

Abstract
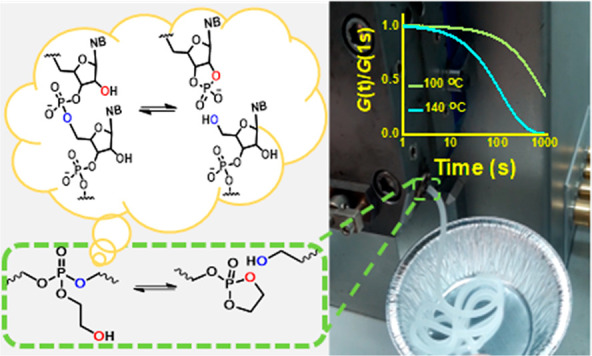
Bond exchange via neighboring group-assisted reactions in dynamic covalent networks results in efficient mechanical relaxation. In Nature, the high reactivity of RNA toward nucleophilic substitution is largely attributed to the formation of a cyclic phosphate ester intermediate via neighboring group participation. We took inspiration from RNA to develop a dynamic covalent network based on β-hydroxyl-mediated transesterifications of hydroxyethyl phosphate triesters. A simple one-step synthetic strategy provided a network containing phosphate triesters with a pendant hydroxyethyl group. 31P solid-state NMR demonstrated that a cyclic phosphate triester is an intermediate in transesterification, leading to dissociative network rearrangement. Significant viscous flow at 60–100 °C makes the material suitable for fast processing via extrusion and compression molding.
Introduction
Dynamic covalent networks (DCNs) are covalently cross-linked polymers that undergo bond exchange under an external stimulus such as heat or light. Thermally activated DCNs show mechanical properties comparable to those of thermosets at low temperatures while at elevated temperatures, they become reprocessable like thermoplastics via bond exchange-mediated network rearrangement.1−6 Such properties have resulted in extensive efforts to develop a wide range of dynamic covalent chemistries for use in DCNs with various applications.7 Based on the network rearrangement mechanism, DCNs have been classified into associative and dissociative networks. In a dissociative DCN, the dissociation of an existing bond precedes the formation of a new bond and therefore the rearrangement occurs via an intermediate with lower cross-link density. In an associative DCN, the new bond formation precedes dissociation of existing bonds and the cross-link density does not decrease during the network rearrangement process.8,9 The first reported DCN was based on a Diels–Alder reaction, which allows network rearrangement via a dissociative process.10 Subsequently, many DCNs have been designed based on Diels–Alder and other pericyclic reactions among which triazolinedione (TAD) chemistry is notable because of its fast network rearrangement kinetics.11 Leibler and co-workers were the first to report associative DCNs, also known as “vitrimers”, based on catalyzed transesterification in polyester-polyols.12 A series of contributions on transesterification-based vitrimers followed, mainly focused on tuning material properties by varying the transesterification catalyst.13−15 In view of the problems related to catalyst leaching, current interest has shifted toward vitrimers which show bond exchange without the aid of external catalysts. Dynamic chemistries such as oxime-ester,16 boronic ester,17,18 silyl ether19 exchange, vinylogous urethane transamination,20 and transthioetherification21 have been implemented to obtain catalyst free DCNs with excellent thermomechanical properties. Our group has recently introduced catalyst-free phosphate triester transesterification as a dynamic covalent chemistry in polymeric networks.22 Stress relaxation of the network at high temperatures was comparable to that of previously reported carboxylate ester networks but without the need for a catalyst. Subsequent papers have reported flame retardant DCNs based on phosphate esters,23 including an associative network based on efficient β-hydroxy phosphate exchange,24 although the chemistry and the mechanism of rearrangement were not studied in detail.
A recent development in DCN research is the use of neighboring group-assisted exchange reactions.25−28 These reactions lead to relatively fast topological rearrangement via a dissociative mechanism without the aid of external catalysts. The rearrangement occurs through a high energy cyclic intermediate such that the equilibrium lies almost completely toward the associated state and thus the material shows vitrimer-like properties. The network integrity is maintained during processing while viscous flow in the material is relatively restricted. On the other hand, dissociative DCNs with strongly temperature-dependent equilibrium constants are easily reprocessed because network dissociation leads to low viscosity at high temperature.29 However, network re-formation on cooling is often slow and therefore re-establishment of the network integrity can be an issue.8 A dissociative DCN with a moderate temperature-dependence of the equilibrium constant, with respect to the above extremes, might show significant viscous flow under processing conditions while maintaining a decent network integrity. These properties are highly desirable for fast reprocessing of DCNs. To achieve these properties in a catalyst-free DCN based on neighboring group-assisted phosphate transesterification, we turned to biological phosphate chemistry for inspiration.
A classic example of neighboring group participation in biochemical reactions is the β-hydroxyl-mediated transesterification of the phosphate diester in RNA, yielding a 2′,3′ cyclic phosphate diester. RNA ligation occurs via subsequent transesterification on the 2′,3′ cyclic phosphate diester to establish a 2′-5′ or 3′-5′ linkage (Figure 1a).30 These reactions occur enzymatically on phosphate diesters at relatively low temperatures. On the basis of this transesterification mechanism, we designed a DCN based on β-hydroxyl-mediated transesterification in phosphate triesters (Figure 1b), which has a higher reactivity than that of diesters in nucleophilic substitutions.31 Recently, hydrolysis of β-hydroxy phosphate triester containing polymers was shown to be very fast.32 We use a simple one-step approach to synthesize a polycaprolactone network incorporating phosphate triesters with β-hydroxyl functionality (Figure 2a). The flow properties of the network are studied in detail with shear rheology. The material shows rapid rearrangement at relatively low temperatures of 60–120 °C, making the material eligible for fast processing techniques such as extrusion. The mechanism of network rearrangement is studied in detail with 31P solid-state NMR (31P SSNMR) at variable temperature (VT).
Figure 1.
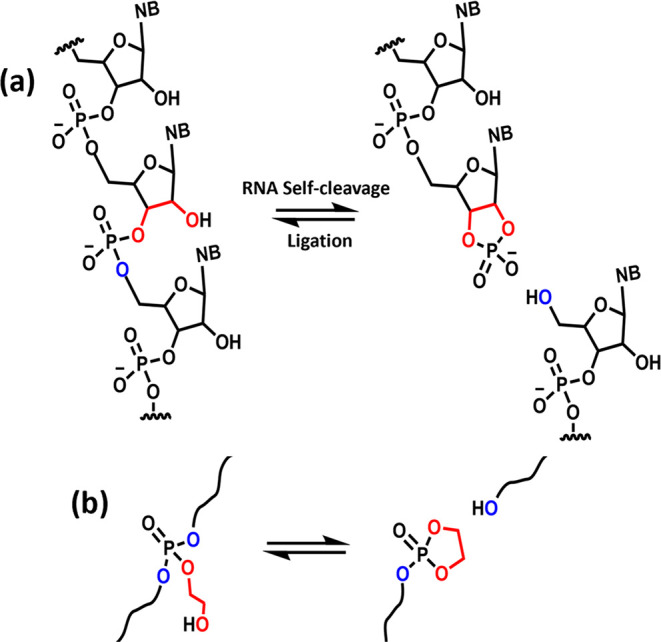
Illustration of (a) RNA self-cleavage through participation of β-hydroxyl functionality, a reaction highly relevant in several biochemical pathways. (b) Polymer chain cleavage at phosphate triester links with β-hydroxyl functionality, a design inspired from RNA structure.
Figure 2.

(a) Reaction scheme for synthesis of PCL-PX. (b) 31P SSNMR spectrum of PCL-PX at room temperature. (c) Image showing PCL-PX reprocessing by compression molding.
Results and Discussion
A polymeric network was synthesized by reacting commercially available polycaprolactone (PCL) triol (Mn = 1.93 kg/mol) with ethylene glycol chlorophosphate in toluene in a 2:3 molar ratio (Figure 2a). The reaction mixture was heated to 45 °C for 30 min followed by 60 °C for 2 h, and residual HCl and toluene were removed under vacuum at 100 °C for 18 h. The product (PCL-PX) had a gel fraction, determined by extraction with THF, of 75% and a swelling ratio of 4.3 by weight. The static 31P SSNMR spectrum of PCL-PX at room temperature had a single, relatively broad peak centered at −1.35 ppm which is in the characteristic chemical shift region of aliphatic phosphate triesters (Figure 2b). A second, smaller peak around–7.28 ppm may be assigned to mixed anhydride moieties formed in side reactions. The 31P SSNMR spectrum acquired at 75 °C has more resolved peaks due to enhanced chain mobility (see Supporting Information) and shows that chemically nonequivalent phosphate triesters are present, presumably different phosphate triesters with a varied number of ethylene glycol and PCL ester links. The part of the peak above 0 ppm indicates the presence of some phosphate diesters with a β-hydroxyl group that can form as a result of hydrolysis.
The material was shaped into circular disks and rectangular bars by compression molding at 100 °C for 25 min at a pressure of 10 MPa followed by slow cooling to room temperature under the same pressure (Figure 2c). The reshapability of the network is a qualitative indication that the material shows active bond exchange at higher temperatures. Isothermal thermogravimetric analysis (TGA) on PCL-PX at 100 °C shows that the material remains thermally robust for processing at this temperature (see Supporting Information).
Dynamic mechanical thermal analysis (DMTA) was performed on PCL-PX under elongational deformation to study its thermomechanical properties. The DMTA plot showed three transitions (Figure 3a). The transition around −40 °C is attributed to the glass transition. A second transition around 45 °C corresponds to melting of PCL crystalline domains within the network.33 These assignments have also been confirmed with differential scanning calorimetry (DSC) (see Supporting Information). Finally, above 110 °C a gradual drop in the storage modulus indicates lowering of the cross-link density, a typical feature for dissociative DCNs, which is attributed to a shift of the equilibrium toward the dissociated state. The reversibility of network dissociation was investigated with DMTA under shear. The network was heated from 100 °C to 140 °C at 0.5 °C/min and then cooled back to 100 °C following the same temperature ramp down. Upon heating, the rubbery plateau modulus dropped from 5.4 × 104 Pa to 4.5 × 104 Pa and within the experimental time was almost completely recovered on cooling, to 5.3 × 104 Pa (Figure 3b).
Figure 3.
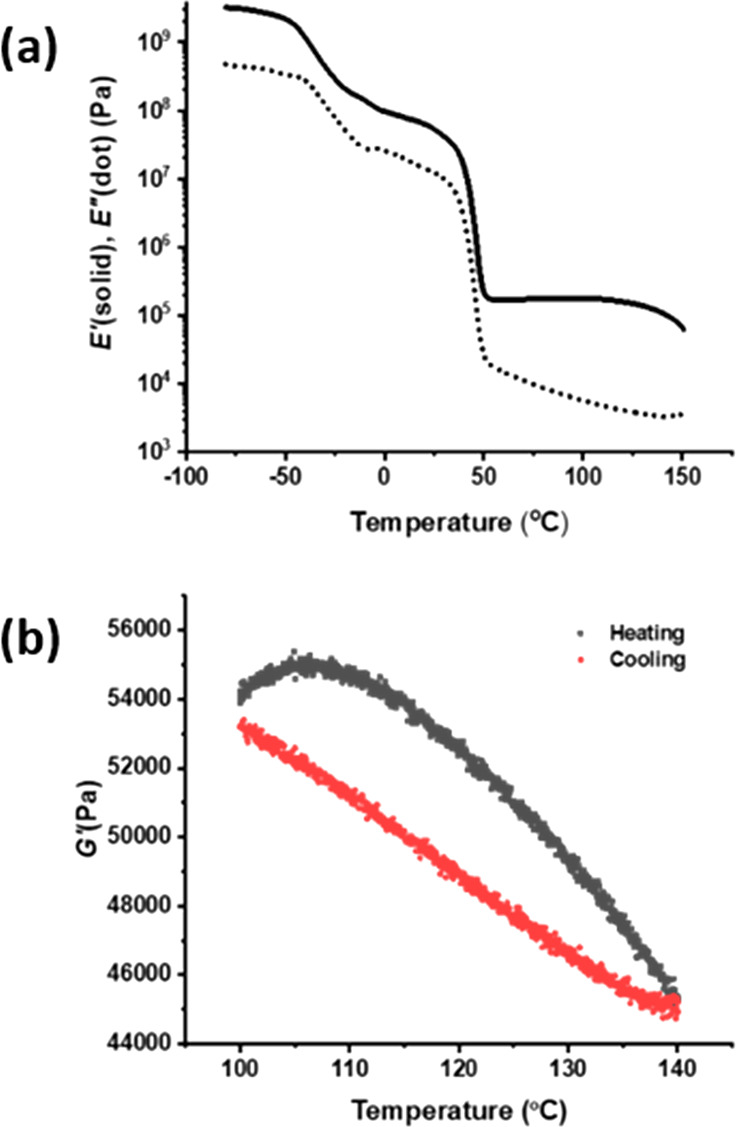
(a) DMTA on compression-molded PCL-PX sample under elongation rheology at 3 °C/min and 1 Hz frequency. (b) DMTA with heating and cooling run at 0.5 °C/min and 1 Hz frequency on compression-molded PCL-PX sample under shear rheology.
The network rearrangement kinetics were probed further with both step transient and oscillatory shear rheology. Stress relaxation experiments were performed on compression-molded PCL-PX samples at temperatures ranging from 80 °C to 160 °C under 1% strain (Figure 4a). At 160 °C, the attained stress was completely relaxed within 300 s. Stress relaxation was fitted satisfactorily with a stretched exponential function normalized at 1 s (eq 1) (fit parameters in Table 1).34 The temperature dependence of the characteristic relaxation time, obtained from the fit, follows Arrhenius behavior with an activation energy of 68 kJ/mol (Figure 4b), significantly lower than in the associative phosphate triester network previously published by us, where the activation energy is 104.5 kJ/mol.22 The value of the stretch parameter β lies between 0.85 and 0.9 and indicates a similar narrow distribution of relaxation times at all temperatures. Frequency sweep experiments under oscillatory shear were performed at temperatures between 100 °C and 160 °C to probe the effects of bond exchange on viscoelastic properties. The plateau storage modulus dropped significantly from 0.1 MPa at 100 °C to 0.04 MPa at 160 °C, indicative of network dissociation due to a shift in equilibrium toward the dissociated state. Furthermore, at 140 °C and 160 °C, the storage moduli become frequency dependent at lower frequencies and reach a crossover point with the loss modulus around 0.02 rad·s–1 at 160 °C (Figure 4c). This indicates that under those conditions, network rearrangement via bond exchange gives rise to predominantly viscous behavior.
Figure 4.

Shear rheology experiments on compression-molded PCL-PX samples. (a) Stress relaxation experiments under 1% strain with temperature variation. Relaxation plots fitted with stretched exponential decay (eq 1) (yellow dashed lines). (b) Arrhenius plot using characteristic relaxation times (τ) obtained from fit in plot a. (c) Frequency sweep experiments at different temperatures. (d) Creep-recovery experiments at room temperature (magnified in the inset), 60 °C (∼15 °C above melting temperature) and 100 °C (processing temperature).
Table 1. Parameters Obtained from Fitting the Stress Relaxation Plots of PCL-PX with Stretched Exponential Functiona.
| temperature (°C) | τ (s) | β |
|---|---|---|
| 80 | 3204 | 0.85 |
| 100 | 957 | 0.9 |
| 120 | 349 | 0.89 |
| 140 | 122 | 0.87 |
| 160 | 42 | 0.85 |
τ: Characteristic relaxation time, β: stretch factor, indicative of the distribution of relaxation times.
| 1 |
Creep was tested both at low temperatures and at the processing temperature using a parallel plate geometry under 104 Pa for 103 s (Figure 4d). A recovery step was also introduced after creep to allow the material to recover from the elastic strain. Therefore, the residual strain accounted for the permanent deformation of the material which can be attributed to the topology rearrangement. At 25 °C, PCL-PX is a semicrystalline solid and shows a negligible creep with a permanent deformation of 0.04%. Creep-recovery experiments were also performed at 60 °C to test flow properties above the melting point of PCL-PX. The material shows highly enhanced elastic strain; however, the residual strain after recovery is low at 1.4%. At 100 °C, which is also the material processing temperature, PCL-PX shows considerable creep, reaching 21% permanent deformation within 103 s under 104 Pa stress with a strain rate of 0.023% s–1.
The thermomechanical properties indicate that in this material, network rearrangement is dominated by a dissociative process. Chemical insight into the network rearrangement process was obtained from the network material directly. Often, mechanistic studies on network rearrangements are performed using model systems. However, the kinetic and thermodynamic parameters for the bond exchange process in a network may differ significantly from their values in model compounds. We studied the exchange process in PCL-PX in the bulk material with in situ variable temperature (VT) 31P SSNMR spectroscopy. Upon heating from 60 °C to 120 °C, two new peaks appear around 17 ppm next to the main peak at −1.35 ppm. The peak at 16.84 ppm can be attributed to the formation of cyclic phosphate triesters.35 A smaller peak at 17.96 ppm probably represents formation of a small amount of cyclic phosphate diester.36 At higher temperatures, these peaks become larger (Figure 5a) and their intensity drops again on cooling (Figure 5b). These results show that a dynamic equilibrium exists between cyclic phosphate esters and ring-opened phosphate esters. The cyclic phosphate triester formation temporarily removes a cross-link from the network and represents a dissociative mechanism for network rearrangement (Figure 5c). Formation of the cyclic phosphate ester is an endothermic process which is favored at high temperatures. The concentration ratio between the cyclic phosphate esters ([CP]) and ring-opened phosphate ester ([OP]) was calculated based on the VT 31P SSNMR spectra obtained during the heating run (see Supporting Information). The contribution from the phosphate diesters is expected to be insignificant, and hence it was considered that the [CP]:[OP] ratio is a fair representation of the relative number of dissociated chains at a given temperature. Considering this, ΔHo for the dissociation process was determined from the temperature dependence of [CP]:[OP] using the Van’t Hoff equation (see Supporting Information for details) and found to be 35 kJ/mol.
Figure 5.

VT 31P SSNMR spectra of PCL-PX during (a) heating and (b) cooling, showing reversible formation of cyclic phosphate triester at higher temperatures. (c) Schematic illustration depicting the equilibrium between the associated state and the dissociated state of PCL-PX. (d) Temperature dependence of the ratio between cyclic phosphate ester and ring-opened phosphate ester.
Because the network rearrangement in PCL-PX is fast, reprocessing via extrusion is possible at elevated temperatures. PCL-PX granules were loaded in a twin screw compounder–extruder. During the compounding, the torque dropped significantly from 15.1 N m to 7.2 N m when the temperature was increased from 70 °C to 120 °C (see Supporting Information). The extrusion was performed at 120 °C under 50 rpm screw speed (Figure 6a-image i). The extrudate was then cooled to room temperature (Figure 6a-image ii) and sonicated in THF (a good solvent for PCL) for 30 min in an ultrasonic bath. The extrudate maintained its shape after sonication in THF which indicates that under the given condition, the network particles underwent efficient sintering (Figure 6a-image iii). Moreover, the gel fraction of the material was retained (∼75% in THF at 20 °C) after the extrusion process. 31P NMR was performed on the pristine material and the extrudate by swelling the network in d6-DMSO. The results show no significant change in the chemical nature of the network material except for some exchange among the phosphate triesters (Figure 6b).
Figure 6.
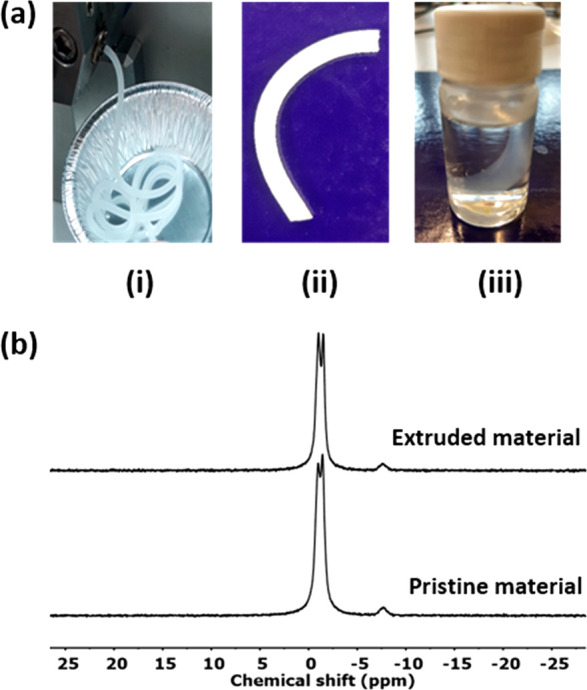
(a) Images showing (i) reprocessing via extrusion of PCL-PX at 120 °C, (ii) the extrudate, (iii) extrudate retains shape after sonication in THF for 30 min. (b) 31P NMR of pristine PCL-PX and extrudate swollen in d6-DMSO.
As previously discussed, the network is reprocessable at 100 °C and the material is thermally stable at this temperature. The limits of thermal stability and the reprocessing efficacy were studied by performing cycles of stress relaxation on the same PCL-PX sample. The results show identical stress relaxation rates at least up to five cycles at 100 °C (Figure 7a). However, repeated cycles of stress relaxation at 160 °C show a gradual decrease in stress relaxation rates in the consecutive cycles (Figure 7b). In our previous report on phosphate triester-based dynamic covalent networks, we showed that transesterification via distant OH groups is significant at temperatures around 160 °C. The slower stress relaxation on the subsequent cycles in PCL-PX at 160 °C can be attributed to loss of ethylene glycol from the network via distant OH-mediated transesterification (Figure 7c). Loss of ethylene glycol from the network implies that some of the phosphate triester links are devoid of β-hydroxyl functionality so neighboring assisted transesterification is not possible anymore. Further, the expulsion of ethylene glycol transforms a chain extension link into a cross-link thus increasing the cross-link density. This hypothesis was supported by a oscillatory shear experiment at 160 °C, that showed a gradual increase in storage modulus over time (Figure 7d) and with concomitant proportional weight loss as observed in isothermal TGA measurements at 160 °C (see Supporting Information). This observation re-emphasizes the importance of the β-hydroxyl functionality for the fast network rearrangement.
Figure 7.

(a) Repeated cycles of stress relaxation on same PCL-PX sample at (a) 100 °C and (b) 160 °C. (c) Schematic illustration of distant OH-mediated transesterification resulting in expulsion of ethylene glycol. (d) Isothermal oscillatory shear rheology on compression-molded PCL-PX sample at 160 °C.
Conclusions
The use of β-hydroxyl-assisted transesterification in phosphate triesters, inspired by RNA self-cleavage, results in a dissociative dynamic network that shows an excellent balance between mechanical integrity and processability. In contrast to the associative dynamic network of Feng et al.,24 where the β-hydroxyl functionality is part of the network, the pendant hydroxyethyl group in PCL-PX results in a dissociative network rearrangement via a cyclic phosphate triester intermediate.
As a result of the neighboring group participation, exchange kinetics, as probed through stress relaxation, are more than 100 times faster than in the phosphate triester network previously published by us,22 while the activation energy for stress relaxation drops from 104.5 kJ/mol in the associative phosphate network to 68 kJ/mol in the dissociative system. Fast kinetics ensure that after processing the cross-link density is recovered rapidly on cooling.
At processing temperatures, the dissociation of cross-links is limited as the material shows Arrhenius behavior up to at least 160 °C, implying that relaxation is still determined by the bond exchange kinetics under these conditions. On the other hand, dissociation is sufficient to allow extrusion at a relatively low temperature of 120 °C.
The activation energy of transesterification in PCL-PX (68 kJ/mol) is similar to other fast-exchanging dynamic covalent chemistries such as vinylogous urethane transamination (60 kJ/mol), dioxaborolane metathesis (77 kJ/mol), and transthioetherfication (63 kJ/mol). Fast exchanging networks often show significant creep at application temperatures. Due to the presence of the PCL crystalline domains below 45 °C, PCL-PX has a high modulus and shows negligible creep at room temperature. Even at 60 °C, well above its melting point, creep is limited. Therefore, this chemistry does provide a relatively wide temperature window for applications requiring low creep.
Incorporation of organophosphorus moieties imparts interesting properties to polymeric materials, such as flame retardancy,37−39 antifouling nature,40 and biodegradability.41,42 The current work shows that neighboring group participation in phosphate triester DCNs results in efficient network rearrangement and provides materials with excellent processability. In combination with attractive hydrolytic properties of β-hydroxy phosphate triesters and potential biocompatibility, these networks provide opportunities for product fabrication with additive manufacturing techniques such as fused deposition modeling and selective laser sintering in biomedical applications.
Acknowledgments
We thank Shrestha Banerjee, Radboud University for fruitful discussions.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.macromol.1c01504.
Materials, synthesis, instrumentation, and methods of characterization. Additional results from NMR studies, DSC, TGA, and rheology supporting data. Comparison of moduli, torque–temperature relationship in a compounder–extruder (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This work was funded by the Dutch Research Council (NWO), project no. 731.016.202.
The authors declare no competing financial interest.
Supplementary Material
References
- Wojtecki R. J.; Meador M. A.; Rowan S. J. Using the Dynamic Bond to Access Macroscopically Responsive Structurally Dynamic Polymers. Nat. Mater. 2011, 10 (1), 14–27. 10.1038/nmat2891. [DOI] [PubMed] [Google Scholar]
- Kloxin C. J.; Scott T. F.; Adzima B. J.; Bowman C. N. Covalent Adaptable Networks (CANs): A Unique Paradigm in Cross-Linked Polymers. Macromolecules 2010, 43 (6), 2643–2653. 10.1021/ma902596s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloxin C. J.; Bowman C. N. Covalent Adaptable Networks: Smart, Reconfigurable and Responsive Network Systems. Chem. Soc. Rev. 2013, 42 (17), 7161–7173. 10.1039/C3CS60046G. [DOI] [PubMed] [Google Scholar]
- Denissen W.; Winne J. M.; Du Prez F. E. Vitrimers: Permanent Organic Networks with Glass-like Fluidity. Chem. Sci. 2016, 7 (1), 30–38. 10.1039/C5SC02223A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Zee N. J.; Nicolaÿ R. Vitrimers: Permanently Crosslinked Polymers with Dynamic Network Topology. Prog. Polym. Sci. 2020, 104, 101233. 10.1016/j.progpolymsci.2020.101233. [DOI] [Google Scholar]
- Zou W.; Dong J.; Luo Y.; Zhao Q.; Xie T.. Dynamic Covalent Polymer Networks: From Old Chemistry to Modern Day Innovations. Adv. Mater. 2017, 29 ( (14), ), 1606100. 10.1002/adma.201606100. [DOI] [PubMed] [Google Scholar]
- McBride M. K.; Worrell B. T.; Brown T.; Cox L. M.; Sowan N.; Wang C.; Podgorski M.; Martinez A. M.; Bowman C. N. Enabling Applications of Covalent Adaptable Networks. Annu. Rev. Chem. Biomol. Eng. 2019, 10, 175–198. 10.1146/annurev-chembioeng-060718-030217. [DOI] [PubMed] [Google Scholar]
- Winne J. M.; Leibler L.; Du Prez F. E. Dynamic Covalent Chemistry in Polymer Networks: A Mechanistic Perspective. Polym. Chem. 2019, 10 (45), 6091–6108. 10.1039/C9PY01260E. [DOI] [Google Scholar]
- Scheutz G. M.; Lessard J. J.; Sims M. B.; Sumerlin B. S. Adaptable Crosslinks in Polymeric Materials: Resolving the Intersection of Thermoplastics and Thermosets. J. Am. Chem. Soc. 2019, 141 (41), 16181–16196. 10.1021/jacs.9b07922. [DOI] [PubMed] [Google Scholar]
- Chen X.; Dam M. A.; Ono K.; Mal A.; Shen H.; Nutt S. R.; Sheran K.; Wudl F. A Thermally Re-Mendable Cross-Linked Polymeric Material. Science 2002, 295 (5560), 1698–1702. 10.1126/science.1065879. [DOI] [PubMed] [Google Scholar]
- Billiet S.; De Bruycker K.; Driessen F.; Goossens H.; Van Speybroeck V.; Winne J. M.; Du Prez F. E. Triazolinediones Enable Ultrafast and Reversible Click Chemistry for the Design of Dynamic Polymer Systems. Nat. Chem. 2014, 6 (9), 815–821. 10.1038/nchem.2023. [DOI] [PubMed] [Google Scholar]
- Montarnal D.; Capelot M.; Tournilhac F.; Leibler L. Silica-like Malleable Materials from Permanent Organic Networks. Science 2011, 334 (6058), 965–968. 10.1126/science.1212648. [DOI] [PubMed] [Google Scholar]
- Brutman J. P.; Delgado P. A.; Hillmyer M. A. Polylactide Vitrimers. ACS Macro Lett. 2014, 3 (7), 607–610. 10.1021/mz500269w. [DOI] [PubMed] [Google Scholar]
- Zhou Y.; Goossens J. G. P.; Sijbesma R. P.; Heuts J. P. A. Poly(Butylene Terephthalate)/Glycerol-Based Vitrimers via Solid-State Polymerization. Macromolecules 2017, 50 (17), 6742–6751. 10.1021/acs.macromol.7b01142. [DOI] [Google Scholar]
- Self J. L.; Dolinski N. D.; Zayas M. S.; Read De Alaniz J.; Bates C. M. Brønsted-Acid-Catalyzed Exchange in Polyester Dynamic Covalent Networks. ACS Macro Lett. 2018, 7 (7), 817–821. 10.1021/acsmacrolett.8b00370. [DOI] [PubMed] [Google Scholar]
- He C.; Shi S.; Wang D.; Helms B. A.; Russell T. P. Poly(Oxime-Ester) Vitrimers with Catalyst-Free Bond Exchange. J. Am. Chem. Soc. 2019, 141 (35), 13753–13757. 10.1021/jacs.9b06668. [DOI] [PubMed] [Google Scholar]
- Cromwell O. R.; Chung J.; Guan Z. Malleable and Self-Healing Covalent Polymer Networks through Tunable Dynamic Boronic Ester Bonds. J. Am. Chem. Soc. 2015, 137 (20), 6492–6495. 10.1021/jacs.5b03551. [DOI] [PubMed] [Google Scholar]
- Röttger M.; Domenech T.; Van Der Weegen R.; Breuillac A.; Nicolaÿ R.; Leibler L. High-Performance Vitrimers from Commodity Thermoplastics through Dioxaborolane Metathesis. Science 2017, 356 (6333), 62–65. 10.1126/science.aah5281. [DOI] [PubMed] [Google Scholar]
- Nishimura Y.; Chung J.; Muradyan H.; Guan Z. Silyl Ether as a Robust and Thermally Stable Dynamic Covalent Motif for Malleable Polymer Design. J. Am. Chem. Soc. 2017, 139 (42), 14881–14884. 10.1021/jacs.7b08826. [DOI] [PubMed] [Google Scholar]
- Denissen W.; Rivero G.; Nicolaÿ R.; Leibler L.; Winne J. M.; Du Prez F. E. Vinylogous Urethane Vitrimers. Adv. Funct. Mater. 2015, 25 (16), 2451–2457. 10.1002/adfm.201404553. [DOI] [Google Scholar]
- Ishibashi J. S. A.; Kalow J. A. Vitrimeric Silicone Elastomers Enabled by Dynamic Meldrum’s Acid- Derived Cross-Links. ACS Macro Lett. 2018, 7 (4), 482–486. 10.1021/acsmacrolett.8b00166. [DOI] [PubMed] [Google Scholar]
- Majumdar S.; Zhang H.; Soleimani M.; Van Benthem R. A. T. M.; Heuts J. P. A.; Sijbesma R. P. Phosphate Triester Dynamic Covalent Networks. ACS Macro Lett. 2020, 9 (12), 1753–1758. 10.1021/acsmacrolett.0c00636. [DOI] [PubMed] [Google Scholar]
- Feng X.; Li G. Versatile Phosphate Diester-Based Flame Retardant Vitrimers via Catalyst-Free Mixed Transesterification. ACS Appl. Mater. Interfaces 2020, 12 (51), 57486–57496. 10.1021/acsami.0c18852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng X.; Li G. Catalyst-Free β-Hydroxy Phosphate Ester Exchange for Robust Fire-Proof Vitrimers. Chem. Eng. J. 2021, 417, 129132. 10.1016/j.cej.2021.129132. [DOI] [Google Scholar]
- Delahaye M.; Winne J. M.; Du Prez F. E. Internal Catalysis in Covalent Adaptable Networks: Phthalate Monoester Transesterification as a Versatile Dynamic Cross-Linking Chemistry. J. Am. Chem. Soc. 2019, 141 (38), 15277–15287. 10.1021/jacs.9b07269. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Majumdar S.; Van Benthem R. A. T. M.; Sijbesma R. P.; Heuts J. P. A. Intramolecularly Catalyzed Dynamic Polyester Networks Using Neighboring Carboxylic and Sulfonic Acid Groups. ACS Macro Lett. 2020, 9 (2), 272–277. 10.1021/acsmacrolett.9b01023. [DOI] [PubMed] [Google Scholar]
- Podgórski M.; Mavila S.; Huang S.; Spurgin N.; Sinha J.; Bowman C. N. Thiol-Anhydride Dynamic Reversible Networks. Angew. Chem., Int. Ed. 2020, 59 (24), 9345. 10.1002/anie.202001388. [DOI] [PubMed] [Google Scholar]
- Cuminet F.; Caillol S.; Dantras É.; Leclerc É.; Ladmiral V. Neighboring Group Participation and Internal Catalysis Effects on Exchangeable Covalent Bonds: Application to the Thriving Field of Vitrimer Chemistry. Macromolecules 2021, 54 (9), 3927–3961. 10.1021/acs.macromol.0c02706. [DOI] [Google Scholar]
- Reutenauer P.; Buhler E.; Boul P. J.; Candau S. J.; Lehn J. M. Room Temperature Dynamic Polymers Based on Diels-Alder Chemistry. Chem. - Eur. J. 2009, 15 (8), 1893–1900. 10.1002/chem.200802145. [DOI] [PubMed] [Google Scholar]
- Semlow D. R.; Silverman S. K. Parallel Selections in Vitro Reveal a Preference for 2′-5′ RNA Ligation upon Deoxyribozyme-Mediated Opening of a 2′,3′-Cyclic Phosphate. J. Mol. Evol. 2005, 61 (2), 207–215. 10.1007/s00239-004-0326-y. [DOI] [PubMed] [Google Scholar]
- Kirby A. J.; Mora J. R.; Nome F. New Light on Phosphate Transfer from Triesters. Biochim. Biophys. Acta, Proteins Proteomics 2013, 1834 (1), 454–463. 10.1016/j.bbapap.2012.04.010. [DOI] [PubMed] [Google Scholar]
- Kosarev M. A.; Gavrilov D. E.; Nifant’ev I. E.; Shlyakhtin A. V.; Tavtorkin A. N.; Dyadchenko V. P.; Roznyatovsky V. A.; Ivchenko P. V. Ultrafast Hydrolytic Degradation of 2,3-Dihydroxypropyl Functionalized Poly(Ethylene Phosphates). Mendeleev Commun. 2019, 29 (5), 509–511. 10.1016/j.mencom.2019.09.010. [DOI] [Google Scholar]
- Meng Y.; Jiang J.; Anthamatten M. Body Temperature Triggered Shape-Memory Polymers with High Elastic Energy Storage Capacity. J. Polym. Sci., Part B: Polym. Phys. 2016, 54 (14), 1397–1404. 10.1002/polb.23990. [DOI] [Google Scholar]
- Li L.; Chen X.; Jin K.; Torkelson J. M. Vitrimers Designed Both to Strongly Suppress Creep and to Recover Original Cross-Link Density after Reprocessing: Quantitative Theory and Experiments. Macromolecules 2018, 51 (15), 5537–5546. 10.1021/acs.macromol.8b00922. [DOI] [Google Scholar]
- Procházková E.; Šimon P.; Straka M.; Filo J.; Májek M.; Cigáň M.; Baszczyňski O. Phosphate Linkers with Traceable Cyclic Intermediates for Self-Immolation Detection and Monitoring. Chem. Commun. 2021, 57 (2), 211–214. 10.1039/D0CC06928K. [DOI] [PubMed] [Google Scholar]
- Procházková E.; Šimon P.; Straka M.; Filo J.; Májek M.; Cigáň M.; Baszczyňski O. Phosphate Linkers with Traceable Cyclic Intermediates for Self-Immolation Detection and Monitoring. Chem. Commun. 2021, 57 (2), 211–214. 10.1039/D0CC06928K. [DOI] [PubMed] [Google Scholar]
- Velencoso M. M.; Battig A.; Markwart J. C.; Schartel B.; Wurm F. R. Molecular Firefighting—How Modern Phosphorus Chemistry Can Help Solve the Challenge of Flame Retardancy. Angew. Chem., Int. Ed. 2018, 57 (33), 10450–10467. 10.1002/anie.201711735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markwart J. C.; Battig A.; Urbaniak T.; Haag K.; Koschek K.; Schartel B.; Wurm F. R. Intrinsic Flame Retardant Phosphonate-Based Vitrimers as a Recyclable Alternative for Commodity Polymers in Composite Materials. Polym. Chem. 2020, 11, 4933–4941. 10.1039/D0PY00275E. [DOI] [Google Scholar]
- Markwart J. C.; Battig A.; Velencoso M. M.; Pollok D.; Schartel B.; Wurm F. R. Aromatic vs. Aliphatic Hyperbranched Polyphosphoesters as Flame Retardants in Epoxy Resins. Molecules 2019, 24 (21), 3901. 10.3390/molecules24213901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang L. F.; Jeon S.; Kakihana Y.; Kakehi J.; Zhu B. K.; Matsuyama H.; Zhao S. Improved Antifouling Properties of Polyvinyl Chloride Blend Membranes by Novel Phosphate Based-Zwitterionic Polymer Additive. J. Membr. Sci. 2017, 528, 326–335. 10.1016/j.memsci.2017.01.044. [DOI] [Google Scholar]
- Tee H. T.; Lieberwirth I.; Wurm F. R. Aliphatic Long-Chain Polypyrophosphates as Biodegradable Polyethylene Mimics. Macromolecules 2019, 52 (3), 1166–1172. 10.1021/acs.macromol.8b02474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S. W.; Zhuo R. X. Recent Advances in Polyphosphoester and Polyphosphoramidate-Based Biomaterials. Phosphorus, Sulfur Silicon Relat. Elem. 2008, 183 (2–3), 340–348. 10.1080/10426500701734620. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.