

Summary
Cell fate commitment is controlled by cis-regulatory elements often located in remote regions of the genome. To examine the role of long-range DNA interactions in early development, we generated a high-resolution contact map of active enhancers in avian neural crest cells. This analysis uncovered a diverse repertoire of enhancers that are part of the gene regulatory network underlying specification. We found that neural crest identity is largely regulated by cis-regulatory elements that propagate signaling inputs to network components. These genomic sensors display a combination of optimal and suboptimal TCF/LEF binding sites, which allow cells to respond to Wnt signaling in a position-dependent manner. We propose that, rather than acting as upstream activators, signaling systems feed into regulatory circuits in a hub-and-spoke architecture. These results shed light on the tridimensional organization of the neural crest genome and define how signaling systems provide progenitors with spatial cues that transform their molecular identity.
eTOC Blurb
Neural crest development is controlled by genetic circuits composed of cis- and transregulators. Azambuja and Simoes-Costa use chromatin conformation capture to identify distal enhancers activated by Wnt signaling. Their results uncover cis-regulatory features that underlie signaling responses in neural crest cells and demonstrate how spatial cues drive developmental transitions.
Graphical Abstract
Introduction
Cell state transitions during embryonic development require the coordinated expression of thousands of genes. Such extensive shifts in gene expression are controlled by gene regulatory networks (GRNs), which integrate cell-intrinsic and environmental cues to alter cell identity (Peter and Davidson, 2015, Davidson and Levine, 2008). This paradigm is typified by the genetic program underlying the formation of the neural crest (NC), a migratory stem cell population in vertebrate embryos (Le Douarin and Kalcheim, 1999, Le Lievre and Le Douarin, 1975, Lwigale et al., 2004). NC formation is controlled by a complex GRN that endows ectodermal cells with unique features such as multipotency, stemness, and the ability to migrate (Meulemans and Bronner-Fraser, 2004, Sauka-Spengler and Bronner-Fraser, 2008, Simoes-Costa and Bronner, 2015). After specification, NC cells delaminate from the neural tube to give rise to the facial skeleton, the peripheral nervous system, and the pigmentation of the skin (Le Douarin and Kalcheim, 1999). Due to this migratory behavior, NC cells are a tractable model to investigate how spatial cues affect cell identity.
Like many developmental genetic networks, the NC GRN is initiated by the action of environmental signals. Extracellular inputs, particularly from the Wingless (Wnt) pathway, promote the expression of cell type-specific transcriptional regulators in precise locations in the embryo (Groves and LaBonne, 2014, Simoes-Costa and Bronner, 2015, Garcia-Castro et al., 2002). These factors form transcriptional circuits that orchestrate gene expression by interacting with regulatory regions throughout the genome (Simoes-Costa and Bronner, 2015). As a result, much effort has been placed in the identification and dissection of NC-specific enhancers (Betancur et al., 2010, Barembaum and Bronner, 2013, Simoes-Costa et al., 2012, Vadasz et al., 2013). Recent genomic analyses have increased the number of known NC enhancers (Rada-Iglesias et al., 2012, Williams et al., 2019, Rothstein and Simoes-Costa, 2020). However, a lack of information on the spatial organization of the NC genome has prevented precise integration of epigenomic data in genetic circuits. Since enhancers of developmental genes are often located in remote regions of the genome, the assignment of enhancers to promoters in embryonic progenitors can be challenging (Schoenfelder et al., 2015, Amano et al., 2009). Thus, a contact map of cis-regulatory elements is necessary for the assembly of a reliable model of the NC GRN.
The limitations of the current version of the GRN have prevented us from elucidating regulatory mechanisms central to NC formation, such as the role of signaling systems. Wnt signaling is necessary and sufficient to drive neural specification (Garcia-Castro et al., 2002, Simoes-Costa et al., 2015), and functional studies indicate it acts on multiple levels of the transcriptional network (Yin et al., 2015, Garcia-Castro et al., 2002, Simoes-Costa et al., 2015, Raible and Ragland, 2005). Disruption of the pathway causes loss of the NC in model organisms (Baron and Kneissel, 2013, Brault et al., 2001, Kormish et al., 2010), and Wnt agonists are utilized in protocols for in vitro differentiation (Gomez et al., 2019, Mica et al., 2013, Leung et al., 2016). Despite these findings, we still have a superficial understanding of how Wnts promote NC identity. Enhancer analysis of bona fide NC markers like SOX10, FOXD3, and ETS1 has failed to reveal TCF/LEF motifs, even though these genes are affected by manipulation of the pathway (Betancur et al., 2010, Barembaum and Bronner, 2013, Simoes-Costa et al., 2012). While Wnt-responsive elements have been identified in the loci of AXUD1 (Azambuja and Simoes-Costa, 2021) and SNAI2 (Vallin et al., 2001), several lines of evidence indicate a broader function for canonical Wnt signaling than what is currently known.
To address these questions, we performed H3K27ac-HiChIP to generate a high-resolution contact map of active enhancers in primary NC cells. Our results revealed that NC genes are associated with enhancers that contact promoters in a tissue-specific manner and are often hundreds of kilobases away from their target genes. Quantification of putative enhancer-promoter interactions allowed us to assign thousands of cis-regulatory regions to the components of the NC GRN. This led to the identification of a system of cis-regulatory elements that are directly regulated by Wnt signaling and relay environmental inputs to network components. These enhancers have not previously been identified since they (i) tend to be further away from target genes and (ii) often interact with Wnt nuclear effectors via cryptic, suboptimal TCF/LEF binding sites. Our results provide an comprehensive characterization of the nuclear architecture of NC cells and reveal a pervasive role for environmental signals in the operation of gene regulatory networks.
Results
A high-resolution contact map of active NC enhancers
To examine the nuclear architecture of the premigratory NC, we generated a high-resolution contact map of active enhancers with H3K27ac-HiChIP (Mumbach et al., 2016). NC cells were isolated from HH9 avian embryos via microdissection, which allowed us to obtain samples with high purity (>80% expressing PAX7, Figure S1A). Whole embryo samples from the same developmental stage were processed as controls (Figure S1B–E). The resulting interaction matrices allowed us to define the location of TADs and sub-TADs in the avian genome (Figure 1A). To examine how chromatin architecture is integrated within the epigenomic landscape of NC cells, we examined chromatin accessibility (ATAC-seq), occupancy of the DNA loop regulator CTCF (Rao et al., 2014) and histone H3K27 acetylation levels (H3K27ac) (Bhattacharya et al., 2020) (Figure 1B). These datasets confirmed that our contact map linked accessible H3K27ac+ regions to promoters. Putative enhancer-promoter loops were the most abundant (179,967 loops, 31%), followed by putative enhancer-enhancer (136,607 loops, 23%), and promoter-promoter (1941 loops, 0.3%) interactions. While many of the loop anchors were associated with CTCF, we also found numerous examples of CTCF-independent looping, consistent with previous reports (Handoko et al., 2011, Sanyal et al., 2012).
Figure 1. Mapping of chromatin architecture reveals tissue-specific enhancer-promoter contacts in neural crest cells.
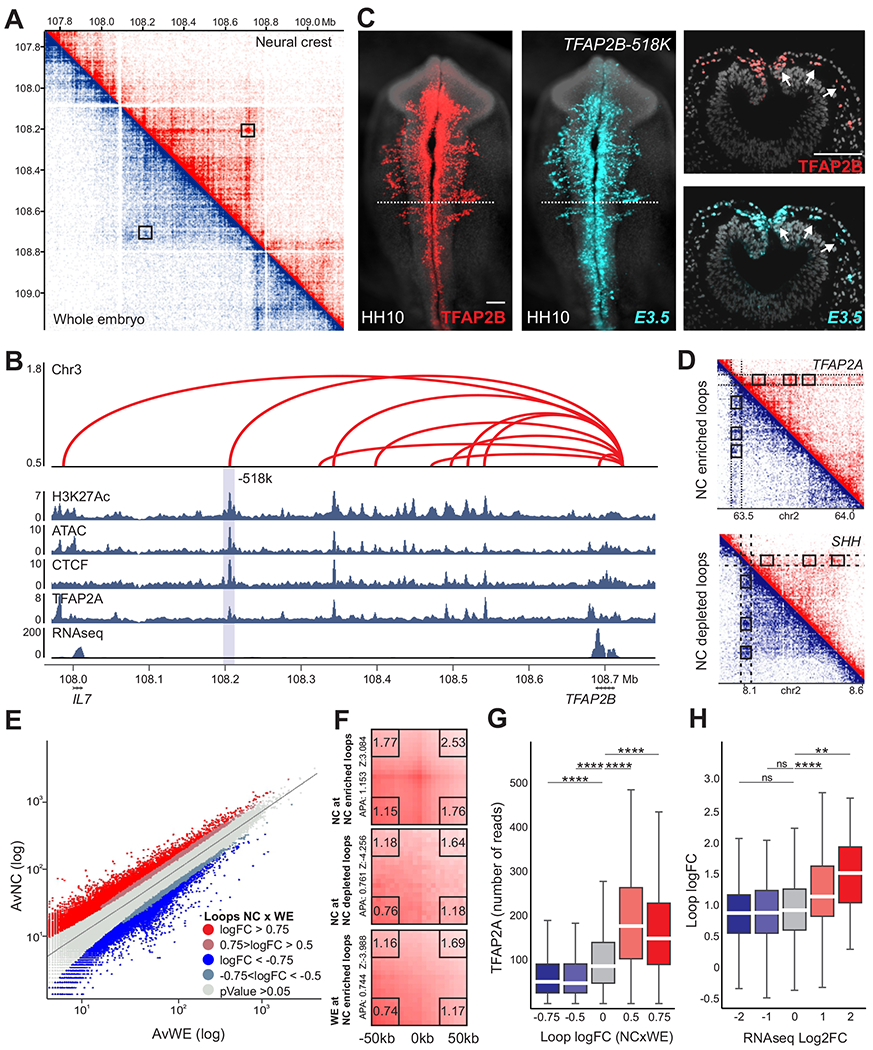
(A) HiChIP contact matrix depicting normalized contact frequencies for NC (red, top right) and Whole Embryo (blue, bottom left) at the TFAP2B TAD. A NC-enriched loop anchored at the TFAP2B promoter is highlighted (TFAP2B-518k).
(B) Enhancer-promoter contacts, ATAC-seq, RNA-seq and CUT&RUN profiles for H3K27ac, CTCF, and TFAP2A at the TFAP2B locus. Y-axis represents loop logFC NC vs. WE. Gray bar highlights the loop indicated in (A).
(C) Immunohistochemistry (whole-mount and transverse sections) for the endogenous TFAP2B protein (red) in a transgenic embryo. Reporter activity of E3.5 indicates robust and specific GFP (turquoise) expression at the migratory NC.
(D) HiChIP matrices depicting normalized contact frequencies for NC (red, top right) and WE (blue, bottom left) in the TFAP2A and SHH loci. NC cells display enrichment (TFAP2A) or depletion (SHH) of putative enhancer-promoter contacts. Dotted boxes highlight the promoter regions of each gene.
(E) Scatter plot displaying NC and whole embryo average counts of all valid chromatin contacts. NC-enriched loops are highlighted in red, while depleted contacts are highlighted in blue.
(F) Aggregate Peak Analysis (APA) illustrates the significant differences of NC and WE samples. APA plots showing aggregated enrichment of NC and WE datasets across the enriched (logFC>0.75, pValue<0.05) and depleted (logFC<−0.75, pValue<0.05) contacts highlighted in (E).
(G) TFAP2A CUT&RUN signal at the genomic regions corresponding to H3K27ac peaks at putative enhancer-promoter contacts binned by logFC (NCxWE).
(H) Boxplot depicting putative enhancer-promoter loops logFC (NCxWE) at genes binned by RNA-seq signal enrichment in crest cells.
Also see Figure S1. H3K27ac-HiChIP performed in 3 biological replicates. HH, Hamburger and Hamilton; NC, neural crest; WE, whole embryo; Mb, megabase. Scale bar, 100um.
**p<0.01, ****p<0.0001.
We next employed our HiChIP maps to examine whether NC cells display cell type-specific nuclear architecture. A comparison between interaction maps obtained from the NC and whole embryo samples did not reveal differences in TAD organization (Figure 1A). Nevertheless, we found a large number of NC-enriched intra-TAD interactions, which connected promoters of NC genes to distal H3K27ac+ regions (Figure 1A–B). Testing a cohort of these regions with transient transgenesis in avian embryos showed that they are often active in NC (Figures 1C and 2). Comparison of interaction frequencies allowed us to annotate putative enhancer-promoter contacts, where 10,531 were significantly enriched in NC (logFC>0.75; pValue<0.05), and 1,464 were depleted (logFC<−0.75; pValue<0.05) when compared to the whole embryo (Figure 1D–E). These results were supported by aggregate peak analysis (APA) plots, which resulted in high scores (APA = 1.153, Zscore = 3.084) for the set of enriched DNA loops (Figure 1F). Putative enhancer-promoter contacts enriched in NC cells displayed abundant binding of TFAP2A (Figure 1G), a pioneer factor associated with active NC enhancers (Rothstein and Simoes-Costa, 2020, Rada-Iglesias et al., 2012). Indeed, 56.6% of enhancer-promoter loops with high NC enrichment (logFC>0.75; pValue<0.05) displayed association with TFAP2A. Notably, genes contacted by NC-enriched loops also displayed higher mRNA enrichment in this cell population (Figure 1H). Taken together, these results indicate that expression of NC genes is regulated by cis-regulatory elements that contact promoters in a tissue-specific manner.
Figure 2. Enrichment and frequency of interaction with promoters are strong predictors of enhancer activity.
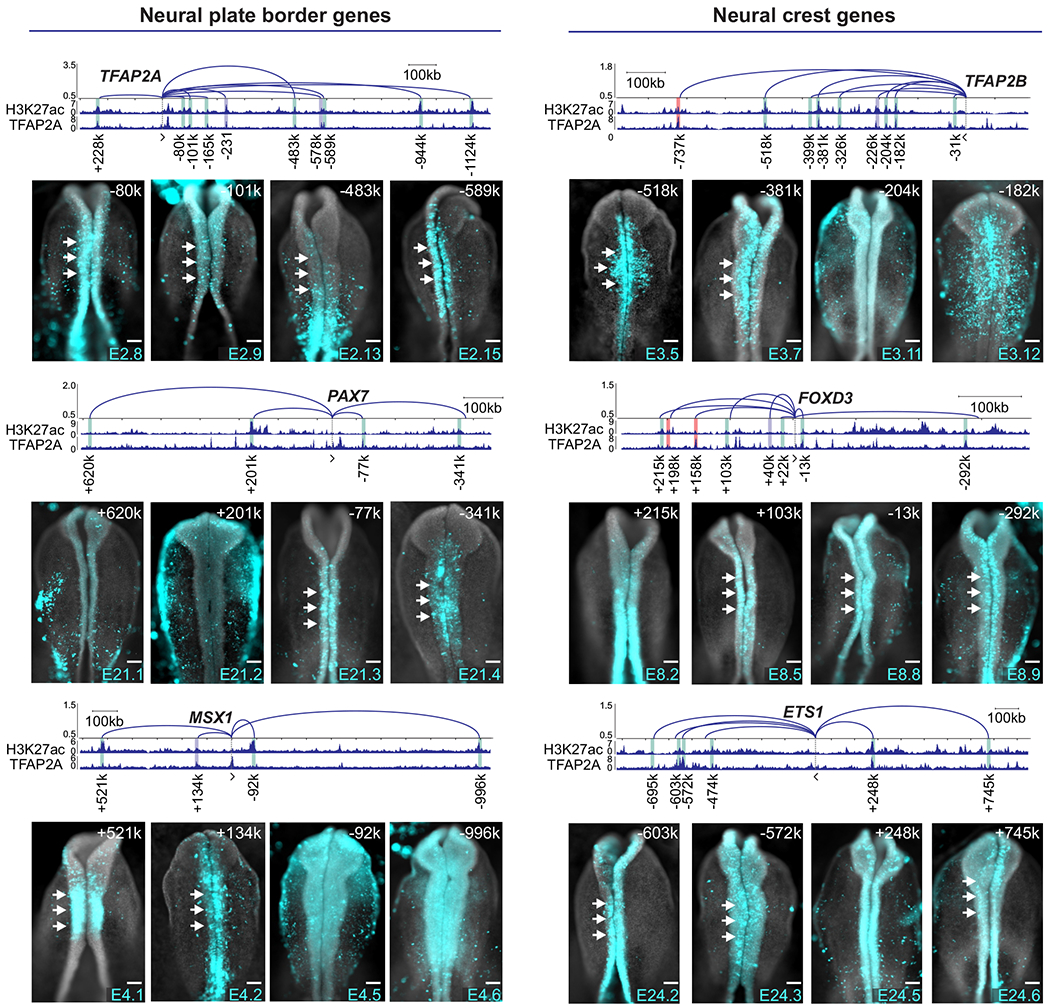
Reporter assays in transgenic embryos show domains of activity of enhancers that interact with promoters of NC genes. y-axis in arc plot represents Z-scores calculated from the mean counts of NC HiChIP replicates. All NC-enriched contacts co-occupied by TFAP2A are presented. Enhancer-promoter contacts are identified by its loop width (+, upstream or −, downstream gene’s promoter). Vertical bars indicate regions tested in enhancer-reporter assays. Representative image of enhancer activity is displayed for four elements per gene (See also Figure S2A). Gray bars represent previously published enhancers, green novel distal elements with cranial activity at HH9-10 stages and red bars represent tested regions with no cranial activity at HH9-10 stages. White arrows indicate activity in the NC. Kb, kilobase. Scale bar, 100um.
Next, we employed our interaction matrix to assign putative NC enhancers, here defined as H3K27ac+ regions contacting components of the NC GRN (Supplemental Tables 1–2). We were also able to link NC cis-regulatory elements identified by previous studies to their respective promoters (Figure S1F, Supplemental Table 2). These elements were identified via mapping of TFAP2A occupancy with ChIP-Seq (Rada-Iglesias et al., 2012), chromatin accessibility (Williams et al., 2019), or occupancy of TFAP2A/B heterodimers (Rothstein and Simoes-Costa, 2020). Gene assignment with HiChIP revealed a high prevalence of promoter associations with distal regulatory elements (Figure S1G) in the NC genome. The average length of the strongest cell type-specific putative enhancer-promoter loop was larger than what we observed in ubiquitous and depleted interactions. NC genes also displayed a higher number of putative enhancer-promoter interactions (Figure S1H), suggesting a high degree of complexity in the cis-regulatory apparatus of tissue-specific transcription factors.
We next tested a selection of cis-regulatory elements using reporter assays (Simoes-Costa et al., 2012). Putative NC enhancers were cloned in pTK-eGFP (Uchikawa et al., 2003) and transfected in chicken embryos. We tested ~70 putative enhancers that (i) displayed a high frequency of interaction with a promoter of a specification gene, (ii) were part of a DNA loop enriched in NC cells and (iii) were associated with TFAP2A (Figures 2 and S2A, Supplemental Table 3). These criteria were strong predictors of enhancer activity as >90% of the tested regions were able to drive reporter expression. This strategy uncovered regulatory elements of core GRN components like TFAP2A, PAX7, MSX1, TFAP2B, FOXD3 and ETS1. While many elements were crest-specific, others were also active in the neural tube and in the lateral ectoderm (Figures 2 and S2A). To test if these cis-regulatory regions are important for gene expression, we targeted them with CRISPRi (Williams et al., 2019) (Figure S2B). This resulted in loss of expression of the associated NC gene (n=17/18, Figure S2C–D), confirming a biological role for the enhancers identified with HiChIP.
Pervasive regulation of the NC GRN by Wnt signaling
Having established the repertoire of enhancers associated with promoters of NC genes, we next set out to identify general trans-regulators of the GRN. We examined the sequences of H3K27ac-bound regions that contacted promoters in a tissue-specific manner. HOMER analysis of NC-enriched vs. depleted putative enhancer-promoter contacts (Figure 3A) identified TCF/LEF as the top enriched motif in these sequences (pValue = 1.0 e-365, Figure 3B). By allowing for precise enhancer-promoter assignment (Figure S2E), our interaction map raised the possibility that Wnt nuclear effectors interact with multiple NC regulatory regions. To test this, we examined the genome-wide occupancy of Wnt nuclear effectors in NC cells. We mapped the genome occupancy of both LEF1, the main NC TCF/LEF paralog (Azambuja and Simoes-Costa, 2021), and CTNNB1 using Cleavage Under Targets and Release Using Nuclease (CUT&RUN)(Skene and Henikoff, 2017) (Figure S3). We were able to identify 1878 binding events for LEF1, 9892 for CTNNB1, and 1398 regions bound by canonical Wnt-signaling (defined by the co-occupation of both regulators, Figure 3C). Regions co-occupied by LEF1 and CTNNB1 were mostly intergenic and displayed high H3K27ac, indicating they are active enhancers (Figure 3D–E). As expected, motif enrichment analysis identified an abundance of TCF/LEF binding sites in these elements (Figure 3F). Consistent with our previous motif enrichment analysis (Figure 3B), we also observed that NC-enriched loops displayed high association with CTNNB1 and LEF1 (Figure 3G).
Figure 3. Genome-wide mapping of nuclear effectors identifies direct targets of canonical Wnt signaling.
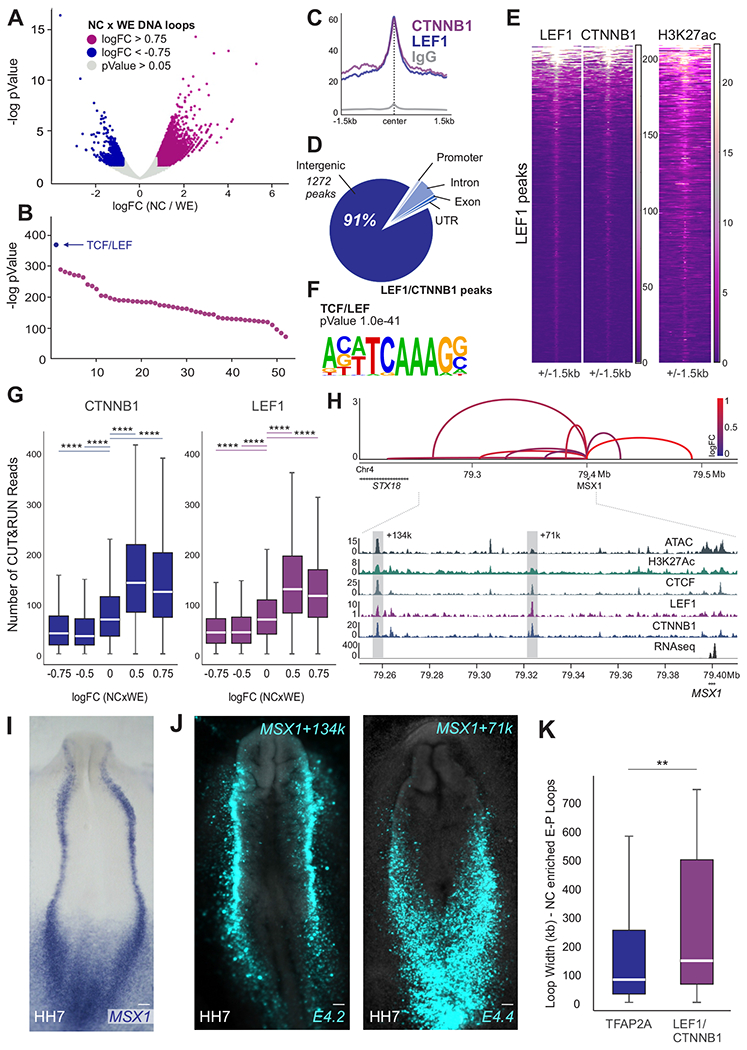
(A) Volcano plot depicting all enhancer-promoter contacts after quality control. Highlighted dots represent NC-enriched (logFC>0.75, pValue<0.05) and depleted (logFC<−0.75, pValue<0.05) contacts.
(B) Transcription factor binding sites identified in NC-enriched vs. depleted putative enhancer-promoter contacts highlighted in (A).
(C) CUT&RUN profiles showing binding of LEF1, CTNNB1 and normal rabbit IgG at LEF1 bound peaks.
(D) Pie chart depicting genomic location of LEF1/CTNNB1 peaks.
(E) Heatmaps displaying LEF1, CTNNB1 and H3K27ac CUT&RUN signal at LEF1 peaks.
(F) Motif enrichment analysis shows enrichment for the TCF/LEF motif in regions co-occupied by LEF1/CTNNB1.
(G) Boxplots displaying read counts of CTNNB1 and LEF1 CUT&RUN at HiChIP loops.
(H) Arc plot depicting putative enhancer-promoter contacts, ATAC-seq, RNA-seq and CUT&RUN profiles for H3K27ac, CTCF, LEF1 and CTNNB1 at the MSX1 locus. y-axis of arc plot represents Z-scores calculated from the mean counts of NC HiChIP replicates. Arc color represents loop enrichment in NC (logFC NCxWE). Dotted boxes highlight enhancers E4.2 (loop MSX1+134k) and E4.4 (loop MSX1+71k).
(I-J) Whole-mount in situ hybridization for MSX1 (I) and activity of enhancer regions E4.2 and E4.4 (J) shown via transient transgenesis. Enhancers E4.2 and E4.4 are active in the neural plate border.
(K) Size (loop width) of the strongest TFAP2A or LEF1/CTNNB1-bound loop per gene. TFAP2A is a marker of active NC enhancers, and thus represents average NC loop size (Figure S1F) while LEF1/CTNNB1 bound loops display more distal contacts.
Also see Figure S3. LEF1 and CTNNB1 CUT&RUN performed in 2 biological replicates. NC, neural crest; WE, whole embryo; HH, Hamburger and Hamilton; kb; kilobase; Mb, megabase. Scale bar, 100um. **p<0.01, ****p<0.0001.
Examination of individual NC genes confirmed the association of LEF1 and CTNNB1 with tissue-specific enhancers. In the MSX1 locus, we identified two regulatory regions 142 and 77kb from the TSS that are associated with the promoter of the gene and display high occupancy by LEF1 and CTNNB1 (Figure 3H). Reporter assays revealed that both regions are active in domains that recapitulate MSX1 endogenous expression (Figure 3I–J). Thus, although MSX1 has been described as target of BMP signaling (Tribulo et al., 2003), here we show that it is also a direct target of canonical Wnt signaling. This regulation takes place via distal enhancer elements that are H3K27ac+ and display specific expression in the neural plate border and NC cells (Figure 3H–J). Wnt-associated elements were often far from their respective promoters; while the median distance between a TFAP2A-bound enhancer and its respective promoter was ~100kb, cis-regulatory regions regulated by canonical Wnts were found to be twice as far away on average (Figure 3K).
Next, we employed transient transgenesis to test the activity of genomic regions regulated by Wnt signaling. We selected 30 genomic regions that interact with promoters of NC genes and are associated with both LEF1 and CTNNB1. These regions were cloned in pTK-eGFP and co-transfected in developing embryos with a NC specific enhancer (Tfap2aE1:mChe) (Rothstein and Simoes-Costa, 2020). While many of the enhancers displayed overt NC expression, we also identified elements that were active in neural, placodal and epidermal progenitors (Figures 4A and S4A). Furthermore, quantification of enhancer strength (eGFP intensity) and specificity (defined by the ratio between double positive cells and eGFP+ cells) revealed a striking variation in the output of Wnt-associated elements (Figures 4B and S4B–C). To examine epigenomic features associated with enhancer strength or specificity, we grouped Wnt-associated elements according to reporter activity (Figure 4B). This analysis revealed that enhancer strength correlates with promoter interaction frequency, H3K27ac levels and binding of CTNNB1 (Figure 4B–C). In contrast, the best predictors for enhancer specificity were loop and H3K27ac enrichment in the NC (Figure 4B,D). Notably, loop strength was inversely correlated with output specificity, suggesting that stable enhancer-promoter interactions are less prone to tissue-specific regulation (Figure 4D).
Figure 4. Wnt signaling directly regulates a diverse set of active enhancers via multiple binding events.
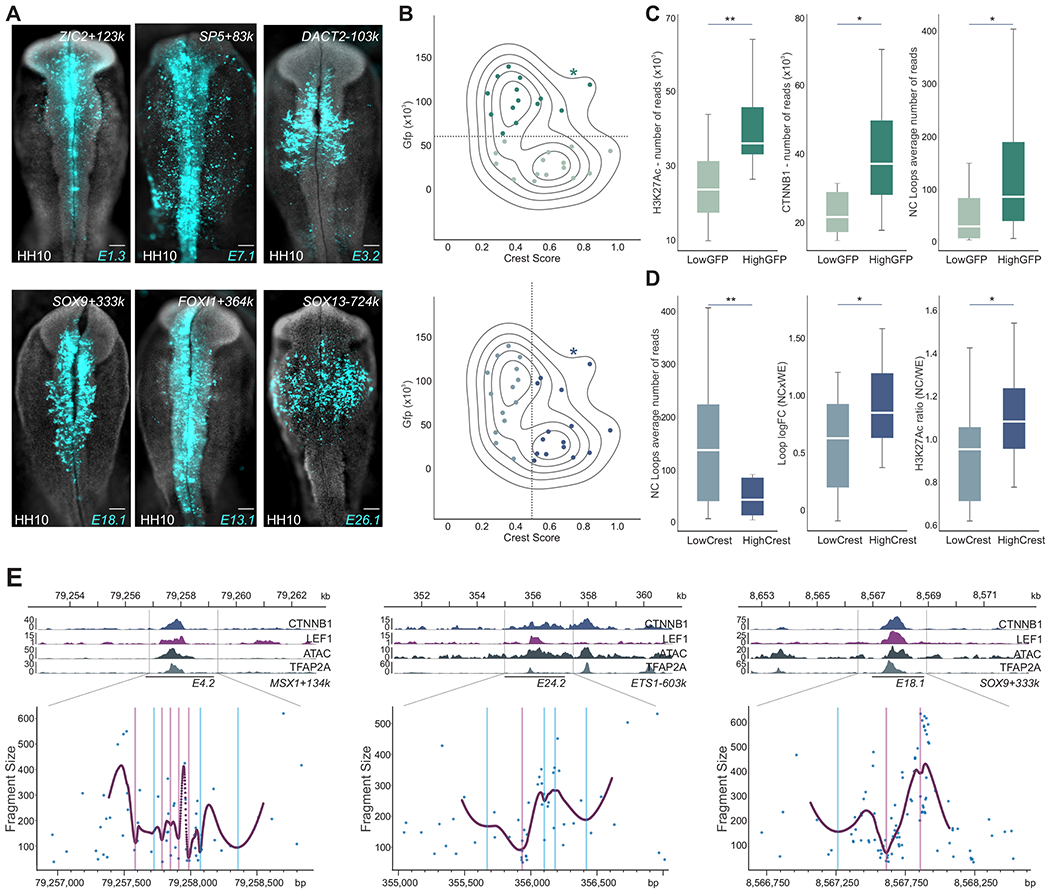
(A) LEF1/CTNNB1-bound enhancers vary in strength (GFP intensity) and specificity. HH10 embryos displaying GFP expression driven by three highly active enhancers, E1.3 (ZIC2+123K), E7.1 (SP5+83k) and E3.2 (DACT2-103k) and three highly crest-specific enhancers, E18.1 (SOX9+333k), E13.1 (FOXI1+364k) and E26.1 (SOX13-724k).
(B) Level plot displaying GFP intensity and specificity score of NC putative enhancers bound by LEF1/CTNNB1. Y-axis represents mean GFP intensity of analyzed cells. X-axis represents a specificity score defined by the ratio of double positive cells (Tfap2aE1:mChe+/GFP+) in the GFP-positive population. *depicts values for the Tfap2aE1 enhancer.
(C) Boxplots showing H3K27ac and CTNNB1 binding, and loop reads for the two subpopulations of enhancers defined by the horizontal dotted line in (B).
(D) Boxplots showing loop reads, logFC, and H3K27ac binding ratio (NC/WE) for the two subpopulations of enhancers defined by the vertical dotted line in (B).
(E) Local minimal DNA protection by transcription factor DNA binding (EChO) reveals multiple LEF1 binding sites across Wnt-regulated enhancers. CTNNB1, LEF1, ATAC and TFAP2A signals and EChO profiles at the putative enhancers E4.2 (MSX1+134k), E24.2 (ETS1-603k) and E18.1 (SOX9+333k). Purple and blue lines represent foci positions associated with high and low score LEF1 motifs, respectively.
Also see Figure S4. HH, Hamburger and Hamilton; NC, neural crest; WE, whole embryo; bp, base pair; kb, kilobase. Scale bar, 100um. *p<0.05, **p<0.01.
The diversity in the activity of Wnt-associated elements led us to survey their interaction with nuclear effectors in more detail. We thus examined the patterns of LEF1 binding in Wnt-target regions using Enhanced Chromatin Occupancy (EChO) analysis. EChO takes advantage of the improved resolution afforded by CUT&RUN to identify binding foci within peaks, which reflect the position occupied by a transcription factor when it is interacting with an enhancer (Meers et al., 2019). This analysis revealed that the typical Wnt-target region displays a high number of LEF1 binding events within each CUT&RUN peak (Figure 4E). By examining TCF/LEF binding sites in proximity to LEF1 foci, we found that some binding events take place via canonical TCF/LEF motifs (Figure 4E), while others utilize suboptimal binding sites. These results suggest that the regulation of Wnt-associated elements involves multiple interactions with nuclear effectors, which take place via both canonical and suboptimal binding motifs.
Wnt signaling controls NC enhancers via optimal and suboptimal TCF/LEF binding sites
To define the regulatory features of Wnt-associated elements, we examined how LEF1 and CTNNB1 interact with Axud1E1, an enhancer directly activated by canonical Wnts (Azambuja and Simoes-Costa, 2021). Axud1E1 is active in NC cells (Figure 5A) and anchors the loop with the highest NC enrichment in this TAD (Figure S5A–B). Other TFAP2A-associated enhancers contacting the AXUD1 promoter were not active in the cranial NC (Figure S5A–C). EChO analysis revealed multiple foci across Axud1E1 indicating binding events associated with both high and low-scoring TCF/LEF motifs (Figure 5B). Analysis of enhancer sequence revealed four high-scoring TCF/LEF motifs located at the 5’ region of the enhancer. To establish the importance of these motifs, we generated an enhancer variant in which they were mutated (Figure 5C) (Axud1E1_500bp_HighMUT). Bilateral tranfection of the wild-type vs the HighMUT construct into embryos (Simoes-Costa et al., 2012) revealed a moderate decrease in enhancer activity (Figure 5D–E).
Figure 5. Nuclear effectors of Wnt signaling interact with neural crest enhancers via optimal and suboptimal binding sites.
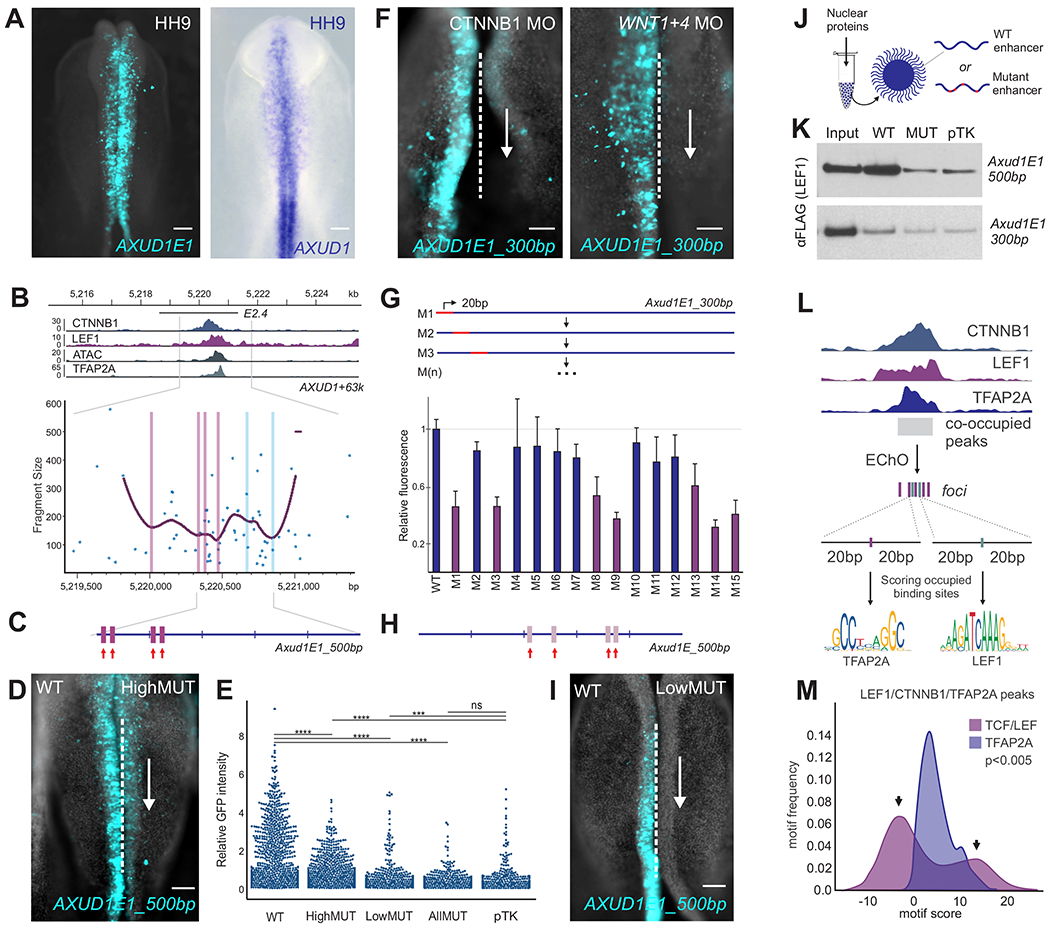
(A) Transgenic avian embryo showing the activity of Axud1E1 (left) and AXUD1 expression (right).
(B) Signal of CTNNB1, LEF1, ATAC and TFAP2A and EChO profiles at the AXUD1 enhancer E2.4 (AXUD1+63k, Axud1E1). Purple and blue lines represent foci positions associated with high and low scoring LEF1 motifs, respectively.
(C) Location of high-scoring TCF/LEF binding sites in Axud1E1_500bp. The mutated motifs are identified by red arrows.
(D) Whole-mount view of an embryo bilaterally electroporated with the wild-type (left) and mutant (HighMUT, right) constructs (n=6).
(E) Scatter plots displaying flow cytometry analysis of enhancer-reporter assays of Axud1E1_500bp wild-type and mutant constructs (n=800 sorted cells). Y-axis represents the GFP/mCherry intensity ratio of HH9 cranial single cells.
(F) Axud1E1_300bp displays Wnt signaling-dependent activity (n=5). Bilateral electroporation of CTNNB1 and WNT1/4 morpholinos disrupted activity of the enhancer.
(G) Quantification of effect of individual 20bp mutations on Axud1E1_300bp (GFP/mCherry). Enhancer variants with enhancer activity below wild-type threshold are highlighted in purple (n=3, 2 ROIs per embryo). Error bar represents ± S.E.M.
(H) Low-score TCF/LEF binding sites identified by mutation analysis. Mutated motifs are identified by red arrows.
(I) Mutation of the four low-scoring TCF/LEF motifs at the Axud1E1_500bp (LowMUT) (H) strongly reduces enhancer activity. Whole-mount view of an embryo bilaterally electroporated with the wild-type (left) and mutant (right) constructs (n=4).
(J) Diagram of enhancer pulldown assays. Biotinylated enhancers and mutant variants were conjugated with streptavidin beads and incubated with nuclear extracts. Protein-DNA interaction was assayed with western blots.
(K) Enhancer pull down of Axud1E1_500bp, Axud1E1_300bp and its mutants variants (_AllMUT and _LowMUT, respectively) showing that suboptimal TCF/LEF binding sites are required for interaction of LEF1 with the Axud1E1 enhancer.
(L) Schematic of foci motif analysis. EChO analysis was performed for LEF1 and TFAP2A foci definition at LEF1/CTNNB1/TFAP2A co-occupied regions. LEF1 and TFAP2A foci were extended to a 40bp window and submitted to FIMO for LEF1 and TFAP2A motif score evaluation.
(M) Histograms displaying LEF1 (pink) and TFAP2A (purple) motif scores around LEF1 and TFAP2A foci, respectively, as described in (L). LEF1 motifs present a bimodal distribution (arrows) indicating higher variability in binding site sequences.
Also see Figure S5. bp, base pair; HH, Hamburger and Hamilton; MO, morpholino. Scale bar, 100um. ***p<0.001, ****p<0.0001.
These results indicate that while the high-scoring TCF/LEF motifs contribute to enhancer output, they are not essential for specific activity. This led us to postulate that suboptimal TCF/LEF motifs also play an important role in the regulation of Wnt-associated elements. To test this, we scrutinized a smaller version of the Axud1E1_500bp enhancer (Axud1E1_300bp), which contains a 5’ 200bp deletion that eliminated the four TCF/LEF high-scoring motifs (Azambuja and Simoes-Costa, 2021). Axud1E1_300bp is active in NC cells, and EChO analysis indicates that it is associated with LEF1 (Figure 5B). To test if Axud1E1_300bp is responsive to Wnt manipulation, we electroporated embryos with morpholinos targeting CTNNB1 or the WNT1/4 ligands (Simoes-Costa et al., 2015). These knockdowns resulted in loss of enhancer activity (Figure 5F). Combined with the EChO results, this indicates that Wnt nuclear effectors interact with Axud1E1 via suboptimal motifs. To determine which suboptimal motifs are important for enhancer activity, we dissected Axud1E1_300bp by individually mutating every 20bp of the enhancer and quantifying the activity of the variants with transient transgenesis (Figure 5G). This mutation screen identified four suboptimal TCF/LEF motifs that contribute to enhancer activity.
We next compared the relative contributions of optimal and suboptimal motifs for Axud1E1 output. We generated two additional versions of Axud1E1_500bp: LowMUT, in which the four low-scoring TCF/LEF sites identified in the screen were mutated (Figure 5H, Supplemental Table 4), and AllMUT, with mutations in all eight TCF/LEF sites (four high and 4 low scoring motifs). Reporter assays showed that the LowMUT construct displayed a marked decrease in enhancer activity (Figure 5E,I), which was stronger than the one observed in HighMUT. The AllMUT construct had no activity above background levels (Figure 5E). To confirm that mutations in TCF/LEF binding sites affected the interaction between Wnt nuclear effectors and the enhancer, we employed DNA pull down assays (Figure 5J). These assays showed that mutation of TCF/LEF binding sites (AllMUT) eliminated LEF1 affinity with the enhancer. Pulldown assays with a variant of Axud1E1_300bp containing mutations in the four suboptimal binding sites also resulted in decreased levels of LEF1 binding (Figure 5K). Taken together, these results indicate that (i) Wnt nuclear effectors interact with cis-regulatory regions via multiple binding sites, (ii) both optimal and sub-optimal TCF/LEF motifs may contribute to enhancer activity, and (iii) sub-optimal motifs may underlie a substantial percentage an enhancer’s output.
To further examine the role of optimal and suboptimal TCF/LEF sites in Axud1E1, we tested the activity of Axud1E1_500bp, HighMUT and LowMUT under different conditions of Wnt activation. We treated explants with different concentrations of the Wnt agonist CHIR99021 (Figure S5D) and quantified reporter gene levels with qPCR. We found that the wild type enhancer exhibited a dose-response relationship with Wnt activation, with an increase in output across the three conditions (Figure S5E). Notably, LowMUT only responded to low levels of Wnt activation, consistent with the higher affinity of optimal binding sites to nuclear effectors. We postulate this reporter construct did not detect the increase of agonist concentration due to saturation of the TCF/LEF binding sites. Alternatively, HighMUT could only respond to higher doses of Wnt agonist (Figure S5E), indicating that higher levels of nuclear LEF1/CTNNB1 are required for binding to suboptimal motifs. Importantly, the wild type enhancer displayed the combined responses of the HighMUT and LowMUT constructs. These results show that the combination of optimal and suboptimal affinity binding sites is necessary for Axud1E1 to act as a Wnt sensor and respond to different levels of the signaling pathway.
Finally, to test if signaling via optimal and suboptimal binding sites is a general feature of Wnt-associated elements, we analyzed the syntax of occupied motifs in enhancers regulated by Wnt nuclear effectors and TFAP2A (Figure 5L). While occupied TFAP2A motifs displayed high confidence scores, TCF/LEF sites have a bimodal distribution in respect to score frequency (Figure 5M), with most binding events taking place via degenerate motifs. Analysis of datasets from other cell types for TCF7L2 (Quaife-Ryan et al., 2020) in and LEF1 (Tsankov et al., 2015) revealed 50% and 32% of binding was associated with regions with only low affinity sites, respectively. In our CUT&RUN datasets, approximately 37% of LEF1 binding and 43% of the LEF1/CTNNB1 binding takes place in cis-regulatory regions that only have suboptimal TCF/LEF motifs. Taken together, these results indicate that Wnt-associated elements are regulated by multiple binding events within the same enhancer. Since optimal and suboptimal TCF/LEF motifs mediate responses to different levels of Wnt activation, we propose that combinations of binding sites allow enhancers to act as genomic sensors by responding to fluctuations in Wnt activity during NC development.
Canonical Wnts regulate the NC GRN in a spatial-specific manner
The cell state transitions inherent to NC development require substantial shifts in transcriptional activity. We hypothesize that this transition is driven by the growing distance between the NC cells and the Wnt-producing dorsal neural tube, which acts as a stem niche (Figure 6A) (Bhattacharya et al., 2018). Indeed, expression of genes like AXUD1 and SOX9 plummets during migration (Figure 6B). Our findings suggest that these changes are mediated by enhancers that act as sensors of nuclear Wnt activity. To test this, we first quantified the changes in Wnt signaling during NC migration with the TOPFLASH12X reporter (Figure S6A). The results show a gradual decrease in Wnt response in NC cells as they migrate ventrally (Figures 6C and S6B). To examine if this is mediated by Wnt nuclear effectors, we performed proximity ligation assays (PLA) for LEF1 and CTNNB1 (Figure S6C). PLA quantification showed that the frequency of interaction between the two proteins decreases during migration (Figures 6D and S6D). These results are consistent with a scenario in which the loss of Wnt activity in migratory cells drives the dissolution of the LEF1/CTNNB1 complex, leading to the subsequent silencing of the specification GRN.
Figure 6. Wnt signaling controls the neural crest gene regulatory network in a position-dependent manner.
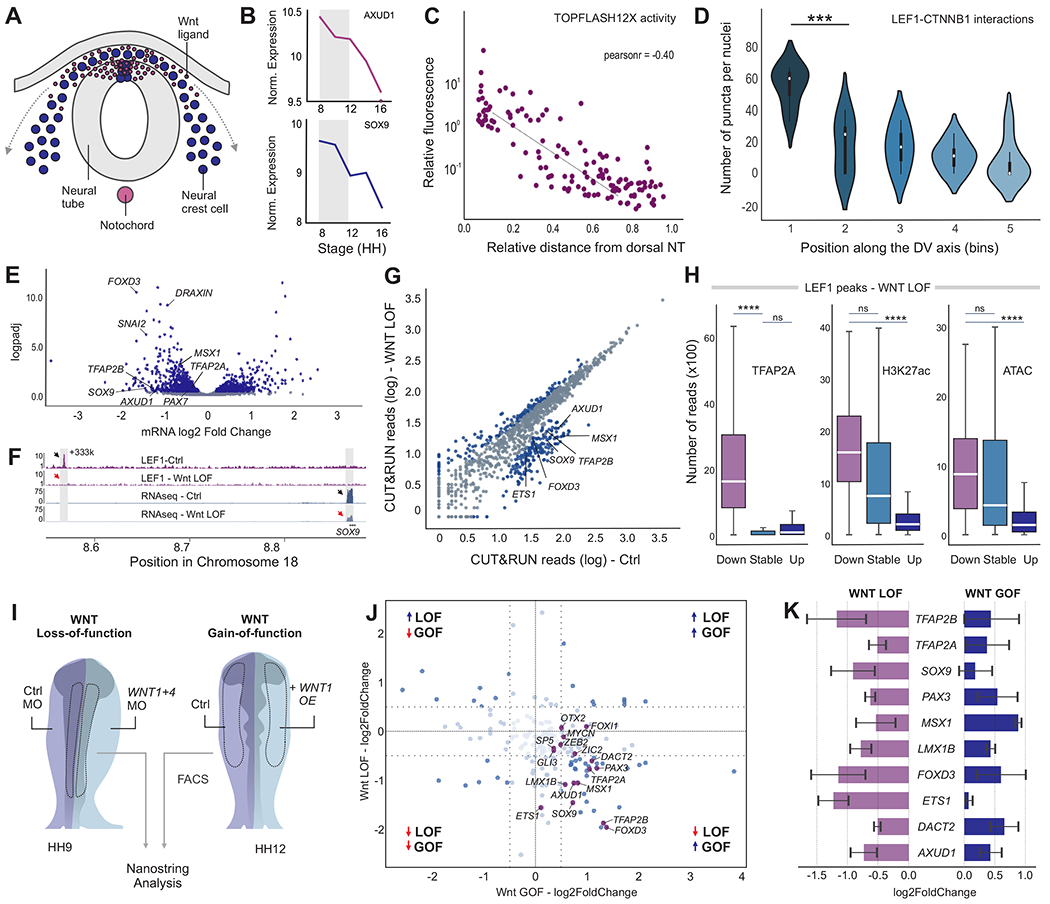
(A) Schematic representation of a transverse section of an avian embryo, depicting the migration of NC cells from the Wnt stem cell niche in the dorsal neural tube.
(B) Expression levels of Wnt target genes AXUD1 and SOX9 decrease during NC migration.
(C-D) Decrease in activity of the Wnt signaling pathway during NC migration. Scatter plot of Wnt reporter fluorescence in NC cells relative to their distance from the dorsal neural tube (C) (n=134 Tfap2AE1:mChe+ cells.) Violin plots of LEF1:CTNNB1 PLA puncta per NC cell across five different dorsal-ventral regions (D) (n=4 embryos).
(E) Volcano plot depicting NC transcripts measured by RNA-seq after Wnt loss-of-function (n=3). Blue dots represent transcripts with pValue<0.05.
(F) LEF1 signal and RNA-seq profiles for control and Wnt loss-of-function conditions at the SOX9 locus. Shaded box highlights the putative enhancer E18.1 tested in Figure 4B.
(G) Scatter plot displaying LEF1 binding at LEF1/CTNNB1-associated peaks in control and upon Wnt knockdown (n=2). Blue dots represent peaks with pValue<0.05 between the two datasets.
(H) Box plots depicting TFAP2A, H3K27ac and ATAC-seq signals at peaks that lose, gain or display stable association with LEF1 upon Wnt knockdown.
(I) Diagram showing the electroporation scheme for assessing spatial-specific effect of Wnt signaling manipulation. Control and WNT1/4 morpholinos were bilaterally electroporated at HH4 and NC cells were microdissected from embryos at the specification stage (n=3). Conversely, sustained Wnt function was obtained from embryos electroporated with a WNT1 overexpression vector. Late NC migratory cells were FACS-sorted (Tfap2aE1:mChe+) from HH12 embryos (n=3).
(J) Representative scatter plot displaying log2FoldChange in a Wnt loss-of-function (y-axis) and a Wnt gain-of-function (x-axis) replicate. Genes contacting Wnt putative enhancers tested in Figure 4 are highlighted (K).
(K) Bar plot showing the effect of Wnt loss-of and gain-of-function on expression of GRN components.
Also see Figure S6. NT, neural tube; DV dorsal-ventral; LOF, loss-of-function; GOF, gain-of-function; MO, morpholino; OE, overexpression; HH, Hamburger and Hamilton; Mb, megabase. Error bar represents ± S.E.M.
To further investigate how Wnt signaling modulates the NC GRN, we knocked down WNT1/4 and compared mRNA expression profiles from control and morphant NC. This analysis identified several genes affected by the disruption of Wnt signaling, including TFAP2A, TFAP2B, PAX7, FOXD3, MSX1 and SOX9 (Figure 6E, Supplemental Table 5). Gene ontology analysis confirmed that epithelial to mesenchymal transition and NC development were the processes most affected by the manipulation (Figure S6E). Next, we surveyed how disruption of Wnt signaling affected the interaction between nuclear effectors and Wnt-associated elements in NC cells. CUT&RUN in WNT1/4 morphant embryos revealed that binding of nuclear effectors was lost upon disruption of the pathway (Figure 6F–G). These shifts in enhancer occupancy were evident in the loci of NC GRN components like SOX9, FOXD3, TFAP2B and AXUD1 (Figures 6F–G and S6F). Thus, inhibition of Wnt signaling prevents interaction between NC enhancers and nuclear effectors, leading to the collapse of the GRN.
We next analyzed how LEF1 genomic occupancy was affected by disruption in Wnt signaling. Cis-regulatory elements that lost interaction with LEF1 following Wnt inhibition displayed features of active NC enhancers, such as association with TFAP2A, high H3K27ac, and increased accessibility (Figure 6H). Surprisingly, the overall binding of the nuclear effector did not significantly change upon WNT1/4 knockdown (Figure S6G). Instead, LEF1 binding was observed in different regions of the genome in the morphant embryos. Motif enrichment analysis revealed that, in the absence of Wnt signals, LEF1 was associated with regions containing the zinc finger 263 motif (Figure S6I). Next, we examined the importance of Wnt signaling in enhancer activity by evaluating accessibility, association with H3K27Ac and eRNA levels (Figure S6G–H) in Wnt loss-of-function conditions. Disruption of the signaling pathway did not alter chromatin accessibility, but we observed a significant loss in eRNA levels following knockdown. This indicates that LEF1 and CTNNB1 are acting as bona fide transcriptional activators in the control of enhancer activity in NC cells.
To test if Wnt signaling modulates the NC GRN in a spatial-specific manner, we manipulated the activity of the pathway. We performed loss-of-function experiments in premigratory cells (HH9), which are close to the dorsal neural tube and receive high levels of Wnts. Conversely, we overactivated the pathway in late migratory cells (HH12), which are far away from the Wnt niche (Figure 6I). In both experiments, NC cells were isolated from the right side of individual embryos, and their transcriptional profile was compared to contralateral control cells using Nanostring (Figure 6I). We found Wnt signaling to be necessary and sufficient for the expression of a large number of NC genes, which are associated with Wnt-associated enhancers (Figure 6J–K). Thus, instead of regulating NC identity via a small number of downstream targets, Wnt signaling directly controls a large part of the genetic circuitry of the GRN. This allows NC cells to display overarching changes in gene expression in response to the signals they encounter before and during migration.
Discussion
Cell type specification involves drastic shifts in gene expression and overarching changes in the spatial organization of the genome. Our high-resolution contact map of active enhancers demonstrates that NC formation involves the establishment of thousands of cell type-specific intra-TAD loops. Examination of cis-regulatory elements that are part of tissue-specific loops uncovered important principles in the spatial regulation of gene regulatory networks. In NC development, Wnt signaling is both necessary and sufficient for specification. As a result, Wnt signaling has been placed at the top of the NC gene regulatory network (Meulemans and Bronner-Fraser, 2004, Simoes-Costa and Bronner, 2015), thought to act as an upstream activator of a transcriptional cascade that results in specification. In this scenario, Wnts would directly promote the expression of a small group of effector genes that relay the signal throughout the gene regulatory network (Simoes-Costa et al., 2015). Our results refute this hierarchical mode of the action of signaling systems, and instead support the existence of an integrated system of signal-responsive regulatory elements that directly regulate network sub-circuits. We propose a model where Wnt signaling is connected to multiple components of the NC gene regulatory network in hub-and-spoke architecture (Figure 7).
Figure 7. Model for position-dependent regulation of the neural crest identity by canonical Wnt signaling.
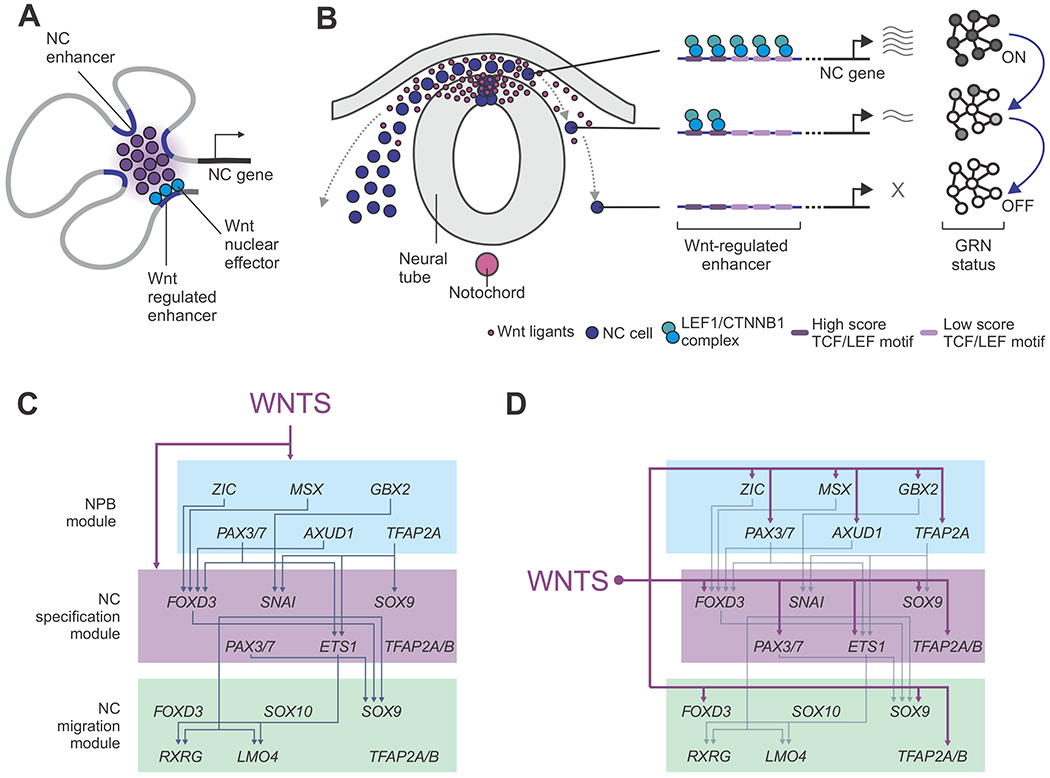
(A) NC genes are regulated by multiple enhancers that contact promoters in a tissue-specific manner. Essential components of the NC GRN are regulated by at least one Wnt-associated element.
(B) Wnt-associated elements act as genomic sensors that respond to environmental inputs. These enhancers possess multiple TCF/LEF binding motifs that allow response to levels of signaling. As cells migrate away from the stem cell niche, these elements lose their interactions with nuclear effectors, resulting in loss of gene expression and inactivation of the GRN.
(C-D) Models for the role of Wnt signaling in the control of the NC GRN. Instead of acting as an upstream regulator (C), our results indicate canonical Wnts simultaneously control multiple components the GRN in a hub-and-spoke architecture (D).
Our results show that NC specification genes possess elements that are associated with Wnt nuclear effectors in their cis-regulatory repertoire. We propose that such elements act as genomic sensors that propagate environmental inputs directly to network components, orchestrating shifts in gene expression. These enhancers would allow the network to quickly respond to changes in cell position during NC migration. Indeed, previous studies show that mRNA expression of NC specification genes changes rapidly as cells move away from the neural tube (Bhattacharya et al., 2018), towards locations that have lower activity of Wnt signaling. Manipulating this spatial distribution of Wnt signaling results in the expected changes in the transcriptome (Figure 6I–J). This is consistent with our previous suggestion that the dorsal neural tube acts as a Wnt niche that promote cellular properties such as stemness and pluripotency (Bhattacharya et al., 2018). Our model (Figure 7) explains how inductive signals are able to rapidly and thoroughly reprogram the regulatory states of target cells.
The Wnt-associated elements we identified in this study possess a number of intriguing regulatory features that set them apart from previously studied NC enhancers. They are often distant from the genes they regulate and display complex syntax, containing multiple TCF/LEF motifs. Our analysis of transcription factor occupancy shows that both optimal and suboptimal motifs interact with Wnt nuclear effectors and contribute to enhancer output. Consistent with previous reports showing that suboptimal binding sites underlie enhancer specificity (Farley et al., 2015), we found that degenerate TCF/LEF motifs are critical for the for the activity of NC enhancers (Figure 5E,I). Notably, we also observed that combinations of optimal and suboptimal binding sites confer enhancers with the ability to respond to different levels of Wnt activation. Such regulatory features allow these cis-regulatory elements to generate a range outputs according to cell-extrinsic cues, and act as genomic sensors of positional information during cell migration.
Finally, our results also indicate that Wnt signaling may play a direct role in the spatial reorganization of chromatin that takes place during NC specification. This is consistent with previous data that shows that HMG domain proteins (like TCF/LEF) can mediate the assembly of higher-order nucleoprotein structures. LEF1 itself has been implicated in folding of DNA (Love et al., 1995) and CTNNB1 interacts with a number of chromatin modifiers that may aid in the formation of the enhanceosome (Mosimann et al., 2009). This points at a mechanism that links a cell’s positional information to its nuclear architecture, explaining how cells that are exposed to different environmental signals may undergo rapid changes in chromatin organization. Indeed, recent studies also have shown that Wnt nuclear effectors may be involved in mediator condensates at super-enhancers (Zamudio et al., 2019). The regulatory features of Wnt-associated elements (e.g. prevalence of long-range enhancer elements, reliance on low-affinity binding sites, clustered binding of nuclear effectors) are consistent with a role for nuclear compartmentalization in the regulation of the NC gene regulatory network. Elucidating the interplay between environmental signals and nuclear organization will have a significant impact on our understanding of the spatial control of cell identity.
Limitations of Study
This study employed H3K27ac-HiChIP to examine the chromatin conformation in NC cells. Since this experimental strategy relies on an antibody to target H3K27ac+ genomic regions, our connectome does not contain interactions between loci that are not associated with this histone mark. Furthermore, our contact map was assembled with premigratory NC cells isolated from HH9 avian embryos. Thus, it does not contain information about the changes in chromatin conformation that take place during NC development. Finally, our H3K27ac-HiChIP and CUT&RUN analyses were performed in bulk samples, which prevented us from surveying the levels of cellular heterogeneity in chromatin conformation and genomic occupancy of transcription factors.
Star Methods
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Marcos Simoes-Costa (simoescosta@cornell.edu).
Materials Availability
This study did not generate new unique reagents
Data and Code availability
The H3K27ac-HiChIP, the CUT&RUN datasets for CTNNB1, LEF1 and CTCF in wild type chick neural crest cells, the CUT&RUN for LEF1 and H3K27ac, ATAC-seq and RNA-seq datasets in WNT loss of function neural crest cells havge been deposited to the Gene Expression Omnibus GSE150006. The CUT&RUN dataset for H3K27ac in wild type chick neural crest cells has been deposited to the Expression Omnibus GSE142101. The TFAP2A CUT&RUN and the ATAC-seq datasets in wild type chick neural crest cells have been deposited to the Expression Omnibus GSE126880.
Experimental Models and subject details
Chick embryo collection and electroporation
Fertilized Leghorn White chicken eggs were obtained from the Department of Animal Science, University of Connecticut. Eggs were incubated at 37°C until embryos reached the desired developmental stage. Embryos were collected and cultured according to the EC protocol (Chapman et al., 2001), and staged according to Hamburger and Hamilton (Hamburger and Hamilton, 1951). Enhancer plasmids, morpholinos and expression vectors were transfected in chick embryos at HH4 by ex ovo electroporation, as previously described (Simoes-Costa et al., 2015). Briefly, constructs were injected between the epiblast and vitelline membrane of embryos at a concentration of 1-2ug/ul and electroporated with platinum electrodes (five 50ms pulses of 5.1V, with an interval of 100ms between pulses). Enhancer reporter assays were performed using whole embryo injections. Mutants, knockdown and overexpression constructs were injected bilaterally with their respective controls. Following electroporation, embryos were cultured in albumin at 37°C until they reached appropriate developmental stages. Embryo survival was >90% and all embryos were screened to ensure uniform electroporation and proper embryo morphology prior to further downstream analysis.
Method details
Enhancers and Mutant Reporter Assays
Putative neural crest enhancers were defined based on the contact of H3K27ac peaks with promoters (HiChIP enhancer-promoter loops) of neural crest genes. Our set of neural crest genes (Supplemental Table 1) was assembled based in two criteria: (1) they were found to be enriched in neural crest cells in transcriptomic studies (Simoes-Costa et al., 2014) and (2) they have been shown to be important to neural crest development via functional studies (Simoes-Costa and Bronner, 2015).
Selected putative enhancers (Supplemental Tables 2–3) were amplified from HH10 chicken genomic DNA and cloned in pTK-EGFP (Uchikawa et al., 2003). To assess enhancer activity, HH4 embryos were co-electroporated with Tfap2aE1:mChe (Rothstein and Simoes-Costa, 2020) and the pTK-EGFP constructs, and cultured until the desired developmental stage. Single cell fluorescence quantification, using the Attune NxT cytometer, was performed to access enhancer intensity and neural crest specificity. Heads from four transgenic embryos were dissected and dissociated in Accumax (Innovative Cell Technologies, #AM105). mCherry and eGFP fluorescence was accessed from 1500 cells. eGFP intensity was used to define the enhancer strength while the specificity (crest score) was defined by the ratio of double-positive cells (eGFP+/mCherry+) in the eGFP+ population.
TCF/LEF1 binding sites in Axud1E1-500bp and Axud1E1-300bp were defined using the JASPAR database of transcription factor binding profiles(Mathelier et al., 2016). Mutant constructs (Supplemental Table 4) were cloned into pTK-EGFP and the mutated enhancer activity was compared to a pTK-mCherry wild-type construct (Axud1E1-500bp:mCherry or Axud1E1-300bp:mCherry) co-transfected in the same embyro. eGFP/mCherry fluorescence was measured in a Attune NxT cytometer as described above. Individual 20bp mutations in Axud1E1-300bp (Supplemental Table 4) were assembled with Gibson assembly. The 15 mutants generated were co-injected with Axud1E1-300bp:mChe. Mutant enhancer activity was accessed by quantification of eGFP/mCherry fluorescence ratio in three ROIs defined in the head of HH9 embryos.
HiChIP
In situ contact libraries were generated according to the HiChIP protocol published by Mumbach and colleagues (Mumbach et al., 2016) with modifications for initial low input optimization. Briefly, neural crest cells were dissected from HH9 embryos (n=120 per replicate) in Ringers and dissociated in Accumax (Innovative Cell Technologies, #AM105). Whole HH9 embryos were prepared in parallel as control samples (n=20 per replicate). After crosslinking, samples were washed in cold PBS and transferred to Hi-C lysis buffer. Lysed cells were incubated in 0.5% SDS at 62°C for 10 min. DNA digestion was performed with MboI (New England Biolabs, #R0147) at 37°C for 2h. After enzyme inactivation and incorporation of biotinylated nucleotides, fragments were re-ligated with T4 DNA ligase for 4h and immediately processed for the ChIP protocol. Ligation solution was replaced by Nuclear Lysis buffer and samples were sonicated with a Diagenode Sonicator (10 cycles of 30s ON, 30s OFF). Samples were then diluted with ChIP Dilution Buffer, precleared in ProteinG dynabeads (Life Technology, #10004D), and incubated overnight with H3K27ac antibody (Abcam, #177178). Samples were then cold washed in a sequence of low-salt, high-salt and LiCl wash buffers. Eluted DNA was treated with 10mg/ml Proteinase K overnight. Sample purification was performed using DNA Clean and Concentrator columns (Zymo Research) and after DNA quantification Streptavidin C-1 beads (Thermo Fisher) were used for recovery of biotin bound fragments. DNA fragments were tagmented with TN5 and PCR amplified. After size selection, libraries were quantified and sequenced with paired-end 75bp reads on an Illumina NextSeq500 instrument.
Immunohistochemistry
For whole-mount immunohistochemistry, embryos were collected at appropriate developmental stages and fixed in 4% PFA-PB for 20 mins at RT. Post fixation, embryos were dissected from the filter paper and washed in TBS containing 0.1% Triton and 1% DMSO (TBTD). Embryos were blocked at RT for 2h in TBTD supplemented with 10% donkey serum and incubated in anti-TFAP2B (Santa Cruz Biotechnology, #sc390119) primary antibody diluted in blocking solution, overnight at 4°C. Following the primary antibody incubation, embryos were washed, blocked for 30mins at RT, and stained with appropriate secondary antibodies for 2h at RT. Secondary antibodies used included donkey anti-mouse/rabbit IgG conjugated with Alexa Fluor 488/568 (Molecular Probes). Following the secondary antibody step, the embryos were washed, stained with DAPI and post-fixed with 4% PFA for 1h, prior to imaging. Whole-mount images were collected using an upright Zeiss Axio Imager fluorescent microscope.
Immunohistochemistry of Cell Suspensions
Immunohistochemistry of dissociated neural crest cells was performed as previously described (Wang et al., 2014). Neural crest cells obtained from 30 HH9 embryos were micro-dissected in Ringers, and dissociated in Accumax Cell dissociation solution for 15-20 mins at RT. Following dissociation, cells were washed in PBS and fixed in 0.4% PFA solution for 15 mins at RT. Post-fixation, cells were permeabilized with PBS+0.3% Triton solution and blocked with 10% donkey serum solution (in PBS+0.1% Tween 20) for 1h at RT. The cells were subsequently incubated overnight with mouse anti Pax7 primary antibody (DSHB) in blocking solution. Following incubation with primary antibody, cells were washed in PBS+0.1% Tween 20 and incubated with the secondary antibody diluted to a concentration of 1:1000 in blocking solution for 1h at RT. The samples were then washed twice in PBS+0.1% Tween 20 and the staining intensity was measured using the Attune Nxt flow cytometer at the Cornell Flow facility. The cytometry data were analyzed using the FCS Express 6 software.
Loss-of-function assays
Wnt knockdown was performed by the combined inhibition of Wnt1 and Wnt4 by morpholinos at 1.25uM each. Enhancer constructs were co-electroporated with control and targeted morpholinos in the left and right sides of the embryos, respectively. For RNA expression levels in Wnt loss-of-function assays, paired single neural crest dissections (left side = control, right side = knockdown) were finely dissected from three embryos and processed individually. Total RNA was isolated with the RNAqueous-Micro Total RNA Isolation Kit (Thermo Fisher Scientific, # AM1931) according to the kit’s protocol. RNA was poly(A) selected using the NEBNext Poly(A) mRNA Magnetic Isolation Module (New England Biolabs, #E7490). TruSeq-barcoded RNA-seq libraries were generated with the 615 NEBNext Ultra II Directional RNA Library Prep Kit (New England Biolabs, #E7760) and sequenced on an Illumina NextSeq 500 instrument with single-end 75bp reads. For eRNA quantification, paired single neural folds were microdissected from control and targeted sides of the embryo, and subsequently lysed in lysis buffer from Power SYBR Green Cells-to-CT Kit. RNA extraction and cDNA preparation were performed according to the kit’s protocol. RT-PCR was performed using Power Sybr Green PCR master mix (Thermo Fisher, 4368577) in an ABI viia7 RT-PCR machine. Ct values of all genes were normalized to reference gene HPRT1 and expressed as a fold change compared to the control sample. The e RNA qPCR primers sequences are listed in Supplemental Table 6.
CRISPR-Cas9 mediated enhancer loss-of-function
We employed a CRISPR-Cas9 system optimized for chick embryos to disrupt the activity of enhancers (Gandhi et al., 2017). gRNAs were designed using online resources (crispor.tefor.net) and cloned downstream of the U6 promoter in the cU6.3 vector (Supplemental Table 6). To assess the effect of endogenous enhancers knockdown, gastrula-stage embryos were electroporated with a pCAG dCas9-KRAB-2A-EGFP (Williams et al., 2019) vector and each enhancer gRNA. A control gRNA was used on the left side of the embryo. Embryos were re-incubated at 37oC. At stage HH9, embryos were screened for robust GFP expression in both sides, and half heads were dissected for control and targeted sides of the embryo. Half heads were then dissociated in Accumax (Innovative Cell Technologies, #AM105) for 30 min. After dissociation, cells were resuspended in HANKS solution supplemented with 0.5% BSA. Control and target GFP+ cell suspensions (75-150 cells) were sorted into 25μl of lysis buffer from the Power SYBR Green Cells-to-CT Kit (ThermoFisher, 4402955) using a BD AriaFusion cell sorter. Samples were processed following the manufacturer’s protocol. Respective genes expression levels were determined using RT-PCR as described above. Ct values of all genes were normalized to reference gene HPRT1 and expressed as a fold change compared to the control sample. The qPCR primers sequences are listed in Supplemental Table 6.
CUT&RUN
Neural crest cells were dissected from HH9 embryos (n=20 per CUT&RUN experiment). Cells were dissociated in Accumax for 20min at RT under mild agitation. CUT&RUN experiments were carried out as previously described (Rothstein and Simoes-Costa, 2020). Cells were immobilized on BioMag Plus Concanavalin A magnetic beads (Bangs Laboratories, BP531) and incubated with rabbit anti-LEF1 (Abcam, #ab137872), anti-CTNNBI (Abcam, #ab32572) or anti-CTCF (Abcam, #ab188408) antibody (1:50) overnight at 4°C. After washing away unbound antibody, protein A-MNase was added to a final concentration of 700ng/mL and incubated for 1h at 4°C. Cells were cooled to 0°C and CaCl2 was added to a final concentration of 2mM to activate the MNase enzyme. MNase digestion was performed for 45min and terminated by the addition of 2XSTOP buffer. The protein-DNA complexes were released by centrifugation and digested with proteinase K for 10 min at 70°C. DNA fragments were isolated via phenol-chloroform extraction and ethanol precipitation. Protein A-MNase and spike-in DNA were kindly provided by Dr. Steven Henikoff (Skene and Henikoff, 2017). To quantify LEF1 binding in a Wnt loss-of-function context, HH4 embryos were electroporated with Wnt1/4 combined morpholinos and cultured until HH9 when neural crest cells were dissected and samples processed following the protocol previously described.
CUT&RUN Library Preparation
CUT&RUN libraries were prepared using the NEBNext Ultra II DNA Library Prep Kit (New England Biolabs, #E7645) following the manufacturers protocol. Fragment analysis was performed with ABI 3730xl DNA Analyzer to perform quality control for the libraries. Equimolar concentrations of the libraries were pooled using the KAPA Library Quantification Kit - ROX Low (Roche, #07960336001) and sequenced with paired-end 37bp reads on an Illumina NextSeq500 instrument.
Enhancer Pulldown
For enhancer pulldown experiments, HH4 embryos were electroporated with a FLAG-tagged LEF1 construct cloned in a pCAGGS-H2B-RFP backbone. Embryos were incubated until stage HH9. After electroporation efficiency was confirmed, embryos were dissected in Ringer’s solution (n=4 embryos per sample). Nuclear protein extracts were obtained as previously described(Simoes-Costa et al., 2015). Wild-type and mutant forms of Axud1E1 were PCR amplified from pTK-eGFP vectors using a biotinylated forward primer (5’-AAAATAGGCTGTCCCCAGTG-3’) and an untagged reverse primer (5’-ATATTTCTTCCGGGGACACC-3’). Immobilization of nucleic acids was performed using Dynabeads MyOne Streptavidin T1 (Invitrogen, #2023-11-30) following manufacturer’s protocol. For enhancer pulldown, nuclear protein extracts were diluted in 10 mM Tris-HCl, 1 mM EDTA, 0.5 mM EGTA, 10% Glycerol, 0.25% NP-40 supplemented with 10 ug Poly(dI-dC) and incubated with biotinylated DNA coated Dynabeads for 90 min in rotation at 4°C. Magnetics beads were washed four times in with Washing Buffer (10 mM Tris-HCl, 1 mM EDTA, 0.5 mM EGTA, 100 mM NaCl, 10% Glycerol, 0.25% NP-40), and proteins eluted in RIPA modified buffer containing 1x Sample Reducing Agent and 1x LDS Sample Buffer (Invitrogen, #B0009 and #B0007) for 15 min at 80°C and 1400 rpm. Proteins were separated by electrophoresis on Bolt 4–12% Bis-Tris Plus mini gels (Invitrogen, #NW04120BOX), followed by immunoblotting on nitrocellulose membranes using a monoclonal anti-FLAG antibody (Sigma, #A8592).
Nanostring Analysis
To assess the spatial effect of Wnt signaling manipulation in neural crest GRN Nanostring analysis was performed for stage-specific Wnt loss- and gain-of-function assays. HH4 embryos were bilaterally injected with a Wnt1/4 morpholino mix or a vector driving Wnt1 expression, respectively. All embryos were co-injected with the Tfap2aE1:mChe enhancer. Knockdown experiments were performed in paired neural crest dissections from HH9 (6 somites) morphant embryos. For overexpression experiments embryos were cultured until HH12 and screened for robust mCherry expression. Control and treated head sides were then dissected and the dorsal neural tube removed to avoid late non-migratory cells still present at the dorsal neural folds. Half heads were separately dissociated and processed for FACS sorting of Tfap2aE1:mChe-positive cells (n=500 cells). Neural crest dissections (knockdown) and FACS sorted cells (overexpression) were lysed in 5ul of Cell-to-CT lysis buffer (Cell-to-CT kit, Thermo Fisher). The cell lysate was hybridized to a Nanostring Probe Set containing 200 probes for neural crest, placode and neural genes (Bhattacharya et al., 2018), at 65°C for 16 hrs. Nanostring data were analyzed using the nSolver software package. Knockdown and overexpression assays were performed in three biological replicates each.
Proximity Ligation Assays (PLA)
For Proximity Ligation Assay (PLA), HH4 embryos were injected with Tfap2aE1:GFP and cultured until HH12. Embryos were screened for strong GFP expression, dissected in Ringers solution and fixed for 20min in phosphate buffer (PB) containing 4% PFA. Embryos were then embedded in gelatin and cryosectioned at 10um. PLA was performed with the Duolink PLA detection kit (Sigma Aldrich, #DUO92101) according to the manufacturer’s protocol. The primary antibodies used were: mouse anti-LEF1 (Millipore, #17–604) and Rabbit Anti-CTNNBI (Abcam, ab32572). For LEF1:CTNNB1 PLA quantification, PLA-positive puncta were quantified in Tfap2aE1:GFP-positive cells across five adjacent regions of interest (ROI) defined on the dorsal-ventral axis. Number of cells (indicated via DAPI staining) and number of puncta were counted within each ROI and used to calculate the ratio puncta/nuclei per dorsal-ventral region.
In situ Hybridization
For in situ hybridization, embryos were fixed in phosphate buffer saline (PBS) containing 4% paraformaldehyde (PFA) for 2 hours at RT or overnight at 4°C. Following fixation, embryos were dissected, washed with PBST, dehydrated and stored in methanol at −20°C. Whole-mount in situ hybridization was performed as previously described(Wilkinson, 1992).
Explant cultures
Neural crest were performed as previously described (Coles et al., 2007). Neural crest explants were derived via microdissection of neural folds of HH9 embryos (8 somites) previously electroporated with the Axud1E1:GFP mutant variants along with the control plasmid pCI-Cherry-Ras. The dissected tissues were then transferred to 96-well cell culture plates coated with fibronectin and containing 100uL of 10%FBS DMEM culture media and the Wnt agonist CHIR99021 (Tocris, 4423) at the final concentrations of 1uM or 3uM (the same volume of vehicle was used for control explants). The explants were incubated for up to 24 hours at 37°C in a CO2 incubator. Explants were lysed with the lysis buffer from the Power SYBR Green Cells-to-CT Kit (ThermoFisher, 4402955) and processed for qPCR as described above using the GFP primers (forward-5’-TGACCCTGAAGTTCATCTGC-3’, reverse – 5’-AAGTCGTGCTGCTTCATGTG-3’) and the mCherry primers (forward - 5’-AGTTCATGCGCTTCAAGGTG-3’, reverse – 5’-TTGGTCACCTTCAGCTTGGC-3’). GFP expression values are presented normalized to mCherry.
Quantification and Statistical Analysis
Description of all datasets used in each analysis, pValues and number of biological replicates are presented in Supplemental Table 7. Student’s t-test (two-tailed) was performed to calculate pValues. Welch’s unpaired two-tailed t-test was performed for unequal sample sized experiments. pValues<0.05 were considered to be significant (*p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001).
HiChIP Data Analysis
Each HiChIP replicate was initially processed with the HiC-Pro pipeline (Servant et al., 2015). Raw fastq (paired-end reads) were aligned to the chicken reference genome (Galgal5) and default settings used to remove duplicate reads, assign reads to Mbol restriction fragments and filter for valid interactions. Preprocessed valid interactions were subsequently submitted to the Hichipper pipeline (Lareau and Aryee, 2018b), a platform developed for bias-corrected peak calling, library quality control and DNA loop calling in HiChIP datasets using a standard MACS2 background model for H3K27ac peak calling and loop anchoring. The framework Diffloop (Lareau and Aryee, 2018a), a R/Bioconductor package (Gentleman et al., 2004), was employed for identification of differential intrachromosomal chromatin interactions comparing neural crest (NC) and whole embryos (WE) datasets (three replicates each). Briefly, after mango algorithm correction with a FDR of 0.05, differential loops (NC vs.WE) were called and annotated to promoters and enhancers (H3K27ac CUT&RUN HH9 dataset). Promoter contacting regions were filtered using a minimum loop width of 1.5kb. Non-prevalent loops were also excluded (minimum of 5 counts in at least 3 replicates). Loops Log Fold Change (logFC) and pValues<0.05 were used to define NC enriched and depleted loops. Contact matrices were generated by dumped hic file outputs from Juicer Tools (Durand et al., 2016). Pairs of matrices were visualized using the Juicebox platform applying the Square Root Vanilla Coverage Normalization. WashU Epigenome Browser was used for concomitant visualization of loop plots, DNA binding profiles and RNAseq datasets. Aggregate Peak Analysis (APA)(Rao et al., 2014) was performed using Juicer Tools(Durand et al., 2016) comparing NC and WE matrices to NC enriched (loop logFC>0.75) and depleted (loop logFC<−0.75) contacts considering a local background of 50kb. P2LL (Peak to Lower Left) values above 1 and above 0 for Zscore were used as indicative of enrichment.
CUT&RUN Data Analysis
Paired-end sequencing reads from the CUT&RUN libraries were trimmed using Cutadapt (Martin, 2010). Reads were filtered for those with a minimum length of 25bp or longer and aligned to the reference chicken Galgal5 assembly using Bowtie2 (Langmead and Salzberg, 2012). Picard MarkDuplicates tool was used to mark duplicate reads and BAM files were filtered with SAMtools to discard unmapped reads (those that were not the primary alignment, reads failing platform/vendor quality checks, and PCR/optical duplicates (-f 2 -F 1804)). Peak calling was performed using MACS version 2.1 with a p value cutoff of 0.01, skipping the shifting model and extending read sizes to 200bp (--nomodel --extsize 200). Representative heatmaps showing the CTNNB1, IgG and H3K27ac at LEF1 bound peaks were generated using the deepTools2 package (Ramirez et al., 2016). To identify transcription factor motifs in the genomic regions occupied by LEF1 and CTNNB1, motif enrichment analysis was performed using the HOMER findMotifsGenome package (Heinz et al., 2010). The GO-category analysis was performed on putative WNT targets by utilizing the Panther Classification System package(Thomas et al., 2003) to assay for over-represented Biological Processes having a p value cut off of 0.05. BEDTools suit, sub-command PairToBed (Quinlan and Hall, 2010) was used to assign peaks to HiChIP contact matrices.
EChO
Enhanced Chromatin Occupancy (EChO) was performed as described in(Meers et al., 2019) and https://github.com/FredHutch/EChO. Briefly, LEF1 and CTNNB1 co-occupied peaks were extended 500bp upstream and downstream from the peak summit and used as LEF1 enriched region file. Then, a matrix of base pair-resolution EChO fragment size values spanning a 400bp window for every entry in the region file was generated for our LEF1 CUT&RUN dataset. The LEF1 locally weighted average fragment-size profile was plotted at Wnt putative enhancer loci for local minima visualization. JASPAR database screening was used to associate TCF/LEF1 profiles in a window of 40bp around each focus. TCF/LEF1 profiles with scores >80% (JASPAR database) where considered high-scoring motifs while scores of >70% where considered low-scoring motifs . For motif score analysis, LEF1 and TFAP2A foci defined at LEF1/CTNNB1/TFAP2A co-occupied peaks were expended to a 40bp window. FIMO (Grant et al., 2011) (Find Individual Motif Occurrences) analysis was performed to estimate a score and a pValue for each LEF1(MA0768.1) and TFAP2A(MA0003.3) motif occurrence independently. Score frequencies of LEF1 an TFAP2A motifs with a pValue<0.005 were plotted.
RNAseq analysis
Reads were trimmed using CutAdapt (Martin, 2010), and aligned to the galGal5 genome using hisat2 (Kim et al., 2015) and quantified using featureCounts on genes from ENSEMBL release 93. Gene counts were processed in R and differentially expressed genes were called using DESeq2 (Love et al., 2014). Lowly-expressed genes were removed (less than 5 counts any two samples) and DESeq2 revealed 254 differentially expressed genes (padj=0.05, Supplemental Table 5). Regularized logarithm counts were exported for additional analysis.
Supplementary Material
Supplemental Table 1. List of neural crest GRN genes. Related to Star Methods.
Supplemental Table 2. Putative enhancer-promoter contacts of components of the neural crest GRN. Related to Star Methods.
Supplemental Table 3. Genomic positions of neural crest enhancers. Related to Star Methods.
Supplemental Table 4. Mutations performed in Axud1E1 variants. Related to Star Methods.
Supplemental Table 5. Differentially expressed genes under Wnt LOF conditions. Related to Star Methods.
Supplemental Table 6. Nucleotide sequences for oligonucleotides or primers used. Related to Star Methods.
Supplemental Table 7. Number of biological replicates and pValues for quantitative experiments. Related to Star Methods.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti H3K27ac | Abcam | Cat#ab177178; RRID: AB_2828007 |
| Mouse monoclonal anti TFAP2B | Santa Cruz Biotechnology | Cat#sc-390119; RRID:AB_2828008 |
| Mouse monoclonal anti Pax7 | DSHB | Cat#PAX7; RRID:AB_2299243 |
| Rabbit monoclonal anti LEF1 | Abcam | Cat#ab137872 |
| Mouse monoclonal anti LEF1 | Millipore | Cat#17–604; RRID:AB_916350 |
| Rabbit monoclonal anti CTNNB1 | Abcam | Cat#ab32572; RRID:AB_725966 |
| Rabbit monoclonal anti CTCF | Abcam | Cat#ab188408; RRID:AB_2819212 |
| Monoclonal anti FLAG M2 (HRP) | Sigma Aldrich | Cat#A8592; RRID:AB_439702 |
| Bacterial and virus strains | ||
| Biological samples | ||
| Chemicals, peptides, and recombinant proteins | ||
| Critical commercial assays | ||
| Duolink PLA detection kit | Sigma Aldrich | Cat#DUO92101 |
| Deposited data | ||
| CTNNB1 CUT&RUN in wild type chick neural crest cells | This paper | GSE150006 |
| LEF1 CUT&RUN in wild type chick neural crest cells | This paper | GSE150006 |
| CTCF CUT&RUN in wild type chick neural crest cells | This paper | GSE150006 |
| LEF1 CUT&RUN in chick neural crest cells (Wnt LOF) | This paper | GSE150006 |
| H3H27ac CUT&RUN in chick neural crest cells (Wnt LOF) | This paper | GSE150006 |
| Chick neural crest cells RNA-seq (Ctrl) | This paper | GSE150006 |
| Chick neural crest cells RNA-seq (Wnt LOF) | This paper | GSE150006 |
| ATAC-seq in chick neural crest cells (Ctrl) | This paper | GSE150006 |
| ATAC-seq in chick neural crest cells (Wnt LOF) | This paper | GSE150006 |
| CTNNB1 CUT&RUN in chick neural crest cells (Ctrl) | This paper | GSE150006 |
| Whole embryo H3K27ac HiChIP | This paper | GSE150006 |
| Neural crest cells H3K27ac HiChIP | This paper | GSE150006 |
| H3K27ac CUT&RUN in wild type chick neural crest cells | Bhattacharya et al, 2020 | GSE142101 |
| TFAP2A CUT&RUN in wild type chick neural crest cells | Rothstein and Simoes-Costa, 2020 | GSE126880 |
| ATAC-seq in wild type chick neural crest cells | Rothstein and Simoes-Costa, 2020 | GSE126880 |
| Experimental models: cell lines | ||
| Experimental models: organisms/strains | ||
| Wild type While Leghorn chicken eggs | University of Connecticut | N/A |
| Oligonucleotides | ||
| Oligos are listed in Table S6 | This paper | N/A |
| Recombinant DNA | ||
| Tfap2E1-mCherry | Rothstein and Simoes-Costa, 2020 | N/A |
| pCAG dCas9-KRAB-2A-EGFP | Addgene | Cat#92396 |
| Software and algorithms | ||
| BEDTools | Quinlan and Hall, 2010 | N/A |
| DESeq2 | Love et al, 2014 | N/A |
| Diffloop | Laureau and Aryee, 2018a | N/A |
| Enhanced Chromatin Occupancy (EChO) | Meers et al, 2019 | N/A |
| FCS express 6 | De novo software | N/A |
| Find Individual Motif Occurrences (FIMO) | Grant et al, 2011 | N/A |
| HiC-Pro | Servant et al, 2015 | N/A |
| Hichipper | Laureau and Aryee, 2018b | N/A |
| HOMER findMotifsGenome package | Heinz et al, 2010 | N/A |
| JASPAR database | Mathelier et al, 2016 | N/A |
| Juicer Tools | Durand et al, 2016 | N/A |
| R/Bioconductor | Gentleman et al, 2004 | N/A |
| nSolver | Nanostring technologies | N/A |
| Other | ||
Highlights.
Chromatin conformation capture identified a cohort of distal neural crest enhancers
Most neural crest genes are directly regulated by Wnt signaling
Enhancers with optimal and suboptimal motifs can respond to distinct levels of Wnts
Wnts modulate the neural crest gene network in a position-dependent manner
Acknowledgments
This work was supported by NIH grant R01DE028576 to M.S.-C.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- AMANO T, SAGAI T, TANABE H, MIZUSHINA Y, NAKAZAWA H & SHIROISHI T 2009. Chromosomal dynamics at the Shh locus: limb bud-specific differential regulation of competence and active transcription. Dev Cell, 16, 47–57. [DOI] [PubMed] [Google Scholar]
- AZAMBUJA AP & SIMOES-COSTA M 2021. A regulatory sub-circuit downstream of Wnt signaling controls developmental transitions in neural crest formation. PLoS Genet, 17, e1009296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BAREMBAUM M & BRONNER ME 2013. Identification and dissection of a key enhancer mediating cranial neural crest specific expression of transcription factor, Ets-1. Dev Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BARON R & KNEISSEL M 2013. WNT signaling in bone homeostasis and disease: from human mutations to treatments. Nat Med, 19, 179–92. [DOI] [PubMed] [Google Scholar]
- BETANCUR P, BRONNER-FRASER M & SAUKA-SPENGLER T 2010. Genomic code for Sox10 activation reveals a key regulatory enhancer for cranial neural crest. Proc Natl Acad Sci U S A, 107, 3570–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BHATTACHARYA D, AZAMBUJA AP & SIMOES-COSTA M 2020. Metabolic Reprogramming Promotes Neural Crest Migration via Yap/Tead Signaling. Dev Cell, 53, 199–211 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BHATTACHARYA D, ROTHSTEIN M, AZAMBUJA AP & SIMOES-COSTA M 2018. Control of neural crest multipotency by Wnt signaling and the Lin28/let-7 axis. Elife, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BRAULT V, MOORE R, KUTSCH S, ISHIBASHI M, ROWITCH DH, MCMAHON AP, SOMMER L, BOUSSADIA O & KEMLER R 2001. Inactivation of the beta-catenin gene by Wnt1-Cre-mediated deletion results in dramatic brain malformation and failure of craniofacial development. Development, 128, 1253–64. [DOI] [PubMed] [Google Scholar]
- CHAPMAN SC, COLLIGNON J, SCHOENWOLF GC & LUMSDEN A 2001. Improved method for chick whole-embryo culture using a filter paper carrier. Dev Dyn, 220, 284–9. [DOI] [PubMed] [Google Scholar]
- COLES EG, TANEYHILL LA & BRONNER-FRASER M 2007. A critical role for Cadherin6B in regulating avian neural crest emigration. Dev Biol, 312, 533–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DAVIDSON EH & LEVINE MS 2008. Properties of developmental gene regulatory networks. Proc Natl Acad Sci U S A, 105, 20063–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DURAND NC, SHAMIM MS, MACHOL I, RAO SS, HUNTLEY MH, LANDER ES & AIDEN EL 2016. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst, 3, 95–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FARLEY EK, OLSON KM, ZHANG W, BRANDT AJ, ROKHSAR DS & LEVINE MS 2015. Suboptimization of developmental enhancers. Science, 350, 325–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GANDHI S, PIACENTINO ML, VIECELI FM & BRONNER ME 2017. Optimization of CRISPR/Cas9 genome editing for loss-of-function in the early chick embryo. Dev Biol, 432, 86–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GARCIA-CASTRO MI, MARCELLE C & BRONNER-FRASER M 2002. Ectodermal Wnt Function As a Neural Crest Inducer. Science, 13, 13. [DOI] [PubMed] [Google Scholar]
- GENTLEMAN RC, CAREY VJ, BATES DM, BOLSTAD B, DETTLING M, DUDOIT S, ELLIS B, GAUTIER L, GE Y, GENTRY J, HORNIK K, HOTHORN T, HUBER W, IACUS S, IRIZARRY R, LEISCH F, LI C, MAECHLER M, ROSSINI AJ, SAWITZKI G, SMITH C, SMYTH G, TIERNEY L, YANG JY & ZHANG J 2004. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol, 5, R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GOMEZ GA, PRASAD MS, WONG M, CHARNEY RM, SHELAR PB, SANDHU N, HACKLAND JOS, HERNANDEZ JC, LEUNG AW & GARCIA-CASTRO MI 2019. WNT/beta-catenin modulates the axial identity of embryonic stem cell-derived human neural crest. Development, 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GRANT CE, BAILEY TL & NOBLE WS 2011. FIMO: scanning for occurrences of a given motif. Bioinformatics, 27, 1017–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GROVES AK & LABONNE C 2014. Setting appropriate boundaries: fate, patterning and competence at the neural plate border. Dev Biol, 389, 2–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HAMBURGER V & HAMILTON HL 1951. A series of normal stages in the development of the chick embryo. J Morphol, 88, 49–92. [PubMed] [Google Scholar]
- HANDOKO L, XU H, LI G, NGAN CY, CHEW E, SCHNAPP M, LEE CW, YE C, PING JL, MULAWADI F, WONG E, SHENG J, ZHANG Y, POH T, CHAN CS, KUNARSO G, SHAHAB A, BOURQUE G, CACHEUX-RATABOUL V, SUNG WK, RUAN Y & WEI CL 2011. CTCF-mediated functional chromatin interactome in pluripotent cells. Nat Genet, 43, 630–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HEINZ S, BENNER C, SPANN N, BERTOLINO E, LIN YC, LASLO P, CHENG JX, MURRE C, SINGH H & GLASS CK 2010. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell, 38, 576–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KIM D, LANGMEAD B & SALZBERG SL 2015. HISAT: a fast spliced aligner with low memory requirements. Nat Methods, 12, 357–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KORMISH JD, SINNER D & ZORN AM 2010. Interactions between SOX factors and Wnt/beta-catenin signaling in development and disease. Dev Dyn, 239, 56–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LANGMEAD B & SALZBERG SL 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods, 9, 357–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LAREAU CA & ARYEE MJ 2018a. diffloop: a computational framework for identifying and analyzing differential DNA loops from sequencing data. Bioinformatics, 34, 672–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LAREAU CA & ARYEE MJ 2018b. hichipper: a preprocessing pipeline for calling DNA loops from HiChIP data. Nat Methods, 15, 155–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LE DOUARIN N & KALCHEIM C 1999. The neural crest, Cambridge, Cambridge University Press. [Google Scholar]
- LE LIEVRE CS & LE DOUARIN NM 1975. Mesenchymal derivatives of the neural crest: analysis of chimaeric quail and chick embryos. J Embryol Exp Morphol, 34, 125–54. [PubMed] [Google Scholar]
- LEUNG AW, MURDOCH B, SALEM AF, PRASAD MS, GOMEZ GA & GARCIA-CASTRO MI 2016. WNT/beta-catenin signaling mediates human neural crest induction via a pre-neural border intermediate. Development, 143, 398–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LOVE JJ, LI X, CASE DA, GIESE K, GROSSCHEDL R & WRIGHT PE 1995. Structural basis for DNA bending by the architectural transcription factor LEF-1. Nature, 376, 791–5. [DOI] [PubMed] [Google Scholar]
- LOVE MI, HUBER W & ANDERS S 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol, 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LWIGALE PY, CONRAD GW & BRONNER-FRASER M 2004. Graded potential of neural crest to form cornea, sensory neurons and cartilage along the rostrocaudal axis. Development, 131, 1979–91. [DOI] [PubMed] [Google Scholar]
- MARTIN M 2010. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnetjournal, 17, 10–12. [Google Scholar]
- MATHELIER A, FORNES O, ARENILLAS DJ, CHEN CY, DENAY G, LEE J, SHI W, SHYR C, TAN G, WORSLEY-HUNT R, ZHANG AW, PARCY F, LENHARD B, SANDELIN A & WASSERMAN WW 2016. JASPAR 2016: a major expansion and update of the open-access database of transcription factor binding profiles. Nucleic Acids Res, 44, D110–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MEERS MP, JANSSENS DH & HENIKOFF S 2019. Pioneer Factor-Nucleosome Binding Events during Differentiation Are Motif Encoded. Mol Cell, 75, 562–575 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MEULEMANS D & BRONNER-FRASER M 2004. Gene-regulatory interactions in neural crest evolution and development. Dev Cell, 7, 291–9. [DOI] [PubMed] [Google Scholar]
- MICA Y, LEE G, CHAMBERS SM, TOMISHIMA MJ & STUDER L 2013. Modeling neural crest induction, melanocyte specification, and disease-related pigmentation defects in hESCs and patient-specific iPSCs. Cell Rep, 3, 1140–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MOSIMANN C, HAUSMANN G & BASLER K 2009. Beta-catenin hits chromatin: regulation of Wnt target gene activation. Nat Rev Mol Cell Biol, 10, 276–86. [DOI] [PubMed] [Google Scholar]
- MUMBACH MR, RUBIN AJ, FLYNN RA, DAI C, KHAVARI PA, GREENLEAF WJ & CHANG HY 2016. HiChIP: efficient and sensitive analysis of protein-directed genome architecture. Nat Methods, 13, 919–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PETER I & DAVIDSON EH 2015. Genomic Control Process: Development and Evolution, Academic Press. [Google Scholar]
- QUAIFE-RYAN GA, MILLS RJ, LAVERS G, VOGES HK, VIVIEN CJ, ELLIOTT DA, RAMIALISON M, HUDSON JE & PORRELLO ER 2020. beta-Catenin drives distinct transcriptional networks in proliferative and nonproliferative cardiomyocytes. Development, 147. [DOI] [PubMed] [Google Scholar]
- QUINLAN AR & HALL IM 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics, 26, 841–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RADA-IGLESIAS A, BAJPAI R, PRESCOTT S, BRUGMANN SA, SWIGUT T & WYSOCKA J 2012. Epigenomic annotation of enhancers predicts transcriptional regulators of human neural crest. Cell Stem Cell, 11, 633–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RAIBLE DW & RAGLAND JW 2005. Reiterated Wnt and BMP signals in neural crest development. Semin Cell Dev Biol, 16, 673–82. [DOI] [PubMed] [Google Scholar]
- RAMIREZ F, RYAN DP, GRUNING B, BHARDWAJ V, KILPERT F, RICHTER AS, HEYNE S, DUNDAR F & MANKE T 2016. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res, 44, W160–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RAO SS, HUNTLEY MH, DURAND NC, STAMENOVA EK, BOCHKOV ID, ROBINSON JT, SANBORN AL, MACHOL I, OMER AD, LANDER ES & AIDEN EL 2014. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell, 159, 1665–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROTHSTEIN M & SIMOES-COSTA M 2020. Heterodimerization of TFAP2 pioneer factors drives epigenomic remodeling during neural crest specification. Genome Res, 30, 35–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SANYAL A, LAJOIE BR, JAIN G & DEKKER J 2012. The long-range interaction landscape of gene promoters. Nature, 489, 109–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAUKA-SPENGLER T & BRONNER-FRASER M 2008. A gene regulatory network orchestrates neural crest formation. Nat Rev Mol Cell Biol, 9, 557–68. [DOI] [PubMed] [Google Scholar]
- SCHOENFELDER S, FURLAN-MAGARIL M, MIFSUD B, TAVARES-CADETE F, SUGAR R, JAVIERRE BM, NAGANO T, KATSMAN Y, SAKTHIDEVI M, WINGETT SW, DIMITROVA E, DIMOND A, EDELMAN LB, ELDERKIN S, TABBADA K, DARBO E, ANDREWS S, HERMAN B, HIGGS A, LEPROUST E, OSBORNE CS, MITCHELL JA, LUSCOMBE NM & FRASER P 2015. The pluripotent regulatory circuitry connecting promoters to their long-range interacting elements. Genome Res, 25, 582–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SERVANT N, VAROQUAUX N, LAJOIE BR, VIARA E, CHEN CJ, VERT JP, HEARD E, DEKKER J & BARILLOT E 2015. HiC-Pro: an optimized and flexible pipeline for HiC data processing. Genome Biol, 16, 259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SIMOES-COSTA M & BRONNER ME 2015. Establishing neural crest identity: a gene regulatory recipe. Development, 142, 242–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SIMOES-COSTA M, STONE M & BRONNER ME 2015. Axud1 Integrates Wnt Signaling and Transcriptional Inputs to Drive Neural Crest Formation. Dev Cell, 34, 544–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SIMOES-COSTA M, TAN-CABUGAO J, ANTOSHECHKIN I, SAUKA-SPENGLER T & BRONNER ME 2014. Transcriptome analysis reveals novel players in the cranial neural crest gene regulatory network. Genome Res, 24, 281–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SIMOES-COSTA MS, MCKEOWN SJ, TAN-CABUGAO J, SAUKA-SPENGLER T & BRONNER ME 2012. Dynamic and differential regulation of stem cell factor FoxD3 in the neural crest is Encrypted in the genome. PLoS Genet, 8, e1003142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SKENE PJ & HENIKOFF S 2017. An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. Elife, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- THOMAS PD, CAMPBELL MJ, KEJARIWAL A, MI H, KARLAK B, DAVERMAN R, DIEMER K, MURUGANUJAN A & NARECHANIA A 2003. PANTHER: a library of protein families and subfamilies indexed by function. Genome Res, 13, 2129–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TRIBULO C, AYBAR MJ, NGUYEN VH, MULLINS MC & MAYOR R 2003. Regulation of Msx genes by a Bmp gradient is essential for neural crest specification. Development, 130, 6441–52. [DOI] [PubMed] [Google Scholar]
- TSANKOV AM, GU H, AKOPIAN V, ZILLER MJ, DONAGHEY J, AMIT I, GNIRKE A & MEISSNER A 2015. Transcription factor binding dynamics during human ES cell differentiation. Nature, 518, 344–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UCHIKAWA M, ISHIDA Y, TAKEMOTO T, KAMACHI Y & KONDOH H 2003. Functional analysis of chicken Sox2 enhancers highlights an array of diverse regulatory elements that are conserved in mammals. Dev Cell, 4, 509–19. [DOI] [PubMed] [Google Scholar]
- VADASZ S, MARQUEZ J, TULLOCH M, SHYLO NA & GARCIA-CASTRO MI 2013. Pax7 is regulated by cMyb during early neural crest development through a novel enhancer. Development, 140, 3691–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VALLIN J, THURET R, GIACOMELLO E, FARALDO MM, THIERY JP & BRODERS F 2001. Cloning and characterization of three Xenopus slug promoters reveal direct regulation by Lef/beta-catenin signaling. J Biol Chem, 276, 30350–8. [DOI] [PubMed] [Google Scholar]
- WANG CC, BAJIKAR SS, JAMAL L, ATKINS KA & JANES KA 2014. A time- and matrix-dependent TGFBR3-JUND-KRT5 regulatory circuit in single breast epithelial cells and basal-like premalignancies. Nat Cell Biol, 16, 345–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WILKINSON DG 1992. In situ hybridization : a practical approach, Oxford ; New York, IRL Press at Oxford University Press. [Google Scholar]
- WILLIAMS RM, CANDIDO-FERREIRA I, REPAPI E, GAVRIOUCHKINA D, SENANAYAKE U, LING ITC, TELENIUS J, TAYLOR S, HUGHES J & SAUKA-SPENGLER T 2019. Reconstruction of the Global Neural Crest Gene Regulatory Network In Vivo. Dev Cell, 51, 255–276 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YIN X, LI J, SALMON B, HUANG L, LIM WH, LIU B, HUNTER DJ, RANSOM RC, SINGH G, GILLETTE M, ZOU S & HELMS JA 2015. Wnt Signaling and Its Contribution to Craniofacial Tissue Homeostasis. J Dent Res, 94, 1487–94. [DOI] [PubMed] [Google Scholar]
- ZAMUDIO AV, DALL’AGNESE A, HENNINGER JE, MANTEIGA JC, AFEYAN LK, HANNETT NM, COFFEY EL, LI CH, OKSUZ O, SABARI BR, BOIJA A, KLEIN IA, HAWKEN SW, SPILLE JH, DECKER TM, CISSE II, ABRAHAM BJ, LEE TI, TAATJES DJ, SCHUIJERS J & YOUNG RA 2019. Mediator Condensates Localize Signaling Factors to Key Cell Identity Genes. Mol Cell, 76, 753–766 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1. List of neural crest GRN genes. Related to Star Methods.
Supplemental Table 2. Putative enhancer-promoter contacts of components of the neural crest GRN. Related to Star Methods.
Supplemental Table 3. Genomic positions of neural crest enhancers. Related to Star Methods.
Supplemental Table 4. Mutations performed in Axud1E1 variants. Related to Star Methods.
Supplemental Table 5. Differentially expressed genes under Wnt LOF conditions. Related to Star Methods.
Supplemental Table 6. Nucleotide sequences for oligonucleotides or primers used. Related to Star Methods.
Supplemental Table 7. Number of biological replicates and pValues for quantitative experiments. Related to Star Methods.
Data Availability Statement
The H3K27ac-HiChIP, the CUT&RUN datasets for CTNNB1, LEF1 and CTCF in wild type chick neural crest cells, the CUT&RUN for LEF1 and H3K27ac, ATAC-seq and RNA-seq datasets in WNT loss of function neural crest cells havge been deposited to the Gene Expression Omnibus GSE150006. The CUT&RUN dataset for H3K27ac in wild type chick neural crest cells has been deposited to the Expression Omnibus GSE142101. The TFAP2A CUT&RUN and the ATAC-seq datasets in wild type chick neural crest cells have been deposited to the Expression Omnibus GSE126880.