

Abstract
Human genetic variation in coding regions is fundamental to the study of protein structure and function. Most methods for interpreting missense variants consider substitution measures derived from homologous proteins across different species. In this study, we introduce human-specific amino acid (AA) substitution matrices that are based on genetic variations in the modern human population. We analyzed the frequencies of >4.8M single nucleotide variants (SNVs) at codon and AA resolution and compiled human-centric substitution matrices that are fundamentally different from classic cross-species matrices (e.g. BLOSUM, PAM). Our matrices are asymmetric, with some AA replacements showing significant directional preference. Moreover, these AA matrices are only partly predicted by nucleotide substitution rates. We further test the utility of our matrices in exposing functional signals of experimentally-validated protein annotations. A significant reduction in AA transition frequencies was observed across nine post-translational modification (PTM) types and four ion-binding sites. Our results propose a purifying selection signal in the human proteome across a diverse set of functional protein annotations and provide an empirical baseline for interpreting human genetic variation in coding regions.
INTRODUCTION
The study of population genetic variation has led to countless scientific and medical applications. Illustrative examples include tracing of ancient migration patterns, estimating the pathogenicity of genetic variants, identifying functional elements in the genome, and assessing the adaptivity and conservation of genes and other genomic regions (1–3). Such inquiries are predicated on robust background models for the dynamics of genetic variation, which are expected to accurately capture the propensities of different genetic variants in various contexts (4,5). For example, a common way to identify functional elements in the genome is to quantify evolutionary conservation, namely a deviation from the expected dynamics of genetic variation. Deciding whether the variants observed in a genomic region deviate from the expected dynamics is dependent on the propensities assigned to these types of variants under the background model. A background model for single-nucleotide variants (SNVs) in coding regions should be able to describe the probabilities of synonymous and nonsense alterations (6,7), as well as all the different types of missense variants (8). Models of genetic variation in coding regions are also useful for estimating the functional damage caused by variants in these regions (9). Indeed, most prediction algorithms that estimate the damage or pathogenicity of variants (e.g. SIFT, Polyphen2 and CADD) rely on such background propensities (10–12). Although variant evaluation tools are heavily used in clinical settings to assess the impact of mutations on human diseases (13–15), the background propensities on which they rely are usually based on long-term cross-species evolution and are not optimized for variants within humans (16–19).
Many models of genetic variation dynamics are based on molecular clocks, which attempt to capture the time spans per mutation events (20,21). However, unlike in other model organisms, time-based mutation rates are challenging to estimate in humans, due to the lack of controlled environments and complex population and reproduction patterns (e.g. migration, admixture, ancestral population-structure, parental age and generation time) (22,23, Rahbari, 2016 #641,24,25). To overcome these challenges, an attempt to estimate human mutation rates was made by counting de novo mutations within parent-child samples. However, the short timescale of such analyses (up to a few generations) mostly ignores the effects of natural selection (26,27). An alternative approach, which circumvents most of these challenges, is to ignore the timescales of mutation events and focus instead on their relative probabilities.
Traditionally, studies of genetic variation dynamics in coding regions make use of amino acid (AA) substitution matrices such as PAM and BLOSUM (28–30). These matrices score AA substitutions by their likelihood (or likelihood ratio), as derived from empirical observations. Specifically, these classic matrices rely on multiple sequence alignment (MSA) of evolutionarily related homologous sequences across species (31) for the deduction of substitution propensities. Notably, a minor error was observed in the compilation of the original BLOSUM matrices (32), and attempts to correct the matrices showed slightly improved performance with respect to homology search (33). Nevertheless, the original BLOSUM62 is still the de facto standard and the default substitution matrix used across protein database searches (e.g. in BLAST), sequence alignment and other bioinformatics tools (31,34–36). In the past 40 years, numerous other substitution matrices have been developed to overcome many of the limitations of PAM and BLOSUM, or to address specific tasks such as the identification of remote homologies (37,38), various genomic or protein regions (e.g. protein domains) (39,40) or different types of proteins (e.g. membrane proteins, enzymes, etc.) (41).
AA substitution matrices also indirectly capture the chemical and biophysical properties of AA and are thus heavily used in the study of protein evolution (42,43). However, since existing matrices are all based on cross-species homology, they are not optimized for human-centric studies. Additionally, current cross-species AA substitution matrices lack directionality (i.e. they do not distinguish between substitution of a first AA to a second AA from a substitution of the second to the first).
The exponential growth in the number of whole-genome and whole-exome sequences, including that of healthy humans (3,44, Fu, 2013 #637), provides an opportunity to form a robust human-specific background model of genetic variation. To this end, we exploited a rich collection of >7M polymorphic sites in the exomes of over 60 000 unrelated, healthy individuals extracted from the Exome Aggregation Consortium (ExAC) (44). From this comprehensive dataset, we constructed a set of human-specific substitution matrices. These matrices provide a solid baseline for genetic and proteomic variation, which may be used for deriving evolutionary and functional insights. We analyze the information contained in our matrices and compare them to the classic matrices. We further used the developed methodology to expose protein-functional constraints, as reflected by post-translational modification (PTM) and ion-binding sites. We provide the community with a generic framework for utilizing aggregated genetic variation data to produce substitution matrices for a broad range of genetic, functional and evolutionary tasks.
MATERIALS AND METHODS
Data
To construct human-specific codon and AA substitution matrices, we combined genetic variation data with annotations of coding genes. Human genetic variants were extracted from ExAC (44), which provides a good combination of quantity and quality of genetic data in the human population. While ExAC is biased towards individuals of European ancestry, it also includes other ethnicities. Specifically, the distribution of ethnic groups in ExAC is: 36% Finnish, 36% Non-Finnish European, 10% South Asian, 7% African or African American, 6% Native American, 5% East Asian and 1% Others. Importantly, the cohort of ExAC was chosen to minimize bias of pathogenic variants, by excluding individuals with rare genetic diseases. We treat each variant as a substitution from the major allele, defined as the most frequent allele in the population, to the minor allele, which is any other allele observed in the specific exomic location.
Functional gene annotations were based on the UniProt database and the GENCODE project. The exact procedure of combining genetic and proteomic annotations is described in (45). Briefly, we used version 19 of GENCODE (compatible with version GRCh37 of the human reference genome, which was used by ExAC). We recovered the DNA sequences of the genes annotated in GENCODE using UCSC’s reference genome. We considered only the protein-coding regions of genes (annotated as ‘CDS’ in GENCODE). Protein sequences and protein annotations (e.g. PTMs) were taken from UniProt for all 20 168 reviewed human proteins (from the SwissProt section). We only considered the GENCODE gene isoforms identical to the primary UniProt protein sequences. We discarded genes that failed this exact one-to-one mapping, ending up with 18 115 successfully mapped genes. These combined genetic-proteomic gene entities allowed us to determine the protein-level consequences of genetic variants (e.g. synonymous, missense or nonsense). This pipeline is available as an independent open-source Python library (https://github.com/nadavbra/geneffect).
ExAC recorded 8 307 864 high-quality genetic variants (with ethnic distribution as detailed above). From this dataset, 427 491 indels were removed. Of the remaining variants, 326 369 genomic positions contained multiallelic variants, which were counted as 631 985 nucleotide substitutions, contributing 305 616 additional variants. From this dataset, 82 991 nonsense variants were discarded. Of the 8 102 999 nucleotide substitutions, 4 693 538 were within the coding regions of the 18 115 mapped protein-coding genes and were interpreted as observed codon substitutions. Each substitution of codons differing by exactly one nucleotide was observed ∼9000 times on average (and at least 573 times across all codon pairs; the total counts per each codon pair is reported in Supplementary Dataset S1). The high number of observations, even for the rarest codon substitutions, ensures the robustness of the estimated codon-substitution probabilities to random noise.
PTMs and ion-binding site annotations
PTMs and ion-binding site annotations were extracted from UniProt, except for ubiquitination annotations, which were obtained from PhosphoSitePlus (46). For a small number of cases, PTM proteomic locations of variants mapped to locations on the reference genome coding for inappropriate AA. We discarded these instances. Importantly, both UniProt and Phosphosite annotations are supported by experimental evidence and literature support.
Constructing probabilistic substitution matrices from genetic variation in the human population
HC1 (Human Codon substitution matrix to the power of 1) represents the estimated substitution probability of each pair of the 61 coding codons (i.e. excluding the three stop codons). Each non-diagonal entry of HC1 was calculated by:
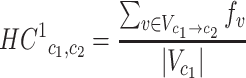 |
(1) |
Where 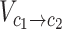 denotes the set of all variants substituting codon
denotes the set of all variants substituting codon 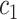 to
to 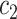 (
(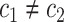 ),
),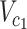 the set of all variants substituting
the set of all variants substituting 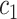 to any codon (including a self-substitution), and
to any codon (including a self-substitution), and 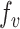 the frequency of the
the frequency of the 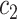 allele in a variant
allele in a variant 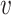 substituting
substituting 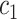 to
to 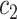 (as derived from ExAC).
(as derived from ExAC).
The diagonal values were calculated by:
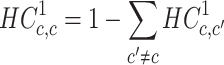 |
(2) |
We assumed the directionality of each variant to be from the major to the minor allele. In particular, we always have that 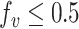 . Note that the major allele is not always the reference allele (i.e. the allele matching the human reference genome). Specifically, this is not the case in ∼8% of the ∼5M processed variants. Since ExAC’s only offers aggregated data about each exomic position independently, this construction ignores possible dependences between variants.
. Note that the major allele is not always the reference allele (i.e. the allele matching the human reference genome). Specifically, this is not the case in ∼8% of the ∼5M processed variants. Since ExAC’s only offers aggregated data about each exomic position independently, this construction ignores possible dependences between variants.
The non-substitution (diagonal) values of the matrix complement the sum of each row to 1. This, along with the normalization in Equation (1), yields a row-stochastic matrix, meaning that each row 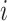 of the matrix can be interpreted as the distribution of conditional substitution probabilities (from the
of the matrix can be interpreted as the distribution of conditional substitution probabilities (from the 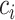 codon to every other codon). Each entry, in turn, may be interpreted as the conditional probability of observing
codon to every other codon). Each entry, in turn, may be interpreted as the conditional probability of observing 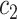 in a genomic position where
in a genomic position where 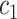 is the major allele.
is the major allele.
The amino acid substitution matrix HA1 (Human Amino acid substitution matrix to the power of 1) is derived from HC1 by considering codon frequencies:
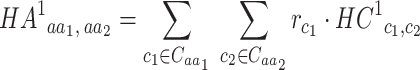 |
(3) |
Where 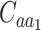 and
and 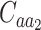 denote the sets of codons that code for the AA
denote the sets of codons that code for the AA 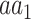 and
and 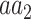 , respectively, and
, respectively, and 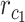 is the frequency in which
is the frequency in which 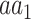 is coded by the
is coded by the 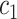 codon, relative to all the codons of
codon, relative to all the codons of 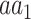 (i.e.
(i.e. 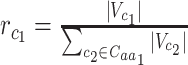 ).
).
As with HC1, an entry of HA1 may be conceived as the conditional probability of observing AA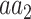 in a proteomic position in which
in a proteomic position in which 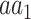 is the major allele.
is the major allele.
As HC1 and HA1 were generated through statistical analysis of single-nucleotide variations, substitutions between codons that differ in more than one nucleotide cannot be directly inferred. Since most codon pairs differ by more than one nucleotide, both HC1 and HA1 are sparse matrices. To estimate substitution frequencies between pairs of codons requiring multiple consecutive substitution events to transition between them, we treat HC1 and HA1 as the transition matrices of Markov chains. For every number of consecutive substitutions k, the k-th power matrices HCk (Human Codon substitution matrix to the power of k) and HAk (Human Amino acid substitution matrix to the power of k) represent the Markov chains that are the result of repeating the original Markov chains k times. To obtain a complete substitution matrix (i.e. with non-zero substitution probabilities for all possible substitutions), we chose to take HA1 to the power of 3, since 3 is the lowest number of nucleotide substitutions required between each pair of coding-codons. The resulting matrix is denoted HA3.
Comparing the AA substitution model to a nucleotide substitution model
To examine to what extent our substitution matrix reflects evolutionary signals at the AA level, we compared it to a matrix derived from nucleotide substitution frequencies, HAN1 (Human Amino Acid substitution matrix based on Nucleotide substitution probabilities to the power of 1), which we used as a simple background model. We first constructed a 4×4 nucleotide substitution matrix, HN1 (Human Nucleotide substitution matrix to the power of 1), derived from the same set of variants used to construct HC1. We then used this nucleotide-level matrix to derive an expected 61×61 codon substitution matrix, by considering the probability of a codon substitution to be the product of the probabilities of the single-nucleotide substitutions involved in that codon (e.g. the substitution of CTG to TTA was assigned the probability of C to T multiplied by the probability of T to T and the probability of G to A). This codon-level substitution matrix was then projected into a 20×20 AA substitution matrix, through the same process used to convert HC1 to HA1 (see previous section). The resulting AA-level matrix reflects the expected probabilities of AA substitutions given only the substitution preferences of single nucleotides while assuming a lack of evolutionary pressure at the level of codons or AA. By dividing the empirical HA1 with this background model (entry-wise), we obtained the observed-to-expected probability ratios of all AA substitutions and could determine which AA substitutions show a substantial deviation from the background model. Additionally, we tested for statistical significance of each of the deviations by using the entries of HAN1 as a background model for the observed probabilities in HA1. Specifically, for each AA substitution, we considered the probability of that substitution (given the source AA) based on the corresponding entry in HAN1. Based on this binomial distribution, we calculated the probability of obtaining the observed number of substitutions underlying HA1 (i.e. 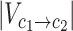 ) or more extreme deviations from the expected probability, resulting the reported p-values.
) or more extreme deviations from the expected probability, resulting the reported p-values.
Deviation from symmetry
To quantify the strength of directionality observed in the substitution matrix, we measured the deviation of AA substitutions from symmetry by calculating the ratios between the conditional probabilities of substitutions to the conditional probabilities of the opposite substitutions. For example, the directionality of the substitution of lysine (K) to arginine (R) under HA3is measured by 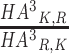 . In other words, we divided HA3, entry-wise, by (HA3)T. To enhance readability, the matrix shown in Figure 3A is lower triangular, and its entries are transformed by log2. This means that positive entries signify substitutions that are preferred over their opposite substitution.
. In other words, we divided HA3, entry-wise, by (HA3)T. To enhance readability, the matrix shown in Figure 3A is lower triangular, and its entries are transformed by log2. This means that positive entries signify substitutions that are preferred over their opposite substitution.
Figure 3.

Deviation from symmetry. (A) The asymmetry of an AA substitution is measured by the probability-ratio between each substitution and its opposite (shown in log2 scale). Probabilities are derived from a version of HA3 which does not include synonymous probabilities (so that each row represents the conditional probabilities of non-synonymous substitutions). (B) Network representation of AA substitutions that substantially deviate from symmetry (defined by a ratio of at least 5). For example, serine (S) is 14 times more likely to substitute into tryptophan (W) than the other way around. Wider arrows signify stronger asymmetry (i.e. higher ratios).
Comparison to BLOSUM and PAM
To compare our substitution models to the score matrices of BLOSUM, Spearman’s correlation coefficient (ρ) was measured for each row of HA3 with each corresponding row of the examined BLOSUM matrix (Figure 4). For each BLOSUM matrix, its average correlation with HA3 was used to summarize these measurements. Furthermore, to examine the effect of the power of the HA1 matrix on these correlations, the average correlation coefficient was calculated for each BLOSUM version and different powers of HA1. We compared our models to the original (uncorrected) BLOSUM matrices, since they are the standard matrices used in the field. Similar analyses were repeated for the PAM matrices.
Figure 4.

Comparison to the BLOSUM and PAM substitution matrices. (A) Spearman’s correlation coefficients (ρ) between the rows of HA3 and the corresponding rows of different versions of the BLOSUM matrices. For example, the rightmost bar refers to the consistency between the substitution probabilities of glycine (G) according to HA3 and the corresponding substitution scores according to BLOSUM100. Dotted lines signify the average correlation coefficients across all AA. (B) Spearman’s correlation coefficients (ρ) between the rows of HA3 and the corresponding rows of different versions of the PAM matrices. (C) The average correlation coefficients between BLOSUM matrices and different powers of HA1. Increasing powers of HA represent an increasing number of consecutive substitutions (which signify longer evolutionary timescales). (D) Average correlation coefficients between PAM matrices and different powers of HA1.
Functional annotation analysis
To demonstrate the capacity of our human-specific substitution model to reflect protein functional annotations (Tables 1 and 2, Figure 5), we examined nine major PTMs (acetylation, hydroxylation, disulfide bond formation, methylation, N-linked glycosylation, O-linked glycosylation, phosphorylation, succinylation and ubiquitination; Table 1) and three types of ion-binding sites (zinc, magnesium and iron; Table 2). For each PTM or ion-binding site, we considered the set of variants at the annotated proteomic locations, and generated new codon and AA substitution matrices corresponding to that subset of variants (e.g. HA1iron-binding, HC3ubiquitination etc.). These matrices were generated through the same method by which the global matrices (e.g. HA1 and HC3) were constructed, and they differ only by the subset of used variants (within the ExAC dataset). To highlight the unique aspects of the substitution profiles for these functionally annotated sites, compared to unannotated sites (Figure 5), each annotation-specific matrix was divided by its corresponding non-annotation matrix, element-wise (i.e. the matrix derived from all the other variants).
Table 1.
Statistically significant AA substitutions in PTM sites
| PTM annotation | From AA | To AA | # sub. w/ annotation | # sub. w/o annotation | # variants FDR q-value | AF FDR q-value | Trenda |
|---|---|---|---|---|---|---|---|
| Acetylation | A | A | 212 | 1 54 569 | 1.94E-02 | E | |
| A | T | 51 | 91 640 | 4.15E-10 | 1.27E-02 | D/D | |
| A | V | 122 | 84 602 | 3.16E-02 | D | ||
| K | K | 398 | 48 774 | 6.89E-03 | E | ||
| K | N | 128 | 22 414 | 2.35E-02 | D | ||
| K | Q | 63 | 10 543 | 4.75E-02 | D | ||
| Disulfide bond | C | C | 2014 | 25 117 | 9.70E-32 | E | |
| C | F | 311 | 5700 | 6.68E-03 | D | ||
| C | G | 238 | 4429 | 8.13E-03 | 2.62E-02 | D/D | |
| C | R | 648 | 11 634 | 3.93E-04 | 6.99E-05 | D/D | |
| C | S | 412 | 8104 | 2.56E-06 | 1.01E-02 | D/D | |
| C | W | 175 | 3205 | 4.18E-02 | D | ||
| C | Y | 870 | 16 183 | 6.68E-08 | 5.26E-05 | D/D | |
| Hydroxylation | K | K | 105 | 49 091 | 7.65E-05 | E | |
| Methylation | K | K | 54 | 49 140 | 2.36E-03 | E | |
| R | R | 154 | 86 468 | 3.05E-02 | E | ||
| N-linked Glycosylation | N | D | 657 | 16 080 | 1.79E-02 | D | |
| N | H | 274 | 7345 | 6.70E-03 | D | ||
| N | N | 2441 | 51 886 | 2.25E-02 | E | ||
| O-linked Glycosylation | S | S | 82 | 1 48 408 | 1.74E-02 | E | |
| T | T | 105 | 1 42 197 | 1.44E-02 | E | ||
| Phosphorylation | S | A | 285 | 9066 | 4.08E-02 | 2.68E-03 | D/D |
| S | F | 1054 | 25 354 | 4.47E-04 | E | ||
| S | L | 1176 | 30 503 | 3.41E-02 | D | ||
| S | G | 706 | 21 817 | 6.68E-03 | D | ||
| S | S | 5403 | 1 43 087 | 1.96E-02 | E | ||
| S | T | 669 | 19 274 | 3.39E-04 | D | ||
| T | T | 1471 | 1 40 831 | 3.41E-02 | D | ||
| Y | H | 85 | 15 654 | 1.38E-02 | D | ||
| Y | Y | 454 | 50 960 | 3.06E-06 | E | ||
| Succinylation | K | E | 66 | 28 051 | 2.08E-02 | D | |
| K | K | 106 | 49 066 | 2.08E-02 | D | ||
| K | R | 86 | 35 506 | 2.08E-02 | E | ||
| Ubiquitination | K | E | 4091 | 24 020 | 3.40E-10 | D | |
| K | I | 324 | 2060 | 1.47E-03 | D | ||
| K | K | 7536 | 41 630 | 1.67E-06 | 3.47E-07 | E/D | |
| K | M | 422 | 2752 | 4.78E-02 | 5.41E-03 | D/D | |
| K | N | 2980 | 19 562 | 3.29E-10 | 7.44E-09 | D/D | |
| K | Q | 1477 | 9129 | 4.78E-02 | D | ||
| K | R | 5420 | 30 168 | 1.30E-03 | 2.47E-09 | E/D | |
| K | T | 1589 | 9119 | 1.40E-07 | D |
aTrends of significant differences between annotated and unannotated residues. Symbols indicate enrichment (E) or depletion (D). When both statistical tests, based on the number of variants (left) and allele frequency (right), are significant (q-value < 0.05), then two distinct symbols are shown. Synonymous substitutions that maintain the same AA are shown in bold.
Table 2.
Statistically significant AA substitutions in ion-binding sites
| Ion-binding annotation | From AA | To AA | # sub. w/ annotation | # sub. w/o annotation | # variants FDR q-value | Trend |
|---|---|---|---|---|---|---|
| Iron | H | H | 70 | 45 629 | 8.67E-04 | E |
| Magnesium | D | D | 73 | 69 334 | 1.72E-04 | E |
| Zinc | H | H | 182 | 45 517 | 2.25E-06 | E |
| H | R | 56 | 27 688 | 1.29E-03 | D | |
| C | C | 132 | 27 001 | 2.25E-06 | E |
Figure 5.
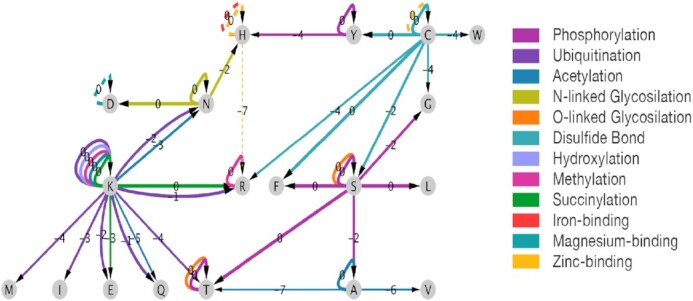
Enrichment of AA substitutions in PTM and ion-binding sites. Each edge represents a substitution between a source and a target AA in the context of a specific annotation (indicated by edge color). Dashed arrows indicate ion-binding annotations. Edges are annotated with the log2 ratio of the substitution probabilities between annotated and unannotated sites. Arrow widths signify the magnitude of these ratios.
To test the significance of each annotation-specific substitution (e.g. lysine [K] to proline [P] in ubiquitination sites), we examined two complementary aspects of significance, based on either (i) the number of annotated variants or (ii) their allele frequencies (AF). In terms of the number of variants, a substitution may exhibit a significantly higher or lower number of variants in sites annotated by that PTM or ion binding. To test whether a substitution 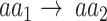 is significantly associated with an annotation in terms of the number of variants, we considered the set of all variants whose major allele is
is significantly associated with an annotation in terms of the number of variants, we considered the set of all variants whose major allele is 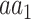 , and used Fisher’s exact test to determine if the subset of these variants that substitute into
, and used Fisher’s exact test to determine if the subset of these variants that substitute into 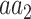 is enriched with the subset of variants with the tested annotation. Likewise, to test differences in AF, we used Mann–Whitney U test (two-tailed) to compare the AF of the variants inducing the tested substitution which are in annotated versus unannotated sites (e.g. lysine [K] to proline [P] variants in ubiquitinated versus non-ubiquitinated sites). In both tests, we required a sample size of at least 50 annotated variants. To control the false discovery rate, Benjamini–Hochberg FDR was applied for each of the two types of tests, across all annotation-specific substitutions, with a significance threshold of 0.05. In this work we show the annotation-specific substitutions that are FDR-significant according to at least one of the two tests (Tables 1 and 2 and Figure 5). Significant annotation-specific substitutions are labeled as either enriched (E) or depleted (D) for each of the two tests. With respect to the number of variants, we consider it to be enriched if the odds-ratio is >1. With respect to the AF test, we consider it to be enriched when the average AF of annotated variants is greater than that of unannotated variants.
is enriched with the subset of variants with the tested annotation. Likewise, to test differences in AF, we used Mann–Whitney U test (two-tailed) to compare the AF of the variants inducing the tested substitution which are in annotated versus unannotated sites (e.g. lysine [K] to proline [P] variants in ubiquitinated versus non-ubiquitinated sites). In both tests, we required a sample size of at least 50 annotated variants. To control the false discovery rate, Benjamini–Hochberg FDR was applied for each of the two types of tests, across all annotation-specific substitutions, with a significance threshold of 0.05. In this work we show the annotation-specific substitutions that are FDR-significant according to at least one of the two tests (Tables 1 and 2 and Figure 5). Significant annotation-specific substitutions are labeled as either enriched (E) or depleted (D) for each of the two tests. With respect to the number of variants, we consider it to be enriched if the odds-ratio is >1. With respect to the AF test, we consider it to be enriched when the average AF of annotated variants is greater than that of unannotated variants.
RESULTS
Constructing human-specific coding substitution matrices
To construct an AA substitution matrix that is specific to the human population (Figure 1A), we merged data from two complementary sources: (i) the ExAC population database, which reports on >7M high-quality single nucleotide variants (SNVs) from the exome sequences of 60 706 non-related individuals (44) and (ii) genomic annotations for all human coding genes. By projecting the >7M SNVs on the gene annotations, we inferred >4.8M observed codon substitutions, found in 37% of all codons in the human coding genome. Overall, 65% of the substitutions are missense, 33% are synonymous and 2% are stop-gain (nonsense) variants.
Figure 1.

Producing human-specific substitution matrices from population genetic variation. (A) We combined 4.8M SNVs with 18K gene annotations to construct a human-specific codon substitution matrix, denoted HC1, in which each entry represents a codon substitution probability. To capture codon substitutions that differ by more than a single nucleotide, we extended the sparse HC1 into a complete HC3matrix through a Markovian process. We further derive corresponding matrices at amino acid resolution, HA1and HA3, in which each entry represents the probability of an amino acid substitution. (B) HC3 (log10 scale). (C) HA3 (log10 scale).
From these codon substitutions and their corresponding allele frequencies (AFs), we constructed a 61×61 codon substitution matrix, denoted HC1 (standing for Human Codon substitutions). The rows and columns of HC1represent all codons (excluding the three stop codons), with rows representing source codons and columns representing the target codons of substitutions. Rows are normalized so that each entry represents the conditional probability of a codon substitution.
To provide substitution probabilities at AA resolution, we transformed HC1into a 20×20 AA substitution matrix, denoted HA1. As our construction relies on SNVs, HC1can capture only pairs of codons that differ by up to a single letter. As a result, 84% of all codon substitutions in HC1 are assigned zero probability. Likewise, 57.5% of HA1values are zero. To model the propensities of all replacements, we considered three consecutive transitions of HC1, thereby deriving a complete codon substitution matrix denoted HC3 (Figure 1B) and a corresponding AA substitution matrix HA3 (Figure 1C). The numeric values of HC1, HA1, HC3, and HA3, are provided in Supplementary Dataset S2.
Patterns of amino acid substitutions in the human population
We examined whether the observed 20×20 AA substitution frequencies are exclusively determined by the transition propensities of nucleotide substitutions. To this end, we constructed a 4×4 single-nucleotide substitution matrix derived from all the reported SNVs, denoted HN1 (Figure 2A; Supplementary Dataset S3, Supplementary Figure S1). This matrix accounts for all 16 single-nucleotide replacement frequencies observed across coding regions in the human population. From HN1, we derived a 20×20 AA substitution matrix, denoted HAN1, which represents the expected AA substitution probabilities under the background model of single-nucleotide substitution propensities described by HN1. We then compared the expected values of HAN1to the empirically observed values of HA1through an element-wise division of the two matrices (Figure 2B). We observe that substitutions of tryptophan (W) to cysteine (C) or serine (S) are roughly 8-fold lower than expected by this naive nucleotide-based background model. Similarly, a substitution from isoleucine (I) to lysine (K) is 16-fold lower than expected. Overall, we reveal that the expected-to-observed ratios are >2 (or <0.5) for 11% of AA pairs, and >1.5 (or <0.66) for 20% of AA pairs. By considering HAN1 as a background model and testing the likelihood of the observed probabilities in HA1, we validated that the differences between the matrices across all AA substitutions (as reported above) are indeed robust and could not have been produced by chance (P-value < 1E-300; see Materials and Methods). These results expose evolutionary signals, even at the low resolution of AA.
Figure 2.
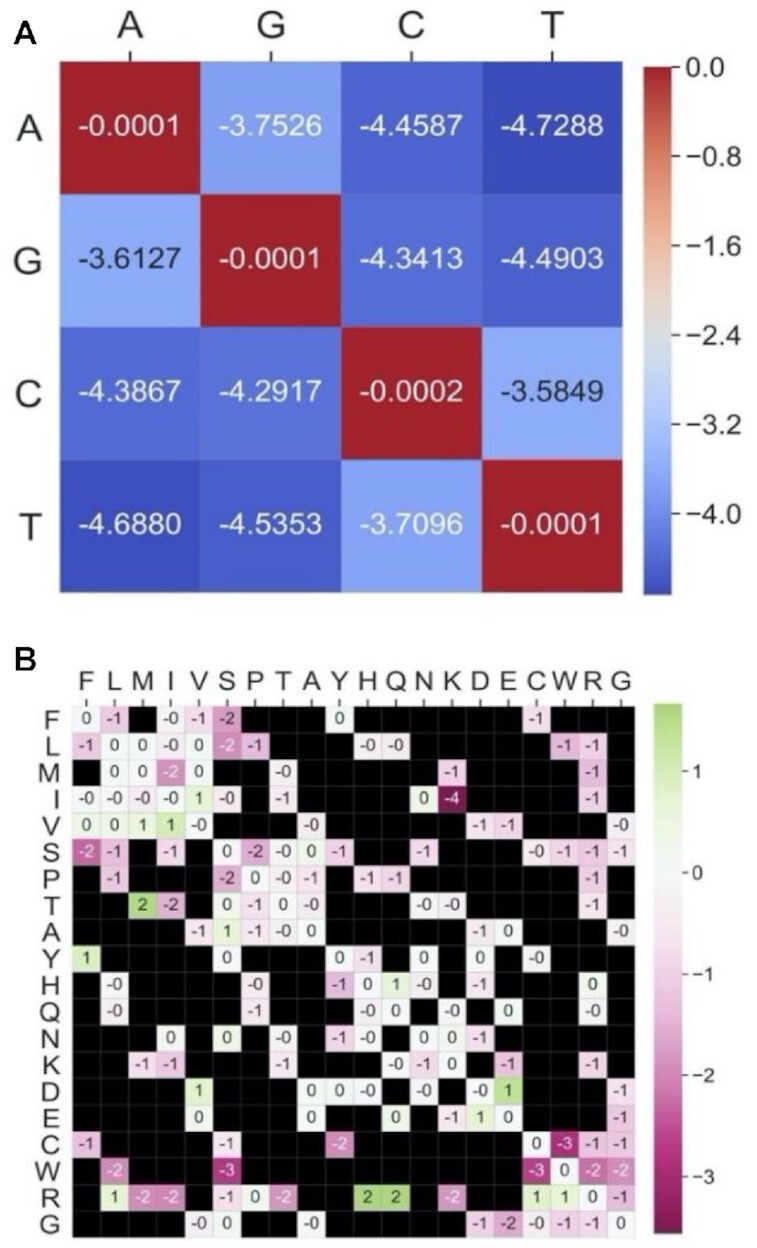
Deviation from single-nucleotide propensities. (A) Values of HN1, the human-specific single-nucleotide substitution matrix capturing the propensities of nucleotide transitions (log10 scale). (B) Observed-to-expected ratios of the entries in HA1 (log2 scale), based on a background model derived from the 4×4 frequencies of nucleotide substitutions described in (A). A log-ratio close to 0 signifies a substitution whose observed frequency is as expected by the substitution tendencies of the nucleotides in the involved codons. Negative log-ratios (colored pink) represent substitutions that are less common than would be expected by their nucleotide composition, while positive log-ratios (green) represent frequencies that are higher than expected.
A probabilistic substitution model is substantially enriched by considering the directionality replacements. We utilized information on the allele frequencies (AF) of genetic variants to deduce the most likely direction of each observed substitution. We measured the asymmetries of substitutions by the ratio between each AA replacement to its opposite (Figure 3A) and highlighted extreme signals of asymmetry (Figure 3B). Some AA substitutions show an order-of-magnitude stronger tendency in one direction compared to the other direction. The AA with the most extreme directionality is tryptophan (W). Specifically, most AA are more likely to substitute into this AA than the other way around. Notably, serine (S) exhibits a 14-times higher tendency to substitute into tryptophan (W) than the opposite substitution. Tryptophan is a biochemically distinct AA, signified by maximal hydrophobicity, bulkiness and the lowest solvation potential (47). From a structural perspective, tryptophan has been implicated in lipid anchoring of membranous proteins (48), binding hotspots and substrate-binding sites (49). Whereas tryptophan (W) emerges as a target hub, valine (V) and isoleucine (I) tend to be source hubs (Figure 3B).
Examining the directionality of AA substitutions (Figure 3) at codon resolution reveals that the directional signal is often dominated by a specific codon substitution (Supplementary Figure S2). For example, In the case of isoleucine (I) to lysine (K), it is the AAA (K) to ATA (I) codon substitution which dominantly defines the asymmetry of the AA substitution, by occurring 8 times more frequently in that direction than the opposite (Supplementary Dataset S3). Similarly, we observe that valine (V) is more likely to substitute into tyrosine (Y) in the context of the target codon TAT (Y), while an opposite tendency is in fact observed for the second codon of tyrosine (TAC). Note that the substitution of valine (V) to tyrosine (Y) requires more than one step of nucleotide replacements.
Human-specific and cross-taxa substitution matrices capture different signals
To highlight the unique features of the human-specific AA substitution matrices, we compared HA3 with the (original) BLOSUM and PAM matrices (Figure 4). We observe that the correlation in substitution propensities between HA3 and the BLOSUM matrices is similar for BLOSUM62 and BLOSUM100 (average across all 20 AA: ρ = 0.52; Figure 4A) but lower for BLOSUM30 (ρ = 0.45). As BLOSUM30 captures longer evolutionary distances, it is expected to be more different from the human-specific matrix. BLOSUM30 also shows greater variability across different AA. Interestingly, isoleucine (I) and tryptophan (W) exhibit higher correlations for the scores of BLOSUM30 than those of BLOSUM62 and BLOSUM100. It should be noted, however, that AA-specific correlations are prone to noise, as they are based on only 20 values (while the overall trend, based on 400 values, are much more robust).
A similar conformity analysis was performed for a set of PAM matrices (Figure 4B). The observed correlations in the case of the PAM matrices (average ρ of 0.61–0.68) are higher than those observed for the BLOSUM matrices. The most substantial conformity of HA3 is noted for PAM10, which represents substitution probabilities between highly similar protein sequences. A correlation analysis was also performed for PAM1, yielding the highest conformity (ρ = 0.69; Supplementary Figure S3). Notably, tyrosine (Y) and cysteine (C) register consistently low conformity between the human-specific and cross-taxa matrices (BLOSUM and PAM). Similar analyses were also conducted based on Pearson’s (rather than Spearman’s) correlation, showing the same trends. However, the correlations in the case of the BLOSUM matrices (average ρ of 0.75–0.80) are higher than those observed for the PAM matrices (Supplementary Figure S4).
HA3 represents the expected pairwise substitution probabilities following three consecutive single-nucleotide substitutions. We inquired whether the correlations reported for HA3 are sensitive to the selection of exactly three transitions in the Markovian process. We repeated the comparison between HAk and BLOSUM (Figure 4C) or PAM (Figure 4D) for different values of k, namely following different numbers of single-nucleotide transitions. We observe that the degree of conformity is steady along many consecutive substitutions. We conclude that these results are robust to the choice of k = 3, which is the minimal number of single-nucleotide substitutions required to obtain all possible AA replacements. HC3 and HA3 have the distinct advantage of avoiding ad-hoc numerical manipulations to pull codon substitution probabilities away from zero, such as adding a small epsilon term (for codons that differ by more than one nucleotide). Instead, the Markov chain framework rigorously imputes probabilities for all codon substitutions.
The human-specific substitution model exposes protein function preservation
We investigated how AA substitution tendencies change in the context of protein functional sites. To this end, we examined nine prominent PTMs (acetylation, disulfide bond, hydroxylation, methylation, N- and O- glycosylation, phosphorylation, succinylation and ubiquitination) and three types of ion-binding sites (zinc, magnesium and iron). Altogether, we examined 92 625 experimentally validated functional sites covering 12 746 unique proteins. We tested for differences in AA substitution propensities between annotated and unannotated sites according to two complementary aspects of significance: (i) the number of variants and (ii) their allele frequencies (AFs). We found 41 substitutions with significantly different propensities in the context of specific PTM sites (Table 1). For example, alanine (A) to threonine (T) substitutions are significantly depleted in acetylated sites with respect to both the number of observed variants (FDR q-value = 4.2E-10) and their allele frequencies (FDR q-value = 1.3E-02). The most significant association is the conservation of cysteine (C) residues involved in disulfide bonds (FDR q-value = 9.7E-32). We attribute this result to the fundamental importance of disulfide bonds in stabilizing the structural fold of a protein. Generally, we find residues to be preserved in the majority of PTMs, while variants causing AA replacement (i.e. missense variants) are generally depleted. For ion-binding residues, 5 AA substitutions were found to be significantly different in annotated sites (Table 2). A complete summary of all AA substitutions with respect to all PTM and ion-binding sites is available in Supplementary Dataset S4.
Having determined which AA substitutions exhibit significant changes in functional sites, we applied our model to examine these differences in the context of substitution propensities (Figure 5 and Supplementary Figure S5). Specifically, we considered the entry-wise ratios of HA3 between annotated and unannotated sites by reconstructing the matrix for each of the two states (e.g. the substitution propensity ratio between phosphorylated serine [S] to alanine [A] and non-phosphorylated serine [S] to alanine [A]). AA substitutions that appear particularly unfavored in specific functional contexts are acetylated lysine (K) to glutamine (Q), zinc-binding histidine (H) to arginine (R), and hydroxylated alanine (A) to valine (V) or threonine (T). Lysine (K) can undergo many types of PTMs, several of which display a strong selection signal under our model. Specifically, lysine (K) residues modified by ubiquitin, acetyl, and succinyl show a high preservation tendency compared to unmodified lysine residues. Even though we considered each lysine (K) independently, it should be noted that the same lysine residues can be used for different PTM types, depending on cell state. For example, the majority of succinyl-modified lysine residues overlap with acetylation sites (50). We did not analyze less prevalent types of lysine modifications (e.g. sumoylation) due to limited experimental evidence.
Collectively, these results are best explained by a signal of negative selection associated with major functional sites in the human proteome. We conclude that a human-centric model of AA substitution propensities based on genetic variation in the human population is a powerful and intuitive tool to study AA across functional contexts such as PTMs and ion binding.
DISCUSSION
In this study, we have presented a set of data-driven novel substitution matrices at codon and AA resolutions based on the natural occurrence of genetic variation in the healthy human population. This human-centric approach reflects the short evolutionary timescale of modern humans (51), while the classic BLOSUM and PAM (as well as many others) substitution matrices are based on MSA sequences from organisms whose common ancestor is dated back many millions of years. The purpose of this work is to highlight patterns in human genetic variation that are not captured by the traditional substitution matrices, which were designed for remote homology searches. Despite this fundamental difference, HA3 still shows a moderate correlation with the classic matrices (Figure 4). In particular, matrices that reflect shorter evolutionary distances (e.g. BLOSUM100 and PAM10) exhibit higher similarity to the human-centric matrix.
In this work, we used a Markov chain model to derive the non-sparse matrices HC3 and HA3 from the empirical HC1 and HA1 matrices. The construction of these matrices through a Markov chain process assumes that multiple-nucleotide codon changes are derived from independent single-nucleotide substitutions. In reality, complex codon changes can sometimes occur as a single indel event, for example through a series of alterations by the DNA repair systems (52). Indeed, codon substitution frequencies show substantial variability between species due to differences in DNA repair pathways (53). This is one reason why generalization of human-centric substitution models to other taxa would be inappropriate. As future work, richer models could be utilized to create more accurate matrices.
Notable differences in substitution propensities between the cross-species and human-centric evolutionary contexts are observed for cysteine (C), tyrosine (Y) and tryptophan (W) (Figure 4). These AA are specified by unique biochemical, functional and structural features (43). Specifically, these three AA have a strong interface propensity and a negative solvation potential (54), and contribute uniquely to folding and protein-protein interaction interfaces in the human proteome (55). Furthermore, these AA are the most likely to occur in rare mutations causal of human disease (56).
We argue that a human-centric model is more appropriate for assessing the impact of human mutations in coding genes, a practice central to genetic consulting and the identification of causal mutations in human diseases. For the task of non-synonymous mutation inference, dozens of prediction tools and algorithms have been developed. With few exceptions (e.g. FIRM, (45)), the vast majority of these tools (e.g. Panther, PhD‐SNP, PolyPhen2, SIFT, SNAP, and SNPs&GO MutPred, nsSNPAnalyzer) incorporate long-range evolutionary information from cross-species conservation into the underlying model (15–17,57–59). We anticipate that the incorporation of short-term, human-centric models such as HA3 could be beneficial to mutation impact evaluation.
Key processes in multicellular organisms are mediated by a network of PTMs (e.g. differentiation, cell division, inflammation and metabolism). In humans, most proteins are subject to multiple PTMs, which greatly increase the proteome's functional repertoire (60,61). Even though ∼200 types of PTMs have been detected by mass spectrometry, only a handful have been systematically studied (62,63). In this study we focused on 9 common types of PTMs and 3 types of ion binding (which may only occur in 10 specific AA), limiting our analysis to experimentally-validated sites. Notably, PTM detection is condition-specific and quite sensitive to experimental protocols. Despite this inherent noise, we were able to establish the enrichment or depletion of certain AA replacements in the context of functional annotations with high confidence (Tables 1 and 2). A particularly strong signal was detected for phosphorylation sites (phosphotyrosine, phosphoserine and phosphothreonine), ion-binding sites, and cysteine residues that form disulfide bonds. Indeed, many human diseases result from missense mutations in codons of cysteine which destroy essential disulfide bridges (64,65). Thanks to our human-specific substitution models, we were also able to quantify the enrichment of AA substitutions across PTM and ion-binding sites (Figure 5).
Interestingly, even the short-term view explored in this study exposes a robust signal of negative natural selection at codon and AA resolution (Figure 2 and Supplementary Figure S2). While most homologues showing high conservation across species are under negative selection, this conservation is typically restricted to folded domains, while loops, intrinsically disordered regions, interdomain linkers and protein tails exhibit relatively low sequence similarity across homologues. Nonetheless, such regions of low conservation are actually the preferred targets for PTMs (e.g. N'-acetylation in protein tails, or phosphorylation and ubiquitination in loops and disordered regions). Due to the lack of cross-species conservation in those sites and differences in cellular contexts (e.g. species-specific kinome), the question of whether human PTM sites are under neutral, negative, or positive selection could not be resolved with cross-species data and remained to be tested (66,67). Our analyses provide evidence for purifying selection in unstructured protein regions (Tables 1 and 2). Another advantage of studying the selection of PTMs from a human-centric perspective is that exact PTM sites are often not conserved between species. Indeed, the conservation signal for most PTMs is negligible across species, and sometimes even shows positive selection (68,69). It was proposed that the amounts of modifiable sites, rather than their exact positions, is the conserved property in many proteins (70,71).
An essential property of the constructed matrices we present is their asymmetry (Figure 3), while AA substitutions matrices from cross-species MSA are usually symmetric by design. Indeed, we have detected substantial asymmetry for some AA (Figure 3B). Specifically, tryptophan (W) and valine (V) appear to be central hubs of such directional tendencies. We found that tryptophan (W), and, to a lesser extent, glutamic acid (E) and glutamine (Q), are common substitution targets. Valine (V), and, to a lesser extent, serine (S) and isoleucine (I), act as substitution sources. Overall, we categorized all 20 AA as substitution sources, targets or a mixture of the two (Supplementary Figure S5). Interestingly, the AA marked as sources include all 6-codon AA (R, S, L) and most 4-codon ones. Inspecting the substitution matrices at codon resolution confirms that some codons may dominate the signal at the AA level (e.g. the ATA to AAA substitution dominates the asymmetric replacement of isoleucine to lysine). Under this view, the 6 serine (S) codons exhibit distinct substitution patterns, leading to broad functional implications (72). Specifically, regions that are subjected to accelerated evolution tend to substitute within a specific set of serine (S) codons, while conserved regions in the same proteins tend to substitute within the complementary set of serine codons. The specific DNA (i.e. codon) alterations dominating some of the AA substitutions, as observed in this work, might be attributed to evolutionary processes at the DNA (rather than protein) level, which could be shaped by cellular mechanisms such as DNA repair (53,73).
In summary, we have constructed human-specific substitution matrices and characterized their unique properties. Given the robustness and interpretability of these matrices, we encourage their use as a baseline model for codon and AA replacement in the human population. An example of such analysis was demonstrated in the context of PTMs and ion-binding sites (Figures 4 and 5), where our model provided a robust baseline allowing the exposure of selection signal. This was allowed by partitioning of the protein residue space within the human proteome into two groups (e.g. those subject to a specific PTM and those that lack evidence for it), and comparing the set of genetic variants occurring in the human populations between these two groups with respect to our AA substitution model (HA3). Similarly, our model could be used to study differences with respect to other protein or residue partitions (e.g. extracellular proteins, protein–protein interfaces, proteins expressed in the brain or those related to the immune system). To allow the extension of this methodology to other datasets, including specific human subpopulations, we provide the source code of our methods (see Materials and Methods). Likewise, we anticipate that our methodology could be easily applied to other organisms with rich genetic variation data [e.g. mouse (74) and primates (21,75)].
DATA AVAILABILITY
The ExAC dataset can be downloaded from https://console.cloud.google.com/storage/browser/_details/gnomad-public/legacy/exacv1_downloads/release0.3/ExAC.r0.3.sites.vep.vcf.gz. The substitution matrices, codon counts, and further results are available in the supplementary data. The entire source code of this study is available at http://www.github.com/tairsha/taxa-specific-substitution-matrix.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Nathan Linial (The Hebrew University of Jerusalem) for his support and suggestions throughout this research. We thank Liran Carmel (The Hebrew University of Jerusalem) and Uri Hirshberg (Haifa University) for their advice and useful comments. The research was partially supported by ISF grant 2753/20.
Contributor Information
Tair Shauli, The Rachel and Selim Benin School of Computer Science and Engineering, The Hebrew University of Jerusalem, Jerusalem, 91904, Israel.
Nadav Brandes, The Rachel and Selim Benin School of Computer Science and Engineering, The Hebrew University of Jerusalem, Jerusalem, 91904, Israel.
Michal Linial, Department of Biological Chemistry, Institute of Life Sciences, The Hebrew University of Jerusalem, Jerusalem, 91904, Israel.
SUPPLEMENTARY DATA
Supplementary Data are available at NARGAB Online.
FUNDING
Israel Science Foundation [2753/20].
Conflict of interest statement. None declared.
REFERENCES
- 1.Rogers J., Gibbs R.A.. Comparative primate genomics: emerging patterns of genome content and dynamics. Nat. Rev. Genet. 2014; 15:347–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Casillas S., Barbadilla A.. Molecular population genetics. Genetics. 2017; 205:1003–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grossman S.R., Andersen K.G., Shlyakhter I., Tabrizi S., Winnicki S., Yen A., Park D.J., Griesemer D., Karlsson E.K., Wong S.H.et al.. Identifying recent adaptations in large-scale genomic data. Cell. 2013; 152:703–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Steiner C.C., Putnam A.S., Hoeck P.E., Ryder O.A.. Conservation genomics of threatened animal species. Annu. Rev. Anim. Biosci. 2013; 1:261–281. [DOI] [PubMed] [Google Scholar]
- 5.Harris K.Evidence for recent, population-specific evolution of the human mutation rate. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:3439–3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hunt R., Sauna Z.E., Ambudkar S.V., Gottesman M.M., Kimchi-Sarfaty C.. Silent (synonymous) SNPs: should we care about them?. Single Nucleotide Polymorphisms. 2009; 578:23–39. [DOI] [PubMed] [Google Scholar]
- 7.Cartegni L., Chew S.L., Krainer A.R.. Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat. Rev. Genet. 2002; 3:285–298. [DOI] [PubMed] [Google Scholar]
- 8.Schork N.J., Fallin D., Lanchbury J.S.. Single nucleotide polymorphisms and the future of genetic epidemiology. Clin. Genet. 2000; 58:250–264. [DOI] [PubMed] [Google Scholar]
- 9.Cargill M., Altshuler D., Ireland J., Sklar P., Ardlie K., Patil N., Shaw N., Lane C.R., Lim E.P., Kalyanaraman N.et al.. Characterization of single-nucleotide polymorphisms in coding regions of human genes. Nat. Genet. 1999; 22:231–238. [DOI] [PubMed] [Google Scholar]
- 10.Kumar P., Henikoff S., Ng P.C.. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009; 4:1073–1081. [DOI] [PubMed] [Google Scholar]
- 11.Rentzsch P., Witten D., Cooper G.M., Shendure J., Kircher M.. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019; 47:D886–D894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R.. A method and server for predicting damaging missense mutations. Nat. Methods. 2010; 7:248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mort M., Evani U.S., Krishnan V.G., Kamati K.K., Baenziger P.H., Bagchi A., Peters B.J., Sathyesh R., Li B., Sun Y.. In silico functional profiling of human disease-associated and polymorphic amino acid substitutions. Hum. Mutat. 2010; 31:335–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shihab H.A., Gough J., Cooper D.N., Stenson P.D., Barker G.L., Edwards K.J., Day I.N., Gaunt T.R.. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden markov models. Hum. Mutat. 2013; 34:57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thusberg J., Olatubosun A., Vihinen M.. Performance of mutation pathogenicity prediction methods on missense variants. Hum. Mutat. 2011; 32:358–368. [DOI] [PubMed] [Google Scholar]
- 16.Gnad F., Baucom A., Mukhyala K., Manning G., Zhang Z.. Assessment of computational methods for predicting the effects of missense mutations in human cancers. BMC Genomics. 2013; 14(Suppl.3):S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dong C., Wei P., Jian X., Gibbs R., Boerwinkle E., Wang K., Liu X.. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum. Mol. Genet. 2015; 24:2125–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tavtigian S.V., Greenblatt M.S., Lesueur F., Byrnes G.B., Group I.U.G.V.W.. In silico analysis of missense substitutions using sequence-alignment based methods. Hum. Mutat. 2008; 29:1327–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hecht M., Bromberg Y., Rost B.. Better prediction of functional effects for sequence variants. BMC Genomics. 2015; 16(Suppl.8):S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nachman M.W., Crowell S.L.. Estimate of the mutation rate per nucleotide in humans. Genetics. 2000; 156:297–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moorjani P., Amorim C.E.G., Arndt P.F., Przeworski M.. Variation in the molecular clock of primates. Proc. Natl Acad. Sci. 2016; 113:10607–10612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lynch M.Rate, molecular spectrum, and consequences of human mutation. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:961–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burgess R., Yang Z.. Estimation of hominoid ancestral population sizes under bayesian coalescent models incorporating mutation rate variation and sequencing errors. Mol. Biol. Evol. 2008; 25:1979–1994. [DOI] [PubMed] [Google Scholar]
- 24.Campbell C.D., Chong J.X., Malig M., Ko A., Dumont B.L., Han L., Vives L., O’Roak B.J., Sudmant P.H., Shendure J.. Estimating the human mutation rate using autozygosity in a founder population. Nat. Genet. 2012; 44:1277–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tajima F.The amount of DNA polymorphism maintained in a finite population when the neutral mutation rate varies among sites. Genetics. 1996; 143:1457–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ségurel L., Wyman M.J., Przeworski M.. Determinants of mutation rate variation in the human germline. Annu. Rev. Genomics Hum. Genet. 2014; 15:47–70. [DOI] [PubMed] [Google Scholar]
- 27.Samocha K.E., Robinson E.B., Sanders S.J., Stevens C., Sabo A., McGrath L.M., Kosmicki J.A., Rehnström K., Mallick S., Kirby A.. A framework for the interpretation of de novo mutation in human disease. Nat. Genet. 2014; 46:944–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Muller T., Spang R., Vingron M.. Estimating amino acid substitution models: a comparison of dayhoff's estimator, the resolvent approach and a maximum likelihood method. Mol. Biol. Evol. 2002; 19:8–13. [DOI] [PubMed] [Google Scholar]
- 29.Henikoff S., Henikoff J.G.. Performance evaluation of amino acid substitution matrices. Proteins. 1993; 17:49–61. [DOI] [PubMed] [Google Scholar]
- 30.Mount D.W.Comparison of the PAM and BLOSUM amino acid substitution matrices. CSH Protoc. 2008; 2008:pdb.ip59. [DOI] [PubMed] [Google Scholar]
- 31.Altschul S.F.Amino acid substitution matrices from an information theoretic perspective. J. Mol. Biol. 1991; 219:555–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Styczynski M.P., Jensen K.L., Rigoutsos I., Stephanopoulos G.. BLOSUM62 miscalculations improve search performance. Nat. Biotechnol. 2008; 26:274. [DOI] [PubMed] [Google Scholar]
- 33.Hess M., Keul F., Goesele M., Hamacher K.. Addressing inaccuracies in BLOSUM computation improves homology search performance. BMC Bioinformatics. 2016; 17:189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McGinnis S., Madden T.L.. BLAST: at the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res. 2004; 32:W20–W25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mooney S.Bioinformatics approaches and resources for single nucleotide polymorphism functional analysis. Brief. Bioinform. 2005; 6:44–56. [DOI] [PubMed] [Google Scholar]
- 36.Pearson W.R.Selecting the right similarity-scoring matrix. Curr. Protoc. Bioinform. 2013; 43:3.5.1–3.5.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jones D.T., Taylor W.R., Thornton J.M.. The rapid generation of mutation data matrices from protein sequences. Bioinformatics. 1992; 8:275–282. [DOI] [PubMed] [Google Scholar]
- 38.Arvestad L.Efficient methods for estimating amino acid replacement rates. J. Mol. Evol. 2006; 62:663–673. [DOI] [PubMed] [Google Scholar]
- 39.Le S.Q., Lartillot N., Gascuel O.. Phylogenetic mixture models for proteins. Philos. Trans. R. Soc. B: Biol. Sci. 2008; 363:3965–3976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brown C.J., Johnson A.K., Daughdrill G.W.. Comparing models of evolution for ordered and disordered proteins. Mol. Biol. Evol. 2010; 27:609–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Leluk J.Regularities in mutational variability in selected protein families and the markovian model of amino acid replacement. Comput. Chem. 2000; 24:659–672. [DOI] [PubMed] [Google Scholar]
- 42.Tomii K., Kanehisa M.. Analysis of amino acid indices and mutation matrices for sequence comparison and structure prediction of proteins. Protein. Eng. 1996; 9:27–36. [DOI] [PubMed] [Google Scholar]
- 43.Harms M.J., Thornton J.W.. Evolutionary biochemistry: revealing the historical and physical causes of protein properties. Nat. Rev. Genet. 2013; 14:559–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lek M., Karczewski K.J., Minikel E.V., Samocha K.E., Banks E., Fennell T., O’Donnell-Luria A.H., Ware J.S., Hill A.J., Cummings B.B.. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016; 536:285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brandes N., Linial N., Linial M.. Quantifying gene selection in cancer through protein functional alteration bias. Nucleic Acids Res. 2019; 47:6642–6655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hornbeck P.V., Zhang B., Murray B., Kornhauser J.M., Latham V., Skrzypek E.. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 2015; 43:D512–D520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vacic V., Uversky V.N., Dunker A.K., Lonardi S.. Composition profiler: a tool for discovery and visualization of amino acid composition differences. BMC Bioinform. 2007; 8:211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ridder A.N., Morein S., Stam J.G., Kuhn A., de Kruijff B., Killian J.A.. Analysis of the role of interfacial tryptophan residues in controlling the topology of membrane proteins. Biochemistry. 2000; 39:6521–6528. [DOI] [PubMed] [Google Scholar]
- 49.Samanta U., Chakrabarti P.. Assessing the role of tryptophan residues in the binding site. Protein Eng. 2001; 14:7–15. [DOI] [PubMed] [Google Scholar]
- 50.Weinert B.T., Scholz C., Wagner S.A., Iesmantavicius V., Su D., Daniel J.A., Choudhary C.. Lysine succinylation is a frequently occurring modification in prokaryotes and eukaryotes and extensively overlaps with acetylation. Cell Rep. 2013; 4:842–851. [DOI] [PubMed] [Google Scholar]
- 51.Reyes-Centeno H., Hubbe M., Hanihara T., Stringer C., Harvati K.. Testing modern human out-of-Africa dispersal models and implications for modern human origins. J. Hum. Evol. 2015; 87:95–106. [DOI] [PubMed] [Google Scholar]
- 52.Khodaverdian V.Y., Hanscom T., Yu A.M., Yu T.L., Mak V., Brown A.J., Roberts S.A., McVey M.. Secondary structure forming sequences drive SD-MMEJ repair of DNA double-strand breaks. Nucleic Acids Res. 2017; 45:12848–12861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Baer C.F., Miyamoto M.M., Denver D.R.. Mutation rate variation in multicellular eukaryotes: causes and consequences. Nat. Rev. Genet. 2007; 8:619–631. [DOI] [PubMed] [Google Scholar]
- 54.Jones S., Thornton J.M.. Analysis of protein-protein interaction sites using surface patches. J. Mol. Biol. 1997; 272:121–132. [DOI] [PubMed] [Google Scholar]
- 55.David A., Sternberg M.J.. The contribution of missense mutations in core and rim residues of protein-protein interfaces to human disease. J. Mol. Biol. 2015; 427:2886–2898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vitkup D., Sander C., Church G.M.. The amino-acid mutational spectrum of human genetic disease. Genome Biol. 2003; 4:R72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hassan M.S., Shaalan A.A., Dessouky M.I., Abdelnaiem A.E., ElHefnawi M.. A review study: computational techniques for expecting the impact of non-synonymous single nucleotide variants in human diseases. Gene. 2019; 680:20–33. [DOI] [PubMed] [Google Scholar]
- 58.Grimm D.G., Azencott C.A., Aicheler F., Gieraths U., MacArthur D.G., Samocha K.E., Cooper D.N., Stenson P.D., Daly M.J., Smoller J.W.et al.. The evaluation of tools used to predict the impact of missense variants is hindered by two types of circularity. Hum. Mutat. 2015; 36:513–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Miosge L.A., Field M.A., Sontani Y., Cho V., Johnson S., Palkova A., Balakishnan B., Liang R., Zhang Y., Lyon S.et al.. Comparison of predicted and actual consequences of missense mutations. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:E5189–E5198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Prabakaran S., Lippens G., Steen H., Gunawardena J.. Post-translational modification: nature's escape from genetic imprisonment and the basis for dynamic information encoding. Wiley Interdiscip. Rev. Syst. Biol. Med. 2012; 4:565–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Woodsmith J., Kamburov A., Stelzl U.. Dual coordination of post translational modifications in human protein networks. PLoS Comput. Biol. 2013; 9:e1002933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Huang K.-Y., Su M.-G., Kao H.-J., Hsieh Y.-C., Jhong J.-H., Cheng K.-H., Huang H.-D., Lee T.-Y.. dbPTM 2016: 10-year anniversary of a resource for post-translational modification of proteins. Nucleic Acids Res. 2015; 44:D435–D446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wilhelm M., Schlegl J., Hahne H., Gholami A.M., Lieberenz M., Savitski M.M., Ziegler E., Butzmann L., Gessulat S., Marx H.. Mass-spectrometry-based draft of the human proteome. Nature. 2014; 509:582. [DOI] [PubMed] [Google Scholar]
- 64.Bechtel T.J., Weerapana E.. From structure to redox: the diverse functional roles of disulfides and implications in disease. Proteomics. 2017; 17:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wong J.W., Ho S.Y., Hogg P.J.. Disulfide bond acquisition through eukaryotic protein evolution. Mol. Biol. Evol. 2011; 28:327–334. [DOI] [PubMed] [Google Scholar]
- 66.Reimand J., Wagih O., Bader G.D.. Evolutionary constraint and disease associations of post-translational modification sites in human genomes. PLoS Genet. 2015; 11:e1004919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yang Y., Peng X., Ying P., Tian J., Li J., Ke J., Zhu Y., Gong Y., Zou D., Yang N.et al.. AWESOME: a database of SNPs that affect protein post-translational modifications. Nucleic Acids Res. 2019; 47:D874–D880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Duan G., Walther D. The roles of post-translational modifications in the context of protein interaction networks. PLoS Comput. Biol. 2015; 11:e1004049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tan C.S., Pasculescu A., Lim W.A., Pawson T., Bader G.D., Linding R.. Positive selection of tyrosine loss in metazoan evolution. Science. 2009; 325:1686–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Beltrao P., Bork P., Krogan N.J., van Noort V.. Evolution and functional cross-talk of protein post-translational modifications. Mol. Syst. Biol. 2013; 9:714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Levy E.D., Michnick S.W., Landry C.R.. Protein abundance is key to distinguish promiscuous from functional phosphorylation based on evolutionary information. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2012; 367:2594–2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schwartz G.W., Shauli T., Linial M., Hershberg U.. Serine substitutions are linked to codon usage and differ for variable and conserved protein regions. Sci. Rep. 2019; 9:17238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Eisen J.A., Hanawalt P.C.. A phylogenomic study of DNA repair genes, proteins, and processes. Mutat. Res. 1999; 435:171–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fairfield H., Gilbert G.J., Barter M., Corrigan R.R., Curtain M., Ding Y., D’Ascenzo M., Gerhardt D.J., He C., Huang W.et al.. Mutation discovery in mice by whole exome sequencing. Genome Biol. 2011; 12:R86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Navarro F.C., Galante P.A.. A genome-wide landscape of retrocopies in primate genomes. Genome Biol. Evol. 2015; 7:2265–2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The ExAC dataset can be downloaded from https://console.cloud.google.com/storage/browser/_details/gnomad-public/legacy/exacv1_downloads/release0.3/ExAC.r0.3.sites.vep.vcf.gz. The substitution matrices, codon counts, and further results are available in the supplementary data. The entire source code of this study is available at http://www.github.com/tairsha/taxa-specific-substitution-matrix.