

Abstract
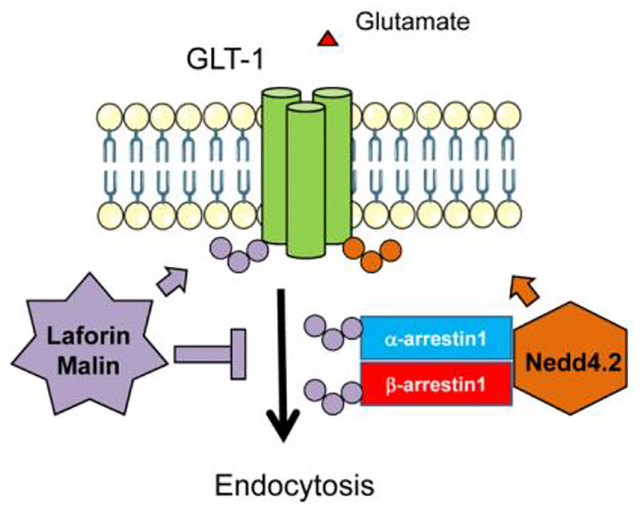
Lafora disease (LD) is a fatal rare type of progressive myoclonus epilepsy that appears during early adolescence. It is caused by mutations in EPM2A or EPM2B genes, which encode laforin, a glucan phosphatase, and malin, an E3-ubiquitin ligase, respectively. Although the exact roles of laforin and malin are still not well understood, it is known that they work as a complex in which laforin recruits targets that will be ubiquitinated by malin. Recently, we suggested that the type of epilepsy that accompanies LD could be due to deficiencies in the function of the astrocytic glutamate transporter GLT-1. We described that astrocytes from LD mouse models presented decreased levels of GLT-1 at the plasma membrane, leading to increased levels of glutamate in the brain parenchyma. In this work, we present evidence indicating that in the absence of a functional laforin/malin complex (LD cellular models) there is an alteration in the ubiquitination of GLT-1, which could be the cause of the reduction in the levels of GLT-1 at the plasma membrane. On the contrary, overexpression of the laforin/malin complex promotes the retention of GLT-1 at the plasma membrane. This retention may be due to the direct ubiquitination of GLT-1 and/or to an opposite effect of this complex on the dynamics of the Nedd4.2-mediated endocytosis of the transporter. This work, therefore, presents new pieces of evidence on the regulation of GLT-1 by the laforin/malin complex, highlighting its value as a therapeutic target for the amelioration of the type of epilepsy that accompanies LD.
Keywords: Lafora disease, GLT-1, ubiquitination, endocytosis, glutamate, Nedd4.2, arrestins
Graphical abstract
INTRODUCTION
Lafora disease (LD; OMIM #254780) is a fatal rare type of progressive myoclonus epilepsy that appears during late childhood or early adolescence. Although conventional anti-epileptic drugs are used as initial treatment, seizures soon become refractory and the death of the patient occurs ten years after the appearance of the first symptoms. The disease is caused by mutations in EPM2A [(Minassian et al., 1998), (Serratosa et al., 1999)] or EPM2B (Chan et al., 2003) genes, which encode laforin, a glucan phosphatase, and malin, an E3-ubiquitin ligase, respectively. Patients with mutations in either laforin or malin are phenotypically indistinguishable (Gómez-Abad et al., 2005). Although the exact roles of laforin and malin are still not well understood, it is known that they work as a complex in which laforin recruits the targets that will be ubiquitinated by malin. The laforin-malin complex has been described to modulate different cellular processes such as glycogen synthesis, autophagy, and oxidative stress [reviewed in (Garcia-Gimeno, Knecht, & Sanz, 2018)]. LD is characterized by the accumulation of insoluble, poorly branched polyglucosan inclusions, known as Lafora bodies, in the brain and peripheral tissues. Since its description in 1911 the disease was thought to be caused by an accumulation of Lafora bodies in neurons, but recent reports from different laboratories, including ours, have shown that most of them are found in astrocytic processes [(Rubio-Villena et al., 2018), (Auge et al., 2018)], suggesting that astrocytes may play an important role in the disease. This idea is reinforced by the fact that in brains from Epm2a−/− and Epm2b−/− mice there is an accumulation of reactive astrocytes and pro-inflammatory mediators that appears as early as three months of age [(Lopez-Gonzalez, Viana, Sanz, & Ferrer, 2017), (Lahuerta et al., 2020)], the moment at which the pathophysiological phenotype initiates. As the mice get older (16 months of age) there is an exacerbated neuroinflammatory response (Lahuerta et al., 2020), which could lead to neurodegeneration and epilepsy (Vezzani, Balosso, & Ravizza, 2019).
This reactive astrogliosis can cause the loss of the functions carried out normally by the astrocytes (Liddelow & Sofroniew, 2019), suggesting that these cells may take an active part in the disease mechanisms. One of the functions performed normally by astrocytes is the clearance of the glutamate from the synaptic cleft [(Danbolt, 2001), (Robinson, 2006)], carried out by the glutamate transporter-1, GLT-1, in mice (excitatory amino acid transporter 2, EAAT2, in humans), which is responsible for the removal of up to 90% of the glutamate. Glutamate is the main excitatory neurotransmitter in the brain and, therefore, its levels have to be tightly regulated to avoid hyperexcitation of the neurons, as this may cause excitotoxicity and neuronal cell death. A decrease in GLT-1 levels is observed in several neurodegenerative diseases, such as amyotrophic lateral sclerosis (ALS) (Rothstein, Van Kammen, Levey, Martin, & Kuncl, 1995), Parkinson’s disease (Zhang et al., 2017), Rasmussen’s encephalitis (He et al., 2020), and Alzheimer’s disease, among others [reviewed in (Pajarillo, Rizor, Lee, Aschner, & Lee, 2019)]. In addition, a decrease in GLT-1 content by chronic administration of antisense oligonucleotides has shown to produce excitotoxicity and neurodegeneration (Rothstein et al., 1996), and mice deficient in GLT-1 are more susceptible to the generation of seizures, dying soon after birth due to uncontrolled seizures (Tanaka et al., 1997). Moreover, lethal spontaneous seizures occur when GLT-1 is specifically deleted from astrocytes (Petr et al., 2015).
Along with this idea, we have recently shown that there are decreased levels of the glutamate transporter GLT-1 at the plasma membrane of the astrocytes from LD mouse models. We hypothesized that this could be one of the causes of the type of epilepsy present in this disorder (Munoz-Ballester, Berthier, Viana, & Sanz, 2016) since we reported that this reduction observed in LD primary astrocytic cultures led to a decreased competence of these astrocytes in the uptake of glutamate, and correlated with an accumulation of glutamate in vivo in the brain parenchyma [(Munoz-Ballester et al., 2016), (Munoz-Ballester et al., 2019)]. Interestingly, LD animal models did not show a decrease in the total amount of GLT-1 protein or mRNA (Munoz-Ballester et al., 2019), but a decreased localization of the transporter at the plasma membrane of astrocytes (Munoz-Ballester et al., 2016). This is important to be highlighted because GLT-1 needs to be located at the plasma membrane to perform its function, being the high density of the transporter at this location what determines the efficiency of glutamate uptake [(Murphy-Royal et al., 2015), (Murphy-Royal, Dupuis, Groc, & Oliet, 2017)]. In fact, trafficking of GLT-1 to and from the plasma membrane constitutes a mechanism that controls its activity (Robinson, 2006), being this a highly regulated process. Both constitutive and regulated endocytosis, the latter dependent on protein kinase C (PKC) activation, and subsequent degradation of the transporter depends on the ubiquitination of its C-terminal lysine residues [(Sheldon, Gonzalez, Krizman-Genda, Susarla, & Robinson, 2008), (Gonzalez-Gonzalez, Garcia-Tardon, Gimenez, & Zafra, 2008), (Martinez-Villarreal, Garcia Tardon, Ibanez, Gimenez, & Zafra, 2012)]. It has been shown that the E3-ubiquitin ligase Nedd4.2 is responsible for GLT-1 ubiquitination, and also for the modification of the human orthologue EAAT2 [(Boehmer et al., 2006), (Garcia-Tardon et al., 2012)]. Once inside the cell, ubiquitinated GLT-1 can be targeted to lysosomes for degradation, or it can return to the plasma membrane, being this process dependent on the deubiquitination of GLT-1 by UCH-L1 deubiquitinase (Martinez-Villarreal et al., 2012). Therefore, an ubiquitination/deubiquitination cycle is responsible for maintaining the pool of transporter at the plasma membrane (Rauen, 2016).
Since the laforin/malin complex works as an E3-ubiquitin ligase, and we have found an alteration in the homeostasis of the GLT-1 transporter in LD mice, we hypothesize that the complex could be participating in the correct localization of this transporter at the plasma membrane. In this work, we wanted to get a deep insight into the mechanism of action of the laforin/malin complex on the dynamics of GLT-1. We present evidence indicating that in the absence of a functional laforin/malin complex (as in LD cellular models) there is an alteration in the ubiquitination of GLT-1, which could be the cause of the reduction in the levels of the transporter at the plasma membrane. On the contrary, we show that the laforin/malin complex promotes the retention of GLT-1 at the plasma membrane, leading to a reduction in its rate of endocytosis. This is due to a direct ubiquitination of GLT-1 by laforin and malin and/or to changes in the dynamics of its Nedd4.2-mediated endocytosis, which is assisted by specific adaptors (α- and β-arrestins). In fact, we demonstrate that the laforin/malin complex interacts and ubiquitinates these adaptors. Therefore, this work presents new pieces of evidence on the regulation of GLT-1 by the laforin/malin complex, highlighting its value as a therapeutic target for the amelioration of the type of epilepsy that accompanies Lafora disease.
MATERIALS AND METHODS
Mammalian Cell culture
Human embryonic kidney (HEK293) (HPA Culture Collection #851820602), COS-7 (Public Heath England #87021302), and human osteosarcoma U2OS cells (Public health England #92022711) were used for transfection experiments. Human LD fibroblasts were obtained from a patient bearing a mutation in the laforin gene (R241X/R241X) (a gift from Dr. José M. Serratosa, Fundación Jiménez Díaz, Madrid, Spain) with the corresponding informed consent for experimentation. As in the family of the LD patient, there were no unaffected siblings of the same age, control fibroblasts matched by sex and age were obtained from Coriell Institute for Medical Research (Camden, New Jersey, USA). Experiments were carried out at passages number 6 to 9.
Cells were grown in Dulbecco’s modified medium (Lonza, Barcelona, Spain), supplemented with 10% inactivated fetal bovine serum (FBS) (Invitrogen, Madrid, Spain), 1% L-glutamine, 100 units/ml penicillin and 100 μg/ml streptomycin. All cell types were grown in a humidified atmosphere at 37°C and 5% (v/v) CO2 except otherwise indicated.
Mouse primary astrocytes
Mouse primary astrocytes from Epm2b−/− and Epm2b+/+ mice were obtained from P0-P1 mice cortices. Briefly, cortices including the hippocampus were dissected, the meninges were removed and the tissue was homogenized using the Neural Tissue Dissociation kit and the GentleMACS dissociator from Mylteny Biotec (Madrid, Spain). Once obtained, microglia contamination was removed using CD11b Microbeads in a magnetic field (Mylteny Biotec, Madrid, Spain). The purity of the collected astrocytes was assessed by flow cytometry using anti-ACSA2 antibodies (Mylteny Biotec, Madrid, Spain) and was always higher than 95%. For transfecting astrocytes, we used Lipofectamine 2000 (ThermoFisher, Madrid, Spain), following the manufactureŕs instructions.
Flow cytometry analyses
When indicated, cells were treated before the staining with 100 μM Dynasore (D7693, Sigma, Madrid, Spain) during 1 hour to block endocytosis. All the staining was performed at 4°C to avoid the internalization of membrane proteins. Briefly, cells were trypsinized, washed twice with cold phosphate buffer saline (PBS), and incubated with FcR Blocking Reagent (MACS Miltenyi Biotec). Then cells were stained using mouse anti-GLT-1 (sc-365634, Santa Cruz Biotech, Madrid, Spain) at 4°C for 30 min. Cells were washed again and incubated with a phycoerythrin-conjugated anti-mouse antibody for 30 min. Cells were washed again and fixed with 4% paraformaldehyde (PFA) in PBS for 20 min. Fluorescence was monitored in a BD FACSCanto flow cytometer (BD Biosciences, Madrid, Spain) and analyzed using FlowJo software (BD Biosciences, Madrid, Spain).
Plasmid constructs
Plasmids coding for Flag-tagged or HA-tagged forms of laforin and malin and pEGFP-malin have been previously described (Sanchez-Martin, Lahuerta, Viana, Knecht, & Sanz, 2020). Plasmid pEGFP-malin P69A is described in (Couarch et al., 2011). Plasmid pCDNA3-EAAT2 was a generous gift from Dr. Peter Dodd (School of Chemistry and Molecular Biosciences, University of Queensland, Queensland, Australia). Plasmid pCMV-HA-Nedd4.2 (containing isoform2 of Nedd4.2) was from Dr. Lynne Yenush (Instituto de Biología Molecular y Celular de Plantas, CSIC, Valencia, Spain). Plasmids expressing HA-tagged GLT-1 WT and 7KR mutant, the NG1 fusion protein, containing the extracellular and the transmembrane domains of p75 fused to the amino-terminus of GLT-1, and plasmids ARRB1-GFP (expressing β-arrestin1-GFP) and ARRB2-GFP (expressing β-arrestin2-GFP) were kindly provided by Dr. Francisco Zafra (CMSO-CSIC, Madrid) (Martinez-Villarreal et al., 2012). Plasmids ARRDC1-GFP (expressing α-arrestin1-GFP) and plasmid VSVG-ts045-GFP were from Addgene (#38320 and #11912, respectively); the latter plasmid encodes a thermosensitive version of the viral glycoprotein ts045 of the vesicular stomatitis virus fused to GFP (Presley et al., 1997). Plasmid pCMV-6xHis-Ubiquitin was kindly provided by Dr. Manuel Rodríguez (Proteomics Unit, CIC-BioGUNE, Vizcaya, Spain).
VSVG-ts045-GFP transport
Control and LD fibroblasts were transfected with the VSVG-ts045-GFP expressing plasmid and 24 hours later, cells were reseeded on coverslips and were grown for another 24 hours. Cells were shifted to 40°C overnight to accumulate the VSVG-ts045-GFP protein in the ER. During the last hour of incubation, cells were treated with 350 μM cycloheximide to prevent de novo protein synthesis. Then, cells were moved to 32°C for different times as indicated. Cells were fixed with 4% paraformaldehyde in PBS for 20 min, nuclei counterstained with DAPI, and coverslips mounted for microscopy to monitor the movement of the protein throughout the different compartments of the secretory pathway. Images were acquired with a Leica TCS SP8 confocal microscope (Leica, Wetzlar, Germany) and processed using FIJI software (NIH, Bethesda, MD). Between 75 and 100 cells were analyzed in each condition.
RNA interference
For RNA interference we used the ON-Target Pool siRNAs from Dharmacon targeting Nedd4.2 (L-007187-00; Dharmacon, Lafayette, Colorado), and the ON-TargetPlus control pool as a negative control (D-001810-10-05). We transfected a concentration of 50 nM of each siRNA into HEK293 cells, using Lipofectamine RNAiMax (Invitrogen, Waltham, Massachusetts). The efficiency of Nedd4.2 knockdown was measured by Western blot 72 hours after transfection.
Antibody feeding assay
The antibody feeding assay has been carried out using the chimeric protein NG1, corresponding to the extracellular and transmembrane domains of p75 fused to the amino-terminus of GLT-1 (Martinez-Villarreal et al., 2012). Briefly, COS-7 cells were transfected with NG1 in the presence or absence of laforin and malin. One day after transfection, cells were collected and reseeded on glass coverslips. After 24h, surface staining was performed at 4°C using an anti-p75 antibody (1:500; AB-N01; Advanced Targeting Systems, San Diego). After washing with Hank’s balanced salt solution (HBSS), cells were incubated with anti-rabbit AlexaFluor 568 (A10042, ThermoFisher Scientific, Madrid, Spain) 30 min at 4°C. After washing, cells were placed in a 37°C incubator and, at the selected times, were fixed with 4% PFA.
Immunofluorescence analyses
Cells were fixed with 4% PFA in PBS for 20 minutes. For the detection of Flag-laforin, cells were permeabilized with 0.5% Triton X-100 for 15 minutes and then blocked with 1% bovine serum albumin (BSA) + 0.05% Triton X-100 in PBS during 2 hours at room temperature. Next, coverslips were incubated with primary antibodies against Flag (F3165, Sigma, Madrid, Spain) overnight at 4°C. The next day, coverslips were washed and incubated with the secondary antibody (anti-mouse AlexaFluor 633; A-21050, ThermoFisher Scientific, Madrid, Spain) for two hours at room temperature. Coverslips were extensively washed and nuclei were counterstained with DAPI (Sigma, Madrid, Spain). Then, they were mounted using Fluoromount-G (ThermoFisher Scientific, Madrid, Spain). Images were acquired with a Leica TCS SP8 confocal microscope (Leica, Wetzlar, Germany). Five stacks of 0.5 μm were obtained. Images were processed using FIJI software (NIH, Bethesda, MD). At least 50 cells were counted for each time point in five independent experiments.
Ubiquitination experiments
HEK293 cells and primary astrocytes were transfected with the plasmids indicated in each experiment using X-treme GENE HP transfection reagent according to the manufacturer’s protocol (Roche Diagnostics, Barcelona, Spain) and Lipofectamine 2000 (ThermoFisher, Madrid, Spain), respectively. When indicated, during the last 4 hours of incubation cells were treated with inhibitors of protein degradation (20 mM NH4Cl, 100 μM leupeptin, and 10 μM MG132). After 24–48 hours, cells were lysed in 8 M guanidinium hydrochloride (to avoid the action of deubiquitinases) with or without 1% NP-40. Ubiquitinated proteins were purified by TALON metal affinity chromatography (Clontech, Madrid, Spain) as previously described (Kaiser & Tagwerker, 2005). Crude extracts (30 μg) and bound proteins were analyzed by Western blot.
GFP-trap analysis
HEK293 cells were transfected with the indicated plasmids and the GFP-trap assay was performed following the manufactureŕs instructions (GFP-Trap Agarose, Chromothek, Planegg-Martinsried, Germany). The incubation of the cells extracts with the resin was allowed to take place for 10 to 15 minutes. As a control, a plasmid coding for GFP (pEGFP-N1; #6085–1 Clontech) was used.
Western blot analyses
Cell homogenates (30 μg protein) were subjected to SDS/PAGE and transferred into a PVDF membrane. Membranes were blocked with 5% non-fat milk in Tris-buffered saline containing 0.1% Tween-20 (TBS-T) and incubated overnight at 4°C with the corresponding antibodies: guinea pig anti-GLT-1 (AB1783, Millipore); mouse anti-EAAT2 (sc-365634, Santa Cruz Biotechnologies); rabbit anti-Nedd4.2 (#4013, Cell Signaling); mouse anti-HA (H9658, Sigma); mouse anti-Flag (F3165, Sigma) and rabbit anti-GFP (210-PS-1GFP, Inmunokontackt). Mouse anti-Gapdh (sc-32233, Santa Cruz Biotechnologies) and mouse anti-tubulin (T6199, Sigma) were used as loading controls. After washing, membranes were incubated with the corresponding HRP-conjugated secondary antibodies. When indicated, anti-HA-HRP was used (H6533, Sigma). Images were obtained with a FujiLAS400 (GE Healthcare, Barcelona, Spain) using Lumilight Western Blotting Substrate (Roche Applied Science, Barcelona, Spain). Quantification of the protein bands was carried out using the software Image Studio version 5.2 (LI-COR Biosciences, Germany). Values of the ubiquinated forms were normalized to the levels of the corresponding proteins in the crude extract and referred to those found in control levels, which were set to 100.
Statistical analysis.
Results are shown as means +/− standard error of the mean (SEM) of at least three independent experiments. Differences between samples were analyzed by unpaired two-tailed Student’s t-tests using Graph Pad Prism version 5.0 statistical software (La Jolla, CA, USA). P-values have been considered as *p<0.05, **p<0.01 and ***p< 0.001.
RESULTS
Anterograde transport of GLT-1 to the plasma membrane is not affected in Lafora disease cellular models.
We have previously shown that LD mouse models have decreased levels of GLT-1 at the plasma membrane of primary astrocytes, but the same total amount of protein [(Munoz-Ballester et al., 2016), (Munoz-Ballester et al., 2019)]. The amount of transporter located at the cell surface at a particular moment depends on the balance between insertion and removal from the plasma membrane. Therefore, the decrease in the levels of GLT-1 at the cell surface previously observed in mouse models of LD could be due to a defect in one of such mechanisms. We first studied whether the insertion of the protein from the intracellular compartments to the plasma membrane was affected in cellular models of LD. Once synthesized at the endoplasmic reticulum (ER), integral plasma membrane proteins move from the ER to the Golgi, from where they are delivered to the plasma membrane. To visualize ER-to-Golgi transport, we took advantage of the thermosensitive fusion protein VSVG-ts045-GFP, which consists of GFP protein fused to the thermosensitive viral glycoprotein ts045 of the vesicular stomatitis virus (Presley et al., 1997). Being a thermosensitive protein, VSVG-ts045-GFP is retained at the ER at 40°C, and when the cells are shifted back to the permissive temperature, proper folding of the protein is allowed, enabling its exit from the ER and its movement to the Golgi.
To analyze whether ER-to-Golgi transport is affected in LD, we used human primary fibroblasts that bear the mutation R241X in the laforin gene or fibroblasts from a healthy donor, as control. Fibroblasts from a human patient were the only human cellular model we had to check the effect of a dysfunctional laforin on a particular pathway. In this way, we could assess whether the absence of a functional laforin affected the export of the protein. Cells were transfected with a plasmid expressing VSVG-ts045-GFP and incubated at 40°C to retain the fusion protein at the ER. Then, cells were treated with 350 μM cycloheximide for 1 hour to prevent de novo protein synthesis, shifted to 32°C, and the movement of the fusion protein through the secretory pathway was monitored at different times, by fixing the cells and observing the localization of the VSVG-ts045-GFP protein. As shown in Fig.1, control fibroblasts showed an ER-like distribution of the VSVG-ts045-GFP reporter when growing at 40°C (panel A), but after 30 min at 32°C, most of the cells showed the VSVG-ts045-GFP fusion in a Golgi-like localization (Fig. 1, panel C). A similar movement pattern was observed in LD fibroblasts (panels D-F). In both cases, VSVG-ts045-GFP reached the Golgi after 30 min in about 80% of the cells (Fig. 1G). These results indicated that LD fibroblasts did not have altered machinery to synthesize and export the reporter up to the Golgi.
Figure 1. Anterograde transport is not affected in Lafora disease fibroblasts.
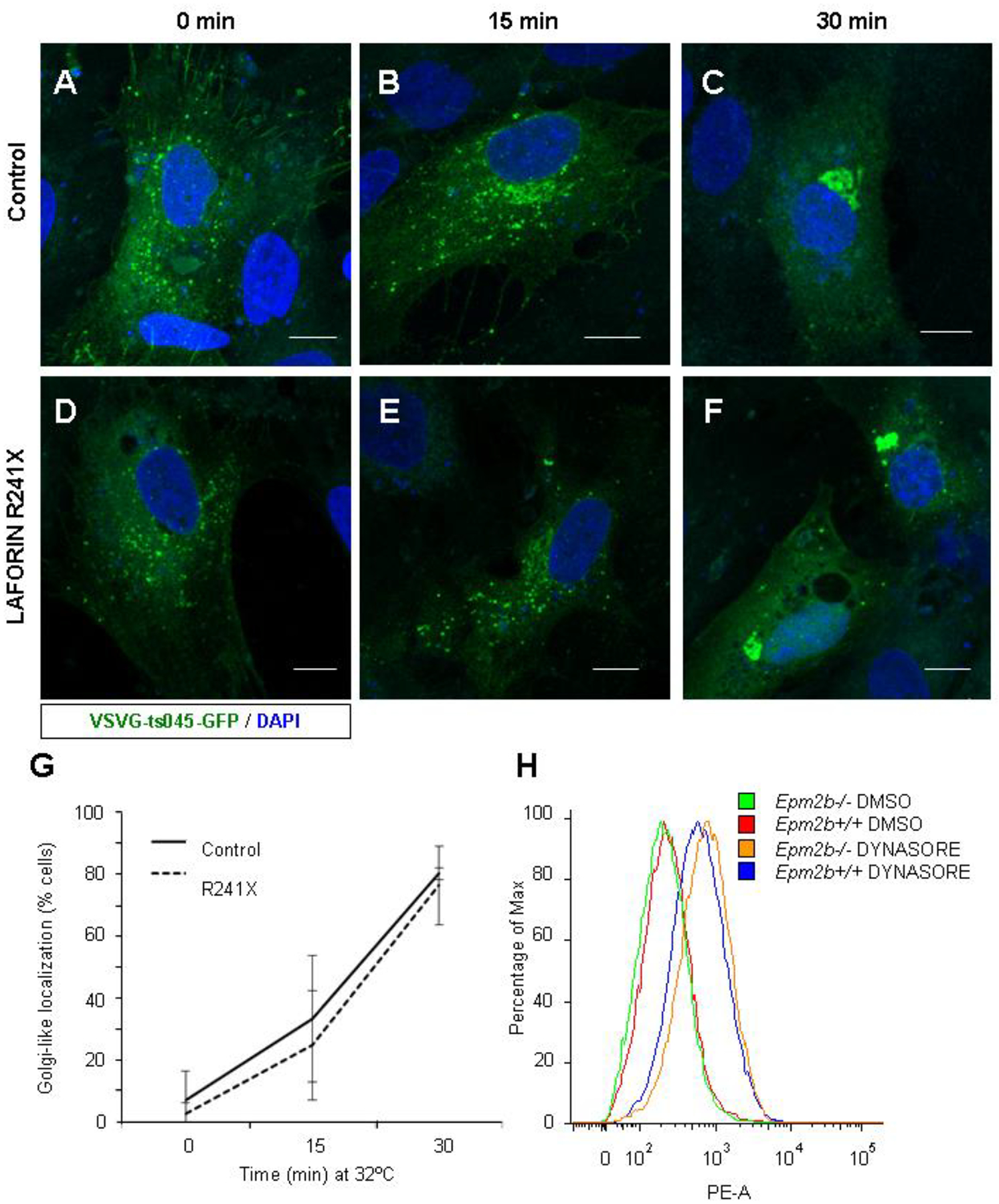
Control (A-C) or LD (D-F) fibroblasts grown on coverslips were transfected with VSVG-ts045-GFP expressing plasmid and incubated overnight at 40°C to accumulate VSVG-ts045-GFP protein in the ER. Cells were then treated with 350 μM cycloheximide for 1 hour and shifted to 32°C At the indicated times, cells were fixed and nuclei were counterstained with DAPI. Coverslips were mounted and the movement of the protein along the different compartments of the secretory pathway was monitored using a confocal microscope. Bar: 10 μm. (G) More than 75 cells from each genotype and each time point of three independent experiments were counted and the mean percentage and standard deviation of cells with a Golgi-like localization are represented. (H) Analysis of the levels of GLT-1 at the plasma membrane by flow cytometry. Primary astrocytes from control and Epm2b−/− mice were obtained as described in Materials and Methods, treated or not with 100 μM Dynasore for one hour and GLT-1 at the surface was stained with anti-GLT-1 antibodies. The histogram represents the amount of phycoerythrin (PE)-signal respect to the maximum number of cells for each sample. A representative histogram of three independent experiments is shown.
To study whether the insertion of GLT-1 to the plasma membrane was affected, we extended our studies to primary astrocytes from an LD mouse model (Epm2b−/− mice). In this way, we could analyze the dynamics of endogenous GLT-1 protein. It has been described that endocytosis of GLT-1 is mainly mediated by clathrin-dependent mechanisms (Susarla & Robinson, 2008). By blocking the endocytosis of the transporter, we can study the insertion of the protein to the plasma membrane since under these conditions any difference in the amount of transporter at the plasma membrane should be produced by differences in the insertion of the protein. With this in mind, we treated the cells with Dynasore, a dynamin inhibitor that prevents endocytosis (Macia et al., 2006), and checked the presence of endogenous GLT-1 at the plasma membrane by using an anti-GLT-1 antibody (see Methods). As shown in Fig. 1H, in both Epm2b+/+ wild type and Epm2b−/− deficient astrocytes, the blockade of endocytosis by Dynasore produced a similar relative accumulation of GLT-1 at the plasma membrane, as measured by flow cytometry (mean fluorescence intensity increases from 684 to 975 in the Epm2b+/+ astrocytes and from 513 to 997 in the Epm2b−/− astrocytes, after Dynasore treatment). Taken together, these results show no defect in the insertion of GLT-1 to the plasma membrane in LD cellular models.
Endocytosis of GLT-1 is delayed by Laforin and Malin.
Having shown that the decrease in GLT-1 concentration at the plasma membrane in LD astrocytes was not due to a delayed insertion, we hypothesized that it could be due to altered endocytosis. To analyze the effect of laforin and malin on the rate of endocytosis of the transporter, we performed an antibody-feeding assay using a chimeric GLT-1 fused to the extracellular and the transmembrane domains of the p75 receptor (Martinez-Villarreal et al., 2012). This chimeric protein is fully functional in glutamate transport, indicating that it reaches the cell surface and it is correctly folded (Martinez-Villarreal et al., 2012). The chimeric protein, named NG1, was expressed in COS-7 cells that were overexpressing or not laforin and different forms of GFP-tagged malin (wild type or the pathogenic-related form P69A). COS-7 cells were used in the antibody feeding experiments because they were large enough to allow the observation of different subcellular locations of the analyzed protein. The NG1 fusion located at the cell surface was stained in live cells at 4°C, temperature at which membrane trafficking is blocked. Next, cells were washed and shifted back to 37°C allowing the internalization of the labeled NG1 chimera. The localization of the labeled transporter was followed throughout the different endocytic compartments at different times at 37°C. As shown in Figure 2, in COS-7 cells expressing NG1 and an empty GFP plasmid, at time 0 min, NG1 was located at the plasma membrane (Fig. 2A). After 15 min at 37°C, the transporter was located in endocytic vesicles with few forms of the transporter remaining at the plasma membrane (Fig. 2B) (only 30 ± 15% of the cells showed some residual surface staining; Fig. 2J). After 30 min, the transporter was located in intracellular vesicles close to the nucleus, which may correspond to the late endosomal fraction (Fig. 2C) (only 13 ± 4% of the cells showed some residual surface staining at this time; Fig. 2J). However, in cells expressing NG1 together with laforin and malin (selected by the presence of Flag-laforin and GFP-malin derived fluorescence), there was a delay in the endocytosis of the receptor. After 15 min at 37°C, around 60% (58 ± 13%) of cells still showed NG1 at the plasma membrane (Fig. 2E and J), and after 30 min, although most of the transporter was already located in endocytic vesicles, it was dispersed throughout the cell (Fig. 2F). At this time, 23 ± 8% of the cells still showed surface staining. Quantification of the number of cells that contained NG1 in the perinuclear late endosomal fraction revealed that when a functional laforin/malin complex was present, only 22 ± 4% of the cells showed this localization compared to 57 ± 3% when there was no overexpression of the laforin/malin complex (Fig. 2K).
Figure 2. The laforin/malin complex promotes a delay in the endocytosis of GLT-1.
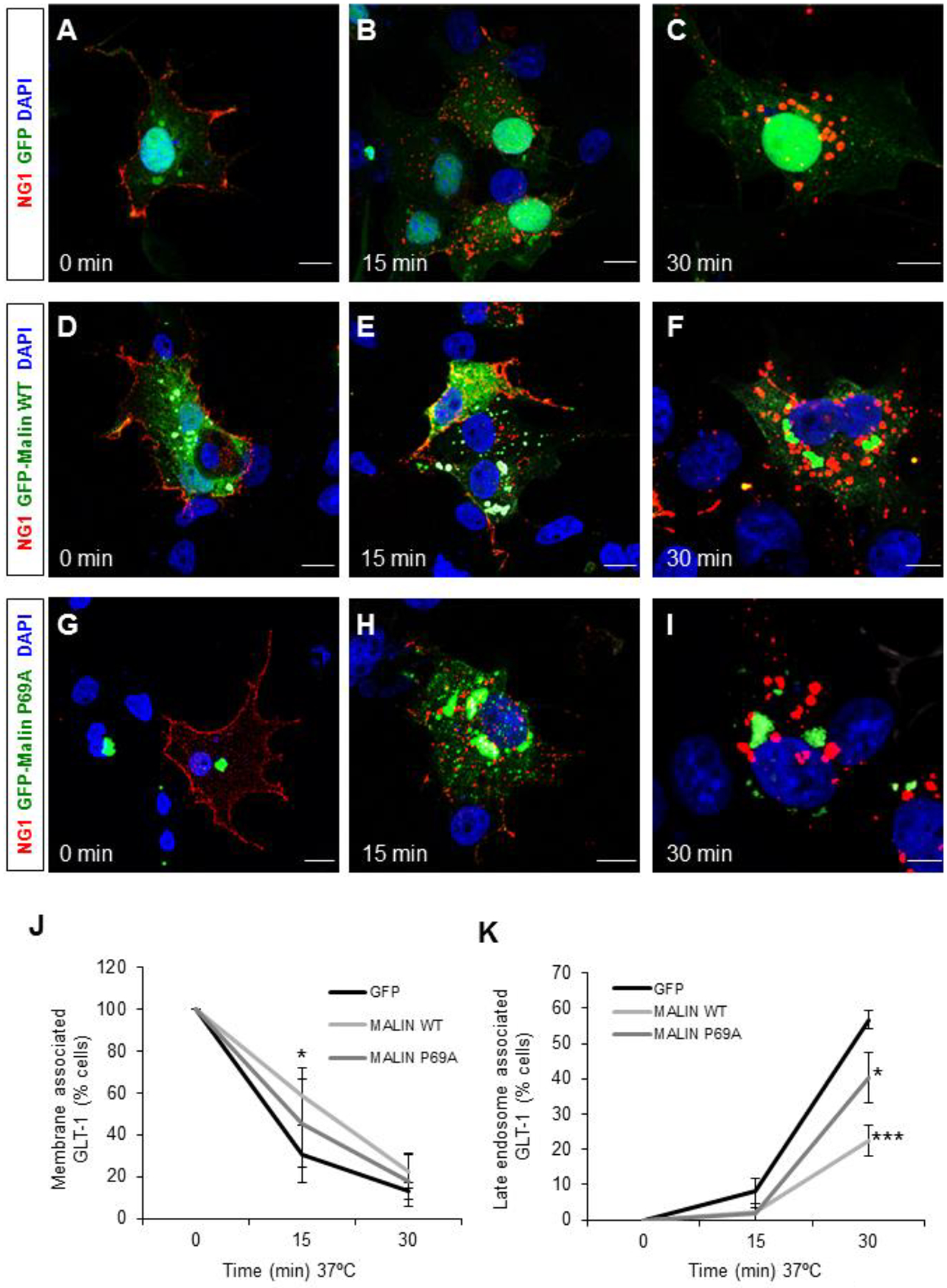
(A-I) Time course of GLT-1 endocytosis. COS-7 cells were transfected with a plasmid expressing the NG1 chimeric protein, containing the extracellular and transmembrane domains of the p75 NGF receptor and the full-length GLT-1, and GFP-empty plasmid as control (A-C), or plasmids expressing Flag-laforin and GFP-malin WT (D-F), or plasmids expressing Flag-laforin and GFP-malin P69A (G-I). Cells were labeled at 4°C with anti-p75R and AlexaFluor 568-secondary antibody, washed, and subsequently incubated at 37°C. Cells were fixed at the indicated times, and immunolabelled with anti-Flag antibodies (to visualize laforin; in white). Images were captured with a confocal microscope, and five stacks of 0.5 μm each were obtained and merged using the FIJI software. Representative images are shown. GLT-1 labeling is shown in red. GFP-empty and GFP-Malin are shown in green. DAPI stained nuclei are shown in blue. Bar: 10 μm. (J and K) Quantification of the number of cells presenting GLT-1 associated with the plasma membrane (J) or with late endosomes (K) at different times. At least 50 cells expressing at the same time Flag-laforin and GFP-malin forms were counted in each case. Results are the mean of more than 4 independent experiments. Bars indicate SEM. Statistical significance between cells expressing GFP-empty and GFP-malin forms is indicated as *p<0.05, ***p<0.001.
To verify whether the delay in the endocytosis of GLT-1 was due to the presence of a functional laforin/malin complex, we co-transfect COS-7 cells with NG1, Flag-laforin, and the most prevalent pathological mutant of malin, in which the proline 69 of the RING domain is mutated to alanine (Malin P69A) [(Gómez-Abad et al., 2005), (Turnbull et al., 2016)]. This mutant showed no defect in expression (Supplementary. Fig. S1) and had the same subcellular distribution as the wild type protein (Couarch et al., 2011); however, its interaction with laforin was highly decreased and its E3-ubiquitin ligase activity was severely compromised [(Couarch et al., 2011), (Sanchez-Martin et al., 2020)]. We observed that in the case of the presence of the malin P69A mutant, after 15 min at 37°C there was an increase in the number of cells in which the protein was intracellular (Fig. 2H), in comparison to cells expressing wild type malin (Fig. 2E). After 30 min at 37°C, in cells expressing the malin P69A mutant, the amount of NG1 that had reached the late endosomal compartment was significantly higher (41 ± 7% of the cells presenting NG1 at this localization) compared to the values observed in the presence of wild type malin, and was closer to the amount present in cells expressing GFP alone (Fig 2I and 2K). These results suggested that the activity of malin was crucial for the retention of GLT-1 at the plasma membrane. Similar results of delayed endocytosis of NG1 by the action of a functional laforin/malin complex were obtained using a different cell line, U2OS (another large enough cell line to allow the observation of different subcellular locations of the analyzed protein), indicating that this is a general process and that it is not due to a cell-specific effect (Supplementary Fig. S2). Taking together these results suggest that a functional laforin/malin complex is required to maintain GLT-1 at the plasma membrane (Fig. 2).
We tried to analyze the dynamics of GLT-1 endocytosis in mouse primary astrocytes, using the antibody-feeding assay based on the endogenous levels of GLT-1. However, due to the lower levels at the plasma membrane of the endogenous transporter in Epm2b−/− astrocytes, we were unable to reach any conclusion. The difficulties in the analysis of the homeostasis of endogenous GLT-1 transporter in primary astrocytes have also been reported by other authors, who asserted that GLT-1 expression in primary astrocytes were highly heterogeneous and therefore were not optimal for immunofluorescence studies (Martinez-Villarreal et al., 2012).
Laforin/Malin complex mediates the ubiquitination of GLT-1.
As previously described, the localization of GLT-1 at the plasma membrane is controlled by a ubiquitination/deubiquitination cycle [(Boehmer et al., 2006), (Sheldon et al., 2008), (Gonzalez-Gonzalez et al., 2008), (Martinez-Villarreal et al., 2012), (Garcia-Tardon et al., 2012)]. Namely, endocytosis of GLT-1 is mediated by ubiquitination of lysine residues located at its carboxy terminus by the E3-ubiquitin ligase Nedd4.2 (Garcia-Tardon et al., 2012). Since as described above the laforin/malin complex promotes the retention of GLT-1 at the plasma membrane, we sought to investigate whether in the presence of laforin/malin the ubiquitination of the transporter was altered. Butchbach et al. (2004) described that EAAT2/GLT-1 is associated with cholesterol-rich lipid raft microdomains and that this localization is required for the proper function of the transporter (Butchbach, Tian, Guo, & Lin, 2004). They also showed that EAAT2 in the lipid rafts is soluble in Triton X-100, but not in Brij-58. Having in mind the differences in the solubility of the transporter depending on the presence of different detergents, we analyzed the effect of the laforin/malin complex on the ubiquitination of EAAT2, including a detergent in the lysis buffer to solubilize the plasma membrane forms of EAAT2. We used NP-40, because of its similarity to Triton X-100 and because it is compatible with the subsequent manipulation of the extracts (lysis in guanidinium hydrochloride and TALON metal affinity chromatography; see below). To carry out these experiments we used HEK293 cells co-transfected with plasmids expressing EAAT2, Nedd4.2, and/or laforin and malin, together with a modified form of ubiquitin, tagged with 6xHis residues, which allowed the purification of ubiquitinated proteins. HEK293 cells were chosen in these experiments because they are easy to transfect with a combination of plasmids. The plasmid expressing EAAT2 was used because of its superior performance in ubiquitin assays in comparison to plasmids expressing GLT-1 [both proteins have a 95% identity, with lysine residues in the same positions along the sequence (Supplementary Fig. S3)]. Thirty-six hours after transfection, cells were lysed in guanidinium hydrochloride (that prevents the activity of deubiquitinases and therefore improves the recovery of ubiquitinated proteins) in the presence of 1% NP-40 to solubilize the membranes. Ubiquitinated proteins were purified by metal affinity chromatography and the presence of EAAT2 in the ubiquitinated fraction was examined by Western blot using specific anti-EAAT2 antibodies. As shown in Figure 3A, overexpression of laforin and malin promoted the ubiquitination of EAAT2 (Fig. 3A, lane 3) (*p<0.05), and this ubiquitination was maximal when laforin and malin were expressed together with Nedd4.2 (Fig. 3A, lane 4) (*p<0.05), suggesting that EAAT2 could be a substrate of both the laforin/malin complex and Nedd4.2.
Figure 3. The laforin/malin complex promotes the ubiquitination of GLT-1/EAAT2.
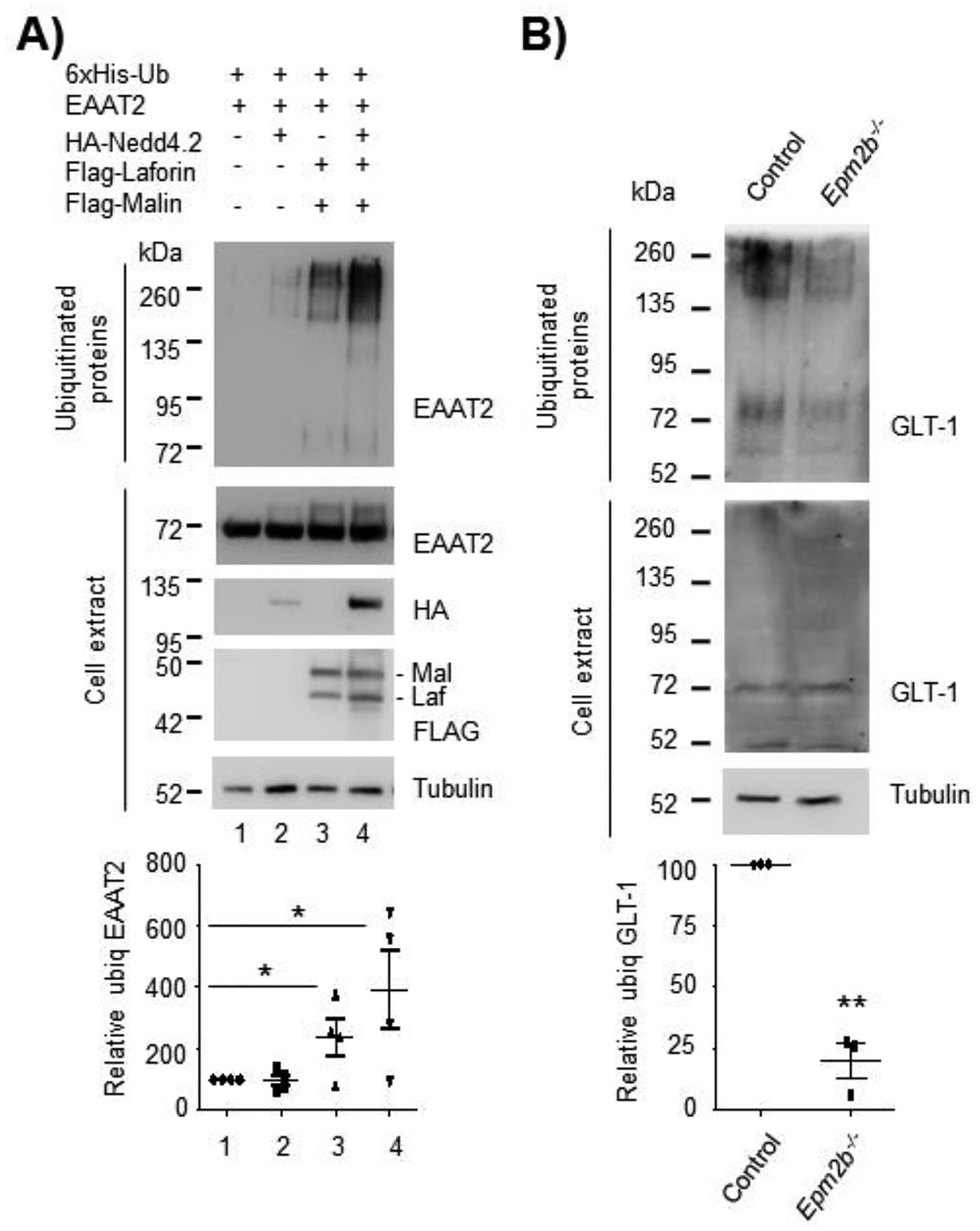
(A) Laforin/malin promotes the ubiquitination of EAAT2. HEK293 cells were transfected with the indicated plasmids and the ubiquitination of EAAT2 was analyzed as described in Materials and Methods. Proteins retained in the TALON metal affinity resin (representing the ubiquitinated fraction) and crude cell extracts (30 μg) were analyzed by Western blot using the indicated antibodies. (B) Decreased ubiquitination of GLT-1 in LD primary astrocytes. Control or Epm2b−/− primary astrocytes were transfected with a plasmid expressing 6xHis-ubiquitin, and 36 hours later cell extracts were obtained and the ubiquitinated proteins purified as above. Proteins present in the bound fraction and in crude cell extract (30 μg) were analyzed by Western blot using the indicated antibodies. A representative blot of 3–4 independent experiments is shown. Molecular weight markers are indicated in kDa on the left of the blots. The bottom panels in (A) and (B) indicate the quantification of the ubiquitinated bands as described in Materials and Methods. Statistical significance between the samples is indicated as *p<0.05 and **p<0.01 [n:4 in (A) and n:3 in (B)].
Next, we wanted to analyze the ubiquitination status of GLT-1 in primary astrocytes derived from Epm2b+/+ or Epm2b−/− mice. Since the laforin/malin complex promotes the ubiquitination of EAAT2 (see above), we expected that astrocytes from LD mouse models had a decreased ubiquitination of the transporter. That was the case since as indicated in Figure 3B, Epm2b−/− astrocytes showed a reduction in the number of ubiquitinated forms of GLT-1 (**p<0.01). The decrease of GLT-1 ubiquitination in LD astrocytes, together with the increase in ubiquitination by the laforin/malin complex shown in Figure 3A, indicates that the laforin/malin complex promotes the ubiquitination of GLT-1 transporter.
The ubiquitination of GLT-1 by laforin/malin is independent of Nedd4.2.
To date, the only E3-ubiquitin ligase described to ubiquitinate GLT-1 is Nedd4.2 [(Boehmer et al., 2006), (Garcia-Tardon et al., 2012)]. In fact, knock-down of Nedd4.2 produces a decrease in the ubiquitination of GLT-1 accompanied by increased levels of the transporter at the plasma membrane and glutamate uptake capacity, probably due to an impairment in the endocytosis of the transporter (Zhang et al., 2017). To verify whether the ubiquitination of EAAT2 by laforin and malin was dependent or not on the presence of Nedd4.2, we repeated the ubiquitination experiments in HEK293 cells where the expression of Nedd4.2 was silenced. We used the ON-Target Pool from Dharmacon that confers a 75–80% reduction of the amount of protein in only 24h (Supplementary Fig. S4). 48h after transfection with 50 nM of siNedd4.2 or non-targeting siRNA as control, cells were transfected with plasmids coding for EAAT2, 6xHis-ubiquitin and laforin and malin, and incubated 24 hours more. Cell extracts were prepared as above and the ubiquitination assay was performed. As shown in Figure 4A, after 72 hours of knock-down, the levels of Nedd4.2 were almost negligible (lane 4 in crude extract). However, the ubiquitination of EAAT2 by the laforin/malin complex remained unaltered (**p<0.01), indicating that this ubiquitination was independent of the presence of Nedd4.2.
Figure 4. The ubiquitination of GLT-1 by the laforin/malin complex is not related to the action of Nedd4.2.
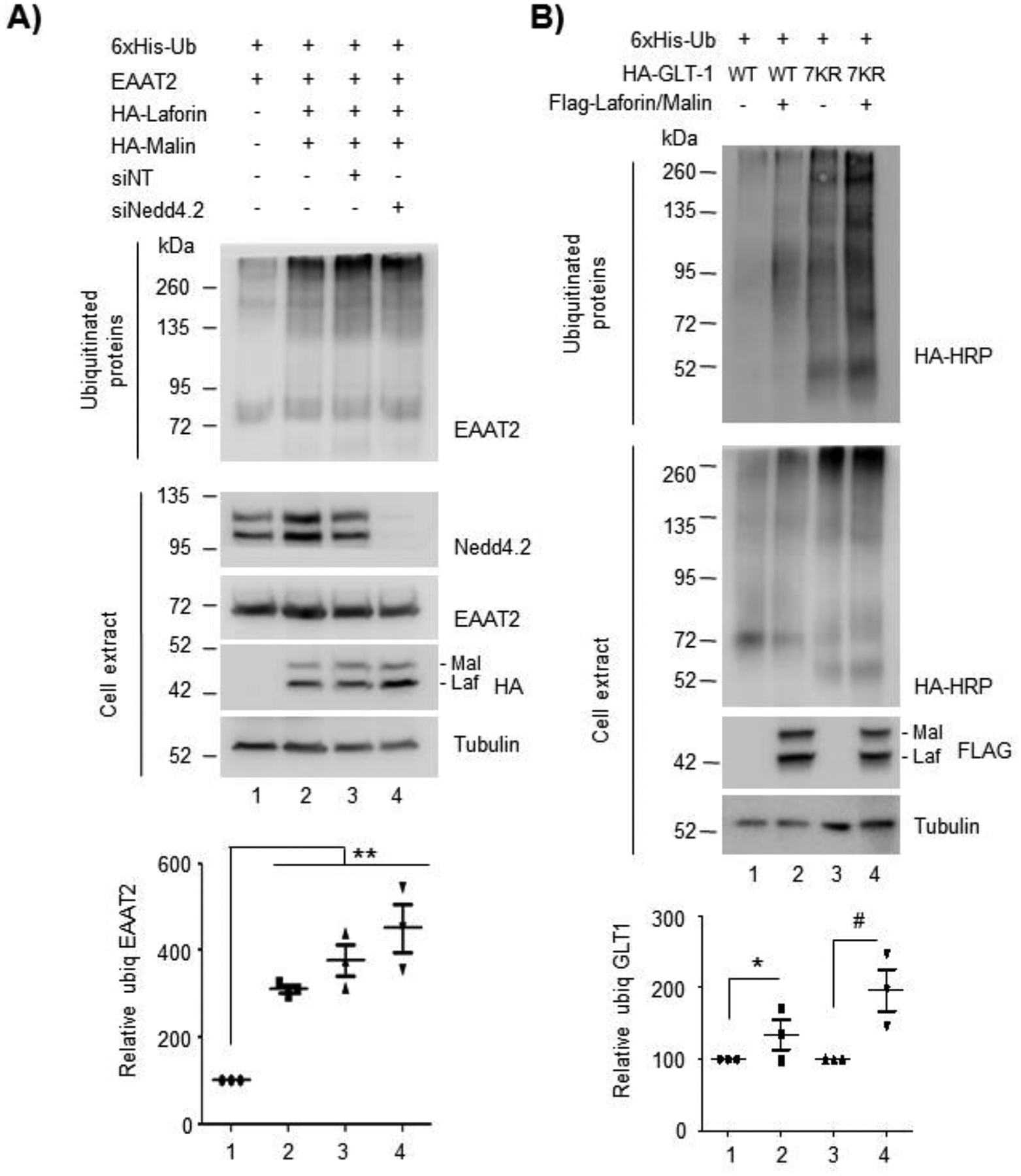
(A) The ubiquitination of GLT-1 by the laforin/malin complex occurs in the absence of Nedd4.2. HEK293 cells were transfected with siRNA for Nedd4.2 or the non-targeting control as indicated. 48 hours later cells were transfected with the indicated plasmids and after 24 hours, cell extracts were obtained and subjected to the purification of ubiquitinated proteins. The fraction retained in the TALON metal affinity resin (representing the ubiquitinated fraction) and crude cell extracts (30 μg) were analyzed by Western blot using the indicated antibodies. A representative blot of 3 independent experiments is shown. (B) The ubiquitination of GLT-1 by the laforin/malin complex does not take place at the C-terminus part of the protein. HEK293 cells were transfected with plasmids expressing GLT-1 (WT) or the mutant form with the 7 lysines of the C-terminal portion mutated to arginines (7KR), together with a plasmid expressing 6xHis-Ubiquitin. When indicated, cells were also transfected with plasmids expressing Flag-laforin/Flag-malin or empty plasmids. 36–48 hours after transfection cells were lysed and ubiquitinated proteins purified. The fraction retained in the TALON metal affinity resin (representing the ubiquitinated fraction) and crude cell extract (30 μg) were analyzed by Western blot using the indicated antibodies. Molecular weight markers are indicated in kDa on the left of the blots. A representative blot of 3 independent experiments is shown. The bottom panels in (A) and (B) indicate the quantification of the ubiquitinated bands as described in Materials and Methods. Statistical significance between the samples is indicated as *p<0.05, #p<0.05 and **p<0.01 (n:3).
Next, we wanted to investigate where the ubiquitination of EAAT2/GLT-1 by the laforin/malin complex could take place. As described previously, the use of a mutant form of GLT-1 in which the seven lysine residues of the carboxy terminus were mutated to arginines (GLT-1 7KR mutant) allowed the definition that the endocytosis of the transporter was mediated by the ubiquitination of the C-terminal portion of the protein (Sheldon et al., 2008). However, mutation of all the C-terminal lysine residues (GLT-1 7KR mutant), although impaired endocytosis, did not prevent the ubiquitination of the protein (Sheldon et al., 2008), suggesting that other lysine residues in the N-terminal part of the protein were also ubiquitinated and could play other regulatory functions (see Supplementary Fig. S3). With this information in mind, we used the mutant GLT-1 7KR and analyzed the ubiquitination of this form by laforin and malin. As observed in Figure 4B, the presence of laforin and malin promoted the ubiquitination of the GLT-1 7KR mutant (compare lane 3 and lane 4) (#p<0.05), indicating that other lysine residues different from the ones present at the C-terminus were ubiquitinated by the laforin/malin complex.
Laforin and malin interact and ubiquitinate α- and β-arrestins.
So far, our results show that the laforin/malin complex promotes the retention of GLT-1 at the plasma membrane, probably due to the ubiquitination of the transporter at sites different from the lysine residues at the C-terminus, and, for this ubiquitination, the laforin/malin complex does not require the presence of Nedd4.2. As it has been described that endocytosis of GLT-1 is mediated by Nedd4.2, we sought to investigate whether laforin and malin could, in addition, interfere with the Nedd4.2-mediated endocytosis of GLT-1. Previous work in our lab had shown that the laforin/malin complex did not interact, nor ubiquitinate Nedd4.2 and that Nedd4.2 levels were not altered in brain samples from mouse models of Lafora disease (Munoz-Ballester et al., 2016). Thus, it seems that the laforin/malin complex does not affect Nedd4.2 directly. Regularly, Nedd4.2 binds to targets containing PPXY domains to promote the ubiquitination of the corresponding substrate; however, there are examples where the targets of Nedd4.2 lack these domains. In these cases, adaptor molecules mediate the binding of Nedd4.2 to the substrates. As GLT-1 does not contain PPXY domains, Nedd4.2 needs to assemble with β-arrestin1 to promote the endocytosis of the transporter (Ibanez, Diez-Guerra, Gimenez, & Zafra, 2016). Therefore, we analyzed whether laforin and malin could affect indirectly Nedd4.2 function by interacting with β-arrestin1. We analyzed also the effect of laforin and malin on β-arrestin2, as it has been shown that this adaptor interacts also with GLT-1, although to a lesser extent (Ibanez et al., 2016). First, we studied the physical interaction of the β-arrestins with laforin and malin by GFP-trap analysis. As shown in Figure 5A, β-arrestin1 could interact with both laforin and malin, independently of these proteins forming or not part of a complex, while in the case of β-arrestin2, we could only detect a direct interaction with laforin (Figure 5B). We then analyzed whether these β-arrestins could be ubiquitinated by the laforin/malin complex. Figure 6 shows that both β-arrestin1 and 2 were able to be ubiquitinated by the laforin/malin complex (Fig. 6A lane 2, and 6B lane 2, respectively) (*p<0.05 in both cases). To investigate the outcome of this ubiquitination, we performed the ubiquitination study in the presence or absence of protein degradation inhibitors. Accumulation of ubiquitinated proteins upon inhibition of protein degradation could indicate that this ubiquitination may promote the degradation of the protein. As observed in Figure 6A, ubiquitinated β-arrestin1 was accumulated upon inhibition of protein degradation (compare lanes 2 and 4) (&p<0.05), indicating that laforin and malin could promote the degradation of β-arrestin1. In the case of β-arrestin2, we did not observe accumulation of this protein upon inhibition of protein degradation (Figure 6B, lanes 2 and 4).
Figure 5. Laforin interacts with β-arrestins 1 and 2.
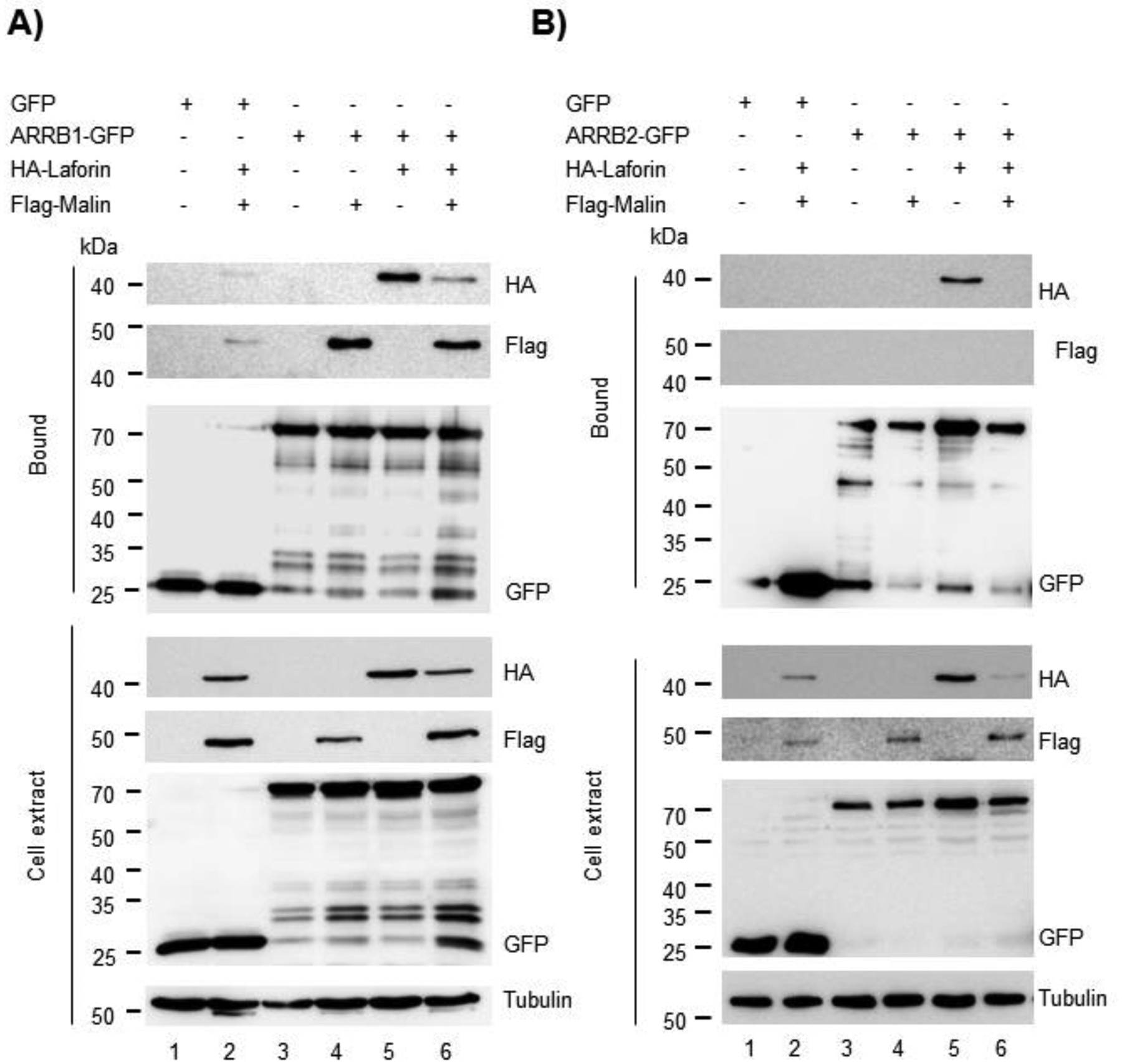
HEK293 cells were co-transfected with the plasmids expressing HA-Laforin, Flag-Malin, and GFP-tagged β-arrestin1 (ARRB1-GFP) (A) or β-arrestin2 (ARRB2-GFP) (B) or GFP as control. Cells were lysed and 1.5 mg of protein in the crude extracts were analyzed as indicated in Materials and Methods. Bound fractions and crude cell extracts were analyzed by Western blot using the indicated antibodies. In A and B, a representative blot of three independent experiments is shown. Molecular weight markers are indicated in kDa on the left of the blots.
Figure 6. The laforin/malin complex can ubiquitinate both β-arrestin 1 and β-arrestin 2.
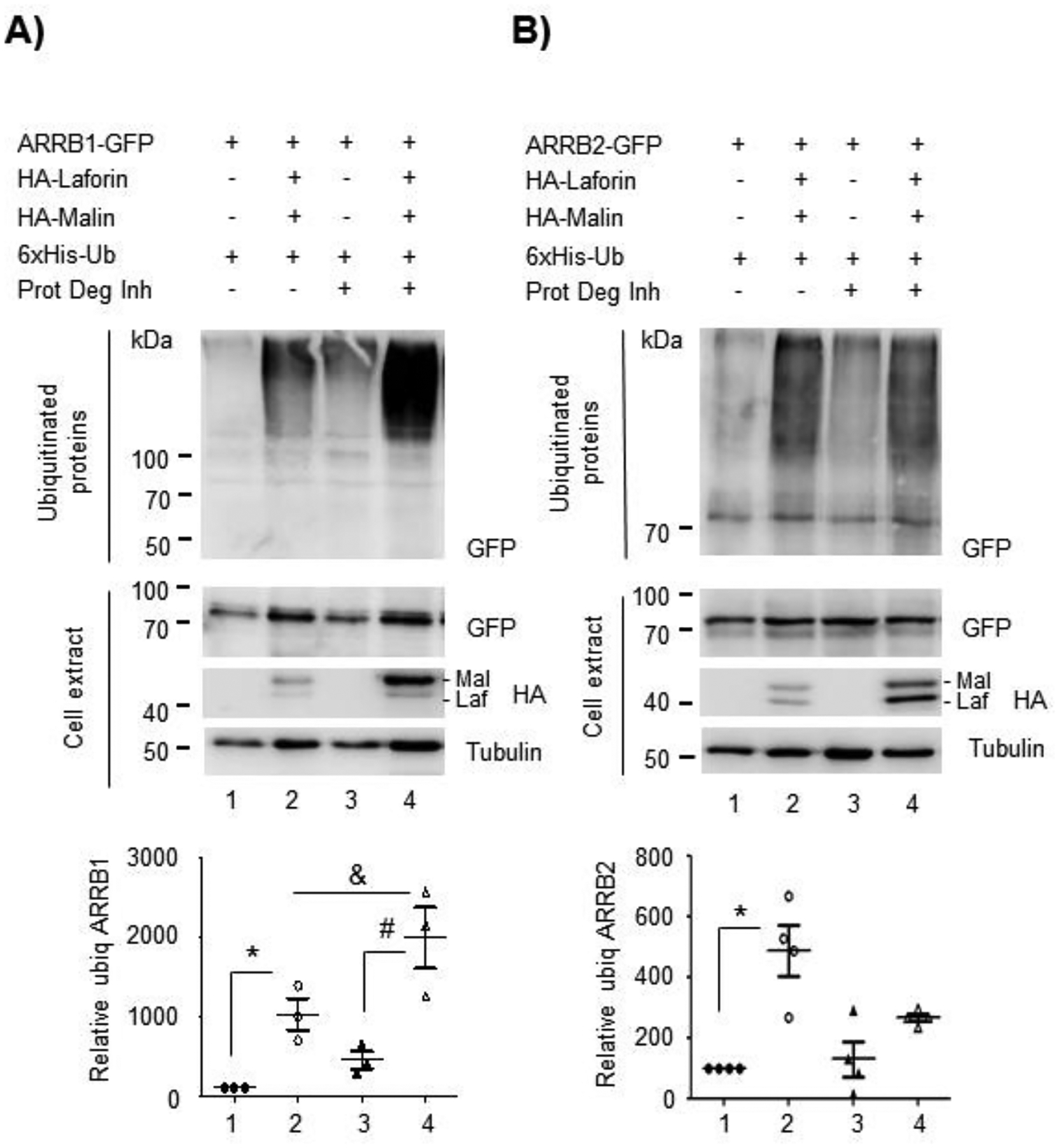
HEK293 cells were transfected with the indicated plasmids and the ubiquitination of β-arrestin1 (A) and β-arrestin2 (B) were analyzed as described in Materials and Methods. The ubiquitinated fractions and crude cell extracts (30 μg) were analyzed by Western blot using the indicated antibodies. A representative blot of 3–4 independent experiments is shown. Molecular weight markers are indicated in kDa on the left of the blots. The bottom panels in (A) and (B) indicate the quantification of the ubiquitinated bands as described in Materials and Methods. Statistical significance between the samples is indicated as *p<0.05, #p<0.05 and &p<0.05 [n:3 in (A) and n:4 in (B)].
As β-arrestins1 and 2 also lack PPXY motifs, Ibañez et al. (2016) elegantly showed that Nedd4.2 needs to bind to a third adaptor to allow the recruitment of Nedd4.2 to β-arrestin1 and GLT-1 (Ibanez et al., 2016). They propose as candidate α-arrestin1. So, we analyzed whether the laforin/malin complex could affect this protein. By GFP-trap we observed that α-arrestin1 interacted with both laforin and malin, alone or in combination (Figure 7A). We also performed a ubiquitination assay and observed that α-arrestin1 was also able to be ubiquitinated by the laforin/malin complex (Figure 7B, lane 2) (*p<0.05), but this ubiquitination did not increase upon inhibition of the protein degradation systems (Figure 7B, lane 4).
Figure 7. The laforin malin complex interacts with α-arrestin 1 and promotes its ubiquitination.
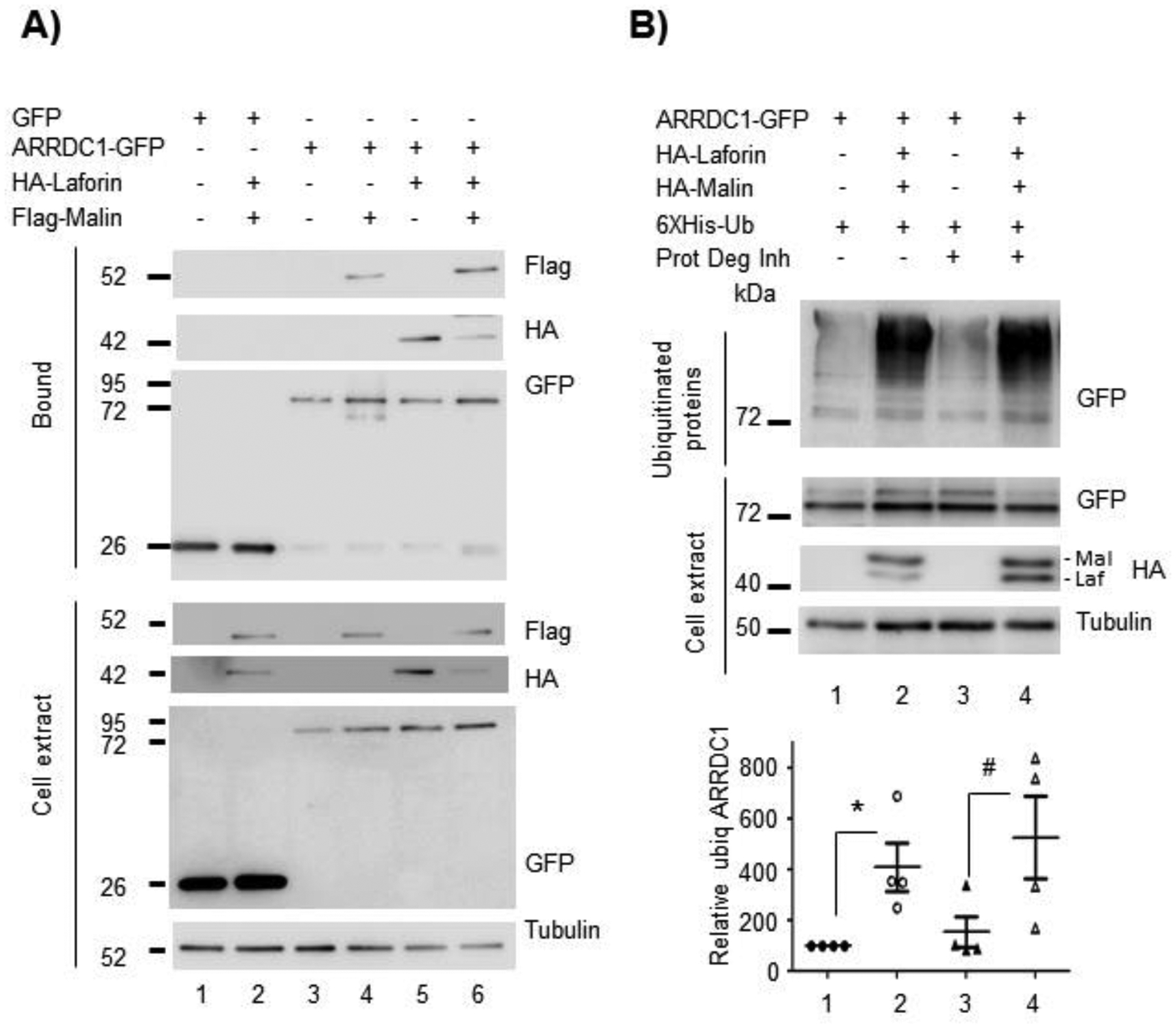
(A) HEK293 cells were co-transfected with the plasmids expressing HA-Laforin, Flag-Malin, and GFP-tagged α-arrestin1 (ARRDC1-GFP) or GFP as control. Cells were lysed and 1.5 mg of protein in the crude extracts were analyzed as indicated in Materials and Methods. Bound fractions or crude cell extracts were analyzed by Western blot using the indicated antibodies. (B) HEK293 cells were transfected with the indicated plasmids and the ubiquitination of α-arrestin1 was analyzed as described in Materials and Methods. The ubiquitinated fractions and crude cell extracts (30 μg) were analyzed by Western blot using the indicated antibodies. In A and B, a representative blot of four independent experiments is shown. Molecular weight markers are indicated in kDa on the left of the blots. The bottom panel in (B) indicates the quantification of the ubiquitinated bands as described in Materials and Methods. Statistical significance between the samples is indicated as *p<0.05 and #p<0.05 (n:4).
Taken together, all these results show that the laforin/malin complex interacts with and ubiquitinates different adaptors (α-arrestin1 and β-arrestins1 and 2) of the Nedd4.2 system. This could interfere with the function of the Nedd4.2-mediated endocytosis of GLT-1.
DISCUSSION
Lafora disease is a fatal rare neurodegenerative disorder that causes the death of the patient irrevocably within 10 years after the appearance of the first symptoms. Regretfully, available treatment is only symptomatic, although great efforts are being made trying to find a cure for this devastating disease, including the generation of a Natural History Study that will be crucial for the development of future clinical trials. To date, four therapeutic approaches are being developed to treat LD [for the latest update see (Gentry et al., 2020)]. These include the use of antisense oligonucleotides targeting glycogen synthase to avoid the formation of Lafora bodies, enzyme therapy directed to degrade them once formed (Brewer et al., 2019), the use of small-molecule inhibitors of glycogen synthase (Tang et al., 2020), and the use of repurposing drugs such as metformin (Berthier et al., 2016), although none of them have been translated to the clinic yet. It is therefore of great importance to continue investigating the molecular mechanisms of the disease to find a good therapeutic target and an effective treatment that could be used in clinical trials.
We have recently shown that there are decreased levels of the glutamate transporter GLT-1 at the plasma membrane of the astrocytes from LD mouse models and hypothesized that this could be one of the causes of the type of epilepsy present in this disorder (Munoz-Ballester et al., 2016). The glutamate transporter GLT-1 has been proposed as an interesting therapeutic target because of its implication in different neurological diseases (Peterson & Binder, 2019). In some of these diseases, the defect in glutamate uptake is produced by a decreased expression of GLT-1, as seen in models of Huntington’s disease (Sari, Prieto, Barton, Miller, & Rebec, 2010), Parkinson, or Amyotrophic lateral sclerosis (Takahashi, Foster, & Lin, 2015). In this situation, a therapy directed to activate the transporter, either at a transcriptional, translational, or functional level will be desired. Conversely, there are other conditions in which the total levels of the protein are maintained, but the protein is not efficiently located at the plasma membrane. For example, the YAC128 mouse model of Huntington’s disease has a reduced uptake of glutamate due to a decreased palmitoylation of the transporter (Huang et al., 2010). In addition, Scimemi et al. reported in an in vitro model of Alzheimer’s disease decreased levels of GLT-1 in the surface without a change in the total protein levels (Scimemi et al., 2013). This is also the case of Lafora disease, where we have previously shown a decreased glutamate uptake in mouse models of LD due to a lower presence of the glutamate transporter GLT-1 at the plasma membrane, with no changes in total protein or mRNA levels [(Munoz-Ballester et al., 2016), (Munoz-Ballester et al., 2019)]. In these latter conditions, increasing the transcription or the translation of GLT-1 will not improve the health of the patients. Therefore, it is essential to understand the molecular mechanism that leads to the dysfunction of GLT-1 in each particular disorder in order to design an effective therapy that allows the recovery of its functionality.
Since the homeostasis of GLT-1 depends on the ubiquitination/deubiquitination cycle, the goal of the present study was to determine whether the laforin/malin complex could be affecting the localization of the transporter at the plasma membrane. Our results indicate that this complex promoted the ubiquitination of GLT-1/EAAT2 transporter, demonstrated by an increase in its ubiquitination when the laforin/malin complex was overexpressed, and by the fact that primary astrocytes derived from LD mouse models presented a decreased ubiquitination of the transporter. Although we have not identified yet the exact lysine residues ubiquitinated by the laforin/malin complex, our results indicated that this modification should be taken place at lysine residues different from the ones present in the C-terminal tail of the protein, since a GLT-1 7KR mutant, with all the lysine residues located at the C-terminus, changed to arginines, was still able to be ubiquitinated by the laforin/malin complex. GLT-1 contains 4 lysine residues in the N-terminal portion and 5 more lysines distributed in the internal loops 1 to 3 (Palacin, Estevez, Bertran, & Zorzano, 1998) (see Supplementary Fig. S3). Thus, we presume that the laforin/malin complex might be ubiquitinating any of these alternative lysine residues of the transporter. In any case, our results define the laforin/malin complex as an alternative E3-ubiquitin ligase system involved in the ubiquitination of GLT-1, in addition to Nedd4.2 [(Boehmer et al., 2006), (Garcia-Tardon et al., 2012)]. As there are several examples in the literature of proteins that are ubiquitinated at different positions with opposite effects (Ball et al., 2016), ubiquitination of GLT-1 by the laforin/malin complex could have an opposite effect to the Nedd4.2-mediated ubiquitination of the transporter. In the case of Nedd4.2, it has been described that the modification that it introduces in GLT-1 accelerates the endocytosis of the transporter, decreasing its levels and functionality in the membrane [(Garcia-Tardon et al., 2012), (Zhang et al., 2017). Therefore, the laforin/malin complex and Nedd4.2 could affect the endocytosis of GLT-1 transporter in opposite ways: Nedd4.2 would induce the endocytosis of GLT-1, whereas the laforin/malin complex would prevent this process.
We then studied whether the laforin/malin complex could have a direct effect on the activity of Nedd4.2. Previous work in our lab had shown that the laforin/malin complex did not affect Nedd4.2 directly. However, in this work, we provide evidence that the laforin/malin complex promotes the stability of GLT-1 by interacting with and ubiquitinating the adaptor molecules needed by Nedd4.2 to mediate the endocytosis of GLT-1, i.e., β-arrestins. Perhaps, this laforin/malin-mediated ubiquitination of these adaptors could influence negatively the activity of Nedd4.2 on the homeostasis of the GLT-1 transporter. Moreover, we observed that the laforin/malin complex interacted with and ubiquitinated α-arrestin1, an additional adaptor required for the function of Nedd4.2 on GLT-1 endocytosis, reinforcing the hypothesis that the laforin/malin complex influenced negatively the functionality of Nedd4.2 on GLT-1 homeostasis. We also analyzed the possible functional interaction of the laforin/malin complex on additional members of the α-arrestin family, such as α-arrestin2 and α-arrestin3 [the most abundant α-arrestin in astrocytes; (Zhang et al., 2014)], but we could not observe any interaction between any of these proteins and the laforin/malin complex (data not shown).
In conclusion, in this work, we present the molecular mechanism by which GLT-1 homeostasis is altered in Lafora disease, namely, altered recycling leading to glutamate uptake impairment, and propose this process as a novel therapeutic target that could be used to treat this devastating disease, as well as other diseases where there is a defect in the homeostasis of GLT-1 transporter.
Supplementary Material
Main points:
Laforin/malin complex ubiquitinates GLT-1 glutamate transporter.
Laforin/malin complex modulates the endocytosis of GLT-1 negatively.
Laforin/malin complex opposes the effect of Nedd4.2 on GLT-1, by interacting and ubiquitinating α- and β-arrestins.
Acknowledgments:
We want to thank Dr. Peter Dodd, Dr. Lynne Yenush, Dr. Francisco Zafra, and Dr. Manuel Rodríguez for plasmids. This work was supported by grants from the Spanish Ministry of Economy and Competitiveness SAF2017-83151-R, a grant from Fundación Ramón Areces (CIVP18A3935), and a grant from the National Institute of Health (NIH-NINDS) P01NS097197, which established the Lafora Epilepsy Cure Initiative (LECI), to P.S.
Footnotes
Data Sharing policy: The data that support the findings of this study are available from the corresponding author upon reasonable request.
Conflict of interest: Authors declare that none of them has any type of conflict of interest.
REFERENCES
- Auge E, Pelegri C, Manich G, Cabezon I, Guinovart JJ, Duran J, & Vilaplana J (2018). Astrocytes and neurons produce distinct types of polyglucosan bodies in Lafora disease. Glia, 66(10), 2094–2107. doi: 10.1002/glia.23463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball KA, Johnson JR, Lewinski MK, Guatelli J, Verschueren E, Krogan NJ, & Jacobson MP (2016). Non-degradative Ubiquitination of Protein Kinases. PLoS Comput Biol, 12(6), e1004898. doi: 10.1371/journal.pcbi.1004898 PCOMPBIOL-D-16–00023 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthier A, Paya M, Garcia-Cabrero AM, Ballester MI, Heredia M, Serratosa JM, … Sanz P (2016). Pharmacological Interventions to Ameliorate Neuropathological Symptoms in a Mouse Model of Lafora Disease. Mol Neurobiol, 53(2), 1296–1309. doi: 10.1007/s12035-015-9091-8 10.1007/s12035-015-9091-8 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehmer C, Palmada M, Rajamanickam J, Schniepp R, Amara S, & Lang F (2006). Post-translational regulation of EAAT2 function by co-expressed ubiquitin ligase Nedd4-2 is impacted by SGK kinases. J Neurochem, 97(4), 911–921. doi:JNC3629 [pii] 10.1111/j.1471-4159.2006.03629.x [DOI] [PubMed] [Google Scholar]
- Brewer MK, Uittenbogaard A, Austin GL, Segvich DM, DePaoli-Roach A, Roach PJ, … Gentry MS (2019). Targeting Pathogenic Lafora Bodies in Lafora Disease Using an Antibody-Enzyme Fusion. Cell Metab, 30(4), 689–705 e686. doi:S1550–4131(19)30375–4 [pii] 10.1016/j.cmet.2019.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butchbach ME, Tian G, Guo H, & Lin CL (2004). Association of excitatory amino acid transporters, especially EAAT2, with cholesterol-rich lipid raft microdomains: importance for excitatory amino acid transporter localization and function. J Biol Chem, 279(33), 34388–34396. doi: 10.1074/jbc.M403938200 M403938200 [pii] [DOI] [PubMed] [Google Scholar]
- Couarch P, Vernia S, Gourfinkel-An I, Lesca G, Gataullina S, Fedirko E, … Baulac S (2011). Lafora progressive myoclonus epilepsy: NHLRC1 mutations affect glycogen metabolism. J Mol Med (Berl), 89(9), 915–925. doi: 10.1007/s00109-011-0758-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan EM, Young EJ, Ianzano L, Munteanu I, Zhao X, Christopoulos CC, … Scherer SW (2003). Mutations in NHLRC1 cause progressive myoclonus epilepsy. Nat Genet, 35(2), 125–127. [DOI] [PubMed] [Google Scholar]
- Danbolt NC (2001). Glutamate uptake. Prog Neurobiol, 65(1), 1–105. doi:S0301–0082(00)00067–8 [pii] 10.1016/s0301-0082(00)00067-8 [DOI] [PubMed] [Google Scholar]
- Garcia-Gimeno MA, Knecht E, & Sanz P (2018). Lafora Disease: A Ubiquitination-Related Pathology. Cells, 7(8), 8. doi:E87 [pii] 10.3390/cells7080087 cells7080087 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Tardon N, Gonzalez-Gonzalez IM, Martinez-Villarreal J, Fernandez-Sanchez E, Gimenez C, & Zafra F (2012). Protein kinase C (PKC)-promoted endocytosis of glutamate transporter GLT-1 requires ubiquitin ligase Nedd4-2-dependent ubiquitination but not phosphorylation. J Biol Chem, 287(23), 19177–19187. doi: 10.1074/jbc.M112.355909 M112.355909 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentry MS, Afawi Z, Armstrong DD, Delgado-Escueta A, Goldberg YP, Grossman TR, … Serratosa JM (2020). The 5th International Lafora Epilepsy Workshop: Basic science elucidating therapeutic options and preparing for therapies in the clinic. Epilepsy Behav, 103(Pt A), 106839. doi:S1525–5050(19)31266–1 [pii] 10.1016/j.yebeh.2019.106839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez-Abad C, Gómez-Garre P, Gutiérrez-Delicado E, Saygi S, Michelucci R, Tassinari CA, … Serratosa JM (2005). Lafora disease due to EPM2B mutations. A clinical and genetic study. Neurology, 64, 982–986. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Gonzalez IM, Garcia-Tardon N, Gimenez C, & Zafra F (2008). PKC-dependent endocytosis of the GLT1 glutamate transporter depends on ubiquitylation of lysines located in a C-terminal cluster. Glia, 56(9), 963–974. doi: 10.1002/glia.20670 [DOI] [PubMed] [Google Scholar]
- He X, Chen F, Zhang Y, Gao Q, Guan Y, Wang J, … Li T (2020). Upregulation of adenosine A2A receptor and downregulation of GLT1 is associated with neuronal cell death in Rasmussen’s encephalitis. Brain Pathol, 30(2), 246–260. doi: 10.1111/bpa.12770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang K, Kang MH, Askew C, Kang R, Sanders SS, Wan J, … Hayden MR (2010). Palmitoylation and function of glial glutamate transporter-1 is reduced in the YAC128 mouse model of Huntington disease. Neurobiol Dis, 40(1), 207–215. doi: 10.1016/j.nbd.2010.05.027 S0969–9961(10)00189–0 [pii] [DOI] [PubMed] [Google Scholar]
- Ibanez I, Diez-Guerra FJ, Gimenez C, & Zafra F (2016). Activity dependent internalization of the glutamate transporter GLT-1 mediated by beta-arrestin 1 and ubiquitination. Neuropharmacology, 107, 376–386. doi:S0028–3908(16)30114–9 [pii] 10.1016/j.neuropharm.2016.03.042 [DOI] [PubMed] [Google Scholar]
- Kaiser P, & Tagwerker C (2005). Is this protein ubiquitinated? Methods Enzymol, 399, 243–248. [DOI] [PubMed] [Google Scholar]
- Lahuerta M, Gonzalez D, Aguado C, Fathinajafabadi A, Garcia-Gimenez JL, Moreno-Estelles M, … Sanz P (2020). Reactive Glia-Derived Neuroinflammation: a Novel Hallmark in Lafora Progressive Myoclonus Epilepsy That Progresses with Age. Mol Neurobiol, 57(3), 1607–1621. doi: 10.1007/s12035-019-01842-z 10.1007/s12035-019-01842-z [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddelow SA, & Sofroniew MV (2019). Astrocytes usurp neurons as a disease focus. Nat Neurosci, 22(4), 512–513. doi: 10.1038/s41593-019-0367-6 10.1038/s41593-019-0367-6 [pii] [DOI] [PubMed] [Google Scholar]
- Lopez-Gonzalez I, Viana R, Sanz P, & Ferrer I (2017). Inflammation in Lafora Disease: Evolution with Disease Progression in Laforin and Malin Knock-out Mouse Models. Mol Neurobiol, 54(5), 3119–3130. doi: 10.1007/s12035-016-9884-4 10.1007/s12035-016-9884-4 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macia E, Ehrlich M, Massol R, Boucrot E, Brunner C, & Kirchhausen T (2006). Dynasore, a cell-permeable inhibitor of dynamin. Dev Cell, 10(6), 839–850. doi:S1534–5807(06)00163–8 [pii] 10.1016/j.devcel.2006.04.002 [DOI] [PubMed] [Google Scholar]
- Martinez-Villarreal J, Garcia Tardon N, Ibanez I, Gimenez C, & Zafra F (2012). Cell surface turnover of the glutamate transporter GLT-1 is mediated by ubiquitination/deubiquitination. Glia, 60(9), 1356–1365. doi: 10.1002/glia.22354 [DOI] [PubMed] [Google Scholar]
- Minassian BA, Lee JR, Herbrick JA, Huizenga J, Soder S, Mungall AJ, … Scherer SW (1998). Mutations in a gene encoding a novel protein tyrosine phosphatase cause progressive myoclonus epilepsy. Nat Genet, 20(2), 171–174. [DOI] [PubMed] [Google Scholar]
- Munoz-Ballester C, Berthier A, Viana R, & Sanz P (2016). Homeostasis of the astrocytic glutamate transporter GLT-1 is altered in mouse models of Lafora disease. Biochim Biophys Acta, 1862(6), 1074–1083. doi:S0925–4439(16)30054–0 [pii] 10.1016/j.bbadis.2016.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz-Ballester C, Santana N, Perez-Jimenez E, Viana R, Artigas F, & Sanz P (2019). In vivo glutamate clearance defects in a mouse model of Lafora disease. Exp Neurol, 320, 112959. doi:S0014–4886(19)30105–0 [pii] 10.1016/j.expneurol.2019.112959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy-Royal C, Dupuis J, Groc L, & Oliet SHR (2017). Astroglial glutamate transporters in the brain: Regulating neurotransmitter homeostasis and synaptic transmission. J Neurosci Res, 95(11), 2140–2151. doi: 10.1002/jnr.24029 [DOI] [PubMed] [Google Scholar]
- Murphy-Royal C, Dupuis JP, Varela JA, Panatier A, Pinson B, Baufreton J, … Oliet SH (2015). Surface diffusion of astrocytic glutamate transporters shapes synaptic transmission. Nat Neurosci, 18(2), 219–226. doi: 10.1038/nn.3901 nn.3901 [pii] [DOI] [PubMed] [Google Scholar]
- Pajarillo E, Rizor A, Lee J, Aschner M, & Lee E (2019). The role of astrocytic glutamate transporters GLT-1 and GLAST in neurological disorders: Potential targets for neurotherapeutics. Neuropharmacology, 161, 107559. doi:S0028–3908(19)30080–2 [pii] 10.1016/j.neuropharm.2019.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palacin M, Estevez R, Bertran J, & Zorzano A (1998). Molecular biology of mammalian plasma membrane amino acid transporters. Physiol Rev, 78(4), 969–1054. doi: 10.1152/physrev.1998.78.4.969 [DOI] [PubMed] [Google Scholar]
- Peterson AR, & Binder DK (2019). Post-translational Regulation of GLT-1 in Neurological Diseases and Its Potential as an Effective Therapeutic Target. Front Mol Neurosci, 12, 164. doi: 10.3389/fnmol.2019.00164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petr GT, Sun Y, Frederick NM, Zhou Y, Dhamne SC, Hameed MQ, … Rosenberg PA (2015). Conditional deletion of the glutamate transporter GLT-1 reveals that astrocytic GLT-1 protects against fatal epilepsy while neuronal GLT-1 contributes significantly to glutamate uptake into synaptosomes. J Neurosci, 35(13), 5187–5201. doi: 10.1523/JNEUROSCI.4255-14.2015 35/13/5187 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presley JF, Cole NB, Schroer TA, Hirschberg K, Zaal KJ, & Lippincott-Schwartz J (1997). ER-to-Golgi transport visualized in living cells. Nature, 389(6646), 81–85. doi: 10.1038/38001 [DOI] [PubMed] [Google Scholar]
- Rauen T, Tanui R, Grewer C (2016). Structural and functional dynamics of excitatory amino acid transporters (EAAT). AIMS Molecular Science, 1(3), 99–125. [Google Scholar]
- Robinson MB (2006). Acute regulation of sodium-dependent glutamate transporters: a focus on constitutive and regulated trafficking. Handb Exp Pharmacol(175), 251–275. doi: 10.1007/3-540-29784-7_13 [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW, … Welty DF (1996). Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron, 16(3), 675–686. doi:S0896–6273(00)80086–0 [pii] 10.1016/s0896–6273(00)80086–0 [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Van Kammen M, Levey AI, Martin LJ, & Kuncl RW (1995). Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann Neurol, 38(1), 73–84. doi: 10.1002/ana.410380114 [DOI] [PubMed] [Google Scholar]
- Rubio-Villena C, Viana R, Bonet J, Garcia-Gimeno MA, Casado M, Heredia M, & Sanz P (2018). Astrocytes: new players in progressive myoclonus epilepsy of Lafora type. Hum Mol Genet, 27(7), 1290–1300. doi: 10.1093/hmg/ddy044 4835225 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Martin P, Lahuerta M, Viana R, Knecht E, & Sanz P (2020). Regulation of the autophagic PI3KC3 complex by laforin/malin E3-ubiquitin ligase, two proteins involved in Lafora disease. Biochim Biophys Acta Mol Cell Res, 1867(2), 118613. doi:S0167–4889(19)30221–6 [pii] 10.1016/j.bbamcr.2019.118613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sari Y, Prieto AL, Barton SJ, Miller BR, & Rebec GV (2010). Ceftriaxone-induced up-regulation of cortical and striatal GLT1 in the R6/2 model of Huntington’s disease. J Biomed Sci, 17, 62. doi: 10.1186/1423-0127-17-62 1423-0127-17-62 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scimemi A, Meabon JS, Woltjer RL, Sullivan JM, Diamond JS, & Cook DG (2013). Amyloid-beta1-42 slows clearance of synaptically released glutamate by mislocalizing astrocytic GLT-1. J Neurosci, 33(12), 5312–5318. doi: 10.1523/JNEUROSCI.5274-12.2013 33/12/5312 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serratosa JM, Gomez-Garre P, Gallardo ME, Anta B, de Bernabe DB, Lindhout D, … de Cordoba SR (1999). A novel protein tyrosine phosphatase gene is mutated in progressive myoclonus epilepsy of the Lafora type (EPM2). Hum Mol Genet, 8(2), 345–352. [DOI] [PubMed] [Google Scholar]
- Sheldon AL, Gonzalez MI, Krizman-Genda EN, Susarla BT, & Robinson MB (2008). Ubiquitination-mediated internalization and degradation of the astroglial glutamate transporter, GLT-1. Neurochem Int, 53(6–8), 296–308. doi: 10.1016/j.neuint.2008.07.010 S0197–0186(08)00126–5 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susarla BT, & Robinson MB (2008). Internalization and degradation of the glutamate transporter GLT-1 in response to phorbol ester. Neurochem Int, 52(4–5), 709–722. doi:S0197–0186(07)00252–5 [pii] 10.1016/j.neuint.2007.08.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Foster JB, & Lin CL (2015). Glutamate transporter EAAT2: regulation, function, and potential as a therapeutic target for neurological and psychiatric disease. Cell Mol Life Sci, 72(18), 3489–3506. doi: 10.1007/s00018-015-1937-8 10.1007/s00018-015-1937-8 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, … Wada K (1997). Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science, 276(5319), 1699–1702. [DOI] [PubMed] [Google Scholar]
- Tang B, Frasinyuk MS, Chikwana VM, Mahalingan KK, Morgan CA, Segvich DM, … Hurley TD (2020). Discovery and Development of Small-Molecule Inhibitors of Glycogen Synthase. J Med Chem, 63(7), 3538–3551. doi: 10.1021/acs.jmedchem.9b01851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbull J, Tiberia E, Striano P, Genton P, Carpenter S, Ackerley CA, & Minassian BA (2016). Lafora disease. Epileptic Disord, 18(S2), 38–62. doi:epd.2016.0842 [pii] 10.1684/epd.2016.0842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vezzani A, Balosso S, & Ravizza T (2019). Neuroinflammatory pathways as treatment targets and biomarkers in epilepsy. Nat Rev Neurol, 15(8), 459–472. doi: 10.1038/s41582-019-0217-x 10.1038/s41582-019-0217-x [pii] [DOI] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, … Wu JQ (2014). An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci, 34(36), 11929–11947. doi: 10.1523/JNEUROSCI.1860-14.2014 34/36/11929 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, He X, Meng X, Wu X, Tong H, Zhang X, & Qu S (2017). Regulation of glutamate transporter trafficking by Nedd4-2 in a Parkinson’s disease model. Cell Death Dis, 8(2), e2574. doi: 10.1038/cddis.2016.454 cddis2016454 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.