

Early in the firstwave of the COVID‐19 pandemic, one might be forgiven for suggesting that SARS‐CoV‐2 mutations were not a major cause for concern; there were other more pressing issues such as designing and testing new vaccines (Grubaugh et al. 2020). Unfortunately, recent events have challenged this optimistic take. Variant lineages with multiple mutations in spike protein and elsewhere in the genome, notably Alpha (B.1.1.7, first sequenced in the UK), Beta (B.1.351, first sequenced in South Africa), Gamma (P.1, first sequenced in Brazil) and now Delta (B.1.657.2, first sequenced in India) have rapidly expanded (Peacock et al. 2021b; Rambaut et al. 2020; Faria et al. 2021; Tegally et al. 2021). Worryingly, some of these variants appear to have an increase in mortality (Davies et al. 2021) or a reduction in vaccine effectiveness (Madhi et al. 2021). The increased transmissibility (estimates suggest an increase of 50‐70% for Alpha variant (Davies et al. 2021; Volz et al. 2021) and a further similar increase above Alpha for Delta (Public Health England 2021)) meant that nonpharmaceutical control measures to limit virus spread have failed to prevent a second and third wave of the pandemic in the United Kingdom. Opinion has been split among evolutionary biologists about whether the emergence of more transmissible variants was an unexpected development or an obvious prediction. Did current theories of viral evolution not support that more transmissible variants of SARS‐CoV‐2 would evolve? Or does evolution of viruses during a pandemic differ from normal evolution of viruses making the emergence of these variants inevitable?
It seems possible, indeed likely, that viruses that are already endemic evolve differently from those that have newly emerged. Seasonal influenza viruses such as H3N2 influenza continually adapt to humans resulting in gradual antigenic changes, necessitating vaccine updates but without other dramatic phenotypic changes: the fundamental properties of H3N2, how it transmits and the disease it causes, remain the same. Despite generating large diversity within an individual and strong selective pressure on antigenic variants at the population level, H3N2 mutations take a relatively long time to emerge at the population level (Xue et al. 2017; Han et al. 2019; Morris et al. 2020). This is partly because of the extreme bottleneck present at transmission – very few viruses infect an individual (Fig. 1A) – and there will not be immediate selective pressure caused by antibodies within a host (Han et al. 2019). Thus, mutants arising de novo within a single host do not grow to a large population size within the duration of the acute infection. If an advantageous mutation does not occur at the earliest stage of replication within a host, it will be diluted out by other viruses and is unlikely to transmit to a new host (Fig. 1A). Recent studies have suggested that there is relatively low intrahost diversity and a tight bottle neck in SARS‐CoV‐2 transmission (Lythgoe et al. 2021; Valesano et al. 2021). Perhaps experience of seasonal influenza evolution coupled with the proofreading capacity of the SARS‐CoV‐2 polymerase led scientists to believe that SARS‐CoV‐2 would evolve relatively slowly. However, with hindsight, it seems evolving during a pandemic allowed SARS‐CoV‐2 to overcome these restrictive bottlenecks seen in seasonal viruses and evolve differently from influenza.
Figure 1.
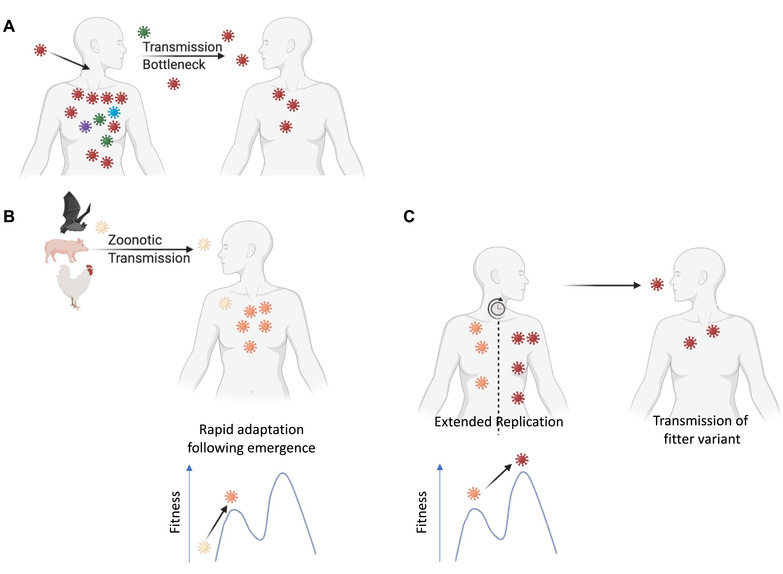
(A) Following infection of a seasonal virus, diversity is generated within a host. However, due to early transmission and a tight bottleneck, transmission of mutated viruses is rare. (B) Following emergence from a zoonotic source, adaptation within human host is rapid as mutations with a large fitness effect are easily gained. (C) Extended replication within a host can lead to multiple mutations and evolution of a fitter virus, which can subsequently transmit and spread. Mutations with epistatic effects could include compensatory mutations allowing the virus to cross fitness valleys. Figure created using BioRender.
How does Viral Evolution Differ in a Pandemic?
A pandemic might lead to a different tempo of viral evolution for the following three interrelated reasons: (1) The virus is not yet optimally adapted to humans. (2) There are a large number of susceptible hosts. (3) These hosts include a proportion of immunocompromised individuals in whom viral replication will be prolonged.
When a virus switches host, there is no clear way to predict how it will evolve (Geoghegan and Holmes 2018; Visher et al. 2021); the virus can become more or less pathogenic provided it maximizes transmission. As viruses are error‐prone and have such high population sizes, it has been suggested that every possible single mutation will be generated in an individual (Sanjuán and Domingo‐Calap 2016). Therefore, once a virus enters a new population, the virus will quickly become adapted and further changes will be gradual (Fig. 1B). This is seen when H7N9 avian influenza is transmitted to humans where the adaptive PB2 E627K mutation gives a large fitness advantage and is quickly amplified following a zoonotic event (Fonville et al. 2013; Liu et al. 2020). At the beginning of a pandemic, a virus may not yet have optimized all its interactions with human host factors such as receptors, antiviral restriction factors, and the innate immune system. A virus that has been transmitting for decades in humans will have more thoroughly explored the local sequence space than a virus that has only recently jumped into humans. SARS‐CoV‐2 was a zoonotic virus and likely maladapted to human factors when it first emerged. Thus, it had considerable potential to increase its transmissibility through evolution in its new host.
In a pandemic, the large number of infections increases the probability of rare events and means that more of the viral sequence space can be explored (Burch et al. 2021). The vast majority of individuals are susceptible that means that every infected person has the potential to spread their mutated virus to others.
Finally, as millions of people will be infected in a pandemic, this will include a larger number of immunocompromised people. Such individuals may not effectively clear the infection allowing extended evolution of the virus within a chronically infected host. The longer period of time for selection to act can potentially lead to fitter variants with multiple mutations especially when mutations interact epistatically. There can be selective pressure on the virus to avoid any functioning immune elements and to maximize growth within a host (Grenfell et al. 2004). In addition, an extended infection allows for recombination between viruses within an individual, which would increase the probability of linking multiple beneficial mutations on a single genome. Combining multiple mutations could allow the virus to find compensatory mutations to overcome potential fitness valleys caused by the costs of an individual mutation, which might not happen during a shorter infection (Fig. 1C). Extended evolution also happens when immunocompromised people are infected by seasonal viruses and remarkably the mutations seen in individual hosts mirror those that later appear fixed in the population (Eden et al. 2017; Xue et al. 2017; Morris et al. 2020). During a pandemic, the extended replication of a maladapted virus within immunocompromised people may be responsible for rare events leading to relatively large jumps in sequence space as opposed to the gradual accumulation of mutations normally seen in viral evolution (Fig. 1C). The Alpha variant rapidly became the major circulating strain in the United Kingdom and subsequently Europe (O'Toole et al. 2021). Alpha has 23 mutations and no closely related viruses with a subset of these mutations have been detected. Alpha also shares several mutations that have been observed in multiple immunocompromised patients (Choi et al. 2020; Karim et al. 2021; Peacock et al. 2021b). It is therefore possible that Alpha evolved during an extended infection of an immunocompromised patient (Rambaut et al. 2020).
Could We have Predicted More Transmissible Variants?
Could experience from previous pandemics have predicted the possibility of more transmissible SARS‐CoV‐2 variants? Large phenotypic changes have occasionally been observed when viruses have crossed between non‐human species (Brault et al. 2007; Kerr et al. 2017; Geoghegan and Holmes 2018): the insertion of a multi‐basic cleavage site causes a switch from low pathogenesis to high pathogenesis when wild avian influenza enters domestic poultry (Monne et al. 2014); a deletion in swine coronavirus TGEV (transmissible gastroenteritis virus) switched the virus from a primarily enteric virus to a solely respiratory virus with reduced virulence (Rasschaert et al. 1990; Kim et al. 2000); and after canine parvovirus jumped from cats to dogs, antigenic variants spread worldwide in the first years following emergence (Parrish 1999). Yet, there have been few historical human pandemics with enough data to determine how the virus evolved during the first years in a human host. Previous zoonotic viruses that caused outbreaks (e.g., SARS‐CoV‐1, Ebola) gained mutations suggested to increase transmission, although this has been difficult to demonstrate experimentally (Grubaugh et al. 2020). These mutations did not change the viral phenotype enough to alter control of the outbreak, but the number of people infected by SARS‐CoV‐1 in 2003 and Ebola in 2014 were orders of magnitude smaller than the current pandemic. In a more apposite comparison, in the influenza pandemic of 1918, the rate of mortality in the second and third wave was greater than in the first wave (Taubenberger and Morens 2006). While the cause for this increased mortality is unknown, there is evidence of virological changes from early in the pandemic (Glaser et al. 2005; Patrono et al. 2021) and this should have been a warning that such changes are possible. Evolutionary virologists were therefore naïve in their belief that variants were unlikely to change the trajectory of the COVID‐19 pandemic – we should have predicted this possibility. The unique conditions of a pandemic allow for larger viral phenotypic changes and a different evolutionary trajectory from that observed for endemic pathogens.
Future Research to Characterize and Predict Variants in the Event of a New Pandemic
Human transmission is determined by a combination of virological, ecological, and human behavioral factors that are difficult to recreate in a laboratory‐based environment. Although we have yet to pinpoint the exact reasons why some SARS‐CoV‐2 variants are more transmissible, several lines of research made promising predictions and suggest a pathway for research in future pandemics. When viruses emerge into humans, receptor binding is usually not optimized for a human receptor and therefore, enhanced receptor binding could cause increased transmissibility due to more efficient cellular entry. Deep mutagenesis scanning revealed that SARS‐CoV‐2 Spike binding to the human ACE2 receptor could be significantly strengthened and highlighted sites (e.g., N501) which subsequently have evolved in multiple variants (Starr et al. 2020). An additional approach could be to examine properties of the virus that differ from close relatives (Escalera‐Zamudio et al. 2021). For SARS‐CoV‐2, it was quickly identified that a multi‐basic furin cleavage site in spike was an essential difference between SARS‐CoV‐2 and both SARS‐CoV and other closely related bat coronaviruses (Coutard et al. 2020; Hoffmann et al. 2020). This cleavage site was necessary for efficient infection but was not an optimal cleavage site in the first wave virus, which suggested that further evolution could lead to increased cleavage (Peacock et al. 2021a). Indeed mutations in the cleavage site that likely increase cleavage have now been seen across variants (Hodcroft et al. 2021) most notably P681R in the Delta variant (Peacock et al. 2021c).
The human innate immune system places a strong selection pressure on zoonotic viruses and this effect can be measured by the degree to which viruses stimulate and are affected by interferon. HIV needed to evolve to counteract human restriction factors before becoming a pandemic (Sharp and Hahn 2011). Recent preprints have suggested that the Alpha variant may be more resistant to interferon, which could partially explain the increase in transmission and the increased severity of this variant (Guo et al. 2021; Thorne et al. 2021). Documenting the types of adaptive change in SARS‐CoV‐2 successful variants will guide our ability to identify variants faster in future pandemics.
Current Research Needs on Emerging Variants
The key questions concerning variants of SARS‐CoV‐2 are whether emerging variants will lead to vaccine escape and whether there will be further increase of pathogenesis and transmissibility. Evidence of antigenic evolution from other coronaviruses shows that antigenic drift is a possibility in SARS‐CoV‐2 (De Wit et al. 2011; Eguia et al. 2021). Passaging viruses in the presence of human sera or the use of deep mutagenesis scanning can indicate whether escape from vaccine‐induced immunity is possible as well as identifying which mutations have the potential to cause escape and should be monitored (Andreano et al. 2020; Greaney et al. 2021; Starr et al. 2021). It remains to be seen whether a universal coronavirus vaccine can be developed that will encompass future variants or whether global monitoring and updating of the SARS‐CoV‐2 vaccine will be required as with influenza. The methods discussed above can be used to predict whether current variants have the potential to become more transmissible. In vitro evolution of the receptor binding domain of spike has shown the potential of even stronger binding to ACE2 to evolve (Zahradnik et al. 2021) and it remains to be seen if these mutations will occur in future variants. However, it is noteworthy that current variants of concern have multiple mutations that appear to be positively selected rather than single mutations (Peacock et al. 2021b; Otto et al. 2021). The need for multiple mutations might explain why these variants arose later in 2020, unlike D614G in spike that increased viral fitness and arose quickly in the first months of the pandemic (Plante et al. 2021). Disentangling how these mutations interact epistatically will be difficult and will not always be possible with approaches such as deep mutagenesis scanning. Multiple mutations present in variants have occurred in immunocompromised patients and one approach might be the use of immunocompromised animal models to attempt to recreate/forecast this evolution (van der Vries et al. 2013). In addition, multiple mutations could be studied rationally through structural analysis or through combining mutations occurring in different variants into a single virus. The emergence of new variants has sent countries scrambling to put into place pipelines to identify and examine variants in a systematic manner. As the number of people naïve to SARS‐CoV‐2 dwindles, a multi‐pronged approach will be needed to understand whether evolving variants are likely to have prolonged infection, be more transmissible, and/or evade immunity (Otto et al. 2021).
Virus biodiversity and the potential for virus evolution remains vast and unexplored. Given the paucity of pandemics, virologists have had little experience of how a virus evolves immediately after jumping into humans; mutations may cause viruses to change in ways that have significant impacts on their epidemiology as well as on disease. An increase in transmissibility of 50% for SARS‐CoV‐2 was surprising but should have been foreseen. Further evolution of SARS‐CoV‐2 (e.g., the rise of the Delta variant) and evolution in future pandemics will now be closely monitored. It has become clear that limiting the spread of a pandemic may not only be necessary to prevent infections but also to prevent the evolution of strains with the potential to cause deadlier outbreaks (Burch et al. 2021). In addition, we should be warier of the continued potential for change in currently circulating viruses in both animals and humans. For example, emergence of arboviruses such as Chikungunya and Zika with enhanced transmission and pathogenesis potential is likely to reoccur. Moreover, we do not understand the sudden recent outbreaks of EV‐D68 and why it causes Acute Flaccid Myelitis (Imamura and Oshitani 2015). How many mutations is this virus away from becoming the next polio? The potential for a handful of mutations to turn a benign virus into a deadly virus has never been clearer.
AUTHOR CONTRIBUTIONS
D.G. and W.B. conceived and wrote the manuscript.
CONFLICT OF INTEREST
The authors have no conflicts of interest.
Associate Editor: R. Kassen
Handling Editor: T. Chapman
ACKNOWLEDGMENTS
The authors would like to thank Valerie Morley, Thomas Peacock, Elisa Visher and Brian Wasik for helpful comments and discussion. This work was supported by Wellcome Trust grant 205100.
LITERATURE CITED
- Andreano, E. , Piccini G., Licastro D., Casalino L., Johnson N. V., Paciello I., Monego S. D., Pantano E., Manganaro N., Manenti A., et al. 2020. SARS‐CoV‐2 escape in vitro from a highly neutralizing COVID‐19 convalescent plasma. bioRxiv:2020.2012.2028.424451. [DOI] [PMC free article] [PubMed]
- Brault, A. C. , Huang C. Y., Langevin S. A., Kinney R. M., Bowen R. A., Ramey W. N., Panella N. A., Holmes E. C., Powers A. M., and Miller B. R.. 2007. A single positively selected West Nile viral mutation confers increased virogenesis in American crows. Nat. Genet. 39:1162‐1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burch, C. , Weinreich D. M., Raynes Y., and Decurzio J. M.. 2021. Perspective: The evolutionary dangers of high COVID case counts. EcoEvoRxiv.
- Choi, B. , Choudhary M. C., Regan J., Sparks J. A., Padera R. F., Qiu X., Solomon I. H., Kuo H. H., Boucau J., Bowman K., et al. 2020. Persistence and evolution of SARS‐CoV‐2 in an immunocompromised host. N Engl J Med 383:2291‐2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutard, B. , Valle C., de Lamballerie X., Canard B., Seidah N. G., and Decroly E.. 2020. The spike glycoprotein of the new coronavirus 2019‐nCoV contains a furin‐like cleavage site absent in CoV of the same clade. Antiviral Res 176:104742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies, N. G. , Abbott S., Barnard R. C., Jarvis C. I., Kucharski A. J., Munday J. D., Pearson C. A. B., Russell T. W., Tully D. C., Washburne A. D., et al. 2021. Estimated transmissibility and impact of SARS‐CoV‐2 lineage B.1.1.7 in England. Science 372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Wit, J. , Cook J. K., and Van der Heijden H. M.. 2011. Infectious bronchitis virus variants: a review of the history, current situation and control measures. Avian pathology 40:223‐235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eden, J. S. , Chisholm R. H., Bull R. A., White P. A., Holmes E. C., and Tanaka M. M.. 2017. Persistent infections in immunocompromised hosts are rarely sources of new pathogen variants. Virus Evol 3:vex018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eguia, R. T. , Crawford K. H. D., Stevens‐Ayers T., Kelnhofer‐Millevolte L., Greninger A. L., Englund J. A., Boeckh M. J., and Bloom J. D.. 2021. A human coronavirus evolves antigenically to escape antibody immunity. PLoS Pathog 17:e1009453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escalera‐Zamudio, M. , Pond S. L. K., de la Vina N. M., Gutierrez B., Theze J., Bowden T. A., Pybus O. G., and Hulswit R. J. G.. 2021. Identification of site‐specific evolutionary trajectories shared across human betacoronaviruses. bioRxiv:2021.2005.2024.445313.
- Faria, N. R. , Mellan T. A., Whittaker C., Claro I. M., Candido D. D., Mishra S., Crispim M. A. E., Sales F. C., Hawryluk I., McCrone J. T., et al. 2021. Genomics and epidemiology of the P.1 SARS‐CoV‐2 lineage in Manaus, Brazil. Science 372:815‐+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonville, J. M. , Burke D. F., Lewis N. S., Katzelnick L. C., and Russell C. A.. 2013. Quantifying the fitness advantage of polymerase substitutions in Influenza A/H7N9 viruses during adaptation to humans. PLoS One 8:e76047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geoghegan, J. L. , and Holmes E. C.. 2018. The phylogenomics of evolving virus virulence. Nat Rev Genet 19:756‐769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaser, L. , Stevens J., Zamarin D., Wilson I. A., Garcia‐Sastre A., Tumpey T. M., Basler C. F., Taubenberger J. K., and Palese P.. 2005. A single amino acid substitution in 1918 influenza virus hemagglutinin changes receptor binding specificity. J Virol 79:11533‐11536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greaney, A. J. , Starr T. N., Gilchuk P., Zost S. J., Binshtein E., Loes A. N., Hilton S. K., Huddleston J., Eguia R., Crawford K. H. D., et al. 2021. Complete mapping of mutations to the SARS‐CoV‐2 Spike Receptor‐Binding Domain that Escape Antibody Recognition. Cell Host & Microbe 29:44‐+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grenfell, B. T. , Pybus O. G., Gog J. R., Wood J. L., Daly J. M., Mumford J. A., and Holmes E. C.. 2004. Unifying the epidemiological and evolutionary dynamics of pathogens. Science 303:327‐332. [DOI] [PubMed] [Google Scholar]
- Grubaugh, N. D. , Petrone M. E., and Holmes E. C.. 2020. We shouldn't worry when a virus mutates during disease outbreaks. Nat. Microbiol. 5:529‐530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, K. , Barrett B. S., Mickens K. L., Hasenkrug K. J., and Santiago M. L.. 2021. Interferon Resistance of Emerging SARS‐CoV‐2 Variants. bioRxiv:2021.2003.2020.436257. [DOI] [PMC free article] [PubMed]
- Han, A. X. , Maurer‐Stroh S., and Russell C. A.. 2019. Individual immune selection pressure has limited impact on seasonal influenza virus evolution. Nature Ecology & Evolution 3:302‐+. [DOI] [PubMed] [Google Scholar]
- Hodcroft, E. B. , Domman D. B., Snyder D. J., Oguntuyo K., Van Diest M., Densmore K. H., Schwalm K. C., Femling J., Carroll J. L., Scott R. S., et al. 2021. Emergence in late 2020 of multiple lineages of SARS‐CoV‐2 Spike protein variants affecting amino acid position 677. medRxiv:2021.2002.2012.21251658. [Google Scholar]
- Hoffmann, M. , Kleine‐Weber H., and Pohlmann S.. 2020. A Multibasic Cleavage Site in the Spike Protein of SARS‐CoV‐2 Is Essential for Infection of Human Lung Cells. Molecular Cell 78:779‐+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamura, T. , and Oshitani H.. 2015. Global reemergence of enterovirus D68 as an important pathogen for acute respiratory infections. Reviews in medical virology 25:102‐114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karim, F. , Moosa M. Y. S., Gosnell B. I., Cele S., Giandhari J., Pillay S., Tegally H., Wilkinson E., San J. E., Msomi N., et al. 2021. Persistent SARS‐CoV‐2 infection and intra‐host evolution in association with advanced HIV infection. medRxiv:2021.2006.2003.21258228.
- Kerr, P. J. , Cattadori I. M., Liu J., Sim D. G., Dodds J. W., Brooks J. W., Kennett M. J., Holmes E. C., and Read A. F.. 2017. Next step in the ongoing arms race between myxoma virus and wild rabbits in Australia is a novel disease phenotype. Proceedings of the National Academy of Sciences of the United States of America 114:9397‐9402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, L. , Hayes J., Lewis P., Parwani A. V., Chang K. O., and Saif L. J.. 2000. Molecular characterization and pathogenesis of transmissible gastroenteritis coronavirus (TGEV) and porcine respiratory coronavirus (PRCV) field isolates co‐circulating in a swine herd. Arch Virol 145:1133‐1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, W. J. , Li J., Zou R., Pan J., Jin T., Li L., Liu P., Zhao Y., Yu X., Wang H., et al. 2020. Dynamic PB2‐E627K substitution of influenza H7N9 virus indicates the in vivo genetic tuning and rapid host adaptation. Proc Natl Acad Sci U S A 117:23807‐23814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lythgoe, K. A. , Hall M., Ferretti L., de Cesare M., MacIntyre‐Cockett G., Trebes A., Andersson M., Otecko N., Wise E. L., Moore N., et al. 2021. SARS‐CoV‐2 within‐host diversity and transmission. Science 372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhi, S. A. , Baillie V., Cutland C. L., Voysey M., Koen A. L., Fairlie L., Padayachee S. D., Dheda K., Barnabas S. L., Bhorat Q. E., et al. 2021. Efficacy of the ChAdOx1 nCoV‐19 Covid‐19 Vaccine against the B.1.351 Variant. N Engl J Med 384:1885‐1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monne, I. , Fusaro A., Nelson M. I., Bonfanti L., Mulatti P., Hughes J., Murcia P. R., Schivo A., Valastro V., Moreno A., et al. 2014. Emergence of a highly pathogenic avian influenza virus from a low‐pathogenic progenitor. J Virol 88:4375‐4388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris, D. H. , Petrova V. N., Rossine F. W., Parker E., Grenfell B. T., Neher R. A., Levin S. A., and Russell C. A.. 2020. Asynchrony between virus diversity and antibody selection limits influenza virus evolution. Elife 9:e62105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Toole, A. , Hill V., Pybus O. G., A. Watts Bogoch, II , Khan K., J. P. Messina, C.‐G. U. consortium, A. Network for Genomic Surveillance in South, U. K. C. G. N. Brazil , Tegally H., Lessells R. R., Giandhari J., Pillay S., et al. 2021. L. National Virus Reference, C. S. Seq, C. Danish Covid‐19 Genome, N. Communicable Diseases Genomic, S.‐C.‐s. p. Dutch National, D. Division of Emerging Infectious, T. de Oliveira, N. Faria, A. Rambaut, and M. U. G. Kraemer. Tracking the international spread of SARS‐CoV‐2 lineages B.1.1.7 and B.1.351/501Y‐V2. Wellcome Open Res 6:121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto, S. P. , Day T., Arino J., Colijn C., Dushoff J., Li M., Mechai S., Van Domselaar G., Wu J., Earn D. J. D., et al. 2021. The origins and potential future of SARS‐CoV‐2 variants of concern in the evolving COVID‐19 pandemic. Current Biology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish, C. R. 1999. Host range relationships and the evolution of canine parvovirus. Vet Microbiol 69:29‐40. [DOI] [PubMed] [Google Scholar]
- Patrono, L. V. , Vrancken B., Budt M., Düx A., Lequime S., Boral S., Gilbert M. T. P., Gogarten J. F., Hoffmann L., Horst D., et al. 2021. Archival influenza virus genomes from Europe reveal genomic and phenotypic variability during the 1918 pandemic. bioRxiv:2021.2005.2014.444134. [DOI] [PMC free article] [PubMed]
- Peacock, T. P. , Goldhill D. H., Zhou J., Baillon L., Frise R., Swann O. C., Kugathasan R., Penn R., Brown J. C., Sanchez‐David R. Y., et al. 2021a. The furin cleavage site in the SARS‐CoV‐2 spike protein is required for transmission in ferrets. Nat Microbiol 6:899‐909. [DOI] [PubMed] [Google Scholar]
- Peacock, T. P. , Penrice‐Randal R., Hiscox J. A., and Barclay W. S.. 2021b. SARS‐CoV‐2 one year on: evidence for ongoing viral adaptation. J Gen Virol 102:001584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peacock, T. P. , Sheppard C. M., Brown J. C., Goonawardane N., Zhou J., Whiteley M., de Silva T. I., and Barclay W. S.. 2021c. The SARS‐CoV‐2 variants associated with infections in India, B.1.617, show enhanced spike cleavage by furin. bioRxiv:2021.2005.2028.446163.
- Plante, J. A. , Liu Y., Liu J. Y., Xia H. J., Johnson B. A., Lokugamage K. G., Zhang X. W., Muruato A. E., Zou J., Fontes‐Garfias C. R., et al. 2021. Spike mutation D614G alters SARS‐CoV‐2 fitness (vol 592, pg 116, 2021). Nature 592:116‐121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Public Health England . 2021. SARS‐CoV‐2 variants of concern and variants under investigation in England Technical briefing 16. 18 June 2021. www.gov.uk.
- Rambaut, A. , Loman N., Pybus O., Barclay W., Barrett J., Carabelli A., Connor T., Peacock T., Robertson D. L., Volz E., et al. 2020. Preliminary genomic characterisation of an emergent SARS‐CoV‐2 lineage in the UK defined by a novel set of spike mutations, virological.org.
- Rasschaert, D. , Duarte M., and Laude H.. 1990. Porcine respiratory coronavirus differs from transmissible gastroenteritis virus by a few genomic deletions. Journal of General Virology 71:2599‐2607. [DOI] [PubMed] [Google Scholar]
- Sanjuán, R. , and Domingo‐Calap P.. 2016. Mechanisms of viral mutation. Cellular and molecular life sciences 73:4433‐4448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp, P. M. , and Hahn B. H.. 2011. Origins of HIV and the AIDS pandemic. Cold Spring Harbor perspectives in medicine 1:a006841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr, T. N. , Greaney A. J., Addetia A., Hannon W. W., Choudhary M. C., Dingens A. S., Li J. Z., and Bloom J. D.. 2021. Prospective mapping of viral mutations that escape antibodies used to treat COVID‐19. Science 371:850‐854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr, T. N. , Greaney A. J., Hilton S. K., Ellis D., Crawford K. H. D., Dingens A. S., Navarro M. J., Bowen J. E., Tortorici M. A., Walls A. C., et al. 2020. Deep Mutational Scanning of SARS‐CoV‐2 Receptor Binding Domain Reveals Constraints on Folding and ACE2 Binding. Cell 182:1295‐+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taubenberger, J. K. , and Morens D. M.. 2006. 1918 influenza: the mother of all pandemics. Emerging Infectious Diseases 12:15‐22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tegally, H. , Wilkinson E., Giovanetti M., Iranzadeh A., Fonseca V., Giandhari J., Doolabh D., Pillay S., San E. J., Msomi N., et al. 2021. Detection of a SARS‐CoV‐2 variant of concern in South Africa. Nature 592:438‐443. [DOI] [PubMed] [Google Scholar]
- Thorne, L. G. , Bouhaddou M., Reuschl A. K., Zuliani‐Alvarez L., Polacco B., Pelin A., Batra J., Whelan M. V. X., Ummadi M., Rojc A., et al. 2021. Evolution of enhanced innate immune evasion by the SARS‐CoV‐2 B.1.1.7 UK variant. bioRxiv:2021.2006.2006.446826.
- Valesano, A. L. , Rumfelt K. E., Dimcheff D. E., Blair C. N., Fitzsimmons W. J., Petrie J. G., Martin E. T., and Lauring A. S.. 2021. Temporal dynamics of SARS‐CoV‐2 mutation accumulation within and across infected hosts. Plos Pathogens 17:e1009499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Vries, E. , Stittelaar K. J., van Amerongen G., Veldhuis Kroeze E. J., de Waal L., Fraaij P. L., Meesters R. J., Luider T. M., van der Nagel B., Koch B., et al. 2013. Prolonged influenza virus shedding and emergence of antiviral resistance in immunocompromised patients and ferrets. PLoS Pathog 9:e1003343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visher, E. , Evensen C., Guth S., Lai E., Norfolk M., Rozins C., Sokolov N. A., Sui M., and Boots M.. 2021. The Three Ts of Pathogen Evolution During Zoonotic Emergence. EcoEvoRxiv. [DOI] [PMC free article] [PubMed]
- Volz, E. , Mishra S., Chand M., Barrett J. C., Johnson R., Geidelberg L., Hinsley W. R., Laydon D. J., Dabrera G., O'Toole A., et al. 2021. Assessing transmissibility of SARS‐CoV‐2 lineage B.1.1.7 in England. Nature 593:266‐269. [DOI] [PubMed] [Google Scholar]
- Xue, K. S. , Stevens‐Ayers T., Campbell A. P., Englund J. A., Pergam S. A., Boeckh M., and Bloom J. D.. 2017. Parallel evolution of influenza across multiple spatiotemporal scales. Elife 6:e26875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahradnik, J. , Marciano S., Shemesh M., Zoler E., Chiaravalli J., Meyer B., Dym O., Elad N., and Schreiber G.. 2021. SARS‐CoV‐2 RBD in vitro evolution follows contagious mutation spread, yet generates an able infection inhibitor. bioRxiv.