

Abstract
Although hormonal therapy (HT) inhibits the growth of hormone receptor-positive (HR+) breast (BrCa) and prostate (PrCa) cancers, HT resistance frequently develops within the complex metastatic microenvironment of the host organ (often the bone), a setting poorly recapitulated in 2D culture systems. To address this limitation, we cultured HR+ BrCa and PrCa spheroids and patient-derived organoids in 3D extracellular matrices (ECM) alone or together with bone marrow stromal cells (BMSC). In 3D monocultures, antiestrogens and antiandrogens induced anoikis by abrogating anchorage-independent growth of HR+ cancer cells but exhibited only modest effects against tumor cells residing in the ECM niche. In contrast, BMSC induced hormone-independent growth of BrCa and PrCa spheroids and restored lumen filling in the presence of HR-targeting agents. Molecular and functional characterization of BMSC-induced hormone independence and HT resistance in anchorage-independent cells revealed distinct context-dependent mechanisms. Cocultures of ZR75–1 and LNCaP with BMSC exhibited paracrine IL-6-induced HT resistance via attenuation of HR protein expression, which was reversed by inhibition of IL-6 or JAK signaling. Paracrine IL-6/JAK/STAT3-mediated HT resistance was confirmed in patient-derived organoids cocultured with BMSC. Distinctly, MCF7 and T47D spheroids retained ER protein expression in cocultures but acquired redundant compensatory signals enabling anchorage independence via ERK and PI3K bypass cascades activated in a non-IL-6-dependent manner. Collectively, these data characterize the pleiotropic hormone-independent mechanisms underlying acquisition and restoration of anchorage-independent growth in HR+ tumors. Combined analysis of tumor and microenvironmental biomarkers in metastatic biopsies of HT-resistant patients can help refine treatment approaches.
Introduction
Systemic endocrine therapies, which encompass HR blockade with small molecule antagonists (e.g. antiestrogens and antiandrogens for ER and PR, respectively), can prolong disease-free and overall survival for HR+ BrCa/PrCa patients. However, some initially sensitive HR+ tumors acquire HT resistance and others exhibit de novo resistance (reviewed in (1–3)).
Potential HT resistance mechanisms in BrCa include the deregulation of ER co-activators/co-repressors, modulation of ER activity by growth factor receptor pathways, and deregulation of cell cycle signaling molecules (1,4,5). Castration-resistant PrCa (CRPC) can involve multiple non-mutually exclusive mechanisms including AR overexpression though amplification of the AR gene (6) and/or its enhancer region (7), suppression of miRNAs downregulating AR expression (8,9), AR splice variants (10), and increased expression or activation of AR binding partners (11,12). Another common mechanism of HT resistance involves mutations in ER or AR genes (13,14). Second generation HR antagonists (e.g. fulvestrant and enzalutamide) can overcome HT resistance mediated by incomplete blockade of hormone signaling (15,16). However, acquisition of hormone independence remains a common mechanism of HT resistance. Alternative cancer cell growth signals, including amplification/overexpression of HER2 and acquisition of activating mutations in the PI3K/Akt signaling pathway or PTEN loss, are frequently observed in HT-resistant BrCa/PrCa (17–20). These pre-clinical and clinical observations raise the possibility that loss of HR expression or attenuation of its activity may serve as alternate means of escaping HR-targeted therapies, plausibly through concomitant activation of compensatory signaling pathways.
As new targeted therapies are being developed against HT-resistant BrCa and PrCa tumors (21–23), identifying response predictors is essential for their rational use. However, putative biomarkers in primary tumors may not account for potential HT resistance mechanisms operating metastatic tumors. Furthermore, preclinical model of HT resistance mechanisms has principally invovled in in vitro cultures of cancer cells on 2D plastic surfaces: these conventional systems do not necessarily recapitulate the biological properties of HR+ cancer cells growing in 3D anchorage-independent conditions and their evasion of anoikis, a key property acquired during malignant transformation (24,25). Distinctly from 2D cultures, 3D cultures and coculture systems, which involve nonmalignant cells of the metastatic milieu, can better recapitulate the cancer architecture and the heterotypic cellular interactions within the physical space of the tumor microenvironment (26), which can putatively affect the response of cancer cells to HT.
Bone is the most common metastatic site for HR+ BrCa and PrCa. To assess how the cellular, structural and paracrine elements of the bone metastatic milieu can simultaneously interact with the tumor cells and affect their sensitivity to HT, we developed 3D cocultures of BrCa/PrCa spheroids and bone marrow stromal cells (BMSCs). We observed that HR-targeting agents have modest effect against cancer cells in the extracellular matrix (ECM) niche, but specifically abrogate the anchorage-independent growth of HR+ BrCa/PrCa cells, a hallmark of tumorigenesis. Importantly, BMSCs restore the ability of HR+ tumor cells to evade anoikis in the presence of HT through distinct and context-dependent mechanisms. Our results highlight that tumor-stroma interactions in the metastatic microenvironment are a potential source of HT resistance in BrCa/PrCa patients.
Material and Methods
Cell lines and reagents:
Cancer cell lines (MCF7, T47D, ZR75–1 and LNCaP) and immortalized non-malignant cells lines HS5, HS27A, HOBIT, hFOB, THLE3 and SVGP12 were originally obtained from ATCC. Lenti-X-293T cells were purchased from Clontech (Mountain View, CA). The MCF7 cells stably expressing doxycycline-inducible mutant ER (Y537S) and the respective WT control were established by the lab of Dr. Myles Brown, as previously described (27). The cells were used for up to 15 passages after thawing. The identity of the cell lines was confirmed by short tandem repeat (STR) analysis. The cells were last tested for the presence of mycoplasma in May 2019, using the MycoAlert mycoplasma detection kit (Lonza). The culture conditions and reagents used in this study are further described in the Supplemental Materials.
3D cultures:
The BrCa and PrCa cells were cultured in 3D conditions as previously described (28). Briefly, cells were suspended in 80% matrigel or in rat tail collagen (1mg/ml; prepared according to manufacturer’s instructions), and liquid gels were casted in either 12-well (for histological and molecular analyses) or multi-well plates (for cell viability assays; described below). Gels were incubated for 1h at 37°C to allow for solidification, supplemented with medium, and used for further downstream applications (see other sections of Methods). To extract cells from 3-D organoids, gels were first disrupted mechanically in cold PBS; next, spheroids were isolated by centrifugation and incubated in trypsin for 20–30 min to generate single-cell solution. When applicable, separation of tumor and stromal cell subpopulations was performed using mesenchymal cells depletion microbeads (cat.# 130–050-601; Miltenyi, Bergisch Gladbach, Germany) and magnetic columns, according to manufacturer’s instructions.
Cell viability assays:
Cell lines MCF-7, T47D, ZR-75–1, LNCaP, and PrCa patient-derived cells MSK-PCa1, MSK-PCa2 and MSK-PCa3 were stably transfected with lentiviral vectors expressing the luciferase gene, allowing the use of the emitted bioluminescence to longitudinally and non-disruptively measure cancer cell viability in the presence of luciferase-negative non-malignant cells (compartment-specific bioluminescence imaging, CS-BLI) (29). For 2-D culture cell viability assays, the cells were seeded in 384-well plates (500–2000 cells/well), in supplemented media (50uL-100uL/well) and incubated for 24h prior to addition of compound/s at indicated concentrations. For 3-D spheroid cell viability assays, cells were suspended in matrigel, collagen, or mixture of the two matrices, at a density of 105 cells/ml, and plated in 384-well plates. After 24h antiestrogens were added in the medium at the indicated final concentrations. At each indicated time point either luciferin (for the luciferase-positive cells; concentration per manufacturer’s instructions) or CTG reagent (per manufacturer’s instructions) was added to each well, and the plates were read using a microplate reader (BioTek Synergy 2). Cell viability assays were performed in quadruplicates and error bars represent standard error of mean.
Apoptosis assay:
The rate of cell apoptosis in 2D and 3D cultures was estimated by luminescence measurement, using the RealTime-Glo™ Annexin V Apoptosis assay (Promega, Madison, WI) following the manufacturer’s instructions. Briefly, the cells were incubated with two annexin V fusion proteins (Annexin V-LgBiT and Annexin V-SmBiT) which contain complementary subunits of NanoBiT® Luciferase; and a time-released luciferase. The cell surface levels of membrane phosphatidylserine in apoptotic cells brings the Annexin V-LgBiT and Annexin V-SmBiT luciferase subunits into complementing proximity, which is reflected by the strength of bioluminescence signal emitted in culture (quadruplicate measurements).
Histological assays:
Cells suspended in matrigel were casted in 12-well plates (1.5 ml/well) and gels were let to solidify for 1h at 37°C. After gel solidification, the 3-D cultures were supplemented with medium and incubated for 7–8 days to allow for organoid formation and antiestrogen/antiandrogen treatment as indicated. Gels were then scooped out of the culture vessel and fixed in 10% formalin. Subsequently, gels were processed for paraffin embedding and immunohistochemistry (slide sections), as previously described (28).
Genome-scale CRISPR screen:
A CRISPR/Cas9-based gene editing screen of MCF7 cells in the presence vs. absence of 4-OHT was performed similarly to prior studies (30) (further details in the Supplemental Material).
Two-photon microscopy:
Two-photon excited fluorescence (TPEF) imaging of 3D spheroids was performed as previously described (31) (further details in the Supplemental Material).
Animal studies:
The in vivo experiments were conducted in accordance with the guidelines of the DFCI Institutional Animal Care and Use Committee. MCF7 cells alone or mixed with HS5 cells were suspended in medium containing 50% matrigel and injected subcutaneously in the flank of NU(NCr)- Foxn1nu mice implanted with estradiol pellets (5×106 cells of each type per injection). When the average tumor diameter reached approximately 1cm, mice were separated into treatment vs. control groups as indicated and treated with either tamoxifen (3mg/kg in corn oil, biweekly s.c. injections) or vehicle. Tumor growth and response to treatment was quantified by in vivo bioluminescence measurements. Statistical analyses (GraphPad Prism 8.2.0) compared all time-matched mean bioluminescence values between treatment vs. vehicle cohorts via two-way analysis of variance (ANOVA) with correction for multiple comparisons (Sidak test, default option of the statistical package).
Sample collection and gene expression analysis:
The cells in 2-D cultures were collected by scraping in cold cell recovery solution, pelleted by centrifugation and stored at −80°C. The 3-D gels were cultured for 8–10 days after seeding to allow for spheroid formation, followed by mechanical disruption using cold PBS and centrifugation to collect the cells. The BrCa cells were separated from BMSCs as described above (BrCa cells from monocultures were also subjected to the same protocol, as controls), counted and stored at −80°C. The RNA was isolated using Qiagen RNA isolation kit (experimental triplicates). The RNA isolated from collagen gels was analyzed by hybridization in Affymetrix Prime View 3’ microarrays at the Molecular Biology Core Facility (Dana-Farber Cancer Institute) and further evaluated for differential gene expression analysis for microarray data of each cell line in 2D and 3D organoid models using the bioconductor LIMMA packageA. The samples isolated from matrigel cultures were supplemented with ERCC RNA spike-in mix to adjust for the cell number and analyzed by RNA sequencing using Illumina NextSeq 500 Next Gen at the Molecular Biology Core Facility (Dana-Farber Cancer Institute), followed by differential gene expression analysis for the RNA-seq data using the bioconductor DEseq2 package. The data have been deposited in the Gene Expression Omnibus database (GEO accession number GSE152312).
Gene set enrichment analysis (GSEA):
GSEA analysis was performed using the GSEA software version 3.0 (gsea-3.0.jar) based on the preranked option. For each differential gene expression analysis, we used the per gene fold-change for gene ranking. For the analysis of microarray data sets, if a gene was represented by multiple probes, we used the fold-change of the probe with the smallest nominal p-value. For visualizations, we used the GSEA enrichment scores (ES) and the gene set size normalized enrichment scores (NES). For genes with transcripts upregulated (log2FC>2) in MCF7 spheroids cocultured in matrigel with BMSCs (vs. monoculture) and with sgRNA significantly (p<0.05) depleted in CRISPR-Cas9 screen in the presence of 4-OHT, we queried the C6 Oncogenic Signatures (representing 189 gene sets deregulated in cancer) of the Molecular Signatures Database (MSigDB https://www.gsea-msigdb.org/gsea/msigdb/annotate.jsp; last accessed on Feb. 12th 2020).
Correlation of BMSC-induced transcriptional signature of MCF-7 cells with patient outcome:
To assess the translational relevance of our 3D co-culture models, we used the transcriptional profiles of MCF7 spheroids co-cultured with BMSCs in matrigel or collagen gels (vs. respective monocultures). We initially established a consensus molecular signature comprised of genes that were commonly upregulated (log2FC >1) or commonly downregulated (log2FC < −1) in both matrigel and collagen co-cultures (53 and 27 genes respectively). These differentially expressed genes were initially used to stratify the n=1445 ER+ patients in the METABRIC dataset (last accessed on May 28th 2019) and examine potential differences in clinical outcome between the patients with high vs low BMSC-induced MCF7 molecular signature. Next, the correlation of MCF7 spheroid gene profiling data with BrCa patient outcome was also confirmed using the online meta-analysis tool KM-plotter (which aggregates data from several clinical trials (32); http://kmplot.com/analysis/) for ER+ BrCa patients. To assess the effects of genes of interest in KM-plotter (last accessed on March 2nd, 2020), we used the mean expression of the upregulated or the downregulated genes in co-cultures to analyze data from n = 355 patients with ER+ cancer that received systemic endocrine therapy; quality control and other tool options were used at default setting; and compared the relapse-free survival of patients of the upper tertile vs. lower two tertiles of expression of the respective gene signatures. For the METABRIC data, we compared the disease-specific survival of patients in the upper tertile vs. lower two tertiles of expression of the respective gene signatures using log-rank (Mantel-Cox) test (GraphPad Prism 8.2.0). Similar results were obtained in KM-plotter and METABRIC databases by analyses of additional stroma-induced signatures from the Matrigel or collagen co-cultures; and/or with use of additional cutoff points to stratify patients with low vs. high expression of these respective signatures.
Results
HR blockade abrogates the acquisition of anchorage independence by HR+ cancer cells.
To assess how HR signaling affects anchorage-independent cancer cell growth, we cultured ER+ BrCa cells in ECM, where tumor cells grow as 3D spheroids by evading anoikis, a form of apoptosis in matrix-detached epithelial cells that enables lumen formation (24,33). Each spheroid was formed by one initial cell, with the outer cellular layer attached to the ECM and the centrally-localized cells growing in matrix-detached conditions. Exposure of MCF7 3D spheroids to antiestrogens prevented anchorage-independent growth, leading to lumen clearance and acinar differentiation (Fig. 1A). Antiestrogens also induced apoptosis in 3D (but not 2D) cultures (Fig. 1B), corroborating the morphological observations. Antiestrogen treatment had only modest effect on the peripheral ECM-attached BrCa cells, suggesting that cell adhesion mediated signaling attenuates the effect of antiestrogens in BrCa cells. After antiestrogen washout, the peripheral cells resumed lumen filling, indicating that the malignant potential is retained in the ECM niche during treatment (Fig. 1A). The survival of anchorage-independent cancer cells depends on suppression of redox stress by oncogenic signals (33). To measure the redox stress within BrCa spheroids we used two-photon excited fluorescence metabolic imaging (31). Antiestrogens significantly increased redox stress in the interior of 3D spheroids within 24h (Fig. 1C and D), suggesting that ER contributes in redox homeostasis during the acquisition of anchorage independence by cancer cells. Indeed, N-Acetyl Cysteine (NAC), a ROS-neutralizing antioxidant that can prevent anoikis in the center of acini (33), could rescue the MCF7 cells treated with fulvestrant in 3D cultures, but had negligible effect in 2D cultures (Fig. 1E). These observations suggest that ER signaling is necessary for lumen filling by ER+ BrCa cells, and that HT induces anoikis through selective cytotoxic effect on matrix-detached tumor cells.
Figure 1. Anchorage-independent growth of BrCa cells is abrogated by antiestrogens and rescued by BMSCs during drug exposure.

a) MCF7 3D monocultures treated with DMSO, fulvestrant (300nM; 7 days) and 7 days after fulvestrant washout; H&E, magn x400. b) Fulvestrant-treated MCF7 2D and 3D cultures (Annexin V-based apoptosis assay; bioluminescence readout). c) Two-photon excited fluorescence imaging of fulvestrant (24h; 300nM)- vs. DMSO-treated MCF7 spheroids. d) Redox ratio slope from the peripheral cellular layers towards the center of MCF7 spheroids and degree of mitochondrial clustering, in monocultures or cocultures with BMSCs (fulvestrant 300nM 24hrs). e) Effect of N-Acetyl Cysteine on fulvestrant sensitivity in MCF7 2D and 3D cultures; 6 days. f) MCF7 spheroids in monocultures or cocultures with BMSCs, +/− fulvestrant (300nM; 7 days); H&E, magn. x200.
Interaction with non-malignant accessory cells of the metastatic microenvironment restores anchorage-independent growth of cancer cells treated with hormone antagonists:
Bone metastases are frequent in HR+ BrCa and PrCa. To model these lesions, we cocultured BrCa cells and non-malignant cells of the bone microenvironment in 3D conditions in the presence vs. absence of antiestrogens. We adapted our previously developed compartment-specific cell bioluminescence imaging (CS-BLI) approach (29) which involves a bioluminescence-based viability method to selectively quantify the cancer cell viability in the presence vs. absence of non-malignant cells from the bone milieu (e.g. osteoblasts and bone marrow stromal cells [BMSCs]). Some of these non-malignant cells (e.g. HS5 BMSCs) mitigated the antiestrogen-mediated suppression of BrCa growth in 3D cultures (Suppl. Fig. 1A). Notably, BMSCs abrogated the effect of antiestrogen treatment on inducing redox stress of anchorage-independent BrCa cells (Fig. 1D) and acinar-like differentiation of cancer spheroids (Fig. 1F). CS-BLI assays confirmed that BMSCs attenuated the effect of antiestrogens on BrCa spheroid growth in matrigel (Fig. 2A), concordant with the morphological observations about BMSC-induced rescue of ECM-detached cancer cells in the presence of antiestrogens (Fig. 1F). To account for any putative Matrigel-borne growth factors potentially affecting antiestrogen responses, we also confirmed these results in separate, collagen-based, 3D cocultures (Suppl. Fig. 1B). To further assess the translational relevance of this antiestrogen resistance model, we compared the antiestrogen responses of BrCa 3D spheroids cocultured with primary BMSCs isolated from the bone metastatic lesion of a cancer patient vs. from the bone marrow of a normal donor. Notably, the metastasis-derived, but not donor-derived, BMSCs induced antiestrogen resistance in BrCa spheroids (Fig. 2B and Suppl. Fig. 1C); consistent with the notion that non-malignant accessory cells within the 3D architecture of the bone metastatic environment can confer HT resistance to BrCa cells. Furthermore, similar co-cultures of LNCaP cells with BMSCs abrogated the inhibitory effect of antiandrogens on PrCa spheroid growth (Fig. 2C), supporting the generalizability of this microenvironmentally-induced HT resistance phenotype in hormone-dependent cancers.
Figure 2. The response of HR+ cancer 3D spheroid cultures to hormone antagonists is attenuated by BMSCs.

a-d) Antiestrogen response of BrCa (a, b) or PrCa (c) cell lines, or MCF7 spheroids stably expressing mutant ER (d) in 3D monocultures vs. cocultures with HS5 or primary BMSCs cells from bone metastasis or normal donor; 8 days assays.
We examined whether HR mutations associated with HT resistance in in vitro models and in patients (13,27) can supersede the microenvironmentally-induced endocrine resistance phenotype in 3D cultures. We thus quantified the antiestrogen sensitivity of MCF7 3D spheroids harboring the ER mutation Y537S, in the presence vs. absence of BMSCs. The Y537S ER mutation induced antiestrogen resistance in MCF7 3D spheroid monocultures, confirming previous results in 2D cultures (27). However, the antiestrogen response of Y537S MCF7 spheroids was even further attenuated in 3D cocultures with BMSCs (Fig. 2D and Suppl. Fig. 1D), suggesting that nonmalignant cells of the bone metastatic microenvironment can enhance HT resistance in BrCa, even in the presence of ER mutations.
Coculturing BrCa cells with non-malignant “accessory” cells from other frequently-colonized organs (e.g. brain and liver) also tended to attenuate the inhibitory effect of antiestrogens on BrCa spheroid growth (Suppl. Fig. 1, E and F), suggesting that microenvironmentally-induced HT resistance mechanisms may operate in other metastatic sites beyond the bone, the focus of the present study.
To validate our findings in vivo we implanted immuno-compromised mice with MCF7 cells alone (monotypic xenografts) or together with BMSCs (heterotypic xenografts). Engraftment was higher for heterotypic xenografts (Suppl. Fig. 2A). Tamoxifen treatment delayed tumor growth in monotypic xenografts, but not their heterotypic counterparts without affecting ER expression (Suppl. Fig. 2A, B, C), consistent with further the in vitro findings about BMSC-induced antiestrogen resistance in BrCa spheroids.
Distinct effects of BMSCs on the ER expression in BrCa cells:
Coculture with BMSCs induced in BrCa spheroids (compared to their monoculture counterparts) transcriptional changes that were highly concordant across cell lines (Fig. 3A). The transcriptomes of BrCa spheroids in cocultures reflected decreased estrogenic signaling (Fig. 3B and C and Suppl. Fig 3A, B and C). Because attenuation of HR expression can be associated with hormone independence and HT resistance, we examined the ER gene expression in BrCa 3D cultures. BMSCs induced downregulation (but not complete abrogation) of ER transcript levels in BrCa spheroids (Fig. 3D). Next, we examined the ER protein expression in BrCa 3D monocultures and cocultures (Fig. 3E and F and Suppl. Fig. 3D). Immunohistochemistry (IHC) confirmed the antiestrogen-induced acinar differentiation in MCF7, T47D and ZR75–1 3D spheroid monocultures. The ECM-attached BrCa monolayer cells in all monoculture models retained ER protein expression during the antiestrogen-induced acinar differentiation. Additionally, MCF7 and T47D spheroids in coculture with BMSCs were similar in size and number to the respective monocultures and retained ER protein expression (Fig. 3E and Suppl. Fig. 3D). Congruously, ER protein was expressed in vivo in heterotypic xenografts (Suppl. Fig. 2B), and has also been shown to remain expressed in the bone metastases of at least a proportion of BrCa patients (34) (example shown in Suppl. Fig. 2C). In contrast to MCF7 and T47D cocultures, ER protein was lost in ZR75–1 spheroids cocultured with BMSCs (Fig. 3F). Furthermore, the overall number of ZR75–1 spheroids in cocultures was smaller and their overall growth was delayed (compared to the respective monoculture). Because ER transcriptional changes were similar across the three BrCa coculture models (Fig. 3D), we examined whether protein degradation via the ubiquitin-proteasome system contributes to the distinct ER protein levels in 3D cocultures of ZR75–1 vs. cocultures of MCF7 or T47D cells. Exposure of BrCa 3D monocultures and cocultures to the proteasome inhibitor MG132 confirmed that BMSCs-conditioned medium induced proteasome-depended degradation of ER in ZR75–1 (but not in MCF7) 3D cultures (Suppl. Fig. 3E).
Figure 3. BMSCs attenuate estrogen-induced gene expression in BrCa spheroids independently of preservation or loss of ER expression.

a) Transcriptional changes in BrCa 3D spheroids in cocultures (vs. respective monocultures; genes with significant changes in all cell lines are shown). b-c) Estrogen-responsive gene set in in MCF7 (b) and ZR75–1 (c) spheroids cocultured with BMSCs (vs. respective monocultures). d) ER transcript levels in BrCa spheroid cultures +/− BMSCs. e-f) ER immunostaining of MCF7 (e) or ZR75–1 (f) spheroid cultures +/− BMScs cells, +/− 4-OHT (300nM; 7 days), x200. (8 day cultures in panels a-d).
To assess how the bone microenvironment may affect the HR expression in PrCa, we also examined LNCaP spheroids in 3D monocultures vs. coculture with BMSCs. IHC indicated that coculture with BMSCs induced loss of AR protein expression in LNCaP cells (Suppl. Fig. 3F) and tended to decrease the number and size of the formed spheroids; a pattern similar to ZR75–1 cocultures. Interestingly, the AR-negative LNCaP spheroids formed in coculture with BMSCs also expressed higher levels of neuroendocrine markers (Suppl. Fig. 3G).
BMSC-secreted IL6 induces hormone-independent growth in BrCa and PrCa 3D spheroids via activation of JAK/STAT pathway.
Prior secretome profiling of various stromal cell types, including immortalized HS5 BMSCs (35) (reanalyzed in Suppl. Fig. 4A) identified a multitude of cytokines, including high levels of IL-6, a ligand known to activate several intracellular signaling cascades including JAK/STAT, PI3K and MAPK/ERK. We observed strong enrichment for IL6/JAK/STAT3 pathway target genes in the examined BrCa spheroids cocultured with BMSCs (Fig. 4A). We hypothesized that BMSC-secreted IL-6 induces activation of alternative pathways and estrogen-independent growth in BrCa spheroids. Indeed, exposing ZR75–1 3D monocultures to recombinant IL-6 induced proteasome-depended degradation of ER (Suppl. Fig. 3E) and slower ZR75–1 spheroid growth (similar to cocultures with BMSCs) and attenuation of antiestrogen response (Suppl. Fig. 4B), phenocopying the respective coculture of ZR75–1 with BMSC. Concordantly, antibodies against IL-6 itself or IL-6 receptor, or the JAK/STAT pathway inhibitor ruxolitinib, restored ER expression (Fig. 4B) and attenuated the BMSC-induced antiestrogen resistance (Fig. 4C) in ZR75–1 cocultures with BMSCs. Similarly, blocking IL6/JAK/STAT signaling also restored antiandrogen sensitivity in the LNCaP 3D cocultures with BMSCs (Suppl. Fig. 4C), suggesting that similar IL-6-dependent microenvironmental signaling mechanism may operate in the bone metastatic microenvironment to induce androgen independence in PrCa.
Figure 4. BMSCs induce antiestrogen resistance in BrCa spheroids via paracrine IL-6 or in IL-6-independent manner.
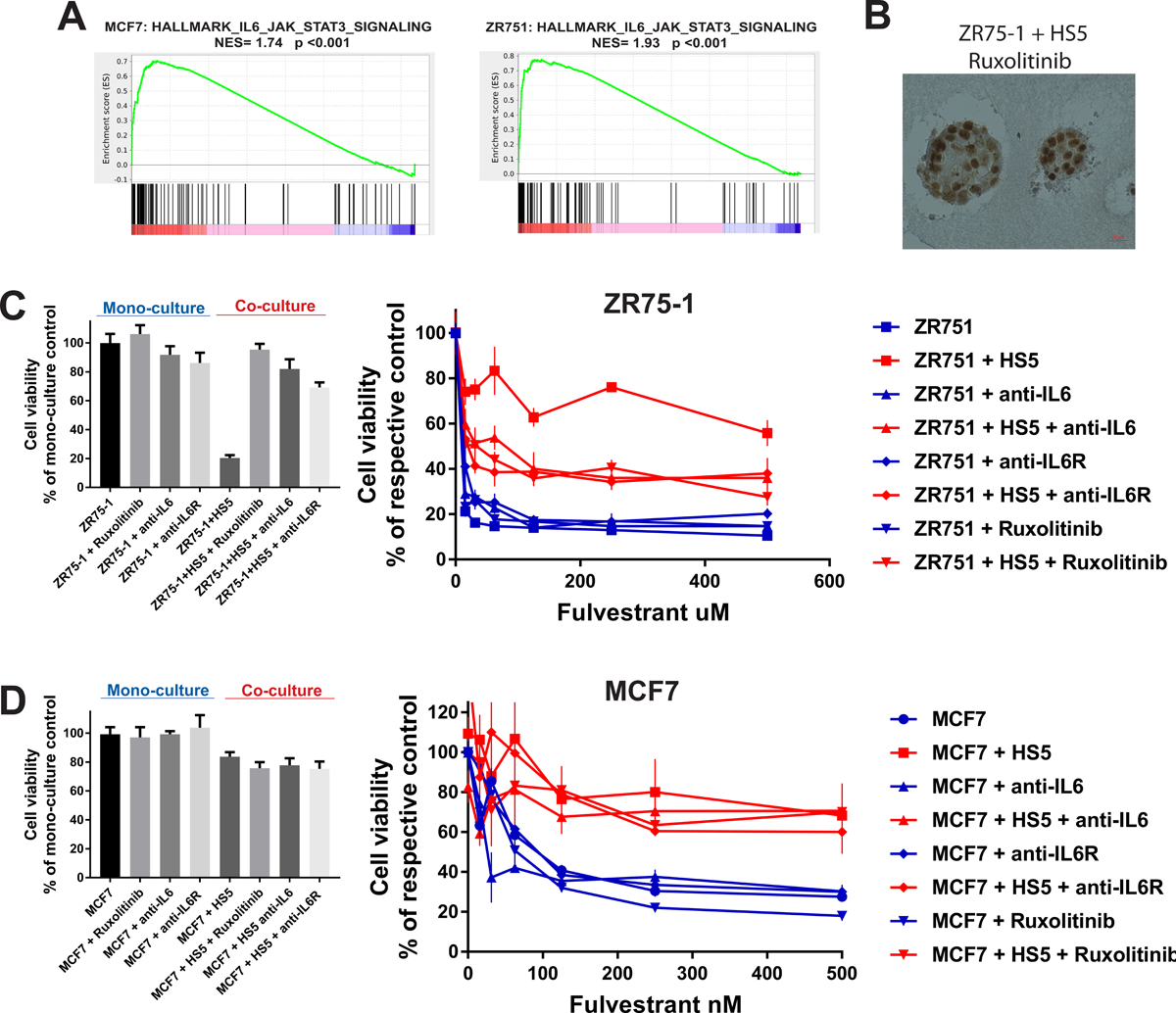
a) Enrichment of IL6-JAK-STAT3 target genes in MCF7 and ZR75–1 3D cocultures with BMSCs (vs. respective monocultures). b) ER immunostaining in ZR75–1 cocultures treated with ruxolitinib (100nM) (respective control shown in Fig. 3F). c-d) CS-BLI assays for growth and fulvestrant sensitivity of ZR75–1 (c) or MCF7 (d) cells, in 3D monoculture or coculture with BMSCs +/− anti-IL6 or anti-IL6R antibodies (1 μg/ml) or ruxolitinib (100nM). (8 day cultures in a-d)
BMSCs induce IL-6-independent hormone therapy resistance in BrCa 3D spheroids via alternate activation of ERK and PI3K/Akt signaling cascades.
In contrast to the results with ZR75–1, recombinant IL-6 had only limited effect in MCF7 and T47D 3D spheroid growth (Suppl. Fig. 4B). Inhibition of IL6/JAK/STAT signaling pathway did not have substantial impact on antiestrogen sensitivity in MCF7 and T47D spheroid cocultures with BMSCs (Fig. 4D and Suppl. Fig. 4D). Therefore, BMSC-induced HT resistance in MCF7/T47D spheroids may be driven by distinct, IL-6-independent, paracrine signals.
To assess the translational relevance of these putative IL-6-independent microenvironmental models, we examined transcriptional signatures derived from MCF7 spheroids cultured in the presence of BMSCs (see Methods). In the METABRIC study, ER+ BrCa patients with shorter disease-specific survival had in their pre-treatment tumors low levels of transcripts downregulated in MCF7 spheroids in coculture (Fig. 5A, left) or, conversely, high levels of transcripts upregulated in MCF7 spheroids in coculture (Fig. 5A, right). This association with clinical outcome was also confirmed in additional publicly-available molecular profiling data, aggregated from several clinical trials, of ER+ BrCa tumors (32) (Fig. 5B), but not ER-negative BrCa patients (Suppl. Fig. 5). Concordantly, genes upregulated in MCF7 spheroids cocultured with BMSCs were significantly enriched for genes upregulated in tamoxifen-resistant BrCa patient-derived xenografts (36) (Fig. 5C).
Figure 5. BMSC-induced molecular signature in MCF7 spheroids is associated with antiestrogen resistance and activation of growth factor signaling.
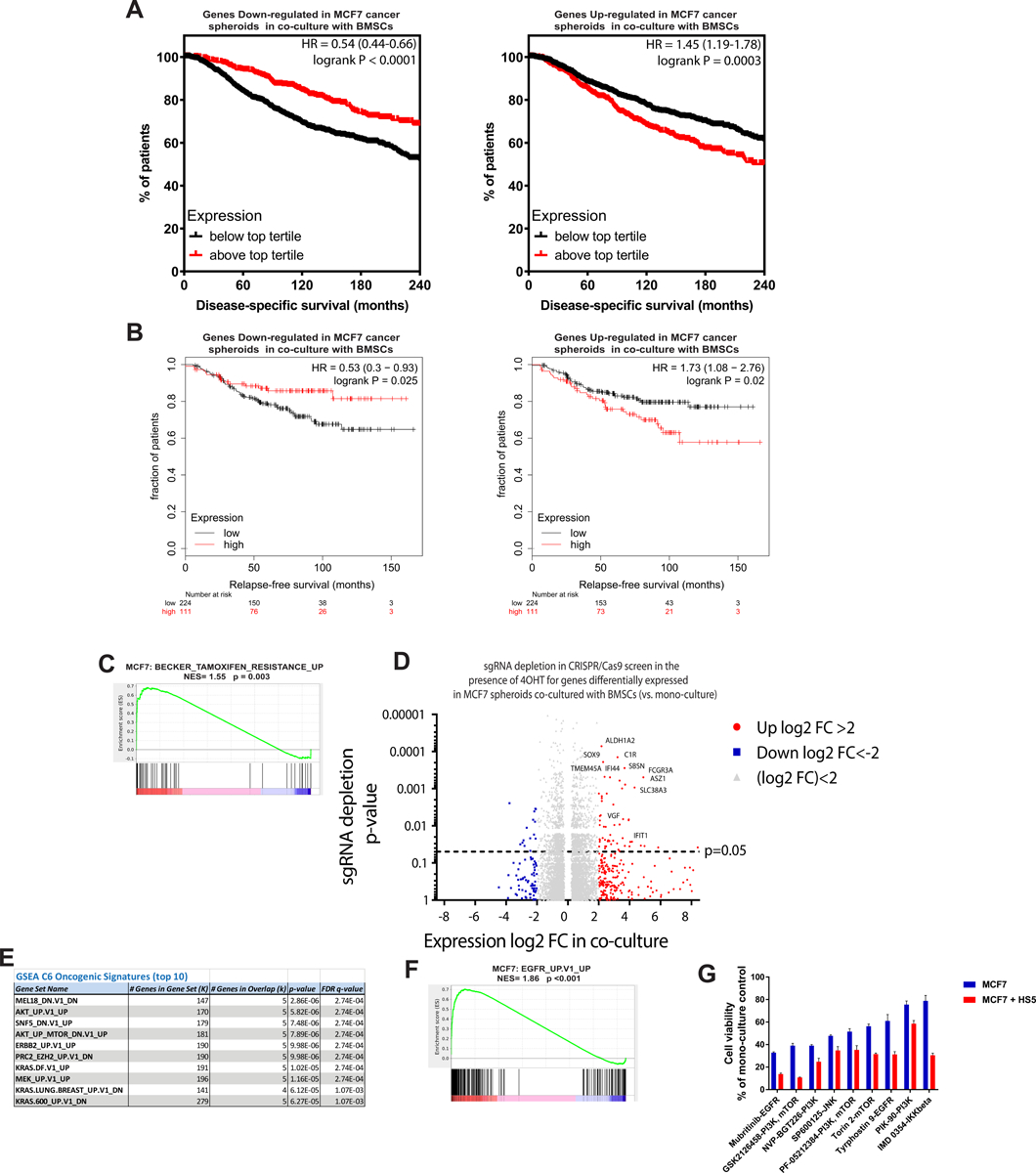
a-b) Genes upregulated or downregulated in MCF7 spheroids cocultured with HS5 (vs. monoculture; 8 days) have higher or lower, respectively, levels in ER+ BrCa tumors of patients with (a) shorter disease-specific survival (METABRIC study) and (b) shorter relapse-free survival (aggregate data from several clinical studies; KM-plotter meta-analysis (32); also see Methods). c) Genes associated with antiestrogen resistance are upregulated in MCF7 spheroids cocultured with BMSCs (vs. monoculture). d) Depletion of sgRNAs (p-values) in genome-scale CRISPR screen in MCF7 cells (+/− 4-OHT, 300nM) for those genes with differential expression (FDR<0.05, log2-fold change in x-axis) in MCF7 3D spheroids cocultured with BMSCs (vs. monocultures); see Methods. e) Top 10 oncogenic signature genesets enriched for the genes upregulated by log2FC>2 in MCF7 spheroid cocultures (vs. monoculture) and sgRNA-depleted (p<0.05) in CRISPR screen of 4-OHT (300nM)-treated MCF7 cells. f) Enrichment of genes associated with growth factor signaling in MCF7 spheroid cocultures (vs. monoculture). g) Examples of increased sensitivity of MCF7 spheroids cocultures (10 days) with BMSCs to selected kinase inhibitors; 100nM, 72h (Suppl. Fig. 6 depicts the complete phenotypic screen).
To identify functional mediators of antiestrogen-resistance in 3D cocultures with BMSC, we conducted a genome-scale CRISPR-Cas9 screen of MCF7 cells in 2D monocultures in the presence vs. absence of 4-hydroxytamoxifen (4-OHT); and compared the patterns of sgRNA changes for genes differentially expressed in MCF7 spheroids cocultured with BMSCs (Fig. 5D). Genes upregulated in cocultures with BMSCs tended to have their respective sgRNAs depleted in the CRISPR/Cas9 screen in the presence of 4-OHT (Wilcoxon signed rank rest for depletion rank p<0.0001), including genes previously associated with tamoxifen resistance in MCF7 cells (e.g. SOX9 (37)). These functional data support the notion that BMSCs induce in BrCa spheroids genes which, individually or in concert, contribute to decreased antiestrogen sensitivity. Gene set enrichment analysis for the collection of genes that were upregulated in the 3D cocultures and were also associated with 4-OHT resistance in the genome-scale CRISPR/Cas9 screen, indicated an overall transcriptional signature consistent with stimulation of MAPK and PI3K/Akt pathways or upstream growth factor receptors in MCF7 cells (Fig. 5E and F).
Given the complex secretome of BMSCs (Suppl. Fig. 4A) and the redundancy of BMSC-induced upregulation of genes and signaling pathways associated with reduced sensitivity to antiestrogens in cancer cells (Fig. 5D, E, and F), we postulated that the antiestrogen resistance in 3D cocultures could be mediated by pleiotropic effects of BMSCs on the MCF7 spheroids. The results of our genome-scale CRISPR/Cas9 screen of MCF7 cells in the presence of antiestrogens suggest that single gene by gene approaches may not be powerful enough to reveal the therapeutic vulnerabilities in the setting of a redundant network. To identify therapeutically-targetable pathways driving MCF7 spheroid anchorage-independent growth in coculture with BMSCs we turned to the alternative approach of functional phenotypic screening, and examined the sensitivity of BrCa spheroids in 3D monocultures and cocultures to a set of tool chemical compounds including 416 kinase inhibitors targeting collectively ~300 kinases (Fig. 5G and Suppl. Fig. 6A). We identified increased sensitivity of MCF7 + BMSCs 3D cocultures (vs. the respective monocultures) to several inhibitors of ERK and PI3K/MTOR signaling pathway components (Fig. 5G and Suppl. Fig. 6B), but not to other drug classes (Suppl. Fig. 6C). Overall, these observations are concordant with the molecular data in the 3D cocultures of MCF7 with BMSCs, indicating that acquisition of hormone independence by anchorage-independent BrCa cells in cocultures is mediated, at least in part, through activation of ERK/PI3K pathways; a finding concordant with previous studies in cell-autonomous models (1).
Translational relevance of the BMSC-induced resistance to hormonal therapy.
To further explore the translational relevance of our in vitro 3D models of microenvironmentally-induced HT resistance, we studied tumor organoids established from bone metastases of three patients with antiandrogen-refractory PrCa, namely MSK-PCa1, MSK-PCa2 and MSK-PCa3; characterized in prior studies (38). We observed in 3D monocultures that MSK-PCa1 organoids were AR-negative and refractory to antiandrogens. MSK-PCa3 organoids were AR-positive, but insensitive to antiandrogen treatment, suggesting a cell-autonomous antiandrogen resistance model attributable to putative activation of alternative signaling pathways. Interestingly, MSK-PCa2 organoids expressed AR and were sensitive to antiandrogen treatment, indicating that the clinical refractoriness may be due to microenvironmental mechanisms. To test this hypothesis, we compared the characteristics of MSK-PCa2 cells in 3D monoculture vs. coculture with BMSCs. In cocultures, the MSK-PCa2 organoids acquired distinct morphology marked by diffuse cells invading the ECM instead of spheroids. Congruously, BMSCs attenuated the sensitivity of MSK-PCa2 cells to enzalutamide (Fig. 6). Similarly to studies with ZR75–1 and LNCaP spheroids described above, exposing cocultures to ruxolitinib or neutralizing antibodies against IL-6 and IL-6R restored antiandrogen sensitivity in MSK-PCa2 organoids (Fig. 6). Overall, these observations support the translational relevance of the 3D coculture models as a system to simulate preclinically the context of HT resistance of BrCa and PrCa bone metastatic lesions.
Figure 6. BMSC-secreted IL6 induces antiandrogen resistance in patient-derived PrCa organoids.

Growth and enzalutamide response of MSK-PCa2 3D cultures +/− BMSCs, +/− anti-IL6 or anti-IL6R antibodies (1 μg/ml) or ruxolitinib (100nM); 15 days, CS-BLI assay.
Discussion
The preclinical efforts to dissect BrCa/PrCa HT resistance have so far focused mostly on cell-autonomous mechanisms modeled in conventional 2D cultures (1,3). Clinical studies have also reflected this preclinical emphasis, by focusing mostly on characterizing the molecular features of tumor cells (typically from the primary tumor) which eventually are associated with development of more rapid endocrine resistance and metastasis. Moreover, studies to identify genomic drivers of metastatic BrCa (39) or PrCa (40) do not address whether cell non-autonomous signals can also operate as parallel or alternate resistance mechanisms. To study the potential contribution of cell non-autonomous mechanisms to endocrine resistance, we examined the responses of BrC and PrCa cells to HT in 3D culture conditions (where clonal expansion of each initial cell forms a distinct spheroid) in the presence vs. absence of non-malignant accessory cells of the microenvironment of key metastatic sites. We reasoned that it is biologically plausible for BMSCs to play such a role in the bone milieu, a main site for metastases of HR+ BrCa and PrCa, because extensive studies in hematologic neoplasias have documented that BMSCs can confer to them cell non-autonomous resistance against diverse pharmacological and immune-based therapies (29,41,42). While our study focuses on modeling the interactions between tumor cells and BMSCs in the 3D microenvironment, other cell populations in the bone marrow (e.g. osteoblasts, immune cells etc.) may also function as “accessories” to the tumor cells in distinct contexts. Thus, microenvironment-mediated endocrine resistance may be determined by the aggregate effects of these different nonmalignant cell populations. Adapting our 3D co-culture systems to include these additional tumor-adjacent cell types will improve the biological relevance of these microenvironmental models for BrCa and PrCa metastases.
Here, we observed that cell non-autonomous mechanisms emerging within the local metastatic microenvironment can indeed profoundly affect the response of HR+ tumor cells to HR-targeting agents. We identified two cellular phenotypes that occur within the 3D tumor architecture and are distinct with regard to tumor cell sensitivity to HT. Hormone antagonists had limited effect on the viability of BrCa/PrCa cells residing in the ECM-attached niche. In contrast, antiestrogens/antiandrogens abrogated the ability of BrCa/PrCa cancer cells to grow in anchorage-independent conditions and induced acinar-like differentiation in 3D cultures. Notably, cocultures with BMSCs rescued the anchorage independence of BrCa/PrCa cells despite their exposure to HT.
Normal epithelial cells require attachment to the basement membrane for survival (24,28,33). The activation of oncogenic signals enable cancer cells to evade anoikis and survive outside their normal ECM niches, a key step towards transformation of glandular epithelial cells (25). For instance, HER2 overexpression in normal mammary acini rescues the metabolic defects caused by matrix detachment through stabilization of EGFR/PI3K activation, resulting in lumen filling (33). Our results indicate that in hormone-dependent cancers, HR is directly involved in the evasion of anoikis, while HR blockade compromises redox homeostasis of ECM-detached HR+ cells and their ability for anchorage-independent growth (e.g. Fig. 1A–E). The ability of ECM-attached HR+ cancer cells to survive and grow in the presence of HR-targeting agents indicates that cell adhesion signals are also important in HT resistance, suggesting that alternative signaling axes (e.g., integrin/FAK/SRC kinase) could be targeted to overcome HT resistance. Maintenance of lumen clearance and acinar morphology in BrCa spheroids was dependent on continuation of antiestrogen treatment. After antiestrogen washout the peripheral monolayer cells repopulated the lumen (Fig. 1A), indicating that the ECM niche can preserve a reservoir of latent HR+ cancer cells that can re-initiate tumor growth if HT is interrupted. These results are congruent with the clinical observation that prolonging adjuvant HT treatment beyond 5 years has survival benefit for patients with hormone-dependent BrCa (43).
The suppression of anchorage-independent growth of BrCa/PrCa cells by HT was dramatically attenuated in 3D cocultures with BMSCs. Interestingly, although the global transcriptional signatures induced in BrCa spheroids by BMSCs (e.g. suppression of estrogenic signals and activation of IL6/JAK/STAT3 pathway) were similar for the different BrCa cell lines examined (e.g. Fig. 3A–C), the mechanisms of antiestrogen resistance were distinct, namely IL-6-mediated vs. IL-6-independent. High serum IL-6 concentrations are associated with poor prognosis in metastatic ER+ BrCa patients (44,45). In an in vivo BrCa model of acquired adaptive HT resistance, ER blockade led to increased IL-6 cytokine levels and ER-independent, IL6/Notch3-driven, cancer stem cell growth via a feed-forward loop (46). Distinctly from the findings of this latter study, IL-6-mediated antiestrogen resistance was not associated with concomitant IL-6-stimulated cell growth in our microenvironmental HT resistance model of ZR75–1 3D spheroids (Suppl. Fig. 4B); indicating that other putative changes occurring during prolonged antiestrogen exposure in cell-autonomous models of acquired HT resistance may exert additional effects on function of IL-6/Notch signaling which are not pertinent to our 3D coculture models of de novo resistance. Concordantly, in both these IL-6-dependent models of acquired (46) and de novo (in our study) HT resistance, blocking IL-6 signaling using IL6-targetting antibodies (and also confirmed with JAK inhibitors, in our study) restored HR expression and HT sensitivity.
ERK and PI3K/Akt signaling cascades are crucial for survival of transformed cancer cells during displacement from the normal ECM niche and anchorage-independent growth (33). The PI3K/Akt and ERK pathways can be indirectly activated by HR (1,3), which could explain the dependence of BrCa/PrCa cells on HR signaling for anoikis evasion in our 3D monocultures. These growth factor signaling cascades seem to be further activated through putative paracrine factors in cocultures with BMSCs (e.g. Fig. 5E). We hypothesize that BMSC-induced paracrine activation of ERK/PI3K/Akt axes in ECM-detached MCF7 cells bypasses their dependence on ER signaling for anchorage-independent growth; therefore providing an alternative mechanism of HT resistance in the 3D metastatic microenvironment. Interestingly, although MCF7 spheroids have functional IL-6 receptor and BMSC-secreted IL-6 can potentially activate the ERK and PI3K/Akt pathways in cancer cells, HT resistance in this coculture setting was not abrogated by IL-6 inhibition, indicating either dependency on alternate BMSC-secreted cytokine/s or redundancy of paracrine extracellular signals in this context. Conversely, although IL6/JAK/STAT3 pathway was upregulated in MCF7 spheroid cocultures with BMSCs, antiestrogen resistance was not associated with ER expression loss (distinct from ZR75–1 cocultures) and was not reversed by JAK inhibition, indicating that alternate IL-6-independent mechanisms confer endocrine resistance to MCF7 spheroids.
Although extensive preclinical studies have used a multitude of informative, but mostly cell-autonomous, models to identify actionable mechanisms of HT resistance in BrCa and PrCa, a comprehensive panel of clinical biomarkers is needed for the efficient deployment of therapeutic agents targeting these mechanisms in patients. Our study shows that the use of metastatic biopsies to identify and/or validate biomarkers related to tumor-stroma interactions in the metastatic lesion can help to distinguish between the various forms of microenvironmentally-induced HT resistance (which may operate in the absence of, or concurrently with, relevant genetic lesions) and to refine the treatment strategy. For instance, HT-refractory metastatic tumors that retain expression of HR and have increased ERK/PI3K/Akt activity though paracrine microenvironmental signal could be sensitive to kinase inhibitors targeting EGFR/HER2 and/or PI3K/Akt/MTOR. Conversely, loss of HR expression along with high local IL-6 levels could indicate IL-6-induced hormone independence; in this case the use of agents targeting IL-6 or JAK should perhaps be administered concomitantly with HT, because targeting IL6/JAK/STAT alone may not be effective and could also fuel HR-dependent tumor growth.
The metastatic microenvironment is complex in structure and heterogeneous in cellular and paracrine elements. The recapitulation of these microenvironmental features in vitro, using 3D coculture systems, projects a complex landscape of various, possibly co-existing, forms of HT resistance in BrCa and PrCa. Further development of these preclinical models of metastasis, ideally using patient-derived tumor and stromal cells, could help the identification of therapeutic targets to overcome HT resistance and of biomarkers to stratify patients likely to benefit from these therapies.
Supplementary Material
Statement of significance.
This study uncovers a previously underappreciated dependency of tumor cells on HR signaling for anchorage-independent growth and highlights how the metastatic microenvironment restores this malignant property of cancer cells during hormone therapy.
Acknowledgements
We thank Zach Herbert and the members of DFCI Molecular Biology Core Facilities for the transcriptional analyses. This study was supported by the Breast Cancer Alliance Young Investigator Award (E.D., C.S.M), Claudia Adams Barr Program for Innovative Cancer Research (E.D., C.S.M.), Hellenic Women’s Club (C.S.M., E.D.), Terri Brodeur Breast Cancer Foundation Grant (E.D.), Avon Foundation Breast Cancer Research Program (C.S.M., E.D.), Elsa U. Pardee Foundation Grant (C.S.M., E.D.), Department of Defense grant W81XWH-15-1-0012 (A.C., C.S.M.) and National Cancer Institute Grant U54-CA233223 (N.M.). Raw CRISPR and RNA-seq sequencing data was processed on the Orchestra High Performance Compute Cluster at Harvard Medical School (grant NCRR 1S10RR028832-01, http://rc.hms.harvard.edu).
Conflict of interest:
The authors declare no potential conflict of interest relevant to the content of this specific manuscript. Outside the direct scope of this work, C.S.M. discloses employment of a relative with Takeda; consultant/honoraria from Fate Therapeutics, Ionis Pharmaceuticals, FIMECS; and research funding from Janssen/Johnson & Johnson, TEVA, EMD Serono, Abbvie, Karyopharm, Sanofi, Arch Oncology and Novartis.
References
- 1.Osborne CK, Schiff R. Mechanisms of endocrine resistance in breast cancer. Annual review of medicine 2011;62:233–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Clarke R, Tyson JJ, Dixon JM. Endocrine resistance in breast cancer--An overview and update. Molecular and cellular endocrinology 2015;418Pt 3:220–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Crawford ED, Schellhammer PF, McLeod DG, Moul JW, Higano CS, Shore N, et al. Androgen Receptor Targeted Treatments of Prostate Cancer: 35 Years of Progress with Antiandrogens. The Journal of urology 2018;200:956–66 [DOI] [PubMed] [Google Scholar]
- 4.Schiff R, Massarweh S, Shou J, Osborne CK. Breast cancer endocrine resistance: how growth factor signaling and estrogen receptor coregulators modulate response. Clinical cancer research : an official journal of the American Association for Cancer Research 2003;9:447s–54s [PubMed] [Google Scholar]
- 5.Lavinsky RM, Jepsen K, Heinzel T, Torchia J, Mullen TM, Schiff R, et al. Diverse signaling pathways modulate nuclear receptor recruitment of N-CoR and SMRT complexes. Proceedings of the National Academy of Sciences of the United States of America 1998;95:2920–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Linja MJ, Savinainen KJ, Saramaki OR, Tammela TL, Vessella RL, Visakorpi T. Amplification and overexpression of androgen receptor gene in hormone-refractory prostate cancer. Cancer research 2001;61:3550–5 [PubMed] [Google Scholar]
- 7.Takeda DY, Spisak S, Seo JH, Bell C, O’Connor E, Korthauer K, et al. A Somatically Acquired Enhancer of the Androgen Receptor Is a Noncoding Driver in Advanced Prostate Cancer. Cell 2018;174:422–32.e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coarfa C, Fiskus W, Eedunuri VK, Rajapakshe K, Foley C, Chew SA, et al. Comprehensive proteomic profiling identifies the androgen receptor axis and other signaling pathways as targets of microRNAs suppressed in metastatic prostate cancer. Oncogene 2016;35:2345–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fletcher CE, Sulpice E, Combe S, Shibakawa A, Leach DA, Hamilton MP, et al. Androgen receptor-modulatory microRNAs provide insight into therapy resistance and therapeutic targets in advanced prostate cancer. Oncogene 2019;38:5700–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Paschalis A, Sharp A, Welti JC, Neeb A, Raj GV, Luo J, et al. Alternative splicing in prostate cancer. Nature reviews Clinical oncology 2018;15:663–75 [DOI] [PubMed] [Google Scholar]
- 11.Gregory CW, Fei X, Ponguta LA, He B, Bill HM, French FS, et al. Epidermal growth factor increases coactivation of the androgen receptor in recurrent prostate cancer. The Journal of biological chemistry 2004;279:7119–30 [DOI] [PubMed] [Google Scholar]
- 12.Culig Z, Hobisch A, Cronauer MV, Radmayr C, Trapman J, Hittmair A, et al. Androgen receptor activation in prostatic tumor cell lines by insulin-like growth factor-I, keratinocyte growth factor, and epidermal growth factor. Cancer research 1994;54:5474–8 [PubMed] [Google Scholar]
- 13.Jeselsohn R, Bergholz JS, Pun M, Cornwell M, Liu W, Nardone A, et al. Allele-Specific Chromatin Recruitment and Therapeutic Vulnerabilities of ESR1 Activating Mutations. Cancer cell 2018;33:173–86.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tilley WD, Buchanan G, Hickey TE, Bentel JM. Mutations in the androgen receptor gene are associated with progression of human prostate cancer to androgen independence. Clinical cancer research : an official journal of the American Association for Cancer Research 1996;2:277–85 [PubMed] [Google Scholar]
- 15.Dowsett M, Nicholson RI, Pietras RJ. Biological characteristics of the pure antiestrogen fulvestrant: overcoming endocrine resistance. Breast cancer research and treatment 2005;93Suppl 1:S11–8 [DOI] [PubMed] [Google Scholar]
- 16.Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science (New York, NY) 2009;324:787–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miller TW, Rexer BN, Garrett JT, Arteaga CL. Mutations in the phosphatidylinositol 3-kinase pathway: role in tumor progression and therapeutic implications in breast cancer. Breast cancer research : BCR 2011;13:224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gutierrez MC, Detre S, Johnston S, Mohsin SK, Shou J, Allred DC, et al. Molecular changes in tamoxifen-resistant breast cancer: relationship between estrogen receptor, HER-2, and p38 mitogen-activated protein kinase. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2005;23:2469–76 [DOI] [PubMed] [Google Scholar]
- 19.Shoman N, Klassen S, McFadden A, Bickis MG, Torlakovic E, Chibbar R. Reduced PTEN expression predicts relapse in patients with breast carcinoma treated by tamoxifen. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc 2005;18:250–9 [DOI] [PubMed] [Google Scholar]
- 20.Carver BS, Chapinski C, Wongvipat J, Hieronymus H, Chen Y, Chandarlapaty S, et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer cell 2011;19:575–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yardley DA, Noguchi S, Pritchard KI, Burris HA 3rd, Baselga J, Gnant M, et al. Everolimus plus exemestane in postmenopausal patients with HR(+) breast cancer: BOLERO-2 final progression-free survival analysis. Advances in therapy 2013;30:870–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Toren P, Kim S, Cordonnier T, Crafter C, Davies BR, Fazli L, et al. Combination AZD5363 with Enzalutamide Significantly Delays Enzalutamide-resistant Prostate Cancer in Preclinical Models. European urology 2015;67:986–90 [DOI] [PubMed] [Google Scholar]
- 23.Baker SJ, Reddy EP. CDK4: A Key Player in the Cell Cycle, Development, and Cancer. Genes & cancer 2012;3:658–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Debnath J, Mills KR, Collins NL, Reginato MJ, Muthuswamy SK, Brugge JS. The role of apoptosis in creating and maintaining luminal space within normal and oncogene-expressing mammary acini. Cell 2002;111:29–40 [DOI] [PubMed] [Google Scholar]
- 25.Frisch SM, Screaton RA. Anoikis mechanisms. Current opinion in cell biology 2001;13:555–62 [DOI] [PubMed] [Google Scholar]
- 26.Vidi PA, Bissell MJ, Lelievre SA. Three-dimensional culture of human breast epithelial cells: the how and the why. Methods in molecular biology (Clifton, NJ) 2013;945:193–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jeselsohn R, Yelensky R, Buchwalter G, Frampton G, Meric-Bernstam F, Gonzalez-Angulo AM, et al. Emergence of constitutively active estrogen receptor-alpha mutations in pretreated advanced estrogen receptor-positive breast cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 2014;20:1757–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dhimolea E, Maffini MV, Soto AM, Sonnenschein C. The role of collagen reorganization on mammary epithelial morphogenesis in a 3D culture model. Biomaterials 2010;31:3622–30 [DOI] [PubMed] [Google Scholar]
- 29.McMillin DW, Delmore J, Weisberg E, Negri JM, Geer DC, Klippel S, et al. Tumor cell-specific bioluminescence platform to identify stroma-induced changes to anticancer drug activity. Nature medicine 2010;16:483–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science (New York, NY) 2014;343:84–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Quinn KP, Bellas E, Fourligas N, Lee K, Kaplan DL, Georgakoudi I. Characterization of metabolic changes associated with the functional development of 3D engineered tissues by non-invasive, dynamic measurement of individual cell redox ratios. Biomaterials 2012;33:5341–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nagy A, Lanczky A, Menyhart O, Gyorffy B. Validation of miRNA prognostic power in hepatocellular carcinoma using expression data of independent datasets. Scientific reports 2018;8:9227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schafer ZT, Grassian AR, Song L, Jiang Z, Gerhart-Hines Z, Irie HY, et al. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature 2009;461:109–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Idirisinghe PK, Thike AA, Cheok PY, Tse GM, Lui PC, Fook-Chong S, et al. Hormone receptor and c-ERBB2 status in distant metastatic and locally recurrent breast cancer. Pathologic correlations and clinical significance. American journal of clinical pathology 2010;133:416–29 [DOI] [PubMed] [Google Scholar]
- 35.Straussman R, Morikawa T, Shee K, Barzily-Rokni M, Qian ZR, Du J, et al. Tumour microenvironment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 2012;487:500–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Becker M, Sommer A, Kratzschmar JR, Seidel H, Pohlenz HD, Fichtner I. Distinct gene expression patterns in a tamoxifen-sensitive human mammary carcinoma xenograft and its tamoxifen-resistant subline MaCa 3366/TAM. Molecular cancer therapeutics 2005;4:151–68 [PubMed] [Google Scholar]
- 37.Jeselsohn R, Cornwell M, Pun M, Buchwalter G, Nguyen M, Bango C, et al. Embryonic transcription factor SOX9 drives breast cancer endocrine resistance. Proceedings of the National Academy of Sciences of the United States of America 2017;114:E4482–E91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gao D, Vela I, Sboner A, Iaquinta PJ, Karthaus WR, Gopalan A, et al. Organoid cultures derived from patients with advanced prostate cancer. Cell 2014;159:176–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Razavi P, Chang MT, Xu G, Bandlamudi C, Ross DS, Vasan N, et al. The Genomic Landscape of Endocrine-Resistant Advanced Breast Cancers. Cancer cell 2018;34:427–38.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Abida W, Cyrta J, Heller G, Prandi D, Armenia J, Coleman I, et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proceedings of the National Academy of Sciences of the United States of America 2019;116:11428–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McMillin DW, Delmore J, Negri JM, Vanneman M, Koyama S, Schlossman RL, et al. Compartment-Specific Bioluminescence Imaging platform for the high-throughput evaluation of antitumor immune function. Blood 2012;119:e131–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McMillin DW, Negri JM, Mitsiades CS. The role of tumour-stromal interactions in modifying drug response: challenges and opportunities. Nature reviews Drug discovery 2013;12:217–28 [DOI] [PubMed] [Google Scholar]
- 43.Davies C, Pan H, Godwin J, Gray R, Arriagada R, Raina V, et al. Long-term effects of continuing adjuvant tamoxifen to 10 years versus stopping at 5 years after diagnosis of oestrogen receptor-positive breast cancer: ATLAS, a randomised trial. Lancet (London, England) 2013;381:805–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bachelot T, Ray-Coquard I, Menetrier-Caux C, Rastkha M, Duc A, Blay JY. Prognostic value of serum levels of interleukin 6 and of serum and plasma levels of vascular endothelial growth factor in hormone-refractory metastatic breast cancer patients. British journal of cancer 2003;88:1721–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Salgado R, Junius S, Benoy I, Van Dam P, Vermeulen P, Van Marck E, et al. Circulating interleukin-6 predicts survival in patients with metastatic breast cancer. International journal of cancer 2003;103:642–6 [DOI] [PubMed] [Google Scholar]
- 46.Sansone P, Ceccarelli C, Berishaj M, Chang Q, Rajasekhar VK, Perna F, et al. Self-renewal of CD133(hi) cells by IL6/Notch3 signalling regulates endocrine resistance in metastatic breast cancer. Nature communications 2016;7:10442. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.