Target protease specificity of the viral serpin CrmA. Analysis of five caspases. J. Biol. Chem. 1997;272:7797–7800. doi: 10.1074/jbc.272.12.7797. [DOI] [PubMed] [Google Scholar]
- 99.Chiou S.-K., Tseng C.-C., Rao L., White E. Functional complementation of the adenovirus E1B 19-kilodalton protein with Bcl-2 in the inhibition of apoptosis in infected cells. J. Virol. 1994;68:6553–6566. doi: 10.1128/jvi.68.10.6553-6566.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Henderson S., Huen D., Rowe M., Dawson C., Johnson G., Rickinson A. Epstein-Barr virus-coded BHRF1 protein, a viral homologue of Bcl-2, protects human B cells from programmed cell death. Proc. Natl. Acad. Sci. 1993;90:8479–8483. doi: 10.1073/pnas.90.18.8479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kvansakul M., Wei A.H., Fletcher J.I., Willis S.N., Chen L., Roberts A.W., et al. Structural basis for apoptosis inhibition by Epstein-Barr virus BHRF1. PLoS Pathog. 2010;6 doi: 10.1371/journal.ppat.1001236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Delanghe T., Dondelinger Y., Bertrand M.J.M. RIPK1 Kinase-Dependent Death: A Symphony of Phosphorylation Events. Trends Cell Biol. 2020;30:189–200. doi: 10.1016/j.tcb.2019.12.009. [DOI] [PubMed] [Google Scholar]
- 103.Newton K., Wickliffe K.E., Dugger D.L., Maltzman A., Roose-Girma M., Dohse M., et al. Cleavage of RIPK1 by caspase-8 is crucial for limiting apoptosis and necroptosis. Nature. 2019;574:428–431. doi: 10.1038/s41586-019-1548-x. [DOI] [PubMed] [Google Scholar]
- 104.Lalaoui N., Boyden S.E., Oda H., Wood G.M., Stone D.L., Chau D., et al. Mutations that prevent caspase cleavage of RIPK1 cause autoinflammatory disease. Nature. 2020;577:103–108. doi: 10.1038/s41586-019-1828-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Orozco S., Yatim N., Werner M.R., Tran H., Gunja S.Y., Tait S.W., et al. RIPK1 both positively and negatively regulates RIPK3 oligomerization and necroptosis. Cell Death Differ. 2014;21:1511–1521. doi: 10.1038/cdd.2014.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wang H., Sun L., Su L., Rizo J., Liu L., Wang L.-F., et al. Mixed Lineage Kinase Domain-like Protein MLKL Causes Necrotic Membrane Disruption upon Phosphorylation by RIP3. Mol. Cell. 2014;54:133–146. doi: 10.1016/j.molcel.2014.03.003. [DOI] [PubMed] [Google Scholar]
- 107.Oberst A., Dillon C.P., Weinlich R., McCormick L.L., Fitzgerald P., Pop C., et al. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature. 2011;471:363–367. doi: 10.1038/nature09852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Upton J.W., Kaiser W.J., Mocarski E.S. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe. 2012;11:290–297. doi: 10.1016/j.chom.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Cho Y.S., Challa S., Moquin D., Genga R., Ray T.D., Guildford M., et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137:1112–1123. doi: 10.1016/j.cell.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Daniels B.P., Snyder A.G., Olsen T.M., Orozco S., Oguin T.H., 3rd, Tait S.W.G., et al. RIPK3 Restricts Viral Pathogenesis via Cell Death-Independent Neuroinflammation. Cell. 2017;169 doi: 10.1016/j.cell.2017.03.011. 301–13.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Daniels B.P., Kofman S.B., Smith J.R., Norris G.T., Snyder A.G., Kolb J.P., et al. The Nucleotide Sensor ZBP1 and Kinase RIPK3 Induce the Enzyme IRG1 to Promote an Antiviral Metabolic State in Neurons. Immunity. 2019;50:64–76.e4. doi: 10.1016/j.immuni.2018.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Koehler H., Cotsmire S., Langland J., Kibler K.V., Kalman D., Upton J.W., et al. Inhibition of DAI-dependent necroptosis by the Z-DNA binding domain of the vaccinia virus innate immune evasion protein, E3. Proc. Natl. Acad. Sci. U. S. A. 2017;114:11506–11511. doi: 10.1073/pnas.1700999114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Upton J.W., Kaiser W.J., Mocarski E.S. Virus inhibition of RIP3-dependent necrosis. Cell Host Microbe. 2010;7:302–313. doi: 10.1016/j.chom.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Liu Z., Nailwal H., Rector J., Rahman M.M., Sam R., McFadden G., et al. A class of viral inducer of degradation of the necroptosis adaptor RIPK3 regulates virus-induced inflammation. Immunity. 2021;54 doi: 10.1016/j.immuni.2020.11.020. 247–58.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Man S.M., Hopkins L.J., Nugent E., Cox S., Glück I.M., Tourlomousis P., et al. Inflammasome activation causes dual recruitment of NLRC4 and NLRP3 to the same macromolecular complex. Proc. Natl. Acad. Sci. U. S. A. 2014;111:7403–7408. doi: 10.1073/pnas.1402911111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Orning P., Weng D., Starheim K., Ratner D., Best Z., Lee B., et al. Pathogen blockade of TAK1 triggers caspase-8–dependent cleavage of gasdermin D and cell death. Science. 2018;362:1064–1069. doi: 10.1126/science.aau2818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Sagulenko V., Thygesen S.J., Sester D.P., Idris A., Cridland J.A., Vajjhala P.R., et al. AIM2 and NLRP3 inflammasomes activate both apoptotic and pyroptotic death pathways via ASC. Cell Death Differ. 2013;20:1149–1160. doi: 10.1038/cdd.2013.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Tsuchiya K., Nakajima S., Hosojima S., Thi Nguyen D., Hattori T., Manh Le T., et al. Caspase-1 initiates apoptosis in the absence of gasdermin D. Nat. Commun. 2019;10:2091. doi: 10.1038/s41467-019-09753-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zheng M., Kanneganti T.D. Newly Identified Function of Caspase-6 in ZBP1-mediated Innate Immune Responses, NLRP3 Inflammasome Activation, PANoptosis, and Host Defense. J. Cell Immunol. 2020;2:341–347. doi: 10.33696/immunology.2.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zheng M., Kanneganti T.D. The regulation of the ZBP1-NLRP3 inflammasome and its implications in pyroptosis, apoptosis, and necroptosis (PANoptosis) Immunol. Rev. 2020;297:26–38. doi: 10.1111/imr.12909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Huang Z., Wu S.-Q., Liang Y., Zhou X., Chen W., Li L., et al. RIP1/RIP3 Binding to HSV-1 ICP6 Initiates Necroptosis to Restrict Virus Propagation in Mice. Cell Host Microbe. 2015;17:229–242. doi: 10.1016/j.chom.2015.01.002. [DOI] [PubMed] [Google Scholar]
- 123.Fletcher-Etherington A., Nobre L., Nightingale K., Antrobus R., Nichols J., Davison A.J., et al. Human cytomegalovirus protein pUL36: A dual cell death pathway inhibitor. Proc. Natl. Acad. Sci. 2020;117:18771–18779. doi: 10.1073/pnas.2001887117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Carter J.J., Nemeno J.G.E., Oh J.J., Houghton J.E., Dix R.D. Atypical cytomegalovirus retinal disease in pyroptosis-deficient mice with murine acquired immunodeficiency syndrome. Exp. Eye Res. 2021;209 doi: 10.1016/j.exer.2021.108651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Carter J., Richardson Q., Jasso G., Alston C., Dix R.D. Pyroptosis Inducer, Gasdermin D, is Stimulated Intraocularly in Mice with Retrovirus-Induced Immunosuppression (MAIDS) During Experimental Murine Cytomegalovirus (MCMV) Retinitis. Invest. Ophthalmol. Vis. Sci. 2017;58:3626. [Google Scholar]
- 126.Malireddi R.K.S., Tweedell R.E., Kanneganti T.D. PANoptosis components, regulation, and implications. Aging. 2020;12:11163–11164. doi: 10.18632/aging.103528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Place D.E., Kanneganti T.D. The innate immune system and cell death in autoinflammatory and autoimmune disease. Curr. Opin. Immunol. 2020;67:95–105. doi: 10.1016/j.coi.2020.10.013. [DOI] [PubMed] [Google Scholar]
- 128.Samir P., Malireddi R.K.S., Kanneganti T.-D. The PANoptosome: A deadly protein complex driving pyroptosis, apoptosis, and necroptosis (PANoptosis) Front. Cell. Infect. Microbiol. 2020;10 doi: 10.3389/fcimb.2020.00238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Briard B., Malireddi R.K.S., Kanneganti T.D. Role of inflammasomes/pyroptosis and PANoptosis during fungal infection. PLoS Pathog. 2021;17 doi: 10.1371/journal.ppat.1009358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lee S, Karki R, Wang Y, Nguyen LN, Kalathur RC, Kanneganti TD. AIM2 forms a complex with pyrin and ZBP1 to drive PANoptosis and host defense. Nature. 2021;597(7876):415–419. doi: 10.1038/s41586-021-03875-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Wang Y, Kanneganti TD. From pyroptosis, apoptosis and necroptosis to PANoptosis: A mechanistic compendium of programmed cell death pathways. Comput Struct Biotechnol J. 2021;19:4641–4657. doi: 10.1016/j.csbj.2021.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Broz P, von Moltke J, Jones JW, Vance RE, Monack DM. Differential requirement for Caspase-1 autoproteolysis in pathogen-induced cell death and cytokine processing. Cell Host Microbe. 2010;8(6):471–483. doi: 10.1016/j.chom.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Van Opdenbosch N, Gurung P, Vande Walle L, Fossoul A, Kanneganti TD, Lamkanfi M. Activation of the NLRP1b inflammasome independently of ASC-mediated caspase-1 autoproteolysis and speck formation. Nat Commun. 2014;5(3209) doi: 10.1038/ncomms4209. [DOI] [PMC free article] [PubMed] [Google Scholar]