

Abstract
Background/Aim
SARS‐CoV‐2 is one of the coronavirus families that emerged at the end of 2019. It infected the respiratory system and caused a pandemic worldwide. Fluoroquinolones (FQs) have been safely used as antibacterial agents for decades. The antiviral activity of FQs was observed. Moreover, substitution on the C‐7 position of ciprofloxacin enhanced its antiviral activity. Therefore, this study aims to investigate the antiviral activity of 7‐(4‐(N‐substituted‐carbamoyl‐methyl)piperazin‐1yl)‐chalcone in comparison with ciprofloxacin against SARS‐CoV‐2 main protease (Mpro).
Materials and methods
Vero cells were infected with SARS‐CoV‐2. After treatment with ciprofloxacin and the chalcone at the concentrations of 1.6, 16, 160 nmol/L for 48 h, SARS‐CoV‐2 viral load was detected using real‐time qPCR, SARS‐CoV‐2 infectivity was determined using plaque assay, and the main protease enzyme activity was detected using in vitro 3CL‐protease inhibition assay. The activity of the chalcone was justified through molecular docking within SARS‐CoV‐2 Mpro, in comparison with ciprofloxacin.
Results
The new chalcone significantly inhibited viral load replication where the EC50 was 3.93 nmol/L, the plaque formation ability of the virus was inhibited to 86.8% ± 2.47. The chalcone exhibited a significant inhibitory effect against SARS‐CoV‐2 Mpro in vitro in a dose‐dependent manner. The docking study into SARS‐CoV‐2 Mpro active site justified the importance of adding a substitution to the parent drug. Additionally, the assessment of the drug‐likeness properties indicated that the chalcone might have acceptable ADMET properties.
Conclusion
The new chalcone might be useful and has new insights for the inhibition of SARS‐CoV‐2 Mpro.
Keywords: ciprofloxacin‐chalcone, fluoroquinolones, molecular docking, protease enzyme, SARS‐CoV‐2
1. INTRODUCTION
Fluoroquinolones (FQs) are a group of approved compounds by the FDA for their antibacterial activity; they target bacterial topoisomerase and DNA gyrase enzymes [1]. FQs are considered a multi‐faceted family, they showed antiprotozoal, antiparasitic, antifungal, anticancer and antiviral activities [2, 3, 4, 5, 6]. A previous study indicated the effectiveness of FQs against papovavirus, human cytomegalovirus and varicella‐zoster virus [7]. Others indicated the efficacy of some FQs including ciprofloxacin, ofloxacin, levofloxacin and gatifloxacin in the treatments of single‐stranded RNA hepatitis C virus (HCV); they suppressed the growth and proliferation of hepatoma Huh‐7 and Huh‐8 cells of HCV by targeting HCV helicase activity [8, 9]. Ciprofloxacin was demonstrated to inhibit BK viral replication in a dose‐dependent manner [10]. Additionally, ciprofloxacin inhibited African swine fever virus. A study by Mottola et al., examined thirty different members of FQs family against African swine fever virus, six members severely reduced the cytopathic effect of the virus. The genome of the virus was completely undetectable after 7 days of treatment [11]. A recent study by Scroggs et al. showed that FQs were effective in the inhibition of flaviviruses zika viral replication, where enoxacin inhibited the virus in an intermediate step during viral life span [12]. FQs were also demonstrated to inhibit dengue virus [12] and rhinovirus [13]. Clinical trials data revealed that FQs exerted activity against DNA and RNA viruses [14]. Ali et al. demonstrated that FQs can inhibit simian virus 40 (SV40) growth and DNA replication through inhibition of SV40 helicase activity [14].
Fluoroquinolones are considered a central scaffold and an endless source of production of various bioactive molecules. The ease and the flexibility of their chemical synthesis led to the development of several thousands of derivatives in literature, where many could exert potent activity towards different diseases, in particular, viral diseases. A structural modification in the C‐7 position of the parent drug, ciprofloxacin, to obtain new derivatives has been shown to enhance its potency, spectrum, pharmacokinetic, antiviral and antibacterial activity against ciprofloxacin‐resistant pathogen with improved antianaerobic activity [15, 16]. It was reported that the presence of a carboxylic group at C‐3 and bulky substituent on C‐7 position and N1 position of the quinolone moiety contributed to the enhancement of the antiviral activity [16]. Many quinolones with different substituents were investigated as antiviral agents against human immunodeficiency virus (HIV)‐1, which could be used in Acquired Immune Deficiency Syndrome (AIDS) [16]. Substitution on position 7 was observed to inhibit retroviruses such as HIV‐1 and HIV‐2 transcription with no effect on the viability and proliferation of the host cell [17]. Hagihara et al. demonstrated that substitution on piperazine ring seems necessary for anti‐HIV‐1 activity, where different substitution exerted variable selectivity towards HIV‐1 [18]. Also, sulphamidomethyl substitution on a piperazine ring yielded an active derivative against influenza H1N1, H3N2 and H5N1 viruses [19]. FQs substituted with (2‐hydroxyethoxy) methyl fragment at N‐1 showed activity against herpes virus with IC50 2.3 μmol/L [20]. While tricyclic FQs were found to exert a high activity against hepatitis B virus where the IC50 is 0.1 μmol/L [21].
Coronaviruses are a large family of viruses that infected the liver, digestive system, central nervous system and respiratory system, in both humans and animals [22]. SARS‐CoV‐2, a new strain that emerged at the end of 2019, caused a pandemic worldwide [23]. Remdesivir was approved by the FDA for COVID‐19 treatment; however, it is only administrated intravenously to hospitalized patients over the age of 12 years [24]. These limitations desperately urge scientists worldwide to investigate potential therapies. The main protease (Mpro) of SARS‐CoV‐2 offers a frequent target for developing specific anti‐SARS‐CoV‐2 agents due to its considerable role in the viral life cycle, gene replication and expression [25, 26, 27].
All previous data of the antiviral activity of FQs are prompted this study. Herein, we proposed the possibility of ciprofloxacin activity against SARS‐CoV‐2. Also, since structural alteration at the C‐7 position has enhanced the antiviral activity of FQs against various viral diseases, we hypothesized that structure modification and incorporation of an extension at C‐7 position of the parent drug, ciprofloxacin, could heighten its effectiveness towards the viral infection, SARS‐CoV‐2. Therefore, the present study aims to investigate the antiviral activity of 7‐(4‐(N‐substituted carbamoyl methyl) piperazin‐1 yl)‐ chalcone (the chalcone) against SARS‐CoV‐2 Mpro in vitro, in comparison with the parent drug ciprofloxacin, supported by a molecular docking study.
2. MATERIALS AND METHODS
2.1. Chemistry
Ciprofloxacin‐chalcone was prepared from a reaction between the appropriate amine and bromoacetyl bromide using potassium carbonate as a base in dichloromethane followed by alkylation of ciprofloxacin (#17850, Sigma‐Aldrich, Inc, St Louis, MO, USA) with 2‐bromoacetamide derivatives in acetonitrile in the presence of triethylamine (TEA) [28, 29]. The structure of ciprofloxacin and the chalcone is shown in Figure 1, where the incorporated substitution to C‐7 position is highlighted. This chalcone was identified by 1HNMR, 13C‐NMR and mass spectrometry as previously reported [29, 30].
FIGURE 1.
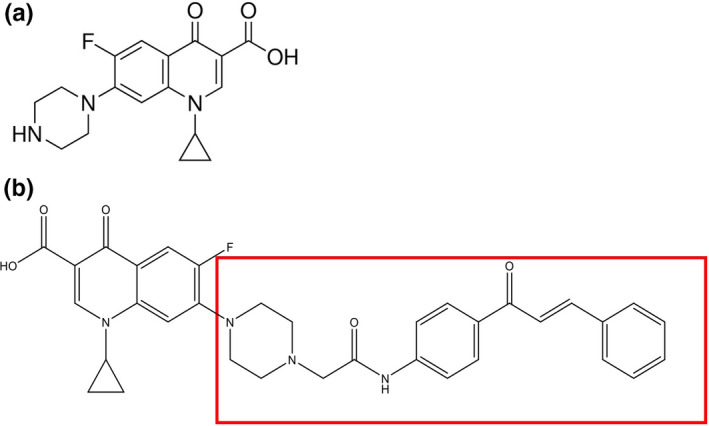
Chemical structure of (a) ciprofloxacin and (b) 7‐(4‐(N‐substituted carbamoyl methyl) piperazin‐1 yl) chalcone of ciprofloxacin, where the incorporated substitution to C‐7 position is highlighted
2.2. Cell culture, viral infection and drug treatment
Vero cells were obtained from American type culture collection (#CCL‐81, ATCC, Manassas, VA, USA). Fresh Dulbecco's Modified Eagle's Medium (#D5030, DMEM, Sigma‐Aldrich, Inc) was used as a culture medium, augmented with 10% foetal bovine serum (#FBS‐12A, BioSolutions International, Melbourne, Australia), 1% penicillin‐streptomycin mixture (#15140148, Invitrogen, Grand Island, NY, USA) and 1% L‐glutamine (#G7513, Sigma‐Aldrich, Inc) in a humidified 5% CO2 atmosphere at 37°C. Cells were seeded into 24‐well tissue culture plates for 24 h, followed by transfection with SARS‐CoV‐2 (ATCC, nCoV‐WA1‐2020) at Multiplicity of infection (MOI) of 0.01 for 24 h. The inoculum was discarded and replaced with fresh media containing 1.6, 16, 160 nmol/L of ciprofloxacin and the new chalcone, while Remdesivir (#GS5734, Sigma‐Aldrich, Inc) was used as positive control [31] followed by incubation for 48 h. The experiment was done in triplicate.
2.3. Viral RNA isolation and real‐time qPCR assay
Viral RNA was extracted from cell pellet using QIAamp Viral RNA Mini Handbook (#52904, Qiagen, Hilden, Germany) according to manufacturer's instruction, then the viral load of SARS‐CoV‐2 was detected using abTES™ COVID‐19 qPCR I Kit (#300142, AITbiotech, Singapore) according to manufacturer's instruction. This kit enables simultaneous detection of two SARS‐CoV‐2‐specific signature regions from its non‐structure polypeptide (orf1a) in a single reaction. It also includes detection of human housekeeping gene, GAPDH, as an Internal Control (IC) to identify possible PCR inhibitions from sample processing. Real‐time qPCR assays were performed on an Applied Biosystems ABI 7500 Fast real‐time PCR system (Applied Biosystems, Foster City, CA, USA) using cycling conditions with an initial reverse transcription at 59°C for 10 min for 1 cycle, then 95°C for 2 min for 1 cycle, followed by 95°C for 10 s, 57.5°C for 30 s for 45 cycles. Fluorescence was measured using FAM and Texas Red for NS1 and NS2, respectively, while using (HEX/VIC) for the internal control, GAPDH. Viral RNA concentration equivalent per mL of SARS‐CoV‐2 was determined using RT‐PCR [32].
2.4. Plaque assay
Vero cells were seeded (106 cells /well) into 6‐well plate and left overnight at 37°C with 5% CO2. SARS‐CoV‐2 was diluted with complete DMEM and added to each well (200 μL/well), the plate was incubated for 1 h at 37°C with 5% CO2. Different concentrations of ciprofloxacin and the chalcone 1.6, 16, 160 nmol/L was added to the sample wells. Remdesivir was used as positive control. The overlay media was prepared (1 mL PBS, 31.5 2× MEM (#M5650, minimum essential medium, Sigma‐Aldrich), 17.5 mL Oxid agar (#LP0011B, ThermoFischer Scientific)) and added to each well (2 mL/well), then incubated at 37°C for 72 h. The plate was then fixed with 5% formaldehyde (1.5 mL/well) and left overnight to ensure virus inactivation. The plate was stained with crystal violet for 1 h. Number of plaques was determined using ImageJ software, virus titre (PFU/ml) was determined. The experiment was done in triplicate.
2.5. In vitro 3CL‐protease inhibition assay
To understand the mechanism of action of the chalcone on the inhibition of viral replication of SARS‐CoV‐2, 3CL‐ Protease inhibition (Mpro) was examined using 3CL‐ Protease (SARS‐CoV‐2) Assay Kit (#78042, Bioscience, San Diego, CA, USA) according to manufacturer's instructions [33], 3CL inhibitor #GC376 was used as positive inhibitor. 0.5 M of dithiothreitol (DTT) was resuspended with 3CL‐Protease assay buffer to a final concentration of 1 mmol/L. The 3CL‐Protease was diluted with 1mM DTT at 3–5 ng/μL. 30 μL diluted 3CL‐Protease enzyme solution was added to all wells, followed by10 µL GC376 was added to the positive control wells, 10 μL no inhibitor buffer was added to the negative control wells, and the new chalcone was used at different concentrations (0.016, 0.16, 1.6, 16) μmol/L to the sample wells. The experiment was done in triplicate. Plate was sealed and incubated overnight. Fluorescence intensity was measured at 450 nm in a microtiter plate‐reading fluorimeter (ROBONIK P2000 ELISA READER, India).
2.6. Cell viability assay
Cell viability assay was achieved using MTT reagent [3‐(4, 5‐dimethyl thiazol‐2yl)‐2, 5‐diphenyltetrazolium bromide] (#11465007001, Sigma‐Aldrich). Vero cells (10 000 cells/well) were seeded in 96‐well plates and allowed to grow in fresh DMEM medium for 24 h. Then, medium was changed with fresh DMEM containing different concentrations (0.016, 0.16, 1.6, 16, 160 μmol/L) of ciprofloxacin and the chalcone. After incubation for 24, 10 μL of MTT (5 μg/mL) was added per well and incubated in the dark for 3 h at 37°C. To dissolve the Formazan crystals that were formed, 100 μl of DMSO were used and absorbance was measured using an ELISA reader at 450 nmol/L [34].
2.7. Molecular docking
Molecular modelling and visualization processes were performed on COVID‐19 Mpro using Molecular Operating Environment (MOE) 2019.0102. The co‐crystal structure was retrieved from the RCSB Protein Data Bank (PDB ID: 6LU7). Ciprofloxacin and its chalcone were prepared with the standard protocol in MOE 2019 and the energy of the docked compounds was minimized with gradient RMS of 0.0001 kcal/mol. [35] Then, protein structure was prepared by using the MOE QuickPrep protocol. The native ligand N3 was re‐docked into the active site using the same set of parameters as described above to validate the present docking study at the active site. The root mean square deviation (RMSD) of the best‐docked pose was 0.9010 Å and the energy score was −9.3 Kcal/mol, thus validating the docking study with MOE software (RMSD is the measure of the average distance of the docked conformation compared to the reference conformation). The two compounds were docked in the active site using the method of Alpha triangle placement with Amber10: EHT forcefield. Refinement was performed with Forcefield and scored using the Affinity dG scoring system.
2.8. Statistical analysis
Results were obtained from at least three independent experiments. Data were expressed as mean ± standard deviation. Differences were normalized and analysed by Student's t test after one‐way analysis of variance (ANOVA), with the use of GraphPad Prism 9 statistical software (GraphPad Software, San Diego, CA, USA). Differences were considered significant when the probability values (P) were less than 0.05.
3. RESULTS
3.1. The chalcone suppressed SARS‐CoV‐2 viral load in Vero cells
Ciprofloxacin and the new chalcone were investigated for their efficacy in inhibiting viral replication of SARS‐CoV‐2 in Vero cells. The infected cells were treated with both compounds for 48 hours at different concentrations, while Remdesivir was used as a positive control. As shown in Figure 2, both compounds suppressed viral load in a dose‐dependent manner. At the concentration of 160 nmol/L, SARS‐CoV‐2 viral load in cells treated with ciprofloxacin and the chalcone was reduced (P < 0.001) to 398 782 ± 32 059 IU/mL and 133 172 ± 12 873 IU/mL, respectively, where the viral titre was 1 948 834 IU/mL in infected untreated cells. The percentage of inhibition of ciprofloxacin and the chalcone against viral SARS‐CoV‐2 was 79.5 ± 3.81% and 93.1 ± 2.92%, respectively, while it was 100% for Remdesivir at the same concentration. The EC50 of ciprofloxacin, the chalcone and Remdesivir was calculated using GraphPad Prism 9 software as presented in Table 1.
FIGURE 2.
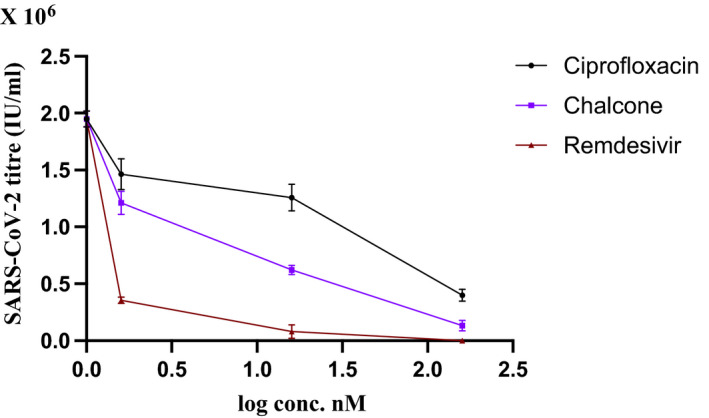
Antiviral activity of different concentrations of ciprofloxacin and the chalcone against SARS‐CoV‐2 viral replication in Vero cells after 48 treatment. Data represents mean ± SD. Remdesivir was used as positive control
TABLE 1.
EC50 values for ciprofloxacin, the chalcone and Remdesivir against SARS‐CoV‐2 viral replication in Vero cells
| Virus | Drug | EC50 |
|---|---|---|
| SARS‐CoV‐2 | Ciprofloxacin | 50.07 nmol/L |
| The chalcone | 3.93 nmol/L* | |
| Remdesivir | 1.55 nmol/L* |
P < 0.001, when compared to ciprofloxacin.
3.2. Plaque assay
To better examine the infectivity of the virus after treatment with ciprofloxacin and the chalcone, plaque assay was established. As shown in Figure 3, the plaque formation ability of SARS‐CoV‐2 was reduced in a concentration‐dependent manner. When compared to infected untreated cells, it was significantly reduced (P < 0.001) after treatment with ciprofloxacin and the chalcone at the concentration of 160 nmol/L to 1 388 000 ± 140 171 PFU/mL and 452 000 ± 43 714 PFU/mL, respectively, where it was 3 811 000 PFU/mL in infected untreated Vero cells. The percentage of plaque formation inhibition of ciprofloxacin and the chalcone was 63.5 ± 3.1% and 88.1 ± 4.4%, respectively. Remdesivir was used as positive control.
FIGURE 3.
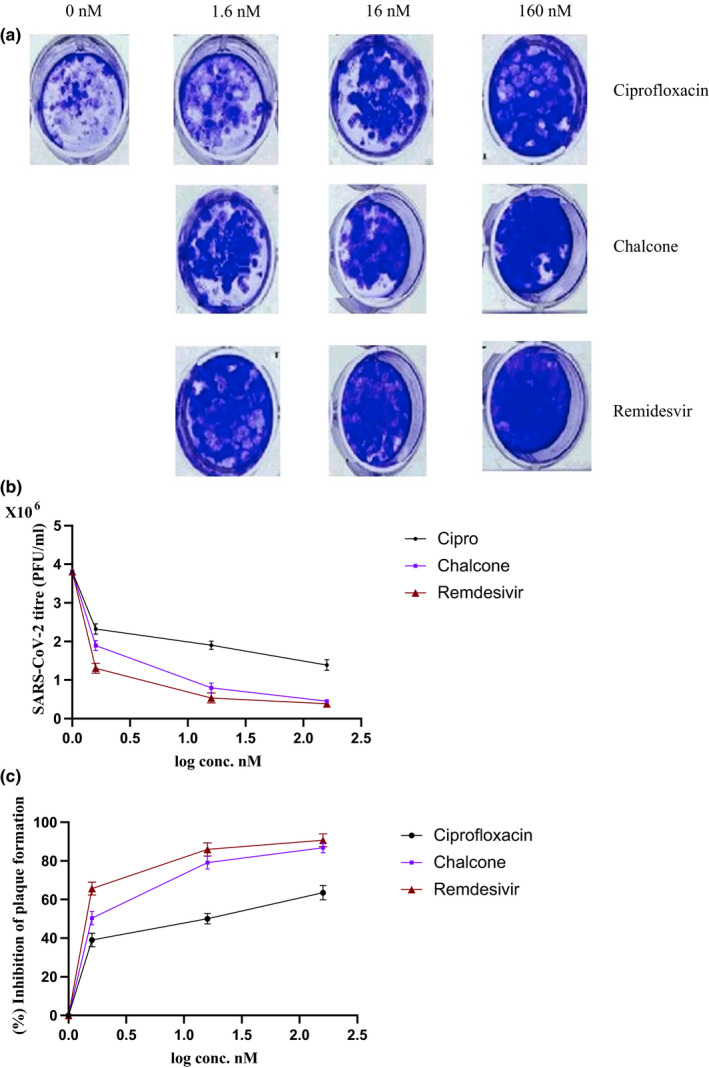
Plaque formation ability of SARS‐CoV‐2 after treatment with ciprofloxacin and the chalcone. Remdesivir was used as positive control. (a) Representative photos of plaque formation after treatment with different concentrations of ciprofloxacin and the chalcone. (b) The plaque formation ability (PFU/ml) of SARS‐CoV‐2 after treatment with different concentrations of ciprofloxacin and the chalcone. (c) Percentage of inhibition of SARS‐CoV‐2 plaque formation after ciprofloxacin and the chalcone treatment at different concentrations. Data represents mean ± SD. Significant difference was analysed by one‐way ANOVA test and student t‐test. **P < 0.01, ***P < 0.001, compared to infected untreated cells
3.3. Inhibition of viral 3CL‐protease activity
For better understanding of the biochemical basis underlying the activity of ciprofloxacin and the chalcone against SARS‐CoV‐2, we investigated their activity against the virus Mpro. As shown in Figure 4, ciprofloxacin and the chalcone inhibited (P < 0.001) SARS‐CoV‐2 Mpro by 62.65% ± 2.51 and 81.6% ± 4.50, respectively, at the concentration of 1.6 μmol/L, where 100% represents complete inhibition of the virus Mpro. Remdesivir was used as a positive control. The Mpro IC50 (inhibition concentration 50 value against SARS‐CoV‐2 main protease enzyme) of ciprofloxacin, the chalcone and Remdesivir was calculated using GraphPad Prism 9 software as presented in Table 2.
FIGURE 4.
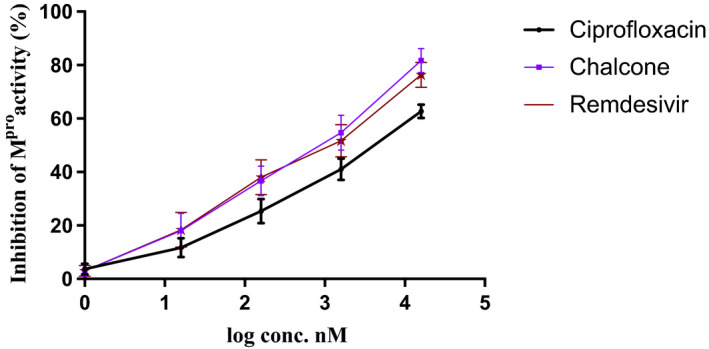
The inhibitory activity of ciprofloxacin and the chalcone against SARS‐CoV‐2 Mpro enzyme activity. Data represent mean ± SD. Remdesivir was used as positive control
TABLE 2.
Inhibition concentration 50 values for ciprofloxacin, the chalcone and Remdesivir against SARS‐CoV‐2 main protease enzyme (Mpro IC50)
| Virus | Drug | Mpro IC50 |
|---|---|---|
| SARS‐CoV−2 | Ciprofloxacin | 5.13 ± 0.19 μmol/L |
| The chalcone | 0.6 ± 0.05 μmol/L* | |
| Remdesivir | 1.01 ± 0.97 μmol/L* |
P < 0.001, when compared to ciprofloxacin.
3.4. Cell viability assay
To examine the safety of the chalcone on Vero cells, cell viability assay was performed. After 24 h treatment with wide range of different concentrations of ciprofloxacin and the chalcone against Vero cells, there was no change in cell viability at the concentrations of 16, 160, 1600 nmol/L, as shown in Figure 5. Only the chalcone exerted non‐significant reduction in cell viability to 93.3% ± 4.16 at the concentration of 16 μmol/L. While at the concentration of 160 μmol/L, cytotoxic activity of ciprofloxacin and the chalcone was observed where cell viability was significantly reduced (P < 0.001) after treatment with ciprofloxacin and the chalcone to 81.6% ± 4.72 and 63% ± 3.6, respectively.
FIGURE 5.
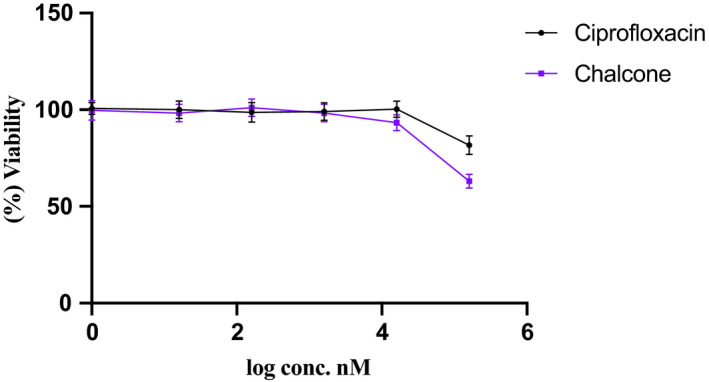
Survival of Vero cells after 24‐h treatment with different concentrations of ciprofloxacin and the chalcone. Data represent mean ± SD. Significant difference was analysed by one‐way ANOVA test, compared to untreated cells
3.5. Docking study
Ciprofloxacin and the chalcone were docked into SARS‐CoV‐2 Mpro active site (PDB code: 6LU7) to gain insight into their potential binding mode [36]. To evaluate the docking performance, the native ligand N3 was self‐docked into the Mpro active site showing an acceptable RMSD value of 0.9010 Å (Figure S1). [37] The native ligand N3 is a peptidomimetic inhibitor that interacts irreversibly within the Mpro active site, resulting in blockage of the active site [38]. The chalcone showed more interactions to occupy the Mpro active site and possessed a comparable interaction energy compared to the native ligand N3 (−8.9 and −9.3 Kcal/mol, respectively), as shown in Table 3 & Figure 6. Our chalcone possessed several important interactions inside the active site; the carbonyl oxygen in the amide linker formed two hydrogen bonds with Gly143 and the key amino acid Cys145 with bond distance of 3.64 and 3.58 Å, respectively (Figure 7). The piperazine ring showed a H…pi interaction with His41 amino acid (bond length = 3.85 Å). The carbonyl oxygen presents in the quinoline moiety formed a hydrogen bond with Thr190 amino acids. Also, the quinoline ring showed H…pi interactions with Met165 and Gln189 amino acids. It is worth noticing that this new chalcone was able to make the same interactions as that of the native ligand N3 (Table 3).
TABLE 3.
Energy scores (kcal/mol) and receptor interactions for ciprofloxacin and its potent chalcone, compared to the native ligand N3, within the COVID‐19 Mpro binding site
| Compound | Energy score (S) (kcal/mol) | Interacting residues |
|---|---|---|
| Ciprofloxacin | −7.0 | Cys145, Met49, Met165, Glu166 |
| Chalcone | −8.9 | Cys145, His41, Gln189, Met49, Met165, Asn142, Thr190, Gly143 |
| N3 | −9.3 | Cys145, His163, Gln189, Met49, Glu166, Gly143, Thr25, Thr190, Leu141 |
FIGURE 6.
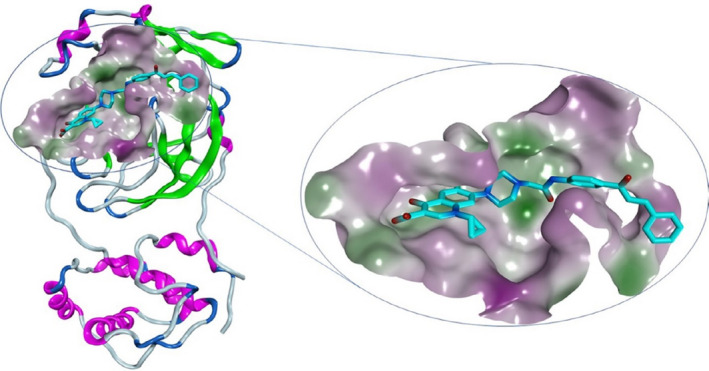
3D representation of the new ciprofloxacin‐derivative occupying COVID‐19 Mpro active site, PDB code: 6LU7
FIGURE 7.
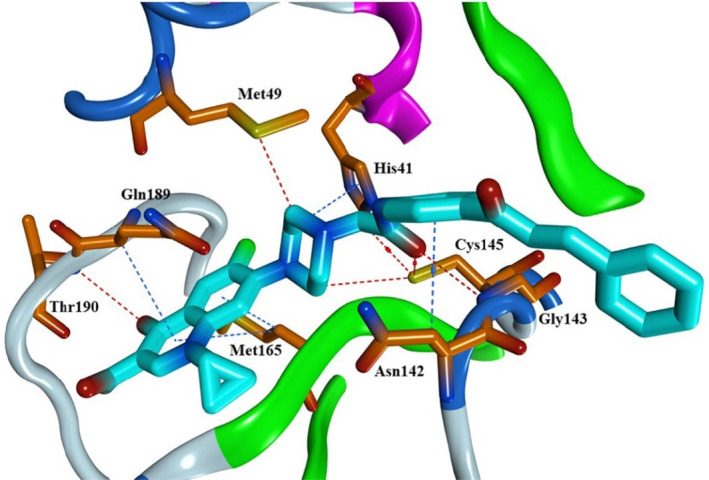
Binding mode in 3D representation of the new ciprofloxacin‐derivative inside the Mpro active site, PDB code: 6LU7
The docking pose of ciprofloxacin revealed the formation of several hydrogen bonds with Cys145, Met49, Met165 and Glu166 amino acids (Figure 8).
FIGURE 8.
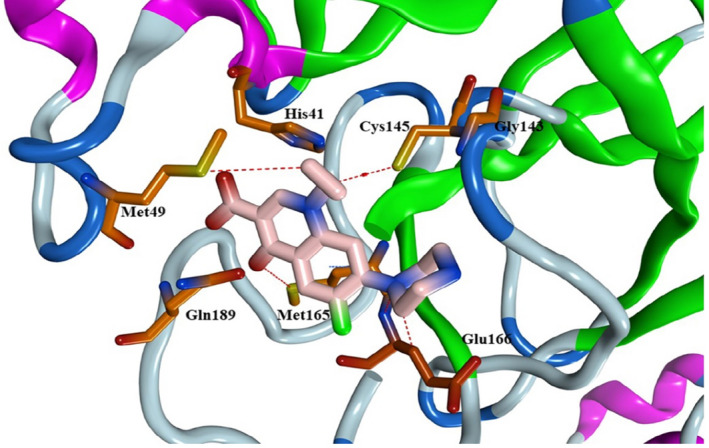
Binding mode in 3D representation of ciprofloxacin (pink) inside the Mpro active site, PDB code: 6LU
3.6. Drug‐likeness properties
Lipinski rule parameters and drug‐likeness properties were calculated by the Swiss ADME predictor (http://www.swissadme.ch/) and PreADMET server (https://preadmet.bmdrc.kr/adme). [39, 40, 41, 42] The entry of the structures of the ciprofloxacin and its chalcone was through their smiles. These properties comprise molecular weight (MW), number of hydrogen‐bond acceptors (HBA) and donors (HBD), rotatable bonds (nrotb), lipophilicity (ilogP) and topological polar surface area (TPSA). Besides, the blood‐brain barrier (BBB) permeability, cytochrome‐P2D6 inhibitor (CYP2D6), human intestinal absorption and aqueous solubility were investigated. Prediction of the abovementioned ADMET properties assists in predicting the transport properties of the molecules within the membranes such as BBB and gastrointestinal tract. As shown in Table 4, both ciprofloxacin and its chalcone were marked by having H‐bond acceptor and donor atoms less than 10 and 5, respectively. The chalcone displayed 10 rotatable bonds, which might lead to an advanced adaptation in the Mpro active site. Also, both compounds possessed convenient TPSA (less than 140 Å2), along with substantial bioavailability scores (F > zero) that collectively propose a higher possibility of biological activities. The log P values of both compounds are less than 5. It is worth mentioning that the chalcone derivative showed only one violation through the increased molecular weight (>500). However, it still obeys the Lipinski rule. The distribution properties of ciprofloxacin and its chalcone revealed that they have no ability to cross BBB and showed high intestinal absorption (96% and 97%, respectively). Favourably, none of the two compounds are classified as Pan‐Assay Interference Compounds (PAINS). Simply put, both ciprofloxacin and its chalcone are predicted to interact with specific biological sites instead of binding to random targets, and hence, likely to have fewer off‐target effects.
TABLE 4.
Lipinski Parameters of ciprofloxacin and the chalcone
| Compound | MW (g/mol) | HBA | HBD | Nrotb | TPSA (Ų) | iLogP | F | BBB | CYP2D6 inhibitor | HIA % | Log S | PAINS |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ciprofloxacin | 331.34 | 5 | 2 | 3 | 74.57 | 2.24 | 0.55 | No | No | 96 | −1.32 | No |
| The chalcone | 594.63 | 7 | 2 | 10 | 111.95 | 2.70 | 0.55 | No | No | 97 | −4.53 | No |
BBB, blood‐brain barrier permeability; CYP2D6 inhibitor hepatotoxicity, central nervous system toxicity; F, Abbott bioavailability scores; HBA, H‐bond acceptor; HBD, H‐bond donor; HIA, human intestinal absorption; iLogP, n‐octanol/water partition coefficient; Log S, aqueous solubility scale: Insoluble < ‐ 10 < Poorly < ‐ 6 < Moderately < ‐ 4 < Soluble < ‐ 2 < Very Soluble < 0 < Highly soluble; MW, Molecular weight; Nrotb, no. of rotatable bonds; PAINS, Pan‐Assay Interference Compounds; TPSA, topological polar surface area.
4. DISCUSSION
Repurposing drugs and screening agents for alternative activities [43, 44, 45, 46, 47] and new therapeutic goals [48, 49, 50, 51, 52] have become an attractive approach. FQs are known to target and inhibit the replication of RNA viruses; however, the fundamental basis of their antiviral activity is not fully understood. A mechanism was proposed that FQs could interfere and inhibit viral helicase enzymes [53]. FQs, including ciprofloxacin, are approved by the FDA as an antibacterial agent and their antiviral activity against SARS‐CoV‐2 has been previously reported. Marciniec et al., proposed that ciprofloxacin and moxifloxacin could interact with SARS‐CoV‐2 main protease through in silico analysis, they revealed that ciprofloxacin binds to Mpro with many hydrogen bonding and hydrophobic interactions [54]. Their findings suggested the potential activity of ciprofloxacin and moxifloxacin against SARS‐CoV‐2 main protease. Another in silico study by Kumar identified the hypothetical activity of enoxacin against SARS‐CoV‐2 [55]. Touret et al., evaluated the inhibitory activity of many FDA approved drugs against SARS‐CoV‐2 in vitro, their findings identified acceptable inhibitory activity of some FQs such as enoxacin and levofloxacin [56]. Bradfute et al. investigated ambroxol and ciprofloxacin against SARS‐CoV‐2 in Vero cells, their findings revealed antiviral inhibitory activity of ambroxol and ciprofloxacin against SARS‐CoV‐2 at clinically relevant concentrations. They proposed that the activity of ciprofloxacin was resulted from its interaction with the virus RNA and certain viral proteins such as helicase [57]. These investigations are in accordance with our results, where the parent drug, ciprofloxacin, exerted an acceptable antiviral activity towards SARS‐CoV‐2 viral replication. Our findings revealed that ciprofloxacin inhibited SARS‐CoV‐2 RNA replication and the plaque formation ability.
However, Scroggs et al., evaluated four FQ members such as enoxacin, ciprofloxacin, levofloxacin and moxifloxacin in Vero cells and A549 cells overexpressing ACE2, their findings indicated that all four FQs inhibited the replication of SARS‐CoV‐2 at high micromolar concentration. They concluded that the moderate antiviral activity of FQs was not convincing to warrant the in vivo studies [58]. Ciprofloxacin showed promising scaffold [59]. As mentioned, there are numerous available studies about structure modification of FQs to enhance their antiviral activity. Herein we examined a chalcone derived from ciprofloxacin, which exerted an enhanced activity against SARS‐CoV‐2.
The results of the present study, for the first time, showed significant inhibition of the new 7‐(4‐(N‐substituted carbamoyl methyl) piperazin‐1 yl)‐ chalcone against the RNA replication of SARS‐CoV‐2 at the concentration of 160 nmol/L by 93.1%, where the EC50 was 3.93 nmol/L. This was further confirmed by the plaque assay, where the chalcone inhibited plaque formation ability of the virus by 88.1%. Interestingly, the ease of synthesizing this chalcone, which exhibited a significantly high percentage of inhibition in viral load replication than the parent drug, could help with the recent pandemic COVID‐19. Although ciprofloxacin has a safety profile, it has been known to be toxic at high molar concentrations, we examined the toxic activity of ciprofloxacin and the chalcone against Vero cell at wide range of different concentrations to eliminate any other possible factor affecting SARS‐CoV‐2 viral load. Our findings revealed that the tested concentrations of ciprofloxacin and the chalcone against SARS‐CoV‐2 did not affect cell viability, which confirm that the inhibition in viral load and plaque forming unit occurred solely by the effect of ciprofloxacin and the chalcone, and cell cytotoxicity started to occur at the concentration of 160 μmol/L.
To get more insights about the mechanism of action on the inhibition of SARS‐CoV‐2 replication, the in vitro binding capacity towards SARS‐CoV‐2 Mpro 3CL was investigated, using a wide range of concentrations (0.016, 0.16, 1.6, 16) μmol/L of ciprofloxacin and the new chalcone, supported by a molecular docking study.
SARS‐CoV‐2 Mpro, also known as 3‐chymotrypsin‐like protease—3CLpro, is considered a potential target for viral replication inhibition. The polyprotein was cleaved by the 3CLpro at 11 different sites to obtain many non‐structural proteins, which are critical for the replication of the virus [60]. Our results showed, for the first time, that the chalcone exhibited an efficient inhibitory effect against SARS‐CoV‐2 Mpro (3CL pro) in vitro and in silico, which can be explained by the marked ability of the new chalcone to occupy the active site of the virus Mpro, thus, blocking the enzyme activity to a greater extent. Moreover, the docking pose of new chalcone revealed its ability to form several crucial interactions. The addition of an extension to ciprofloxacin (acetamido chalcone moiety) allowed the chalcone to make additional binding interfaces within the active site. Moreover, this extension enabled it to be oriented roughly the same as the native ligand with favourable interaction energy. These promising interactions would warrant for the potent activity of this new chalcone. It is worth noticing that our new chalcone was able to make most of the interactions as the native ligand N3.
While, the parent drug, ciprofloxacin revealed the formation of several essential hydrogen bonds. However, it did not interact with the key amino acid His41 and was not able to occupy the whole Mpro active site that might explain the decreased activity of ciprofloxacin compared to its chalcone.
Providentially, the obtained calculations by the Swiss ADME predictor indicated that the new ciprofloxacin‐chalcone obeyed the Lipinski rule and met the essential bioavailability requirements. Favourably, both ciprofloxacin and its chalcone were not listed as PAINS (compounds that interact non‐specifically with various biological targets) [41].
5. CONCLUSION
For the first time, the present study investigated the in vitro antiviral activity of 7‐(4‐(N‐substituted carbamoyl methyl) piperazin‐1 yl)‐ chalcone against SARS‐CoV‐2 Mpro, supported by a molecular docking study. The new ciprofloxacin‐chalcone exhibited significant inhibition in SARS‐CoV‐2 viral replication and virus Mpro enzyme in vitro and in silico in a dose‐dependent manner. Docking analysis justified the potent inhibitory activity of the chalcone against SARS‐CoV‐2 Mpro, when compared to ciprofloxacin. Docking pose indicated the importance of structural alteration and modification to enhance the antiviral activity. These bewildering findings may provide a novel perspective into the therapeutic properties of this new chalcone, suggesting a further investigation of ciprofloxacin‐containing chalcones to curb the COVID‐19 crisis. Further research is required to improve understanding of the molecular mechanism that underlies the antiviral activity of this chalcone against SARS‐CoV‐2.
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
AUTHOR CONTRIBUTIONS
R.A. was involved in conceptualization, methodology, data curation, resources, software and writing—original draft. M.M was involved in validation, visualization, software and writing—original draft, G.A was involved in project administration and supervision. M.F. was involved in methodology, supervision, formal analysis and writing—review and editing.
Supporting information
Figure S1
Alaaeldin R, Mustafa M, Abuo‐Rahma GE‐DA, Fathy M. In vitro inhibition and molecular docking of a new ciprofloxacin‐chalcone against SARS‐CoV‐2 main protease. Fundam Clin Pharmacol. 2022;36(1):160–170. 10.1111/fcp.12708
Funding information
This research did not receive any specific grant from funding agencies in the public, commercial or not‐for‐profit sectors.
REFERENCES
- 1. Correia S, Poeta P, Hébraud M, Capelo JL, Igrejas G. Mechanisms of quinolone action and resistance: where do we stand? J Med Microbiol. 2017;66:551‐559. [DOI] [PubMed] [Google Scholar]
- 2. Dalhoff A. Antiviral, antifungal, and antiparasitic activities of fluoroquinolones optimized for treatment of bacterial infections: a puzzling paradox or a logical consequence of their mode of action? Eur J Clin Microbiol Infect Dis. 2015;34:661‐668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Alaaeldin R, Nazmy MH, Abdel‐aziz M, Abuo‐rahma G‐D, Fathy M. Cell cycle arrest and apoptotic effect of 7‐(4‐(N‐substituted carbamoylmethyl) piperazin‐1‐yl) ciprofloxacin‐derivative on HCT 116 and A549 cancer cells. Anticancer Res. 2020;40:2739‐2749. [DOI] [PubMed] [Google Scholar]
- 4. Fathy M, Sun S, Zhao Q‐L et al. A new ciprofloxacin‐derivative inhibits proliferation and suppresses the migration ability of hela cells. Anticancer Res. 2020;40:5025‐5033. [DOI] [PubMed] [Google Scholar]
- 5. Kuhlmann J, Dalhoff A, Zeiler H‐J. Quinolone antibacterials. Vol. 127. Springer Science & Business Media. 2012. [Google Scholar]
- 6. Miranda C, Silva V, Capita R, Alonso‐Calleja C, Igrejas G, Poeta P. Implications of antibiotics use during the COVID‐19 pandemic: present and future. J Antimicrobial Chemother. 2020;75:3413‐3416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sara R et al. Antiviral properties of quinolone‐based drugs. Curr Drug Targets Infect Disord. 2004;4:111‐116. [DOI] [PubMed] [Google Scholar]
- 8. Takada A, Takase S, Tsutsumi M, Sawada M. Effects of ofloxacin for type C hepatitis. Int Hepatol Commun. 1993;1:272‐277. [Google Scholar]
- 9. Kojima H, Kaita KDE, Hawkins K, Uhanova J, Minuk GY. Use of fluoroquinolones in patients with chronic hepatitis C virus‐induced liver failure. Antimicrob Agents Chemother. 2002;46:3280‐3282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sharma BN, Li R, Bernhoff E, Gutteberg TJ, Rinaldo CH. Fluoroquinolones inhibit human polyomavirus BK (BKV) replication in primary human kidney cells. Antiviral Res. 2011;92:115‐123. [DOI] [PubMed] [Google Scholar]
- 11. Mottola C, Freitas FB, Simões M, Martins C, Leitão A, Ferreira F. In vitro antiviral activity of fluoroquinolones against African swine fever virus. Vet Microbiol. 2013;165:86‐94. [DOI] [PubMed] [Google Scholar]
- 12. Scroggs SLP, Andrade CC, Chinnasamy R et al. Old drugs with new tricks: efficacy of fluoroquinolones to suppress replication of flaviviruses. Viruses. 2020;12:1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yamaya M, Nishimura H, Hatachi Y et al. Levofloxacin inhibits rhinovirus infection in primary cultures of human tracheal epithelial cells. Antimicrob Agents Chemother. 2012;56:4052‐4061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ali SH, Chandraker A, DeCaprio JA. Inhibition of Simian virus 40 large T antigen helicase activity by fluoroquinolones. Antivir Ther. 2007;12:1. [PubMed] [Google Scholar]
- 15. Fedorowicz J, Sączewski J. Modifications of quinolones and fluoroquinolones: hybrid compounds and dual‐action molecules. Monatshefte für Chemie ‐ Chemical Monthly. 2018;149:1199‐1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Richter S et al. Antiviral properties of quinolone‐based drugs. Current drug targets. Infect Disord. 2004;4:111‐116. [DOI] [PubMed] [Google Scholar]
- 17. Baba M, Okamoto M, Makino M et al. Potent and selective inhibition of human immunodeficiency virus type 1 transcription by piperazinyloxoquinoline derivatives. Antimicrob Agents Chemother. 1997;41:1250‐1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hagihara M, Kashiwase H, Katsube T et al. Synthesis and anti‐HIV activity of arylpiperazinyl fluoroquinolones: A new class of anti‐HIV agents. Bioorg Med Chem Lett. 1999;9:3063‐3068. [DOI] [PubMed] [Google Scholar]
- 19. Selvam P, Rathore P, Karthikumar S et al. Synthesis and antiviral studies of novel N‐sulphonamidomethyl piperazinyl fluoroquinolones. Indian J Pharm Sci. 2009;71:432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lucero BDA, Gomes CRB, Frugulhetti ICdPP et al. Synthesis and anti‐HSV‐1 activity of quinolonic acyclovir analogues. Bioorg Med Chem Lett. 2006;16:1010‐1013. [DOI] [PubMed] [Google Scholar]
- 21. Charushin VN et al. Fluoroquinolones: Synthesis and Application. In: Nenajdenko V, ed. Fluorine in Heterocyclic Chemistry Volume 2: 6‐Membered Heterocycles. Cham: Springer International Publishing; 2014:111‐179. [Google Scholar]
- 22. Luo X, Zhou G‐Z, Zhang Y, Peng L‐H, Zou L‐P, Yang Y‐S. Coronaviruses and gastrointestinal diseases. Military Med Res. 2020;7:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Aleissa MM, Silverman EA, Paredes Acosta LM, Nutt CT, Richterman A, Marty FM. New perspectives on antimicrobial agents: remdesivir treatment for COVID‐19. Antimicrob Agents Chemother. 2020;65:e01814‐e1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Beigel JH, Tomashek KM, Dodd LE et al. Remdesivir for the treatment of Covid‐19 — final report. N Engl J Med. 2020;383:1813‐1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mousavizadeh L, Ghasemi S. Genotype and phenotype of COVID‐19: Their roles in pathogenesis. J Microbiol Immunol Infect. 2020;54:159‐163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ogando NS, Dalebout TJ, Zevenhoven‐Dobbe JC et al. SARS‐coronavirus‐2 replication in Vero E6 cells: replication kinetics, rapid adaptation and cytopathology. J Gen Virol. 2020;101:925‐940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bhavana V, Thakor P, Singh SB, Mehra NK. COVID‐19: Pathophysiology, treatment options, nanotechnology approaches, and research agenda to combating the SARS‐CoV2 pandemic. Life Sci. 2020;261:118336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yadav V, Varshney P, Sultana S, Yadav J, Saini N. Moxifloxacin and ciprofloxacin induces S‐phase arrest and augments apoptotic effects of cisplatin in human pancreatic cancer cells via ERK activation. BMC Cancer. 2015;15:581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Abdel‐Aziz AA, El‐Azab AS, Alanazi AM et al. Synthesis and potential antitumor activity of 7‐(4‐substituted piperazin‐1‐yl)‐4‐oxoquinolines based on ciprofloxacin and norfloxacin scaffolds: in silico studies. J Enzyme Inhib Med Chem. 2016;31:796‐809. [DOI] [PubMed] [Google Scholar]
- 30. Mohammed HHH, Abd El‐Hafeez AA, Abbas SH, Abdelhafez E‐S, Abuo‐Rahma G‐D. New antiproliferative 7‐(4‐(N‐substituted carbamoylmethyl)piperazin‐1‐yl) derivatives of ciprofloxacin induce cell cycle arrest at G2/M phase. Bioorg Med Chem. 2016;24:4636‐4646. [DOI] [PubMed] [Google Scholar]
- 31. Wang M, Cao R, Zhang L et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019‐nCoV) in vitro. Cell Res. 2020;30:269‐271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Drosten C, Günther S, Preiser W et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med. 2003;348:1967‐1976. [DOI] [PubMed] [Google Scholar]
- 33. He J, Hu L, Huang X et al. Potential of coronavirus 3C‐like protease inhibitors for the development of new anti‐SARS‐CoV‐2 drugs: Insights from structures of protease and inhibitors. Int J Antimicrob Agents. 2020;56:106055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Goel A, Prasad AK, Parmar VS, Ghosh B, Saini N. Apoptogenic effect of 7,8‐diacetoxy‐4‐methylcoumarin and 7,8‐diacetoxy‐4‐methylthiocoumarin in human lung adenocarcinoma cell line: role of NF‐kappaB, Akt, ROS and MAP kinase pathway. Chem Biol Interact. 2009;179:363‐374. [DOI] [PubMed] [Google Scholar]
- 35. Mustafa M, Abuo‐Rahma G‐D, Abd El‐Hafeez AA et al. Discovery of antiproliferative and anti‐FAK inhibitory activity of 1,2,4‐triazole derivatives containing acetamido carboxylic acid skeleton. Bioorg Med Chem Lett. 2021;40:127965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jin Z, Jin Z, Du X et al. Structure of M pro from SARS‐CoV‐2 and discovery of its inhibitors. Nature. 2020;582:289‐293. [DOI] [PubMed] [Google Scholar]
- 37. Cavasotto CN. In silico drug discovery and design: theory, methods, challenges, and applications. London: Taylor & Francis. 2015. [Google Scholar]
- 38. Griffin JWD. SARS‐CoV and SARS‐CoV‐2 main protease residue interaction networks change when bound to inhibitor N3. J Struct Biol. 2020;211:107575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lipinski CA. Drug‐like properties and the causes of poor solubility and poor permeability. J Pharmacol Toxicol Methods. 2000;44:235‐249. [DOI] [PubMed] [Google Scholar]
- 40. Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug‐likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7:42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mustafa M, Mostafa YA. Antimicrobial pyridazines: synthesis, characterization, cytotoxicity, substrate promiscuity, and molecular docking. Chem Biodivers. 2020;17:e2000100. 10.1002/cbdv.202000100 [DOI] [PubMed] [Google Scholar]
- 42. Khadse SC, Amnerkar ND, Dave MU et al. Quinazolin‐4‐one derivatives lacking toxicity‐producing attributes as glucokinase activators: design, synthesis, molecular docking, and in‐silico ADMET prediction. Fut J Pharm Sci. 2019;5:11. [Google Scholar]
- 43. Nagura S, Otaka S, Koike C et al. Effect of exogenous Oct4 overexpression on cardiomyocyte differentiation of human amniotic mesenchymal cells. Cell Reprogram. 2013;15:471‐480. [DOI] [PubMed] [Google Scholar]
- 44. Naseem M, Othman EM, Fathy M et al. Integrated structural and functional analysis of the protective effects of kinetin against oxidative stress in mammalian cellular systems. Sci Rep. 2020;10:13330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Oba J, Okabe M, Yoshida T et al. Hyperdry human amniotic membrane application as a wound dressing for a full‐thickness skin excision after a third‐degree burn injury. Burns Trauma. 2020;8:tkaa014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Okabe M, Yoshida T, Suzuki M et al. Hyperdry human amniotic membrane (HD‐AM) is supporting aciclovir included device of Poly‐N‐p‐vinylbenzyl‐D‐lactonamide (PVLA) sphere for treatment of HSV‐1 infected rabbit keratitis model. J Biotechnol Biomater. 2017;7:251. [Google Scholar]
- 47. Otaka S, Nagura S, Koike C et al. Selective isolation of nanog‐positive human amniotic mesenchymal cells and differentiation into cardiomyocytes. Cell Reprogram. 2013;15:80‐91. [DOI] [PubMed] [Google Scholar]
- 48. Fathy M, Fawzy MA, Hintzsche H, Nikaido T, Dandekar T, Othman EM. Eugenol exerts apoptotic effect and modulates the sensitivity of HeLa cells to cisplatin and radiation. Molecules. 2019;24:3979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Fathy M, Khalifa E, Fawzy MA. Modulation of inducible nitric oxide synthase pathway by eugenol and telmisartan in carbon tetrachloride‐induced liver injury in rats. Life Sci. 2019;216:207‐214. [DOI] [PubMed] [Google Scholar]
- 50. Abd El‐Baky RM, Hetta HF, Koneru G et al. Impact of interleukin IL‐6 rs‐1474347 and IL‐10 rs‐1800896 genetic polymorphisms on the susceptibility of HCV‐infected Egyptian patients to hepatocellular carcinoma. Immunol Res. 2020;68:118‐125. [DOI] [PubMed] [Google Scholar]
- 51. Fathy M, Okabe M, Saad Eldien HM, Yoshida T. AT‐MSCs antifibrotic activity is improved by eugenol through modulation of TGF‐beta/smad signaling pathway in rats. Molecules. 2020;25:348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Othman EM, Fathy M, Bekhit AA et al. Modulatory and toxicological perspectives on the effects of the small molecule kinetin. Molecules. 2021;26:670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Scroggs SLP, Offerdahl DK, Flather DP et al. Fluoroquinolone antibiotics exhibit low antiviral activity against SARS‐CoV‐2 and MERS‐CoV. Viruses. 2021;13:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Marciniec K, Beberok A, Pęcak P, Boryczka S, Wrześniok D. Ciprofloxacin and moxifloxacin could interact with SARS‐CoV‐2 protease: preliminary in silico analysis. Pharmacol Rep. 2020;72:1553‐1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kumar D et al. In silico identification of potent FDA approved drugs against Coronavirus COVID‐19 main protease: A drug repurposing approach. Chem Biol Lett. 2020;7:166‐175. [Google Scholar]
- 56. Touret F, Gilles M, Barral K et al. In vitro screening of a FDA approved chemical library reveals potential inhibitors of SARS‐CoV‐2 replication. Sci Rep. 2020;10:1‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bradfute SB, Ye C, Clarke EC, Kumar S, Timmins GS, Deretic V. Ambroxol and Ciprofloxacin Show Activity Against SARS‐CoV2 in Vero E6 Cells at Clinically‐Relevant Concentrations. BioRxiv. 2020.
- 58. Scroggs SL et al. Fluoroquinolone antibiotics exhibit low antiviral activity against SARS‐CoV‐2 and MERS‐CoV. Viruses. 2021;13:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Adefurin A, Sammons H, Jacqz‐Aigrain E, Choonara I. Ciprofloxacin safety in paediatrics: a systematic review. Arch Dis Childhood. 2011;96:874‐880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Anand K. Coronavirus main proteinase (3CLpro) structure: basis for design of anti‐SARS drugs. Science. 2003;300:1763‐1767. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1