

Abstract
Previous studies have shown that Dnmt2-null sperm block the paternal transmission (through sperm) of certain acquired traits, e.g., high-fat diet–induced metabolic disorders or white tails due to a Kit paramutation. Here, we report that DNMT2 is also required for the transmission of a Kit paramutant phenotype (white tail tip) through the female germline (i.e., oocytes). Specifically, ablation of Dnmt2 led to aberrant profiles of tRNA-derived small RNAs (tsRNAs) and other small noncoding RNAs (sncRNAs) in sperm, which correlate with altered mRNA transcriptomes in pronuclear zygotes derived from wild-type oocytes carrying the Kit paramutation and a complete blockage of transmission of the paramutant phenotype through oocytes. Together, the present study suggests that both paternal and maternal transmissions of epigenetic phenotypes require intact DNMT2 functions in the male germline.
Keywords: epigenetics, paramutation, tRNA fragments, sperm, oocytes, fertilization, reprogramming, epigenetic inheritance, acquired trait
Dnmt2-null sperm block maternal transmission of epimutations, suggesting sperm-borne tsRNAs may be required for transmission of epimutations through the female germline.
Introduction
DNA methylation, as a major epigenetic modification in the genome of higher eukaryotes, is catalyzed by DNA methyltransferases (DNMTs) by addition of a methyl group to the 5th carbon position of cytosine, resulting in 5-methylcytosine (5mC) [1, 2]. DNMT1 is the most abundant DNA methyltransferase in mammalian cells, which mainly methylates hemi-methylated CpG dinucleotides as a maintenance methyltransferase [3]. Both DNMT3a and DNMT3b are able to methylate previous unmethylated CpG sequences and, thus, are called de novo methyltransferases [1, 2]. In addition to DNMT1, DNMT3a, and DNMRT3b, there are two noncanonical family members, DNMT2 and DNMT3L, neither of which has DNA methyltransferase activity [4, 5]. DNMT3L appears to interact with DNMT3a and DNMT3b to regulate their DNA methyltransferase activities [6], whereas DNMT2 has been found to act as a tRNA methyltransferase, responsible for catalyzing methylation of cytosine 38 (C38) in the anticodon loop of tRNAAspGUC [7]. DNMT2-dependent methylation appears to protect the anticodon loop of tRNAs from cleavage in flies and mice, and a lack of m5C C38 of three tRNAs (tRNAAspGUC, tRNAGlyGCC, and tRNAValAAC) induces fragmentation of these tRNAs, leading to the production of tRNA fragments, called tRNA-derived small RNAs (tsRNAs) [7, 8]. It has been reported that tsRNAs are highly abundant in sperm [9]. More interestingly, the tsRNA profiles are altered in sperm of the male mice with high-fat diet–induced metabolic disorders, and injection of small RNA fractions containing tsRNAs purified from these male mice into the wild-type zygotes recapitulates the metabolic disorder phenotype in offspring [10]. A related study also reports that the sperm may have gained a large number of the tsRNAs from the epididymal exosomes (called epididymosomes) secreted by the epididymal epithelial cells during sperm transit through the epididymis, and sperm tsRNA profiles get altered in sperm from male mice on a protein-restricted diet [11]. These data strongly suggest that sperm-borne tsRNAs may function as an epigenetic messenger to transmit acquired traits to offspring [12]. Therefore, it is critical to understand how tsRNAs are produced and how these tRNA fragments act to convey epigenetic information during fertilization and early embryonic development.
Recent reports have shown that Dnmt2 inactivation in male mice blocks, to a large extent, the paternal (i.e., sperm-mediated) transmission of acquired phenotypes, including the white tails in Kit paramutant mice (genetically wild-type mice with one of the two kit alleles derived from a Kit+/lacZ parent) and HFD-induced metabolic disorders [13, 14]. While these studies suggest a critical role of DNMT2 in epigenetic inheritance, many questions remain, e.g., how does ablation of DNMT2 affect sperm-borne tsRNA contents? What is the impact of DNMT2 ablation on the profiles of other sperm-borne sncRNAs? Are normal sperm-borne tsRNAs also required for efficient transmission of a maternal, in addition to paternal, epigenetic phenotype through oocytes/eggs? How do sperm-borne tsRNAs and other sncRNAs act to affect gene expression during zygotic genome activation in pronuclear embryos? The present study attempted to address these fundamental questions at least partially.
We adopted a Kit paramutation mouse model because the phenotype induced by the paramutant wild-type allele, i.e., white tail tip (WTT), is easy to spot and measure [15]. Using WTT as a phenotypic readout, we set out and tested the effects of Dnmt2-null sperm on the maternal transmission efficiency of the WTT phenotype. To our surprise, our breeding data revealed that DNMT2 ablation in sperm almost completely abolished maternal transmission of the WTT phenotype. The blocked maternal transmission of the paramutant phenotype by sperm correlated with altered small RNA profiles in Dnmt2-null sperm and aberrant mRNA profiles in zygotes derived from wild-type or Kit+/copGFP oocytes injected with Dnmt2-null or wild-type sperm.
Methods and materials
Animal use and care
All mice used in this study were on C57Bl6/J background and housed in a temperature- and humidity-controlled, specific pathogen-free facility under a light–dark cycle (12:12 light–dark) with food and water ad libitum. Animal use protocol was approved by Institutional Animal Care and Use Committee (IACUC) of the University of Nevada, Reno, and is in accordance with the “Guide for the Care and Use of Experimental Animals” established by National Institutes of Health (1996, revised 2011). The Kit+/copGFP mice were generated as described [15, 16]. Dnmt2 knockout mice were purchased from the Jackson Laboratory (Stock#:006240) and were backcrossed for six generations onto C57Bl6/J background before experiments were carried out. The Kit paramutant mice were those that are genetically wild-type, but one of the two Kit alleles was derived from a Kit+/copGFP parent, as reported previously [15].
Sperm collection
Mature sperm were isolated from the cauda epididymis of male mice as described [13]. In brief, cauda epididymal sperm were allowed to swim into 1 mL HTF medium incubated at 37 C, and an aliquot of 300 μL of swim-up sperm was collected after 1 h of incubation. Sperm were centrifuged at 100 × g for 10 min and resuspended in 200 μL of Tris–HCL medium (pH = 8.0). The resuspended sperm were then aliquoted into tubes (50 μL/tube), which were then snap frozen in liquid nitrogen followed by storage in −80 C.
Intracytoplasmic sperm injection (ICSI) and collection of oocytes and zygotes
Adult (6–8 weeks of age) virgin female WT and KitcopGFP/+ mice were selected as oocyte donors for ICSI. Superovulation was performed by intraperitoneal injection with 5 IU PMSG at 19:30 and intraperitoneal injection with hCG 48 h later. Oocytes were collected 13–14 h after hCG administration. ICSI was performed according to the established protocol [17]. PN3 or PN4 stage zygotes were collected 8 h after ICSI, as described previously [18].
Small noncoding RNA bisulfite sequencing (sncRNA-BS-seq)
RNAs were isolated from frozen sperm using mirVana miRNA Isolation Kit (Invitrogen). Bisulfite conversion of sncRNAs was conducted using the EZ RNA Methylation Kit (ZYMO Research). Small RNA libraries were constructed using the NEBNext Small RNA Library Prep Set for Illumina (NEB). The sncRNA libraries were sequenced on an Illumina HiSeq4000 sequencer (SE50).
Bioinformatic analyses of sncRNA-BS-seq data
The sncRNA-BS-seq data have been deposited into the NCBI SRA database (accession#: PRJNA516832). The low-quality sncRNA bisulfite sequencing reads and reads shorter than 15 nt were trimmed by Cutadapt [19]. Then, clean reads were C→T converted. The AASRA pipeline [20] with default parameters was used to map the remaining reads to known mouse sncRNA, consisting of mature miRNA (miRBase, release 21), precursor miRNA (miRBase, release 21), tRNA (Genomic tRNA Database), piRNA (piRBase), rRNA (ENSEMBL, release 76), snRNA, snoRNA, and mitochondrial RNA (ENSEMBL, release 76). The sncRNA references are also C→T converted prior to alignment. Considering tRNA fragmentation caused by bisulfite treatment, all the reads aligned to tRNA reference sequences are recognized as tRNA reads. The cytosine on tRNA reads is recorded and percentage of m5C is calculated after alignment (methylated count/all read counts, at each tRNA nucleotide). tRNA alignment positions were tracked, with insertion/deletion and mismatch considered. Then, nucleotide position density was plotted to reflect tRNA nucleotide density from 5′ to 3′. To determine small RNA expression levels, the aligned reads were counted by featureCounts [21]. Read count outputs from AASRA was normalized and analyzed by DESeq2 [22]. Similar analyses were conducted using small RNA-seq datasets of oocytes and early embryos (Accession#: SRR15386553 and 1538552) from a previous study (Yang et al., 2016).
Single-zygote genotyping
Since we only wished to study the zygotes with wild-type Kit, we genotyped single zygotes based on first observation of copGFP followed by PCR-based detection of copGFP using cDNA libraries constructed for RNA-seq. Specific primers that detect copGFP cDNAs were used, as previously described [16], and Gapdh was used as an internal positive control. For primer sequences, please see Supplemental Table S12. Zygotes with Gapdh but without copGFP expression (Kit+/+) were chosen for sequencing, whereas those with both Gapdh and copGFP expression (Kit+/copGFP) were excluded. A total of 80 single zygotes (20 for each group) were sequenced, and 34 that passed the quality control were used for bioinformatic analyses to identify differentially expressed genes (DEGs).
Single-zygote RNA-seq
The zygotes of PN3-PN4 stages collected were stored in −80 C prior to library construction. The single-cell cDNA libraries were made using SMART-Seq v4 Ultra Low Input RNA Kit (Takara). The cDNAs were fragmentized and Illumina Sequencing adaptors were added using Nextera DNA Library Preparation Kit. The single-zygote libraries were sequenced on an Illumina NextSeq500 sequencer (PE75).
Bioinformatic analyses of single-zygote mRNA-Seq data
The single-zygote RNA-seq data have been deposited into the NCBI SRA database (accession#: PRJNA516834). The raw data were processed by Trimmomatic (v0.33) [24] to remove adaptor sequences and filter out the bad-quality reads. The clean reads were then aligned to UCSC MM9 mouse genome by HiSAT2 (v2.1.0) [25]. The raw gene count data were generated by featureCounts [21] and analyzed by the R package Scater [26] to filter out biased samples and get library-size and spike-in normalized gene expression values. Transcripts were assembled by Cufflinks (v2.2.1) [27]. The enriched genes were sent to gene ontology analysis performed by Gene Ontology Consortium with PANTHER Classification System using default parameters [28].
Results
Inhibition of maternal transmission of the Kit paramutant phenotype by Dnmt2-null sperm
Insertion of a reporter gene cassette, either LacZ or copGFP, into exon1 of Kit has been shown to induce coat color changes in heterozygous mice of C57Bl/6J strain, characterized by white patches in the abdomen and the tail [15, 29]. Interestingly, wild-type (WT) offspring derived from heterozygous (Kit+/copGFP) parents tend to display white tail tips (WTT) that are highly variable in length [15]. The white tail tips represent a specific phenotype induced by the paramutant Kit allele, i.e., the WT allele derived from the Kit+/copGFP parent [15, 29]. The WTT phenotype can be transmitted via both paternal and maternal germlines and maintained across multiple generations [15]. In our WT C57Bl6/J mouse breeding colonies (totally unrelated to the KitcopGFP line), ~30% of the mice displayed WTTs, but the accumulative length was usually shorter than 5 mm (Figure 1A). A similar distribution pattern of the WTT phenotype was observed among WT Kit offspring derived from the breeding pairs of Dnmt2−/− males and WT females, both of which had black tail tips (BTT) (Figure 1B), suggesting that Dnmt2 deficiency has no or minor effects on the baseline penetrance and severity of the WTT phenotype. When WT BTT males were bred with Kit+/copGFP females, ~59% of the WT offspring had WTTs, including 31% with longer (>3 mm) and 28% with shorter WTTs (Figure 1C), suggesting that the WT paramutant Kit allele (the maternal WT Kit allele–bearing epimutations) can be transmitted to offspring through the maternal germline (oocytes), and the WTT phenotype induced by the Kit paramutant allele is often more severe (>5 mm in length). Intriguingly, when the WT BTT males were switched to Dnmt2-null BTT males (Dnmt2−/− males × Kit+/copGFP females), the maternal transmission of the paramutation-induced WTT phenotype was largely abolished, as evidenced by the significantly reduced penetrance of the WTT phenotype (59 → 35%) and decreased severity (31 → 3%) in Kit wild-type offspring (Figure 1D); both the phenotypic penetrance and severity were similar to those in offspring of WT BTT breeding pairs (Figure 1A). Although the F1s studied were all wild-type in Kit locus, those fathered by Dnmt2-null males are heterozygous in Dnmt2 locus (Dnmt2+/−), raising the possibility that the reduced penetrance of the WTT phenotype may be due to the Dnmt2 heterozygosity (Dnmt2+/−). In other words, one allele of Dnmt2 in the WT Kit early embryos may not be sufficient to support efficient transmission of the WTT phenotype carried by the maternal WT Kit allele. To test this possibility, we set up breeding pairs using Dnmt2+/− males and Kit+/copGFP females to examine the penetrance of the WTT phenotype in offspring that are WT in both Kit and Dnmt2 loci, as well as offspring that are WT in Kit locus, but heterozygous in Dnmt2 locus. Interestingly, the distribution of the phenotypic penetrance in both Dnmt2+/+; Kit+/+ and Dnmt2+/−; Kit+/+ F1s was similar to that in F1s derived from WT males and Kit+/copGFP females, all showing increased proportions of the longer WTT (Supplementary Figure S1). This finding strongly suggests that Dnmt2 heterozygosity in early WT Kit embryos does not affect the efficiency of maternal transmission of the WTT phenotype carried on the maternal WT Kit allele because both Dnmt2+/+ and Dnmt2+/− F1s displayed the same enhanced penetrance of the longer WTT phenotype. Thus, it is highly likely that Dnmt2-null sperm, rather than Dnmt2 heterozygosity of F1 embryos, are responsible for the inhibitory effects on maternal transmission of the WTT phenotype.
Figure 1 .
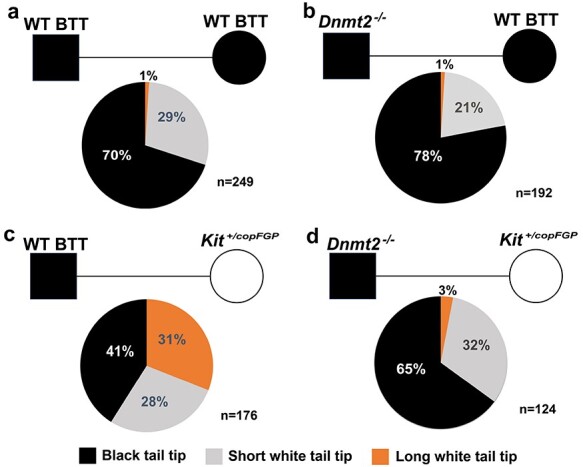
Blockage of maternal transmission of a Kit paramutant phenotype by Dnmt2-null sperm. Pie charts showing the penetrance of the white tail tip (WTT) phenotype in wild-type (WT, Kit+/+) offspring derived from four breeding schemes, including Kit+/+ males × Kit+/+ females with black tail tips (BTT) (a), Dnmt2−/−;Kit+/+ males × Dnmt2+/+;Kit+/+ females with BTT (b), Dnmt2+/+;Kit+/+ males with BTT × Dnmt2+/+;Kit+/copGFP females (c), and Dnmt2−/−;Kit+/+ males with BTT × Dnmt2+/+;Kit+/copGFP females (d). In the present study, longer and shorter white tail tips were defined by the cumulative length of WTT >5 and <5 mm, respectively. The number (n) of mice observed is marked. Black square and circle represent male and female mice, respectively.
Altered tsRNA profiles characterized by decreased m5C levels at C38, increased tsRNA levels, and global shortening of tsRNAs in Dnmt2-null sperm
Unlike DNMT1 and DNMT3, DNMT2 catalyzes the 5′ cytosine methylation (m5C) at position 38 (C38) of three tRNAs: ValAAC, AspGUC, and GlyGCC [7] (Figure 2A). Proper C38 methylation protects these tRNAs from degradation into tsRNAs [5, 8]. Thus, DNMT2 is believed to be a critical regulator of tsRNA biogenesis [4, 7]. To determine tRNA 5mC levels in Dnmt2-null sperm, we performed high-throughput small RNA bisulfite sequencing (sRNA-BS-Seq). The cytosine on tRNA reads were identified first and the percentage of m5C (methylated counts/total read counts for each tRNA) was calculated. We found that the 5mC levels on C38 of all seven isodecoders of the three DNMT2 substrates (ValAAC, AspGUC, and GlyGCC) were drastically reduced in Dnmt2-null sperm than in WT sperm (Figure 2B, the left panel). The sRNA-BS-Seq assays did not detect full-length tRNAs due to bisulfite-induced tRNA fragmentation. While the normalized counts of reads that were aligned to the full-length reference tRNAs were used to reflect total tRNA levels, the normalized counts of reads aligned to the 5′ and 3′ 26 nt reference tRNAs were employed to represent the 5′ and 3′ tsRNAs, respectively. Consistent with the notion that loss of 5mC on C38 facilitates tRNA fragmentation [4, 7], total levels of the seven isodecoders of the three DNMT2 substrates (ValAAC, AspGUC, and GlyGCC) appeared to be down-regulated in Dnmt2-null sperm compared to WT sperm (Figure 2B, the middle panel). Consistent with previous data [7], as a non-DNMT2 substrate, tRNA GluUUC showed no significant changes in either methylation levels at C38 or total read counts in Dnmt2-null sperm (Figure 2B, the middle panel), validating our quantitative analyses. While tRNA degradation appeared to be enhanced, levels of tsRNAs derived from DNMT2 substrate tRNAs were increased in Dnmt2-null sperm (Figure 2B, the right panel). Moreover, the average length of tsRNAs derived from the three DNMT2 substrate tRNAs appeared to be significantly decreased in Dnmt2-null sperm compared to WT sperm (Figure 2C and Supplementary Figure S2). Although the length reduction was the most prominent in tsRNAs derived from the DNMT2 substrate tRNAs, those from non-DNMT2 substrate tRNAs, e.g., GluUUC, also displayed a minor length reduction despite comparable abundance (Figure 2C). Further analyses of other tRNAs revealed a global shortening in tsRNAs in Dnmt2-null sperm (Figure 2D). To determine underlying mechanism, we analyzed the abundance of individual nucleotides by plotting the normalized percentage abundance of each nucleotide against each position along the full-length tRNAs (Figure 2E). Remarkably, a stretch of nucleotides around the anticodon region (27-45 nt) of tRNAs was underrepresented (arrow in Figure 2E), suggesting that the enhanced hydrolysis of tRNAs at the anticodon loop is responsible for the global shortening of tRNAs.
Figure 3 .
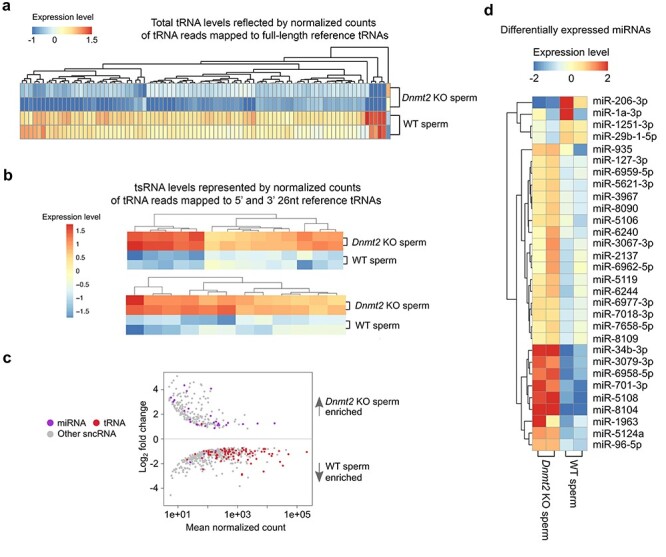
Altered profiles of other sperm-borne sncRNAs in Dnmt2-null sperm. (a) Heatmap showing differentially expressed total tRNA levels between WT and Dnmt2 KO sperm (P < 0.05). The relative expression levels are presented by the mean-centered and normalized log-expression values. (b) Heatmap showing differentially expressed tsRNA levels between WT and Dnmt2 KO sperm (P < 0.05). (c) MA plots showing differentially expressed (P < 0.05) miRNAs, tRNAs, and other sncRNAs between WT and Dnmt2 KO sperm. Mean normalized counts represent the mean value of DESeq2 normalized counts (>5) between WT and Dnmt2 KO sperm. Log2 fold change was calculated by the Log2 sncRNA counts of Dnmt2 KO sperm/WT sperm. (d) Heatmap showing differentially expressed miRNAs between WT and Dnmt2 KO sperm (P < 0.05). The relative expression levels are presented by the mean-centered and normalized log-expression values. sncRNAs-Seq data were from two biological replicates with two technical repeats (n = 2).
Figure 2 .
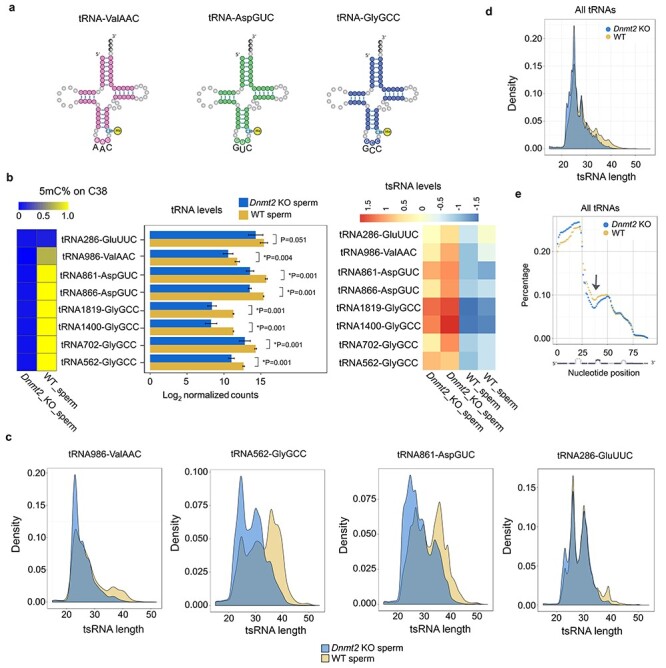
Altered tsRNA profiles in Dnmt2-null sperm characterized by decreased m5C levels on C38 of substrate tRNAs, reduced total substrate tRNA levels, increased tsRNA levels, and global shortening of total tRNAs. (a) Schematics of the three confirmed substrate tRNAs of DNMT2 (tRNA-ValAAC, AspGUC, and GlyGCC) in mice. (b) Levels of 5mC on C38 (left panels), total tRNAs (middle panels), and tsRNAs of the seven isodecoders of three substrate tRNAs in Dnmt2 KO and wild-type (WT) sperm. Heatmap (left panels) shows the mean percentage of m5C on C38 of tRNAs, and the bar graphs (middle panels) indicate the log2 values of normalized counts of tRNA reads mapped to full-length reference tRNAs, which represent the total levels of the three substrate tRNAs in Dnmt2 KO and WT sperm. The relative levels of substrate tRNAs-derived tsRNAs are reflected by the counts of reads mapped to 5′ and 3′ 26 nt reference tRNAs normalized against total tRNA counts (heatmap in the right panels). A non-DNMT2 substrate tRNA (tRNA286-GluUUC) was used as a control. The data were presented as means ± SD from biological replicates, n = 2. Sums of two technical repeats in each of the biological replicates were used for analyses. *: P < 0.05. (c) Density plots showing the length distribution of tsRNAs derived from three substrate tRNAs (tRNA986-ValAAC, tRNA562-GlyGCC, and tRNA861-AspGUC) and one non-DNMT2 substrate (tRNA286-GluUUC) in Dnmt2 KO and WT sperm. (d) Density plots showing the length distribution of the tsRNAs derived from both substrate and nonsubstrate tRNAs. (e) Distribution of individual nucleotides along the entire length of tRNAs. The normalized percentage abundance (y axis) was plotted against positions of each nucleotide (x axis). Note that a stretch of nucleotides around the anticodon region (27–45 nt) of tRNAs was drastically underrepresented (arrow) and that 5′ tsRNAs appeared to be more abundant than 3′ tsRNAs in WT sperm, and this bias appeared to be more evident in Dnmt2-null sperm.
Moreover, 5′ tsRNAs appeared to be more abundant than 3′ tsRNAs in WT sperm, and this bias became more prominent in Dnmt2-null sperm (Figure 2E). To corroborate this finding, we analyzed the small RNA-Seq data from another publication [23] and obtained similar results showing 5′ tsRNAs were dominant in both sperm and zygotes in mice (Supplementary Figure S3). This finding is consistent with the notion that the 3′ regions of tRNAs are commonly underrepresented in NGS data because of the RNA modification-related biases in cDNA library preparation [30]. Together, out data support the role of DNMT2 as a methyltransferase responsible for the C38 5mC of tRNAs, and a lack of this mark leads to enhanced hydrolysis of not only the three substrate tRNAs but also other tRNAs around the anticodon loop region (between positions 27 and 45 nt).
Expression profiles of other sncRNAs are also altered in Dnmt2-null sperm
To explore the effect of Dnmt2 inactivation on other sncRNA expression, we annotated eight other sncRNA species, including endo-siRNAs, miRNAs, mitochondrial DNA-encoded rRNA (mt rRNA) and tRNAs (mt tRNA), piRNAs, rRNAs-derived small RNAs (rsRNAs), snoRNAs, and snRNAs using AASRA [20], and compared their normalized total counts between Dnmt2-null and WT sperm. The counts of tRNA reads mapped to the full-length reference tRNAs were down-regulated (Figure 3A), whereas the counts of tRNA reads mapped to 5′ and 3′ 26 nt reference tRNA fragments, which were derived from both substrate and nonsubstrate tRNAs, were all up-regulated in Dnmt2-null sperm, as compared to WT sperm (Figure 3B). The tRNA fragments analyzed are listed in Supplementary Table S1. Interestingly, while total tRNA levels were down-regulated, miRNAs were mostly up-regulated in Dnmt2-null sperm (Figure 3C). Indeed, 26 out of 30 miRNAs that were differentially expressed between Dnmt2-null and WT sperm were up-regulated (Figure 3D). To validate the dysregulation of miRNAs, we analyzed levels of eight DNMT2-dependent miRNAs (miR-34b-3p, 701-3p, 96-5p, 5108, 8104, 5106, 6240, and 6958-5p) in WT, Dnmt2+/−, and Dnmt2−/− sperm using qPCR and found that levels of these eight miRNAs were similar in both WT and Dnmt2+/− sperm, but much higher in Dnmt2-null sperm (Supplementary Figure S4). Expression profiles of other sncRNAs, including piRNAs, endo-siRNAs, snoRNAs, snRNAs, and rsRNAs, were also altered (Supplementary Figure S5 and Supplementary Table S2). Together, sperm of the Dnmt2 KO males contain altered sncRNAs profiles, characterized by lowered levels of total tRNAs and increased levels of tsRNAs, as well as mostly up-regulated miRNAs.
Transcriptomic alterations upon zygotic genome activation in PN3/PN4 prenuclear embryos derived from Kit+/copGFP oocytes injected with either Dnmt2-null or WT sperm
The major difference between the two breeding schemes (i.e., Dnmt2-null males × Kit+/copGFP females vs. WT males × Kit+/copGFP females) lies in the sperm (Dnmt2-null vs. WT). Therefore, the cause for failed maternal transmission of the paramutant WTT phenotype must be hidden within the Dnmt2-null sperm. Given that DNMT2 methylates tRNAs and the profiles of tsRNAs and other sncRNA species are altered in Dnmt2-null sperm, it is plausible to hypothesize that the Dnmt2-null sperm, due to their altered sncRNA profiles, exert effects on the transcriptome of early embryos and, thus, block the maternal transmission of the WTT phenotype. To test this hypothesis, we injected WT or Dnmt2-null sperm into the oocytes from either Kit+/copGFP or WT mice and collected zygotes at PN3 and PN4 stages for single-cell RNA-seq analyses (Figure 4A). We chose to analyze the transcriptome of zygotes at pronuclear PN3/PN4 stages because the paternal genome has been activated with greater transcriptional activities than the maternal genome at these stages [18]. When WT and Dnmt2-null sperm were injected into Kit+/copGFP oocytes, 50% of the resulting embryos, in theory, would be heterozygous for Kit (Kit+/copGFP), which were excluded based on microscopic observation of copGFP expression and PCR detection of copGFP transcripts using total cDNAs prepared from single zygotes. Only Kit+/+ zygotes were selected for single-cell RNA-seq analyses.
Figure 4 .
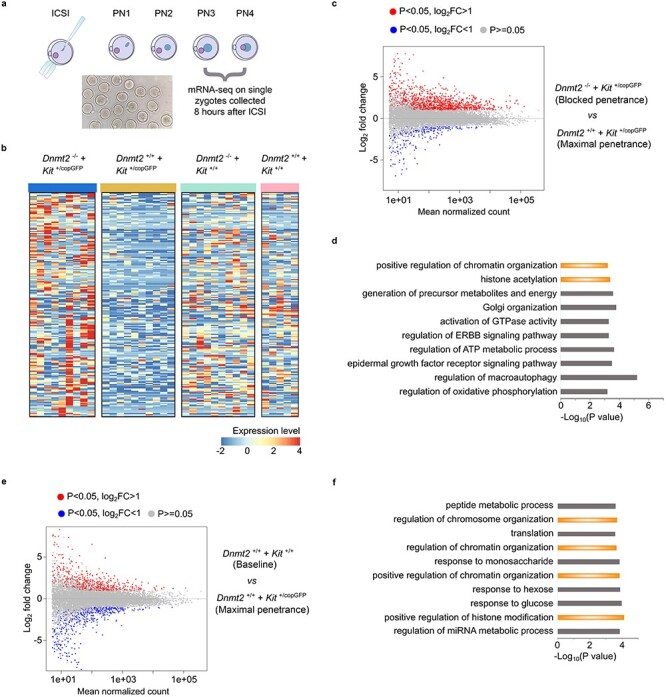
Dynamic transcriptomic changes in the pronuclear zygotes derived from Kit+/copGFP oocytes and Dnmt2-null sperm. (a) Schematics showing the workflow of transcriptomic analyses on single zygotes of pronuclear stages 3 and 4 (PN3/PN4) produced by intracytoplasmic sperm injection (ICSI). (b) Heatmap showing the top 200 differentially expressed genes (DEGs) (P < 0.05, Log2_fold change) between Dnmt2+/− zygotes from Kit+/copGFP oocytes injected with Dnmt2-null sperm and WT zygotes from Kit+/copGFP oocytes injected with WT sperm (left two panels). The expression levels of these 200 DEGs in Dnmt2+/− zygotes from WT oocytes injected with Dnmt2-null and WT zygotes from WT oocytes injected with WT sperm (right two panels). The four types of zygotes mirror the four types of F1 mice used to analyze the penetrance of the WTT phenotype, as shown in Figure 1C. (c) MA plots showing the DEGs in zygotes derived from Kit+/copGFP oocytes injected with Dnmt2-null (n = 9) or WT (n = 10) sperm. (d) Gene ontology (GO) analyses on genes dysregulated in zygotes from Kit+/copGFP oocytes injected with Dnmt2-null sperm compared to those from Kit+/copGFP oocytes injected with WT sperm. Top 10 enriched gene ontology terms are shown in bar graphs with −Log10 P values indicated. (e) MA plots showing the DEGs in zygotes derived from Kit+/copGFP (n = 9) or WT (n = 5) oocytes injected with WT sperm. (f) Gene ontology (GO) analyses on genes dysregulated in zygotes derived from WT oocytes injected with WT sperm compared to those from Kit+/copGFP oocytes injected with WT sperm. Top 10 enriched gene ontology terms are shown in bar graphs with −Log10 P values indicated.
A total of four types of single PN3/PN4 stage zygotes were used for RNA-seq: Dnmt2+/−; Kit+/+ PN3/4 embryos from Kit+/copGFP oocytes injected with Dnmt2-null sperm, Dnmt2+/+; Kit+/+ PN3/4 embryos from Kit+/copGFP oocytes injected with WT sperm, Dnmt2+/−; Kit+/+ PN3/4 embryos from WT oocytes injected with WT sperm, and Dnmt2+/+; Kit+/+ PN3/4 embryos from WT oocytes injected with WT sperm. The four types of pronuclear embryos mirror the F1 offspring from the four breeding schemes used (Figure 1). A total of 80 single zygotes (20 for each group) were sequenced, and 34 that passed the quality control were used for bioinformatic analyses to identify differentially expressed genes (DEGs) (Supplementary Figure S6). By calibrating against the median expression values among the four types of single zygotes, we chose the top 200 dysregulated genes (P < 0.05, Log2_fold change) between zygotes from the two mating groups (Dnmt2+/−; Kit+/+ PN3/4 embryos from Kit+/copGFP oocytes injected with Dnmt2-null sperm, and WT PN3/4 embryos from Kit+/copGFP oocytes injected with WT sperm) and created heatmaps to show the DEG patterns (Figure 4B, left two panels, and Supplementary Table S3). As a comparison, we also generated the heatmaps for the other two groups of zygotes (Dnmt2+/−; Kit+/+ PN3/4 embryos from WT oocytes injected with WT sperm, and WT PN3/4 embryos from WT oocytes injected with WT sperm) (Figure 4B, right two panels). In general, similar DEG profiles were observed between zygotes derived from WT oocytes injected with either WT or Dnmt2-null sperm (Figure 4B, right two panels), whereas the DEG profiles were drastically different between zygotes from the Kit+/copGFP oocytes injected with WT and Dnmt2-null sperm (Figure 4B, left two panels). Moreover, the majority of the top 200 DEGs appeared to be up-regulated in the zygotes from Kit+/copGFP oocytes injected with Dnmt2-null sperm. In contrast, the majority of the mRNAs were mostly down-regulated in the zygotes from Kit+/copGFP oocytes injected with WT sperm. These data suggest that zygotes-derived WT oocytes are more resistant than those from Kit+/copGFP oocytes to the impact of sperm genotype (WT vs. Dnmt2-null).
The Dnmt2+/− zygotes derived from Dnmt2−/− sperm and Kit+/copGFP oocytes and WT zygotes derived from WT sperm and Kit+/copGFP oocytes resemble the two breeding schemes that showed the totally blocked transmission of the WTT phenotype and the maximal transmission of the WTT phenotype (Figures 1C and 4C). Between these two types of zygotes, a total of 1948 dysregulated genes were identified, including 1378 up-regulated and 570 down-regulated in Dnmt2+/− zygotes (Figure 4C and Supplementary Table S4). GO term enrichment analyses revealed that these dysregulated genes were involved in multiple biological functions, including epigenetic regulation (e.g., histone modifications and chromosome/chromatin organization), energy metabolism, cell signaling, and autophagy (Figure 4D).
To reveal the differences in gene expression between zygotes derived from WT (baseline levels of WTT phenotype) and Kit+/copGFP oocytes (maximal penetrance of the WTT phenotype) injected with WT sperm, we also compared their transcriptomes and found a total of 1453 dysregulated genes, including 675 up-regulated and 778 down-regulated ones (Figure 4E and Supplementary Table S5). Of interests, the GO term enrichment analyses identified that these dysregulated genes, similar to those dysregulated genes between the Dnmt2+/− zygotes from Kit+/copGFP oocytes injected with Dnmt2-null and WT sperm (Figure 4D), are mostly involved in epigenetic regulation, e.g., histone modifications and chromosome/chromatin organization, as well as energy metabolism. More interestingly, among all the DEGs between the two groups, a total of 264 genes were shared, and these shared genes function in multiple cellular processes including histone modifications, signaling, and metabolism (Supplementary Figure S7).
Discussion
The phenotypes caused by genetic mutations are relatively homogenous, whereas those caused by epimutations tend to be highly variable [31, 32]. In our Kit paramutation mouse model, the WTT phenotype is highly variable with the WTT length ranging from millimeters to centimeter. Also, even in the pure WT C57Bl6/J breeding colonies, ~30% of the WT mice display the WTT phenotype, but the vast majority are short in length (<5 mm). In the breeding scheme for maternal transmission of the WTT phenotype, Kit+/copGFP females are mated with pure WT males with black tail tips, and the penetrance of the WTT phenotype among the WT offspring almost double (from ~30% baseline levels to ~59%). More interestingly, the increased penetrance predominantly comes from mice with more severe phenotype (>5 mm of WTT), suggesting that longer WTTs likely represent the paramutant phenotype specifically induced by the Kit paramutation on the maternal WT allele. The fact that the increased penetrance of the longer WTT phenotype was abolished in Kit+/+ offspring derived from Kit+/copGFP females mated with Dnmt2-null males strongly suggests that Dnmt2-null sperm lack factors required for the transmission of the Kit paramutation carried on the WT maternal allele through oocytes. Alternatively, this effect may result from Dnmt2 heterozygosity because these mice, although WT in Kit locus, are heterozygous in Dnmt2 locus (Dnmt2+/−). However, it is highly unlikely that Dnmt2 heterozygosity contributed to the inhibitory effects on the maternal transmission of the WTT paramutation phenotype observed because of the following: (1) Sperm sncRNA profiles are similar in sperm from Dnmt2+/− and WT males (Supplementary Figure S5). (2) When Dnmt2+/− males were bred with Kit+/copGFP females, both Dnmt2 WT (Dnmt2+/+) and heterozygous (Dnmt2+/−) offspring with WT Kit displayed a distribution pattern of the WTT phenotype similar to that of offspring from WT male and Kit+/copGFP female breeding pairs (Supplementary Figure S1). Therefore, the drastic inhibition of transmission of the WTT phenotype must be due to the lack of DNMT2-regulayted sncRNAs in Dnmt2-null sperm and the action should take place during the early zygotic stages when zygotic genome activation just gets started. Moreover, the slightly decreased baseline penetrance of the WTT phenotype in offspring (Dnmt2+/−; Kit+/+) from Dnmt2-null males and WT females is indicative of a suppressive effect on the transmission of the WTT phenotype, which is consistent with notion that Dnmt2-null sperm are capable of blocking maternal transmission of the paramutant WTT phenotype in the WT C57Bl6/J colonies. Earlier reports have shown that DNMT2 is required for the paternal transmission of the HFD-induced metabolic disorder and the Kit paramutation-induced white tail phenotype fails to be transmitted to offspring when either parent is on Dnmt2-null background [13, 14]. Our finding here strongly suggests that Dnmt2-null sperm block the maternal transmission of the Kit paramutation-induced WTT phenotype to offspring.
Our sncRNA-BS-seq analyses further support the role of DNMT2 as a methyltransferase responsible for the 5mC modification on C38 of three major tRNA substrates [7]. Earlier studies have also shown that Dnmt2 inactivation results in increased tRNA breakage and consequently enhanced tsRNA production [4]. However, it remains unclear whether tRNA breakage happens randomly or in a site-specific manner. Our data demonstrate that ablation of DNMT2 causes enhanced breakage of not only the three substrate tRNAs at the position of C38 but also other non-DNMT2 substrate tRNAs around the anticodon loop region (position of 27-45 nt). This is an intriguing finding because it suggests that in addition to 5mC at C38, DNMT2 may affect other types of modifications at other positions and, consequently, their stability. Indeed, decreased m2G in tRNAs has been shown as a secondary effect of Dnmt2 inactivation in sperm [13]. Although a lack of m5C at C38 of certain DNMT2 substrates affects translation of a group of proteins in a tRNA-specific manner [33], we noticed that while DNMT2 ablation induces global shortening of tsRNAs, the overall levels of total tRNAs appear to be only slightly reduced. Consequently, the overall translation function of tRNAs may not be affected significantly, which may explain why Dnmt2-null mice show no obvious phenotype [7]. Consistent with a previous report [34], an inverse relationship between miRNA and tRNA expression profiles in sperm was observed, suggesting that biogenesis of miRNAs and tRNAs/tsRNAs may be interrelated. Indeed, it has been shown that production of certain tsRNAs, just like miRNA biogenesis, is Dicer-dependent [35]. Thus, competing for Dicer binding and processing may lead to such an inverse relationship in both WT and Dnmt2-null sperm.
If sperm-borne sncRNAs have an epigenetic role during early embryonic development, they should act prior to or during zygotic genome activation in pronuclear zygotes in mice [18]. Once zygotic genome is activated, many of the sperm-borne RNAs, including tsRNAs and other sncRNAs, can be, in theory, produced by the zygotic genome. Besides, the massive degradation of both maternal and paternal transcripts after zygotic genome activation would not allow them to continue to exist and to act for an extended period of time [36]. Therefore, we chose to use ICSI to produce PN3-PN4 zygotes that mirror the F1 offspring in the four breeding schemes used for studying the penetrance of the paramutant WTT phenotype in live mice. The single-zygote RNA-seq allowed us to explore the underlying molecular mechanism through which Dnmt2-null sperm block the transmission of maternal transmission of the WTT paramutant phenotype. Hundreds of mRNAs are dysregulated between Kit+/+ zygotes from Kit+/copGFP oocytes injected with either Dnmt2-null or WT sperm. Interestingly, the top 200 most dysregulated genes are mostly up-regulated in Kit+/+ zygotes from Kit+/copGFP oocytes injected with Dnmt2-null sperm, whereas these genes displayed similar expression patterns between Kit+/+ zygotes from WT oocytes injected with either Dnmt2-null or WT sperm. These data suggest that the sources of both sperm (Dnmt2-null or WT males) and oocytes (from Kit+/copGFP females) have great influence on the gene expression patterns in PN3-PN4 zygotes. Of great interest, genes involved in histone modifications and chromatin configuration stand out as the potential targets of dysregulated tsRNAs and miRNAs in Dnmt2-null sperm, providing a clue on the molecular mechanism underlying the function of the Dnmt2-dependent sncRNAs as the potential carrier of epigenetic information.
In summary, our data support DNMT2 as a methyltransferase responsible for the 5mC modification on C38 of the substrate tRNAs, and a lack of this mark leads to enhanced hydrolysis of not only the three substrate tRNAs but also other tRNAs around the anticodon loop region between positions 27 and 45 nt. Intact function of DNMT2 in the male germline is required for maternal transmission of a Kit paramutation-induced phenotype (i.e., WTT). Future investigation is warranted to uncover the molecular actions of tsRNAs and to test whether this phenomenon occurs in other models of epigenetic inheritance.
Authors’ contribution
WY designed the research. TY performed sncRNA-BS-seq and single-zygote RNA-seq. YX and CT conducted bioinformatic analyses; YW performed ICSI and collected all of the PN3/PN4 zygotes; SY and HZ conducted all of the breeding and phenotypic analyses; WY wrote the manuscript.
Supplementary Material
Acknowledgements
We thank Dr. Qi Chen at University of California, Riverside, for critical reading of our manuscripts and for his helpful comments.
Conflict of Interest: The authors declare no competing interests.
Grant Support:This work was supported by grants from the NIH (HD098593, HD0085506, and HD099924 to WY) and the Templeton Foundation (PID: 61174 to WY).
Contributor Information
Tian Yu, Department of Physiology and Cell Biology, Reno School of Medicine, University of Nevada, Reno, NV, USA.
Yeming Xie, Department of Physiology and Cell Biology, Reno School of Medicine, University of Nevada, Reno, NV, USA.
Chong Tang, Department of Physiology and Cell Biology, Reno School of Medicine, University of Nevada, Reno, NV, USA.
Yue Wang, Department of Physiology and Cell Biology, Reno School of Medicine, University of Nevada, Reno, NV, USA.
Shuiqiao Yuan, Department of Physiology and Cell Biology, Reno School of Medicine, University of Nevada, Reno, NV, USA.
Huili Zheng, Department of Physiology and Cell Biology, Reno School of Medicine, University of Nevada, Reno, NV, USA.
Wei Yan, Department of Physiology and Cell Biology, Reno School of Medicine, University of Nevada, Reno, NV, USA; The Lundquist Institute for Biomedical Innovation at Harbor-UCLA Medical Center, Torrance, CA, USA; Department of Medicine, David Geffen School of Medicine at UCLA, Los Angeles, CA, USA.
Data and materials availability
Datasets from single-zygote RNA-seq and sncRNA-BS-seq have been deposited into the NCBI SRA database with accession numbers PRJNA516834 and PRJNA516832.
References
- 1.Schmitz RJ, Lewis ZA, Goll MG. DNA methylation: shared and divergent features across eukaryotes. Trends Genet 2019; 35:818–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cooper DN. Eukaryotic DNA methylation. Hum Genet 1983; 64:315–333. [DOI] [PubMed] [Google Scholar]
- 3.Gowher H, Stockdale CJ, Goyal R, Ferreira H, Owen-Hughes T, Jeltsch A. De novo methylation of nucleosomal DNA by the mammalian Dnmt1 and Dnmt3A DNA methyltransferases. Biochemistry 2005; 44:9899–9904. [DOI] [PubMed] [Google Scholar]
- 4.Jeltsch A, Ehrenhofer-Murray A, Jurkowski TP, Lyko F, Reuter G, Ankri S, Nellen W, Schaefer M, Helm M. Mechanism and biological role of Dnmt2 in nucleic acid methylation. RNA Biol 2017; 14:1108–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schaefer M, Lyko F. Lack of evidence for DNA methylation of Invader4 retroelements in drosophila and implications for Dnmt2-mediated epigenetic regulation. Nat Genet 2010; 42:920–921 author reply 921. [DOI] [PubMed] [Google Scholar]
- 6.Veland N, Lu Y, Hardikar S, Gaddis S, Zeng Y, Liu B, Estecio MR, Takata Y, Lin K, Tomida MW, Shen J, Saha D et al. DNMT3L facilitates DNA methylation partly by maintaining DNMT3A stability in mouse embryonic stem cells. Nucleic Acids Res 2019; 47:152–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goll MG, Kirpekar F, Maggert KA, Yoder JA, Hsieh CL, Zhang X, Golic KG, Jacobsen SE, Bestor TH. Methylation of tRNAAsp by the DNA methyltransferase homolog Dnmt2. Science 2006; 311:395–398. [DOI] [PubMed] [Google Scholar]
- 8.Tuorto F, Herbst F, Alerasool N, Bender S, Popp O, Federico G, Reitter S, Liebers R, Stoecklin G, Grone HJ, Dittmar G, Glimm H et al. The tRNA methyltransferase Dnmt2 is required for accurate polypeptide synthesis during haematopoiesis. EMBO J 2015; 34:2350–2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peng H, Shi J, Zhang Y, Zhang H, Liao S, Li W, Lei L, Han C, Ning L, Cao Y, Zhou Q, Chen Q et al. A novel class of tRNA-derived small RNAs extremely enriched in mature mouse sperm. Cell Res 2012; 22:1609–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen Q, Yan M, Cao Z, Li X, Zhang Y, Shi J, Feng GH, Peng H, Zhang X, Zhang Y, Qian J, Duan E et al. Sperm tsRNAs contribute to intergenerational inheritance of an acquired metabolic disorder. Science 2016; 351:397–400. [DOI] [PubMed] [Google Scholar]
- 11.Sharma U, Conine CC, Shea JM, Boskovic A, Derr AG, Bing XY, Belleannee C, Kucukural A, Serra RW, Sun F, Song L, Carone BR et al. Biogenesis and function of tRNA fragments during sperm maturation and fertilization in mammals. Science 2016; 351:391–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen Q, Yan W, Duan E. Epigenetic inheritance of acquired traits through sperm RNAs and sperm RNA modifications. Nat Rev Genet 2016; 17:733–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang Y, Zhang X, Shi J, Tuorto F, Li X, Liu Y, Liebers R, Zhang L, Qu Y, Qian J, Pahima M, Liu Y et al. Dnmt2 mediates intergenerational transmission of paternally acquired metabolic disorders through sperm small non-coding RNAs. Nat Cell Biol 2018; 20:535–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kiani J, Grandjean V, Liebers R, Tuorto F, Ghanbarian H, Lyko F, Cuzin F, Rassoulzadegan M. RNA-mediated epigenetic heredity requires the cytosine methyltransferase Dnmt2. PLoS Genet 2013; 9:e1003498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yuan S, Oliver D, Schuster A, Zheng H, Yan W. Breeding scheme and maternal small RNAs affect the efficiency of transgenerational inheritance of a paramutation in mice. Sci Rep 2015; 5:9266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ro S, Park C, Jin J, Zheng H, Blair PJ, Redelman D, Ward SM, Yan W, Sanders KM. A model to study the phenotypic changes of interstitial cells of Cajal in gastrointestinal diseases. Gastroenterology 2010; 138:1068–1078, e1061-1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yoshida N, Perry AC. Piezo-actuated mouse intracytoplasmic sperm injection (ICSI). Nat Protoc 2007; 2:296–304. [DOI] [PubMed] [Google Scholar]
- 18.Schultz RM. The molecular foundations of the maternal to zygotic transition in the preimplantation embryo. Hum Reprod Update 2002; 8:323–331. [DOI] [PubMed] [Google Scholar]
- 19.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J 2011; 17:10. [Google Scholar]
- 20. C. Tang, Y. Xie, M. Guo, W. Yan, AASRA: An Anchor Alignment-Based Small RNA Annotation Pipeline. Biol Reprod 2021. doi: 10.1093/biolre/ioab062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014; 30:923–930. [DOI] [PubMed] [Google Scholar]
- 22.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014; 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang Q, Lin J, Liu M, Li R, Tian B, Zhang X, Xu B, Liu M, Zhang X, Li Y, Shi H, Wu L. Highly sensitive sequencing reveals dynamic modifications and activities of small RNAs in mouse oocytes and early embryos. Sci Adv 2016; 2:e1501482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014; 30:2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods 2015; 12:357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McCarthy DJ, Campbell KR, Lun AT, Wills QF. Scater: pre-processing, quality control, normalization and visualization of single-cell RNA-seq data in R. Bioinformatics 2017; 33:1179–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and cufflinks. Nat Protoc 2012; 7:562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thomas PD, Campbell MJ, Kejariwal A, Mi H, Karlak B, Daverman R, Diemer K, Muruganujan A, Narechania A. PANTHER: a library of protein families and subfamilies indexed by function. Genome Res 2003; 13:2129–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rassoulzadegan M, Grandjean V, Gounon P, Vincent S, Gillot I, Cuzin F. RNA-mediated non-mendelian inheritance of an epigenetic change in the mouse. Nature 2006; 441:469–474. [DOI] [PubMed] [Google Scholar]
- 30.Kim HK. Transfer RNA-derived small non-coding RNA: dual regulator of protein synthesis. Mol Cells 2019; 42:687–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rakyan VK, Beck S. Epigenetic variation and inheritance in mammals. Curr Opin Genet Dev 2006; 16:573–577. [DOI] [PubMed] [Google Scholar]
- 32.Richards EJ. Inherited epigenetic variation—revisiting soft inheritance. Nat Rev Genet 2006; 7:395–401. [DOI] [PubMed] [Google Scholar]
- 33.Shanmugam R, Fierer J, Kaiser S, Helm M, Jurkowski TP, Jeltsch A. Cytosine methylation of tRNA-asp by DNMT2 has a role in translation of proteins containing poly-asp sequences. Cell Discov 2015; 1:15010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shigematsu M, Kirino Y. tRNA-derived short non-coding RNA as interacting partners of argonaute proteins. Gene Regul Syst Bio 2015; 9:27–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Durdevic Z, Mobin MB, Hanna K, Lyko F, Schaefer M. The RNA methyltransferase Dnmt2 is required for efficient Dicer-2-dependent siRNA pathway activity in drosophila. Cell Rep 2013; 4:931–937. [DOI] [PubMed] [Google Scholar]
- 36.Sha QQ, Zhang J, Fan HY. A story of birth and death: mRNA translation and clearance at the onset of maternal-to-zygotic transition in mammalsdagger. Biol Reprod 2019; 101:579–590. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.