

Abstract
Translation of human pluripotent stem cell (hPSC)-derived therapies to the clinic demands scalable, cost-effective methods for cell expansion. Culture media currently used for hPSC expansion rely on high concentrations and frequent supplementation of recombinant growth factors due to their short half-life at physiological temperatures. Here, we developed a biomaterial strategy using mineral-coated microparticles (MCMs) to sustain delivery of basic fibroblast growth factor (bFGF), a thermolabile protein critical for hPSC pluripotency and proliferation. We show that the MCMs stabilize bFGF against thermally induced activity loss and provide more efficient sustained release of active growth factor compared to polymeric carriers commonly used for growth factor delivery. Using a statistically driven optimization approach called Design of Experiments, we generated a bFGF-loaded MCM formulation that supported hPSC expansion over 25 passages without the need for additional bFGF supplementation to the media, resulting in greater than 80% reduction in bFGF usage compared to standard approaches. This materials-based strategy to stabilize and sustain delivery of a thermolabile growth factor has broad potential to reduce costs associated with recombinant protein supplements in scalable biomanufacturing of emerging cell therapies.
1. Introduction:
Cell therapies are poised to significantly impact public health over the coming decades, as evidenced by over 350 clinical trials since 2016 (>70 currently active) using cell-based products to treat cardiovascular disease, diabetes, and cancer (http://www.clinicaltrials.gov). Stem cells provide a rich source from which to derive cellular therapies, due to their unlimited expansion capacity and ability to differentiate into a variety of clinically relevant cell types1,2. However, scaling up established stem cell culture methods for biomanufacturing cell therapies remains a significant challenge. For example, the number of cells needed for human pluripotent stem cell (hPSC)-derived therapies range from 105–1010 cells/patient3, and recent reports project that commercialization of these therapies will need to accommodate treatment of 10,000–100,000 patients per year (i.e., production of 1011–1014 cells per year)4,5.
Culture media constitute a significant portion of the costs of cell biomanufacturing, as cell expansion and differentiation protocols often rely on media containing high concentrations of costly recombinant growth factors and cytokines. In addition, culture media must be replenished frequently to offset limited stability of these components6,7. As a result, it has been estimated that growth factors and cytokines account for 25–58% of the costs of producing stem cell-based therapies at a commercial scale4,8,9. The need for basic fibroblast growth factor (bFGF) in hPSC culture provides an illustrative example, as a threshold level of bFGF signaling is critical for hPSC survival, proliferation, and self-renewal10. Due to the poor stability of bFGF6,11, standard commercial media such as Essential 8 (E8) and TeSR6,12 contain high concentrations of recombinant bFGF (100 ng/ml). Even with these high concentrations, daily media changes are required to provide sufficient active bFGF and prevent spontaneous differentiation events that can quickly compromise hPSC quality7,10,13.
Previously, we have shown that calcium phosphate-based mineral coatings can bind various growth factors out of aqueous media via electrostatic interactions, and can be tuned to control protein release kinetics via calcium dissolution14,15. Additionally, we have shown that proteins sequestered in the nanoporous ionic structures of the mineral coatings can preserve the activity of growth factors against insults such as degradation due to proteases and denaturation due to organic solvents14–17. As such, we hypothesized that these mineral coatings could sustain the delivery of active bFGF in hPSC culture and therefore significantly reduce the amount of growth factor required to maintain pluripotency. Our approach uses a synthetic material amenable to protein loading in conditions devoid of organic solvents and detergents and requires no modification of the growth factor itself. In contrast to previously explored strategies for stabilizing bFGF, which include using poorly defined biological materials such as heparin18, carrier materials that require protein-denaturing solvents during loading19,20, or creation of mutant FGF isoforms11 that may have poorly characterized effects on cell signaling.
Here, we report the use of mineral coatings applied to microparticles (MCMs) as carriers to stabilize bFGF and sustain its delivery in order to reduce the amount of the growth factor needed for long-term expansion of hPSCs. The use of MCMs reduced the total bFGF required for hPSC culture by more than 80%. In addition, bFGF-loaded MCMs (bFGF-MCMs) eliminated the need for supplemental addition of soluble bFGF during long-term, chemically defined expansion of hPSCs. Together, these findings demonstrate that a biomaterial that both stabilizes and sustains delivery of a thermolabile growth factor can aid in cost-effective expansion of stem cells. As growth factors are critical for both expansion and differentiation of stem cells, the approach may be broadly applicable in chemically defined biomanufacturing of emerging cell therapies.
2. Materials and Methods:
2.1. Fabrication and characterization of MCMs:
Hydroxyapatite powder (Plasma Biotal Limited) was used as the microparticle core material. The core material was suspended at concentrations of 1 mg/mL in modified simulated body fluid (mSBF) formulated as follows: 141 mM NaCl, 4.0 mM KCl, 0.5 mM MgSO4, 1.0 mM MgCl2, 100 mM NaHCO3, 20.0 mM HEPES, 5.0 mM CaCl2, and 2.0 mM KH2PO4 with the pH was adjusted to 6.80. The suspension was rotated at 37°C for 24 hrs, at which point the microparticles were centrifuged at 2,000g for 2 min, and the supernatant decanted and replaced with freshly made mSBF. We repeated this process daily for 5 days, at which point the MCMs were washed three times with 50 mL deionized water, filtered through a cell strainer (40 μm pore size), suspended in 15 mL distilled water, flash frozen in liquid nitrogen, and lyophilized for 48 hrs. The lyophilized MCMs were then analyzed by SEM and calcium release assays as previously described17,21. MCMs were sputter-coated with gold and imaged on a LEO 1530 scanning electron microscope (Gemini) at 3kv. Ca2+ release was measured by incubating MCMs in 0.02 M Tris base buffer (pH 7.4) with gentle rotation, and centrifuging MCMs, collecting supernatant, and replacing with fresh buffer daily. The supernatant Ca2+ concentration was measured by mixing 50 μL of supernatant with 150 μL of assay working solution containing 0.4 mM Arsenazo III (MP Biomedicals, Solon, OH). The absorbance of this solution was measured at 650 nm to determine Ca2+ concentration using a standard curve.
2.2. Generation of bFGF-loaded MCMs:
MCMs were spread onto a flat surface and sterilized via UV irradiation for 30 min, then aliquotted into sterile tubes and stored until use. For MCM loading, sterilized MCMs were suspended in sterile PBS to make a 1 mg/mL stock solution, and appropriate volumes of recombinant human basic fibroblast growth factor (bFGF, carrier-free; R&D Systems) and MCM stock solutions were combined to achieve the desired final concentration of each in the loading solution (e.g., 1.0 μg/mL bFGF, 1.0 mg/mL MCMs for non-optimized loading; 0.456 μg/mL bFGF, 0.375 mg/mL MCMs for optimized loading). The MCM-bFGF solution was incubated at 37°C under constant rotation for 1 h and protein binding was mediated through ionic interactions between the mineral coating and protein as previously described16. The suspension was then centrifuged at 2000g for 3 min to pellet the MCMs. For cell culture experiments, the MCMs were resuspended in E7 media and used immediately unless otherwise stated. bFGF-loaded MCMs were prepared fresh every three passages for Transwell culture studies (unless otherwise specified) and prepared fresh at each passage in the direct culture studies. Protein binding efficiency was determined indirectly by Quantikine bFGF ELISA, by measuring the amount of bFGF in original loading solution and subtracting the amount of bFGF in the supernatant after MCM loading.
2.3. General culture of hPSCs:
H1 human embryonic stem cells (WA01-DL-12, WiCell) or WTc11 human induced pluripotent stem cells (kindly provided by Dr. Bruce Conklin, Gladstone Institute of Cardiovascular Disease) were maintained in Essential 8 medium in 6-well plates coated with Matrigel (8.7 μg/cm2) at 37°C and 5% CO2 in a humidified incubator. Media were exchanged daily, and cells were passaged onto new Matrigel-coated plates every 3 to 4 days by standard protocols22 using Versene (Life Technologies). Media used in hPSC maintenance experiments were Essential 8 (E8, Life Technologies) and E7 (Essential 6 media (Life Technologies) supplemented with 1.76 ng/mL human transforming growth factor beta 1 (TGF-β1, carrier free; R&D Systems)).
2.4. Transwell culture method:
MCMs or bFGF-MCMs were prepared as described above and added to polycarbonate Transwells (0.4 μm pore size) in 12-well culture plates (Corning). hPSCs were cultured on Matrigel as described above, in either E8 or E7 media as indicated in each figure. The Transwell inserts were removed while the cells were imaged via phase microscopy and the base media (i.e., without bFGF-MCMs) were exchanged daily. At each passage, the Transwell inserts were removed while the cells were passaged with Versene as described above. The cells were allowed to adhere to new Matrigel-coated plates for 30 min prior to adding Transwell inserts to the culture wells. Unless otherwise stated, the same Transwells (containing original bFGF-MCMs) were used throughout the entirety of an experiment and transferred to new wells at each passage.
2.5. Direct culture method:
MCMs or bFGF-MCMs were prepared as described above and added directly to hPSCs cultured in 12-well culture plates on Matrigel, in either E8 or E7 media as indicated in each figure. The base media were exchanged daily (unless otherwise indicated) and the cells were imaged via phase microscopy. At each passage, the cells were passaged with Versene as described in the general hPSC culture method section. The cells were allowed to adhere to new Matrigel-coated plates for 30 min prior to adding fresh bFGF-loaded MCMs directly into the new wells.
2.6. DOE design and modeling:
We designed the surface-response design of experiments (DOE) using JMP software (SAS). The design was rotatable around a single center point with axial positions for two factors: bFGF concentration and MCM concentration during MCM loading. The factor levels were set at log2 steps (step sizes of −/+1 for the surface positions and −/+ 1.41 for the axial positions) away from the center point concentrations of 1 μg bFGF and 1 mg MCMs per mL, as specified by JMP. The DOE was carried out in triplicate and %Oct4+/Nanog+ at passage 3 was used as the response variable for all conditions. The model was generated with least squares fitting of linear and squared dependences for bFGF and MCM (bFGF, MCM, bFGF2, and MCM2) and crossed dependences for bFGF and MCM (bFGF*MCM) as factors for the Oct4+/Nanog+ response. The Factor Profiler in JMP was used to select the DOE-optimized MCM loading conditions using the desirability plot to identify concentrations of bFGF and MCMs that achieved 95% Oct4+/Nanog+ while minimizing total bFGF.
2.7. Quantification of Oct4+/Nanog+ populations by flow cytometry:
Briefly, hPSCs were collected, washed with PBS, and incubated with 0.25% trypsin/EDTA for 8–10 min at 37°C followed by pipetting to dissociate. Trypsin activity was quenched by adding 2X volume of 20% FBS in RPMI supplemented with 5 μM Y-27632. Samples were centrifuged at 200g for 5 min and pelleted samples were fixed with 1% paraformaldehyde for 20 min at room temperature, permeabilized with ice-cold 90% methanol for 15 min at 4°C, and stored at −20°C until processing. Samples were washed twice with Flow Buffer 1 (0.5% BSA in PBS) to remove residual methanol, incubated for 1 hr at room temperature with primary antibodies in Flow Buffer 2 (0.5% BSA + 0.1% Triton X-100 in PBS), washed with Flow Buffer 2, and incubated in the dark for 30 min at room temperature with secondary antibodies in Flow Buffer 2. Samples were washed twice with Flow Buffer 2, resuspended in Flow Buffer 1, and stored on ice prior to analysis. Data were collected on a MACSQUANT flow cytometer (Miltenyi Biotec) and analyzed using FlowJo software. Primary antibodies and dilutions used were mouse anti-human Oct3/4 (Santa Cruz Biotechnology, sc-5279, 1:400), rabbit anti-human Nanog (Cell Signaling Technology, 4903S, 1:200). Secondary antibodies and dilutions used were goat anti-mouse AlexaFluor 488 and goat anti-rabbit AlexaFluor 647 (Thermo Fisher Scientific, 1:1000 each).
2.8. Immunocytochemistry:
Samples were fixed with 10% neutral buffered formalin for 15 min, permeabilized with 0.2% Triton X-100 in PBS for 5 min, blocked with 1% BSA in PBS for 30 min, and stained with primary antibodies (dilutions made in 1% BSA in PBS) for 1 hr at room temperature. Samples were washed three times with 0.05% Tween-20 in PBS and stained with secondary antibodies (dilutions made in PBS) for 1 hr at room temperature or overnight at 4°C. Nuclei were counterstained with DAPI. Primary antibodies and dilutions used: mouse anti-human Oct3/4 (Santa Cruz Biotechnology, sc-5279, 1:100), rabbit anti-human Nanog (Cell Signaling Technology, 4903S, 1:100), mouse IgG2a anti-βIII tubulin (R&D, MAB1195, 1:200), mouse IgG2b anti-alpha fetoprotein (R&D, MAB1369, 1:200), rabbit anti-alpha smooth muscle actin (Abcam, ab124964, 1:200), mouse IgG1 anti-PECAM-1 (EMD Millipore, MAB2184, 1:50). Secondary antibodies and dilutions used were goat anti-mouse AlexaFluor 488, goat anti-mouse IgG2a AlexaFluor 488, goat anti-rabbit or anti-mouse AlexaFluor 568, and goat anti-mouse IgG2b AlexaFluor 647 (Thermo Fisher Scientific, 1:1000 for all secondaries).
2.9. Spontaneous differentiation of embryoid bodies:
To form spontaneous EBs, hPSCs in standard E8 culture (6-well format) were washed with PBS and incubated in pre-warmed Dispase (Gibco, 1 U/mL) for 3 min at 37°C. Dispase was removed by aspiration and 2 mL PBS was added per well. Cells were dislodged from the culture surface using a cell scraper and the cell suspension was added to a tube and centrifuged for 200g/5 min. The cell pellet from each individual well was resuspended in 2.5 mL of E8 supplemented with 5 μM Y-27632 and the cell suspension was added to a 6-well ultra-low adhesion culture plate (Corning). The time of initial seeding was designated as “day −3.” EBs were subjected to spontaneous differentiation as previously described23. Briefly, EBs were transitioned from E8 to differentiation medium (DM) from day 0 to day 3; EBs were placed in 75:25 E8:DM at day 0, 50:50 E8:DM at day 1, 25:75 E8:DM at day 2, and 100% DM at day 3, and maintained in DM thereafter. DM consisted of 20% Knockout Serum Replacement in Knockout-DMEM with 0.1 mM β-mercaptoethanol, 1% non-essential amino acids, and 1% L-glutamine. At day 4, EBs were transferred to Matrigel-coated plates and allowed to form adherent outgrowths. Media was changed on adherent EBs every 2 days. Adherent EBs were fixed with 10% neutral buffered formalin at day 14 for analysis.
2.10. Directed differentiation toward ectoderm, mesoderm, and endoderm lineages:
H1 hESCs and WTc11 hiPSCs were subjected to directed differentiation following established protocols. Neural progenitor differentiation (adapted from24): Starting from 60% confluency, hPSCs were passaged via Accutase and seeded in SB medium (DF3S containing 10 μg/mL transferrin, 5 μg/mL insulin, and 10 μM SB431542) supplemented with 100 ng/mL basic fibroblast growth factor (bFGF) and 10 μM Y-27632 at a density of 5×104 cells/well in 12-well Matrigel-coated plates. DF3S medium consisted of DMEM/F-12, L-ascorbic acid-2-phosphate magnesium (64 μg/mL), sodium selenium (14 ng/mL), and NaHCO3 (543 μg/mL). The cells were transitioned to medium without Y-27632 the following day. The adherent cells were then transitioned to either SB medium for hindbrain specification or SBNog medium (SB medium containing 100 ng/mL Noggin) for forebrain specification and maintained in the respective media for 6 days with daily media exchange. On day 9, the cells were passaged with Accutase at a 1:6 ratio and seeded onto Matrigel-coated plates in Neural Expansion Medium (DF3S medium containing N-2 supplement, B-27 supplement, and 5 ng/mL bFGF). Neural Expansion Medium was changed every 2–3 days and the cells were passaged 1:6 with Accutase every 6–8 days. The cells were maintained in Neural Expansion Medium for a total of 24 days prior to fixing for immunocytochemistry.
Endothelial differentiation (adapted from25):
hPSCs at 80% confluency were dissociated with Accutase (Invitrogen) and passaged 1:4 onto Matrigel-coated plates. hPSCs were differentiated into mesoderm by culturing for 2 days in E8BAC medium (Essential 8 with 5 ng/mL bone morphogenetic protein (BMP)-4, 25 ng/mL Activin A, and 1 μM CHIR-99021), with 10 μM Y-27632 included in the media on the first day to improve cell survival. Mesoderm was then cultured for 4 days in E7Vi medium (Essential 6 with 100 ng/mL bFGF, 50 ng/mL vascular endothelial growth factor (VEGF)-A165, and 5 μM SB431542) before fixing for immunocytochemistry.
Hepatocyte differentiation (adapted from26):
One day before starting differentiation, hPSCs were dissociated with Accutase and seeded onto Matrigel-coated plates at 5 × 104 cells/cm2 in E8 supplemented with 10 μM Y-27632. To initiate differentiation, media was changed to RPMI containing 1X B-27 minus insulin and 100 ng/mL Activin A on the day after seeding. Cells were maintained in this differentiation medium for a total of three days. Media was then changed to RPMI + B-27 minus insulin supplemented with 30 ng/mL FGF-4 and 20 ng/mL BMP-2 for five days. Finally, cells were maintained in RPMI + B-27 minus insulin supplemented with 20 ng/mL hepatocyte growth factor (HGF) for an additional five days, prior to fixing for immunocytochemistry.
2.11. Karyotyping and teratoma assays:
Samples were provided to WiCell (Madison, WI) for teratoma formation assays and G-banded karyotyping with analysis and interpretation by the Cytogenetics Lab at WiCell. Histology from teratoma assays was analyzed and interpreted by the Comparative Pathology Laboratory of the University of Wisconsin-Madison.
2.12. ELISAs:
ELISAs for measuring bFGF (Quantikine basic FGF ELISA, R&D Systems, DFB50), phosphoERK and total ERK (Abcam, ab176660) were performed following the manufacturer’s recommended protocol. MCMs were confirmed not to interfere with bFGF Quantikine ELISA readouts (Supplementary Fig. S14). For phosphoERK and total ERK assays, 10 μg total protein was loaded per well of the ELISA based on protein concentration of cell lysates as quantified by microBCA.
2.13. bFGF biological activity:
For experiments correlating bFGF ELISA readout to ERK phosphorylation, hPSCs were starved of bFGF (in E7) for 24 hrs, followed by 2 hr restimulation with E7 containing different concentrations of fresh bFGF in solution (created by mixing E8 and E7 media at different ratios). For experiments correlating loss of bFGF bioactivity to ERK phosphorylation, E8 media was incubated for 1, 6, or 24 hr at 37°C or 65°C and allowed to cool before restimulating bFGF-starved hPSCs as described above. bFGF content in restimulation media was measured by Quantikine bFGF ELISA. Relative phosphoERK content in restimulated hPSCs was determined by phosphoERK and total ERK ELISAs on cell lysates. Briefly, cells were washed with PBS and resuspended in ice-cold RIPA buffer containing 1X Halt Protease/Phosphatase Inhibitor Cocktail. Samples were agitated for 15 min at 4°C and spun at 12,000g for 15 min at 4°C. The supernatants from samples were collected and total protein in lysates was quantified by microBCA assay (Thermo Fisher Scientific). For experiments comparing thermal stability of free bFGF vs. MCM-bound bFGF, solutions containing 100 ng/mL bFGF in E7 or 62 μg/mL optimized bFGF-MCMs in E7 (to match usage in cell culture experiments) were stored at 4°C or incubated for different durations at 37°C prior to assessing active bFGF content via Quantikine bFGF ELISA.
2.14. Comparison of bFGF-MCMs and PLGA microspheres:
bFGF-releasing PLGA microspheres (StemBeads® FGF2) were purchased from StemCultures. For all comparison studies, bFGF-MCMs and PLGA microspheres were resuspended to concentrations equivalent to those used in cell culture experiments (62 μg/mL for optimized bFGF-MCMs; 8 μL/mL for PLGA microspheres, following manufacturer’s recommendations).
Active and total bFGF release.
bFGF-MCMs and PLGA microspheres were suspended in DF3S medium (Thermo Fisher Scientific), as TGF-β1 in E7 was found to contribute substantial background to the total protein measurement. The resulting solutions were incubated and rotated at 37°C with daily collection of releasate over 4 days. To measure active bFGF release, a fraction of the homogeneous MCM and microsphere suspensions was first collected. To measure total bFGF release, the MCM or microsphere suspensions were then centrifuged at 22,000g/2 min and the supernatant was collected and fresh DF3S medium replaced. The amount of active and total bFGF released were determined by Quantikine bFGF ELISA (R&D Systems) and NanoOrange Protein Quantitation kit (Thermo Fisher Scientific), respectively. Cumulative daily values of active bFGF release were extrapolated from bFGF content in releasates during the 2 hr sample incubation step of the bFGF Quantikine ELISA (Supplementary Fig. S14). Quantitation of total protein content by NanoOrange was performed after concentrating releasates with Amicon 3 kDa MWCO Centrifugal Filter Units (EMD Millipore).
2.15. Cell expansion and doubling rate:
H1 hESCs or WTc11 hiPSCs were cultured in E8 or in direct culture with optimized bFGF-MCMs and passaged at a 1:8 split ratio every four days (96 hours) as described above. At each passage, 80% of the passaged cell suspension was collected and frozen at −80°C. Total DNA content in collected cell samples was measured by Quant-iT PicoGreen dsDNA Assay Kit (Thermo Fisher Scientific) following manufacturer’s instructions. Cell number at each passage was calculated from total DNA content using a cell standard containing known numbers of singularized hPSCs.
2.16. Figure generation and statistics:
All schematic graphics were created by the authors using Adobe Illustrator and Photoshop except for the microparticle graphic, which was commissioned from a professional graphic designer. The DOE graphs and models were generated in JMP while all other graphs were generated in GraphPad Prism. Statistics between Oct4+/Nanog+ population percentages were done in GraphPad Prism. The degree of significance for each comparison is denoted in each figure/caption.
3. Results:
3.1. MCMs efficiently bound and released bFGF to maintain Oct4 and Nanog expression in hPSCs in a dose-dependent manner.
Incubation of hydroxyapatite powder in modified simulated body fluid produced microparticles with inorganic nanostructured, plate-like coatings capable of sustaining delivery of bFGF in hPSC culture (Supplementary Fig. S1A). These coatings demonstrated a high capacity for bFGF binding, with 80.1 ± 6.5% (0.97 μg bFGF/mg MCM) bound after incubation for one hour at room temperature (Fig. 1A, Supplementary Fig. S1B). We have previously demonstrated that growth factor release from mineral coatings occurs with dissolution of the mineral coating, and consequently that calcium release from coating dissolution over time correlates well with protein release profiles14–17. The mineral coatings on our MCMs in the current study gradually dissolved at physiological pH, as indicated by sustained calcium dissolution in pH 7.4 Tris-buffered saline over 7 days (Fig. 1B, Supplementary Fig. S1C). In Transwell culture, MCMs released bFGF without directly contacting cells, and bFGF-MCM-containing Transwells were carried over for continued sustained release throughout the duration of culture. In the direct culture format, bFGF-MCMs were prepared fresh for each passage and added directly to wells containing hPSCs (Fig. 1C). The MCMs interacted closely with the cell membrane but were not internalized, as evidenced by the clear nanostructured features that remained observable by SEM after direct culture of MCMs with hPSCs (Fig. 1D).
Figure 1. MCMs bind and release bFGF for sustained bFGF presentation in hPSC culture.
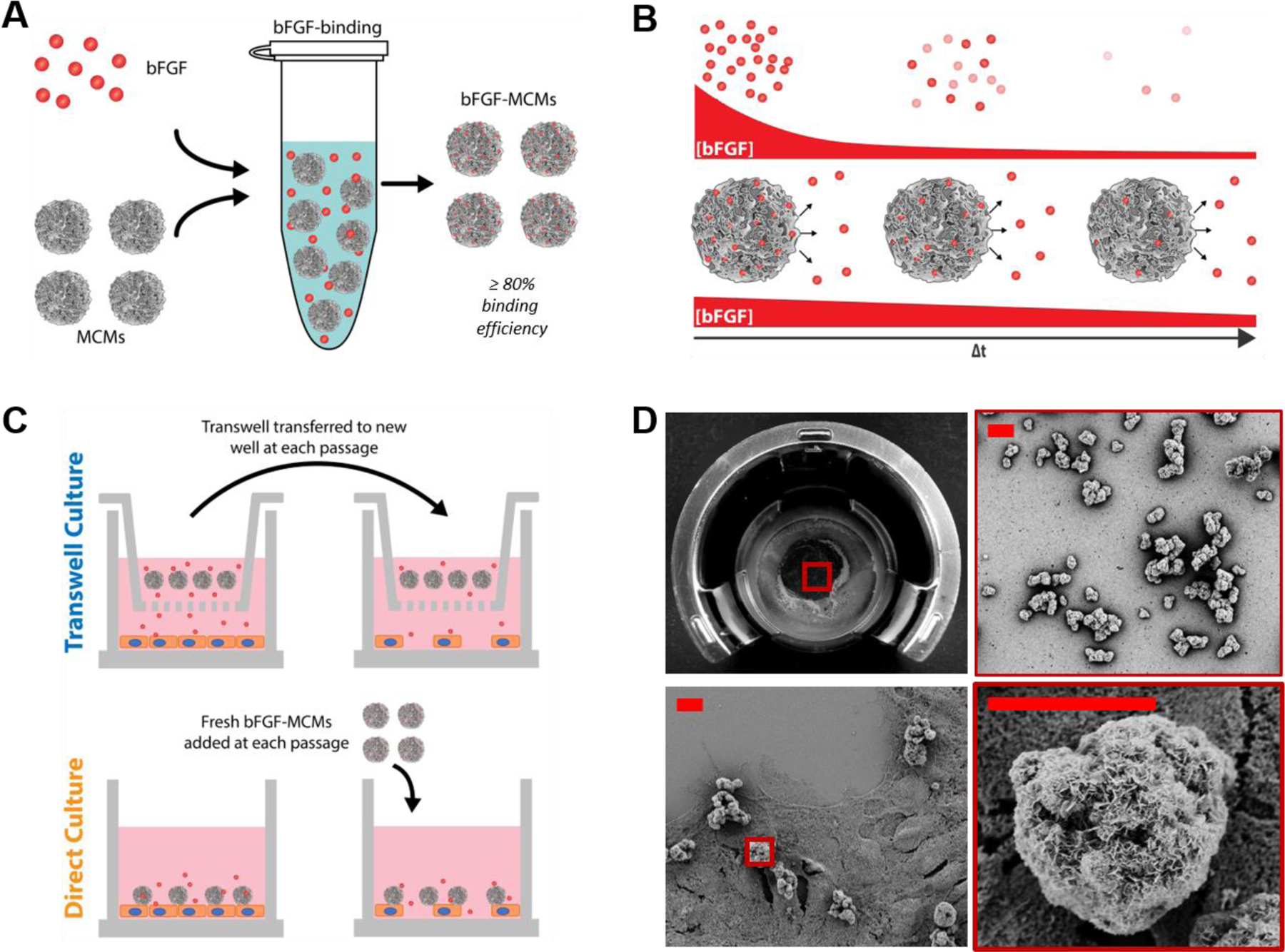
(A) Schematic for binding of bFGF to mineral-coated microparticles (MCMs) in solution. (B) Proposed model for presentation and presence of active bFGF over time in (top) conventional chemically-defined hPSC culture or (bottom) culture with bFGF-loaded MCMs (bFGF-MCMs). bFGF loses activity over time when delivered as soluble protein via daily media changes in culture, while MCMs stabilize and allow sustained release of active bFGF. (C) Schematic representation of two culture formats employed in this study: Transwell culture, in which MCMs release bFGF from Transwells, and direct culture, in which MCMs releasing bFGF are added directly to cells. (D) Photographs and scanning electron micrographs of bFGF-MCMs in (top) 12-well Transwell format and (bottom) direct culture format with hPSCs. Scale bar = 10 μm.
Sustained bFGF release from MCMs maintained hPSC expression of pluripotency markers Oct4 and Nanog in a dose-dependent manner in chemically defined media devoid of bFGF (Essential 8 media minus bFGF, hereafter referred to as “E7”). Culture with bFGF-MCMs maintained hPSC colonies with epithelial-like morphologies, similar to those of cells in E8. The appearance of spontaneously differentiating mesenchymal-like cells was inversely correlated with bFGF-MCM dose (Fig. 2A). hPSCs cultured in E7 alone underwent spontaneous differentiation by passage 2, as evidenced by the appearance of cells with mesenchymal-like morphology and concomitant loss of Oct4 and Nanog expression (21.2 ± 10.5% Oct4+/Nanog+ by passage 2; 14.0 ± 1.7% by passage 3) (Fig. 2A–B). Increasing amounts of bFGF-MCMs (mass of MCMs per well = 0.1 mg/“low”, 0.5 mg/“med”, or 1.0 mg/“high”) resulted in 50.0 ± 1.4%, 76.1 ± 5.6%, and 88.8 ± 3.1% Oct4+/Nanog+ hPSCs at passage 3, respectively (Fig. 2B–C). Importantly, the presence of unloaded MCMs did not significantly affect pluripotency marker expression of hPSCs cultured in Transwell or direct culture formats over 10 passages (40 days) in E8 medium (Supplementary Fig. S2). In comparison to standard culture of hPSCs in E8 (which consistently maintained ~95% Oct4+/Nanog+ cells with 100 ng/mL bFGF per day), the high dose of bFGF-MCMs reduced bFGF consumption by 16.7% (Fig 2B, Supplementary Table 1), but was unable to maintain undifferentiated hPSCs beyond passage 3 (Supplementary Fig. S3).
Figure 2. bFGF-loaded MCMs (bFGF-MCMs) in Transwell culture have a dose-dependent effect on the percentage of hPSCs expressing Oct4 and Nanog.

(A) Representative images of H1 hPSCs at passage 3 (day 12) of culture in E8 (control) or E7 with varying doses of bFGF-MCMs (scale bar = 250 μm). Colonies with normal stem cell morphology were observed in the E8 control and E7 + high bFGF-MCM conditions, while varying degrees of spontaneous differentiation were seen in the E7, E7 + low bFGF-MCM, and E7 + med bFGF-MCM conditions (white arrows). (B) Quantification of Oct4/Nanog expression in hPSCs grown with or without bFGF-MCMs in Transwell culture for 3 passages (n = 3, error bars = s.d.), as assessed by flow cytometry. n = 3, error bars = s.d. bFGF utilization as denoted on the right y-axis was calculated based on amount of bFGF used to maintain a single well of hPSCs in a 12-well plate format (1 mL media/well) for three passages, relative to E8 control (100%). (C) Representative flow cytometry plots of hPSCs after Transwell culture with E7 containing low, medium, or high doses of bFGF-MCMs for 3 passages.
3.2. A Design of Experiments (DOE)-based optimization of bFGF-MCM fabrication afforded maintenance of >95% Oct4+/Nanog+ hPSCs while minimizing bFGF required.
We constructed a rotatable response surface design to determine the effects of MCM and bFGF concentrations in the MCM loading solution on hPSC pluripotency. The two-factor, five-level design generated nine unique MCM loading conditions that we tested in Transwell culture for their capacity to maintain pluripotency marker expression for three passages (Fig. 3A). These conditions maintained a range of 44.5 ± 5.6% to 98.1 ± 0.4% Oct4+/Nanog+ hPSCs. Four of the nine MCM loading conditions maintained >95% Oct4+/Nanog+ hPSCs with undifferentiated colony morphology at passage 3, comparable to control hPSCs maintained in E8 media (Fig. 3B–C and Supplementary Fig. S4). Of these four conditions, only (--) reduced the amount of bFGF utilized in comparison to E8 by 16.7% (Supplementary Table 1).
Figure 3. Design of experiments (DOE) optimization of bFGF binding solution identifies MCM conditions that maintain hPSC pluripotency marker expression while minimizing bFGF usage.

(A) Representation of a five-level DOE experimental space. The levels were centered around the point (0,0) of 1 mg/mL of MCMs 1 μg/mL bFGF, with 2-fold based steps above (+) and below (−), and 2.67-fold axial points above (A) and below (a) the center point. The concentrations of bFGF and MCMs in the binding solution were varied while the total amount of bFGF-MCMs used in Transwell culture was held constant. %Oct4+/Nanog+ hPSCs at passage 3 was measured as the response variable. (B) Flow cytometry quantification of %Oct4+/Nanog+ hPSCs cultured with bFGF-MCMs from each DOE condition for 3 passages. n = 3, error bars = s.d. (C) Representative images of hPSCs in E8 medium, E7 medium, and the a0 DOE condition in E7. (D) Results of the DOE-generated model. The model allowed for identification of an optimized binding solution that minimizes bFGF utilization with a predicted 95% Oct4+/Nanog+ cell population at passage 3 of Transwell culture. (E) Comparison of the performance of DOE-optimized MCMs vs. non-optimized MCMs in maintaining hPSC pluripotency in Transwell (left) and direct (right) culture formats. For each culture format, four bFGF-MCM doses were tested, with total bFGF utilization matched between optimized and non-optimized MCMs for each respective dose. bFGF utilization as denoted on the x-axis was calculated based on amount of bFGF used to maintain a single well of hPSCs in a 12-well plate format (1 mL media/well) for three passages (see Supplementary Table S2). n = 3, error bars = s.d.; **** p < 0.0001, ** p < 0.01; two-way ANOVA. “n.d.” denotes a condition for which P3 flow cytometry data were not collected due to inability of the hPSCs to be effectively passaged at the corresponding dose of non-optimized bFGF-MCMs.
Multivariate analysis of the experimental data resulted in a model with goodness of fit F < 0.0001 and statistically significant dependence (P < 0.0001) of %Oct4+/Nanog+ hPSCs on both MCM and bFGF concentration in the MCM loading solution (Fig. 3D, Supplementary Fig. S5). By defining desirability as “minimizing the amount of bFGF utilized while achieving 95% Oct4+/Nanog+ population,” we used the model to determine an optimized MCM loading solution of 0.375 mg/mL MCMs and 0.456 μg/mL bFGF (Fig. 3D). We tested the model by comparing bFGF-MCMs fabricated using the optimized loading condition to non-optimized bFGF-MCMs, across a range of doses in which total bFGF utilization was matched between the two conditions (Supplementary Table 2). All optimized conditions outperformed non-optimized conditions over three passages in the Transwell culture format. Specifically, MCM doses of 0.6, 0.9, and 1.2 μg bFGF maintained >90% Oct4+/Nanog+ hPSCs using optimized MCMs, while corresponding doses of the non-optimized formulations maintained only 54–64% Oct4+/Nanog+ (Fig. 3E, Supplementary Fig. S6, and Supplementary Table 2). In the direct culture format, all four tested doses of optimized MCMs (61–246 μg bFGF-MCMs per well) successfully maintained >97% Oct4+/Nanog+ hPSCs in E7 media over 3 passages, while corresponding doses of the non-optimized formulations maintained 83–93% Oct4+/Nanog+ (Fig. 3E, Supplementary Fig. S6, and Supplementary Table 2). In addition, the optimized MCMs reduced total bFGF usage by 25.0% (Transwell) and 81.2% (direct) compared to standard hPSC culture in E8 medium with daily feeding (100 ng bFGF/mL per day) while maintaining >95% Oct4+/Nanog+ hPSCs (Table 1, Supplementary Table 2).
Table 1.
Summary of bFGF utilization vs. E8, for optimized Transwell and optimized direct culture bFGF-MCMs.
| Culture Method | hPSC Maintenance | Use in Extended Culture | bFGF Reduction vs. E8 |
|---|---|---|---|
|
Passage 3 (Day 12) = 95.0% Oct4+/Nanog+ | Add new bFGF-MCMs every 12 days | 25.0% |

|
Passage 3 (Day 12) = 97.7% Oct4+/Nanog+ | Add new bFGF-MCMs every passage | 81.2% |
3.3. Long-term direct culture with optimized bFGF-MCMs maintained hPSC pluripotency while reducing bFGF by >80%.
Direct culture with optimized bFGF-MCMs maintained pluripotency of two hPSC lines (H1 hESCs and WTc11 hiPSCs) in E7 for 25 passages (>3 months), while reducing the amount of required bFGF by 81.2% compared to standard culture in E8 (Table 1 and Supplementary Table 2). Direct culture bFGF-MCMs yielded colonies with typical undifferentiated morphology and uniform expression of Oct4 and Nanog for both hPSC lines over 25 passages (Fig. 4A and Supplementary Fig. S7A–B), and with no significant changes in cell expansion or growth rate with respect to culture in E8 (Supplementary Fig. S8A–B). In addition, hPSCs in direct culture were able to be transitioned back to standard culture conditions (E8 media/Matrigel) with no apparent MCM carryover after 2 passages (Fig. 4B), likely due to EDTA-mediated passaging resulting in dissolution and removal of the MCMs (Supplementary Fig. S9).
Figure 4. Direct culture with optimized bFGF-MCMs maintains hPSC pluripotency and normal karyotype during long-term culture.

(A) Comparison of colony morphology in H1 hESCs at passages 3 and 25 with bFGF-MCMs. (B) hPSCs in direct culture with bFGF-MCMs can be transitioned back to E8/Matrigel with minimal MCM carryover within 2 passages, and display normal hPSC colony morphology. Scale bars = 100 μm. (C) hPSCs in direct culture with bFGF-MCMs for 25 passages maintain robust expression of pluripotency markers Oct4 and Nanog. Scale bar = 100 μm. (D) G-banded karyotyping of hPSCs maintained in direct culture with optimized bFGF-MCMs for 25 passages. (E) hPSCs in direct culture with bFGF-MCMs retain the potential to spontaneously differentiate into derivatives of the three primary germ layers. EBs were formed, allowed to spontaneously differentiate and adhere to Matrigel-coated dishes, and stained for markers of (i) ectoderm (beta-III tubulin), (ii) mesoderm (alpha smooth muscle actin), and (iii) endoderm (alpha-fetoprotein) lineages. Scale bars = 50 μm. (F) Histological analysis of teratomas generated from hPSCs after long-term (25 passages) direct culture with bFGF-MCMs. Differentiation into all three germ layers is shown: (i) ectoderm (neuroepithelium, pigmented retinal tissue), (ii) mesoderm (cartilage) and (iii) endoderm (liver). Images shown in (E) and (F) are for H1 hESCs. Scale bar = 50 μm.
Long-term expansion with bFGF-MCMs maintained robust expression of pluripotency markers in both hPSC lines, as determined by immunofluorescence staining and flow cytometry (Fig. 4C, Table 2), and both lines displayed a normal karyotype (Fig. 4D). Following 25 passages in direct culture with bFGF-MCMs, hPSCs from both lines retained the ability to generate derivatives of ectoderm, mesoderm, and endoderm in vitro and in teratoma assays. Specifically, differentiated cells expressed βIII-tubulin (ectoderm), alpha smooth muscle actin (mesoderm), and alpha-fetoprotein (endoderm) in spontaneously differentiating embryoid bodies in vitro (Fig. 4E, Supplementary Fig. S10) and each of the hPSC lines generated teratomas containing neuroectoderm, cartilage, and liver tissue in nude mice (Fig. 4F, Supplementary Fig. S11). In addition, we transitioned hPSCs back to E8 after 25 passages in bFGF-MCM-E7, carried out directed differentiation toward neural, endothelial, and hepatic lineages, and observed generation of βIII-tubulin+ neurons, PECAM-1+ endothelial cells, and AFP+ hepatocyte-like cells, respectively (Supplementary Fig. S12). Taken together, these results demonstrate that direct culture with bFGF-MCMs maintains hPSC pluripotency, while reducing the amount of bFGF needed by >80% when compared to standard hPSC media.
Table 2.
Percentage of Oct4+/Nanog+ hPSCs vs. passage number during long-term direct culture with bFGF-MCMs.
| Passage # | P6 | P10 | P20 | P25 |
|---|---|---|---|---|
| H1 hESC | 98.8% | 95.6% | 94.7% | 95.0% |
| WTc11 hiPSC | 96.8% | 92.4% | 93.1% | 92.9% |
3.4. Local bFGF delivery correlated with higher growth factor activity at the culture surface and increased biological response.
bFGF-MCMs in direct culture significantly outperformed Transwell culture by maintaining Oct4/Nanog expression at >95% with lower quantities of input bFGF required (Fig. 3E and Supplementary Fig. S6, Table 1). Indeed, the amount of bFGF-MCMs sufficient for long-term hPSC maintenance in direct culture was unable to maintain Oct4/Nanog expression in the Transwell culture format, even when bFGF-MCMs were added fresh at every passage (Fig. 5A and Supplementary Fig. S13). As this suggested that localization of the growth factor in close proximity to cells may enhance the biological response, we utilized an enzyme-linked immunosorbent assay (ELISA) that detects only active bFGF (Supplementary Fig. S14 and S15) to assess why localized delivery of bFGF-MCMs improved pluripotency maintenance. In this cell-free assay, we observed a 9-fold increase in the amount of active bFGF detected in direct culture relative to Transwell culture when comparing equivalent amounts of input bFGF-MCMs (Fig. 5B). This indicated that direct culture with bFGF-MCMs resulted in a higher local bioavailability of active bFGF at the culture surface compared to Transwell culture.
Figure 5. Local delivery increases growth factor biological potency.

(A) %Oct4/Nanog expression of hPSCs cultured with the same amount of optimized bFGF-MCMs in either Transwell or direct culture. In this experiment, fresh MCMs were replaced at each passage (i.e., every 4 days) in both Transwell and direct culture formats. n = 3, error bars = s.d.; **** p < 0.0001, two-way ANOVA. (B) Local delivery (i.e., direct culture with bFGF-MCMs) amplifies growth factor activity at the culture surface, as measured in a cell-free bFGF bioactivity assay using the Quantikine bFGF ELISA kit. n = 3, error bars = s.d.; * p < 0.05, t-test.
3.5. MCMs stabilized bFGF against activity loss at physiological temperatures.
Experiments in which media were exchanged only at the time of cell passaging (i.e., every 4 days) showed that bFGF-MCMs better maintained Oct4/Nanog expression in hPSCs when compared to “bolus” conditions, in which a matched amount of soluble bFGF was spiked into E7 media. Under these conditions, bFGF-MCMs in direct culture maintained Oct4/Nanog expression in hPSCs for three passages (96.3 ± 0.1% Oct4+/Nanog+ at P3), while bolus incorporation of soluble bFGF failed to maintain Oct4/Nanog after just one passage (78.0 ± 0.9% Oct4+/Nanog+ at P2) (Fig. 6A and Supplementary Fig. S16). After 8 passages, bFGF-MCMs produced a substantially higher percentage of Oct4+/Nanog+ hPSCs (75.9 ± 0.5%) than the bolus soluble bFGF (41.2 ± 2.3%) (Fig. 6A and Supplementary Fig. S16). To quantify bFGF activity, we again used a bFGF ELISA assay that gave a linear response to active bFGF concentration (Supplementary Fig. S15A–B). Importantly, we demonstrated that the assay detected thermally induced loss of bFGF activity in E8 media. bFGF activity loss in media pre-incubated at 37°C and 65°C correlated with decreases in ERK phosphorylation in hPSCs treated with the pre-incubated media (Supplementary Fig. S15C–D). Using the bFGF ELISA, we observed that while only 5.37 ± 2.72% and 1.16 ± 0.31% of soluble bFGF remained active after 1 and 4 days of incubation at 37°C, respectively, bFGF-MCMs maintained 15.84 ± 3.33% and 5.36 ± 1.61% bFGF activity over these same periods (Fig. 6B).
Figure 6. Binding to MCMs improves bFGF thermal stability.
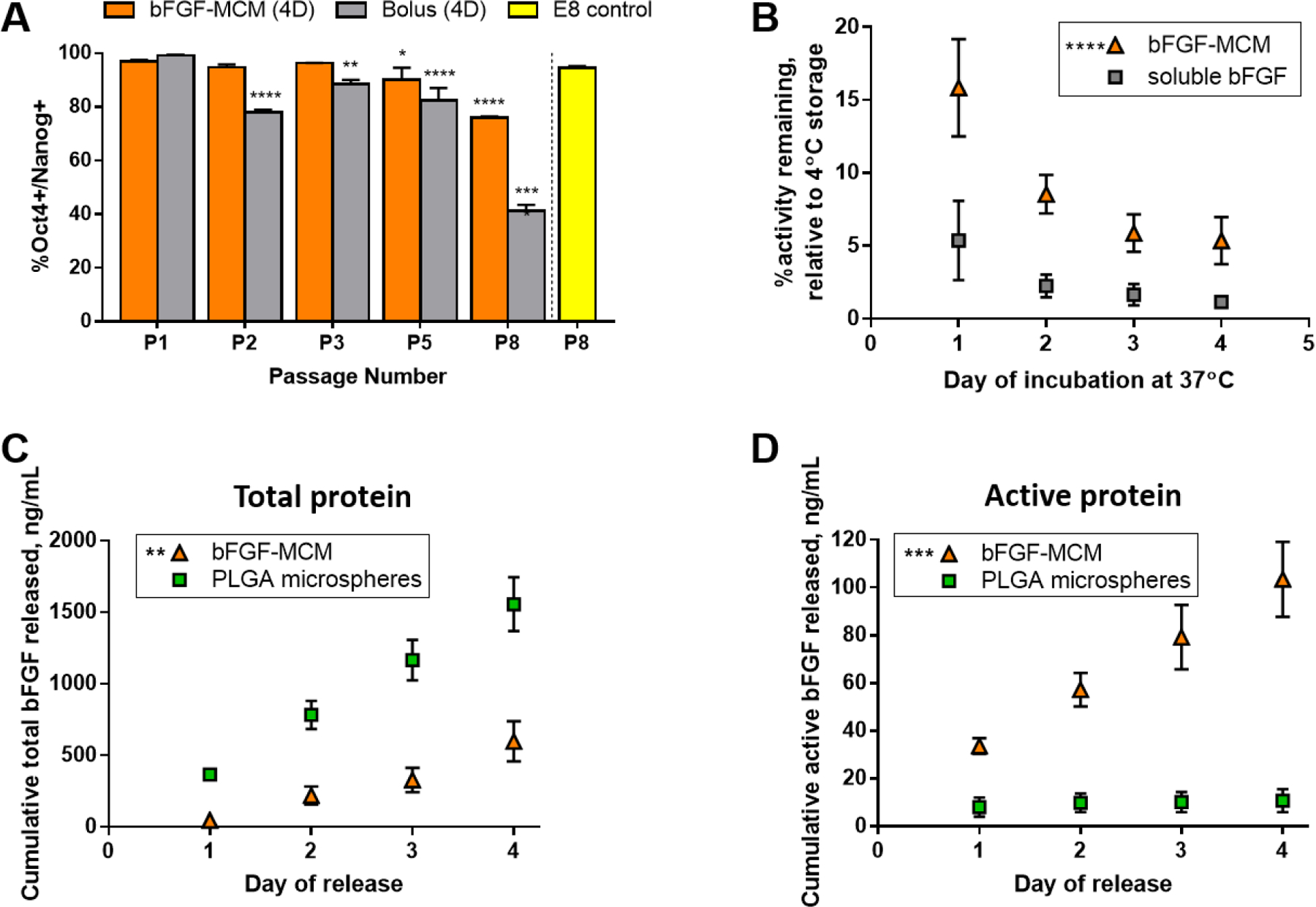
(A) bFGF bound to and released from MCMs maintains hPSC pluripotency marker expression more effectively than a matched amount of soluble bFGF (“Bolus”). “4D” denotes media changes every 4 days (i.e., only at the time of passaging). (B) bFGF bound to MCMs is stabilized against activity loss during incubation at physiological temperatures, as measured by Quantikine bFGF ELISA. “%activity remaining” is expressed relative to 4°C storage of each respective condition. (C) Comparison of total bFGF protein release from bFGF-MCMs vs. PLGA microspheres at 37°C. (D) Comparison of active bFGF protein release from bFGF-MCMs vs. PLGA microspheres. Values for cumulative daily release were extrapolated based on 2 hr release in the Quantikine bFGF ELISA for each time point assessed. Asterisks indicate statistically significant difference compared to (A) E8 control, (B) E7+soluble bFGF, (C-D) PLGA microspheres. n = 3, error bars = s.d.; p < 0.05 (*), 0.01 (**), 0.001 (**), or 0.0001 (****), two-way ANOVA.
MCMs released active bFGF over four days at 37°C more efficiently than commonly used and commercially available polymer microspheres designed for bFGF release in hPSC culture. Specifically, PLGA microspheres with encapsulated bFGF released significantly more total protein (1556 ± 188 ng/mL cumulative) over the course of four days compared to MCMs (597 ± 140 ng/mL) (Fig. 6C). However, MCMs released nearly 10-fold more active bFGF over four days (103.5 ± 15.8 ng/mL cumulative) compared to PLGA microspheres (10.8 ± 4.8 ng/mL), which only released detectable active bFGF at days 1 and 2 (Fig. 6D).
4. Discussion:
In the present study, we report the development of bFGF-loaded mineral-coated microparticles (bFGF-MCMs) that both stabilized and sustained release of bFGF to markedly reduce the amount of growth factor needed in stem cell culture. Sustained bFGF release from MCMs maintained long-term expansion of undifferentiated hPSCs in the absence of supplemental bFGF, while reducing the total amount of bFGF needed by >80% relative to contemporary culture methods. This is the first reported bFGF delivery approach that is synthetic, requires no supplemental bFGF beyond what is delivered from the material, and can be incorporated into existing workflows using chemically defined media for hPSC expansion and downstream directed differentiation. In addition, as protein binding and release from the mineral coatings is based on promiscuous charge-charge interactions27 and has been used for release of myriad other proteins14,15,17,27–30, this approach can be potentially extended to other costly proteins in stem cell culture.
We focused on bFGF due to its role in promoting the expansion, maintenance, and differentiation of numerous cell types, including hPSCs31,32. In hPSC culture, bFGF is a critical growth factor that promotes cell adhesion and proliferation, and works in concert with TGFβ/Activin signaling to regulate self-renewal33–36. Thus, inclusion of bFGF is a commonality across several commercial hPSC media used for chemically-defined, feeder-free culture12,37–41. We chose Essential 8 (E8) medium as a basis for comparison throughout our study, as it is minimally complex (eight supplemental components, of which two are growth factors), is chemically defined and xeno-free, and has already seen success in translating to scalable hPSC culture in bioreactors42,43. The short half-life of bFGF (t½ ~ 7–10 hrs at 37°C44–46) necessitates high concentrations of the growth factor in cell culture and frequent media changes that contribute considerably to the cost of expanding hPSCs. Heparin, a natural stabilizer of bFGF, has been included in some hPSC media formulations to ameliorate this issue in research applications. However, as sources of heparin are almost exclusively of xenogeneic origin47,48, its introduction into cell culture poses issues of batch variability, risk of zoonotic contamination, and potential supply chain limitations. Thus, synthetic materials that improve protein stability in cell culture may be particularly attractive alternatives in large-scale biomanufacturing of clinical-grade stem cells and their derivatives.
Although the concept of sustained delivery of bFGF in stem cell culture is straightforward, identifying materials that do not intrinsically interfere with stem cell pluripotency in long-term culture is not trivial, as stem cells are known to be sensitive to their microenvironment49–52. Notably, MCMs alone – without bFGF – had no detrimental effects on pluripotency when implemented in either indirect (Transwell) or direct co-culture with hPSCs in E8 (Supplementary Fig. S2), and were compatible with enzyme-free passaging workflows22 (Supplementary Figs. S2 and S9). Loading MCMs with bFGF afforded sustained release of the growth factor in hPSC culture via gradual dissolution of the mineral coating (Fig. 6C–D, Supplementary Fig. S1C), a mechanism described in our previous work wherein the rate of coating dissolution corresponded to protein release kinetics17.
While it is known that bFGF is required for hPSC maintenance, the relationship between bFGF signaling kinetics and the pluripotent state is poorly understood, which precludes a rational design of dosage and release kinetics for a controlled bFGF release platform. Thus, following initial experiments demonstrating the capacity of sustained bFGF release from MCMs to maintain hPSC pluripotency in a dose-dependent manner (Fig. 2), we used Design of Experiments (DOE) methodology to optimize MCM-bFGF formulations that would improve maintenance of hPSCs. Our DOE experiment identified a non-intuitive relationship between MCM:bFGF ratio and their absolute concentrations, which significantly influenced maintenance of Oct4+/Nanog+ hPSCs by bFGF-MCMs (Fig. 3D and Supplementary Fig. S5). It was unsurprising that MCM-bFGF loading formulations with a high ratio of bFGF to MCMs performed well in maintaining hPSC pluripotency (Supplementary Table 2), as we previously observed a positive correlation between protein concentration in the MCM binding solution (with constant MCM mass) and the amount of MCM-bound protein27,53. These results indicated that increasing the bFGF:MCM ratio would increase the total bFGF dose delivered from MCMs. However, there is a known tradeoff between the total mass of protein bound and the MCM-protein binding efficiency53, so DOE was valuable in determining optimal binding formulations that simultaneously maintained pluripotency and reduced the amount of bFGF required. A two-factor, five-level factorial design was sufficient to develop bFGF-MCM formulations that significantly outperformed non-optimized bFGF-MCMs in both Transwell and direct culture (Fig. 3E) for at least 25 passages (>100 days), at which point the expanded cells retained both a normal karyotype and the capacity to spontaneously differentiate in vitro and form teratomas in vivo (Fig. 4).
In this study, we tested serial dilutions of DOE-optimized bFGF-MCMs in both Transwell and direct culture to determine the minimum amount of optimized bFGF-MCMs required for long-term hPSC expansion and identified a condition that reduced bFGF usage by >80% compared to standard culture in E8. Future studies could employ additional iterations of DOE optimization with factors such as bFGF-MCM dose and total media volume in culture to further reduce the amount of growth factor required to maintain hPSCs, as well as use MCMs to deliver each of the protein components of E8 in order to further reduce the costs of cell manufacturing. In addition, further mechanistic insight into why optimized bFGF-MCMs outperformed non-optimized may be of value. Such studies may elucidate optimal growth factor release and signaling kinetics for hPSC pluripotency and exploit these relationships to drive process improvements in stem cell biomanufacturing.
MCMs offered advantages over existing biomaterials for controlled protein release, such as amenability to protein loading in gentle processing conditions devoid of organic solvents and detergents (Fig. 1A and Supplementary Fig. S1B). Importantly, we demonstrated here, and in a previous study29, that MCMs can bind and release a wide range of proteins, and that nanostructured features of the mineral coatings can offer protection against protein instability in the face of stressors such as organic solvents, proteases, and thermal challenge (Fig. 6). This contrasts with traditional controlled protein release approaches based on biodegradable polymers, wherein loss of protein activity from formulation and processing conditions (e.g., high heat, agitation, exposure to detergents and non-aqueous solvents) is a common issue29,19. Loss of biological activity in cell culture is a particular concern for many FGF family members, including bFGF, due to an intrinsic instability at physiological temperatures6,11. In the current study, we showed that binding of bFGF to MCMs stabilized the protein against thermally induced activity loss at physiological temperatures. In cell-free assays, bFGF-MCMs demonstrated 3- to 4-fold higher preservation of bFGF biological activity during incubation at 37°C when compared to free soluble bFGF (Fig. 6B). This protein stabilization likely depends on a physical interaction of the protein with the nanostructured coating that confines bFGF to its native conformation. This proposed hypothesis is in line with previous studies from our group29 and others54–58 showing that stabilization of protein conformation structures at material interfaces can enhance protein bioactivity. Taken together, the unique advantages of MCMs’ ease of protein loading and protein stabilization may be of critical value in the biomanufacturing of cell therapies.
MCMs sustained delivery of biologically active growth factor more efficiently than a commonly used and commercially available polymer-based approach developed for hPSC culture7. bFGF-MCMs demonstrated nearly linear kinetics of active protein release over 4 days (8.9 ng/mL/day) at 37°C, whereas PLGA microspheres with encapsulated bFGF failed to release detectable quantities of active bFGF beyond the first 2 days at 37°C. This result is remarkable, as the PLGA microspheres released more total protein than MCMs during the same time period (Fig. 6C–D). While this observed difference between MCMs and PLGA microspheres can be attributed in part to the stabilization of bFGF by MCMs, degradation of PLGA to create an acidic microclimate that promotes protein unfolding, aggregation, and peptide chain hydrolysis may have also contributed to the compromised bioactivity of bFGF released from PLGA microspheres19,20. Taken together, these results demonstrate that MCM-mediated bFGF delivery enabled more efficient sustained protein delivery in culture, with a higher fraction of the released protein remaining active and capable of stimulating downstream FGF receptor (FGFR) signaling, compared to PLGA-based delivery.
Localized delivery enhanced the cellular response to bFGF released from MCMs. Direct co-culture of hPSCs with bFGF-MCMs enabled a 4-fold reduction in the total bFGF needed to maintain hPSC pluripotency marker expression when compared to Transwell culture (Table 1). When comparing equivalent bFGF-MCM dosages, direct culture maintained >2.8-fold higher %Oct4+/Nanog+ hPSCs at passage 3 compared to Transwell culture (Fig. 5). This increase in apparent biological potency of a protein delivered in close proximity to cells has previously been demonstrated in several in vitro and in vivo studies (reviewed in59–61). In the current study, the stabilization of bFGF by MCMs likely further accentuated the effect of local growth factor delivery. In a cell culture model in which exogenously delivered bFGF exists in three spatial domains - sequestered in the mineral coating, soluble in the media, and bound to FGF receptors - bFGF delivered to cells from a distant source takes longer to transport to the cell surface, during which the growth factor would be expected to undergo continuous activity loss in solution prior to reaching cell surface FGFRs. Local release of bFGF would be expected to decrease the time it spends in the soluble domain, where it is more subject to activity loss. Experiments in which we separated the soluble releasate from the remaining MCM-bound bFGF after 4 days of release support this hypothesis. After 4 days, the releasate showed no detectable levels of bFGF activity (Supplementary Fig. S17), indicating that the ability of bFGF to withstand long-term thermal challenge in our study depends on the protein remaining bound to the mineral coating.
Technologies that control protein release kinetics offer a potential avenue for tailoring soluble factor signaling and increasing the efficiency and yield of stem cell biomanufacturing. Here, we incorporated bFGF-MCMs into standard hPSC culture workflows, with minimal modification of cell culture or passaging protocols. This amenability to conventional workflows may represent a general advantage of this approach in cell biomanufacturing. One could envision a generalizable workflow, in which biomaterials provide sustained delivery of recombinant proteins from a solid matrix while remaining media components (e.g., amino acids, vitamins, lipids, carbohydrates) and metabolic waste products are exchanged by batch or perfusion feeding. This type of approach would allow cells to receive sufficient nutrients for cell survival and growth via frequent media changes, while conserving the proteins needed to maintain or establish cell state/function. As an example, we tested the effect of less frequent media exchanges on hPSCs in direct culture with bFGF-MCMs and observed a decrease in %Oct4+/Nanog+ cells when media were exchanged every 4 days, which could be rescued by either doubling the volume of E7 or increasing the frequency of E7 exchange (Supplementary Fig. S18). As the amount of bFGF was the same in all conditions, these results suggest that bFGF depletion was not the only factor limiting hPSC maintenance and provide a proof-of-concept for how combining scalable factors like basal media volume with controlled release of expensive growth factors and signaling molecules may broadly facilitate cost reduction in cell biomanufacturing.
5. Conclusions:
Advancing stem cell therapies is critically dependent on efficiently scaling up culture methods and reducing the costs of cell biomanufacturing. Here, we describe a scalable, materials-based approach to reduce by >80% the amount of growth factor required for manufacturing human pluripotent stem cells. MCMs enabled this reduction by protecting bFGF from degradation while providing sustained, localized release of the active growth factor. Adoption of this mineral-based material, combined with statistically driven optimization methodologies such as DOE, has the potential to significantly improve scale-up and cost reduction in biomanufacturing of therapeutically relevant cell types.
Supplementary Material
Significance statement:
Economical approaches for cell biomanufacturing are essential for clinical translation of cell therapies. However, cell expansion and differentiation rely on costly growth factors that are unstable in culture. Here, we describe a biomaterial strategy for stabilizing and delivering thermally labile growth factors for efficient biomanufacturing of human pluripotent stem cells (hPSCs). Mineral-coated microparticles (MCMs) protected basic fibroblast growth factor (bFGF) activity at physiological temperatures and sustained bFGF delivery to reduce the amount needed for maintaining pluripotency. Additionally, we demonstrate the utility of statistically driven optimization strategies such as Design of Experiments in materials formulation, as the approach enabled long-term hPSC expansion without additional bFGF supplementation, reducing total usage by >80% compared to standard approaches. This work demonstrates the potential of a biomaterial strategy to greatly reduce the need for growth factors and should aid in overcoming challenges in cost-effective biomanufacturing.
6.1. Acknowledgments:
The authors acknowledge support from staff and the use of equipment at the Materials Science Center (NSF DMR-1121288), the UW Carbone Cancer Center Flow Cytometry Laboratory (Support Grant P30 CA014520), and the UW Comparative Pathology Laboratory at UW-Madison. This work was supported by funding from the National Institutes of Health (R01HL093282 to W.L.M.; Biotechnology Training Program NIGMS 5 T32-GM08349 to A.W.X. and A.S.K.), the U.S. Environmental Protection Agency (STAR grant no. 83573701 to W.L.M.), and the National Science Foundation (DGE-1256259 to A.W.X. and A.S.K.; DMR-1306482 to W.L.M.)
Footnotes
Declaration of Interests: W.L.M. is a Co-Founder and Chief Science Officer at Stem Pharm, Inc. and Dianomi Therapeutics. A.S.K is a consultant to Stem Pharm, Inc. A.W.X is now an employee of Catalent Pharma Solutions. The authors declare no remaining competing financial interests.
6.3. Data availability:
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
- 1.Fox IJ et al. Use of differentiated pluripotent stem cells in replacement therapy for treating disease. Science (80-. ). 345, 1247391–1247391 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Trounson A & DeWitt ND Pluripotent stem cells progressing to the clinic. Nat. Rev. Mol. Cell Biol 17, 194–200 (2016). [DOI] [PubMed] [Google Scholar]
- 3.Serra M, Brito C, Correia C & Alves PM Process engineering of human pluripotent stem cells for clinical application. Trends Biotechnol. 30, 350–359 (2012). [DOI] [PubMed] [Google Scholar]
- 4.Carmen J, Brindley DA, Davie NL & Smith D in Stem Cells in Regenerative Medicine 49–68 (John Wiley & Sons, Ltd, 2016). doi: 10.1002/9781118846193.ch4 [DOI] [Google Scholar]
- 5.Pigeau GM, Csaszar E & Dulgar-Tulloch A Commercial Scale Manufacturing of Allogeneic Cell Therapy. Front. Med 5, 1–8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen G, Gulbranson DR, Yu P, Hou Z & Thomson JA Thermal Stability of Fibroblast Growth Factor Protein Is a Determinant Factor in Regulating Self-Renewal, Differentiation, and Reprogramming in Human Pluripotent Stem Cells GUOKAI. Stem Cells 30, 623–630 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lotz S et al. Sustained Levels of FGF2 Maintain Undifferentiated Stem Cell Cultures with Biweekly Feeding. PLoS One 8, 1–10 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lopes AG, Sinclair A & Frohlich B Cost Analysis of Cell Therapy Manufacture: Autologous Cell Therapies, Part 1. Bioprocess Int. 16, S3–S8 (2018). [Google Scholar]
- 9.Lipsitz YY et al. A roadmap for cost-of-goods planning to guide economic production of cell therapy products. Cytotherapy 19, 1383–1391 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Levenstein ME et al. Basic Fibroblast Growth Factor Support of Human Embryonic Stem Cell Self-Renewal. Stem Cells 24, 568–574 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Buchtova M et al. Instability restricts signaling of multiple fibroblast growth factors. Cell. Mol. Life Sci 72, 2445–2459 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ludwig TE et al. Derivation of human embryonic stem cells in defined conditions. Nat. Biotechnol 24, 185–187 (2006). [DOI] [PubMed] [Google Scholar]
- 13.Horiguchi I, Urabe Y, Kimura K & Sakai Y Effects of glucose, lactate and basic FGF as limiting factors on the expansion of human induced pluripotent stem cells. J. Biosci. Bioeng 125, 111–115 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Lee JS, Suarez-Gonzalez D & Murphy WL Mineral Coatings for Temporally Controlled Delivery of Multiple Proteins. Adv. Mater 23, 4279–4284 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Suárez-González D et al. Controllable mineral coatings on PCL scaffolds as carriers for growth factor release. Biomaterials 33, 713–721 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu X et al. Nanostructured Mineral Coatings Stabilize Proteins for Therapeutic Delivery. Adv. Mater 29, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu X, Khalil A, Dang PN, Alsberg E & Murphy WL Multilayered inorganic microparticles for tunable dual growth factor delivery. Adv. Funct. Mater 24, 3082–3093 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Furue MK et al. Heparin promotes the growth of human embryonic stem cells in a defined serum-free medium. Proc. Natl. Acad. Sci. U. S. A 105, 13409–14 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fu K, Klibanov AM & Langer R Protein stability in controlled-release systems. Nat. Biotechnol 18, 24–25 (2000). [DOI] [PubMed] [Google Scholar]
- 20.Zhu G, Mallery SR & Schwendeman SP Stabilization of proteins encapsulated in injectable poly (lactide-co- glycolide). Nat. Biotechnol 18, 52–57 (2000). [DOI] [PubMed] [Google Scholar]
- 21.Choi S, Yu X, Jongpaiboonki L, Hollister SJ & Murphy WL Inorganic coatings for optimized non-viral transfection of stem cells. Sci. Rep 3, 1567–93 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Beers J et al. Passaging and colony expansion of human pluripotent stem cells by enzyme-free dissociation in chemically defined culture conditions. Nat. Protoc 7, 2029–2040 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xie AW et al. Controlled Self-assembly of Stem Cell Aggregates Instructs Pluripotency and Lineage Bias. Sci. Rep 7, 1–15 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chambers SM et al. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol 27, 275–280 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang J et al. A Genome-wide Analysis of Human Pluripotent Stem Cell-Derived Endothelial Cells in 2D or 3D Culture. Stem Cell Reports 8, 907–918 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sengupta S et al. Aggregate culture of human embryonic stem cell-derived hepatocytes in suspension are an improved in vitro model for drug metabolism and toxicity testing. Toxicol. Sci 140, 236–45 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jongpaiboonkit L, Franklin-Ford T & Murphy WL Mineral-Coated Polymer Microspheres for Controlled Protein Binding and Release. Adv. Mater 21, 1960–1963 (2009). [Google Scholar]
- 28.Lee JS, Lu Y, Baer GS, Markel MD & Murphy WL Controllable protein delivery from coated surgical sutures. J. Mater. Chem 20, 8894–8903 (2010). [Google Scholar]
- 29.Yu X et al. Nanostructured Mineral Coatings Stabilize Proteins for Therapeutic Delivery. Adv. Mater 29, 1701255 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Clements AEB, Groves ER, Chamberlain CS, Vanderby R & Murphy WL Microparticles Locally Deliver Active Interleukin-1 Receptor Antagonist In Vivo. Adv. Healthc. Mater 7, 1–8 (2018). [DOI] [PubMed] [Google Scholar]
- 31.Bikfalvi A, Klein S, Pintucci G & Rifkin DB Biological roles of fibroblast growth factor-2. Endocr. Rev 18, 26–45 (1997). [DOI] [PubMed] [Google Scholar]
- 32.Xu C et al. Basic Fibroblast Growth Factor Supports Undifferentiated Human Embryonic Stem Cell Growth Without Conditioned Medium. Stem Cells 23, 315–323 (2005). [DOI] [PubMed] [Google Scholar]
- 33.Vallier L Activin/Nodal and FGF pathways cooperate to maintain pluripotency of human embryonic stem cells. J. Cell Sci 118, 4495–4509 (2005). [DOI] [PubMed] [Google Scholar]
- 34.Xiao L, Yuan X & Sharkis SJ Activin A Maintains Self-Renewal and Regulates Fibroblast Growth Factor, Wnt, and Bone Morphogenic Protein Pathways in Human Embryonic Stem Cells. Stem Cells 24, 1476–1486 (2006). [DOI] [PubMed] [Google Scholar]
- 35.Greber B, Lehrach H & Adjaye J Fibroblast Growth Factor 2 Modulates Transforming Growth Factor β Signaling in Mouse Embryonic Fibroblasts and Human ESCs (hESCs) to Support hESC Self-Renewal. Stem Cells 25, 455–464 (2007). [DOI] [PubMed] [Google Scholar]
- 36.Singh H & Brivanlou AH The Molecular Circuitry Underlying Pluripotency in Embryonic Stem Cells and iPS Cells. Principles of Regenerative Medicine 3, (Elsevier Inc., 2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Greber B, Lehrach H & Adjaye J Control of Early Fate Decisions in Human ES Cells by Distinct States of TGFβ Pathway Activity. Stem Cells Dev. 17, 1065–1078 (2008). [DOI] [PubMed] [Google Scholar]
- 38.Yao S et al. Long-term self-renewal and directed differentiation of human embryonic stem cells in chemically defined conditions. Proc. Natl. Acad. Sci. U. S. A 103, 6907–12 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lu J, Hou R, Booth CJ, Yang S-H & Snyder M Defined culture conditions of human embryonic stem cells. Proc. Natl. Acad. Sci. U. S. A 103, 5688–5693 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Akopian V et al. Comparison of defined culture systems for feeder cell free propagation of human embryonic stem cells. Vitr. Cell. Dev. Biol. - Anim 46, 247–258 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen G et al. Chemically defined conditions for human iPS cell derivation and culture. Nat. Methods 8, 424–429 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang Y et al. Scalable expansion of human induced pluripotent stem cells in the defined xeno-free E8 medium under adherent and suspension culture conditions. Stem Cell Res. 11, 1103–1116 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Badenes SM et al. Defined Essential 8™ Medium and Vitronectin Efficiently Support Scalable Xeno-Free Expansion of Human Induced Pluripotent Stem Cells in Stirred Microcarrier Culture Systems. PLoS One 11, e0151264 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shiba T et al. Modulation of mitogenic activity of fibroblast growth factors by inorganic polyphosphate. J. Biol. Chem 278, 26788–26792 (2003). [DOI] [PubMed] [Google Scholar]
- 45.Beenken A & Mohammadi M The FGF family: Biology, pathophysiology and therapy. Nat. Rev. Drug Discov 8, 235–253 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dvorak P et al. Computer-assisted engineering of hyperstable fibroblast growth factor 2. Biotechnol. Bioeng 1–13 (2018). doi: 10.1002/bit.26531 [DOI] [PubMed] [Google Scholar]
- 47.Keire D et al. Diversifying the global heparin supply chain: Reintroduction of bovine heparin in the united states? BioPharm Int. 28, 36–42 (2015). [Google Scholar]
- 48.Oduah EI, Linhardt RJ & Sharfstein ST Heparin: Past, present, and future. Pharmaceuticals 9, 1–12 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Derda R et al. High-throughput discovery of synthetic surfaces that support proliferation of pluripotent cells. J. Am. Chem. Soc 132, 1289–1295 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mei Y et al. Combinatorial development of biomaterials for clonal growth of human pluripotent stem cells. Nat. Mater 9, 768–78 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brafman D a et al. Long-term human pluripotent stem cell self-renewal on synthetic polymer surfaces. Biomaterials 31, 9135–44 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Villa-Diaz LG et al. Synthetic polymer coatings for long-term growth of human embryonic stem cells. Nat. Biotechnol 28, 581–583 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Clements AEB, Leiferman EM, Chamberlain CS, Vanderby R & Murphy WL Addition of Mineral-Coated Microparticles to Soluble Interleukin-1 Receptor Antagonist Injected Subcutaneously Improves and Extends Systemic Interleukin-1 Inhibition. Adv. Ther 1800048, 1800048 (2018). [Google Scholar]
- 54.Mozhaev VV, Sergeeva MV, Belova AB & Khmelnitsky YL Multipoint attachment to a support protects enzyme from inactivation by organic solvents: α‐Chymotrypsin in aqueous solutions of alcohols and diols. Biotechnol. Bioeng 35, 653–659 (1990). [DOI] [PubMed] [Google Scholar]
- 55.Takahashi H et al. Catalytic activity in organic solvents and stability of immobilized enzymes depend on the pore size and surface characteristics of mesoporous silica. Chem. Mater 12, 3301–3305 (2000). [Google Scholar]
- 56.Kim J & Grate JW Single-enzyme nanoparticles armored by a nanometer-scale organic/inorganic network. Nano Lett. 3, 1219–1222 (2003). [Google Scholar]
- 57.Wang P Nanoscale biocatalyst systems. Curr. Opin. Biotechnol 17, 574–579 (2006). [DOI] [PubMed] [Google Scholar]
- 58.Giri J, Li WJ, Tuan RS & Cicerone MT Stabilization of proteins by nanoencapsulation in sugar-glass for tissue engineering and drug delivery applications. Adv. Mater 23, 4861–4867 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen RR & Mooney DJ Polymeric growth factor delivery strategies for tissue engineering. Pharm. Res 20, 1103–1112 (2003). [DOI] [PubMed] [Google Scholar]
- 60.Zisch AH, Lutolf MP & Hubbell JA Biopolymeric delivery matrices for angiogenic growth factors. Cardiovasc. Pathol 12, 295–310 (2003). [DOI] [PubMed] [Google Scholar]
- 61.Ansorge M & Pompe T Systems for localized release to mimic paracrine cell communication in vitro. J. Control. Release 278, 24–36 (2018). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.